

Graphical abstract

Keywords: Modulation of protein–protein interactions, Drug discovery, Interface hot-spot residues, Protein docking simulations, Cavity detection, Molecular dynamics
Abstract
Protein-protein interactions play an essential role in many biological processes, and their perturbation is a major cause of disease. The use of small molecules to modulate them is attracting increased attention, but protein interfaces generally do not have clear cavities for binding small compounds. A proposed strategy is to target interface hot-spot residues, but their identification through computational approaches usually require the complex structure, which is not often available. In this context, pyDock energy-based docking and scoring can predict hot-spots on the unbound proteins, thus not requiring the complex structure. Here, we have devised a new strategy to detect protein–protein inhibitor binding sites, based on the integration of molecular dynamics for the generation of transient cavities, and docking-based interface hot-spot prediction for the selection of the suitable cavities. This integrative approach has been validated on a test set formed by protein–protein complexes with known inhibitors for which complete structural data of unbound molecules and complexes is available. The results show that local conformational sampling with short molecular dynamics can generate transient cavities similar to the known inhibitor binding sites, and that docking simulations can identify the best cavities with similar predictive accuracy as when knowing the real interface. In a few cases, these predicted pockets are shown to be suitable for protein–ligand docking. The proposed strategy will be useful for many protein–protein complexes for which there is no available structure, as long as the the unbound proteins do not deviate dramatically from the bound conformations.
1. Introduction
Protein-protein interactions (PPIs) are involved in the majority of diseases, and thus are essential to understand, prevent and correct pathological situations. Indeed, experimental studies show that many pathological mutations can affect protein–protein interactions, either by disrupting the entire interaction network of the mutated protein, or by specifically affecting some interactions [1], [2], [3]. In this context, PPIs emerge as attractive targets for drug discovery, and the field is shifting the focus of target identification and characterization from individual proteins to interaction networks [4]. Recent examples of protein–protein interfaces as potential therapeutic targets are the mitochondria–endoplasmic reticulum contact sites (MERCs), which are involved in different neurodegenerative and metabolilc disorders and cancer [5], or the disassembling of the trimeric structure of SARS-CoV-2 spike glycoprotein, which has been proposed as a therapeutic strategy against COVID-19 [6]. Protein-protein interactions have been successfully modulated with different apporaches, such as peptidomimetics [7], peptide aptamers [8], nucleic acid aptamers [9], or nanobodies [10]. However, modulation of PPIs with small molecules is perhaps the most sought way to contribute to new therapeutic developments, and several strategies have been reported for this purpose [11]. A variety of small-molecule inhibitors of PPIs have been identified, most of them by optimization of peptides or natural ligands, fragment-based, high-throughput screening, or rational design [12], [13], [14]. However, the path from a hit to a therapeutic drug is challenging. Indeed, few small-molecule PPI inhibitors have already been approved by FDA or are in clinical trials [2], [15].
Computational identification of small molecules that can modulate PPIs faces important challenges. A major difficulty is the absence of natural pockets in protein–protein interfaces that are ready to bind a small molecule. Contrary to standard ligand design, which usually aims to target a protein cavity (e.g. enzyme active site) that is often well defined and characterized from the structural and even energetic point of view, small-molecule modulators of protein–protein interactions need to target a protein–protein interface that does not usually contain obvious cavities [16], [17]. Indeed, analysis of available structures of PPI inhibitors bound to one of the partner proteins shows that these PPI inhibitor binding sites are less clearly defined than those of enzyme inhibitors [18]. Moreover, in the majority of cases these cavities need to be identified in the 3D structure of the unbound proteins or in that of the protein–protein complex, which is even more challenging, since these pockets may show large conformational rearrangements to adapt their shape and physicochemical environment to a potential inhibitor and thus remain mostly hidden in the unbound or bound-to-protein states. This imposes a real limitation to the use of ligand identification tools that are based on single protein structures, such as small-molecule docking or virtual screening, which usually have a rather limited exploration of the protein conformational space. Therefore, a dynamic view of protein–ligand recognition is essential here. Indeed, small-molecule ligands can show a large conformational diversity to maximize complementarity with different surface pockets, which in turn can exhibit significant variability in their geometrical and physicochemical environments [19], [20], [21]. This claims for the necessity of a thorough conformational analysis of the interacting molecules when analyzing potential ligand-binding sites in protein–protein interaction surfaces. In this context, conformational sampling with computational molecular dynamics (MD) in water and other solvent mixtures has been reported to identify transient cavities at protein–protein surfaces that are similar to the known PPI inhibitor binding sites [16], [22], [23]. More recently, biased simulations were able to generate protein fluctuations at specific surface sites leading to PPI-inhibitor pockets [24].
However, among the different transient cavities generated across a given protein–protein interface, it could be difficult to select a suitable cavity for ligand binding, which can be optimally located to compete with a protein interaction [17], [25]. Protein-protein complex structures usually have large interfaces [26] that show high geometrical and physicochemical complementarity [27], [28], and the problem is to identify which transient cavity could be optimal for binding a small molecule capable of disrupting this network of tightly bound interface side-chains [29], [30], [31]. Regarding this, it has been shown that residue composition of protein–protein interfaces is not homogeneous. There are usually a few amino acids (so-called “hot-spots”) that contribute to most of the free energy of binding [32]. Targeting such hot-spot residues with a small-molecule could have significant impact on a protein–protein interaction, which can be exploited for the discovery of PPI modulators [33]. Indeed, the knowledge of such hot-spots has been shown to help identifying transient pockets in interleukin-2 complexes [34]. But the identification of hot-spots is not trivial. There is experimental information on the energetic impact of mutations for a limited number of cases [35], but for large-scale applications a variety of bioinformatics approaches have been reported, from statistical analyses to molecular modeling and energetic calculations. Several studies found that hot-spots are enriched in arginine, tyrosine, and tryptophan, whereas leucine, serine, threonine, and valine are less frequent [36], [37]. The total number of hot-spots in a complex is proportional to the interface size, and they are usually found at the center of the contact interface [17], [37], [38].
Lastly, an important limitation is that the identification of suitable cavities and hot-spots at protein–protein interfaces, as above mentioned, requires the 3D structure of the protein–protein complex and/or the precise location of the interface residues, and this information is not available for the majority of interactions. Indeed, at the Protein Data Bank (PDB) [39] there are available 3D structures for only around 7,500 interactions between human proteins (Interactome3D, https://interactome3d.irbbarcelona.org/, 2020_1 version) [40], a small fraction of the estimated total number of PPIs in human, ranging between 130,000 [41] and 650,000 [42] interactions. In this scenario, protein–protein complexes with no available structure can be computationally modelled by a variety of template-based modeling [43], [44] and in silico docking approaches [45], [46], [47], [48], [49], [50], [51], [52]. Among the docking methods using energy-based scoring, pyDock [53] has shown excellent predictive results in the most recent CASP-CAPRI and CAPRI assessment experiments [54], [55], and can also be used for the identification of interface residues and hot-spots [56], [57]. Actually, this is one of the few methods that can identify hot-spots on two specific interacting proteins without requiring the 3D structure of the protein–protein complex. An alternative approach is computational solvent mapping, which can identify druggable hot-spots in protein–protein interfaces on unbound protein structures with minimal side-chain sampling [58].
Here, we have systematically explored the application of these docking-based interface and hot-spot predictions to the selection of transient cavities generated by local conformational sampling with MD at protein–protein interfaces. The results show that computational hot-spot predictions can help to identify PPI-inhibitor binding sites on protein surfaces in cases in which the structure of the protein–protein complex is not available.
2. Materials and methods
2.1. Benchmark set
Benchmark cases were extracted from structural databases of PPIs with known small-molecule modulators: TIMBAL version 1 [59] and 2P2I version 1 [60]. For each PPI, we defined the receptor as the protein that was known to bind the PPI inhibitor, and the ligand as the partner protein in the PPI. We selected a total of nine non-redundant PPIs with available 3D structure for the complex between the small-molecule modulator and one of the interacting proteins (the receptor), as well as for the protein–protein complex and for the entire unbound proteins (Table 1; see Table 1 SI for extended information). We defined redundant PPIs as those in which both receptor and ligand molecules had sequence identity ≥ 30% with respect to the corresponding receptor and ligand molecules in another PPI. A larger dataset of 14 complexes with known PPI inhibitors and available structure for the unbound proteins has been recently reported [61], but there are two major differences with our set here: in that study a larger sequence identity threshold (>95%) was applied to remove redundancies, and we have removed from our study cases in which the structure of the unbound entire protein (not just a peptide) is not available.
Table 1.
