

A conserved herpesvirus tegument protein recruits host protein phosphatase PPM1G to inactivate STING and MAVS for immune evasion.
Abstract
The adaptor proteins, STING and MAVS, are components of critical pathogen-sensing pathways that induce innate immunity. Phosphorylation of either adaptor results in activation of the type I interferon pathway. How this phosphorylation is regulated and how it is manipulated by pathogens remain largely unknown. Here, we identified host protein phosphatase, Mg2+/Mn2+ dependent 1G (PPM1G) as a negative regulator of innate immune pathways and showed that this host system is hijacked by Kaposi’s sarcoma-associated herpesvirus (KSHV). Mechanistically, KSHV tegument protein ORF33 interacts with STING/MAVS and enhances recruitment of PPM1G to dephosphorylate p-STING/p-MAVS for immunosuppression. Inhibition of PPM1G expression improves the antiviral response against both DNA and RNA viruses. Collectively, our study shows that PPM1G restricts both cytosolic DNA– and RNA–sensing pathways to naturally balance the intensity of the antiviral response. Manipulation of PPM1G by KSHV provides an important strategy for immune evasion.
INTRODUCTION
As the first line of host defense, the innate immune response is rapidly activated upon detection of invading pathogens including viruses. Viral proteins, DNA, and RNA may be sensed as pathogen-associated molecular patterns (PAMPs) by host pattern recognition receptors to activate antiviral immunity. Extensive studies have shown that the cyclic guanosine monophosphate (GMP)–adenosine monophosphate (cGAMP) synthase (cGAS) and retinoic acid–inducible gene I (RIG-I) are the principal sensors for cytosolic DNA and RNA, respectively (1, 2). After sensing viral DNA, cGAS is activated and catalyzes the production of the second messenger cGAMP, which then binds to and activates STING (stimulator of interferon genes, also known as MITA) (2–4). Alternatively, after sensing viral RNA, RIG-I is activated and binds to and activates MAVS (mitochondrial antiviral signaling protein, also known as VISA, Cardif and IPS-1) (5–8). These two adaptor proteins, STING and MAVS, then recruit the protein kinase, TANK-binding kinase 1 (TBK1), which, in turn, phosphorylates either adaptor. Phosphorylated (p-)STING and p-MAVS subsequently recruit interferon regulatory factor 3 (IRF3), which, in turn, is phosphorylated by activated TBK1 (9). Last, phosphorylated IRF3 translocates to the nucleus and drives production of type I interferons (IFNs) to provide broad protection against DNA and RNA viruses (10). Although STING- and MAVS-mediated cytosolic nucleic acid–sensing pathways play crucial roles in antiviral immunity, excessive activation of the system is associated with sometimes fatal inflammatory diseases (11, 12). Thus, the activity of the system, and in particular the activities of the innate immune adaptors, must be precisely regulated to ensure a proper and balanced innate immune homeostasis in infected host cells. Since MAVS, STING, TBK1, and IRF3 all share the requirement for phosphorylation by TBK1 to be activated (9, 13), phosphorylation is essential in regulating the cytosolic nucleic acid–induced type I IFN production pathways. How this phosphorylation is regulated and how it is manipulated by pathogens remain largely unknown.
Kaposi’s sarcoma-associated herpesvirus (KSHV) is a human DNA tumor virus, and the causative agent of Kaposi’s sarcoma, primary effusion lymphoma, and a subset of multicentric Castleman’s disease (14–17). Like other herpesviruses, KSHV virion structure consists of four distinct components: a double-stranded DNA genome, an icosahedral capsid, an envelope on the surface of virus particle, and a tegument layer located between the capsid and the envelope (18–20). The KSHV virion has many tegument proteins that contribute to different stages of viral infection and replication (21). On one hand, as virion components, they are directly delivered into newly infected cells upon de novo infection and can modulate the cellular microenvironment, promote viral gene transcription at the immediate-early phase of viral infection, and regulate host innate immunity (21). On the other hand, as structural proteins newly synthesized during the late phase of viral lytic replication, they play essential roles in virion assembly and egress and are required for virus propagation (22–24).
KSHV ORF33 is a tegument protein that is conserved among α-, β-, and γ-herpesvirinae subfamilies. Its homologs include UL16 from herpes simplex virus type 1 (HSV-1, an α-herpesvirinae member), UL94 from human cytomegalovirus (HCMV, a β-herpesvirinae member), BGLF2 from Epstein-Barr virus (EBV), and ORF33 from KSHV and murine gammaherpesvirus 68 (MHV-68, all are γ-herpesvirinae members). Previous studies have shown that these homologs of ORF33 play pivotal roles in virion assembly process (25–28). In particular, MHV-68 ORF33 is essential for virion assembly and egress (29, 30). However, whether ORF33 carries out additional functions, such as modulating innate immunity, and how it does so, remains unclear. In this study, we demonstrated that KSHV ORF33 negatively regulates the IFNβ production pathway for immune evasion. Mechanistically, we identified a host protein phosphatase, Mg2+/Mn2+ dependent 1G (PPM1G), that dephosphorylates both STING and MAVS and suppresses both cytosolic DNA– and RNA–sensing pathways. Manipulation of this system by ORF33 is an important strategy for immune evasion, which facilitates KSHV propagation.
RESULTS
ORF33-null KSHV stimulates an increased IFNβ production
The KSHV life cycle is composed of two different stages: lytic replication and latency. During lytic infection by KSHV, tegument proteins contained within virus particles are released into the cytoplasm of host cells upon fusion of viral envelope with the host cell plasma membrane. Depending on the setting and cell type, lytic replication may proceed during which tegument proteins are made as structural components of progeny virions. Alternatively, cells may remain latently infected with KSHV, and once appropriately stimulated, they reactivate virus and undergo lytic replication where newly synthesized tegument proteins contribute to virion structure and may also regulate innate immune response. To examine whether endogenous ORF33 protein plays a role in innate immune response, we took advantage of the KSHV bacterial artificial chromosome (BAC) system (31) and constructed an ORF33-null KSHV BAC (ORF33-stop) by inserting triple-frame termination codons into the N terminus of ORF33 on viral genome (fig. S1, A and B). ORF33-null BAC was then transfected into iSLK cells to generate a stable cell line that was induced to produce ORF33-null mutant virus. We then infected THP-1 cells with either wild-type (WT) or ORF33-null mutant virus at the same genome copy number and detected the mRNA level of IFNβ at 6 h post infection (hpi). Reverse transcription quantitative polymerase chain reaction (RT-qPCR) results indicated that ORF33-null virus induced more IFNβ production than WT virus did (Fig. 1A, left). In addition, we checked IFNβ production after reactivation of WT or ORF33-null virus. RT-qPCR and enzyme-linked immunosorbent assay (ELISA) results showed that the IFNβ production in cells latently infected with ORF33-null virus was significantly higher than that in cells latently infected with WT virus at 48 hours after reactivation (Fig. 1A, right, and fig. S1C). Together, these data reveal that KSHV stimulates higher levels of IFNβ in the absence of ORF33, suggesting that ORF33 is necessary for inhibiting host cell IFN production in response to KSHV infection.
Fig. 1. KSHV ORF33 inhibits STING- and MAVS-mediated IFNβ production.
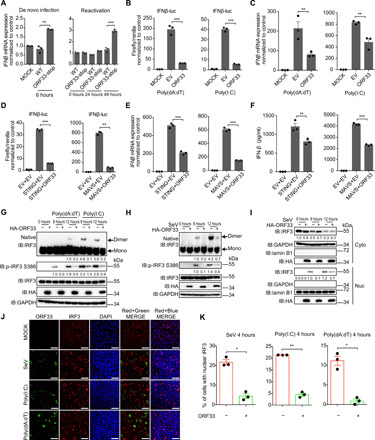
(A) IFNβ mRNA levels during de novo infection and reactivation of ORF33-null KSHV. THP-1 cells were infected with WT or ORF33-null KSHV at 50 genome copies per cell for 6 hours (left). iSLK cells carrying WT KSHV BAC or ORF33-null KSHV BAC were induced for 24 or 48 hours (right). (B to F) Influence of ORF33 on IFNβ production. Human embryonic kidney (HEK) 293 cells were transfected with the indicated expression plasmids (B to F), along with IFNβ-luc and TK-Renilla reporter plasmids (B and D), for 24 hours. Cells were challenged with transfection of poly(dA:dT) (1 μg/ml) (left) or poly(I:C) (1 μg/ml) (right) for 18 hours (B) or for 12 hours (C). Luciferase assays (B and D), RT-qPCR (C and E), and ELISA (F) were conducted. (G to K) Impact of ORF33 on dimerization, phosphorylation, and translocation of IRF3. HEK293 cells were transfected with the indicated expression plasmids for 24 hours. Cells were challenged with poly(dA:dT) or poly(I:C) (1 μg/ml each) or SeV infection (50 HA U/ml) for 12 hours (G to I) or 4 hours (J and K). (G and H) p-IRF3 levels were measured by SDS–polyacrylamide gel electrophoresis (PAGE), and IRF3 dimerization levels were measured by native-PAGE and quantified using ImageJ and normalized to IRF3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (I) Nuclear (Nuc) and cytoplasmic (Cyto) fractions were separated, and IRF3 levels were quantified using ImageJ and normalized to lamin B1 or GAPDH. (J) Cells were fixed and immunostained with anti-HA and anti-IRF3 antibodies. Scale bars, 40 μm. DAPI, 4′,6-diamidino-2-phenylindole. (K) The percentage of cells with nuclear IRF3 in ORF33-expressing or -nonexpressing population under the three different stimulations was calculated. (A to F and K) Data presented are means ± SEM of three independent measurements, representative of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001; Student’s t test, n = 3. See also figs. S1 to S3.
KSHV ORF33 inhibits STING- and MAVS-mediated IFNβ production
Next, we examined whether ORF33 protein alone is sufficient to inhibit IFNβ production. We first used poly(deoxyadenylic-deoxythymidylic) [poly(dA:dT)] or polyinosinic-polycytidylic acid [poly(I:C)] to stimulate cytosolic DNA– or RNA–triggered IFNβ production pathway, respectively. ORF33 significantly inhibited the activation of the IFNβ promoter in reporter assays (Fig. 1B). Consistently, RT-qPCR results showed that ORF33 significantly inhibited the production of IFNβ mRNA under poly(dA:dT) or poly(I:C) stimulation (Fig. 1C). Furthermore, ORF33 homologs in other herpesviruses (HSV-1, HCMV, and EBV) all inhibited IFNβ production in response to either poly(dA:dT) or poly(I:C) stimulation (fig. S2, A and B), indicating that the IFNβ inhibition function of ORF33 is conserved among herpesviruses.
