

1. Introduction
In 1869, Ashworth presented a preliminary analysis of circulating tumor cells (CTCs) and its implications for understanding cancer metastasis (Ashworth 1869). With few exceptions, this observation was ignored until the late twentieth century, in large part because CTCs act at ultra-low concentrations in the bloodstream: in the range of 1 in 109 blood cells. Recent technological developments, however, have now made it possible to identify CTCs from noninvasive liquid biopsies, and while the field is still much in its infancy, CTC frequency has already been prognostically linked to overall survival in metastatic breast, colorectal, and prostate cancer (Budd et al. 2006; Cohen et al. 2008; de Bono et al. 2008). But even if we can detect CTCs, any effort in using them to study cancer progression dynamics has a number of other obstacles. One worth discussion is tumor heterogeneity: the emergence of different cellular phenotypes and enigmatic cellular interactions inflicted in the tumor microenvironment and circulation—posing significant challenges for treatment decision-making. Although there have been waves of CTC detection technologies, each with its own successes, most rely on protein enrichment or physical selection methods (Ozkumur et al. 2013; Yap et al. 2014). Since there is no general CTC biomarker consensus (Phillips et al. 2014; Samson and Baas 2015), such size-specific or protein-based assumptions are dangerous, as potentially relevant cellular events could inadvertently be lost through these conventional assays. Thus, the search for which tumor cells are critical for disease progression may be missing something essential.
In stark contrast to most CTC detection methods, the high-definition single-cell assay (HD-SCA) workflow was designed as an enrichment-free, high-throughput assay for the entire population of cells in a liquid sample, while at the same time being fully compatible with clinical pathology. Founded by the Scripps Physical Sciences-Oncology Center (PS-OC1), the HD-SCA workflow brings together modern methods of immunofluorescence with more sophisticated image processing to rapidly and accurately detect rare tumor cells among the milieu of platelets, erythrocytes, and leukocytes in the peripheral blood. This approach has been established as a reliable and sensitive CTC detection and characterization workflow for metastatic breast, lung, and prostate cancers (Marrinucci et al. 2009, 2012; Nieva et al. 2012; Pecot et al. 2011), with direct clinical applications (Carlsson et al. 2014; Dago et al. 2014; Gross et al. 2015).
The purpose of this chapter is to explain how the HD-SCA technology endows visualization, characterization, and measurement of rare CTCs from liquid biopsies, and what the resulting information teaches us about how cancer spreads through the human body. We introduce the basic concepts of the HD-SCA workflow, focusing on topics relevant to image processing and analysis; show how this technology has been used to investigate and measure the liquid phase of cancer metastasis; and explore how the HD-SCA technology, together with the current standard of care, has implications for improving the precision of cancer diagnostics and interventions.
2. Standardized Blood Cell Preparation and Immunofluorescent Staining
Advancing our knowledge of the spatiotemporal evolution of cancer in the human body requires direct quantitative access to CTCs—potential “seeds” responsible for the metastatic cascade (Chaffer and Weinberg 2011; Scott et al. 2012). Ideally, we would like to measure all the heterogeneous CTCs and their evolution, both as the disease naturally progresses and under treatment pressure. As such, each blood biopsy is treated with erythrocyte lysis and then, with much care, all nucleated blood cells are plated as a biological monolayer onto custom glass slides [as published in Marrinucci et al. (2012)]. The glass slides have a proprietary adhesive coating that enables maximum retention