

Abstract
Developing safe and efficient delivery systems for therapeutic biomacromolecules is a long-standing challenge. Herein, we report a newly developed combinatorial library of cholesteryl-based disulfide bond-containing biodegradable cationic lipidoid nanoparticles. We have identified a subset of this library which is effective for protein and mRNA delivery in vitro and in vivo. These lipidoids showed comparable transfection efficacies but much lower cytotoxicities compared to the Lpf2k in vitro. In vivo studies in adult mice demonstrated the successful delivery of genome engineering protein and mRNA molecules in the skeletal muscle (via intramuscular injection), lung and spleen (via intravenous injection), and brain (via lateral ventricle infusion).
Keywords: lipidoid nanoparticle, stimuli responsive, protein delivery, mRNA delivery, genome engineering
Graphical Abstract
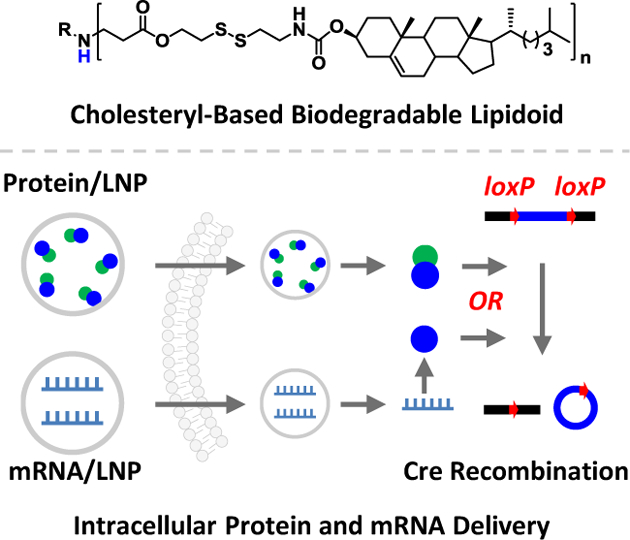
Intracellular delivery: a new combinatorial library of cholesteryl-based and reduction-responsive lipidoid nanoparticles can deliver genome engineering protein and mRNA molecules in vitro and in vivo. Using adult Ai14 mouse model, successful Cre-mediated gene recombination events were observed in skeletal muscle, brain, lung, and spleen through the local and systemic administrations.
Introduction
Nanoparticle-based delivery systems[1] have been demonstrated to be advantageous[2] for solubilizing drugs, improving their stabilities, prolonging blood circulation time, achieving site-specificity and controlled drug release, avoiding drug resistance mechanisms and facilitating cellular uptake.[3] Cumulatively, these effects serve to enhance the bioavailability and reduce the side-effects of the bioactive molecules. Materials including lipids and lipidoids,[4] synthetic polymers and biomacromolecules,[5] carbon and silica nanoparticles,[6] and metal and metal oxide nanoparticles have been developed for drug delivery.[7] Nanoparticles can be fabricated with a variety of sizes, shapes, and chemistries. The advantages as well as limitations of these delivery platforms have been summarized in recent reviews.[2–3, 8] Nevertheless, developing safe and efficient delivery systems for therapeutic agents remains a long-standing challenge.
Among all the nanocarriers, lipid and lipidoid nanoparticles (LNPs) have achieved the most success in research and clinical translation,[4a, 9] with FDA approved lipid formulation drugs such as Doxil[10] and the most recent Patisiran.[11] In the pursuit of developing new and improved LNP drug delivery carriers, the combinatorial library strategy has been demonstrated by us[12] and others,[13] to be an effective method to develop novel materials with unique delivery properties. Combinatorial strategies rely on the ability to generate a library of new materials containing a tremendous diversity of physicochemical properties, based on a relatively small set of starting chemical components.[13d] One advantage of the combinatorial strategy is that the addition of one single new chemical structure can result in dozens of novel delivery vehicles.[12a]
We previously reported a library of biodegradable LNPs for protein and RNA delivery, in which disulfide bond was incorporated into lipidoids’ linear alkyl tails.[14] Others have reported LNPs integrated with naturally-derived molecules (e.g. cholesteryl), covalently and non-covalently, for improved delivery efficacy.[15] As the delivery properties of synthetic LNPs are exquisitely sensitive to the chemical structure of the lipidoid, we thus hypothesized that by changing the lipid tail from a linear to a bulky structure could result in a new class of lipidoids with new self-assembly behaviors and different biological effects.[16] In this study, we synthesized a combinatorial library of cholesteryl- and disulfide bond-containing biodegradable LNPs. The newly developed LNPs outperformed the commercially available gold standard Lipofectamine (Lpf2k), previously reported alkyl linear tail-containing biodegradable lipidoids, and cholesteryl-based non-degradable lipidoids in vitro. We found that these new LNPs can deliver genome engineering protein and mRNA molecules in adult mice in vivo, through local or systemic administration routes. Interestingly, while we found a particular subset of LNPs which are consistently effective as delivery vehicles in vitro, our in vivo experiments have indicated that individual LNPs may have particular tendencies for delivery to particular organs. We present some evidence for this, although future studies will be necessary to further validate and improve this effect.
Results and Discussion
The new lipid tail, termed here as OCholB, which contains both a biodegradable disulfide bond and a cholesteryl moiety, was synthesized though a multi-step reaction (Figure S1). The structures of OCholB and its precursors were confirmed by NMR spectroscopy (Figure S2–5). Lipidoids were synthesized through the Michael addition reaction and named as R-OCholB (R stands for amine number; Figure 1A). New OCholB lipidoids were compared against alkyl linear tail-containing biodegradable lipidoids (R-O16B) and cholesteryl-containing non-degradable lipidoids (R-OChol) (Figure 1A).[14b] The synthetic routes and characterization of OChol and O16B tails are shown in Figure S6–S9. Structures of lipidoids were confirmed by MS (Figure S10–S13).
Figure 1.
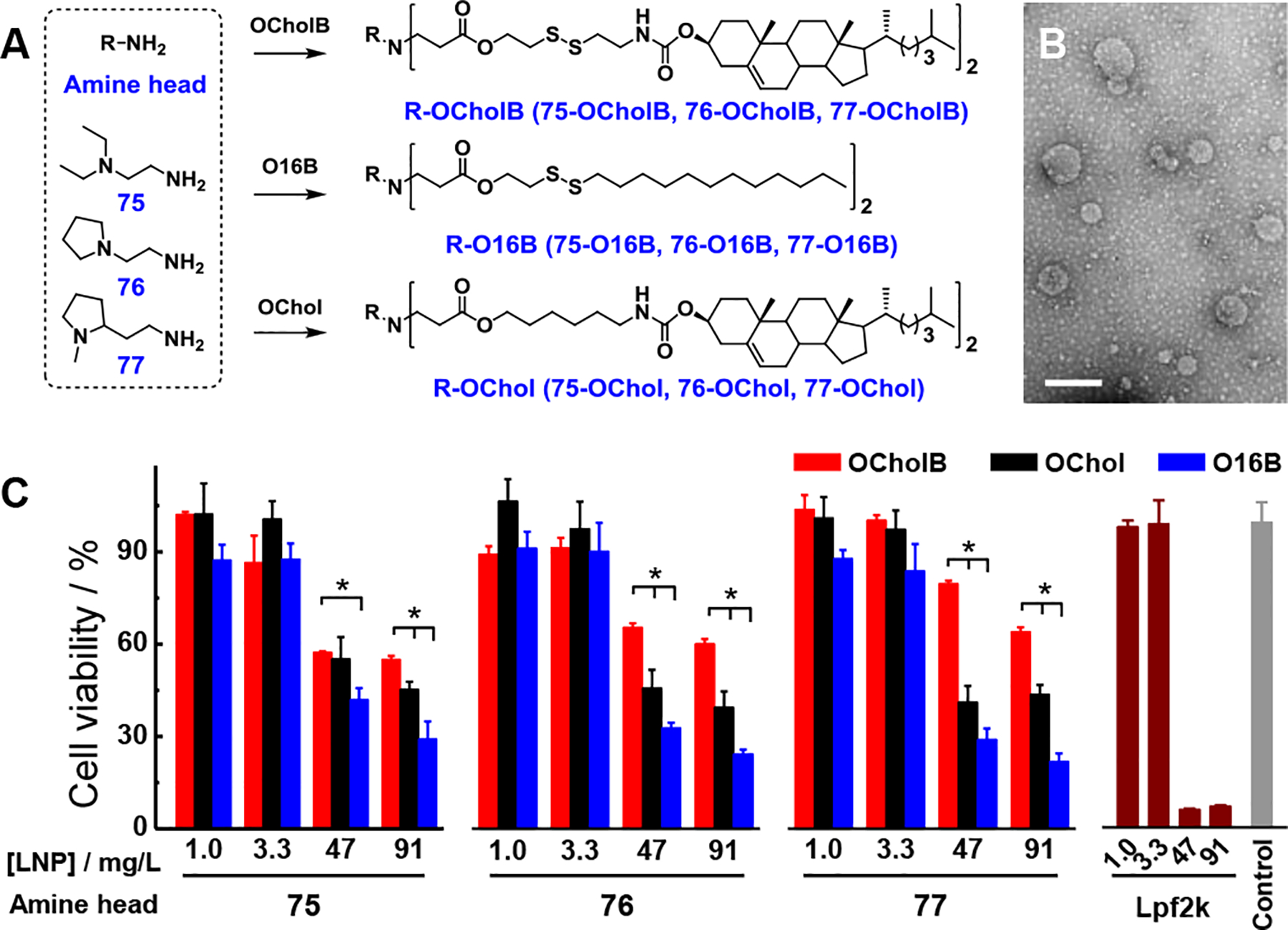
Synthesis, characterization, and cytotoxicity test of combinatorial libraries of lipidoid nanoparticles. (A) Chemical structures and synthetic routes employed for the preparation of OCholB-, O16B-, and OChol-tailed lipidoids. (B) TEM image of 77-OCholB LNPs. Scale bar = 200 nm. (C) Cell viability tests of OCholB, O16B, OChol, and Lpf2k nanoparticles against HeLa cells. N = 4, two-tailed student’s t-test, p < 0.05. Data points at 1.0 and 3.3 mg/L were collected 8 h after dose. Data points at 47 and 91 mg/L were collected 24 h after dose. All data points in this and other figures are shown as mean±SD.
