

Abstract
Haematological malignancies induce important alterations of the immune system, which account for the high frequency of autoimmune complications observed in patients. Cutaneous immune-mediated diseases associated with haematological malignancies encompass a heterogeneous group of dermatoses, including, among others, neutrophilic and eosinophilic dermatoses, autoantibody-mediated skin diseases, vasculitis and granulomatous dermatoses. Some of these diseases, such as paraneoplastic pemphigus, are associated with an increased risk of death; others, such as eosinophilic dermatoses of haematological malignancies, run a benign clinical course but portend a significant negative impairment on a patient’s quality of life. In rare cases, the skin eruption reflects immunological alterations associated with an unfavourable prognosis of the associated haematological disorder. Therapeutic management of immune-mediated skin diseases in patients with haematological malignancies is often challenging. Systemic corticosteroids and immunosuppressive drugs are considered frontline therapies but may considerably augment the risk of serious infections. Indeed, developing a specific targeted therapeutic approach is of crucial importance for this particularly fragile patient population. This review provides an up-to-date overview on the immune-mediated skin diseases most frequently encountered by patients with onco-haematological disorders, discussing new pathogenic advances and therapeutic options on the horizon.
Key Points
| Several immune-mediated diseases of the skin have been shown to occur in patients with haematological malignancies. |
| Treatment of these dermatoses is often challenging and mostly relies on the use of systemic corticosteroids and immunosuppressive drugs. |
| Improved understanding of the mechanisms by which neoplastic cells drive the emergence of these dermatoses can lead to new targeted therapies that can eventually improve a patient’s outcome. |
Haematological Malignancies as a Trigger for Skin Autoimmunity
Haematological malignancies (HMs) account for a highly heterogeneous group of neoplastic disorders. Based on the cell lineage origin, they are divided into myeloid and lymphoid neoplasms. Each group includes tumours with aggressive or indolent biological behaviour, that in turn results in an acute or chronic clinical course and a different prognosis [1, 2]. B-cell chronic lymphatic leukaemia is by far the most common form, with a reported incidence of approximately 5.1/100,000 new cases per year in Western populations [3].
Patients with HMs can experience a broad range of dermatological manifestations. Comorbid diseases affecting the skin are often associated with a significant impairment on the quality of life and, more rarely, with an increased risk of death [4–6]. Dermatological manifestations associated with HMs can be divided into specific and non-specific. Specific manifestations include the massive infiltration of the skin by neoplastic cells, referred to as leukaemia cutis. Non-specific manifestations include: (i) dermatological changes secondary to bone marrow failure, e.g. pallor and ecchymosis; (ii) skin infections; (iii) adverse reactions to anti-neoplastic drugs; and (iv) immune-mediated diseases [4, 5].
Haematological malignancies can trigger both innate and acquired immune-mediated skin diseases. Various mechanisms can be involved and possibly synergise in driving the immune reaction (Table 1). First, in patients with HMs, both central and peripheral T-cell tolerance might be impaired. Central T-cell tolerance is regulated by the thymus through negative selection of self-reactive T cells. A paradigmatic example of altered negative selection in the thymus induced by malignancies occurs in patients with thymoma, which is often accompanied by autoimmune manifestations. It is interesting to note that both thymoma and HMs, with special regard to lymphoproliferative disorders, induce a similar profile of autoimmune diseases, such as paraneoplastic pemphigus (PNP). It is thus arguable that HMs may favour the emergence of autoreactive T-cell clones that escape or bypass negative selection in the thymus, although there is no evidence that this is related to a direct invasion of the thymus by malignant cells [7].
Table 1.
Mechanisms involved in the pathogenesis of immune-mediated skin diseases in patients with haematological malignancies
| Mechanism | Haematological tumour |
|---|---|
| Defective central T-cell tolerance (escape of immature and autoreactive T cells from the negative selection in the thymus) | Lymphoid malignancies |
| Defective peripheral T-cell tolerance (efficient costimulatory signals to self-reactive T cells by neoplastic cells; defective T-reg function) | Myeloid and lymphoid malignancies |
| Production of autoantibodies by neoplastic cells | B-cell lymphoproliferative disorders |
| Th1/Th2 cell imbalance | Acute myeloid leukaemia, B-cell chronic lymphatic leukaemia, Hodgkin’s lymphoma, cutaneous T-cell lymphomas, chronic myeloid leukaemia |
| Increased Th17 cell activity | Hodgkin’s and non-Hodgkin’s lymphomas, acute leukaemia, B-cell chronic lymphatic leukaemia |
| Cytokine deregulation | Myeloid and lymphoid malignancies |
| Haematological treatments, including allogenic stem cell transplantation and immune checkpoint inhibitors (not extensively discussed in the present review) | Myeloid and lymphoid malignancies |
Th T helper cell, T-reg T-regulatory cell
Peripheral T-cell tolerance involves a complex immunologic mechanism that prevents autoreactive T cells to be activated in peripheral tissues and cause pathology. Specifically, when a naïve self-reactive T cell escapes from the negative selection in the thymus, this usually becomes anergic if it does not encounter an antigen-presenting cell offering sufficient co-stimulatory signals. Interestingly, neoplastic cells from B-cell chronic lymphatic leukaemia and non-Hodgkin lymphomas can serve as excellent antigen-presenting cells, owing to up-regulation of class I and II major histocompatibility molecules, β2 microglobulin and co-stimulatory molecules, such as B7. Collectively, these molecules promote expansion and differentiation of naïve T cells into effector cells [7, 8]. As a means to evade the immune surveillance, HM are able to recruit and activate interleukin (IL)-10-producing T-regulatory (T-reg) cells. Although this mechanism would in theory prevent autoimmunity phenomena, a recent study on chronic lymphatic leukaemia demonstrated that activated T-reg cells are also able to produce IL-4 and IL-17, suggesting they may promote both tolerance against the tumour and inflammation in the periphery [9]. It is also remarkable that T-reg cells are more susceptible compared with other T-cell subsets to the effects of chemotherapeutic agents, such as fludarabine [7].