Structural data of PPIs with known modulators (PDB codes and chain IDs).
| PPI | protein–protein complex PDB | receptor PDB | ligand PDB | PPI-inhibitor PDB |
|---|---|---|---|---|
| Bcl-xL/Bak | 1BXL_A:Bc | 1R2D_Aa | 2YV6_A | 2YXJ_A:N3C |
| HPV E2/E1 | 1TUE_B:A | 1R6K_Aa | (2V9P_A)b | 1R6N_A:434 |
| IL-2/IL-2R | 1Z92_A:B | 1M47_A | (1Z92_B)b | 1PY2_A:FRH |
| HIV Integrase/LEDGF | 2B4J_A:D | 3L3U_A | 1Z9E_A | 3LPU_A:976 |
| MDM2/p53 | 1YCR_A:Bc | 1Z1M_A | 2K8F_B | 4ERF_A:0R3 |
| XIAP BIR3/Caspase | 1NW9_A:B | 1F9X_A | 1JXQ_A | 1TFT_A:997 |
| XIAP BIR3/Smac | 1G73_D:A | 1F9X_A | 1FEW_A | 2JK7_A:BI6 |
| TNFR1A/TNF-β | 1TNR_R:A | 1EXT_A | (1TNR) _A b | 1FT4_A:703 |
| ZipA/FtsZ | 1F47_B:Ac | 1F46_A | (2VAW_A)b | 1S1J_A:IQZ |
Missing loops were built with Modeller 9v10.
Structural model built with Modeller (template PDB ID is indicated in brackets).
The structure of the complex between the receptor protein and the ligand protein is not available, but there is structure for a protein-peptide complex (while the structure for the entire unbound ligand is available).
Six of the nine benchmark cases are related to human signaling pathways involved in cell growth and death. Bcl-xL plays a relevant role as a regulator of apoptosis, since its interaction with Bak activates the mitochondrial apoptotic process [62]. Several inhibitors are known for this interaction, many of them with available structure for the protein-inhibitor complex (Table 1 SI). The best known inhibitor is ABT-737 (813.4 Da), with IC50 of 9 nM, which has been approved by FDA for the treatment of cancer [63], [64]. X-linked inhibitor of apoptosis protein (XIAP) is also an important regulator of apoptosis. XIAP BIR3 domain binds caspase 9 to prevent the formation of its active dimeric form, thus inhibiting its enzymatic apoptotic initiator activity. XIAP activity is inhibited by binding of Smac protein, which promotes apoptosis [65], [66]. Several small molecules are known to target XIAP, which can inhibit specifically the interaction with caspase (e.g. PubChem CID: 5388929; 534.7 Da; Kd 5 nM) or with Smac (e.g. BI6; 494.6 Da; Ki 67 nM). MDM2 inhibits p53 mediated cell cycle arrest and apoptosis by binding its transcriptional activation domain, which also promotes the nuclear export of p53 [67]. A known inhibitor is 0R3 molecule (478.4 Da), with IC50 1.1 nM. Another benchmark case involved in apoptosis is the functional homo-trimeric form of TNFR1A (also known as TNFR1; gene TNFRSF1A) bound to the homo-trimeric cytokine tumor necrosis factor-beta TNF-β (also known as TNFB; gene LTA), which also binds to other proteins, such as TNFBR (also known as TNFR2; gene TNFRSF1B) or HVEM (gene TNFRSF14) [68]. A known inhibitor for this interaction is molecule 703 (PubChem CID 4470566, 456.5 Da), with IC50 270 nM. Another case involving cell growth and death is ZipA, an essential cell division protein that binds FtsZ to stabilize it [69]. Several inhibitors are known for this interaction (e.g. WAC, IQZ, WAI, or CL3, with molecular weights ranging from 240.3 to 423.9 Da). Biological activity is only available for WAI (Kd 12.0 μM) and CL3 (Kd 83.1 μM).
The remaining three benchmark cases are related to viral infection and immune response. HIV integrase binds human lens epithelium-derived growth factor (LEDGF), a transcriptional coactivator involved in neuroepithelial stem cell differentiation and neurogenesis, which facilitates the virus replication and survival [70]. Several inhibitors are known for this interaction (Table 1 SI), being compound 976 (353.8 Da) the most active (IC50 1.37 μM). Another case is the E2 from human papillomavirus type 11 (HPV-11), which plays a role in the initiation of viral DNA replication, and interacts with E1 to improve the specificity of E1 DNA binding activity [71]. There is only one inhibitor for this interaction with available complex structure (PDB ID: 1R6N, molecule 434; PubChem CID 5287508), with Kd 40 nM. This is a large molecule (608.5 Da) formed by several aromatic and carbonil groups, 2 chloride groups, and a thiadiazole group. Another case is Interleukine-2 (IL-2), which acts as a central regulator of the immune response, by binding to the hetero-trimeric IL-2 receptor (IL-2R) and stabilizing this functional oligomeric state [72]. A known inhibitor for this interaction (IC50 60 nM) is FRH (662.56 Da), also quite a large and flexible molecule.
2.2. Surface cavity detection
We applied fpocket [73] (http://fpocket.sourceforge.net/) to identify surface cavities on unbound protein structures and MD-based conformational models (see next section). We analyzed the pockets predicted by fpocket, ranked according to the provided score, which are based on five descriptors: the normalised number of alpha spheres, the normalised mean local hydrophobic density, the normalised proportion of apolar alpha sphere, the polarity score, and the alpha sphere density [73]. In addition, the druggability score provided by fpocket, a multiparametric descriptor previously trained as a bimodal predictive model on available structures of known druggable and nondruggable proteins [74], was also used to filter the cavities predicted on MD conformers (see Results). Volumes of the pockets were computed by ICM browser (www.molsoft.com) using the coordinates of the pocket as provided by fpocket.
To evaluate the performance of fpocket for pocket detection, the predicted pocket residues (based on the list of pocket atoms predicted by fpocket) were compared to those in the reference pocket of the complex-inhibitor complex structure (i.e. protein residues within 5 Å from the inhibitor), and different evaluation metrics were calculated: precision or positive predicted value (PPV), and sensitivity or coverage (COV) (eq. (1)). We usually defined as correct predictions (hits) those ones with PPV 40% and COV 40% with respect to the real position of the residues in the inhibitor pocket.
| (1) |
where for each predicted pocket i, true positives (TP) are the predicted pocket residues that are also found in the reference pocket, false positives (FP) are the predicted pocket residues that are not found in the reference pocket, and false negatives (FN) are the real residues in the reference pocket that are not found in the predicted pocket.
2.3. Molecular dynamics and transient cavities detection
For each analyzed protein structure, ten nanoseconds (ns) of molecular dynamics (MD) simulations were carried out using AMBER10, with the purpose of evaluating whether fast local conformational sampling could be useful to generate transient cavities suitable for protein–protein inhibitors. The unbound structures were prepared with pyDock setup module to use the same files in MD and later in pyDock docking, as described in next section [53]. In some cases, protein structure from available PDB (Table 1) was modified for more realistic conditions (see details in Table 1 SI). This setup step removed all hydrogen atoms. The topology and coordinates of the receptors were obtained by using Force Field 99 and general AMBER force field (GAFF). The water molecules were added keeping their coordinates as in the x-ray structures. A fast minimization with cartesian restraints (100 steps) was performed to remove severe clashes, followed by minimization with explicit solvation (100 steps). Each receptor was embedded in a solvated system within a periodic truncated octahedron box (water solvent TIP3P model), and 150 mM NaCl was added into the system. Lastly, a fast solvent minimization (1000 steps) was performed with a restraint mask of waters and ions. Then equilibration was performed at constant volume, using a 12 Å non-bonding cutoff. The equilibration process started by running 120 ps with protein constraints (50 kcal/mol·Å2, from 0 to 300 K) using Langevin dynamics (LD). The next 40 ps restraints were reduced from 50 to 25 kcal/mol·Å2, and the next 40 ps from 25 to 10 kcal/mol·Å2 at constant pressure. Finally, restraints were reduced to 5 kcal/mol·Å2 including backbone atoms, followed by 20 ps with backbone restraints (1 kcal/mol·Å2), and 60 ps with no restraints using LD, at 300 K. After equilibration, 10 ns MD were performed at 300 K at constant pressure, using explicit solvation (in a periodic truncated octahedron box of TIP3P water model), with a collision frequency of 0.2 ps−1 excluding bonds involving hydrogen atoms, and 2 ps of relaxation time. Then for practical purposes, 1000 conformational states were systematically randomly selected out of the 10,000 snapshots from MD trajectories (starting from first MD snapshot, every 10th one was taken), and they were analyzed using fpocket, focusing on the best-scoring pockets predicted for each of these conformations.