As a proof-of-concept, we began with a small library containing only 3 amine head groups, and compared the lipids generated using these head groups with OCholB, O16B, and OChol tails. Nanoparticles were fabricated through a combination of ultrasonication and vortex procedure.[12a] It should be noted that microfluidics, extrusion, and other techniques could be helpful to optimize the size and distribution of LNPs. The average hydrodynamic diameters (<Dh>) and polydispersity indexes (PDI) of LNPs were determined by dynamic light scattering (DLS; Figure S14A–C). These results indicate that both the amine head (75, 76, and 77) and tail structures (OCholB, O16B, and OChol) affected the supramolecular self-assembly behaviors of lipidoids (see Supporting Information). Typical size distribution profiles of OCholB LNPs are shown in Figure S14D and morphologies were examined by TEM (Figure 1B and S15). Spherical vesicular structures (liposomes) were observed for all samples studied.
The in vitro biocompatibility of the new OCholB-based LNPs was examined and compared to that of OChol, O16B, and Lpf2k LNPs. As shown in Figure 1C, at the low dosage/short exposure time conditions ([lipidoid] = 1.0 or 3.3 μg mL−1, exposure time = 8 h), no LNP showed evident cytotoxicity. When the dosage and exposure duration were both increased ([lipidoid] = 47 or 91 μg mL−1, exposure time = 24 h), OCholB LNPs treated cells showed significantly higher viability comparing to those treated with OChol, O16B, and Lpf2k LNPs. The in vitro cytotoxicity test supports our hypothesis that the incorporation of cholesteryl through biodegradable linkage can improve the compatibility of lipidoids.
Inspired by these results, we then expanded the OCholB lipidoids library to include additional amine head groups (Figure 2A). After characterization of the lipidoids (Figure S16), LNPs were prepared (Figure S17 and S18). Except for 87-OCholB and 90-OCholB, liposomes (60–380 nm in size) were observed for all samples. The apparent size and PDI as determined by TEM micrograph appears to contradict the results obtained by DLS measurement, which is likely resulted from the drying/staining procedures in TEM sample preparation.[14b]
Figure 2.
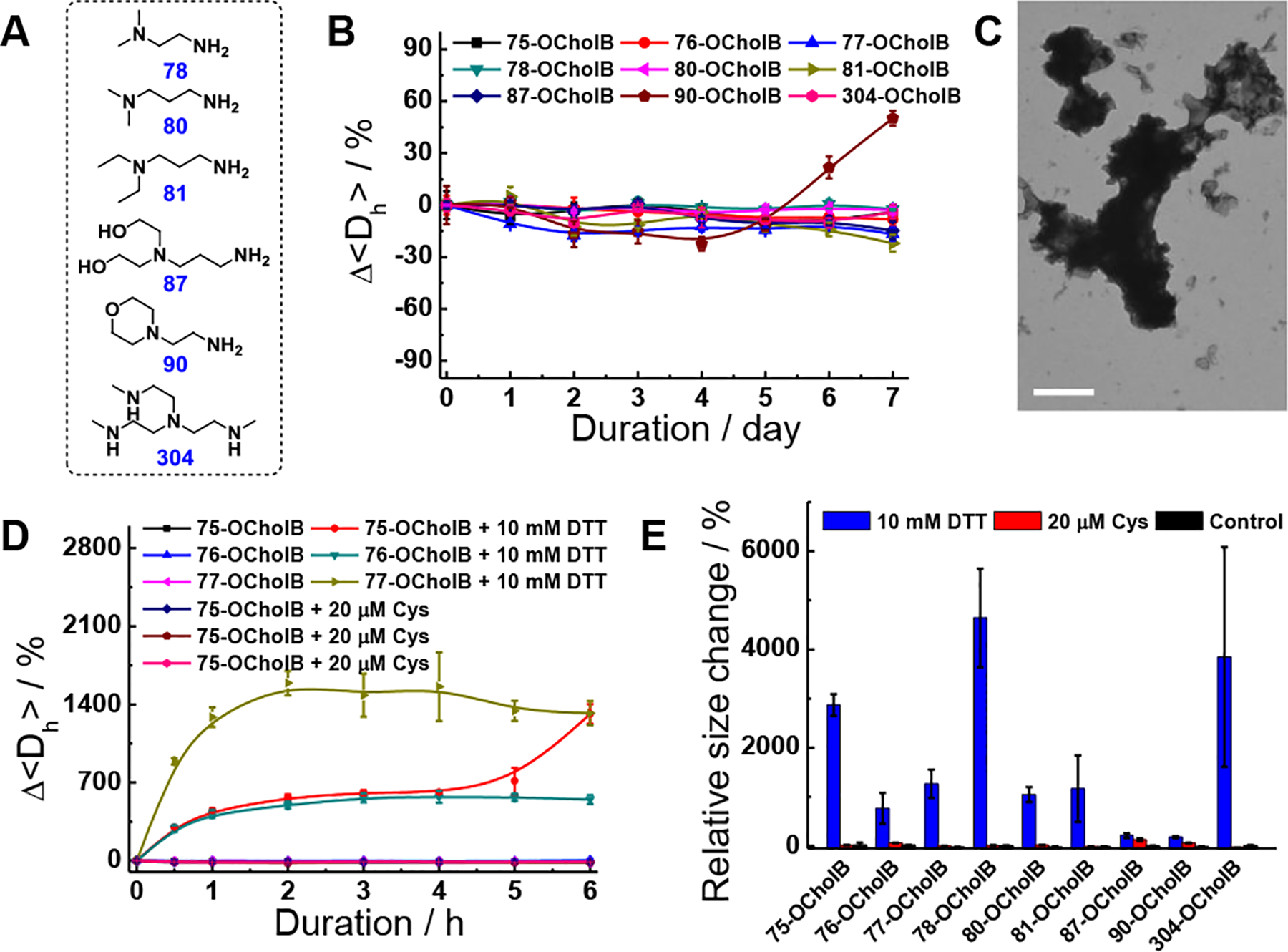
Stability test and reduction responsiveness study of OCholB LNPs. (A) Chemical structures of amine heads. (B) Relative hydrodynamic size variations of OCholB LNPs during storage. (C) TEM image of 77-OCholB LNPs after 10 mM DTT treatment. Scale bar = 600 nm. (D) Time-dependent relative size variations of OCholB LNPs with DTT or Cys treatments. (E) Relative size change of OCholB LNPs after 24 h of DTT or Cys incubation.