There is compelling evidence that neoplastic B cells can recognise various antigens, either foreign antigens or self-antigens. Antigen-dependent stimulation of the B-cell receptor represents a fundamental mechanism that perpetuates B-cell proliferation. In parallel, some neoplastic B cells can still preserve the ability to produce antibodies against the recognised antigen, behaviour that may lead to autoantibody-mediated diseases [10]. Direct production of anti-keratinocyte antigens by tumour cells has been shown to occur in patients with PNP associated with Castleman’s disease [11].
Development of haematological tumours is associated with profound changes in the balance between various T-cell compartments and cytokine deregulation. Remarkably, single nucleotide polymorphisms in several T helper-1/T helper-2 (Th1/Th2) cytokine genes have been associated with an increased risk of non-Hodgkin’s lymphomas [12]. In patients with acute myeloid leukaemia, chronic myeloid leukaemia and B-cell chronic lymphatic leukaemia, there is an increase in the Th2/Th1 ratio that is further augmented during disease progression and in refractory patients [13–15]. Likewise, a shift towards a Th2-type response is observed in patients with Hodgkin’s lymphoma and cutaneous T-cell lymphomas [16]. Hyperactivation of Th2 cells may be responsible for eosinophil recruitment in the skin in eosinophilic dermatosis of haematologic malignancies (EDHM), and for stimulating autoantibody production in HM-associated autoimmune bullous dermatoses. In contrast, studies on chronic myelogenous leukaemia demonstrated increased levels of IL-1 and IL-6, which have been shown to be crucial molecules involved in the pathogenesis of neutrophilic dermatosis [17, 18]. Furthermore, serum levels of IL-17 are elevated in patients with acute myeloid and lymphoid leukaemia compared with healthy donors [19, 20]; similarly, growth and expansion of neoplastic cells in Hodgkin’s lymphoma and B-cell chronic lymphocytic leukaemia are associated with a tumour microenvironment enriched in T helper-17 cells [21, 22]. These cells likely play an important role in several diseases of the skin, including neutrophilic dermatoses and autoimmune bullous dermatoses.
Finally, it should be considered that the emergence of immune-mediated skin diseases can be the consequence of haematological treatments. Examples are patients undergoing allogenic stem cell transplantation, experiencing ‘graft vs host disease’ or those treated with immune checkpoint inhibitors, the latter being associated with an increased risk of developing autoimmune bullous skin diseases, such as bullous pemphigoid (BP) [23, 24].
Based on these findings, it is arguable that the risk of developing an immune-mediated disease of the skin in association with an HM depends on several factors, including genetic and epigenetic factors, the subtype of HM and the impact of the particular treatments [25]. This review provides an up-to-date overview on the main immune-mediated skin diseases associated with HMs. We show that HMs can induce different prototypes of skin inflammation, including neutrophilic and eosinophilic dermatoses and autoantibody-mediated and T-cell-dependent skin diseases.
Neutrophilic Dermatoses
Neutrophilic dermatoses (NDs) are a heterogenous group of disorders hallmarked clinically by polymorphic lesions and histologically by neutrophil-rich tissue infiltrates [26]. Although the pathomechanisms involved in these conditions remain still incompletely defined, an overexpression of proinflammatory cytokines, such as IL-1, IL-8, IL-17 and tumour necrosis factor-α, which amplify neutrophil recruitment and accumulation in the skin, is regarded as the primum movens in the pathophysiological scenario [27, 28]. Neutrophilic dermatoses are currently regarded as polygenic autoinflammatory conditions based on the fact that they share mutations with classic autoinflammatory diseases, which encompass a spectrum of forms characterised by sterile neutrophil-driven inflammation in the affected organs and are due to mutations involving genes regulating the innate immunity [29–31]. The autoinflammatory paradigm is further supported by studies on mouse models of ND [32–36]. Although innate immunity dysfunction is pivotal in the pathogenesis of NDs, the contributing role of adaptive immunity is currently recognised [37–39], mainly based on an overexpression of Th1-related cytokines in the lesional skin of pyoderma gangrenosum (PG) [39].
The association between NDs and HMs is widely acknowledged. The onset of the HM may follow, precede or be concomitant with the diagnosis of ND [40]. Neutrophilic dermatoses most commonly associated with HMs are: (i) PG; (ii) Sweet syndrome (SS); (iii) erythema elevatum diutinum; (iv) subcorneal pustular dermatosis; and (v) neutrophilic eccrine hidradenitis (Fig. 1) [41]. Beyond these entities, other NDs such as neutrophilic panniculitis and immunoglobulin A (IgA) pemphigus [42–44] have been more rarely reported to arise in the context of HMs. Overlap/transitional forms between the different entities as well as extracutaneous localisations have also been reported in association with underlying HMs [45].
Fig. 1.

Spectrum of neutrophilic dermatoses: a pyoderma gangrenosum; b Sweet syndrome; c erythema elevatum diutinum; and d subcorneal pustular dermatosis
Myeloid malignancies, such as acute myeloid leukaemia, chronic myeloproliferative neoplasms, myeloproliferative syndromes and myelomonocytic leukaemia, are the HMs most frequently associated with NDs [46], followed by monoclonal gammopathies and multiple myeloma [47, 48], and, less frequently, chronic or acute lymphoid neoplasms [49, 50]. Increasing evidence indicates that in myeloid neoplasm-associated NDs, skin-infiltrating neutrophils may be clonally related to the neoplastic cells and may have differentiated from them [51–53].
In monoclonal gammopathy-associated NDs, the monoclonal immunoglobulin itself does not seem to be directly responsible for the cutaneous manifestations, while a peculiar inflammatory profile with increased rates of cytokines such as IL-6, vascular endothelial growth factor, epidermal growth factor, intercellular adhesion molecule-1, granulocyte-colony stimulating factor and monocyte chemoattractant protein-1 have been postulated to be either the cause or the consequence of skin symptoms [47]. This is particularly true for paraneoplastic SS, which has been frequently described as associated with high levels of cytokines such as granulocyte-colony stimulating factor, IL-4, IL-6 and interferon-γ, some of which have been reported to be produced by neoplastic cells [54–57]. However, malignancy-associated SS has been also hypothesised to be a hypersensitivity reaction to tumour antigens [58]. Finally, NDs, especially SS but also PG and neutrophilic eccrine hidradenitis, may be induced by drugs, such as azacytidine [59], imatinib [60], lenalidomide and granulocyte-colony stimulating factor [61, 62], used for the treatment of HMs or related conditions [63, 64].