STING and MAVS are the critical adaptor proteins that mediate cytosolic DNA– and RNA–triggered IFNβ production pathway, respectively. We thus examined the effect of ORF33 on STING- and MAVS-mediated IFN production. Results from the reporter assays, RT-qPCR, and ELISA showed that ORF33 significantly inhibited IFNβ production induced by either STING or MAVS (Fig. 1, D to F). In addition, ORF33 also inhibited the transcription of ISG56, an IFN-stimulated gene (ISG) downstream of IFNβ, when induced by STING or MAVS (fig. S3). Together, these results demonstrate that KSHV ORF33 protein by itself is sufficient to inhibit both STING- and MAVS-mediated IFNβ production.
The status of transcription factor IRF3, including its phosphorylation, dimerization, and translocation into the nucleus, is vital to cytosolic DNA– or RNA–triggered IFNβ production. Compared with empty vector, transient expression of ORF33 markedly decreased the level of IRF3 phosphorylation and dimerization when induced by poly(dA:dT) or poly(I:C) stimulation (Fig. 1G) or Sendai virus (SeV) infection (Fig. 1H). Moreover, the result of nuclear and cytoplasmic fractionation experiment showed that transient expression of ORF33 impaired the translocation of IRF3 to the nucleus under the stimulation of SeV infection (Fig. 1I). Consistently, indirect immunofluorescence assay (IFA) showed that the percentage of cells with nuclear IRF3 in the ORF33-expressing population was significantly lower than that in the ORF33-nonexpressing population, under the stimulation of SeV infection, poly(dA:dT) transfection, or poly(I:C) transfection (Fig. 1, J and K). These results demonstrate that ORF33 inhibits the phosphorylation of IRF3 and impairs IRF3 dimerization, resulting in sequestering IRF3 in the cytoplasm and down-regulation of IFNβ production. These results also suggest that ORF33 most likely works upstream of IRF3 to suppress IFNβ production and possibly through targeting the critical adaptor molecules STING and MAVS.
KSHV ORF33 interacts with STING and MAVS
We therefore investigated the possible interactions between ORF33 and STING/MAVS. Coimmunoprecipitation experiments indicated that ORF33 indeed interacts with STING and MAVS (Fig. 2A). Moreover, we expressed and purified glutathione S-transferase (GST)–ORF33-His protein from bacteria and performed in vitro GST pull-down assays. (A C-terminal His tag was added to improve the stability and yield of ORF33 protein.) GST-ORF33-His pulled down hemagglutinin (HA)–STING or HA-MAVS efficiently, demonstrating their direct interactions (Fig. 2B). Moreover, ORF33 expressed from viral genome interacted with endogenous STING and MAVS (Fig. 2C). IFA results showed that KSHV ORF33 and its homologs are localized in not only the nucleus but also the cytoplasm (fig. S4A), consistent with ORF33’s interaction with STING/MAVS. We further performed proximity ligation assay (PLA). In this assay, positive signals (red immunofluorescent dots) appear only when ORF33 (or its homologs) interacts with and therefore is in close proximity to STING/MAVS. The results clearly demonstrated colocalization of ORF33/homologs with STING/MAVS in situ (fig. S4B).
Fig. 2. KSHV ORF33 impairs the recruitment of IRF3 by STING and MAVS.
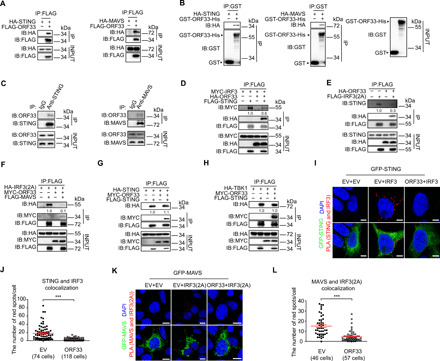
(A to C) Interaction between ORF33 and STING/MAVS. (A) HEK293T cells were transfected with indicated expression plasmids for 36 hours before coimmunoprecipitation. (B) GST-ORF33-His proteins were used to pull down lysates of HEK293T cells expressing HA-STING/HA-MAVS. (C) iSLK.219 cells were induced for 48 hours, and antibodies to STING and MAVS were used to enrich endogenous STING and MAVS, respectively. KSHV ORF33 protein was detected by an anti-ORF33 antibody. IgG, immunoglobulin G. (D to H) Impact of ORF33 on the key molecular events of the IFNβ production pathway. HEK293 cells were transfected with the indicated expression plasmids for 20 hours before coimmunoprecipitation. The gray values are quantified using ImageJ. (I to L) Analysis of the impact of ORF33 on the interaction between STING/MAVS and IRF3 in situ by PLA. HEK293T cells were transfected with the indicated expression plasmids: (I) MYC-ORF33, HA-IRF3, and green fluorescent protein (GFP)–STING or EV control(s); (K) MYC-ORF33, HA-IRF3(2A), and GFP-MAVS or EV control(s). Scale bars, 2.5 μm (I) and 10 μm (K). (J and L) The number of red fluorescent dots in each cell in (I) and (K) was enumerated, respectively. (J and L) Data presented are means ± SEM of three independent measurements, representative of three independent experiments. ***P < 0.001, Student’s t test. See also fig. S4.
To map the domains of STING and MAVS that interact with ORF33, we made deletion constructs of STING and MAVS based on their structural information (6, 32). STING protein has three structural domains, namely, TM (transmembrane region, which enables STING to locate on endoplasmic reticulum), CBD (which binds to c-di-GMP, the second messenger of bacteria, and regulates the dimerization of STING), and CTT (the C-terminal tail, which is responsible for the activation of TBK1 and IRF3). MAVS protein has three domains, namely, CARD (which is responsible for MAVS polymerization), PR (proline enriched area), and TM (transmembrane domain, which enables MAVS to locate on mitochondrial membrane). We found that the CBD domain of STING (fig. S4, C and D) and the TM domain of MAVS (fig. S4, E to F) are required for ORF33 binding, respectively. Collectively, these results demonstrate that KSHV ORF33 interacts with both STING and MAVS, suggesting that ORF33 targets STING and MAVS to negatively regulate IFNβ production pathway.
KSHV ORF33 impairs the recruitment of IRF3 by STING and MAVS
In the process of cytosolic DNA–induced IFNβ production signal transduction, upon stimulation by cGAMP, STING forms a homodimer and then recruits TBK1 and IRF3, leading to phosphorylation and activation of IRF3. Similarly, in response to upstream RIG-I induction upon cytosolic RNA stimulation, MAVS also recruits TBK1 and IRF3, leading to phosphorylation and activation of IRF3. Given the interaction between ORF33 and STING/MAVS, we assessed the effect of such interactions on these key molecular events.
We first tested the effect of ORF33 on recruitment of IRF3 by STING or MAVS. A mutant form of IRF3 [Ser385 → Ala385, Ser386 → Ala386, named IRF3(2A)] was used; this mutant was unable to form homodimers, thereby facilitating detection of its association with STING or MAVS (9, 33). Coimmunoprecipitation experiments indicated that ORF33 inhibited the association between IRF3 and transiently expressed STING (Fig. 2D) or IRF3(2A) and endogenous STING molecules (Fig. 2E). Similarly, ORF33 impaired the association between IRF3(2A) and transiently expressed MAVS (Fig. 2F). However, ORF33 had no obvious effect on the dimerization of STING molecules (Fig. 2G) or the association between STING and TBK1 (Fig. 2H). To further observe these molecular interactions in situ, we used PLA, in which positive signals (red immunofluorescent dots) appear only when STING (or MAVS) interacts with and therefore is in close proximity to IRF3 (9). Enumerating the number of red dots in each cell population demonstrated that ectopic expression of ORF33 severely impaired the association between STING and IRF3 (Fig. 2, I and J) as well as the association between MAVS and IRF3 (Fig. 2, K and L). Collectively, these data reveal that ORF33 impairs the recruitment of downstream IRF3 by STING or MAVS, consistent with the negative effect of ORF33 on the IFNβ production pathway shown above.
KSHV ORF33 decreases the phosphorylation levels of STING and MAVS but not TBK1
One possible mechanism through which ORF33 impairs the recruitment of downstream IRF3 by STING/MAVS is interacting with STING/MAVS molecules and thereby hindering their functions. In both signaling cascades, TBK1 has to be recruited and activated through phosphorylation before it can serve as a kinase to phosphorylate STING and MAVS. Recruitment of IRF3 by STING and MAVS also strictly depends on STING and MAVS phosphorylation. Therefore, the phosphorylation levels of TBK1, STING, and MAVS are critical parameters in determining the outcomes of the IFN signaling pathways (9).
We therefore first examined the effect of ORF33 on the phosphorylation levels of TBK1, STING, and MAVS. Under the stimulation of poly(dA:dT) or ISD (IFN stimulatory DNA), ectopic expression of ORF33 markedly decreased the phosphorylation levels of STING and IRF3; however, ORF33 had no effect on the phosphorylation level of TBK1 (Fig. 3, A and B). The human embryonic kidney (HEK) 293 cells used in this study were examined, and expression of endogenous STING and cGAS was confirmed (Fig. 3A and fig. S5A). Moreover, the expression of IFNβ and ISG56 upon stimulation by poly(dA:dT) was decreased after knocking down cGAS by small interfering RNA (siRNA) (fig. S5, B and C). Similarly, under the stimulation of poly(I:C), ectopic expression of ORF33 markedly decreased the phosphorylation level of IRF3; again, ORF33 had no effect on the phosphorylation level of TBK1 (Fig. 3C). Because of the lack of an anti–p-MAVS antibody, we were unable to detect the change in phosphorylation level of endogenous MAVS. We instead examined the effect of ORF33 on the phosphorylation of MAVS in a cotransfection experiment. Expression plasmids for FLAG-MAVS, HA-TBK1 (or vector control), and HA-ORF33 (or vector control) were cotransfected into HEK293 cells. In the presence of HA-TBK1, the anti-FLAG antibody detected not only a band migrating at the same mobility to that seen in the absence of HA-TBK1 but also slower-migrating bands and smear. These slower-migrating bands and smear disappeared after treatment by lambda protein phosphatase (λpp), indicating their phosphorylated status. Coexpressing ORF33 with FLAG-MAVS and HA-TBK1 decreased the phosphorylation level of MAVS by TBK1 in a dose-dependent manner (Fig. 3D, first five lanes). Cotransfection experiment using expression plasmids for FLAG-STING and HA-TBK1 (or vector control) and HA-ORF33 (or vector control) confirmed that ORF33 also decreased the phosphorylation level of STING by TBK1 in a dose-dependent manner (Fig. 3D, last five lanes). These results indicate that ORF33 does not affect the activation of TBK1 but suppresses the phosphorylation of STING and MAVS, which, in turn, impairs the recruitment of IRF3 and its phosphorylation.