Next, the stability of OCholB LNPs was examined by time-dependent DLS measurement (see Supporting Information). As shown in Figure 2B, with the exception of 90-OCholB, all other LNPs showed a consistent particle size over 7 days of storage (<22% variation in <Dh>). This indicates that most of the OCholB LNPs are stable under long-term storage without obvious aggregation. In all of the following studies, freshly prepared LNPs were used.
The OCholB LNPs were predicted to be degradable in the highly reductive intracellular environment, but not in the blood serum.[17] To verify this, the thiol-triggered degradation of OCholB LNPs was studied in vitro using 10 mM of 1,4-dithiothreitol (DTT) and 20 μM of L-cysteine (Cys) to mimic the reducing environments in the cytoplasm and serum, respectively.[18] The effect of these reducing agents on nanoparticle size and stability was assessed using DLS and TEM. DTT treatment induced the loss of liposomal structures, the formation of large amorphous aggregates at micrometer scale, and a time-dependent increase in OCholB LNP size (Figure 2C, 2D and S19A). On the other hand, Cys treatment resulted in negligible changes in LNP size (Figure 2D). After 24 h of incubation, all LNPs (except for 87- and 90-OCholB) demonstrated dramatic size increases (770–4640%) upon DTT treatment, but negligible effects of Cys treatment (Figure 2E). Furthermore, the responsiveness of OCholB LNPs was tested using β-mercaptoethanol (BME) and L-glutathione (GSH) and the results are shown in Figure S19B–D (see Supporting Information).[16b, 19]
The in vitro intracellular delivery of mRNA using OCholB LNPs was studied using GFP mRNA. The size, PDI, and morphology of mRNA loaded 75-OCholB were first examined by DLS and TEM (Figure S20A and B). The delivery conditions were then optimized using GFP mRNA/75-OCholB in HeLa cells (Figure 3A and S20C). After that, the intracellular delivery efficiencies of all OCholB LNPs were tested in a variety of cell lines (HeLa, B16F10, HEK293, and NIH 3T3), using the optimized delivery conditions (LNP/mRNA = 10/1, w/w; [mRNA] = 0.86 μg mL−1; 24 h exposure). Unless otherwise noted, these conditions were used for all subsequent in vitro mRNA delivery experiments. Lpf2k and naked GFP mRNA were used as positive (98% GFP+ HeLa cells) and negative (2% GFP+ HeLa cells) controls, respectively. As shown in Figure 3B, overall, we identified a subset of the OCholB LNP library that could consistently deliver mRNA to all cell types screened (75-, 77-, 78-, 80-, and 81-OCholB), another subset that was consistently ineffective (87- and 90-OCholB). Cell type-dependent variance was observed (see Supporting Information), which indicates different amine heads resulted in different cellular uptake and warrants further structure-activity study.[13d] Typical fluorescent images of mRNA/LNPs treated cells are shown in Figure S20D.
Figure 3.

OCholB LNPs enabled intracellular mRNA delivery in vitro. (A) LNP/mRNA weight ratio-dependent GFP mRNA transfection efficacy against HeLa cell using 75-OCholB. (B) GFP mRNA transfection efficacy of OCholB LNPs, Lpf2k, and naked mRNA tested on different cell lines. (C) Cytotoxicity of GFP mRNA loaded LNPs against HeLa cell determined by MTT assay.
The MTT assay was conducted to examine the cytotoxicity of mRNA-loaded nanoparticles. After 24 h (Figure 3C and S20C), the mRNA/Lpf2k complex showed significant cytotoxicity against HeLa cells (37.5% cell viability; significant morphological changes were observed as shown in Figure S20E), while the mRNA loaded OCholB LNPs showed negligible cytotoxicities (>84% cell viabilities; no obvious cell morphology change). This result indicate the excellent cell compatibility of OCholB LNPs and the possibility to further increase the delivery efficiencies by increasing the total dosage and/or exposure time of the mRNA/LNPs complexes.
We performed a range of studies in vitro to demonstrate the versatility of OCholB LNPs. The possibilities of using OCholB LNPs to deliver mRNA for genome engineering (Cre-loxP and CRISPR/Cas9 systems; Figure S21A) were examined. For this study, lipids 75-, 76-, and 77-OCholB were selected due to their efficient delivery to both HeLa and HEK cell lines. First, Cre mRNA was complexed with OCholB LNPs and tested against HeLa-DsRed cells, which express DsRed red fluorescent protein only upon Cre-mediated recombination.[12c] After 24 h of incubation with mRNA/LNPs, all three OCholB lipids tested were able to generate red fluorescence, with 50–70% of cells determined to be DsRed+ (Figure S21B). Next, Cas9 mRNA was loaded into LNPs, along with single-guide RNA (sgRNA) that targets a sequence of the GFP gene. GFP-expressing HEK cells (GFP-HEK) were used in this study.[20] Successful Cas9 mRNA and sgRNA delivery would result in a knockout of GFP expression. Interestingly, none of the three tested lipids were capable of inducing a distinct GFP knockout after 48 h (lipidoid/mRNA/sgRNA = 10/1/1; [mRNA] = [sgRNA] = 0.86 μg mL−1) (Figure S21C).
Nevertheless, we hypothesized that Cas9 mRNA/sgRNA delivery would be successful under the correct and optimized LNPs and delivery conditions. Two strategies were tested to improve the Cas9 mRNA delivery, i.e., synthesizing new lipidoids with a single tail rather than full substitution, and adding excipients into fully substituted OCholB LNPs. Single-tailed lipidoids, 75-OCholB-1, 76-OCholB-1, and 76-OCholB-1 were synthesized (Figure S22) and LNPs were fabricated (Figure S23A). The single-tailed LNPs, while effective for GFP and Cre mRNA delivery (Figure S23B), did not improve Cas9 mRNA and sgRNA delivery (Figure S23C). As a result, we returned to use the original fully substituted OCholB lipidoids and single-tailed lipidoids were not used in following studies.
Next, we formulated OCholB lipids (Figure 1A) with the helper lipid DOPE (OCholB lipidoid/DOPE = 1/1, w/w).[20] Formulated LNPs (noted as 75-OCholB-F etc.) were loaded with Cas9 mRNA and sgRNA. As shown in Figure S23D, significant amounts of GFP knockout cells were recorded for 75- and 77-OCholB-F treated GFP-HEK cells. These results indicated that the formulation optimization is useful to achieve improved Cas9 mRNA and sgRNA delivery in vitro (see Supporting Information).
Despite the advantages of DOPE-formulated LNPs in this one particular application (Cas9 mRNA), the original fully substituted and unformulated OCholB LNPs (75-OCholB etc.) achieved acceptable Cre mRNA delivery efficacy (49–73% DsRed+ cells). Taking the overall cost into account, the unformulated OCholB LNPs (i.e., the original LNPs described in Figures 1–3) were prepared and used for all following in vitro and in vivo studies.
Next, we demonstrated the intracellular delivery of genome engineering proteins using OCholB LNPs. (−30)GFP-Cre recombinase and HeLa-DsRed cells were used.[12c] The protein delivery efficacy was determined by the intracellular presence of GFP, and the gene recombination efficacy was determined by DsRed expression. The (−30)GFP-Cre/LNPs complexes were found to be internalized mainly through the clathrin and dynamin-mediated pathways (Figure 4A; see Supporting Information).[12a]
Figure 4.