of live cells, holding ~ 3 × 106 nucleated cells per slide. After plating, all cells are fixed, permeabilized, and immunofluorescently stained with monoclonal antibodies targeting a panel of cytokeratins (1, 4, 5, 6, 8, 10, 13, 18 and 19), an intermediate filament found exclusively in epithelial cells; CD45, a pan-leukocyte-specific marker; 4′,6-diaminido-2-phenylindole (DAPI), a nuclear stain; and—if desired—an additional preselected, disease-specific fourth marker (such as androgen receptor (AR), estrogen receptor, HER2, PDGFRα, VE-cadherin). Finally, each slide is subsequently imaged via a custom fully automated scanning microscopy system to provide a snapshot of the dynamical disease (see Fig. 1 for an overview).
Fig. 1.
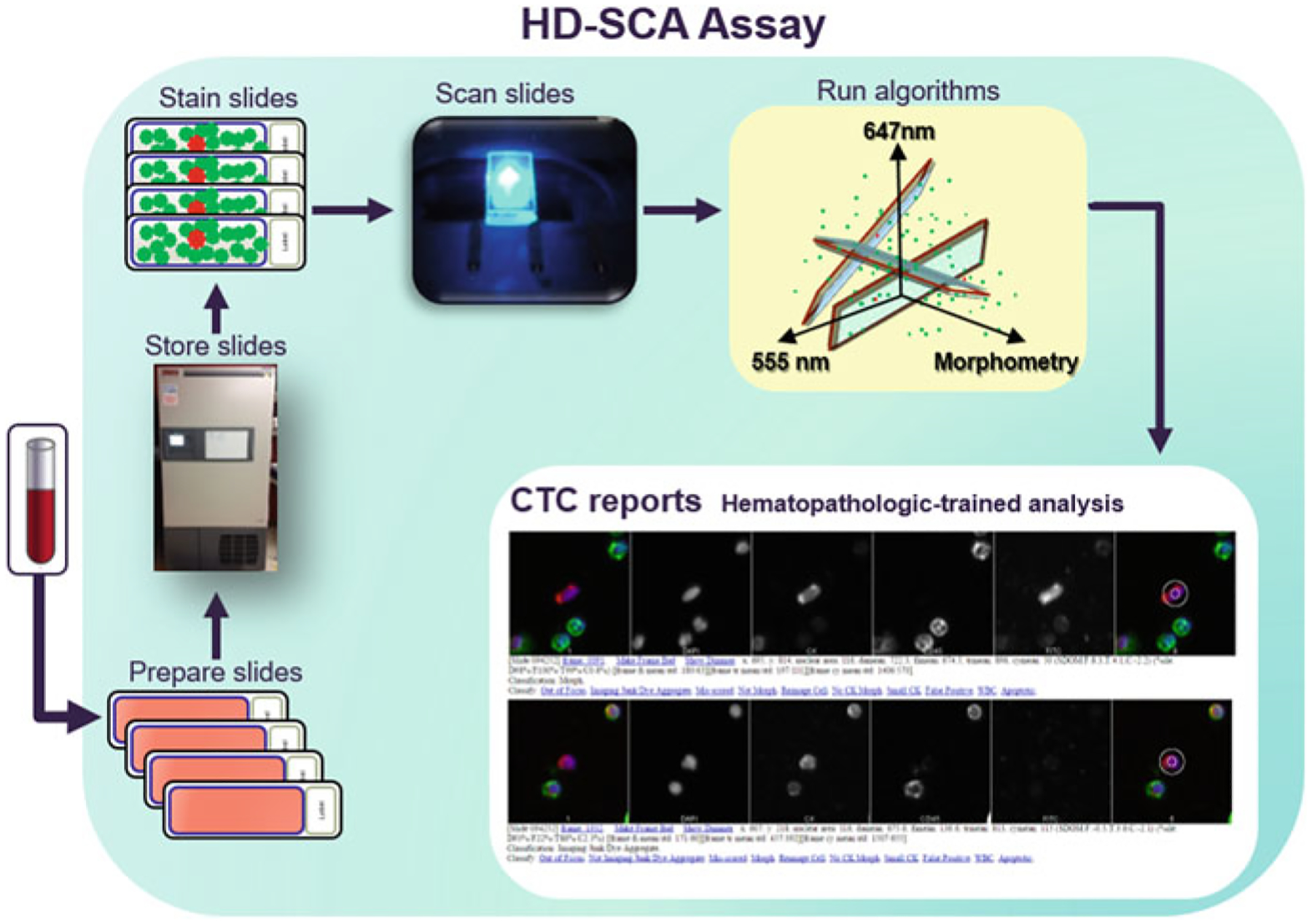
A schematic overview of the HD-SCA workflow. Received patient whole blood is treated with erythrocyte lysis and then plated onto adherent slides. Multiple slides (from the same patient draw) are kept and preserved in a biorepository until analysis is desired. This provides researchers with the ability to assay the same sample using several strategies at any time. When slides are ready to be assayed, they are immunofluorescently stained and imaged via automated scanning microscopy. The resulting images are computationally analyzed to infer candidate CTCs, which are then presented in reports for classification by a hematopathology-trained specialist
Although this approach requires chemically fixed cells, a process with many known limitations (Phillips et al. 2014), it has the advantage of requiring no enrichment step and minimal processing of blood samples. The method can thus be applied to characterize the entire heterogeneous circulating cell population from low volume samples without any selection bias, efficiently and inexpensively. Additionally extensive pre-analytical variable testing has been conducted to ensure accurate and reproducible data output (Rodriguez-Lee 2018; Stephanie accepted)
3. High-Throughput Imaging of CTCs
The advent of fast and reliable automated fluorescence microscopy has granted us measurement of multiple quantitative descriptors over large populations of cells in the peripheral blood, down to the single-cell level (Marrinucci et al. 2012; Nieva et al. 2012; Cho et al. 2012; Kuhn and Bethel 2012; Wendel et al. 2012). In concert with the immunofluorescent staining protocol described above, a custom-designed optical microscope and imaging pipeline make it possible to analyze up to 3 × 105 cells per second. In the following sections, we outline the high-throughput imaging aspects of the HD-SCA workflow, starting with the scanning microscopy system and proceeding through a succession of computational steps on the path to the high-definition single-cell assay (Fig. 2).
Fig. 2.
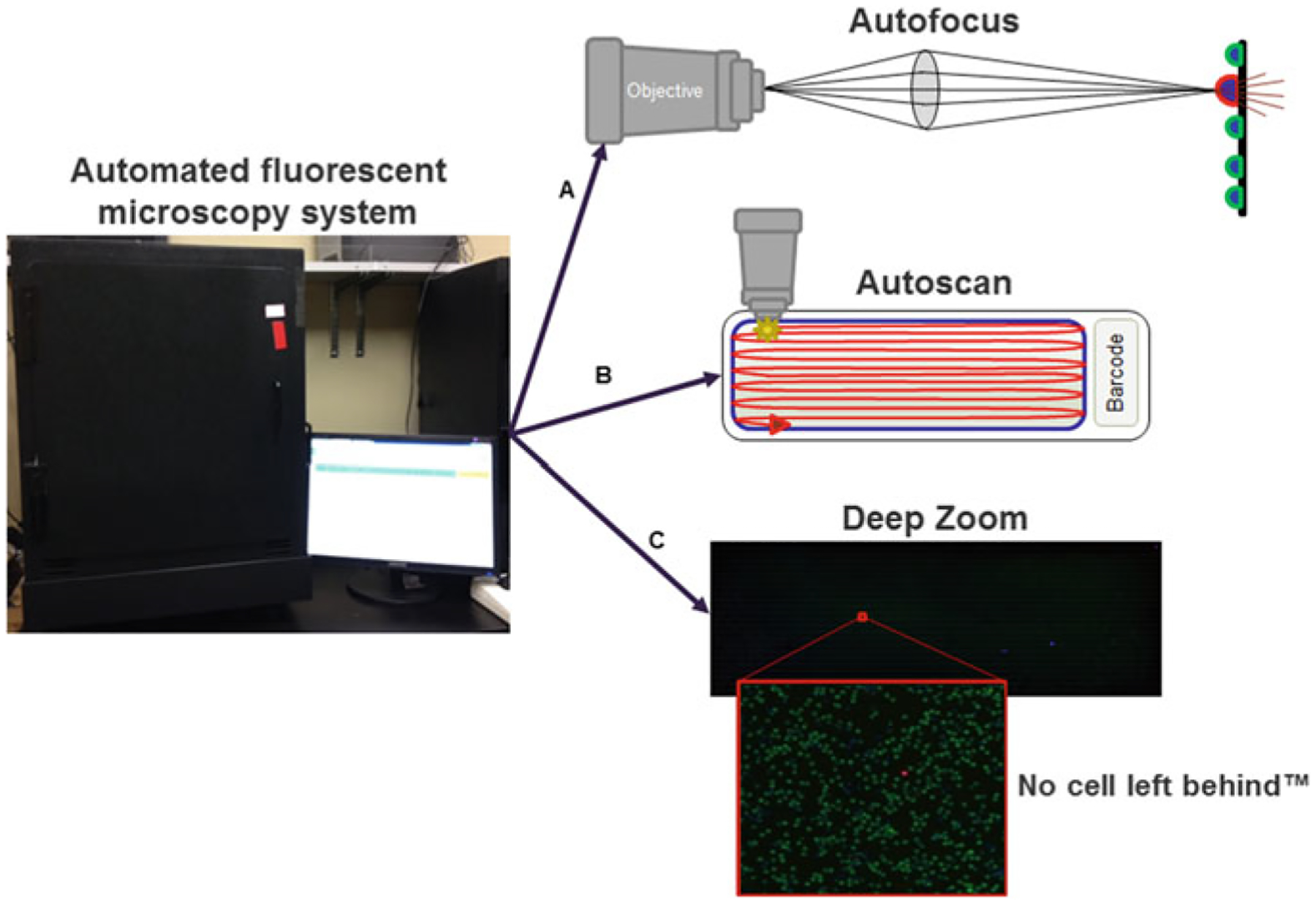
HD-SCA automated fluorescence scanning microscopy system. A First, the focus and exposure (for each fluorescent channel) are automatically set for each slide. B Second, the entire active surface of each slide is automatically scanned, and then, each cell is automatically segmented for extraction of cellular features. C Finally, a deep zoom is created from the collection of scanned images of the slide, allowing researchers to interactively pan around and zoom in/out
4. Automated Fluorescence Imaging
The custom fully automated scanning microscopy system is equipped with a broad-spectrum illuminator; a multiband filter set (optimized for DAPI, fluorescein isothiocyanate (FITC), tetramethylrhodamine-5-isothiocyanate (TRITC), and Cy5, as well as other similar fluorophores); and a 20 MHz, 14-bit camera with an IEEE 1394 interface for high-speed information transmission. Its partner in crime, a homemade imaging analysis pipeline, is an equally important aspect for automatically acquiring and analyzing digital images. Housed with mass computing storage and processing power, this optical setup is poised for high-throughput fluorescence imaging.