Pyoderma Gangrenosum
Pyoderma gangrenosum is a rare inflammatory ND characterised clinically by ulcers with undermined erythematous-violaceous edges (Fig. 1a), and histologically by neutrophil-rich infiltrates. In a retrospective cohort study on 313 patients, Langan et al. reported that haematological disorders had a prevalence of 3.9% [65]. In a systematic review published in 2015 including 823 cases of PG, HMs have been found to be associated with this condition in 12.5% of patients, following inflammatory bowel diseases (n = 537; 65.2%) and polyarthritis (n = 133; 16.1%) [66]. Haematological malignancies associated with PG, which are a major cause of death in this disease [41, 67], are mainly represented by acute and chronic myeloid leukaemia [68, 69], myelodysplastic syndrome [70], multiple myeloma [71], monoclonal gammopathy of undetermined significance [72] and lymphoma [73]. Although IgG isotype gammopathies are overall more frequent, most monoclonal gammopathies associated with PG are of the IgA isotype [47, 74].
In a recent systematic review of 340 cases of HM-associated PG [74], the most frequently observed neoplasm was myelodysplastic syndrome (n = 83; 24.4%), followed by monoclonal gammopathy of undetermined significance (n = 75; 22.1%) and acute myeloid leukaemia (n = 39; 11.5%). In the same systematic review, classic (ulcerative) PG was the most common presentation, while the bullous variant represented the most frequent form observed in the subgroup with acute myeloid leukaemia [74].
This is in contrast to the findings of Bennett et al., who concluded in a study of 86 patients that haematological diseases or malignancies were associated with BP more frequently than with classic PG [75]. The systematic review by Montagnon et al. [74] showed also that the diagnosis of HMs preceded the onset of PG in most cases and that most patients with available data (75%) achieved PG control with systemic corticosteroids, while chemotherapy alone led to healing of PG in only 7.5% of cases. Cyclosporine and TNF-α inhibitors are not recommended in patients with HM-associated PG [41].
Sweet Syndrome
It is acknowledged that SS, a rare skin disease characterised by painful erythematous plaques (Fig. 1b) showing a neutrophilic dermal infiltrate and associated with fever and neutrophilia, is associated with HMs, mostly acute myeloid leukaemia [76, 77] but also myelodysplastic syndrome [78], with a prevalence range of 15–27% according to case series [79, 80]. Chronic myeloid leukaemia, B-cell chronic lymphatic leukaemia, monoclonal gammopathies (mainly immunoglobulin G [IgG] type) and multiple myeloma have also been described as associated with SS. Cutaneous manifestations typically occur months to years before the HM diagnosis (but may also be concomitant with the onset of the HM) and recur after clinical remission during haematological relapses [79]. Idiopathic SS and HM-associated SS share common clinical features, being atypical variants, such as bullous or ulcerated SS, equally frequent in the two forms [81]. However, rare cases of SS presenting with widespread, giant cellulitis-like features have been described mainly in association with HMs [82, 83]. Response to treatment and the rate of recurrences are also similar [81]. Histiocytoid SS is a variant of this disease clinically similar to ‘classic’ SS and histopathopathologically characterised by a dermal infiltrate rich in immature myelomonocytic cells with histiocytoid morphology [84]. This variant has been demonstrated to be more frequent in patients with HMs, in particular those with myelodysplastic syndrome [85]. Therapy of HM-associated SS mainly relies on systemic corticosteroids and control of the underlying HM. The onset of SS in the context of HMs is not a factor of poor prognosis [86].
Erythema Elevatum Diutinum
Erythema elevatum diutinum, a chronic localised form of leukocytoclastic vasculitis typically presenting with red-to-violaceous papules, nodules or plaques on the extensor surfaces of the limbs [87] (Fig. 1c), has been reported as occurring in the context of a wide range of HMs, mainly represented by monoclonal gammopathies, particularly of the IgA isotype [88, 89], and myelodysplastic syndrome [90]. Other HMs more rarely described as associated with erythema elevatum diutinum are multiple myeloma [91], B-cell chronic lymphatic leukaemia [92], chronic myeloid leukaemia [93] and non-Hodgkin’s lymphoma [89, 94]. Dermatologic manifestations usually precede the onset of the HM by years [89]. First-line treatment is represented by oral dapsone.
Subcorneal Pustular Dermatosis
Subcorneal pustular dermatosis is a rare chronic-relapsing pustular eruption characterised by subcorneal flaccid pustules that contain neutrophils on histopathology (Fig. 1d) [95]. It may be associated with monoclonal gammopathies, most often of the IgA isotype, which represent the most common associated HM [96, 97]. Haematological malignancies less frequently reported as related to subcorneal pustular dermatosis are IgA myeloma [98], aplastic anaemia [99], lymphomas [100] and chronic lymphocytic leukaemia [101]. Cutaneous signs may appear years before the diagnosis of the HM and may improve with the treatment of the underlying neoplasm [95]. Oral dapsone remains the mainstay of treatment, while topical corticosteroids, phototherapy, oral corticosteroids and acitretin are second-line therapies [95].
Neutrophil Eccrine Hidradenitis
Neutrophil eccrine hidradenitis is a rare neutrophilic dermatosis characterised clinically by infiltrated or edematous SS-like papules or plaques and histologically by aseptic neutrophilic infiltrates localised around eccrine coils and glands; the lesions are asymptomatic, pruritic or painful, and develop on the trunk, face or extremities [102]. In a literature review of 51 cases, 67% had acute myeloid leukaemia and had received chemotherapy [102]. Other associated haematological neoplasms were B-cell chronic lymphatic leukaemia, chronic myelomonocytic leukaemia, Hodgkin’s lymphoma and non-Hodgkin’s lymphoma [102]. Most patients (84%) had been treated with chemotherapy (particularly cytarabine and anthracyclines) before the onset of neutrophil eccrine hidradenitis [102]. Associated fever, which is frequent, may be in most cases related to neutropenia. Generally, patients with neutrophil eccrine hidradenitis exhibit a self-resolving course within a few days or weeks and no specific treatment is required [102].