Fig. 3. KSHV ORF33 decreases the phosphorylation levels of STING and MAVS but not TBK1.
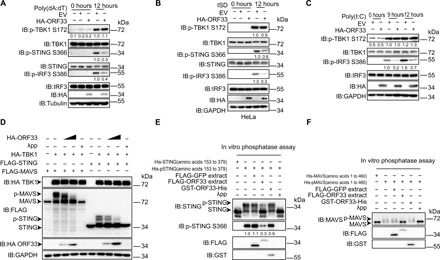
(A to C) Influence of ORF33 on the phosphorylation levels of endogenous TBK1, STING, and IRF3. (A and C) As in Fig. 1G. (B) HeLa cells were transfected with the indicated expression plasmids for 24 hours and then challenged with ISD (2 μg/ml) for 12 hours. The gray values are quantified using ImageJ, and the level of each phosphorylated protein indicated was normalized to the level of corresponding total protein and tubulin/GAPDH. (D to F) Influence of ORF33 on the phosphorylation levels of STING and MAVS. (D) HEK293 cells were cotransfected with TBK1, STING/MAVS expression plasmids, and increasing doses of ORF33 expression plasmid for 24 hours. Cell lysates were either treated with λpp or left untreated as indicated. (E and F) Purified His-STING (amino acids 153 to 379) (1 μg) (E) or His-MAVS (amino acids 1 to 460) (1 μg) (F) was first incubated with FLAG-TBK1 (1 μg) to generate p-STING or p-MAVS (in vitro kinase assay). The influence of FLAG-GFP extract (as a control), FLAG-ORF33 extract, or GST-ORF33-His on p-STING (E) or p-MAVS (F) was examined by in vitro phosphatase assay. See also fig. S5.
Given that ORF33 decreased the levels of both p-STING and p-MAVS but not p-TBK1, we reasoned that ORF33 may affect the levels of p-STING and p-MAVS by dephosphorylating them. One possibility is that ORF33 may serve as a phosphatase to directly dephosphorylate STING and MAVS; however, sequence analysis of ORF33 protein suggested against this hypothesis. To verify this, we first generated p-His-STING (amino acids 153 to 379) using in vitro kinase assay and then performed in vitro phosphatase assay using GST-ORF33-His purified from bacteria. Whereas λpp efficiently dephosphorylated p-His-STING (amino acids 153 to 379), GST-ORF33-His had no effect on the phosphorylation status of p-His-STING (amino acids 153 to 379) (Fig. 3E and fig. S5, D to F). An experiment performed using p-His-MAVS (amino acids 1 to 460) from in vitro kinase assay yielded similar results (Fig. 3F and fig. S5, D to F), demonstrating that ORF33 has no detectable phosphatase activity against p-STING or p-MAVS.
Alternatively, ORF33 may recruit and use a host protein phosphatase to dephosphorylate STING and MAVS. To test this hypothesis, we performed in vitro phosphatase assays using FLAG-ORF33 extracted from HEK293 cells together with p-His-STING (amino acids 153 to 379) or p-His-MAVS (amino acids 1 to 460) (both generated from in vitro kinase assay). Again, λpp efficiently dephosphorylated p-His-STING (amino acids 153 to 379) and p-His-MAVS (amino acids 1 to 460). FLAG-ORF33 extract notably decreased the phosphorylation levels of both STING and MAVS (Fig. 3, E and F). Collectively, these data indicate that ORF33 inhibits the phosphorylation of STING and MAVS not by affecting the activity of the kinase TBK1, but by dephosphorylation of STING and MAVS. ORF33 most likely does so by recruiting and using a host protein phosphatase.
KSHV ORF33 enhances the recruitment of host protein phosphatase PPM1G that dephosphorylates STING and MAVS
To investigate which host protein phosphatase(s) may be recruited by ORF33, we performed coimmunoprecipitation and mass spectrometry analysis to identify host protein phosphatases that interact with ORF33. Several phosphatases were detected from the ORF33 sample but not from the vector control sample, including PPP6C, PPP2CA, PPP1CA, PPP1CB, PPM1G, and PPP3CA (fig. S6A). We examined the effect of these phosphatases on the phosphorylation status of STING and MAVS in a cotransfection experiment. Among these phosphatases, only PPM1G decreased the phosphorylation level of STING and MAVS (fig. S6, B and C). PPM1G, also known as PP2Cγ and PP2CG, is a member of the metal-dependent protein phosphatase family. We expressed and purified PPM1G and a mutant PPM1G deficient in phosphatase activity (D496A) (34) from bacteria (Fig. 4A). In the in vitro phosphatase assays, PPM1G directly dephosphorylated p-STING and p-MAVS, whereas PPM1G D496A failed to do so (Fig. 4, B and C). Coimmunoprecipitation assay confirmed that ORF33 expressed from transiently transfected plasmid and viral genome both interacted with endogenous PPM1G (Fig. 4, D and E), consistent with our hypothesis that ORF33 may recruit and use a host protein phosphatase to dephosphorylate p-STING and p-MAVS.
Fig. 4. KSHV ORF33 recruits PPM1G to dephosphorylate STING and MAVS.
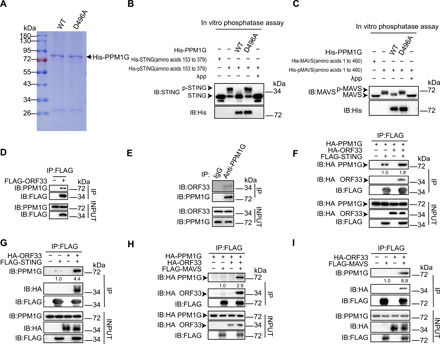
(A) Coomassie Blue–stained gel of purified His-PPM1G and His-PPM1G D496A from bacteria. (B and C) The effect of His-PPM1G and His-PPM1G D496A on the phosphorylation levels of STING and MAVS was examined by in vitro phosphatase assay. (D and E) Interaction between endogenous PPM1G and exogenously expressed ORF33 (D) or ORF33 expressed from viral genome (E) was examined by coimmunoprecipitation (co-IP). (F to I) Enhancement of interaction between PPM1G and STING (F and G) or MAVS (H and I) by ORF33 was examined by co-IP. See also fig. S6.
To examine whether ORF33 could enhance the interaction between STING with PPM1G, we first performed a cotransfection experiment. Results showed that exogenously expressed PPM1G interacted with STING, and this interaction was clearly enhanced in the presence of ORF33 (Fig. 4F). ORF33 also clearly enhanced the interaction between STING and endogenous PPM1G (Fig. 4G). Similarly, ORF33 also markedly enhanced the interaction between MAVS and exogenously expressed PPM1G or endogenous PPM1G (Fig. 4, H and I). Together, these results strongly indicate that ORF33 recruits the host protein phosphatase PPM1G and enhances the interaction between PPM1G and STING/MAVS, resulting in dephosphorylation of p-STING and p-MAVS.
PPM1G negatively regulates antiviral innate immunity
Phosphorylation of STING and MAVS by TBK1 is essential for activation of the antiviral innate immune response induced by cytosolic nucleic acid. Because host protein phosphatase PPM1G can directly dephosphorylate STING and MAVS in vitro (Fig. 4, B and C), we studied the general functional role of PPM1G in DNA- and RNA-induced antiviral innate immune responses. In reporter assays and RT-qPCR assays, using either poly(dA:dT) or poly(I:C) to activate the IFNβ signaling pathway, ectopic expression of PPM1G significantly inhibited activation of the IFNβ promoter as well as the expression of IFNβ mRNA (Fig. 5, A and B). Moreover, PPM1G markedly decreased the level of IRF3 phosphorylation and dimerization under the stimulation of poly(dA:dT) or poly(I:C) (Fig. 5C). Consistently, PPM1G significantly inhibited the expression of downstream ISGs such as ISG56 (fig. S7A).
Fig. 5. PPM1G negatively regulates the antiviral innate immune response.
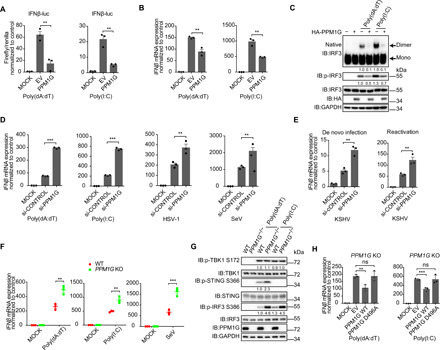
(A and B) Impact of PPM1G on IFNβ production, as described in the legend to Fig. 1 (B and C). (C) Impact of PPM1G on dimerization and phosphorylation levels of IRF3, as described in the legend to Fig. 1G. (D and E) Influence of PPM1G knockdown on IFNβ mRNA levels. (D) HEK293 cells were transfected with indicated siRNA. After 48 hours, cells were challenged as in Fig. 1J, or with infection of HSV-1 [multiplicity of infection (MOI) = 3] for 12 hours. (E) Cells were transfected with the siRNA for 48 hours. iSLK cells were infected with KSHV for 24 hours (left), or iSLK.219 cells were induced for 48 hours (right), and IFNβ mRNA levels were measured. (F) Effect of PPM1G knockout on IFNβ mRNA levels. WT or PPM1G KO HEK293 cells were challenged for 12 hours as described in the legend to Fig. 1J. (G) Effect of PPM1G knockout on phosphorylation levels of IRF3. WT or PPM1G KO HEK293 cells were challenged for 12 hours as described in the legend to Fig. 3 (A and C). (H) The phenotype exhibited by PPM1G knockout cells was reversed by introducing WT but not the catalytically inactive (D496A) PPM1G mutant. PPM1G KO HEK293 cells were transfected with the indicated expression plasmids for 24 hours. Cells were challenged as described in the legend to Fig. 1C. (A, B, D to F, and H) Data presented are means ± SEM of three independent measurements, representative of three independent experiments. **P < 0.01 and ***P < 0.001; ns, not significant; Student’s t test, n = 3. See also fig. S7.