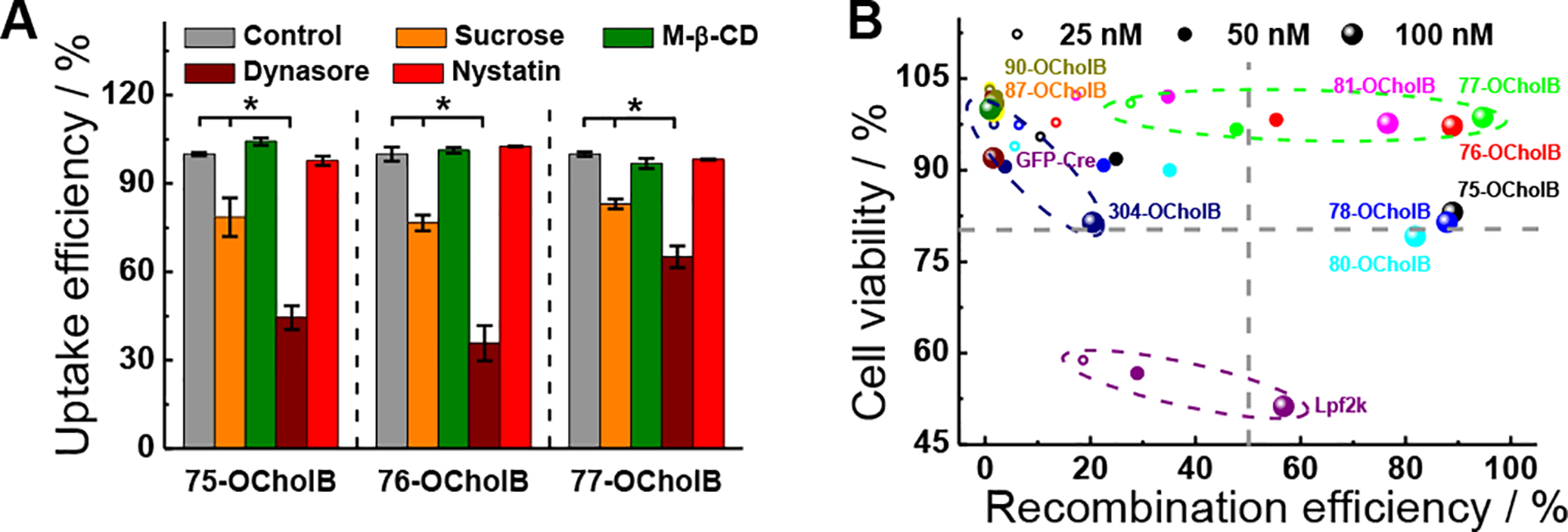
OCholB LNPs enabled intracellular delivery of (−30)GFP-Cre recombinase. (A) Internalization mechanism study of (−30)GFP-Cre/LNPs. N = 3, two-tailed students’ t-test, p < 0.05. (B) (−30)GFP-Cre/LNPs mediated genome engineering efficacy was plotted against corresponding cell viability for each tested condition.
Intracellular protein delivery efficiencies were then determined, with naked (−30)GFP-Cre and (−30)GFP-Cre/Lpf2k used as negative and positive controls (Figure S24). The typical DLS profile and TEM image of (−30)GFP-Cre protein loaded 75-OCholB were shown in Figure S24A and B. A range of protein concentrations (25, 50 and 100 nM), with a consistent lipidoid/protein ratio were examined. Six out of the nine examined OCholB LNPs showed comparable or even higher transfection efficiency than Lpf2k (Figure S24C). The typical fluorescence cell images were shown in Figure S24D. In addition to GFP+ cell count, we analyzed the mean fluorescent intensity (MFI) of green fluorescence emission of each group, which reflects approximately the amount of the intact (−30)GFP-Cre protein delivered per cell. In particular, 76-OCholB resulted in dramatically higher MFI than Lpf2k and other OCholB LNPs, indicating its high effectiveness (Figure S24E). Notably, we analyzed the MFI of the green signal generated by (−30)GFP-Cre protein, which purely reflects the ability of the LNPs to deliver the protein, and does not give any information about subsequent downstream genomic recombination events.
Next, the genome engineering efficiencies were determined after 24 h of incubation (with 8 h of (−30)GFP-Cre/LNPs exposure). Three different concentrations of (−30)GFP-Cre/LNPs were tested (25 nM/1.7 μg mL−1, 50 nM/3.4 μg mL−1, and 100 nM/6.6 μg mL−1). As shown in Figure S24F, naked (−30)GFP-Cre protein cannot induce gene recombination regardless of the protein concentrations; while all the tested nanoparticles including Lpf2k showed a dose-dependent DsRed+ cell percentage pattern. Six of the nine examined OCholB LNPs were comparable or even more efficient than Lpf2k, and genome engineering efficiency (DsRed+ cell population) resembled protein delivery efficiency (GFP+ cell population).
The cytotoxicity profiles of (−30)GFP-Cre/LNPs, (−30)GFP-Cre/Lpf2k and naked (−30)GFP-Cre were measured (HeLa-DsRed cells; 24 h exposure). Both the cell viability (Figure S25A) and morphology studies (Figure S25B) revealed that the tested OCholB LNPs were less toxic than Lpf2k. The DsRed+ cell percentage was then plotted against the corresponding cell viability for all tested conditions (11 samples with 3 different concentrations; Figure 4B), with dotted lines denoting 80% of cell viability and 50% of gene recombination efficacy, respectively. Samples in lower left quadrant are toxic and inefficient; samples in upper right quadrant are non-toxic and efficient. It is clear that Lpf2k (dotted purple circle) at high concentration is relatively efficient but also highly toxic. The OCholB LNPs are all fairly well tolerated, but vary dramatically in terms of gene recombination efficiency, which indicates the biological activity of LNPs depends on their molecular structures. Both high efficacy and excellent tolerability were achieved by using 75-, 76-, and 77-OCholB LNPs (dotted green circle).
Before we moved on to in vivo protein and mRNA delivery, the in vivo toxicity of the OCholB LNPs was examined.[21] Three OCholB LNPs identified from Figure 4B (i.e. 75-, 76-, and 77-OCholB), were selected for initial screening. 4–6 weeks old Balb/c mice (n = 3) were injected with blank OCholB LNPs (2.5 mg/kg of LNPs for each injection) through tail vein at day 1 and day 5, body weights were monitored for 14 days, and blood was collected and analyzed at day 14. As shown in Figure S26A and B, the OCholB LNPs did not induce evident body weight change or severe organ damage. However, it should be noted that the toxicity profile of LNPs may change upon complexation with cargo molecules, and should be examined further.
For the in vivo genome engineering study, transgenic Ai14 mice were used.[22] As shown in Figure 5A, this mouse model has a genetically integrated loxP-flanked STOP cassette that prevents the transcription of tdTomato. Cre recombinase-mediated gene reorganization can excise the STOP cassette, resulting in the expression of tdTomato fluorescent reporter.
Figure 5.