Eosinophilic Dermatoses and Related Skin Manifestations
Eosinophilic Dermatoses of Haematological Malignancies
Eosinophilic dermatoses of HMs encompass a heterogeneous group of cutaneous eruptions that predominantly occur in patients with indolent lymphoproliferative disorders, mostly B-cell chronic lymphatic leukaemia [103]. The condition was first described by Weed in 1965, reporting on ten patients with B-cell chronic lymphatic leukaemia experiencing unusual delayed hypersensitivity reactions to mosquito bites [104]. Until then, several other authors had reported similar clinical manifestations, collectively referred to as “insect bite-like reactions”, in patients with haematological disorders without a clear history of insect bites [105, 106]. The term EDHM can be viewed as an umbrella concept, as these cutaneous eruptions show non-specific clinical findings but share the presence of eosinophil-enriched inflammatory infiltrates on histopathology. The diagnosis is straightforward when other causes of tissue eosinophilia (e.g. drug reactions, scabies and BP) have been excluded [107].
Besides B-cell chronic lymphatic leukaemia, where EDHM is estimated to affect 6–8% of patients, and B-cell lymphoproliferative disorders, EDHM can also occur in association with other HMs, such as T-cell lymphomas, acute leukaemia, chronic myeloproliferative disorders and plasma cell dyscrasias [103, 105, 108, 109]. The disease usually manifests months to years after the diagnosis of the HM; however, a minority of patients experienced the skin eruption up to 10 years prior to the haematological diagnosis [105, 110]. This is a particularly interesting finding because it raises questions as to whether patients affected by ‘idiopathic’ eosinophilic dermatoses should be monitored closely because of the risk of developing a lymphoproliferative disorder. There are contrasting data regarding the association of EDHM with the clinical course of the HM. Large case series failed to identify correlations between skin lesion onset or recurrence and HM activity [103, 105, 110]. Isolated case reports conversely described the occurrence or worsening of EDHM during phases of high tumour burden or transition from indolent to aggressive forms [111]. A lack of prospective studies means it is still uncertain whether patients with haematological disorders with EDHM have a different prognosis compared to patients without EDHM.
On clinical examination, EDHM presents with papules, vesicles, blisters, plaques or nodules that characteristically develop on an erythematous urticarial background. The eruption is often widespread and can affect both photo- and non-photo-exposed areas. Patients always experience intense and often intolerable pruritus. Revision of the published cases allows the identification of at least three main clinical patterns: (i) a bullous pattern, characterised by a predominance of vesiculobullous lesions closely resembling BP (Fig. 2a); (ii) an insect-bite like pattern, characterised by discrete urticarial papules sometimes with overwhelming vesicles resembling papular urticaria (Fig. 2b); and (iii) a cellulitis-like pattern, characterised by thick urticarial plaques or nodules resembling Wells syndrome (Fig. 2c) [112]. In all the cases, histopathology indicates a variably dense, inflammatory dermal infiltrate with a predominance of lymphocytes and eosinophils. Sometimes, flame figures are visible (Fig. 3a, b) [103, 112]. Additional histopathologic features include dermal oedema, dermal-epidermal detachment, eosinophilic spongiosis and parakeratosis [113]. In patients presenting with blisters and histological evidence of dermal-epidermal detachment, direct immunofluorescence of perilesional skin or serum tests usually fail to identify autoantibodies against BP180 or other proteins of the dermal–epidermal junction [114]. This means EDHM can be distinguished from BP or other subepidermal autoimmune bullous disorders. In Asian countries, a distinct presentation of EDHM includes “eosinophilic pustular folliculitis of HM”. It features pruritic, reddish, sterile, follicular papules and pustules; microscopic examination shows an inflammatory infiltrate consisting of lymphocytes and eosinophils around the hair follicles and sebaceous glands, similar to Ofuji disease [115, 116]. Another presentation of EDHM resembling varicella zoster infection has also been reported [117].
Fig. 2.

Main clinical pattern of eosinophilic dermatosis of haematological malignancies: a bullous pemphigoid-like pattern; b insect bite-like pattern; and c eosinophilic cellulitis-like pattern
Fig. 3.

a Histopathological presentation of eosinophilic dermatoses of haematological malignancies, showing intense dermal oedema and a mixed inflammatory infiltrate in the dermis with numerous eosinophils and flame figures; intra-epidermal blistering is also visible (haematoxylin and eosin staining ×20) and b detail of the flame figures (haematoxylin and eosin staining ×40)
The pathogenesis of EDHM is far less clear to date. Many patients refer to a temporal association between the manifestation of EDHM and mosquito bites. Interestingly, experimental studies indicate that mosquito salivary gland extracts activate T-cell proliferation and the release of IL-5 in patients with Wells syndrome [118, 119]. Considering the clinical overlap between Wells syndrome and EDHM, it would be reasonable to think that a similar mechanism may work also for EDHM. Other authors believe that chemotherapeutics may serve as a trigger for EDHM [105]. However, evidence for a direct association between EDHM and drugs comes only from a study by Rossini et al., describing three patients with hairy cell leukaemia treated with cladribine [120].
One of the most intriguing debates concerns the role of leukaemic cells in EDHM. Many studies of patients with B-cell chronic lymphatic leukaemia with EDHM demonstrated the presence of neoplastic B cells within the inflammatory infiltrate of the skin lesions [110, 113, 121]. However, this is not a specific finding of EDHM, as these cells can be similarly detected in many inflammatory skin diseases as well as in close proximity of skin cancers occurring in patients with B-cell chronic lymphatic leukaemia [122]. One hypothesis suggests that leukaemic cells infiltrate the skin and drive either Th2 cells or eosinophil and basophil activation, e.g. by releasing antibodies against the high-affinity immunoglobulin E receptor (FceRI) [123, 124].