2.4. Docking simulations and hot-spot predictions
We applied pyDock docking and scoring method [53] to the unbound structures of the benchmark cases analyzed here. This docking approach consists in a two-step procedure in which docking orientations are generated with the FFT-based approach ZDOCK 2.1 [75], and they are later scored by an energy-based scoring function, composed of desolvation, electrostatics and van der Waals energy terms. The results of docking were used to compute Normalized Interface Propensity (NIP) values for every protein residue, which describes the normalized (expected over random) frequency in which a given residue is found at the docking interface in the best-ranked 100 docking orientations, as previously reported [56], [57]. The predicted hot-spots are defined as those residues with NIP ≥ 0.2. This cutoff was previously found to provide a precision of around 70% for the prediction of interface hot-spots on unbound structures, as benchmarked in a set of experimental hot-spot values [56], [57]. The interacting proteins used in docking were defined according to the biological assembly of the available protein–protein complexes (Table 1 SI). All cases are hetero-dimeric complexes, except the hetero-tetrameric HIV integrase/LEDGF complex, which had 2A:B2 stoichiometry, with two symmetric hetero-dimeric interfaces, and was thus treated as hetero-dimer. According to this, all unbound proteins were treated as monomers independently of their biological unit annotation. We used ICM browser (http://www.molsoft.com) to visualize structures, compute atom distances, select atoms within a given distance, align and superimpose structures.
2.5. Protein-ligand docking
Protein-ligand was performed by Glide XP [76] and rDock [77], in order to evaluate the suitability of the identified inhibitor pockets for their use in docking. Schrödinger software platform (https://www.schrodinger.com/) was used for the preparation of proteins with Protein Preparation Wizard [78], and that of ligands with LigPrep [79] The OPLS2005 [80] forcefield was used for the preparation of ligands in Glide. The same prepared protein and ligand structures were used for the docking executions with rDock. Receptor grids were generated from the inhibitor pockets (either the predicted ones or taken from the protein-inhibitor complexes). In rDock, the center of mass of the residues in a given pocket was used to set the center of the docking box whose side length was defined as the distance between this center to the farthest atom of the pocket, plus 5 Å (radial distance). The side of the cubic OUTER box in Glide was defined as twice the side length of the docking box in rDock, while the INNER box in Glide was kept at the default value (10 Ȧ3).
3. Results
3.1. PPI inhibitor pockets are more difficult to identify than general ligand pockets
Fpocket was previously reported to detect 94% and 92% of the pockets within the three best-ranked predictions (i.e. top 3 performance) on the holo and apo proteins, respectively, in a set of 150 protein–ligand complexes [73]. In order to confirm this performance on protein–ligand complexes, and compare it with that on protein-PPI inhibitor complexes, we applied here fpocket on the bound proteins (holo) of two different datasets: the first one composed of 102 protein–ligand complex structures from DUD-e database (http://dude.docking.org/) [81], and the second one with 264 non-redundant protein-PPI inhibitor complex structures from TIMBAL and 2P2I databases (involving 26 different proteins). For comparison purposes, we initially used the same evaluation metrics as in above mentioned study [73], where the Pocket Picker Criterion (PPC) defined a correctly predicted pocket as that one in which its center of mass lies within 4 Å from at least one atom of the ligand. Using this criterion, a correctly predicted pocket (hit) was found within the three best-ranked predictions in 89% of the DUD-e cases and in 69% of the 2P2I/TIMBAL cases (Fig. 1A). The performance on DUD-e cases is similar to the results described in the above mentioned study (94%). When analyzing only the best-scoring predicted pocket, success rate is 70% for DUD-e and 43% for 2P2I/TIMBAL (Fig. 1A). We note that the top 1 performance thus obtained here for DUD-e is slightly worse than the one obtained in the above mentioned study for the larger set of 150 cases (83%) [73].
Fig. 1.

Prediction of protein–ligand and protein-PPI inhibitor pockets by Fpocket. (A) Performance of fpocket on predicting known protein–ligand (DUD-e database) and protein-PPI inhibitor (TIMBAL/2P2I databases) pockets, considering the best-rank (orange) or 3 best-ranked (blue) predictions. (B) Positive predicted value (PPV) and coverage (COV) of best-scoring predicted pockets on DUD-e (left) and TIMBAL/2P2I (right) databases. The plot shows the best-scoring predicted pocket for each case, with those considered a hit by Pocket Picker Criterion (PPC) represented as * symbol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Since we were concerned that the PPC metrics to define correct pockets might not be too restrictive, we used other evaluation metrics like the PPV and coverage of the predicted pocket residues with respect to the reference ones. As can be seen in Fig. 1B, the large majority of pockets considered good by PPC have PPV and coverage over 40% (both in protein-ligands and in protein-PPI inhibitors). Protein-ligand pockets detected by PPC have better coverage with respect to the reference pockets. Based on the above findings, when using this criterion to define a correctly predicted pocket (PPV and coverage ≥ 40%), the top 1 success rate for DUD-e and 2P2I/TIMBAL is 68% and 53%, respectively, while the top 3 success rate is 82% and 74%, respectively. This follows the same trends as the PPC criterion, and confirms that the PPI inhibitor sites are more difficult to identify than the ligand ones, even when using the protein–ligand and -inhibitor complex structures for test purposes. Previous studies discussed about the importance of the pocket size and accessibility [11], [82]. In this context, PPI inhibitor binding sites tend to be large and flat, making it difficult to identify them with methods initially developed to detect deep cavities. Indeed, the predicted ligand cavities have in average higher fpocket scores than the predicted inhibitor cavities.
To explore the possibility that perhaps in the unsuccessful cases fpocket might have found the correct PPI inhibitor pockets but with poorer score (i.e. ranking > 3), we computed the predictive performance when considering all the predicted pockets for each case, independently of their ranking. In this case, fpocket can identify a correct pocket (i.e. PPV and coverage ≥ 40% with respect to the reference pocket) in 86% of the DUD-e cases, and in 77% of the 2P2I/TIMBAL cases, which is similar to the above shown top 3 performance, thus indicating that no additional correct pockets are found beyond the top 3 predicted pockets in the unsuccessful cases. To further evaluate potential problems in the scoring of PPI inhibitor cavities, we have restricted the predicted pockets to only those at the protein–protein interface (predictions were made on the protein coordinates from the protein–ligand or -inhibitor complexes, and no information from the protein–protein complexes was used until this last test). When selecting only the predicted pockets at the protein–protein interface, the top 1 predictive performance for DUD-e and 2P2I/TIMBAL cases improve up to 79% and 67%, respectively, and the top 3 predictive performance up to 86% and 77%, respectively, obtaining values more similar to those when considering all predicted pockets independently of the ranking. This shows that knowledge of the protein–protein interface might be important to complement fpocket scoring and improve the predictions.
In addition to the above discussed difficulties in detecting PPI inhibitor cavities, there are also problems in the assessment of predictions. Perhaps the definition for a successful predicted pocket used in traditional ligand sites is not optimal for PPI inhibitor cases. Fig. 1 SI shows an example of a predicted pocket that englobes the real PPI inhibitor site. Therefore, it has good coverage, but its PPV is slightly below the cutoff (40%) used to define a correct prediction (PDB ID: 3VNG). In the same figure, there is another case in which PPC indicates unsuccessful prediction, but it is assessed as correct according to PPV and coverage criteria (PDB ID: 3U5L).
3.2. Identifying PPI inhibitor pockets on unbound proteins
After the above described general test on protein–ligand and -inhibitor complex structures, we aimed to perform a more realistic benchmark test by applying fpocket to identify surface cavities on the unbound proteins (apo) of the PPIs of our benchmark set (Table 1) and compare them with the reference pocket in the protein-PPI inhibitor complex. In most of the benchmark cases, there were several available structures with the protein bound to different inhibitors (Table 1 SI). For the initial evaluation of the predictions, we selected as reference the structure of the complex with the inhibitor with the highest experimentally reported inhibitory potency, based on the half maximal inhibitory concentration IC50, which indicates the concentration of compound required to inhibit the target protein–protein interaction by half, or on the inhibitory constant Ki. If these values were not available, the selection criteria was based on the dissociation constant Kd. All these values were taken as annotated in the PDB. As an example, Fig. 2A shows the best-scoring pocket predicted by fpocket in the unbound IL-2 structure, which is not located near the PPI inhibitor with the best IC50, not even at the known protein–protein interface. A second pocket is also predicted for this case, which is not located near the inhibitor site either (Fig. 2A SI). The predicted pockets with the best fpocket score for all the cases are shown in Fig. 3 SI. Only in one case (11% of the benchmark set; see Table 2) we can find a correctly predicted pocket (i.e. PPV and coverage ≥ 40%) (Fig. 4A1 SI). The predicted surface cavities have an average volume of 396 Å3, with values ranging from 193 Å3 (TNFR1A) to 599 Å3 (HIV Integrase). In the only successful case, HPV E2, the predicted cavity has a volume of 412 Å3, close to the average value. For comparison purposes, if we apply fpocket to the proteins bound to these PPI inhibitors (holo), a correctly predicted pocket is found in 33% of the cases (Fig. 4B SI; Table 2), which is better than when using the unbound proteins (11%), but still a poor performance that is comparable to that obtained on TIMBAL/2P2I databases described in the previous section (43%). Interestingly, the predicted inhibitor pockets in the holo structures have an average volume of 352 Å3, similar to the predicted surface cavities in the unbound structures, with values ranging from 132 Å3 (TNFR1A) to 659 Å3 (Bcl-xL). These predictive results confirm that PPI inhibitor pockets are not easy to identify, despite using the holo protein structures, and that they are even more difficult to predict on the unbound proteins. Nevertheless, to disregard the possibility that the inhibitor with the best IC50 might not be the most appropriate to be predicted, we compared the predicted pockets on the unbound proteins with respect to all the other known inhibitors for each system, and the predictive results do not improve (Fig. 4C SI; Table 2). Finally, we have also explored the pocket prediction on the proteins taken from the protein–protein complexes, to check whether they adopt some conformations that could facilitate pocket prediction, but while some cases can be now correctly predicted, the overall predictive rate (33%) is the same as when using the holo proteins bound to the inhibitors (Fig. 4D SI; Table 2). Binding energy calculations on these protein–protein complex structures show tight binding in most of the cases, with high shape (according to van der Waals values) and electrostatic complementarity (Table 2 SI), which can explain the difficulties in finding suitable pockets within the protein–protein interfaces. We should note that here, despite using the protein–protein complex structure for the predictions, we have not restricted the predicted pockets to be located only at the interface region, since our goal was to evaluate the impact of the different conformational states on the predictions.