We next addressed whether knockdown or knockout of PPM1G would enhance antiviral innate immunity. We first used siRNA to knock down PPM1G expression (fig. S7B) and examined the innate immune response of host cell. The results indicated that knockdown of PPM1G significantly increased the IFNβ production under the stimulation of poly(dA:dT) or poly(I:C) or after infection by a DNA virus HSV-1 or an RNA virus SeV (Fig. 5D). In addition, after PPM1G knockdown, the production of both IFNβ and ISG56 was increased after de novo infection by KSHV or reactivation of latently infected KSHV (Fig. 5E and fig. S7C). We further constructed PPM1G knockout cells and found that the IFNβ and ISG56 mRNA levels were significantly higher in PPM1G−/− cells in response to poly(dA:dT) or poly(I:C) transfection, or SeV infection, when compared to WT cells (Fig. 5F and fig. S7D). Consistently, the phosphorylation level of IRF3 was markedly increased in PPM1G−/− cells, compared to that in WT cells (Fig. 5G). Moreover, whereas introducing WT PPM1G into PPM1G−/− cells significantly reduced the expression of IFNβ and its downstream gene ISG56 under the stimulation of poly(dA:dT) or poly(I:C), introduction of the mutant PPM1G D496A had no effect (Fig. 5H and fig. S7E). Together, these results indicate that PPM1G is a negative regulator of cytosolic DNA– and RNA–induced IFNβ signaling, for which the phosphatase activity of PPM1G is essential.
Since IFNβ signaling is critical for host defense against DNA and RNA viruses, we further evaluated the functional consequence of PPM1G knockout on viral infection and replication. WT or PPM1G−/− cells were infected with the DNA virus HSV-1 or the RNA virus SeV and then viral gene expression and genome copy number or viral titer were measured. Compared to WT cells, the viral gene expression levels and genome copy number or viral titer of SeV and HSV-1 were significantly lower in PPM1G−/− cells (Fig. 6, A to D), indicating that PPM1G knockout inhibited viral replication as a result of enhanced host defense against both DNA and RNA viruses. Moreover, knocking down PPM1G in iSLK.219 cells that are latently infected with KSHV severely inhibited KSHV reactivation (Fig. 6, E and F) and expression of viral lytic genes (Fig. 6G), resulting in decreased virus production, as measured by viral genome copy number (Fig. 6H) and viral titer in the supernatant (Fig. 6I, determined in a new round of infection). Collectively, these results indicate that PPM1G negatively regulates antiviral innate immunity by suppressing both STING- and MAVS-mediated IRF3 activation and IFNβ pathway.
Fig. 6. PPM1G knockout or knockdown enhances host defense against both DNA and RNA viruses.
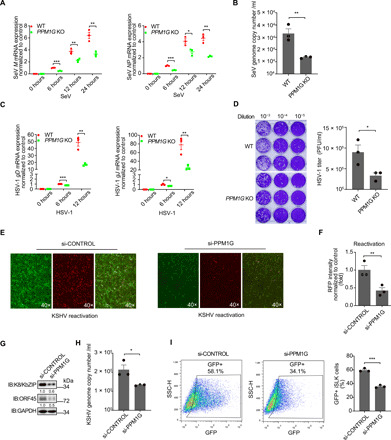
(A and B) Effect of PPM1G knockout on SeV replication. WT or PPM1G KO HEK293 cells were infected with SeV (50 HA U/ml) for the indicated time periods. (A) SeV M and NP mRNA levels were measured by RT-qPCR. (B) SeV genome copy number in culture supernatant at 24 hours was measured by RT-qPCR. (C and D) Effect of PPM1G knockout on HSV-1 replication. WT or PPM1G KO HEK293 cells were infected with HSV-1 (MOI = 3) for the indicated time periods. (C) HSV-1 gD and gJ mRNA levels were measured by RT-qPCR. (D) HSV-1 titers in supernatant at 24 hours were measured. (E to I) Influence of PPM1G knockdown on reactivation levels of KSHV. iSLK.219 cells were transfected with the indicated siRNA for 48 hours and then induced for 48 hours. GFP served as a marker for KSHV latent infection, and red fluorescent protein (RFP) served as a marker for KSHV lytic replication. (F) RFP intensity from (E) was quantified using ImageJ and normalized to control. (G) The expression of K8/KbZIP and ORF45 in iSLK.219 cells was examined with the indicated antibodies. (H) KSHV genome copy number in supernatant was measured by qPCR. (I) Supernatants from (E) were used to infect iSLK cells for 24 hours, and GFP expressed from viral genome was measured by fluorescence-activated cell sorting. (A to D and F to H) Data presented are means ± SEM of three independent measurements, representative of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001; Student’s t test, n = 3.
Inhibition of innate immune response by KSHV ORF33 is dependent on PPM1G
We further investigated whether the inhibitory effect of KSHV ORF33 on STING- and MAVS-mediated IRF3 activation and IFNβ signaling pathway is mediated through PPM1G. In reporter assays, ORF33 and PPM1G synergistically reduced IFNβ-luc activity (fig. S8A). Compared with Flag-ORF33 immunoprecipitated from WT cells, FLAG-ORF33 immunoprecipitated from PPM1G−/− cells failed to dephosphorylate STING and MAVS (fig. S8, B to D), indicating that PPM1G is the primary phosphatase recruited by ORF33 to inactivate STING and MAVS. In WT cells, ORF33 inhibited the production of IFNβ mRNA by more than 50% under either poly(dA:dT) or poly(I:C) stimulation or by SeV and HSV-1 infection, reductions that were both statistically significant. In contrast, in PPM1G−/− cells, IFNβ production decreased only slightly in the presence of ORF33, and such decrease was statistically not significant (Fig. 7A). Consistently, although ORF33 inhibited the phosphorylation of IRF3 in WT cells in response to poly(dA:dT) or poly(I:C) stimulation, ORF33 had no obvious effect on the phosphorylation level of IRF3 in PPM1G−/− cells (Fig. 7B). These results strongly indicate that the immunosuppressive function of ORF33 is mediated by the host protein phosphatase PPM1G.
Fig. 7. The inhibitory effect of ORF33 on innate immune response is mediated by PPM1G.
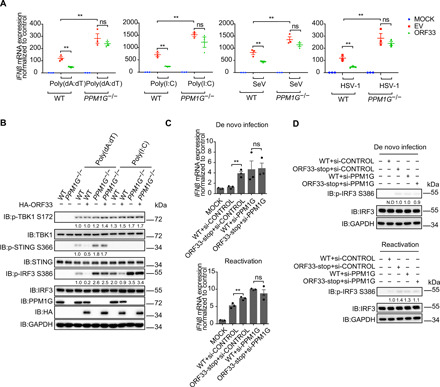
(A) Impact of ORF33 on IFNβ mRNA levels in WT or PPM1G knockout cells. WT or PPM1G KO HEK293 cells were transfected with indicated expression plasmids. After 24 hours, cells were challenged as described in the legend to Fig. 5, and IFNβ mRNA levels were measured. (B) Influence of ORF33 on phosphorylation levels of IRF3 in WT or PPM1G knockout cells. WT or PPM1G KO HEK293 cells were transfected with indicated expression plasmids. After 24 hours, cells were challenged as described in the legend to Fig. 1G. (C and D) IFNβ response in control or PPM1G knockdown cells during de novo infection and reactivation of WT or ORF33-null virus. Cells were transfected with the indicated siRNA for 48 hours. iSLK cells were infected with WT or ORF33-null virus for 24 hours (top). iSLK cells carrying latent KSHV (WT RGB BAC16 or ORF33-null RGB BAC16) were induced for 48 hours (bottom). IFNβ mRNA levels were measured by RT-qPCR (C), and phosphorylated IRF3 levels were measured by immunoblotting; N.D, not detected (D). (A and C) Data presented are means ± SEM of three independent measurements, representative of three independent experiments. **P < 0.01; Student’s t test, n = 3.
Last, we verified the function of PPM1G in immune evasion of KSHV mediated by ORF33. iSLK cells were transfected with siRNA against PPM1G or control siRNA, followed by infection with WT or ORF33-null virus, respectively. In cells transfected with control siRNA, the production of IFNβ stimulated by ORF33-null virus was significantly higher than that by WT virus. However, in PPM1G knockdown cells, no difference was observed in IFNβ production and IRF3 phosphorylation after WT or ORF33-null virus infection (Fig. 7C, top, and Fig. 7D, top). Similar results were obtained after reactivation of WT or ORF33-null virus in PPM1G knockdown or control siRNA knockdown cells (Fig. 7C, bottom, and Fig. 7D, bottom). These results demonstrate that the immune evasion function mediated by ORF33 during KSHV infection and reactivation is dependent on PPM1G.
DISCUSSION
IFN is the host cells’ first line of defense against viral infections. STING and MAVS are key adaptors in cytosolic nucleic acid–sensing pathways. Their phosphorylation by TBK1 is an essential and conserved mechanism that leads to recruitment of IRF3 and subsequent activation of the type I IFN pathway. However, cells must regulate their IFN production carefully to prevent inflammation and autoimmunity. Therefore, the activities of STING and MAVS must be tightly regulated to ensure a proper innate immune homeostasis in infected host cells. Host protein phosphatase PPM1G, as identified in this study, dephosphorylates p-STING and p-MAVS and tones down the response of the cytosolic DNA– and RNA–sensing pathways, thereby contributing to maintenance of a balanced antiviral innate immune response (Fig. 8, left). However, PPM1G is hijacked by KSHV for immune evasion. During KSHV de novo infection, tegument protein ORF33 is released from KSHV virion into the cytoplasm. ORF33 directly interacts with STING/MAVS and recruits PPM1G, resulting in enhanced dephosphorylation of p-STING/p-MAVS, impaired recruitment of IRF3, and suppression of the type I IFN pathway. When KSHV is reactivated from latency, newly synthesized ORF33 is likely to facilitate viral lytic replication by recruiting PPM1G to dephosphorylate p-STING/MAVS, thereby suppressing host antiviral activities (Fig. 8, right).