An alternative hypothesis is that circulating leukaemic cells or the treatment of HMs may cause a persistent skewing of the T-cell repertoire towards the Th2 phenotype, and this may favour T-cell recruitment in the skin and activation of eosinophils in response to environmental triggers, with infiltrating leukaemic cells acting as bystander cells. Both scenarios appear possible [105, 124]. The first is supported by cases of EDHM worsening during phases of high tumour burden, where sometimes important infiltration of the skin by neoplastic elements could be detected. The second scenario is supported by cases, the vast majority where EDHM occurs while the HM is in partial or complete remission, and minimal residual activity of neoplastic cells is expected to sustain the Th2 cell response [124].
Treatment of EDHM is at present challenging, owing to the lack of evidence-based recommendations [125]. Systemic and topical corticosteroids are considered frontline therapies. Immunosuppressants such as methotrexate and azathioprine are also effective and allow a reduction in the dose of systemic corticosteroids needed to control the disease and relieve symptoms [103]. One case report supports the efficacy of lenalidomide [126].
More intriguingly, two cases described successful treatment of EDHM with dupilumab, an anti-IL4 and IL-13 receptor monoclonal antibody approved for use in atopic dermatitis [127]. The efficacy of dupilumab further strengthens the hypothesis that Th2-cell activation is a crucial step toward EDHM pathology and opens the way for a ‘targeted’ approach to EDHM treatment.
T-Cell Papulosis Associated with B-Cell Malignancies
The term “T-cell papulosis associated with B-cell malignancies” was proposed by Visseaux et al. in 2018, reporting 38 cases of cutaneous eruptions occurring in patients with underlying B-cell lymphoproliferative diseases, especially B-cell chronic lymphatic leukaemia. On clinical examination, these cases were characterised by pruritic papules, vesicles, plaques and nodules, similar to EDHM. Like EDHM, the skin rush runs a chronic relapsing course despite treatments; histopathology showed a prominent T-cell infiltration, sometimes resembling cutaneous T-cell lymphoma and, in particular, folliculotropic mycosis fungoides (Fig. 4). Despite EDHM, eosinophils were absent in up to 30% of the cases [110]. It is our clinical experience (unpublished data) that patients with EDHM can present at relapse as “T-cell papulosis” or vice versa, suggesting that both diseases are “different sides of the same coin”.
Fig. 4.

a Clinical presentation of T-cell papulosis associated with B-cell malignancies in a patient with B-cell chronic lymphocytic leukaemia and b–d microscopic examination showed a prominent lymphocytic inflammatory infiltrate with perifollicular distribution resembling folliculotropic mycosis fungoides (haematoxylin and eosin staining ×20)
Need for a Unifying Nomenclature
The observation by Visseaux et al. highlights the need to find a unifying nomenclature that would help clinicians and pathologists to promptly recognise this extremely polymorphic spectrum of cutaneous eruptions. As previously mentioned, different names have been proposed across the years, including exaggerated arthropod bite reactions, insect bite-like reactions and eosinophilic dermatoses of myeloproliferative diseases [124, 125]. More recently, the acronym “He Remained” (Haematologic Related Malignancy-Induced Eosinophilic Dermatoses) has been also proposed to highlight the pathogenetic link between skin manifestations and the presence of the HM [128]. A potential consequence of having a large number of names for the same condition is that they can generate much confusion among clinicians [125]. We respectfully believe that the term “polymorphic eruption of haematologic malignancies” may be best suited to describe this entity. A proposed classification of “polymorphic eruption of haematologic malignancies” is explained in Table 2.
Table 2.
Classification of polymorphic eruption of haematological malignancy
| Eosinophilic | Lymphocytic |
|---|---|
|
Bullous (bullous pemphigoid-like) Panniculitis (Wells syndrome-like) Insect bite-like Eosinophilic pustular folliculitis (Ofuji disease-like) |
T-cell papulosis associated with B-cell malignancies |
Autoimmune Blistering Dermatoses
Paraneoplastic Pemphigus
Paraneoplastic pemphigus is a rare devastating autoimmune disorder of the skin and mucosa, which occurs in patients with an underlying malignancy, although it has been sporadically described even in the absence of tumours [129, 130]. Paraneoplastic pemphigus can occur in association with several types of malignancies. However, the most frequently reported association is with HMs, and in particular, non-Hodgkin’s lymphoma, B-cell chronic lymphatic leukaemia and Castleman’s disease [131–133]. For a long time, PNP has been regarded as a distinct entity within the clinical spectrum of pemphigus, as, like ‘classic’ pemphigus subtypes, it is characterised by suprabasal acantholysis of the skin and mucosa due to IgG autoantibodies targeting adhesion proteins involved in keratinocyte-keratinocyte adhesion [134, 135]. However, there are several clinical, histologic and immunologic features that clearly distinguish PNP from other pemphigus forms: mucosal involvement is generally more severe and recalcitrant to immunosuppressive treatment; skin lesions vary from superficial to tense blisters, resembling pemphigus or BP, respectively, to lesions mimicking erythema multiforme, toxic epidermal necrolysis, lichen planus and graft vs host disease (Fig. 5). Besides acantholysis, PNP shows a lichenoid lymphocytic infiltrate along the dermal–epidermal junction by histopathology [136, 137].
Fig. 5.

Paraneoplastic pemphigus a with extensive haemorrhagic multiform blisters and erosions of the trunk and b extensive fibrinous erosions of the tongue
Serologically, PNP is only rarely associated with IgG antibodies against desmoglein 3 and desmoglein 1, which are the main antigenic targets of pemphigus vulgaris and foliaceus respectively, whereas IgG autoantibodies against envoplakin, periplakin and the protease inhibitor, alpha-2-macroglobulin-like antigen-1 are more frequently detected. Other IgG autoantibodies targeting desmocollins, BP180/BP230, and, occasionally, laminin gamma 1 may be also detected in PNP sera [138–143]. As a result, PNP requires a specific diagnostic approach, e.g. using plakin-rich substrates, such as a rat’s bladder, instead of human skin or a monkey’s esophagus, for indirect immunofluorescence, or dedicated enzyme-linked immunosorbent assay kits for detecting circulating IgG autoantibodies, the latter still only available in highly specialised centres [144, 145].