Fig. 2.

Predicted pockets in IL-2 protein using different conformational states. IL-2 protein is shown in grey surface, with IL-2R partner protein in blue ribbon, and the inhibitor with the best IC50 in green. (A) Predicted pockets (best-scoring in orange, the other one in yellow) on the unbound IL-2. (B) 10 best-scoring pockets predicted on MD side-chain conformers generated from unbound IL-2 (best-scoring pocket in orange, the others in yellow). (C) 10 best-scoring pockets from MD, restricted to the known protein–protein interface (best-scoring pocket in orange, the others in yellow). (D) 10 best-scoring pockets from MD, containing ≥ 3 predicted hot-spots (best-scoring pocket in orange, the others in yellow). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
Success rates for the best-ranked predicted PPI inhibitor pocket by fpocket on different selected protein structures and filtering conditions.
| Protocol | Reference on inhibitor with best IC50 | Reference on all inhibitors |
|---|---|---|
| Unbound protein | 11% | 11% |
| Protein bound to inhibitor | 33% | 33% |
| Protein bound to protein | 33% | 33% |
| MD side-chain conformers | 11% | 11% |
| MD side-chain conformers + interface | 56% | 67% |
| MD side-chain conformers + hot-spots | 44% | 56% |
Overall, these results confirm that x-ray protein structures (either in their unbound states or from protein-protein complexes) do not have surface cavities that can be easy to identify and exploited as binding sites for protein–protein inhibitors. All this suggests that we need to consider conformational variability in the protein structures in order to identify suitable pockets for PPI inhibitors.
3.3. MD simulations can generate transient PPI inhibitor pockets
Molecular dynamics (MD) simulations were previously reported to identify transient pockets in unbound protein surfaces known to be involved in protein–protein interactions, which were not present in the crystal structure of the unbound proteins [16]. We wanted to apply this strategy to the proteins of our benchmark set in order to generate transient pockets that could be suitable for inhibitor binding. With this purpose, we performed local conformational sampling at the side-chain level by generating 10 ns MD trajectories from the unbound protein structures, from which 1,000 conformers were generated (see Methods). Fig. 5 SI shows the evolution of the root mean square deviation (RMSD) for the Cα atoms of protein and inhibitor binding residues with respect to the starting unbound structures along the MD trajectories. Fig. 1 SI shows root mean square fluctuation (RMSF) per residue, computed by CPPTRAJ implemented in AmberTools suite [83]. Binding residues do not seem to be clearly different in their dynamic behaviour with respect to the rest of the protein. Then we used fpocket to identify surface cavities in all these conformers. For each conformer, the pocket with the best fpocket score was selected, as we did for the unbound, holo, or protein–protein complex single structures, but in this case, since we considered all the 1,000 conformers, we obtained a total of around 1000 predicted pockets per protein (for a few conformers, no pocket was predicted). The fpocket score indicates the capacity of the pocket to bind a small-molecule [73], but it does not include the druggability of the pocket. Thus, we took the best-scoring pocket from every conformer, and the resulting set of pockets was filtered to keep only the 100 pockets with the best druggability score as defined by fpocket. Finally from these, we selected the pocket with the best fpocket score. As an example, Fig. 2B shows the 10 best-scoring pockets predicted from the MD-based conformers generated from the unbound IL-2 (predicted pockets detailed in Fig. 2B SI). The best-scoring pocket (in orange) is not located at the PPI inhibitor site, but some of the top 10 pockets are nearby. The performance of pocket prediction on MD-based conformers for all the benchmark cases is shown in Fig. 4E SI, in comparison with all known PPI inhibitors. Correctly predicted pockets are found only in 11% of the cases (Table 2), yielding the same performance as with the unbound protein structures. Despite being able to generate suitable pockets with MD-based local conformational sampling in some of the cases (see for instance Fig. 2B SI), they are not identified with the default fpocket scoring tools.
The problem is that the transient cavities are found accross the entire protein surface, while we are mostly interested on those cavities at the protein–protein interfaces (apart from possible allosteric effects, one would expect that a small-molecule modulator would affect a PPI only if it is bound at the interface). Therefore, we analyzed whether knowing the location of the protein–protein interface could help to improve the predictions. This information is known in the cases of our benchmark, so we filtered the MD-based cavities to select only those in which ≥ 40% of the pocket residues are located at the protein–protein interface (defined as those residues within 5 Å inter-atomic distance from the partner protein). The impact of including the information of the protein–protein interface can be visualized in our IL-2 example, in which now the best-scoring pocket is correctly predicted (Fig. 2C; see more detail in Fig. 2C SI). The benefit of including the protein–protein interface information is clear for many of the benchmark cases: under this assumption, the best-scoring pocket would be correctly located at a known inhibitor binding site in 67% of the cases (Fig. 4F SI; Table 2). This indicates that, although molecular dynamics can generate transient binding pockets that could be suitable for binding PPI inhibitors, such pockets are still difficult to identify unless we know the protein–protein interface location. The main difficulty here is that the 3D structure of the complex is available only for a small fraction of all possible protein–protein interactions (see Introduction), which limits its applicability in real cases. In this context, we will analyze in the next sections whether computational prediction of protein interfaces and structural modeling of protein–protein interactions can help to identify the correct pockets among the many ones predicted from MD side-chain conformers in cases in which the structure of the protein–protein complex is not available.
3.4. Computational docking can identify interface and hot-spot residues
As we discussed above, in realistic situations in which the location of the protein–protein interface is not known, it would be helpful to rely on computational tools for the prediction of the interface residues. Among the several reported methods, we have explored here the use of pyDock, which can identify interface and hot-spot residues on unbound proteins based on docking calculations (see Methods). First, we tested the capabilities of this method for the prediction of interface hot-spots in our benchmark set. For this, docking orientations were generated with pyDock from the unbound proteins for each target PPI, and the 100 lowest-energy orientations were used to calculate NIP values for all residues in each interacting protein of the PPI, as previously described [57]. Fig. 3 shows the docking-based NIP values for the unbound proteins known to bind PPI inhibitors in the benchmark set. The predicted hot-spot residues are defined as those with NIP ≥ 0.2 (see Methods). The number of predicted hot-spots in the unbound proteins of our benchmark set ranges from 0 (ZipA) to 21 (HPV E2) (Table 3). As can be seen in Fig. 3, in many of the cases the predicted hot-spot residues are located at the interface with the partner protein in the target PPI. We have quantified in Table 3 the number of predicted hot-spot residues that are located at the known protein–protein interfaces (i.e. within 10 Å from any atom of the partner protein). In general, 55% of the predicted hot-spots are located at the protein–protein interface. Moreover, in six out of the nine benchmark cases, more than half of the predicted hot-spot residues are located at the protein–protein interface.
Fig. 3.

Docking-based hot-spot predictions. Protein residues are colored by NIP value, resulting from the docking calculations on the unbound proteins known to bind PPI inhibitors. For comparison, the partner protein is shown in green ribbon. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 3.