Fig. 8. Model: PPM1G restricts innate immune signaling mediated by STING and MAVS and is hijacked by KSHV ORF33 for immune evasion.
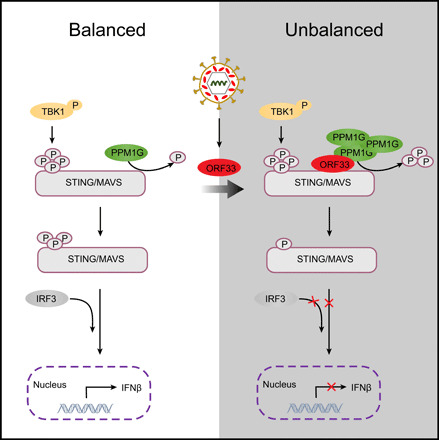
STING and MAVS are key adaptors in cytosolic nucleic acid–sensing pathways during virus infection. Their phosphorylation by TBK1 is an essential and conserved mechanism that leads to recruitment of IRF3 and subsequent activation of the type I IFN pathway. However, cells must regulate their IFN production carefully to prevent inflammation and autoimmunity. Therefore, the activities of STING and MAVS must be tightly regulated to ensure a proper innate immune homeostasis in infected host cells. Host protein phosphatase PPM1G, as identified in this study, dephosphorylates p-STING and p-MAVS and tones down the response of the cytosolic DNA– and RNA–sensing pathways, thereby contributing to maintenance of a balanced antiviral innate immune response (left). However, this system is hijacked by KSHV for immune evasion. During KSHV de novo infection, tegument protein ORF33 is released from KSHV virion into the cytoplasm. ORF33 directly interacts with STING/MAVS and recruits PPM1G, resulting in enhanced dephosphorylation of p-STING/p-MAVS, impaired recruitment of IRF3 and suppression of type I IFN pathway. When KSHV is reactivated from latency, newly synthesized ORF33 is also likely to facilitate viral lytic replication by recruiting PPM1G to dephosphorylate p-STING/MAVS, thereby suppressing host antiviral activities (right).
During both de novo infection and reactivation of a herpesvirus, viral nucleic acid acts as PAMPs to activate both cGAS-STING (35, 36) and RIG-I-MAVS (37, 38) pathways and up-regulates the expression of IFNs. Subsequent activation of ISG leads to a robust host defense against the invading virus (39). To successfully infect host cell and establish long-term latency, KSHV has evolved to use multiple strategies to antagonize host innate immunity. For instance, cytoplasmic isoforms of viral latency-associated nuclear antigen and a tegument protein, ORF52, have been reported to both target cGAS and inhibit cGAS-mediated restriction of viral lytic replication (40, 41). Viral IRF1 targets STING and prevents it from interacting with TBK1, thereby inhibiting STING’s phosphorylation and the DNA-sensing pathway (42). Another tegument protein, ORF64, exbibits deubiquitinase activity and reduces the ubiquitination of RIG-I to inhibit RIG-I–mediated antiviral immune responses (38). Consistent with these reports, we found that there was even a slight (although not statistically significant) decrease in IFNβ production at the early time point of de novo infection or reactivation of WT virus, when compared to mock-infected cells (Fig. 1A). However, in the absence of ORF33, the IFNβ production induced by KSHV infection or reactivation increased significantly, when compared to WT virus–infected cells (Fig. 1A). These data indicated that ORF33 is necessary for inhibiting host cell IFN production in response to KSHV infection and plays a key role in immune suppression in the context of viral infection. Moreover, we showed that ORF33 interacts with both STING and MAVS and antagonizes both DNA- and RNA-sensing pathways (Figs. 1 to 3). Thus, we uncovered a previously unknown strategy for KSHV immune suppression via yet another tegument protein, ORF33.
ORF33 is a tegument protein that is conserved among all herpesviruses. In addition to KSHV ORF33, its homologs from HSV-1, HCMV, and EBV all exhibited potent inhibitory effect on the induction of IFN in response to poly(dA:dT) or poly(I:C) stimulation (fig. S2). Furthermore, all ORF33 homologs colocalized with STING/MAVS in situ (fig. S4, A and B). These results strongly suggested that antagonism against both DNA- and RNA-sensing pathways is a conserved function of ORF33 among all herpesviruses.
As a tegument protein, ORF33 also plays roles in virion assembly (25–30). A previous study has shown that lack of ORF33 expression in KSHV reduced the yield of progeny virion by about 10-fold (28). This observation is undoubtedly consistent with ORF33’s important role in virion assembly. Moreover, the progeny viruses also appeared to be less infectious, and ~16-fold more ORF33-null viral particles than WT viral particles were needed to achieve the same rate of infection (28). We believe that the ability of ORF33 to antagonize STING- and MAVS-mediated DNA- and RNA-sensing pathways for viral immune evasion, as identified in our study, provides additional explanation for the observed deficiency of ORF33-null virus and further demonstrates the functional significance of tegument proteins in viral life cycle.
The adaptors that contribute to cytosolic nucleic acid–induced type I IFN production are regulated by a variety of posttranslational modifications, including ubiquitination, phosphorylation, acetylation, deamidation, and glutamylation (43–49). Among them, phosphorylation plays a fundamental role. Accomplished by opposing activities of kinases and phosphatases, phosphorylation is reversible and therefore constitutes a major form of signaling regulation and must be precisely balanced. In this regard, a number of host factors that regulate the phosphorylation of these critical proteins have been reported (43, 50). For example, the kinase UNC-51–like kinase-1 (ULK1/ATG1) is activated by cGAMPs to phosphorylate STING after autophagy-dependent STING delivery of TBK1 to endosomal/lysosomal compartments, thus serving as a negative-feedback control of STING activity to prevent sustained innate immune signaling (51). The host phosphatase PP1 dephosphorylates the RNA sensors RIG-I and melanoma differentiation–associated gene 5 (MDA5) to activate the signaling pathway, thus working as a positive regulator of the antiviral innate immune responses to several RNA viruses (52). Nonetheless, these balancing or regulatory mechanisms can be sabotaged or hijacked by viruses, and the V protein of either measles virus or Nipah virus has been reported to act as a decoy substrate of PP1 to inhibit the dephosphorylation of MDA5 and RIG-1 for viral immune evasion (53, 54). In addition, two recent studies have separately reported that host protein phosphatase PPM1A targets STING (55) and MAVS and TBK1 (56) for their dephosphorylation. One earlier study identified PPM1B as a TBK1 phosphatase (57). However, in that study, PPM1A was also included in the phosphatase library that the authors screened and showed no effect on TBK1 phosphorylation. It is unclear what accounts for the discrepancy. Li et al. (55) observed that PPM1A failed to remove all the phosphorylation of STING in in vitro or in vivo assays and proposed the existence of other phosphatases that are involved in regulating STING/MAVS dephosphorylation. Consistently, we identified PPM1G in this study, which serves as a phosphatase to directly dephosphorylate not only STING but also MAVS (Fig. 4 and fig. S6).
PPM1G belongs to the PP2C family of serine/threonine (Ser/Thr) protein phosphatases. The reported functions of PPM1G include spliceosome assembly (58, 59), Cajal body localization by dephosphorylating survival motor neurons (34), specific gene expression (60, 61), protein translation and cell growth by dephosphorylating 4E-binding protein 1 (62, 63), cell cycle progression (64, 65), cellular survival and neural development (66), exchange and dephosphorylation of H2A-H2B (67), and DNA damage response (68, 69). Although most of these reported functions of PPM1G are executed in the nucleus, PPM1G is also reportedly localized in the cytoplasm in different cell lines (63), consistent with its ability to directly dephosphorylate STING and MAVS. Consequently, knockdown or knockout of PPM1G remarkably enhanced host innate immune response and inhibited the replication of not only KSHV but also other DNA and RNA viruses (Fig. 6). Moreover, the interaction between PPM1G and STING/MAVS is markedly enhanced by KSHV ORF33 (Fig. 4), and in the context of KSHV infection, the immune evasion function mediated by ORF33 is strictly dependent on PPM1G (Fig. 7). Collectively, these results indicated that under normal physiological conditions, PPM1G, PPM1A, and possibly other unidentified protein phosphatases work to counteract the action of TBK1 and other kinases so as to maintain the phosphorylation of STING and MAVS within an appropriately balanced range. As a result, a normal immune response can be mounted against the invading pathogens, and sustained overactivation of host immunity can be prevented. However, viruses such as KSHV have developed strategies to tip the balance for immune evasion. In particular, KSHV tegument protein ORF33 hijacks PPM1G to enhance its interactions with STING and MAVS molecules, resulting in decreased phosphorylation levels of STING and MAVS, thus inhibiting the normal progression of antiviral immune response and facilitating viral replication. Whether additional viral proteins, in particular KSHV tegument proteins, also target the cytosolic nucleic acid–sensing pathway warrants further investigation. It also remains to be determined how dephosphorylation of STING and MAVS by different phosphatases is regulated and achieved.
In summary, our study identified PPM1G as a previously unidentified negative regulator of both cytosolic DNA– and RNA–sensing pathways and demonstrated that PPM1G balances STING- and MAVS-mediated innate immunity. We also uncovered a previously unknown immune evasion strategy used by KSHV via its tegument protein ORF33 that manipulates PPM1G to promote virus propagation, providing unique insights into virus-host interactions.
MATERIALS AND METHODS
Cell culture and transfection
HEK293, HEK293T, HeLa, iSLK, and iSLK.219 cells (provided by D. Ganem) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and penicillin-streptomycin. THP-1 cells (provided by L. Zhang) were cultured in RPMI with 10% FBS and penicillin-streptomycin. iSLK cells carrying WT RGB BAC16 (provided by J. Jung) and ORF33-null RGB BAC16 were cultured in DMEM with 10% FBS, puromycin (1 μg/ml), G418 (50 μg/ml), hygromycin (500 μg/ml), and penicillin-streptomycin. Transient transfections of plasmids were performed with jetPEI (POLYPLUS TRANSFECTION) or polyethylenimine following standard protocols. Transient transfections of poly(I:C) and poly(dA:dT) were performed with Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol.