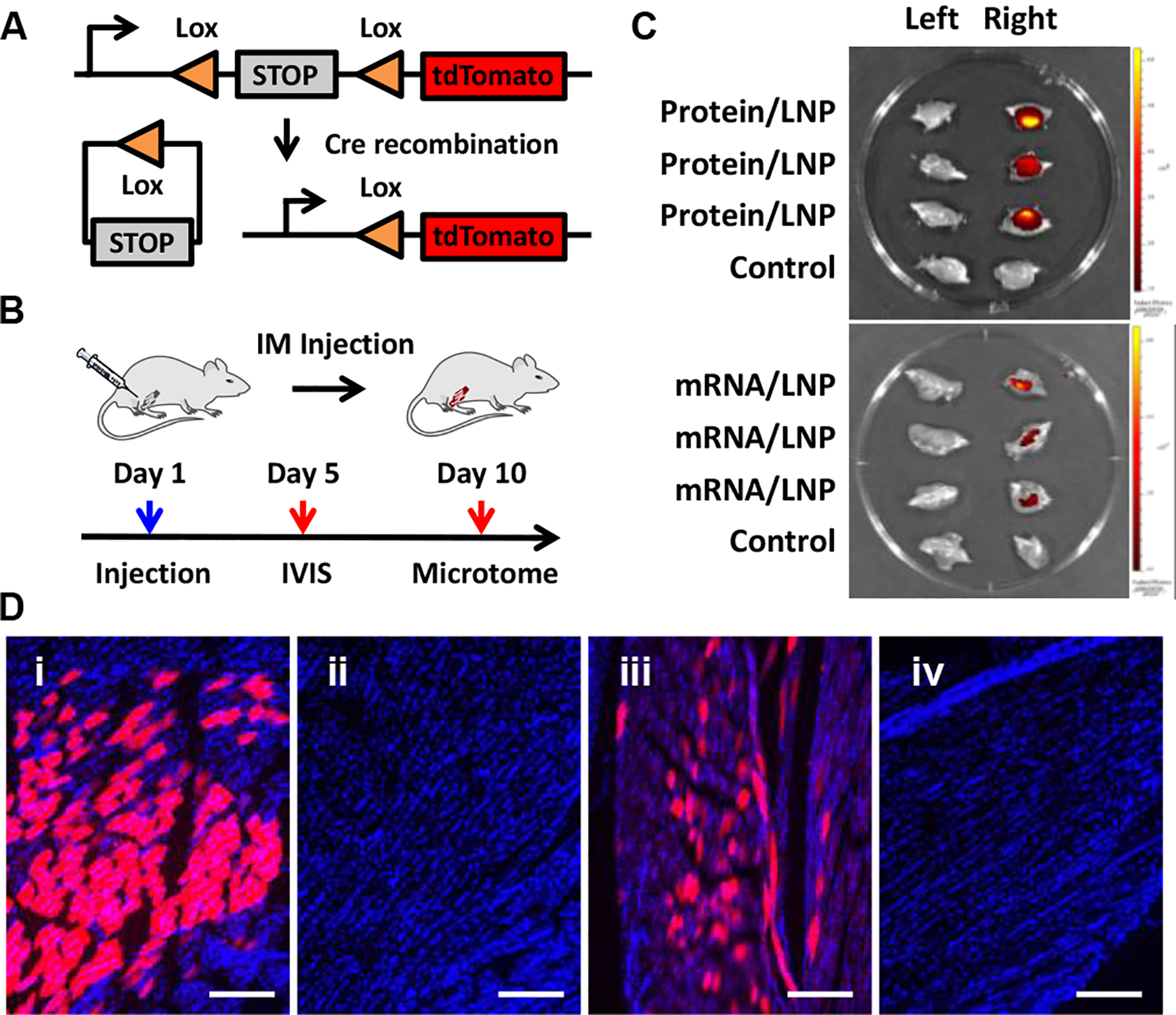
Local (−30)GFP-Cre protein and Cre mRNA delivery for in vivo genome engineering using adult Ai14 mice. (A) Cre-mediated gene recombination. (B) Protocols used for IM injection and analysis. (C) IVIS ex vivo fluorescence images of left (un-injected) and right (protein or mRNA loaded-76-OCholB LNPs injected) hindlimb. Control left and right hindlimb were collected from untreated Ai14 mice. (D) Typical microscope fluorescence images of hindlimb skeletal muscle slices obtained from mice injected (IM injection) with (i) protein and (iii) mRNA loaded 76-OCholB LNPs. (ii) and (iv) are control muscle slices obtained from untreated Ai14 mice. Blue channel, DAPI; Red channel, tdTomato. Scale bar = 250 μm.
We first tried the local delivery through intramuscular injection (IM injection; hindlimb gastrocnemius muscle) using (−30)GFP-Cre protein or Cre mRNA loaded 76-OCholB LNP.[23] 76-OCholB LNP was chosen here because of its high efficacy for protein delivery in vitro (Figure S24C and D). In order to gain insight of the protein and mRNA delivery effects, 76-OCholB was also used for local delivery of Cre mRNA. Adult Ai14 mice (n = 6 per group) received single dose of (−30)GFP-Cre/LNPs (2.5 mg/kg of protein) or Cre mRNA/LNPs (0.5 mg/kg of mRNA) injection on day 1. Two types of controls were included, namely the contralateral (non-injected) limb of treated mice, as well as both limbs from untreated (naïve) mice. Three mice from each group were sacrificed on day 5 for IVIS imaging and other three were sacrificed on day 10 for microtome sectioning and microscope analysis (Figure 5B). As shown in Figure 5C, comparing to the control samples, red fluorescence signals (tdTomato+) were observed from both the (−30)GFP-Cre/LNPs and Cre mRNA/LNP injected right hindlimb of Ai14 mice.[24] Furthermore, the (−30)GFP-Cre/LNPs treated samples showed higher fluorescence intensity than the Cre mRNA/LNP treated samples under the same imaging conditions. As shown in Figure 5D and S27, contrary to untreated control muscles, strong tdTomato signals from both (−30)GFP-Cre/LNPs (image i) and Cre mRNA/LNPs (image iii) injected muscles were recorded. A larger portion of tdTomato+ cells were observed in the protein/LNPs injected muscles than the mRNA/LNPs counterpart, which is consistent with the ex vivo IVIS imaging results (Figure 5C). We hypothesize that the relative instability of mRNA and the additional biochemical steps that are involved in the mRNA delivery process may compromise the overall gene recombination efficiency.
It was reported previously that when mRNA/LNPs were injected intramuscularly, expression of encoded proteins was detected in liver.[25] This could indicate the mRNA/LNPs may be taken up by the muscular vasculature and distributed systemically to unintended targets. In light of this, we examined the heart, liver, spleen, lung, and kidney of the mice injected intramuscularly with protein/LNPs and mRNA/LNPs (see Supporting Information). Typical fluorescence images are shown in Figure S28 and S29, and no tdTomato signal was detected in peripheral organs, indicating the function of cargo/LNPs was highly restricted to the injection site. This could be advantageous for further local genome engineering applications.[23b] Overall, both the protein and mRNA were demonstrated to be successfully delivered into skeletal muscles of the adult mice and induced gene recombination by OCholB LNPs.
We further used OCholB LNPs for systemic delivery.[26] Based on our previous experience and in vitro results indicating that different OCholB lipids may show some preferential delivery to different cell types, we screened the top five OCholB LNPs (75-, 76-, 77-, 78-, and 80-OCholB), each delivering (−30)GFP-Cre protein. Adult Ai14 mice (n = 3 per group) were injected through tail vein (intravenous (IV) injection) with (−30)GFP-Cre/LNPs on day 1 and 5 (2.5 mg/kg of protein for each injection), then sacrificed on day 14 for analysis (Figure 6A). The heart, liver, spleen, lung, and kidney from each group were analyzed. We detected tdTomato signal in the lung of 80-OCholB treated mice (Figure 6B, C, and S30), and in the spleen of 76-OCholB treated mice (Figure S31). As shown in Figure S30 and S31, some weak tdTomato signal was detected in the lung of 75-OCholB and 76-OCholB treated mice, but otherwise minimal fluorescence was detected in any other organs. Additional images with different magnifications can be found in Figure S32 and S33. This indicates that two of our OCholB LNPs can induce functional protein delivery to the lung and spleen, respectively. Notably, most previously-reported systemic lipid nanoparticle delivery systems, including the DLin-MC3-DMA, resulted in accumulation in the liver.[14b, 25, 27] Though liver is an important target for therapeutics development, delivering therapeutic cargo to additional organs can further enable the therapeutic modality. Here, we demonstrated OCholB LNPs can deliver (−30)GFP-Cre protein to the lung and spleen while largely excluding delivery to the liver. Future studies are needed to identify the engineered cell types. From this data it appears as though 80-OCholB demonstrates a slight preferential delivery to the lung, and 76-OCholB, while less preferential, demonstrates delivery to the spleen. Our future studies will further validate and improve the strength of this preference.
Figure 6.