Docking-based prediction of hot-spot residues compared to real interfaces.
| Protein target | Predicted hot-spots (HS) | Predicted HS located at protein–protein interfaces (%)1 | |
|---|---|---|---|
| Bcl-xL | 9 | 7 | (78%) |
| HPV E2 | 21 | 16 | (76%) |
| IL-2 | 4 | 4 | (100%) |
| Integrase | 16 | 0 | (0%) |
| MDM2 | 7 | 7 | (100%) |
| XIAP BIR3/caspase | 18 | 10 | (56%) |
| XIAP BIR3/smac | 19 | 11 | (58%) |
| TNFR1A | 14 | 4 | (29%) |
| ZipA | 0 | 0 | (0%) |
Predicted hot-spot residues located within 10 Å from any atom of the partner protein in the protein–protein complex structure (in brackets, percentage with respect to the total predicted hot-spots).
For some of the predicted hot-spot residues, there is experimental evidence in the literature. For instance, Bcl-xL F146 residue is predicted here as a hot-spot, which is confirmed in previous experimental studies [84], [85], [86]. Several hot-spots have been experimentally identified for E2 (Y19, Q24, E39, Y99 and E100) [87], and we successfully predict two of them (Y19 and E100). In IL-2, we successfully predict three hot-spots (R38, F42 and L72) among the experimentally identified ones (K35, R38, F42, K43, E62 and L72) [88]. Since the docking-based method generated hot-spot predictions on both interacting proteins for a given target PPI, we could also checked the predictive performance on the partner protein (i.e. not the one binding the PPI inhibitor): in some cases, we observed that the interface hot-spots in the partner protein were successfully predicted (data not shown), although they were not used here for the purpose of identifying PPI inhibitor binding sites.
Regarding the type of predicted residues, we find here, in agreement with previous observations, that NIP values are more likely to predict as hot-spots the aromatic phenylalanine, tyrosine and tryptophan residues, the polar lysine, arginine, glutamic acid and threonine residues, and the non-polar leucine residue. NIP values are less likely to predict as hot-spots the non-polar methionine, glycine, valine or alanine residues, and the polar glutamine residue.
3.5. Predicted hot-spot residues are critical to identify PPI inhibitor pockets
We found in previous sections that the use of information on the protein–protein interface is essential to reduce the number of candidate pockets from MD conformers and thus identify the correct pockets with higher precision. Despite this information is not available in the majority of protein–protein complexes, we have studied here whether using the predicted hot-spots from docking simulations could help to improve the identification of the predicted pockets. In our example protein IL-2, when the pockets predicted on MD side-chain conformers from the unbound state are filtered to select only those that contain at least three predicted hot-spot residues, the 10 best-scoring pockets are located at the protein–protein interface, and the best-scoring one is correctly predicted as close to the PPI inhibitor binding site (Fig. 2D; see more detail in Fig. 2D SI). These results are much better than those obtained directly from the MD conformers (Fig. 2B; Fig. 2B SI), and similar to the ones obtained when using the location of the known protein–protein interface (Fig. 2C; Fig. 2C SI). This strategy of selecting only MD-based predicted pockets containing ≥ 3 predicted hot-spots improved the general predictive performance for the benchmark set, obtaining correctly predicted pockets in 56% of the cases (Fig. 4; Table 2). The transient cavities predicted with this strategy have an average volume of 648 Å3, with values ranging from 222 Å3 (IL-2) to 998 Å3 (Bcl-xL). The volume of the transient cavities in the successful cases showed a variety of values: 206 Å3 (MDM2), 222 Å3 (IL-2), 752 Å3 (XIAP BIR3, with respect to smac binding) and 770 Å3 (XIAP BIR3, with respect to caspase binding). The predictive performance obtained by using the information from the predicted hot-spots is comparable with that obtained when using information of the known protein–protein interface (67%, see Section 3.4), which suggests that docking calculations with pyDock can be a helpful tool to locate PPI inhibitor binding sites in the absence of structural information on the protein–protein interfaces. For comparison, we revisited the pocket prediction on the unbound structures, where the selection of pockets based on the best fpocket score was correct only in 11% of the cases (see Section 3.2). We tested on the unbound structures the above mentioned strategy of selecting those pockets containing ≥ 3 predicted hot-spots, independently of their fpocket scores, but the results did not improve (11% success rate). This clearly shows that both MD-based side-chain conformational sampling and hot-spot predictions are required for an optimal predictive strategy.
Fig. 4.
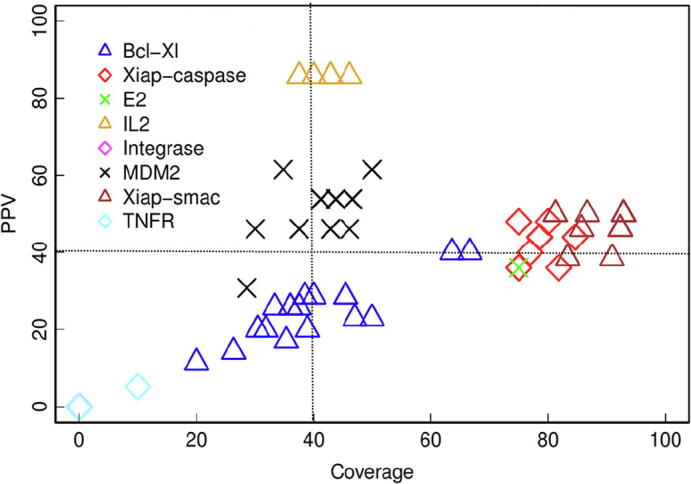
Assessment of the identification of PPI inhibitor pockets by integrating MD simulations and docking-based hot-spot predictions. PPV and coverage of the best-scoring predicted pocket on the MD-based side-chain conformers, which contain at least three predicted hot-spots.
3.6. Assessing the use of the predicted pockets in protein–ligand docking
As above discussed, we have devised a protocol integrating local conformational sampling with MD simulations and docking-based hot-spot predictions to identify transient cavities on protein–protein interfaces suitable for binding of small molecules. We already showed that this approach is able to predict the location of known PPI inhibitor binding sites in 56% of the cases of the benchmark set. Now, we should evaluate whether these predicted pockets are suitable for binding of PPI inhibitors, and can be used in protein–ligand docking simulations. In a recent study on a set of 19 complexes with known PPI inhibitors, low-resolution docking when using protein coordinates from protein-protein complex structures yielded similar success rate as when using the holo proteins, and significantly better than for the unbound proteins [61]. Here we will evaluate the predicted transient pockets with high-resolution docking, a more realistic but challenging scenario.
As an initial test, we aimed to reproduce the known protein-PPI inhibitor binding modes by protein–ligand docking, starting from the structure of the protein bound (holo) to the inhibitor with the best IC50. For the sake of simplicity, the PPI inhibitor conformation was also kept as in the protein-PPI inhibitor complex structure. We used Glide [76] and rDock [77] protein–ligand docking programs, with different parameters. More specifically, we tested several grid size values in order to extend the original grids in up to 10 Ȧ, increased the number of docking poses up to 1000, and tried different force fields: the default Tripos 5.2 force field [89] in rDock, and the OPLS2005 [80] and the newest OPLS3 [90] force fields in Glide. Under these ideal conditions, Glide with OPLS2005 forcefield was the best choice, finding correct docking orientations (i.e. ligand RMSD < 2 Ȧ) within the 5 best-scoring docking models in 7 of the 9 cases, while rDock found correct models in only 3 cases (sampling during docking was extended to up to 1000 poses). According to these results, we decided to assess the use of Glide with the predicted pockets, in realistic conditions.
With this purpose, we applied Glide to the unbound proteins, using the pockets predicted by MD and hot-spot predictions, a more realistic situation. In this case, the performance was much worse. We can get reasonable models (ligand RMSD ≤ 4.0 Ȧ) within the 20 best-scoring docking poses in two of the benchmark cases, but for this we had to use the 10 best-scoring pockets. We should note that ligand RMSD was calculated after superimposing only the inhibitor binding residues, to minimize the effects of global conformational changes on the evaluation of the docked poses (i.e. superposition of the entire protein when the MD conformer and the holo structure have quite different conformation could make the docked ligand to appear in an apparent different orientation to that in the holo structure). However, if the shape of the binding pocket in the MD-based conformer is very different from that in the reference holo structure, the ligand RMSD could be still high due to a different orientation between the docked and the real inhibitor, even in the hypothetical case they have the same binding mode with respect to relevant residues within the pocket. In the case of IL-2, the rank 1 docking model obtained when using the best-scoring pocket from MD and hot-spot prediction (Fig. 5B) has a conformation that is slightly different (ligand RMSD 5.2 Ȧ) from that of the PPI inhibitor in the complex structure (Fig. 5A), but this can be due to the different shape adopted by the transient pocket. When using the pocket ranked 9th from MD and hot-spot prediction, the best-RMSD docking model is closer to the reference (ligand RMSD 3.5 Ȧ) (Fig. 5C). But in general, it seems that the transient pockets generated by MD and selected by the predicted hot-spots, even if they are well located at the PPI inhibitor sites, their conformation is not optimal for ligand docking.