Reagents and antibodies
Rabbit antibodies against human p-TBK1 Ser172 (catalog no. 5483), TBK1 (catalog no. 3504), IRF3 (catalog no. 11904), p-STING Ser366 (catalog no. 85735), and STING (catalog no. 13647) were obtained from Cell Signaling Technology. Rabbit antibodies against human p-IRF3 Ser386 (ab76493), MAVS (ab31334), and green fluorescent protein (GFP) (ab290) were purchased from Abcam. Rabbit antibody against HA was purchased from HUAXINGBIO (HX1820). Mouse antibodies against laminB1 (sc-365962) were purchased from Santa Cruz Biotechnology. Mouse anti-FLAG antibody (M2) (F1804), M2-conjugated agarose (A2220), mouse anti-HA antibody (H3663), and anti-HA–conjugated agarose (A2095) were purchased from Sigma-Aldrich. Rabbit anti-PPM1G antibody (A300-881A) was purchased from Bethyl Laboratories. Poly(I:C) and poly(dA:dT) were purchased from InvivoGen (tlrl-pic, tlrl-patn-1). Mouse anti-ORF33 antibody was provided by F. Zhu (Florida State University, USA). Mouse anti-K8/KbZIP and anti-ORF45 antibodies were provided by Y. Yuan (University of Pennsylvania, USA). Human IFNβ ELISA Kit was purchased from R&D Systems (DIFNB0). Duolink In Situ–Fluorescence kit for PLA was purchased from Sigma-Aldrich (DUO92008, DUO92002, and DUO92004).
Expression plasmid construction
Human cDNAs encoding MAVS, STING, TBK1, IRF3, IRF3(2A), PPP6C, PPP2CA, PPP1CA, PPP1CB, PPM1G, and PPP3CA and human herpesvirus ORF33 homolog cDNAs (HSV-1 UL16, HCMV UL94, EBV BGLF2, and KSHV ORF33) were cloned into three different vectors (pCMV-HA, pCMV-MYC, and pFLAG-CMV2) according to the required tags. GFP-STING was cloned into pEGFP-C1. pGFP-MAVS was provided by Z. Yuan (Beijing Institute of Basic Medical Sciences). DNA sequences for His-STING (amino acids 153 to 379), His-MAVS (amino acids 1 to 460), and His-tagged PPM1G were cloned into pET28a.
Luciferase reporter assays
Firefly luciferase IFNβ reporter plasmid (100 ng) and pRL-TK (Renilla luciferase) plasmid (internal control) (10 ng) were transfected together with the indicated expression plasmids (total 800 ng) into HEK293 cells at a density of 3 × 105 cells per well of 12-well plates. If cotransfected with MAVS or STING expression plasmid, at 24 hours after transfection, then cells were lysed by passive lysis buffer (Promega E1941) for the following luciferase assays. In other cases, transfected cells were cultured for 24 hours, then challenged by transfection of poly(I:C) (1 μg/ml) and poly(dA:dT) (1 μg/ml), and lysed by passive lysis buffer at 18 hours after treatment. The dual luciferase assay kit (Promega E1960) was used to perform luciferase assays, and GloMax-Multi Jr. detection system (Promega) was used to quantify luciferase activity. The relative luciferase activity was calculated by normalizing firefly to Renilla luciferase activity.
Quantitative PCR
HEK293 cells transfected with the indicated expression plasmids were stimulated by transfection of poly(I:C) (1 μg/ml) and poly(dA:dT) (1 μg/ml) or infection of SeV (50 HA U/ml) and HSV-1 (multiplicity of infection = 3) for 12 hours. THP-1 cells were infected with WT KSHV or ORF33-null KSHV for 6 hours. iSLK cells carrying WT RGB BAC16 and ORF33-null RGB BAC16 were induced by doxycycline (2 μg/ml) and sodium butyrate (1 mM) for 24 or 48 hours. RNA was extracted using TRIzol Reagent (Invitrogen 15596-026) according to the manufacturer’s protocol. mRNA was reverse-transcribed to cDNA using PrimeScript RT reagent Kit with genomic DNA Eraser (Takara Biomedical Technology catalog no. RR047A). The relative mRNA levels of human IFNβ and ISG56 were detected by QuantStudio 7 (Life Technologies) using PowerUp SYBR Green Master Mix (Life Technologies A25742) and normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) expression.
qPCR was used to quantify the genome copy number of KSHV. Deoxyribonuclease (DNase) I was added into the medium containing virus (described in the “Virus infection and induction” section) and incubated at 37°C for 1 hour to digest the free viral DNA. Then, the DNase I was inactivated by the addition of 50 mM EDTA and incubation at 70°C for 10 min. Viral DNA was extracted using TIANamp Virus DNA/RNA Kit (TIANGEN, DP315) according to the manufacturer’s protocol. Serially diluted RGB BAC16 DNA (1 to 107 genome copies) was used as standard samples to calculate viral genome copy numbers.
SeV M and NP mRNA levels as well as HSV-1 gD and gJ mRNA levels were measured by RT-qPCR. All the primer sequences are listed in table S1.
Immunofluorescence
To observe the nuclear translocation of endogenous IRF3, HEK293 cells cultured on coverslips in 24-well plates were stimulated by transfection of poly(I:C) (2 μg/ml) and poly(dA:dT) (2 μg/ml) or SeV infection (50 HA U/ml) for 4 hours. The cells were then fixed with 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 7 min at room temperature, blocked with goat serum (Cwbio CW0130S) for 30 min at room temperature, and incubated with primary antibodies (Rabbit anti-HA, 1:500; Mouse anti-FLAG, 1:500; Rabbit anti-IRF3, 1:50) in phosphate-buffered saline, 0.1% Tween 20 (PBST) for 2 hours at room temperature or overnight at 4°C. Last, cells were incubated with respective Alexa Fluor secondary antibodies in PBST for 1 hour and 4′,6-diamidino-2-phenylindole in PBST for 10 min at room temperature. Immunofluorescence images were imaged and analyzed by the Zeiss LSM 700 confocal microscope.
Proximity ligation assay
PLAs were used to detect the interaction of STING-IRF3 and MAVS-IRF3 with Duolink In Situ–Fluorescence kit (Sigma-Aldrich, DUO92008, DUO92002, and DUO92004) according to the manufacturer’s protocol. In Fig. 2, HEK293T cells cultured on coverslips in 24-well plates were cotransfected with expression plasmids for GFP-STING, HA-IRF3 and MYC-EV/MYC-ORF33 (100 ng each), or GFP-MAVS, HA-IRF3, and MYC-EV/MYC-ORF33 (100 ng each) for 20 hours. In fig. S4, HEK293T cells cultured on coverslips in 24-well plates were cotransfected with expression plasmids for GFP-STING and HA-tagged ORF33 homologs or EV (150 ng each) or GFP-MAVS and HA-tagged ORF33 homologs or EV (150 ng each) for 24 hours. Cells were fixed and permeabilized as described in the “Immunofluorescence” section. The cells were then blocked with Duolink blocking buffer in a preheated humidity chamber for 30 min at 37°C and incubated with respective primary antibodies in antibody diluent overnight at 4°C (Rabbit anti-GFP, 1:600; Mouse anti-HA, 1:500). After incubation with probe anti-rabbit PLUS and probe anti-mouse MINUS in a preheated humidity chamber for 60 min at 37°C, the cells were incubated with ligation-ligase solution in a preheated humidity chamber for 30 min at 37°C. At this time, the PLA probes binding to primary antibodies in close proximity would form a closed circle. Then, after samples were incubated with amplification solution in a preheated humidity chamber for 100 min at 37°C, the interaction signal was detected as a distinct red fluorescent spot (excitation, 594 nm; emission, 624 nm) and analyzed by a Zeiss LSM 700 confocal microscope.
Coimmunoprecipitation
HEK293T and HEK293 cells transfected with the indicated expression plasmids in 6-cm plates were lysed in 500 μl of lysis buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM phenylmethanesulfonyl fluoride, EDTA-free protease inhibitor cocktail (Roche 04693), and phosphatase inhibitor (Beyotime P1082)] for 30 min at 4°C. The supernatant of cell lysates was obtained by centrifugation at 13,000 rpm for 15 min (4°C). Fifty microliters of cell lysate supernatant was taken as INPUT. The rest (450 μl) was incubated with anti-FLAG-M2–conjugated agarose (12 μl) or anti-HA–conjugated agarose (12 μl) for 4 to 8 hours at 4°C. The protein-bound beads were then washed five times with lysis buffer at 4°C.
Purification of recombinant proteins
For GST-tagged KSHV ORF33 protein, pGEX-6P-2-GST-ORF33 was transformed into Escherichia coli Rosetta and then cells were induced [OD600 (optical density at 600 nm) ≈ 0.6] with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 16°C for 22 hours to express ORF33 protein. Cells were collected by centrifugation at 4000g for 20 min (4°C), resuspended with PBS [containing 1 mM phenylmethanesulfonyl fluoride, EDTA-free protease inhibitor cocktail (Roche 04693), and lysozyme (1 mg/ml)], and sonicated. The supernatant obtained by centrifugation at 20,000g for 20 min (4°C) was incubated with glutathione-Sepharose resin (CWBIO CW0190) for 2 hours at room temperature. Protein was eluted with 50 mM reduced glutathione in PBS.
For His-tagged STING (amino acids 153 to 379), MAVS (amino acids 1 to 460), and PPM1G proteins, pET28a vector carrying indicated cDNAs was transformed into E. coli Rosetta and then cells were induced (OD600 ≈ 0.6) with 0.5 mM IPTG at 16°C for 22 hours to express proteins. Cells were collected by centrifugation at 4000g for 20 min (4°C), resuspended with lysis buffer [25 mM tris-HCl (pH 7.5), 300 mM NaCl, 1 mM phenylmethanesulfonyl fluoride, EDTA-free protease inhibitor cocktail (Roche 04693), and lysozyme (1 mg/ml)], and sonicated for protein extraction. The supernatant obtained by centrifugation at 20,000g for 20 min (4°C) was incubated with HisPur Ni–nitrilotriacetic acid resin (Thermo Fisher Scientific, 88222) for 2 hours at room temperature. After washing twice with washing buffer [25 mM tris-HCl (pH 7.5), 300 mM NaCl, and 20 mM imidazole], proteins were eluted with elution buffer [25 mM tris-HCl (pH 7.5), 300 mM NaCl, and 250 mM imidazole]. Protein concentrations were measured with Enhanced BCA Protein Assay Kit (Beyotime, P0010).