A fundamental first step for analysis of cellular microscopy-based assays is focusing of nuclei. Accordingly, the HD-SCA imaging pipeline uses an autofocus algorithm based on Vollath’s autocorrelation function (Vollath 1987), which, based on the comparison of 13 focus algorithms for fluorescence microscopy, has been shown to be an optimal (Santos et al. 1997). Second, autoexposure is performed to automatically set the exposure time to within an ideal range for each optical channel (this is in parallel a method for normalizing experimental variance between slides). Altogether, a full scan at 10 × magnification of the entire active area of a slide—that is, 19.3 × 56.9 mm and ~3 × 106 data points—takes 45 min.
The imaging system’s stage is engineered to support 4 slides, ultimately producing over 6900 digital images of upward to 107 cells (Marrinucci et al. 2012). Acquired raw images are then supplied to arrays of hard drives (RAIDs) that have built-in redundancy. The resulting images, which retain fine cellular details of nuclear and cytoplasmic structure, are then fed into a two-part semi-automated algorithm to identify candidate CTCs.
5. Automated Measurement and Detection of CTCs
There are many well-established image processing programs for cell segmentation and feature extraction of microscopy-based cellular assays (for instance, CellPro-filer and ImageJ, as well as programming environments/languages such as Lab-VIEW, MATLAB, Python, and R). The open-source nature and generality of ImageJ have made it an attractive image processing and analysis framework for the HD-SCA workflow. The combination of ImageJ and more advanced algorithms (via Python) enables automated analysis of phenotypic features from thousands of images.
Starting with single-cell segmentation to locate cells and boundaries in images, the algorithm identifies each cell from digital imagery by the DAPI intensity of a given nucleus. Each cell’s center of mass is then computed to generate masks for quantitative descriptors of cellular phenotypes (Marrinucci et al. 2012), including physical characteristics (area, aspect ratio, circularity, roundness, solidity, cluster count) and expression levels (fluorescent signal intensity) for every nucleated cell, in each optical channel (fluorescence excitation wavelength (λex) = 359 nm and fluorescence emission wavelength (λem) = 461 nm for the DAPI fluorophore; λex = 555 nm and λem = 578 nm for the TRITC (cytokeratins) fluorophore; λex = 647 nm and λem = 666 nm for the Cy5 (CD45) fluorophore; λex = 495 nm and λem = 519 nm for the FITC (optional, disease-specific) fluorophore) (Table 1). If further analysis for a cell of interest is desired, the cell’s recorded center of mass, or equivalently, its coordinates on the slide, can easily be mined and thus sequestered on any calibrated system for downstream characterization. As an example, cells of interest can be reimaged at different focal planes with a confocal microscope to obtain 3D information (see Fig. 4; for more examples, see HD-SCA Beyond Fluorescence Section below).
Table 1.
CTC measurements automatically generated with the HD-SCA workflow
| Feature | Description | Units |
|---|---|---|
| Physical characteristics | Physical measurements generated for each nuclei | |
| x | Center of mass x (x-axis) | 16-bit pixel value |
| y | Center of mass y (y-axis) | 16-bit pixel value |
| Roundness | How closely the shape of an object approaches that of a circle | |
| Circularity | Inverse of roundness | |
| Area | Area size | 16-bit pixel value2 |
| Area (local ratio) | Ratio of the area of nuclei of interest to the average area of the surrounding 50 nuclei | |
| Aspect ratio | Relationship between width and height | |
| Solidity | Texture | |
| Cluster count | Individual cell nuclei within a CTC aggregate | Cell nuclei |
| Expression-level measurements | Signal intensities generated for each cell, in each fluorescent channel (DAPI, TRITC, FITC, Cy5) | |
| Mean | Mean pixel intensity of the cell of interest | 16-bit pixel value |
| Standard deviation | Standard deviation of the mean pixel intensities of the cell of interest and the surrounding cells | 16-bit pixel value |
| Standard deviation over the mean | The number of standard deviations over the mean pixel intensity for the cell of interest to the mean pixel intensity of the surrounding 50 cells | 16-bit pixel value |
Fig. 4.
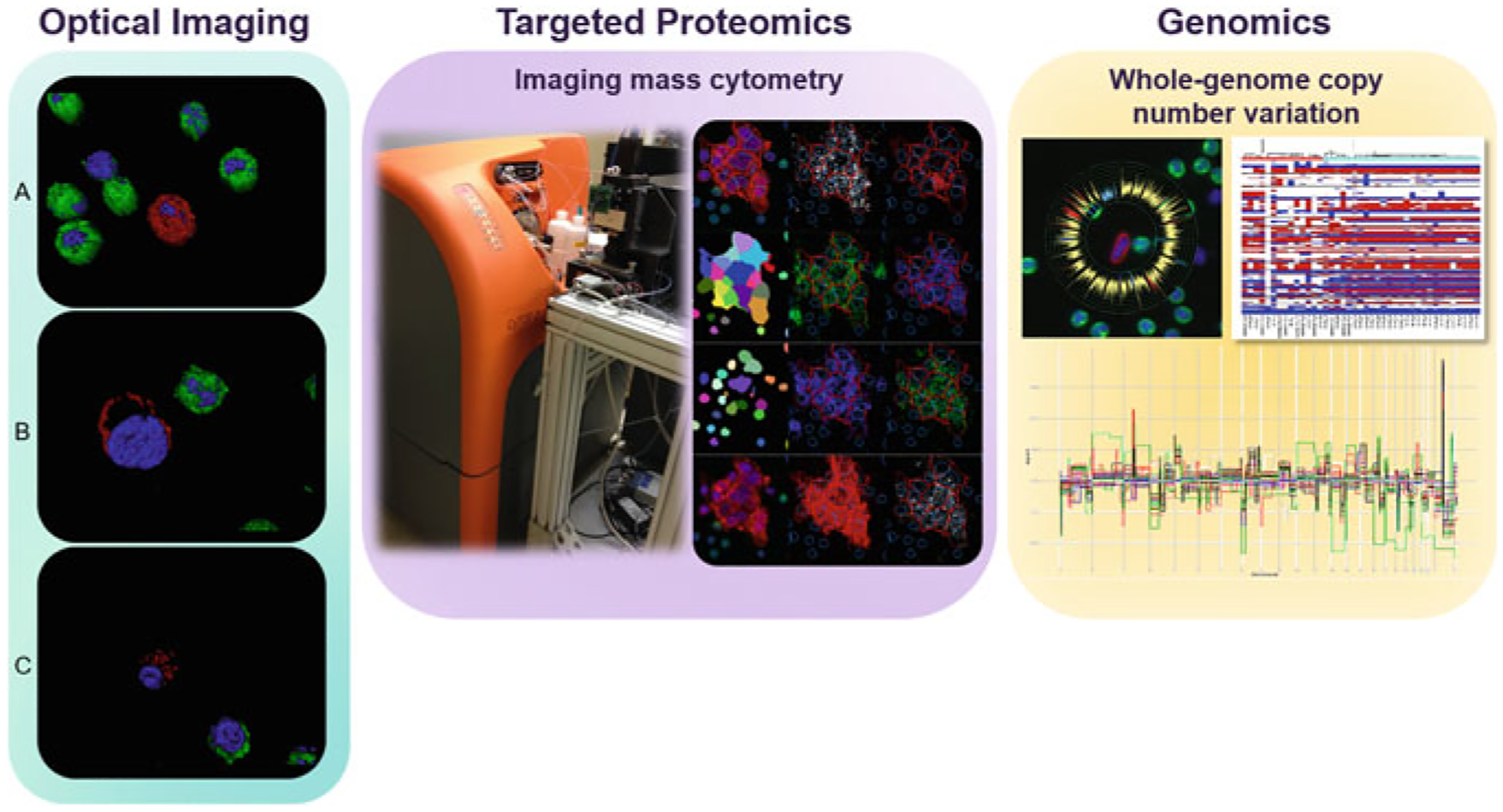