Finally, PNP is characterised by several systemic autoimmune phenomena and sometimes lethal complications, such as bronchiolitis obliterans, which have led researchers to propose the acronym paraneoplastic autoimmune multi-organ syndrome instead of PNP [136]. Besides bronchiolitis obliterans, other systemic manifestations of paraneoplastic autoimmune multi-organ syndrome include myasthenia gravis, ocular complications, such as corneal ulcerations, cicatrizing conjunctivitis, symblepharon, and pterygium, and rarely glomerulosclerosis and paraneoplastic neurological syndrome [146]. Paraneoplastic autoimmune multi-organ syndrome has been rarely described in pediatric patients with HMs, in particular, Castleman’s disease tumours [147–150].
From a pathogenetic point of view, while in pemphigus T-cell activation is of the Th2-cell type and mainly engaged in stimulating IgG autoantibody production by autoreactive desmoglein-specific B cells, PNP demonstrates a mixed Th1- and Th2-type response as well as an activation of CD8+ cytotoxic T cells. These differences may at least partly explain the presence of lichen planus-like lesions, as well as the fact that anti-B-cell therapies, e.g. rituximab, are less effective than in other clinical pemphigus variants [151–153]. Skin and mucosal lesions with lichen planus-like characteristics, mirroring the Th1-type and cytotoxic T-cell response, may present as the predominant clinical feature in patients with PNP who have already received substantial immunosuppressive treatment, which may lead clinicians to overlooking the diagnosis of PNP [154].
Overall, the course of the disease does not strictly follow that of the underlying neoplasm, nonetheless there is a general consensus that early treatment of the neoplasm, especially in the presence of Castleman’s disease or thymoma, followed by rapid immunosuppressive treatment, will dampen PNP symptoms [155, 156]. Based on the evidence of increased levels of IL-6 in PNP sera and the crucial role of IL-6 in Castleman’s disease [157, 158], tocilizumab, a monoclonal antibody targeting IL-6, has been successfully applied as a treatment for PNP in this context, but seems to have limited effect in preventing bronchiolitis obliterans [159, 160]. Rituximab, with or without chemotherapy, represents an effective therapeutic option for PNP [146, 161]; thymoma-associated PNP or multi-organ syndrome also respond well to rituximab treatment, but a significant proportion of these patients still die as a result of infections and sepsis [137].
Paraneoplastic pemphigus is characterised by a high mortality rate (ca. 57%) and most deaths occur within the first year after diagnosis. The presence of anti-envoplakin antibodies, a toxic epidermal necrolysis-like clinical pattern, a BP-like clinical pattern and bronchiolitis obliterans is significantly associated with a poor disease outcome [162].
Autoimmune Bullous Dermatoses Other Than Paraneoplastic Pemphigus
Apart from PNP, other autoimmune bullous dermatoses have been associated with HMs. As opposed to PNP, autoimmune bullous dermatoses occurring in patients with haematological disorders do not show any specific clinical or serologic features as compared to their counterparts occurring in patients without haematological disorders.
With regard to pemphigus, a large population-based study identified a significant correlation with lymphoproliferative disorders, and in particular B-cell chronic lymphatic leukaemia [163]. Likewise, in one meta-analysis study, a significant association emerged between BP and HMs, with B-cell chronic lymphatic leukaemia as again the most represented [164]. In both studies, data were extrapolated by national registries or by analysing case series and small cohort studies, with the obvious limitation of the lack of direct validation of the diagnosis of pemphigus and BP by immunopathology investigations [165]. A potential bias of these studies might be the overestimation of these associations because of an incorrect diagnosis. While this risk is low for pemphigus, it may be considerably higher for BP, given its clinical and pathological overlap with EDHM, which, not by chance, occurs preferentially in patients with B-cell chronic lymphatic leukaemia. Collectively, these findings highlight the need for prospective studies to further validate the association between HMs and both pemphigus and BP, as well as the importance of referring patients with haematological disorders with suspected autoimmune bullous dermatoses to specialised centres, where the diagnosis can be confirmed by appropriate immunopathology parameters.
The association between HMs and subepidermal autoimmune bullous dermatoses other than BP mostly relies on single case reports or small series, and almost always involves lymphoproliferative disorders. Worth mentioning are cases of linear IgA bullous dermatosis associated with B-cell chronic lymphatic leukaemia, acute lymphoblastic leukaemia and Hodgkin’s lymphoma [166–169]; epidermolysis bullosa acquisita with multiple myeloma, mantle cell lymphoma and B-cell chronic lymphatic leukaemia [170–174]; laminin 332-type mucous membrane pemphigoid with non-Hodgkin’s lymphoma; and dermatitis herpetiformis with non-Hodgkin’s lymphoma of the gastrointestinal tract [175].
Miscellaneous
Cutaneous Vasculitis
Cutaneous vasculitis is characterised by inflammation, damage and destruction of blood vessels of different sizes [176]. Cutaneous vasculitis is a quite common disease with an estimated incidence of 39.6/59.8 new cases/million persons/year. Most cases are related to drugs (20%), infections (22%) and connective tissue diseases (12%), while 5% of cutaneous vasculitis occur in patients with underlying malignancies [177–181]. Such cases are classified as paraneoplastic vasculitis. Most of these patients have an underling HM (90%), such as chronic myelomonocytic leukaemia, non-Hodgkin’s lymphoma, Hodgkin’s disease, B-cell chronic lymphatic leukaemia, and multiple myeloma [176, 178, 182, 183]; whereas, paraneoplastic cutaneous vasculitis rarely occurs in patients with solid tumours (10% of cases), the most common being renal, breast and lung carcinomas [184–186].