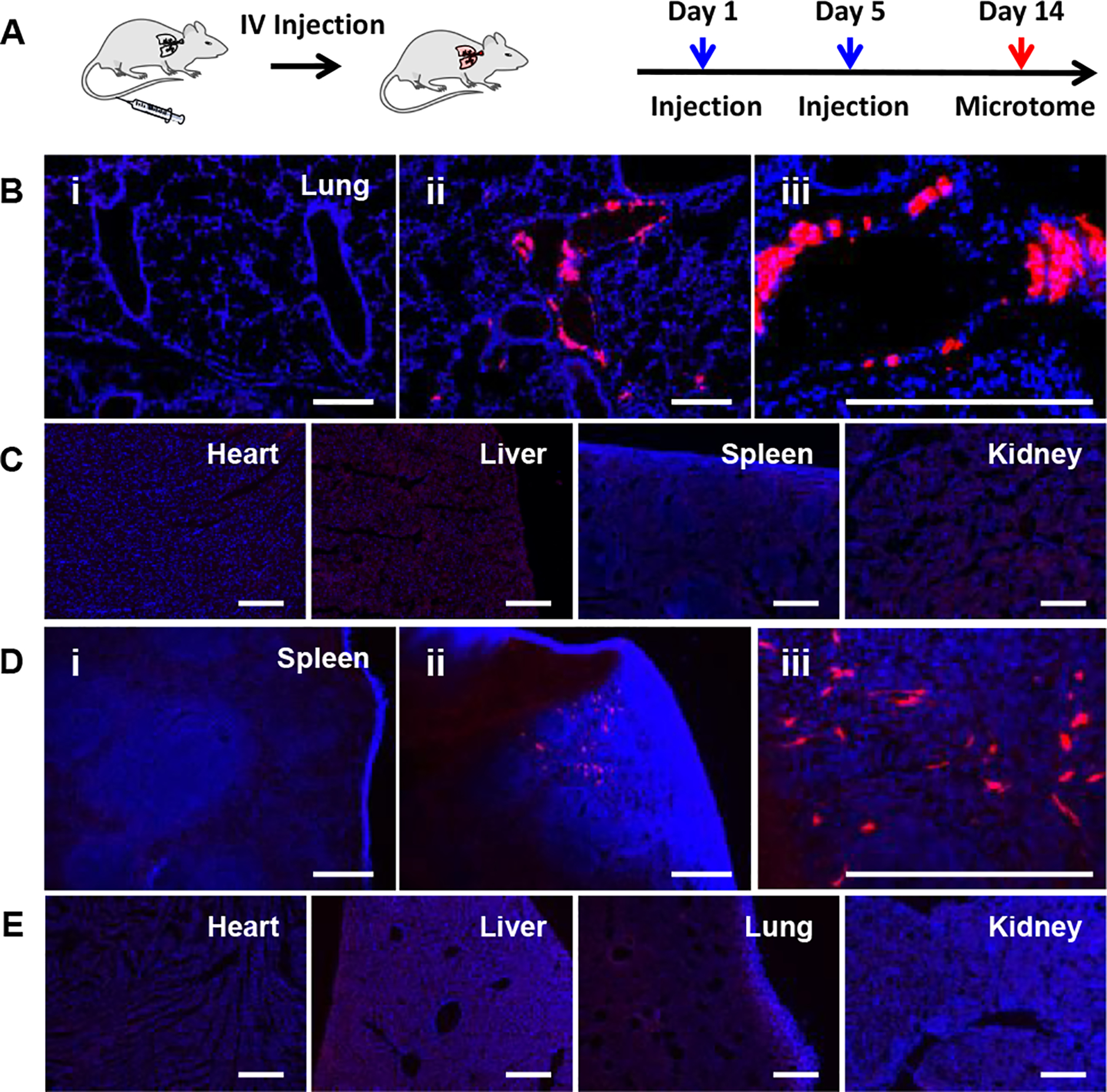
Systemic (−30)GFP-Cre protein and Cre mRNA delivery for in vivo genome engineering using adult Ai14 mice. (A) Protocols used for IV injection and analysis. (B) Typical fluorescence images of lung tissue obtained from (i) untreated control mice, (ii and iii) (−30)GFP-Cre/80-OCholB LNP injected mice under different magnifications. (C) Fluorescence images of heart, liver, spleen, and kidney tissues obtained from (−30)GFP-Cre/80-OCholB LNP injected mice. (D) Typical fluorescence images of spleen tissue obtained from (i) untreated control mice, (ii and iii) Cre mRNA/76-OCholB LNP injected mice under different magnifications. (E) Fluorescence images of heart, liver, spleen, and kidney tissues obtained from Cre mRNA/76-OCholB LNP injected mice. Blue channel, DAPI; Red channel, tdTomato. Scale bar = 250 μm.
Next, in vivo systemic mRNA delivery using OCholB LNPs was studied (Figure 6A). As positive signals were observed in the local Cre mRNA delivery and systemic protein delivery, only 76-OCholB LNP was tested for systemic mRNA delivery. Cre mRNA/76-OCholB LNPs were injected on day 1 and day 5 (0.5 mg/kg of mRNA for each injection), and mice were sacrificed on day 14. The same set of organs was analyzed. As shown in Figure 6D and S33, tdTomato+ cells were observed in the spleen, and no signals were found in other organs (Figure 6E and S34). Again, it is notable that the liver was excluded. It should also be noted that the 76-OCholB LNPs induced successful delivery of both protein and Cre mRNA cargos to the spleen (Figure S33). This indicates that some specific property of a given lipid may influence its biodistribution regardless of the cargo. This observation merits further study.
The systemic delivery was found to be less efficient than local delivery (see Supporting Information). Nevertheless, the successful protein and mRNA delivery to the lung or spleen opens up a wide range of possibilities for novel treatments of diseases.[28] The underlying mechanisms are not clear at this stage. We envision that further structure-activity relationship studies on how LNPs interact with biological systems can shed some light on this issue.
The genome engineering platforms also hold great potential for the mechanistic study and treatment of various neurological disorders.[29] Delivering genome engineering components to certain regions and cells in the brain is crucial to realize this potential, yet remains a technical challenge. We examined the potency of the OCholB LNPs system for mRNA delivery into adult mouse brain for gene recombination. As one of our top LNPs, 76-OCholB, has already been demonstrated to be effective for Cre mRNA delivery locally and systemically, we chose to screen different LNPs (75-OCholB, 78-OCholB, and 80-OCholB) in this study. Adult Ai14 mice (n =3 per group) were implanted with an osmotic pump connected to a brain cannula,[30] and Cre mRNA/LNPs were continuously infused into the lateral ventricle (VL) from day 1 to day 4 (Figure 7A). Mice were then sacrificed on day 9, and brains and spinal cords were collected. The whole brain and spinal cord were sectioned into slices with 15 μm thickness. The sub-regions of brain and spinal cord (Figure S35) were imaged and typical fluorescent images are shown in Figure S36–40 and Figure 7C (images for DAPI and tdTomato separate fluorescence channels were shown in Figure S41). tdTomato+ cells were recorded for Cre mRNA loaded 75-OCholB, 78-OCholB, and 80-OCholB infused mice.
Figure 7.