Downstream characterization with the HD-SCA workflow Left panel: As an example of an optical imaging technique applied to detected cells of interest with the workflow, we present confocal images of A HD-CTC, B cross section of HD-CTC with membrane-bound cytokeratin, and C cross section of CTC-cfDNA producing displaying apoptotic blebbing. Here, images have been rendered with Imaris to visually define each cell and the expression of specific epitopes: Red represents CK expression, green represents CD45 expression, and blue represents DAPI. Middle panel: Targeted proteomic characterization on cells of interest using imaging mass cytometry. Right panel: Whole-genome copy number variation profiling of single cells of interest
Like all cellular assays, the usual caveats apply: Fixation, permeabilization, and staining will inevitably vary. But all of these variables are certainly tractable and manageable by normalizing each rare (CTC) measure with the surrounding nonrare (leukocyte) measures to generate relative CTC metrics within the same experiment. These relative metrics are then computationally analyzed on a cell-by-cell basis, primarily via the fluorescent signal intensity of cytokeratin and CD45, to infer candidate CTCs, which are then passed to a specialist for technical analysis and classification.
6. No Cell Left Behind™
Cancer evolves in the patient from initiation to widespread metastatic disease through a series of subclinical processes that span a wide range of temporal and spatial scales. Broadly speaking, cancer must act in ways that are coherent in the context of Darwinian dynamics and in relation to the biophysical events in the patient’s body. Cancer cells must sense, compute, and make decisions: to find food, to travel through the bloodstream, to evade our immune system, to act collectively (or compete) with other cells, and to proliferate—behavior must be “functional.” In theory, there are many ways in which such functional information could be represented (and thus measured) during the development of cancer metastasis. One possibility is that CTCs represent metastatic seeds with varying degrees of malignancy (Chaffer and Weinberg 2011; Scott et al. 2012). But how much information does a CTC tell us about the disease? What biological signals are important? Answering these questions is crucial because, while there is a general consensus that CTCs express epithelial markers, less is known about cellular states associated with CTCs expressing low epithelial levels, small CTCs, the life span of CTCs, CTC aggregates, and the implications of such phenotypes on disease evolution. These ideas point to the need for detection and characterization of the largest diversity of candidate CTC populations across a wide range of clinical stages, which is made possible with the HD-SCA workflow. This approach has worked exceedingly well for quantifying several morphological and cellular properties of the circulatory phase (Marrinucci et al. 2009, 2012; Nieva et al. 2012; Carlsson et al. 2014; Dago et al. 2014; Gross et al. 2015; Kuhn and Bethel 2012; Wendel et al. 2012; King et al. 2015; Phillips et al. 2012a, b; Ruiz et al. 2015; Marrinucci et al. 2007, 2010; Lazar et al. 2012; Chalfin et al. 2018; Malihi et al. 2018; Williamson et al. 2016; Carlsson et al. 2017; Gross et al. 2015), and technical analysis by a hematopathology-trained specialist has been central to this success. In this section, we turn our focus to the high-definition single-cell assay and classification of candidate CTCs. Throughout, we emphasize a fundamental understanding of CTCs in human cancers is critical for using them as biomarkers.
6.1. High-Definition Single-Cell Assay and Classification
In the second part of the semi-automated algorithm, candidate CTCs are further analyzed by a hematopathology-trained specialist. For contextual comparison, digital images of candidate CTCs are presented with surrounding nonrare cells within the field of view in each optical channel—and overlaid for reference—examples of which are presented in Fig. 3. For quantitative comparison, relative metrics of physical characteristics and expression levels of candidate CTCs are also shown, again for each optical channel. Multiple phase spaces, such as morphometry, cytokeratin intensity, and CD45 intensity, can thus be analyzed simultaneously at the single-cell level. At present, candidate CTCs are classified into four broad categories; see Table 2 for detailed description2 and Fig. 3 for high-definition visualization of each category.
Fig. 3.