GST pull-down assay
The MagneGST glutathione particles (Promega, V861A) were washed three times with PBS for particle equilibration by DynaMag-2 Magnet (Life Technologies, 12321D). GST-ORF33/GST proteins and MagneGST glutathione particles (total 300 μl in PBS) were incubated in a 1.5-ml tube at room temperature on a rotating platform for 1 hour. Then, the GST-ORF33/GST-bound MagneGST glutathione particles were incubated with lysates from HEK293T cells with HA-STING or HA-MAVS overexpression for 4 hours at 4°C. The lysis condition was described in the “Coimmunoprecipitation” section. After five washes with lysis buffer (1 ml) at 4°C by DynaMag-2 Magnet, the protein-bound MagneGST glutathione particles were resolved by 2× SDS loading buffer and boiled for 10 min. The samples were analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) and the indicated antibodies.
In vitro kinase and phosphatase assays
The FLAG-TBK1, FLAG-GFP, and FLAG-ORF33 proteins were immunoprecipitated from HEK293 cell extracts with anti-FLAG-M2–conjugated agarose. After washing three times with lysis buffer (described in the “Coimmunoprecipitation” section), the bound proteins were eluted with 3× FLAG peptide (Sigma-Aldrich, F4799) for 30 min at room temperature on a rotating platform for three times. The collected proteins were concentrated with centrifugal filter units (Millipore, 10 kDa, UFC501008), and the protein concentrations were measured with Enhanced BCA Protein Assay Kit (Beyotime, P0010). For in vitro kinase assay, FLAG-TBK1 (1 μg) was incubated with His-STING (amino acids 153 to 379) (1 μg) or His-MAVS (amino acids 1 to 460) (1 μg) in kinase buffer [25 mM tris-HCl (pH 7.5), 100 mM NaCl, 5 mM adenosine triphosphate, 10 mM dithiothreitol (DTT), and 5 mM MgCl2] for 40 min at 30°C. For in vitro phosphatase assay, p-STING or p-MAVS (obtained above) was concentrated with centrifugal filter units (10 kDa) and the reaction buffer was exchanged with phosphatase buffer (1 mM EGTA, 25 mM MgCl2, 2 mM DTT, 0.1% bovine serum albumin, and 250 mM imidazole). FLAG-ORF33, FLAG-GFP, purified GST-ORF33, or purified individual host protein phosphatase (1 μg each) was added into the phosphatase buffer and incubated for 40 min at 30°C. After the reaction, samples were resolved by 2× SDS loading buffer and boiled for SDS-PAGE analysis.
Genetic manipulation of KSHV BAC genome
KSHV ORF33-null BAC was constructed by inserting triple stop codons at 96 nucleotides of ORF33 coding sequence on RGB-BAC16 (31) using a Red recombination system as described previously (70, 71). The sequence containing Kan/I-Sce I, triple stop codons, and BsiWI site was amplified with the primers listed in table S1.
Virus infection and induction
To make KSHV WT or ORF33-null virus, iSLK-RGB-BAC16 or iSLK-ORF33-null-RGB-BAC16 stable cell lines were constructed as described previously (72). Cells were then induced with doxycycline (2 μg/ml) and 1 mM sodium butyrate for 72 hours, and the supernatants were collected by centrifugation and filtered with a 0.45 μM filter unit (Merck, SLHV033RB). The viruses were concentrated using a centrifugal filter unit (Merck, UFC910096), and viral genome copy numbers were analyzed by qPCR. To infect THP-1 cells, THP-1 cells seeded on a 12-well plate were incubated with WT KSHV or ORF33-null KSHV (50 viral genome copies per cell) and centrifuged at 2000 rpm for 1 hour at 30°C. SeV infection was conducted at 50 HA U/ml.
RNA interference
siRNA against PPM1G [5′-3′: AGGCUACCAUGACUAUUGA (34)] and cGAS [5′-3′: GAUUGAGCUACAAGAAUAU (73)] was synthesized by GenePharma Corporation. Cells were transfected with siRNAs using Lipofectamine RNAiMAX Reagent (Invitrogen, 13778-030) following the manufacturer’s instructions.
CRISPR-Cas9–mediated genome editing
The guide RNAs targeting PPM1G (NM_177983.2) were designed using an online design site (crispr.mit.edu) (74). According to the analysis results, we chose two high score targets, one targeting exon 1 and the other targeting exon 9. After annealing, double-stranded oligos were inserted into puro-Cas9CrisprV2 vector (provided by M. Zhu, Institute of Biophysics, China) after cleavage by Bsm BI. The two puro-Cas9CrisprV2 plasmids containing two different target sites were cotransfected into HEK293 cells and selected using puromycin (1 μg/ml) at 48 hours after transfection. After about 2 weeks, monoclonal PPM1G knockout cells were selected and positive clones were identified by Western blot with antibody against PPM1G. The oligo sequences are listed in table S1.
Supplementary Material
Acknowledgments
We thank J. U Jung for providing RGB-BAC16, D. Ganem for iSLK and iSLK.219 cells, F. Zhu for the antibody to KSHV ORF33, Y. Yan for antibodies to KSHV K8/KbZIP and ORF45, M. Zhu for puro-Cas9CrisprV2 vector, L. Zhang for THP-1 cells, and Z. Yuan for pGFP-MAVS plasmid. We thank E. S. Mocarski for critical reading of this manuscript, and B. He and members of the Deng laboratory for helpful discussions. Funding: This work was supported by grants from the Ministry of Science and Technology (2016YFA0502101), the National Natural Science Foundation of China (nos. 81630059, 81325012, and 31900131), and the Chinese Academy of Sciences (KJZD-SW-L05 and the Strategic Priority Research Program XDB37030205). Author contributions: K.Y. and H.D. conceived the project and analyzed the data. K.Y. performed most of the experiments with help from H.T. K.Y. and H.D. wrote the paper. All authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabd0276/DC1
REFERENCES AND NOTES
- 1.Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T., The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Sun L., Wu J., Du F., Chen X., Chen Z. J., Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhong B., Yang Y., Li S., Wang Y.-Y., Li Y., Diao F., Lei C., He X., Zhang L., Tien P., Shu H.-B., The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa H., Barber G. N., STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K. J., Takeuchi O., Akira S., IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Seth R. B., Sun L., Ea C.-K., Chen Z. J., Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122, 669–682 (2005). [DOI] [PubMed] [Google Scholar]
- 7.McWhirter S. M., tenOever B. R., Maniatis T., Connecting mitochondria and innate immunity. Cell 122, 645–647 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Xu L.-G., Wang Y.-Y., Han K.-J., Li L.-Y., Zhai Z., Shu H.-B., VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 19, 727–740 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., Du F., Ren J., Wu Y.-T., Grishin N. V., Chen Z. J., Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Lin R., Mamane Y., Hiscott J., Structural and functional analysis of interferon regulatory factor 3: Localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 19, 2465–2474 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn J., Gutman D., Saijo S., Barber G. N., STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. U.S.A. 109, 19386–19391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gall A., Treuting P., Elkon K. B., Loo Y.-M., Gale M. Jr., Barber G. N., Stetson D. B., Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma X., Helgason E., Phung Q. T., Quan C. L., Iyer R. S., Lee M. W., Bowman K. K., Starovasnik M. A., Dueber E. C., Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. U.S.A. 109, 9378–9383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang Y., Cesarman E., Pessin M. S., Lee F., Culpepper J., Knowles D. M., Moore P. S., Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Soulier J., Grollet L., Oksenhendler E., Cacoub P., Cazals-Hatem D., Babinet P., d’Agay M. F., Clauvel J. P., Raphael M., Degos L., Sigaux F., Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease [see comments]. Blood 86, 1276–1280 (1995). [PubMed] [Google Scholar]
- 16.Cesarman E., Chang Y., Moore P. S., Said J. W., Knowles D. M., Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332, 1186–1191 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Russo J. J., Bohenzky R. A., Chien M.-C., Chen J., Yan M., Maddalena D., Parry J. P., Peruzzi D., Edelman I. S., Chang Y., Moore P. S., Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U.S.A. 93, 14862–14867 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.F. Liu, Z. H. Zhou, Comparative virion structures of human herpesviruses, in Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley, K. Yamanishi, Eds. (Cambridge Univ. Press, 2007). [PubMed] [Google Scholar]
- 19.Dai W., Jia Q., Bortz E., Shah S., Liu J., Atanasov I., Li X., Taylor K. A., Sun R., Zhou Z. H., Unique structures in a tumor herpesvirus revealed by cryo-electron tomography and microscopy. J. Struct. Biol. 161, 428–438 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rixon F. J., Structure and assembly of herpesviruses. Semin. Virol. 4, 135–144 (1993). [Google Scholar]
- 21.Sathish N., Wang X., Yuan Y., Tegument proteins of Kaposi’s sarcoma-associated herpesvirus and related gamma-herpesviruses. Front. Microbiol. 3, 98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diefenbach R. J., Conserved tegument protein complexes: Essential components in the assembly of herpesviruses. Virus Res. 210, 308–317 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Guo H., Shen S., Wang L., Deng H., Role of tegument proteins in herpesvirus assembly and egress. Protein Cell 1, 987–998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mettenleiter T. C., Klupp B. G., Granzow H., Herpesvirus assembly: An update. Virus Res. 143, 222–234 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Meckes D. G. Jr., Wills J. W., Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J. Virol. 81, 13028–13036 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeh P.-C., Han J., Chadha P., Meckes D. G. Jr., Ward M. D., Semmes O. J., Wills J. W., Direct and specific binding of the UL16 tegument protein of herpes simplex virus to the cytoplasmic tail of glycoprotein E. J. Virol. 85, 9425–9436 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips S. L., Cygnar D., Thomas A., Bresnahan W. A., Interaction between the human cytomegalovirus tegument proteins UL94 and UL99 is essential for virus replication. J. Virol. 86, 9995–10005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu J.-j., Avey D., Li W., Gillen J., Fu B., Miley W., Whitby D., Zhu F., ORF33 and ORF38 of Kaposi’s sarcoma-associated herpesvirus interact and are required for optimal production of infectious progeny viruses. J. Virol. 