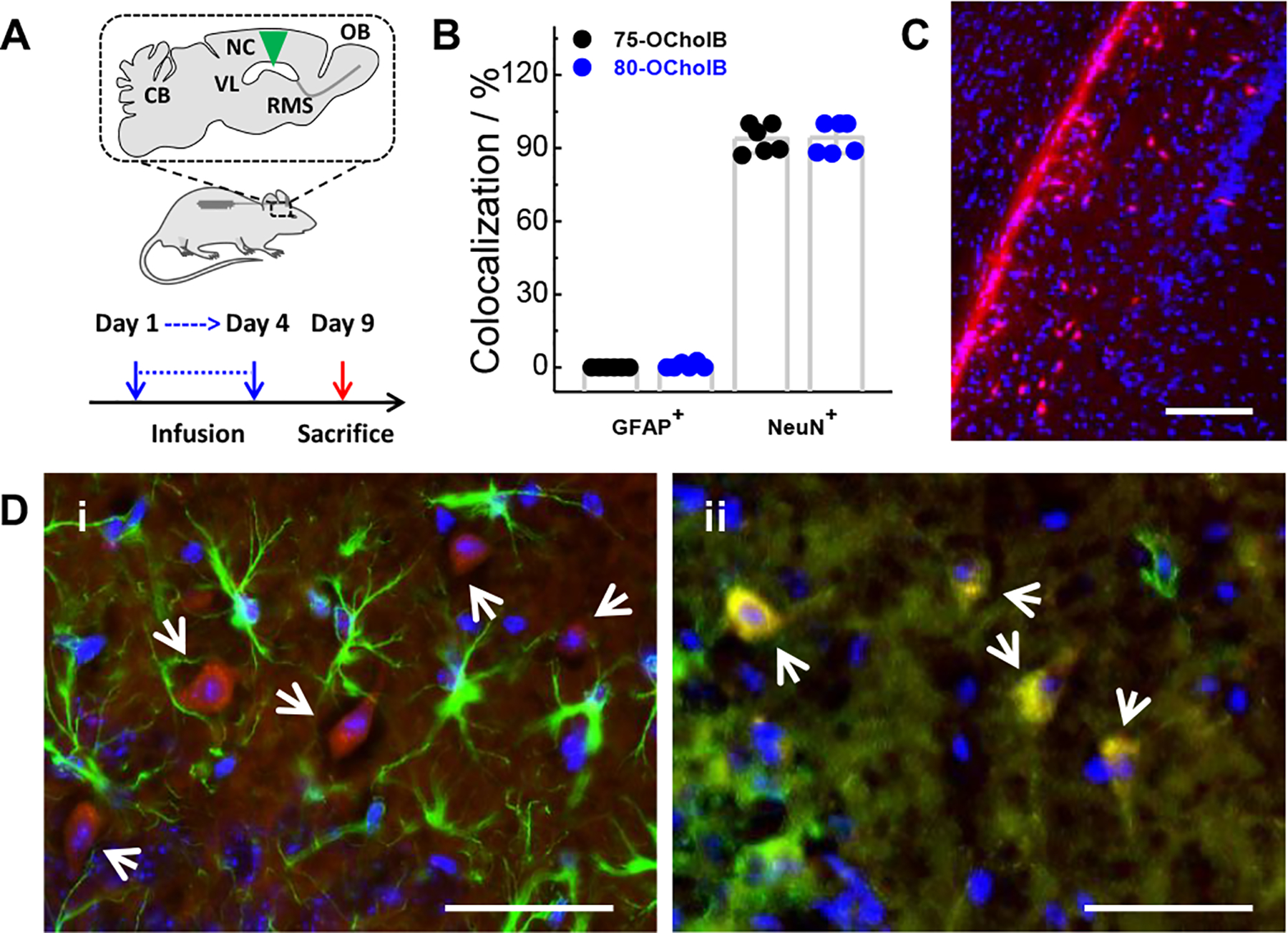
Cre mRNA delivery to adult Ai14 mouse brain enabled by OCholB LNPs. (A) Protocol used for lateral ventricle infusion of Cre mRNA/LNPs. (B) Quantification of the percentages of tdTomato+ cells (located in the Ammon’s horn of HPF) that colocalized with GFAP+ or NeuN+ cells. (C) Overlay fluorescence image of Ammon’s horn of HPF region in the brain slice of Cre mRNA/80-OCholB infused mice. Blue channel, DAPI; Red channel, tdTomato. Scale bar = 150 μm. (D) Identification of transfected cell type in the Ammon’s horn of HPF through immunofluorescence staining. Astrocytes and neurons were stained by (i) anti-GFAP and (ii) anti-NeuN antibodies. Blue channel, DAPI; Green channel, GFAP/NeuN; Red channel, tdTomato. White arrows indicate tdTomato+ cells. Yellow signal indicates colocalization of red and green fluorescence. Scale bar = 50 μm.
Further analysis showed that all tested mRNA/LNPs can induce tdTomato signals in the cells lining the VL (most likely ependymal cells) that borders corpus callosum and hippocampal formation (HPF), as well as in the hippocampus, as shown in Figure S42. Specifically, in the hippocampal region, almost all the tdTomato+ cells were observed in the stratum oriens of Field CA1, CA2 and CA3. All three tested LNPs showed very similar tdTomato+ patterns. All brain samples were sectioned coronally and signals were observed throughout the whole HPF sagittally. Additional fluorescent images of the HPF region from Cre mRNA/LNPs infused and untreated control mice are shown in Figure S43 and S44. It seems reasonable that the infused mRNA/LNPs in the VL migrate along the fiber tracts between HPF and cerebral cortex, and diffused into and transfected cells in the ependymal cells of VL and cells in HPF.
Next, the cell type of tdTomato+ populations located in the Ammon’s horn of HPF from mRNA/75-OCholB and mRNA/80-OCholB infused mice were determined through immunostaining. As shown in Figure 7D and S45, the tdTomato+ cells are largely colocalized with NeuN+ neuronal cells, but not with GFAP+ astrocyte cells. Quantitatively, of all the tdTomato+ cells identified, more than 90% of tdTomato+ cells that are located in the Ammon’s horn of HPF were found to be colocalized with NeuN+ cells (Figure 7B). It should be noted that this data indicated ~90% of tdTomato+ cells in Ammon’s horn express neuronal markers, but notably this does not give any information regarding the overall fraction of neuron cells which express tdTomato. The overall transfection efficacies need to be determined further.
Overall, it is concluded that the Cre mRNA/LNPs can transfect a portion of neuronal cells in the stratum oriens of Field CA1, CA2 and CA3 in Ammon’s horn of HPF, as well as a portion of ependymal cells lining the VL, through VL infusion. The study of overall brain cell delivery efficacy as well as internalization mechanism are underway, and we believe this intriguing property of the OCholB LNPs could contribute to treatment of certain types of neurological disorders in future.
Conclusion
In summary, a new combinatorial library of OCholB lipidoid nanoparticles containing both a cholesteryl and disulfide bond in the tail was synthesized. These lipidoids are effective for intracellular delivery of mRNA and protein both in vitro and in vivo. Successful gene recombination events were observed in skeletal muscle, lung, and spleen through the local and systemic administrations of (−30)GFP-Cre protein or Cre mRNA loaded OCholB LNPs to adult Ai14 mice. A portion of neuronal cells in HPF and ependymal cells in VL can be transfected using the Cre mRNA loaded OCholB LNPs through VL infusion. Future research will explore the structure-activity relationship of these new LNPs, optimization of formulations (size, distribution, and formulation components), and therapeutic applications using diseases-related therapeutic payloads. We believe that the new OCholB LNPs reported here can be a safe and efficient platform for genome engineering therapeutics delivery, both in vitro and in vivo.
Supplementary Material
Acknowledgements
We thank Dr. David Wilbur (Tufts University) for assistance with NMR and MS characterization and Nicki Watson (Whitehead Institute at MIT) for TEM imaging. This work was financially supported by National Institutes of Health (NIH) Grants R01 EB027170-01 and UG3 TR002636-01.
Footnotes
Conflict of interest
Y. Li and Q. Xu are the inventors of a patent filed through Tufts University on the new lipidoid materials described in this study.
Supporting information for this article is given via a link at the end of the document.