Fig. 5.

Docking of inhibitor on the predicted cavities of IL-2. IL-2 is shown in grey surface, and the different binding modes of the inhibitor are shown as ball and sticks. (A) IL-2 bound to FRH inhibitor (PDB ID: 1PY2). (B) Best-scoring docking model by Glide, using the 1st ranked pocket from MD and hot-spot prediction (from a MD snapshot at 2.241 ns). (C) The closest docking model to the reference in terms of ligand RMSD, obtained with the 9th ranked pocket from MD and hot-spot prediction (from a MD snapshot at 2.261 ns).
4. Discussion
4.1. Other possible criteria to select the pockets
In our protocol, we used the hot-spot predictions by pyDock to select the pockets predicted by fpocket from local conformational sampling with MD simulations. This helped to identify the correct pockets. In Fig. 7 SI we show the PPV values for the best-scoring pockets in the 1000 MD conformers as a function of fpocket score. Since this score alone is not able to discriminate the correct pockets in some cases, the addition of a filtering step based on the predicted hot-spots help to improve the predicted pockets.
We wanted to further explore other possible criteria to select the MD-based pockets. For instance, perhaps the size of the pocket in terms of number of residues could help to identify the cavities more suitable for inhibitor binding. In Fig. 8 SI we show the PPV of the MD-based predicted pockets for every case, as a function of the number of residues of each pocket. While in some cases (e.g. XIAP interacting with caspase, or the same protein interacting with smac) the largest pockets are good in terms of coverage, they have PPV < 40%. So in general, the size of the pocket does not help to identify a successful pocket. This is consistent with the fact that PPI inhibitor pockets are in average smaller (15.0 ± 6.2 residues; average and SD computed on pockets in TIMBAL/2P2I databases) than traditional ligand binding sites (23.7 ± 6.4 residues; average and SD computed from pockets in DUD-e set).
In our protocol, we finally sorted the best-scoring predicted pockets from MD by druggability score, selecting only the top 100 pockets according to that score. We also explored the inclusion of druggability score as a filter, with different cutoffs. In Fig. 9 SI we show the effect of using a druggability cutoff of > 0.7%, which is almost not affecting the predictions.
4.2. Lessons from unsuccessful predictions
Unsuccessful cases might be related to conformation and/or oligomerization issues. For instance, the TNFR1A/TNFB complex, besides the difficulty of its large protein–protein interface and the large difference between the unbound conformation and that in the complex with the inhibitor (Table 3 SI), the main challenge is that the functional homo-trimeric form of TNFB is interacting with the homo-trimeric form of TNFR1A in addition to other proteins [68]. Since we docked TNFR1A and TNFB as monomers, and did not consider the rest of interacting proteins, the docking calculations might not be fully accurate (indeed, most of the predicted hot-spots for this case are not located at the studied protein–protein interface, perhaps because they are part of the interface with the other functional interactions). Moreover, the known PPI inhibitors bound to TNFR1A are only partially located at the interface with TNB. We could speculate that the inhibitor may also affect the interaction with the other partner proteins, which indirectly would disrupt TNFR1A/TNFB interaction.
Another unsuccessful case is Bcl-xL. This is another example in which different oligomerization states and conformational arrangements can critically affect the predictions. The structure of this protein in complex with Bak has been determined by NMR (PDB ID: 1BXL), and the unbound structure by x-ray crystallography (PDB ID: 1R2D). In the latter, there is a long missing loop after the N-terminal α-helix, which we modeled based on the NMR structure. In a newer x-ray structure for the Bcl-xL/Bak complex (PDB ID: 5FMK), this N-terminal α-helix has been assigned to a different chain (Fig. 10A SI). The biological assembly assigned by the Protein Data Bank (PDB) is a hetero-tetramer, formed by two copies of Bcl-xL/Bak complex. Since the location of Bak does not seem to affect Bcl-xL homo-dimer interface, this suggests that unbound Bcl-xL could be a homo-dimer (this structure containts a few more Bcl-xL C-terminal residues that might explain the observed different oligomerization). Interestingly, the structure of the homologue Bcl-2 (PDB ID: 5VAX) has the same missing loop and the same N-terminal α-helix as unbound Bcl-xL (PDB ID: 1R2D), and the biological assembly for Bcl-2/peptide interaction is hetero-dimeric. Despite not participating directly in the interaction, this flexible N-terminal α-helix seems to have an essential role in the oligomerization of Bcl-xL. In addition, Bcl-xL shows a large conformational rearrangement when the hinge/turn region between α-helices 5 and 6 is extended, which induces a different dimer orientation (PDB ID: 4PPI) (Fig. 10B SI). Similar dimerization is found by domain swapping, which seems to be important to avoid interaction with p53 [85], [91]. The pocket predicted by our method is far from the known inhibitor of Bcl-xL/Bak interaction (Fig. 10C SI). This could be due to problems derived from the above discussed conformational flexibility of this protein. Indeed, in the unbound protein structure, there could be potential clashes between the inhibitor and the C-term α-helix, which is not present in the protein-PPI inhibitor complex structure (PDB ID: 2YXJ). Another reason is that our approach might be detecting another cavity of biological relevance. Actually, in the structure of Bcl-xL in complex with p53 (PDB ID: 2MEJ), p53 residues 176–193 are found in the predicted pocket region (Fig. 10D SI).
In the case of XIAP, although our approach correctly located the PPI inhibitor binding site, this predicted pocket was not sufficiently open to efficiently bind the small-molecule inhibitor. Perhaps in some cases, the local conformational sampling applied here is not sufficient for finding the relevant cavities, and longer MD simulations would be required. In other cases such as ZipA, TNFR1A or integrase, a major limitation was that the docking calculations provided incorrect hot-spots. In the case of ZipA/FtsZ interaction, the complex structure contained only residues 367–383 of FtsZ, but the entire FtsZ was modelled and used in docking, which perhaps caused problems in the predictions. Finally, the hetero-tetrameric HIV integrase/LEDGF complex was actually treated as hetero-dimer, since its stoichiometry was 2A:B2, with two equal hetero-dimeric interfaces. However, the inhibitor pocket was formed by the two homo-dimer integrase molecules, and the use of only one integrase monomer might have affected the pocket and hot-spot predictions. This shows once again the importance of considering the appropriate oligomerization state of the interacting proteins.
5. Conclusions
In this work we have shown how the integration of structure-based cavity detection, molecular dynamics, and docking-based prediction of hot-spots, can help to identify PPI inhibitor binding sites on the surface of unbound proteins. Two critical aspects here are: i) the use of MD-generated side-chain conformers, which produces thousands of transient cavities accross the protein surface, and ii) the use of protein–protein docking method to predict hot-spots, which can help to locate the interface pockets among all the generated by MD. The main advantage is that this strategy can be applied in the abscence of structural information on the protein–protein complex, a realistic situation in the majority of cases.
One of the limitations is the small size of the benchmark set. There are few cases for which the structure is available for the unbound proteins as well as for the protein–protein complex and the protein-PPI inhibitor complex, which are important for benchmarking new computational approaches. Other problems are related to the existence of large conformational rearrangements, which are not described by the local conformational sampling obtained by the short MD simulations used here, and to the limited knowledge of the possible oligomerization states in some of the proteins. Both are essential for accurate predictions.
Despite the difficulties, we propose here a protocol that can improve the detection of surface cavities for the identification of small-molecule modulators of protein–protein interactions. More work needs to be done in the conformational description of proteins and their interactions, as well as in the optimal use of these predicted pockets in ligand docking protocols, but this study can be helpful towards the goal of targeting PPIs of therapeutic interest.