90, 1741–1756 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo H., Wang L., Peng L., Zhou Z. H., Deng H., Open reading frame 33 of a gammaherpesvirus encodes a tegument protein essential for virion morphogenesis and egress. J. Virol. 83, 10582–10595 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen S., Jia X., Guo H., Deng H., Gammaherpesvirus tegument protein ORF33 is associated with intranuclear capsids at an early stage of the tegumentation process. J. Virol. 89, 5288–5297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brulois K., Toth Z., Wong L.-Y., Feng P., Gao S.-J., Ensser A., Jung J. U., Kaposi’s sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J. Virol. 88, 9335–9349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin Q., Tian Y., Kabaleeswaran V., Jiang X., Tu D., Eck M. J., Chen Z. J., Wu H., Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol. Cell 46, 735–745 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Q., Meng F., Chen S., Plouffe S. W., Wu S., Liu S., Li X., Zhou R., Wang J., Zhao B., Liu J., Qin J., Zou J., Feng X.-H., Guan K.-L., Xu P., Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 19, 362–374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petri S., Grimmler M., Over S., Fischer U., Gruss O. J., Dephosphorylation of survival motor neurons (SMN) by PPM1G/PP2Cγ governs Cajal body localization and stability of the SMN complex. J. Cell Biol. 179, 451–465 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang K., Wang J., Wu M., Li M., Wang Y., Huang X., Mesenchymal stem cells detect and defend against gammaherpesvirus infection via the cGAS-STING pathway. Sci. Rep. 5, 7820 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horan K. A., Hansen K., Jakobsen M. R., Holm C. K., Søby S., Unterholzner L., Thompson M., West J. A., Iversen M. B., Rasmussen S. B., Ellermann-Eriksen S., Kurt-Jones E., Landolfo S., Damania B., Melchjorsen J., Bowie A. G., Fitzgerald K. A., Paludan S. R., Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 190, 2311–2319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiu Y.-H., Macmillan J. B., Chen Z. J., RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inn K.-S., Lee S.-H., Rathbun J. Y., Wong L.-Y., Toth Z., Machida K., Ou J.-H. J., Jung J. U., Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 85, 10899–10904 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider W. M., Chevillotte M. D., Rice C. M., Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 32, 513–545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang G., Chan B., Samarina N., Abere B., Weidner-Glunde M., Buch A., Pich A., Brinkmann M. M., Schulz T. F., Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. U.S.A. 113, E1034–E1043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu J.-j., Li W., Shao Y., Avey D., Fu B., Gillen J., Hand T., Ma S., Liu X., Miley W., Konrad A., Neipel F., Stürzl M., Whitby D., Li H., Zhu F., Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18, 333–344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma Z., Jacobs S. R., West J. A., Stopford C., Zhang Z., Davis Z., Barber G. N., Glaunsinger B. A., Dittmer D. P., Damania B., Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. U.S.A. 112, E4306–E4315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiang C., Gack M. U., Post-translational control of intracellular pathogen sensing pathways. Trends Immunol. 38, 39–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo W.-W., Li S., Li C., Lian H., Yang Q., Zhong B., Shu H.-B., iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 17, 1057–1066 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Ansari M. A., Dutta S., Veettil M. V., Dutta D., Iqbal J., Kumar B., Roy A., Chikoti L., Singh V. V., Chandran B., Herpesvirus genome recognition induced acetylation of nuclear IFI16 is essential for its cytoplasmic translocation, inflammasome and IFN-β responses. PLOS Pathog. 11, e1005019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi S. J., Lee H.-C., Kim J.-H., Park S. Y., Kim T.-H., Lee W.-K., Jang D.-J., Yoon J.-E., Choi Y.-I., Kim S., Ma J., Kim C.-J., Yao T.-P., Jung J. U., Lee J.-Y., Lee J.-S., HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 35, 429–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia P., Ye B., Wang S., Zhu X., Du Y., Xiong Z., Tian Y., Fan Z., Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol. 17, 369–378 (2016). [DOI] [PubMed] [Google Scholar]
- 48.He S., Zhao J., Song S., He X., Minassian A., Zhou Y., Zhang J., Brulois K., Wang Y., Cabo J., Zandi E., Liang C., Jung J. U., Zhang X., Feng P., Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 58, 134–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J., Zhao J., Xu S., Li J., He S., Zeng Y., Xie L., Xie N., Liu T., Lee K., Seo G. J., Chen L., Stabell A. C., Xia Z., Sawyer S. L., Jung J., Huang C., Feng P., Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 24, 234–248.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan Y. K., Gack M. U., Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Konno H., Konno K., Barber G. N., Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wies E., Wang M. K., Maharaj N. P., Chen K., Zhou S., Finberg R. W., Gack M. U., Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38, 437–449 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davis M. E., Wang M. K., Rennick L. J., Full F., Gableske S., Mesman A. W., Gringhuis S. I., Geijtenbeek T. B. H., Duprex W. P., Gack M. U., Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 16, 19–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mesman A. W., Zijlstra-Willems E. M., Kaptein T. M., de Swart R. L., Davis M. E., Ludlow M., Duprex W. P., Gack M. U., Gringhuis S. I., Geijtenbeek T. B. H., Measles virus suppresses RIG-I-like receptor activation in dendritic cells via DC-SIGN-mediated inhibition of PP1 phosphatases. Cell Host Microbe 16, 31–42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z., Liu G., Sun L., Teng Y., Guo X., Jia J., Sha J., Yang X., Chen D., Sun Q., PPM1A regulates antiviral signaling by antagonizing TBK1-mediated STING phosphorylation and aggregation. PLOS Pathog. 11, e1004783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiang W., Zhang Q., Lin X., Wu S., Zhou Y., Meng F., Fan Y., Shen T., Xiao M., Xia Z., Zou J., Feng X.-H., Xu P., PPM1A silences cytosolic RNA sensing and antiviral defense through direct dephosphorylation of MAVS and TBK1. Sci. Adv. 2, e1501889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao Y., Liang L., Fan Y., Sun S., An L., Shi Z., Cheng J., Jia W., Sun W., Mori-Akiyama Y., Zhang H., Fu S., Yang J., PPM1B negatively regulates antiviral response via dephosphorylating TBK1. Cell. Signal. 24, 2197–2204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murray M. V., Kobayashi R., Krainer A. R., The type 2C Ser/Thr phosphatase PP2Cγ is a pre-mRNA splicing factor. Genes Dev. 13, 87–97 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Allemand E., Hastings M. L., Murray M. V., Myers M. P., Krainer A. R., Alternative splicing regulation by interaction of phosphatase PP2Cγ with nucleic acid-binding protein YB-1. Nat. Struct. Mol. Biol. 14, 630–638 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Gudipaty S. A., McNamara R. P., Morton E. L., D’Orso I., PPM1G binds 7SK RNA and Hexim1 to block P-TEFb assembly into the 7SK snRNP and sustain transcription elongation. Mol. Cell. Biol. 35, 3810–3828 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McNamara R. P., McCann J. L., Gudipaty S. A., D’Orso I., Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 5, 1256–1268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu K., Wang L., Feng W., Feng Y., Shu H.-K. G., Phosphatidylinositol-3 kinase-dependent translational regulation of Id1 involves the PPM1G phosphatase. Oncogene 35, 5807–5816 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu J., Stevens P. D., Eshleman N. E., Gao T., Protein phosphatase PPM1G regulates protein translation and cell growth by dephosphorylating 4E binding protein 1 (4E-BP1). J. Biol. Chem. 288, 23225–23233 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suh E.-J., Kim T.-Y., Kim S. H., PP2Cγ-mediated S-phase accumulation induced by the proteasome-dependent degradation of p21WAF1/CIP1. FEBS Lett. 580, 6100–6104 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Sun C., Wang G., Wrighton K. H., Lin H., Songyang Z., Feng X.-H., Lin X., Regulation of p27Kip1 phosphorylation and G1 cell cycle progression by protein phosphatase PPM1G. Am. J. Cancer Res. 6, 2207–2220 (2016). [PMC free article] [PubMed] [Google Scholar]
- 66.Foster W. H., Langenbacher A., Gao C., Chen J., Wang Y., Nuclear phosphatase PPM1G in cellular survival and neural development. Dev. Dyn. 242, 1101–1109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kimura H., Takizawa N., Allemand E., Hori T., Iborra F. J., Nozaki N., Muraki M., Hagiwara M., Krainer A. R., Fukagawa T., Okawa K., A novel histone exchange factor, protein phosphatase 2Cγ, mediates the exchange and dephosphorylation of H2A–H2B. J. Cell Biol. 175, 389–400 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khoronenkova S. V., Dianova I. I., Ternette N., Kessler B. M., Parsons J. L., Dianov G. L., ATM-dependent downregulation of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Mol. Cell 45, 801–813 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beli P., Lukashchuk N., Wagner S. A., Weinert B. T., Olsen J. V., Baskcomb L., Mann M., Jackson S. P., Choudhary C., Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tischer B. K., von Einem J., Kaufer B., Osterrieder N., Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40, 191–197 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Tischer B. K., Smith G. A., Osterrieder N., En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 634, 421–430 (2010). [DOI] [PubMed] [Google Scholar]
- 72.Brulois K. F., Chang H., Lee A. S.-Y., Ensser A., Wong L.-Y., Toth Z., Lee S. H., Lee H.-R., Myoung J., Ganem D., Oh T.-K., Kim J. F., Gao S.-J., Jung J. U., Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86, 9708–9720 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ding S., Diep J., Feng N., Ren L., Li B., Ooi Y. S., Wang X., Brulois K. F., Yasukawa L. L., Li X., Kuo C. J., Solomon D. A., Carette J. E., Greenberg H. B., STAG2 deficiency induces interferon responses via cGAS-STING pathway and restricts virus infection. Nat. Commun. 9, 1485 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., Zhang F., DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/47/eabd0276/DC1