Paraneoplastic cutaneous vasculitis usually presents simultaneously with the associated haematological disease. It shows similar clinical features of non-malignancy-related cutaneous vasculitis, including palpable purpura, ulcers and urticarial vasculitis, with a prominent involvement of the lower limbs (Fig. 6) [187, 188]. However, paraneoplastic cutaneous vasculitis tends to last longer than non-malignancy-related cutaneous vasculitis and is less responsive to glucocorticoid or immunosuppressive treatment. Systemic symptoms such as fever, arthralgia, general malaise and renal involvement may also occur. By histopathology, the usual clues include, like classic cutaneous vasculitis, acute necrotising inflammation of the small vessels, leukocyte perivascular infiltration, nuclear dust and fibrinoid necrosis of wall vessels [189]. As we previously described for EDHM, leukaemic cells, especially in cutaneous vasculitis associated with chronic and acute lymphatic leukaemia, may be observed within the perivascular inflammatory infiltrate. This may pose a relevant diagnostic challenge to leukaemic vasculitis, which is a rare presentation of leukaemia cutis. Distinguishing between both entities is of crucial importance because leukaemic vasculitis is associated with a poor prognosis, reflecting the aggressive biological behaviour of the associated blood disorder [190–192]. The pathogenesis of cutaneous vasculitis in the setting of an HM remains far less clear until now. One possible hypothesis is that neoplastic B cells produce antibodies that may form circulating immune complexes or rather react directly with endothelial antigens in the skin [193]. Of note, HMs augment blood viscosity, which can impair the clearance of normally produced immune complexes, and lead to their accumulation on small blood vessels [178, 184, 194–196].
Fig. 6.
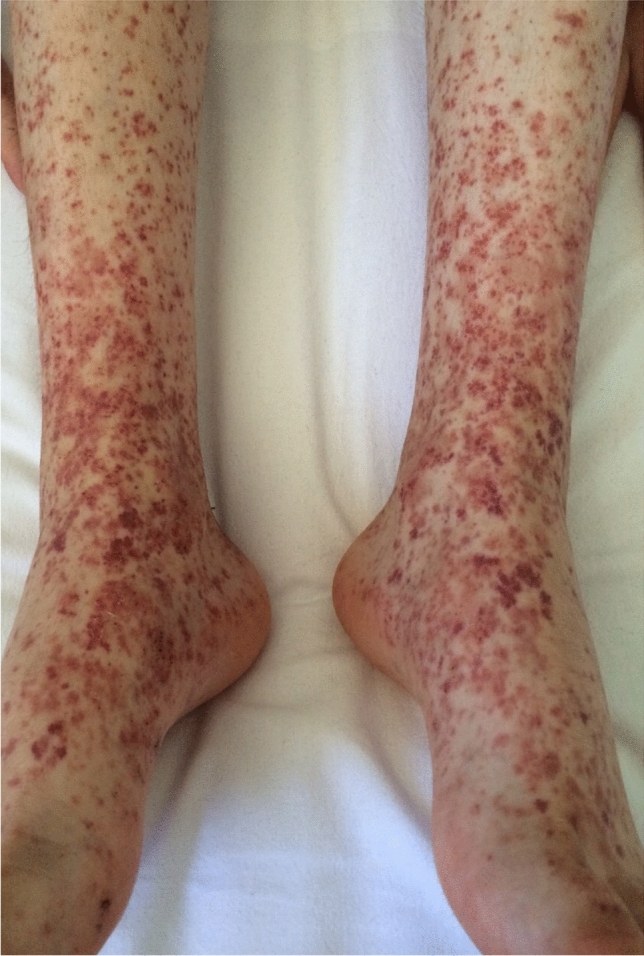
Palpable purpura consistent with cutaneous leukocytoclastic vasculitis in a patient with follicular non-Hodgkin’s lymphoma
It should be also remembered that, besides cutaneous vasculitis, HMs can induce a broad spectrum of systemic vasculitis, which can present with a secondary involvement of the skin, including giant-cell arteritis, large- and medium-vessel vasculitis, anti-neutrophil cytoplasmic antibody-associated vasculitis and Behçet’s disease [192, 197–202].
Connective Tissue Diseases
There is a mutual relationship between connective tissue diseases and HMs. On the one hand, patients with a long-term history of connective tissue diseases including lupus erythematosus (LE), Sjogren’s syndrome and systemic sclerosis demonstrate an increased risk of HMs, with special regard to B-cell lymphoma [203–206]. In patients with systemic LE, the closest association is with diffuse large B-cell lymphoma [207]. On the other hand, marginal zone lymphomas and those associated with mucosal-associated lymphoid-tissue lymphomas are the predominant subtypes observed in patients with Sjogren’s syndrome, where the incidence is up to 44-fold higher than the general population [208, 209]. The pathogenesis of B-cell neoplasms in patients with connective tissue diseases is likely multifactorial, including some shared genetic/environmental factors, chronic inflammation, long-term immunosuppression and persistent high levels of cytokines such as IL-6, BAFF, APRIL and Blc2, which are involved in both autoimmune disease and lymphomagenesis, and, for systemic LE, Epstein–Barr virus infection [204, 207].
However, connective tissue diseases can rarely occur as a paraneoplastic manifestation of an HM. With regard to the various forms of cutaneous LE, subacute cutaneous LE is the most frequently reported as a paraneoplastic phenomenon, but only a few cases of paraneoplastic subacute cutaneous LE have been reported in patients with underlying HMs, including acute myeloid leukaemia and Hodgkin’s lymphoma [210, 211]. Likewise, only sporadic cases do exist about localised or systemic scleroderma as a paraneoplastic manifestation of HMs [212].
Dermatomyositis is widely regarded as facultative paraneoplastic dermatosis. Dermatomyositis is a complex multi-system disease primarily affecting the proximal muscles and the skin. Characteristic skin manifestations include a heliotrope rash (Fig. 7a), shawl and holster signs, Gottron’s papules and erythroderma. Malignancies occur in 13–42% of patients with dermatomyositis, with the highest risk during the first and second year after diagnosis [213]. Although most of the cases of malignancy-associated dermatomyositis occur in patients with solid tumours, both classic and amyopathic dermatomyositis have been shown to rarely occur in patients with HMs, including non-Hodgkin’s lymphomas, Hodgkin’s disease, myeloid leukaemia and myelodysplastic syndromes [214–217]. The presence of erythroderma in dermatomyositis is a clinical hallmark of malignancy, especially of lymphoid origin [213]. In one study, HM-associated dermatomyositis demonstrated a poor prognosis, with only half of the patients still alive after 5 years. In that study, joint and lung involvement and anti-Jo1 antibodies were less commonly exhibited by the patients, suggesting that anti-synthetase syndrome may be a protective factor against HMs in patients with dermatomyositis [215].