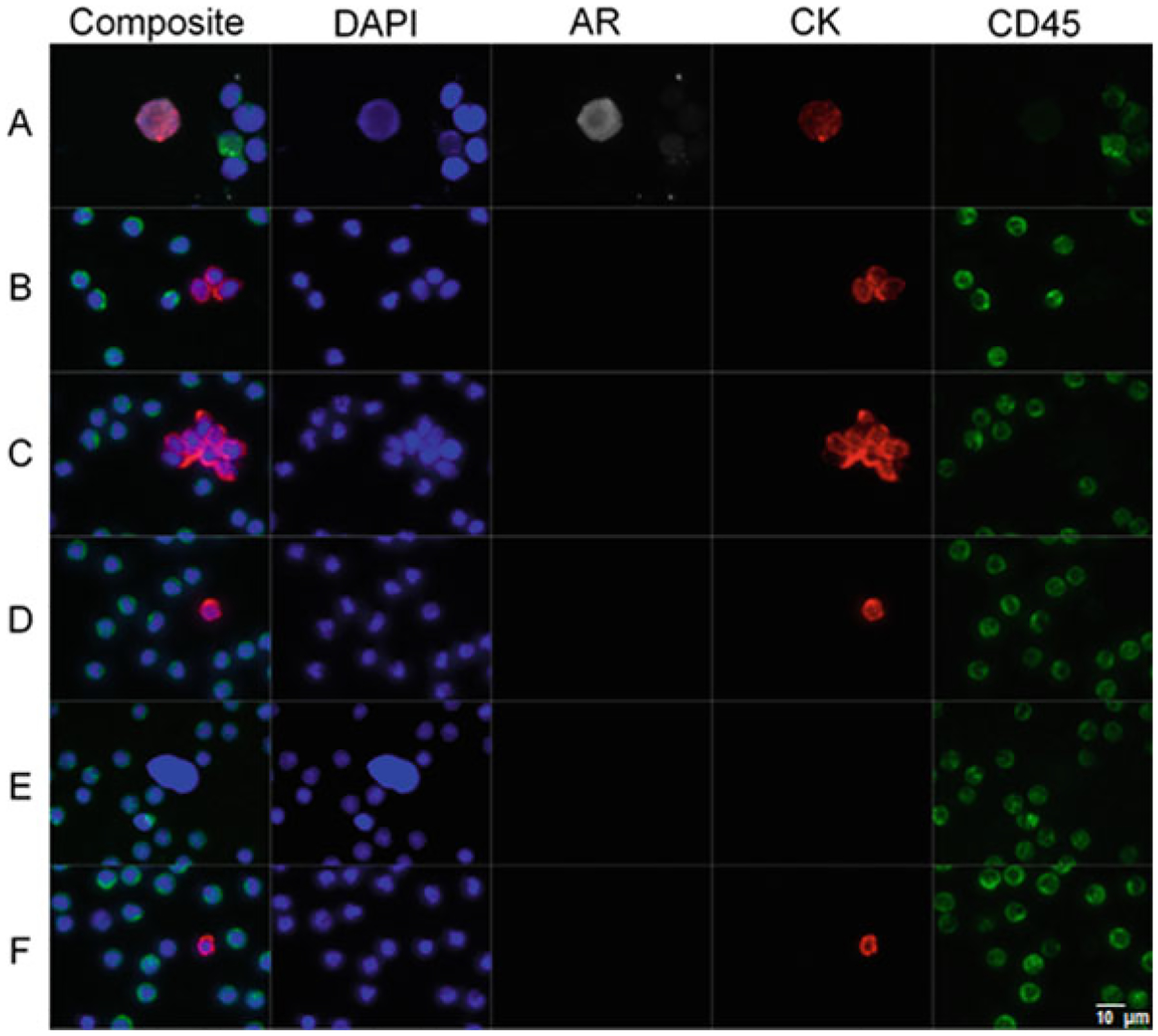
Gallery of representative CTCs detected in the blood of a patient with prostate cancer. Panel A shows a composite image and individual optical channels of an HD-CTC. In addition, this particular tumor cell expresses androgen receptor (AR). HD-CTCs are defined by having an intact nucleus, identified through DAPI (blue); being of epithelial origin, characterized by a bright cytokeratin (red) stain; no CD45 signal (green)—a leukocyte marker; and a distinct morphology. Panel B represents a CTM triplet. Panel C represents a mega-CTM (>5 CTCs). Panel D represents a CTC-small. Panel E represents CTC-low CK. Panel F represents CTC-cfDNA producing. Scale bar = 10 μm
Table 2.
CTC classification schema
| Type | Description |
|---|---|
| HD-CTC | Putative HD-CTCs are defined by having an intact nucleus, identified through DAPI; being of epithelial origin, characterized by a bright cytokeratin (CK) stain; no CD45 signal—a leukocyte marker; and a distinct morphology from the surrounding leukocytes. HD-CTCs must have a clear circumferential cytoplasm containing the entire nucleus. See Panel A in Fig. 3 |
| CTC-small | CTC-small expresses CK at appropriate levels to be a HD-CTC, but has a similar (or smaller) nuclear size in relation to surrounding leukocytes. See Panel D in Fig. 3 |
| CTC-low CK | CTC-low CK expresses an insufficient level of CK to consider a HD-CTC, but has a significantly larger nucleus compared to surrounding leukocytes. This group is potentially associated with cancer stem cells or cells undergoing an epithelial-to-mesenchymal transition. See Panel E in Fig. 3 |
| CTC-cfDNA producing | Candidates with identifiable apoptotic changes, such as cytoplasmic blebbing and/or nuclear fragmentation. See Panel F in Fig. 3 |
7. Morphometry of CTCs
Cellular morphology has been used historically to discern the malignancy of a cell (Tosi et al. 1986). Abnormalities in nuclear size, microstructure (including chromatin organization and mitotic figures), and shape are morphologic hallmarks associated with cancer pathology (Partin et al. 1992; Pienta and Coffey 1991). In the context of the fluid phase of solid tumors, a breakthrough case study of the morphometry of CTCs in a patient with well-differentiated lung adenocarcinoma was found to be strikingly similar to the epithelial cells of the primary tumor: CTCs were larger in size relative to leukocytes and exhibited low nuclear-to-cytoplasmic ratios (Marrinucci et al. 2009). In a larger study of breast and colorectal cancer patients, CTCs shared heterogeneity consistent with cancer cells from other spatial regions (such as the primary and metastatic tumor sites) in the body, including high and low nuclear-to-cytoplasmic ratios, as well as early and late apoptotic changes (Marrinucci et al. 2007, 2010).
While it is necessary to study cancer biology in model systems, it is enormously challenging to address the issues that arise in realistic contexts, e.g., the complexities of fully natural signals in the human with cancer and the dynamic biological processes upon which the disease is acting. To support the cell biology community in translating such studies, the HD-SCA assay was used to morphologically compare CTCs from patients with prostate cancer and prostate tumor cells derived from an LNCaP cell line (Lazar et al. 2012). Lazar et al. demonstrated important differences between the average total cell areas of actual patient CTCs (~90 ± 50 μm2) and LNCaP cells (~140 ± 50 μm2)—as well as differences in the expression levels of cytokeratin and AR—providing translational benchmarks for experiments in classical model systems.
More recently, the HD-SCA workflow was applied to a cohort of metastatic melanoma patients, where circulating melanoma cells (CMCs) were discovered to be, on average, 1.5-fold larger than surrounding nonrare nucleated blood cells (Ruiz et al. 2015), an observation which is also consistent with CTCs detected in patients with prostate cancer (Cho et al. 2012). Overall, relative morphometrics of CTCs have revealed fundamental insight on their biophysical properties and probable pathological origins.
8. Circulating Tumor Microemboli
Perhaps not surprisingly, circulating tumor microemboli (CTM or, equivalently, CTC aggregates) are detected with the HD-SCA workflow in a number of different studies across various cancer types, including breast, non-small-cell lung (NSCL), prostate, and pancreatic cancer (Marrinucci et al. 2012; Carlsson et al. 2014; Cho et al. 2012; Carlsson et al. 2017; Malihi et al. 2018). Intuition tells us that collective cell migration could be a travel strategy for distant metastasis (Friedl and Gilmour 2009; Friedl et al. 2012; Kats-Ugurlu et al. 2009). But how do we make this intuition precise? Early experiments to build clinical metrics for a physical understanding of the potential role of CTM in metastasis showed that individual CTCs within CTM were, on average, smaller in nuclear area (similar or equivalent to surrounding leukocytes) and length (~0.8-fold larger than surrounding leukocytes) than CTCs detected alone (where both nuclear area and length were ~1.5-fold larger than surrounding leukocytes) (Marrinucci et al. 2012; Carlsson et al. 2014; Cho et al. 2012). Subsequent experiments exploited the HD-SCA assay’s sensitivity and found that CTCs were present in a wide range for both early- and late-stage NSCL cancer (Nair et al. 2013). This led to the proposal that CTCs/CTM may be complementary to traditional clinical modalities for risk stratifying large lung nodules in patients and thus aid as a noninvasive diagnostic (Carlsson et al. 2014).