CRediT authorship contribution statement
Mireia Rosell: Investigation, Methodology, Software, Writing - original draft. Juan Fernández-Recio: Conceptualization, Investigation, Funding acquisition, Project administration, Supervision, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was funded by the EU European Regional Development Fund (ERDF) through the Program Interreg V-A Spain-France-Andorra POCTEFA (grant PIREPRED), and by the Spanish Ministry of Science and Innovation (grant PID2019-110167RB-I00). M.R. is recipient of an FPI fellowship from the Severo Ochoa program.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.11.029.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sahni N., Yi S., Taipale M., Fuxman Bass J.I., Coulombe-Huntington J., Yang F. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015;161(3):647–660. doi: 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosell M., Fernández-Recio J. Hot-spot analysis for drug discovery targeting protein-protein interactions. Expert Opin Drug Discov. 2018;13(4):327–338. doi: 10.1080/17460441.2018.1430763. [DOI] [PubMed] [Google Scholar]
- 3.Rolland T., Tasan M., Charloteaux B., Pevzner S.J., Zhong Q., Sahni N. A proteome-scale map of the human interactome network. Cell. 2014;159(5):1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vidal M., Cusick M.E., Barabasi A.L. Interactome networks and human disease. Cell. 2011;144(6):986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magalhães Rebelo A.P., Dal Bello F., Knedlik T., Kaar N., Volpin F., Shin S.H. Chemical modulation of mitochondria-endoplasmic reticulum contact sites. Cells. 2020;9(7) doi: 10.3390/cells9071637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bongini P., Trezza A., Bianchini M., Spiga O., Niccolai N. A possible strategy to fight COVID-19: interfering with spike glycoprotein trimerization. Biochem Biophys Res Commun. 2020;528(1):35–38. doi: 10.1016/j.bbrc.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunningham A.D., Qvit N., Mochly-Rosen D. Peptides and peptidomimetics as regulators of protein-protein interactions. Curr Opin Struct Biol. 2017;44:59–66. doi: 10.1016/j.sbi.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colas P., Cohen B., Jessen T., Grishina I., McCoy J., Brent R. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature. 1996;380(6574):548–550. doi: 10.1038/380548a0. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J., Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16(3):181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caussinus E., Kanca O., Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat Struct Mol Biol. 2011;19(1):117–121. doi: 10.1038/nsmb.2180. [DOI] [PubMed] [Google Scholar]
- 11.Scott D.E., Bayly A.R., Abell C., Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov. 2016;15(8):533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- 12.Ran X., Gestwicki J.E. Inhibitors of protein-protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Current Opin Chem Biol. 2018;44:75–86. doi: 10.1016/j.cbpa.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arkin M.R., Tang Y., Wells J.A. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chemy Biol. 2014;21(9):1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grosdidier S., Totrov M., Fernández-Recio J. Computer applications for prediction of protein-protein interactions and rational drug design. Adv Appl Bioinform Chem. 2009;2:101–123. [PMC free article] [PubMed] [Google Scholar]
- 15.Mabonga L., Kappo A.P. Protein-protein interaction modulators: advances, successes and remaining challenges. Biophys Rev. 2019;11(4):559–581. doi: 10.1007/s12551-019-00570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eyrisch S., Helms V. Transient pockets on protein surfaces involved in protein-protein interaction. J Med Chem. 2007;50(15):3457–3464. doi: 10.1021/jm070095g. [DOI] [PubMed] [Google Scholar]
- 17.Wells J.A., McClendon C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450(7172):1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 18.Pérot S., Sperandio O., Miteva M.A., Camproux A.C., Villoutreix B.O. Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov Today. 2010;15(15–16):656–667. doi: 10.1016/j.drudis.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Stockwell G.R., Thornton J.M. Conformational diversity of ligands bound to proteins. J Mol Biol. 2006;356(4):928–944. doi: 10.1016/j.jmb.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Kahraman A., Morris R.J., Laskowski R.A., Thornton J.M. Shape variation in protein binding pockets and their ligands. J Mol Biol. 2007;368(1):283–301. doi: 10.1016/j.jmb.2007.01.086. [DOI] [PubMed] [Google Scholar]
- 21.Kahraman A., Morris R.J., Laskowski R.A., Favia A.D., Thornton J.M. On the diversity of physicochemical environments experienced by identical ligands in binding pockets of unrelated proteins. Proteins. 2010;78(5):1120–1136. doi: 10.1002/prot.22633. [DOI] [PubMed] [Google Scholar]
- 22.Eyrisch S., Helms V. What induces pocket openings on protein surface patches involved in protein–protein interactions? J Comp-Aid Mol Des. 2009;23(2):73. doi: 10.1007/s10822-008-9239-y. [DOI] [PubMed] [Google Scholar]
- 23.Seco J., Luque F.J., Barril X. Binding site detection and druggability index from first principles. J Med Chem. 2009;52(8):2363–2371. doi: 10.1021/jm801385d. [DOI] [PubMed] [Google Scholar]
- 24.Johnson D.K., Karanicolas J. Druggable Protein Interaction Sites Are More Predisposed to Surface Pocket Formation than the Rest of the Protein Surface. PLOS Comp Biol. 2013;9(3) doi: 10.1371/journal.pcbi.1002951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuller J.C., Burgoyne N.J., Jackson R.M. Predicting druggable binding sites at the protein-protein interface. Drug Discov Today. 2009;14(3–4):155–161. doi: 10.1016/j.drudis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 26.Lo Conte L., Chothia C., Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285(5):2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 27.Lawrence M.C., Colman P.M. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234(4):946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 28.McCoy A.J., Chandana Epa V., Colman P.M. Electrostatic complementarity at protein/protein interfaces. J Mol Biol. 1997;268(2):570–584. doi: 10.1006/jmbi.1997.0987. [DOI] [PubMed] [Google Scholar]
- 29.Basu S., Bhattacharyya D., Banerjee R. Self-complementarity within proteins: bridging the gap between binding and folding. Biophys J. 2012;102(11):2605–2614. doi: 10.1016/j.bpj.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banerjee R., Sen M., Bhattacharya D., Saha P. The jigsaw puzzle model: search for conformational specificity in protein interiors. J Mol Biol. 2003;333(1):211–226. doi: 10.1016/j.jmb.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 31.Yan Y., Huang S.-Y. Pushing the accuracy limit of shape complementarity for protein-protein docking. BMC Bioinf. 2019;20(25):696. doi: 10.1186/s12859-019-3270-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clackson T., Wells J.A. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 33.Geppert T., Bauer S., Hiss J.A., Conrad E., Reutlinger M., Schneider P. Immunosuppressive small molecule discovered by structure-based virtual screening for inhibitors of protein-protein interactions. Angew Chem Int Ed Engl. 2012;51(1):258–261. doi: 10.1002/anie.201105901. [DOI] [PubMed] [Google Scholar]
- 34.Metz A., Pfleger C., Kopitz H., Pfeiffer-Marek S., Baringhaus K.H., Gohlke H. Hot spots and transient pockets: predicting the determinants of small-molecule binding to a protein-protein interface. J Chem Inf Mod. 2012;52(1):120–133. doi: 10.1021/ci200322s. [DOI] [PubMed] [Google Scholar]
- 35.Jankauskaite J., Jiménez-García B., Dapkunas J., Fernández-Recio J., Moal I.H. SKEMPI 2.0: an updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics. 2018;35(3):462–469. doi: 10.1093/bioinformatics/bty635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee G.M., Craik C.S. Trapping Moving Targets with Small Molecules. Science) 2009;324(5924):213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bogan A.A., Thorn K.S. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280(1):1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 38.Grosdidier S., Fernandez-Recio J. Protein-protein docking and hot-spot prediction for drug discovery. Current Pharm Des. 2012;18(30):4607–4618. doi: 10.2174/138161212802651599. [DOI] [PubMed] [Google Scholar]
- 39.Westbrook J., Feng Z., Jain S., Bhat T.N., Thanki N., Ravichandran V. The Protein Data Bank: unifying the archive. Nucl Acids Res. 2002;30(1):245–248. doi: 10.1093/nar/30.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mosca R., Céol A., Aloy P. Interactome3D: adding structural details to protein networks. Nat Met. 2013;10(1):47–53. doi: 10.1038/nmeth.2289. [DOI] [PubMed] [Google Scholar]
- 41.Venkatesan K., Rual J.-F., Vazquez A., Stelzl U., Lemmens I., Hirozane-Kishikawa T. An empirical framework for binary interactome mapping. Na Met. 2009;6(1):83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stumpf M.P., Thorne T., de Silva E., Stewart R., An H.J., Lappe M. Estimating the size of the human interactome. Proc Natl Acad Sci USA. 2008;105(19):6959–6964. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirabello C., Wallner B. InterPred: A pipeline to identify and model protein–protein interactions. Proteins. 2017;85(6):1159–1170. doi: 10.1002/prot.25280. [DOI] [PubMed] [Google Scholar]
- 44.A. Baspinar E. Cukuroglu R. Nussinov O. Keskin A. Gursoy PRISM: a web server and repository for prediction of protein-protein interactions and modeling their 3D complexes Nucl Acids Res 42(Web 2014 Server issue):W285–289. [DOI] [PMC free article] [PubMed]