Reference
- [1].Sun TM, Zhang YS, Pang B, Hyun DC, Yang MX, Xia YN, Angew Chem Int Edit 2014, 53, 12320–12364; [DOI] [PubMed] [Google Scholar]; Angew Chem 2014, 126, 12520. [Google Scholar]
- [2].Shi JJ, Kantoff PW, Wooster R, Farokhzad OC, Nat Rev Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Hubbell JA, Chilkoti A, Science 2012, 337, 303–305; [DOI] [PubMed] [Google Scholar]; b) Lorden ER, Levinson HM, Leong KW, Drug Deliv Transl Re 2015, 5, 168–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Altinoglu S, Wang M, Xu QB, Nanomedicine 2015, 10, 643–657; [DOI] [PubMed] [Google Scholar]; b) Zhang YL, Pelet JM, Heller DA, Dong YZ, Chen DL, Gu Z, Joseph BJ, Wallas J, Anderson DG, Adv Mater 2013, 25, 4641–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Colson YL, Grinstaff MW, Adv Mater 2012, 24, 3878–3886; [DOI] [PubMed] [Google Scholar]; b) Liechty WB, Kryscio DR, Slaughter BV, Peppas NA, Annu Rev Chem Biomol 2010, 1, 149–173; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Miyata K, Nishiyama N, Kataoka K, Chem Soc Rev 2012, 41, 2562–2574. [DOI] [PubMed] [Google Scholar]
- [6].Kostarelos K, Bianco A, Prato M, Nat Nanotechnol 2009, 4, 627–633. [DOI] [PubMed] [Google Scholar]
- [7].Mout R, Moyano DF, Rana S, Rotello VM, Chem Soc Rev 2012, 41, 2539–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Sahoo SK, Labhasetwar V, Drug Discov Today 2003, 8, 1112–1120; [DOI] [PubMed] [Google Scholar]; b) De Jong WH, Borm PJA, Int J Nanomed 2008, 3, 133–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang M, Sun S, Neufeld CI, Perez-Ramirez B, Xu QB, Angewandte Chemie International Edition 2014, 53, 13444–13448; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 13662–13666. [Google Scholar]
- [10].Barenholz Y, J Control Release 2012, 160, 117–134. [DOI] [PubMed] [Google Scholar]
- [11].Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Plante-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB, New Engl J Med 2018, 379, 11–21. [DOI] [PubMed] [Google Scholar]
- [12].a) Li Y, Yang T, Yu Y, Shi N, Yang L, Glass Z, Bolinger J, Finkel IJ, Li W, Xu Q, Biomaterials 2018, 178, 652–662; [DOI] [PubMed] [Google Scholar]; b) Wang XY, Li YM, Li QS, Neufeld CI, Pouli D, Sun S, Yang L, Deng P, Wang M, Georgakoudi I, Tang SQ, Xu QB, J Control Release 2017, 263, 39–45; [DOI] [PubMed] [Google Scholar]; c) Wang M, Zuris JA, Meng FT, Rees H, Sun S, Deng P, Han Y, Gao X, Pouli D, Wu Q, Georgakoudi I, Liu DR, Xu QB, P Natl Acad Sci USA 2016, 113, 2868–2873; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang M, Alberti K, Sun S, Arellano CL, Xu QB, Angewandte Chemie International Edition 2014, 53, 2893–2898; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 2937–2942. [Google Scholar]
- [13].a) Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N, Bacallado SA, Nguyen DN, Fuller J, Alvarez R, Borodovsky, Borland T, Constien R, de Fougerolles A, Dorkin JR, Jayaprakash KN, Jayaraman M, John M, Koteliansky V, Manoharan M, Nechev L, Qin J, Racie T, Raitcheva D, Rajeev KG, Sah DWY, Soutschek J, Toudjarska I, Vornlocher HP, Zimmermann TS, Langer R, Anderson DG, Nat Biotechnol 2008, 26, 561–569; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dong YZ, Love KT, Dorkin JR, Sirirungruang S, Zhang YL, Chen DL, Bogorad RL, Yin H, Chen Y, Vegas AJ, Alabi CA, Sahay G, Olejnik KT, Wang WH, Schroeder A, Lytton-Jean AKR, Siegwart DJ, Akinc A, Barnes C, Barros SA, Carioto M, Fitzgerald K, Hettinger J, Kumar V, Novobrantseva TI, Qin JN, Querbes W, Koteliansky V, Langer R, Anderson DG, Proceedings Of the National Academy Of Sciences Of the United States Of America 2014, 111, 5753–5753; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li B, Luo X, Deng BB, Wang JF, McComb DW, Shi YM, Gaensler KML, Tan X, Dunn AL, Kerlin BA, Dong YZ, Nano Lett 2015, 15, 8099–8107; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Whitehead KA, Dorkin JR, Vegas AJ, Chang PH, Veiseh O, Matthews J, Fenton OS, Zhang YL, Olejnik KT, Yesilyurt V, Chen DL, Barros S, Klebanov B, Novobrantseva T, Langer R, Anderson DG, Nat Commun 2014, 5, 4277; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang XF, Lin B, Luo X, Zhao WY, Jiang J, Zhang CX, Gao M, Chen XF, Dong YZ, Acs Appl Mater Inter 2017, 9, 25481–25487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Wang M, Alberti K, Varone A, Pouli D, Georgakoudi I, Xu QB, Adv Healthc Mater 2014, 3, 1398–1403; [DOI] [PubMed] [Google Scholar]; b) Li Y, Bolinger J, Yu Y, Glass Z, Shi N, Yang L, Wang M, Xu Q, Biomaterials science 2018, 7, 596–606. [DOI] [PubMed] [Google Scholar]
- [15].a) Paunovska K, Da Silva Sanchez AJ, Sago CD, Gan Z, Lokugamage MP, Islam FZ, Kalathoor S, Krupczak BR, Dahlman JE, Adv Mater 2019, 31, e1807748; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Goyal K, Huang L, J Lipos Res 1995, 5, 49–60; [Google Scholar]; c) Lee J, Saw PE, Gujrati V, Lee Y, Kim H, Kang S, Choi M, Kim JI, Jon S, Theranostics 2016, 6, 192–203; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cheng XW, Lee RJ, Adv Drug Deliver Rev 2016, 99, 129–137. [DOI] [PubMed] [Google Scholar]
- [16].a) Sheng RL, Luo T, Zhu YD, Li H, Sun JJ, Chen SD, Sun WY, Cao AM, Biomaterials 2011, 32, 3507–3519; [DOI] [PubMed] [Google Scholar]; b) Koyanagi T, Cifelli JL, Leriche G, Onofrei D, Holland GP, Yane J, Bioconjugate Chem 2017, 28, 2041–2045. [DOI] [PubMed] [Google Scholar]
- [17].Saito G, Swanson JA, Lee KD, Adv Drug Deliver Rev 2003, 55, 199–215. [DOI] [PubMed] [Google Scholar]
- [18].a) Liu Y, Tian Y, Tian YF, Wang YJ, Yang WL, Adv Mater 2015, 27, 7156–7160; [DOI] [PubMed] [Google Scholar]; b) Turell L, Radi R, Alvarez B, Free Radical Bio Med 2013, 65, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Lee MH, Han JH, Lee JH, Choi HG, Kang C, Kim JS, J Am Chem Soc 2012, 134, 17314–17319; [DOI] [PubMed] [Google Scholar]; b) Zhang LW, Duan DZ, Liu YP, Ge CP, Cui XM, Sun JY, Fang JG, J Am Chem Soc 2014, 136, 226–233. [DOI] [PubMed] [Google Scholar]
- [20].Li YM, Li AC, Xu QB, Adv Healthc Mater 2019, 8, e1800996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li F, Li T, Han X, Zhuang H, Nie G, Xu H, ACS Biomaterials Science & Engineering 2017, 4, 1954–1962. [DOI] [PubMed] [Google Scholar]
- [22].Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng HK, Nat Neurosci 2010, 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Glass Z, Li YM, Xu QB, Nature Biomedical Engineering 2017, 1, 854–855; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee K, Conboy M, Park HM, Jiang FG, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, Shobha T, Mehdipour M, Liu H, Huang WC, Lan F, Bray NL, Li, Corn JE, Kataoka K, Doudna JA, Conboy I, Murthy N, Nature Biomedical Engineering 2017, 1, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Kauffman KJ, Oberli MA, Dorkin JR, Hurtado JE, Kaczmarek JC, Bhadani S, Wyckoff J, Langer R, Jaklenec A, Anderson DG, Mol Ther-Nucl Acids 2018, 10, 55–63; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Patel AK, Kaczmarek JC, Bose S, Kauffman KJ, Mir F, Heartlein MW, DeRosa F, Langer R, Anderson DG, Adv Mater 2019, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D, J Control Release 2015, 217, 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mout R, Ray M, Lee YW, Scaletti F, Rotello VM, Bioconjugate Chem 2017, 28, 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Liu J, Chang J, Jiang Y, Meng XD, Sun TM, Mao LQ, Xu QB, Wang M, Adv Mater 2019, 31, e1902575; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sabnis S, Kumarasinghe ES, Salerno T, Mihai C, Ketova T, Senn JJ, Lynn A, Bulychev A, McFadyen I, Chan J, Almarsson O, Stanton MG, Benenato KE, Mol Ther 2018, 26, 1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Sung JC, Pulliam BL, Edwards DA, Trends in biotechnology 2007, 25, 563–570; [DOI] [PubMed] [Google Scholar]; b) Jindal AB, Drug Deliv Transl Re 2016, 6, 473–485. [DOI] [PubMed] [Google Scholar]
- [29].Madigan NN, Staff NP, Windebank AJ, Benarroch EE, Neurology 2017, 89, 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].DeVos SL, Miller TM, Jove-J Vis Exp 2013, 12, e50326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.