To test this idea, Carlsson et al. integrated CTC/CTM data with conventional clinical/imaging information to develop multiple logistic regression models using a case-control design in a training (N = 88 patients; N =71 malignant; and N =17 benign) and test (N = 41 patients; N =33 malignant; and N =8 benign) cohort (N = 129 total eligible patients); and then performed tenfold cross-validation of the entire group (Carlsson et al. 2014). Although the presence of CTCs and CTM is not always related to tumor burden or metabolic activity in the NSCL cancer setting, as measured by fluorodeoxyglucose (FDG) positron emission tomography-computed tomography (PET-CT) (Nair et al. 2013), Carlsson and colleagues demonstrated that patients could be diagnosed more accurately through CTM data combined with clinical/imaging information (where the area under the curve (AUC) = 0.88 and p value = 0.001 for all NSCL cancer patients, and AUC = 0.87 and p value = 0.002 for early-stage NSCL cancer patients), rather than clinical/imaging information alone (where AUC = 0.77 for all NSCL cancer patients as well as early-stage NSCL cancer patients).
Although it has been shown in mouse models that the metastatic potential of CTM is higher than single CTCs (Aceto et al. 2014), there are obvious open questions. Where do CTM come from, and where are they going? Are cells within CTM heterogeneous? What are the biophysical factors that allow CTM to endure the immune response in the circulatory system? Indeed, further technological improvements and multidisciplinary interactions are needed—and ongoing—to investigate the potential role of CTM in the bloodstream and organ arrest.
9. Expression-Level Measurements of CTCs
As mentioned at the start of this chapter, tumor heterogeneity and delineating which circulating cells are malignant across spatial regions in the body are a major challenge in cancer biology and oncology. We would like to know which biological signals in CTCs are relevant to disease progression and hence which CTC populations carry meaningful information about treatment response to ultimately provide clinicians with useful knowledge about the disease in real time. Toward this goal, Dago and coworkers sequentially characterized CTC subcellular AR expression fluctuations and clonal evolution in a patient with castrate-resistant prostate cancer (CRPC) as he progressed through chemotherapy and targeted therapy (Dago et al. 2014). CTCs from liquid biopsies were quantified (at both the phenotypic and the genomic scales) at 4 significant time points, where draw 1 was collected prior to any chemotherapy; draw 2 was collected after chemo- and radiotherapy; draw 3 was collected after 3 weeks of targeted therapy; and draw 4 was collected after 9 weeks of targeted therapy. The patient was showing clinical improvement at the time of draw 3, exhibiting less pain and lower prostate-specific antigen (PSA) levels; but, at the same time, a single clone, which strongly expressed AR, had also emerged. By draw 4, this treatment-resistant clone dominated the CTC population, and the patient exhibited increased pain and PSA levels. The patient died soon after.
Dago and colleagues showed that the dynamic changes across the genomic and phenotypic scales in CTCs, together with clinical information, allow for noninvasive monitoring of therapeutic efficacy. Recent work by Gross et al. examined rapid changes in CTCs immediately following bevacizumab (anti-angiogenic) treatment in CRPC: Blood samples were taken before and within 2 h after bevacizumab administration in 8 patients (Gross et al. 2015). In 6 of the 8 patients that responded, putative CTCs decreased, while apoptotic CTCs increased, suggesting early changes in tumor perfusion as well as which patients would likely benefit from bevacizumab therapy.
The fact that one can follow treatment sensitivity through repeated noninvasive biopsies opens the possibility of a very different approach to cancer treatment. In particular, if we can identify circulating biomarkers in the bloodstream, then we can adapt therapy to control the evolution of cancer in individual patients.
10. HD-SCA Beyond Fluorescence
Because all nucleated cells in a blood sample are assayed with the HD-SCA workflow, any cell of interest can be relocated on the glass slide to further probe the cell’s physical, chemical, and molecular features (see Fig. 4). Such relocation has permitted the characterization of the mass (King et al. 2015; Phillips et al. 2012), volume (Phillips et al. 2012a; b), density (King et al. 2015; Phillips et al. 2012), density fluctuations (King et al. 2015; Phillips et al. 2012), and genome-wide copy number alterations (Dago et al. 2014; Ruiz et al. 2015) of individual disease-derived cells. Dago et al., for example, developed a protocol for isolating DNA from CTCs under conditions suitable for downstream single-cell genomics. In this protocol, individual CTCs are identified, as described above, and subsequently picked off the slide with a micromanipulator for whole-genome amplification of their DNA, followed by library construction for Illumina sequencing. This method, as noted in the previous section, has led to the measurement of sequential clonal changes in CTCs in response to a multi-step therapeutic regime—which culminated in treatment resistance in the patient (Dago et al. 2014). Generalization of this method is straightforward and has been used to profile the copy numbers of CMCs in melanoma patients (Ruiz et al. 2015), thus furthering our ability to genomically characterize CTCs for the development of molecularly targeted therapies and monitoring patients.
In an effort to build (inputs for) blood cell flow models to better understand metastasis, Phillips and collaborators isolated cells of interest from liquid biopsies of patients with breast and ovarian cancer using the HD-SCA workflow and subsequently quantified the volume, density, and dry mass content through quantitative phase microscopy (Phillips et al. 2012a, b). In both cases, the average measured volume of CTCs was (851 ± 45.8 μm3 for breast; 518.3 ± 24.5 μm3 for ovarian) greater than the leukocyte population (234.1 ± 4.1 μm3 for breast; 230.9 ± 78.5 μm3 for ovarian). In the patient with ovarian cancer, the average dry mass content and density of CTCs were found to be 33.6 ± 3.2 pg and 0.065 ± 0.006 pg/fl, respectively. On the other hand, the average mass and density of the leukocyte population were, respectively, 18.7 ± 0.6 pg and 0.085 ± 0.004 pg/fl.
These imaging techniques have also been used to characterize CTM in transit (King et al. 2015). Again, using a combination of the HD-SCA workflow and quantitative phase microscopy, King et al. quantified the physical characteristics and subcellular density organization of CTCs and CTM in a patient with breast cancer. These measures were then used as a translational guide for in vitro and in silico models to investigate the mechanics of CTC transport in the vasculature. The in vitro model system was comprised of microfluidic flow assays to simulate tumor cell adhesion between breast cancer line CTCs/CTM and E-selectin under hemodynamic forces. These experiments found that CTCs exhibit an upward trend in rolling velocity as the number of CTCs/CTM increased, and similarly, the orthogonal displacement of CTM to the applied shear increased with CTM size. This observation was in accord with their numerical simulations of elastic collisions between CTCs and erythrocytes, suggesting that CTCs with more rigid membranes marginate quicker than those with softer membranes, and deformation of the membrane during collisions with erythrocytes can extend the time in which CTCs are flowing in the bloodstream. Taken together, these results provide a translational approach to formulate experiments in classical model systems and the design of numerical models that are grounded in real clinical metrics.