- 45.Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucl Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuntz I.D., Blaney J.M., Oatley S.J., Langridge R., Ferrin T.E. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161(2):269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 47.Katchalski-Katzir E., Shariv I., Eisenstein M., Friesem A.A., Aflalo C., Vakser I.A. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci USA. 1992;89(6):2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritchie D.W., Kemp G.J.L. Protein docking using spherical polar Fourier correlations. Proteins. 2000;39(2):178–194. [PubMed] [Google Scholar]
- 49.Fernández-Recio J., Totrov M., Abagyan R. Soft protein-protein docking in internal coordinates. Protein Sci. 2002;11(2):280–291. doi: 10.1110/ps.19202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dominguez C., Boelens R., Bonvin A.M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125(7):1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 51.Jiménez-García B., Roel-Touris J., Romero-Durana M., Vidal M., Jiménez-González D., Fernández-Recio J. LightDock: a new multi-scale approach to protein–protein docking. Bioinformatics. 2017;34(1):49–55. doi: 10.1093/bioinformatics/btx555. [DOI] [PubMed] [Google Scholar]
- 52.Moal I.H., Bates P.A. SwarmDock and the use of normal modes in protein-protein docking. Int J Mol Sci. 2010;11(10):3623–3648. doi: 10.3390/ijms11103623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng T.M., Blundell T.L., Fernandez-Recio J. pyDock: electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins. 2007;68(2):503–515. doi: 10.1002/prot.21419. [DOI] [PubMed] [Google Scholar]
- 54.Lensink M.F., Brysbaert G., Nadzirin N., Velankar S., Chaleil R.A.G., Gerguri T. Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins. 2019;87(12):1200–1221. doi: 10.1002/prot.25838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lensink MF, Nadzirin N, Velankar S, Wodak SJ: Modeling protein-protein, protein-peptide, and protein-oligosaccharide complexes: CAPRI 7th edition. Proteins 2020, (Epub ahead of print). [DOI] [PubMed]
- 56.Fernández-Recio J., Totrov M., Abagyan R. Identification of Protein-Protein Interaction Sites from Docking Energy Landscapes. J Mol Biol. 2004;335(3):843–865. doi: 10.1016/j.jmb.2003.10.069. [DOI] [PubMed] [Google Scholar]
- 57.Grosdidier S., Fernandez-Recio J. Identification of hot-spot residues in protein-protein interactions by computational docking. BMC Bioinf. 2008;9:447. doi: 10.1186/1471-2105-9-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kozakov D., Hall D.R., Chuang G.-Y., Cencic R., Brenke R., Grove L.E. Structural conservation of druggable hot spots in protein–protein interfaces. Proc Natl Acad Sci USA. 2011;108(33):13528. doi: 10.1073/pnas.1101835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Higueruelo A.P., Schreyer A., Bickerton G.R.J., Pitt W.R., Groom C.R., Blundell T.L. Atomic interactions and profile of small molecules disrupting protein-protein interfaces: the TIMBAL database. Chem Biol Drug Des. 2009;74(5):457–467. doi: 10.1111/j.1747-0285.2009.00889.x. [DOI] [PubMed] [Google Scholar]
- 60.M.J. Basse S. Betzi R. Bourgeas S. Bouzidi B. Chetrit V. Hamon et al. 2P2Idb: a structural database dedicated to orthosteric modulation of protein-protein interactions Nucl Acids Res 41(Database 2013 issue):D824–D827. [DOI] [PMC free article] [PubMed]
- 61.Belkin S., Kundrotas P.J., Vakser I.A. Inhibition of protein interactions: co-crystalized protein-protein interfaces are nearly as good as holo proteins in rigid-body ligand docking. J Comp-Aid Mol Des. 2018;32(7):769–779. doi: 10.1007/s10822-018-0124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chittenden T., Flemington C., Houghton A.B., Ebb R.G., Gallo G.J., Elangovan B. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. Embo J. 1995;14(22):5589–5596. doi: 10.1002/j.1460-2075.1995.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oltersdorf T., Elmore S.W., Shoemaker A.R., Armstrong R.C., Augeri D.J., Belli B.A. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 64.Lee E.F., Czabotar P.E., Smith B.J., Deshayes K., Zobel K., Colman P.M. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Diff. 2007;14(9):1711–1713. doi: 10.1038/sj.cdd.4402178. [DOI] [PubMed] [Google Scholar]
- 65.Shiozaki E.N., Chai J., Rigotti D.J., Riedl S.J., Li P., Srinivasula S.M. Mechanism of XIAP-Mediated Inhibition of Caspase-9. Mol Cell. 2003;11(2):519–527. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 66.Wu G., Chai J., Suber T.L., Wu J.-W., Du C., Wang X. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408(6815):1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 67.Brady M., Vlatković N., Boyd M.T. Regulation of p53 and MDM2 Activity by MTBP. Mol Cell Biol. 2005;25(2):545–553. doi: 10.1128/MCB.25.2.545-553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mauri D.N., Ebner R., Montgomery R.I., Kochel K.D., Cheung T.C., Yu G.L. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8(1):21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- 69.Mosyak L., Zhang Y., Glasfeld E., Haney S., Stahl M., Seehra J. The bacterial cell-division protein ZipA and its interaction with an FtsZ fragment revealed by X-ray crystallography. EMBO J. 2000;19(13):3179–3191. doi: 10.1093/emboj/19.13.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chylack L.T., Jr., Fu L., Mancini R., Martin-Rehrmann M.D., Saunders A.J., Konopka G. Lens epithelium-derived growth factor (LEDGF/p75) expression in fetal and adult human brain. Exp Eye Res. 2004;79(6):941–948. doi: 10.1016/j.exer.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 71.Abbate E.A., Berger J.M., Botchan M.R. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev. 2004;18(16):1981–1996. doi: 10.1101/gad.1220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goudy K., Aydin D., Barzaghi F., Gambineri E., Vignoli M., Ciullini Mannurita S. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol. 2013;146(3):248–261. doi: 10.1016/j.clim.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le Guilloux V., Schmidtke P., Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinf. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmidtke P., Barril X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J Med Chem. 2010;53(15):5858–5867. doi: 10.1021/jm100574m. [DOI] [PubMed] [Google Scholar]
- 75.Chen R., Weng Z. A novel shape complementarity scoring function for protein-protein docking. Proteins. 2003;51 doi: 10.1002/prot.10334. [DOI] [PubMed] [Google Scholar]
- 76.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for Protein−Ligand complexes. J Med Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 77.Ruiz-Carmona S., Alvarez-Garcia D., Foloppe N., Garmendia-Doval A.B., Juhos S., Schmidtke P. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLOS Comp Biol. 2014;10(4) doi: 10.1371/journal.pcbi.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sastry G.M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comp-Aid Mol Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 79.Schrödinger: LigPrep. LLC, New York, NY 2020.
- 80.Banks J.L., Beard H.S., Cao Y., Cho A.E., Damm W., Farid R. Integrated modeling program, applied chemical theory (IMPACT) J Comput Chem. 2005;26(16):1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mysinger M.M., Carchia M., Irwin J.J., Shoichet B.K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pérot S., Regad L., Reynès C., Spérandio O., Miteva M.A., Villoutreix B.O. Insights into an original pocket-ligand pair classification: a promising tool for ligand profile prediction. PLoS ONE. 2013;8(6):e63730. doi: 10.1371/journal.pone.0063730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roe D.R., Cheatham T.E. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theor Comp. 2013;9(7):3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 84.Moroy G., Martin E., Dejaegere A., Stote R.H. Molecular basis for Bcl-2 homology 3 domain recognition in the Bcl-2 protein family: identification of conserved hot spot interactions. J Biol Chem. 2009;284(26):17499–17511. doi: 10.1074/jbc.M805542200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee E.F., Fairlie W.D. The Structural Biology of Bcl-x(L) Intl J Mol Sci. 2019;20(9):2234. doi: 10.3390/ijms20092234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Campbell S.T., Carlson K.J., Buchholz C.J., Helmers M.R., Ghosh I. Mapping the BH3 binding interface of Bcl-xL, Bcl-2, and Mcl-1 using split-luciferase reassembly. Biochemistry. 2015;54(16):2632–2643. doi: 10.1021/bi501505y. [DOI] [PubMed] [Google Scholar]
- 87.Wang Y., Coulombe R., Cameron D.R., Thauvette L., Massariol M.-J., Amon L.M. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J Biol Chem. 2004;279(8):6976–6985. doi: 10.1074/jbc.M311376200. [DOI] [PubMed] [Google Scholar]
- 88.Wilson C.G.M., Arkin M.R. Small-molecule inhibitors of IL-2/IL-2R: lessons learned and applied. Curr Top Microbiol Immunol. 2011;348:25–59. doi: 10.1007/82_2010_93. [DOI] [PubMed] [Google Scholar]
- 89.Clark M., Cramer Iii R.D. Van Opdenbosch N: validation of the general purpose tripos 5.2 force field. J Comp Chem. 1989;10(8):982–1012. [Google Scholar]
- 90.Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J.Y. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput. 2016;12(1):281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 91.O'Neill J.W., Manion M.K., Maguire B., Hockenbery D.M. BCL-XL Dimerization by Three-dimensional Domain Swapping. J Mol Biol. 2006;356(2):367–381. doi: 10.1016/j.jmb.2005.11.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.