11. Conclusion and Outlook
By taking advantage of the HD-SCA workflow’s simplicity and low-level processing of patient blood, intact CTCs can be identified, imaged, and further assayed at the phenotypic level down to the DNA polymer level. The high-throughput imaging workflow we have discussed above makes it clear that liquid biopsies present new opportunities for investigating and characterizing the spatiotemporal dynamics and clinical evolution of cancer. This effort becomes all the more tractable given modern technological developments in the analysis of circulating tumor DNA (Dawson et al. 2013; Forshew et al. 2012; Murtaza et al. 2013) and exosomal microRNAs (Taylor and Gercel-Taylor 2008) from fluid biopsies. In addition, Giesen and colleagues have recently introduced imaging mass cytometry—a combination of CyTOF mass cytometry, labeling of antigens in cells with rare-earth metal isotopes tagged to antibodies, and a high-resolution laser ablation system—for the simultaneous measurement of 32 proteins and posttranslational modifications at a cellular resolution of 1 μm (Giesen et al. 2014). This raises the possibility of extending the HD-SCA workflow for targeted proteomics and a system’s biology approach to understanding the hematogenous dissemination of human cancer (Malihui et al. 2018; Gerdtsson et al. 2018). More concretely, it gives us a path to assess complex, hypothesized cellular states, such as the epithelial-to-mesenchymal transition (Thiery 2002), the mesenchymal-to-epithelial transition (Kalluri and Weinberg 2009), cancer stem cells (Jordan et al. 2006), vasculogenic mimicry (Hendrix et al. 2003; Williamson et al. 2016), cell-to-cell interactions, cellular senescence (Hanahan and Weinberg 2011), tumor cell dormancy (Aguirre-Ghiso 2007), cancer cells in the blood and their pathological links to metastatic spreader or sponge sites (Newton et al. 2013, 2014), and other clinical relationships to (biomarker) expression dynamics. Now that many of these foundational steps have been solidified, we believe that the coming years will see dramatic progress in a comprehensive CTC assay as a real-time, noninvasive liquid biopsy for the development and implementation of patient-specific treatment strategies.
Footnotes
To promote a physical sciences perspective of cancer, the US National Cancer Institute’s PS-OC Program was launched in 2009—initially a Network of 12 Centers, including the Scripps PS-OC—with the aim of converging traditional cancer biology and oncology with the physical and engineering sciences to bring radical new approaches to cancer research (http://physics.cancer.gov).
Some care is needed here, and it is worth noting that CTC classification is under ongoing development and rigorous investigation. Thus, Table 2 is by no means a complete list (!).
Contributor Information
Kevin Keomanee-Dizon, Convergent Science Institute in Cancer, Michelson Center for Convergent Bioscience, Dornsife College of Letters, Arts and Sciences, University of Southern California, 1002 W. Childs Way, Los Angeles 90089-3502, CA, United States; Viterbi School of Engineering, University of Southern California, 1002 W. Childs Way, Los Angeles, CA 90089, United States.
Stephanie N. Shishido, Convergent Science Institute in Cancer, Michelson Center for Convergent Bioscience, Dornsife College of Letters, Arts and Sciences, University of Southern California, 1002 W. Childs Way, Los Angeles 90089-3502, CA, United States
Peter Kuhn, Convergent Science Institute in Cancer, Michelson Center for Convergent Bioscience, Dornsife College of Letters, Arts and Sciences, University of Southern California, 1002 W. Childs Way, Los Angeles 90089-3502, CA, United States; Viterbi School of Engineering, University of Southern California, 1002 W. Childs Way, Los Angeles, CA 90089, United States.
References
- Aceto N, Bardia A, Miyamoto DT et al. (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158(5):1110–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7(11):834–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth TR (1869) A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust Med J 14:146–147 [Google Scholar]
- Budd GT, Cristofanilli M, Ellis MJ et al. (2006) Circulating tumor cells versus imaging-predicting overall survival in metastatic breast cancer. Clin Cancer Res 12(21):6403–6409 [DOI] [PubMed] [Google Scholar]
- Carlsson A, Nair VS, Luttgen MS et al. (2014) Circulating tumor microemboli diagnostics for patients with non-small-cell lung cancer. J Thorac Oncol 9(8):1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Kuhn P, Luttgen MS, Dizon KK, Troncoso P, Corn PG, Kolatkar A, Hicks JB, Logothetis C, Zurita AJ (2017) Paired high-content analysis of prostate cancer cells in bone marrow and Blood characterizes increased androgen receptor expression in tumor cell clusters. Clin Cancer Res 23:1722–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331 (6024):1559–1564 [DOI] [PubMed] [Google Scholar]
- Chalfin HJ, Glavaris SA, Malihi PD, Sperger JM, Gorin MA, Lu C, Goodwin CR, Chen Y, Caruso EA, Dumpit R, Kuhn P, Lang JM, Nelson PS, Luo J, Pienta KJ (2018) Prostate cancer disseminated tumor cells are rarely detected in the bone marrow of localized patientsundergoing radical prostatectomy across multiple rare cell detection platforms. J Urol 199(6):1494–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EH, Wendel M, Luttgen M et al. (2012) Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Phys Biol 9(1):016001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Punt Cj, Iannotti N et al. (2008) Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol 26(19):3213–3221 [DOI] [PubMed] [Google Scholar]
- Dago AE, Stepanskky A, Carlsson A et al. (2014) Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS ONE 9(8):e101777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson SJ, Tsui Dw, Murtaza M et al. (2013) Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 368(13):1199–1209 [DOI] [PubMed] [Google Scholar]
- de Bono JS, Scher HI, Montgomery RB et al. (2008) Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res 14(19):6302–6309 [DOI] [PubMed] [Google Scholar]
- Forshew T, Murtaza M, Parkinson C et al. (2012) Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 4(136):136ra68. [DOI] [PubMed] [Google Scholar]
- Friedl P, Gilmour D (2009) Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10(7):445–457 [DOI] [PubMed] [Google Scholar]
- Friedl P, Locker J, Sahai E et al. (2012) Classifying collective cancer cell invasion. Nat Cell Biol 14 (8):777–783 [DOI] [PubMed] [Google Scholar]
- Gerdtsson E, Pore M, Thiele J-A, Sandstrom Gerdtsson A, Malihi PD, Nevarez R, Kolatkar A, Ruiz Velasco C, Wix S, Singh M, Carlsson A, Zurita AJ, Logothetis C, Merchant AA, Hicks J, Kuhn P (2018) Multiplex protein detection on circulating tumor cells from liquid biopsies using imaging mass cytometry. Converg Sci Phys Oncol 4:015002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesen C, Wang HA, Schapiro D et al. (2014) Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods 11(4):417–422 [DOI] [PubMed] [Google Scholar]
- Gross ME, Dorff TB, Quinn DI, Agus DB, Luttgen M, Bethel K, Kolatkar A, Kuhn P (2015) Rapid changes in circulating tumor cells following anti-angiogenic therapy. Converg Sci Phys Oncol 1(1):015002 [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674 [DOI] [PubMed] [Google Scholar]
- Hendrix MJ, Seftor EA, Hess AR et al. (2003) Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer 3(6):411–421 [DOI] [PubMed] [Google Scholar]
- Jordan CT, Guzman ML, Noble M (2006) Cancer stem cells. N Engl J Med 355(12):1253–1261 [DOI] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119 (1420):1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kats-Ugurlu G, Roodink I, de Weijert M et al. (2009) Circulating tumour tissue fragments in patients with pulmonary metastasis of clear cell renal cell carcinoma. J Pathol 219(3):287–293 [DOI] [PubMed] [Google Scholar]
- King MR, Phillips KG, Mitrugno A et al. (2015) A physical sciences network characterization of circulating tumor cell aggregate transport. Am J Physiol Cell Physiol 308(10):C792–C802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn P, Bethel K (2012) A fluid biopsy as investigating technology for the fluid phase of solid tumors. Phys Biol 9(1):010301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar DC, Cho EH, Luttgen MS et al. (2012) Cytometric comparisons between circulating tumor cells from prostate cancer patients and the prostate-tumor-derived LNCaP cell line. Phys Biol 9 (1):016002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malihi P, Morikado M, Welter L, Liu ST, Miller ET, Cadaneanu RM, Knudsen BS, Lewis MS, Carlsson A, Ruiz Velasco C, Kolatkar A, Rodriguez-Lee M, Garraway IP, Hicks J, Kuhn P (2018) Clonal diversity revealed by morphoproteomic and copy number profiles of singleprostate cancer cells at diagnosis. Converg Sci Phys Oncol 4:015003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrinucci D, Bethel K, Bruce RH et al. (2007) Case study of the morphologic variation of circulating tumor cells. Human Pathol 38(3):514–519 [DOI] [PubMed] [Google Scholar]
- Marrinucci D, Bethel K, Luttgen M et al. (2009) Circulating tumor cells from well-differentiated lung adenocarcinoma retain cytomorphologic features of primary tumor type. Arch Pathol Lab Med 133(9):1468–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrinucci D, Bethel K, Lazar D et al. (2010) Cytomorphology of circulating colorectal tumor cells: a small case series. J Oncol 2010:861341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrinucci D, Bethel K, Kolatkar A et al. (2012) Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys Biol 9(1):016003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaza M, Dawson SJ, Tsui DW et al. (2013) Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497(7447):108–112 [DOI] [PubMed] [Google Scholar]
- Nair VS, Keu KV, Luttgen MS et al. (2013) An observational study of circulating tumor cells and (18)F-FDG PET uptake in patients with treatment-naive non-small cell lung cancer. PLoS ONE 8(7):e67733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton PK, Mason J, Bethel K et al. (2013) Spreaders and sponges define metastasis in lung cancer: a markov chain monte carlo mathematical model. Can Res 73(9):2760–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton PK, Mason J, Hurt B et al. (2014) Entropy, complexity, and Markov diagrams for random walk cancer models. Sci Rep 4:7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieva J, Wendel M, Luttgen MS et al. (2012) High-definition imaging of circulating tumor cells and associated cellular events in non-small cell lung cancer patients: a longitudinal analysis. Phys Biol 9(1):016004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkumur E, Shah AM, Ciciliano JC et al. (2013) Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci Transl Med 5(179):179ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin AW, Steinberg GD, Pitcock RV et al. (1992) Use of nuclear morphometry, gleason histologic scoring, clinical stage, and age to predict disease-free survival among patients with prostate-cancer. Cancer 70(1):161–168 [DOI] [PubMed] [Google Scholar]
- Pecot CV, Bischoff FZ, Mayer JA et al. (2011) A novel platform for detection of CK+ and CK− CTCs. Cancer Discov 1(7):580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KG, Velasco CR, Li J et al. (2012a) Optical quantification of cellular mass, volume, and density of circulating tumor cells identified in an ovarian cancer patient. Front Oncol 2:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KG, Kolatkar A, Rees KJ et al. (2012b) Quantification of cellular volume and sub-cellular density fluctuations: comparison of normal peripheral blood cells and circulating tumor cells identified in a breast cancer patient. Front Oncol 2:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips K, Kuhn P, McCarty OJ (2014) Physical biology in cancer. 2. The physical biology of circulating tumor cells. Am J Physiol Cell Physiol 306(2):C80–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pienta KJ, Coffey DS (1991) Correlation of nuclear morphometry with progression of breast cancer. Cancer 68(9):2012–2016 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lee M, Kolatkar A, McCormick M, Dago AE, Kendall J, Carlsson NA, Bethel K, Greenspan E, Hwang S, Waitman K, Nieva J, Hicks J, Kuhn P (2018) Effect of blood collection tube type and time to processing on the enumeration and high-content characterization of circulating tumor cells using the high-definition single cell assay. Arch Pathol Lab Med 142:198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz C, Li J, Lutthgen MS et al. (2015) Limited genomic heterogeneity of circulating melanoma cells in advanced stage patients. Phys Biol 12(1):016008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson S, Baas C (2015) The great debate: are CTCs ready for prime time? Convergent Science Physical Oncology 1:017001 [Google Scholar]
- Santos A, Ortiz de Solorzano C, Vaquero JJ et al. (1997) Evaluation of autofocus functions in molecular cytogenetic analysis. J Microsc 188(Pt 3):264–272 [DOI] [PubMed] [Google Scholar]
- Scott J, Kuhn P, Anderson AR (2012) Unifying metastasis-integrating intravasation, circulation and end-organ colonization. Nat Rev Cancer 12(7):445–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido SN, Welter L, Rodriguez-Lee M, Kolatkar A, Liya X, Gerdtsson AS, Restrepo-Vassalli S, Anders C, Joe L, Greenspan EJ, Hwang SE, Waitman KR, Nieva J, Bethel K, Hicks J, Peter K. Pre-analytical variables for the genomic assessment of the cellular and acellular fractions of the liquid biopsy in a cohort ofbreast cancer patients. J Mol Diagn (accepted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DD, Gercel-Taylor C (2008) MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol 110(1):13–21 [DOI] [PubMed] [Google Scholar]
- Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2 (6):442–454 [DOI] [PubMed] [Google Scholar]
- Tosi P, Luzi P, Baak JP et al. (1986) Nuclear morphometry as an important prognostic factor in stage I renal cell carcinoma. Cancer 58(11):2512–2518 [DOI] [PubMed] [Google Scholar]
- Vollath D (1987) Automatic focusing by correlative methods. J Microsc-Oxf 147:279–288 [Google Scholar]
- Wendel M, Bazhenova L, Boshuizen R et al. (2012) Fluid biopsy for circulating tumor cell identification in patients with early-and late-stage non-small cell lung cancer: a glimpse into lung cancer biology. Phys Biol 9(1):016005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson S, Metcalf R, Trapani F, Mohan S, Antonello J, Abbott B, Leong H-S, Chester C, Simms N, Polanski R, Nonaka D, Priest L, Fusi A, Carlsson F, Carlsson A, Hendrix M, Seftor R, Seftor E, Rothwell D, Hughes A, Hicks J, Miller C, Kuhn P, Brady G, Simpson K, Blackhall F, Dive C (2016) Vasculogenic mimicry in small cell lung cancer. Nat Commun 7:13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, Lorente D, Omlin A et al. (2014) Circulating tumor cells: a multifunctional biomarker. Clin Cancer Res 20(10):2553–2568 [DOI] [PubMed] [Google Scholar]