Fig. 7.

a Heliotrope rash in a patient with dermatomyositis who developed non-Hodgkin’s lymphoma 2 years after the diagnosis and b flagellate erythema involving the upper arms in a young boy with Hodgkin’s lymphoma receiving a bleomycin-containing regimen
It should be also remembered that dermatomyositis-like eruptions can be observed in patients with chronic myeloproliferative diseases treated with hydroxyurea [218, 219]. Flagellate erythema, an uncommon sign of dermatomyositis, can be also observed in patients receiving bleomycin-based chemotherapeutic regimes, such as in patients with Hodgkin’s disease (Fig. 7b) [220].
Granulomatous Dermatoses
Granulomatous dermatoses comprise a wide range of clinically and aetiologically distinct disorders characterised by a granulomatous dermal inflammatory infiltrate predominantly composed of histiocytes and macrophages [221, 222]. Albeit rare, associations with HMs have been found for various granulomatous dermatoses, including granuloma annulare, cutaneous sarcoidosis, interstitial granulomatosis and palisaded neutrophilic granulomatous dermatitis. While these disorders present with different clinical and histological presentations, the subtypes of associated HMs are similar, and include Hodgkin’s and non-Hodgkin’s lymphoma, myelodysplastic syndromes and chronic myelomonocytic leukaemia [223–229]. Hodgkin’s lymphoma is a unique example of a malignant tumour in which neoplastic cells account for less than 10% of the entire tumoural mass, while the remaining 90% is mostly composed of reactive lymphocytes and macrophages [230]. Interestingly, patients with Hodgkin’s lymphoma can also experience granuloma formation in the lung, liver, lymph nodes and spleen, and this may be a consequence of the ability of Reed–Stemberg cells to produce cytokines and chemokines that also stimulate recruitment of macrophages in peripheral tissues including the skin [231]. The association with myelodysplastic syndromes may be related to the activation of T-reg cells and the downregulation of T helper-17 cells that are characteristically found in the peripheral blood of the patients. The percentage of T-reg cells is negatively correlated with the prognosis of the myelodysplastic syndrome, and coherently, the appearance of granulomatous dermatoses in these patients may be predictive of a poor outcome, sometimes preceding the leukaemic transformation [232–234].
Patients with sarcoidosis harbour a five times higher risk of developing lymphoproliferative disorders, in particular Hodgkin’s lymphoma. Most of these diseases occur within 1–2 years of the diagnosis of sarcoidosis and seem to have a poor prognosis compared with those occurring in patients without sarcoidosis. Remarkably, up to 60% of malignancy-associated sarcoidosis manifest with cutaneous involvement [77, 235–237]. In general, granulomatous dermatoses associated with HMs tend to be widespread (Fig. 8) [235] and less responsive to steroidal treatments.
Fig. 8.

a, b Widespread annular granuloma in a female patient with chronic myelomonocytic leukaemia
Discussion and Concluding Remarks
In this review, we show that HMs can lead to several immune-mediated phenomena affecting the skin, either alone or in the context of systemic diseases. When a dermatologist is faced with such diseases, one major question is whether the cutaneous eruptions harbour any prognostic relevance with respect to the underlying HM. In this regard, we show that some diseases, such as dermatomyositis or granulomatous dermatoses, are clearly linked to a poor outcome of the patient. This possibly reflects the fact that the emergence of the skin disease is related to unfavourable immunological alterations associated with the blood disorder, as observed for granulomatous dermatoses in the context of myelodysplastic syndromes. In other cases, such as PNP, the appearance of the skin disease may be unrelated to the biological behaviour of the associated malignancies, but carries an increased risk of death in the short term, owing to a potentially life-threatening course of the dermatosis itself. For other diseases, such as EDHM or ND, the prognostic meaning remains to be elucidated.
Based on the temporal appearance of the skin complications, we can distinguish at least two groups: diseases that parallel the clinical course of the HM and diseases that do not. One example of the first group is cutaneous vasculitis. Several cases in the literature indicate that cutaneous vasculitis usually resolves upon chemotherapy or chemo-immunotherapy of the associated HM, while recurrence of the skin lesions is usually associated with relapse of the HM [183, 238, 239]. By contrast, examples of the second group include autoimmune bullous dermatoses and PNP, and the vast majority of the cases of HM-associated neutrophilic and eosinophilic dermatoses. For these disorders, chemotherapy may not be a feasible approach; unfortunately, their treatment still relies on the use of systemic and topical corticosteroids and immunosuppressive agents. It should be noted that patients with HMs represent a particularly fragile patient population because of an elevated risk of serious infections. As a result, trying to selectively target the immune pathways leading to a specific skin manifestation is of crucial importance. For autoantibody-mediated diseases, such as PNP and autoimmune bullous dermatoses, rituximab and novel anti-B-cell-targeted therapies, including the Bruton tyrosine kinase inhibitor, represent effective strategies and target both self-reactive and neoplastic B cells [240].
Another feasible approach could be the use of intravenous immunoglobulins, which have shown remarkable efficacy in autoimmune bullous dermatoses and dermatomyositis and some data exist on the potential benefit for ND [241, 242]. Remarkably, they are also used in the haematological setting to replace the pool of circulating immunoglobulins in patients with chemotherapy-induced hypogammaglobulinemia [243].
Finally, we show that IL-4 inhibitors hold major promise as a targeted therapeutic approach in patients with EDHM, with an acceptable safety profile owing to a limited impairment of the immune system [127]. There is a prevailing sentiment that targeting the cytokine milieu of the tumour microenvironment may increase the efficacy of tumour-directed therapies. For example, experimental evidence supports targeting the IL-1-related pathways to augment the efficacy of tyrosine kinase inhibitors in patients with chronic myeloid leukaemia [6]. Likewise, IL-17 has been shown to support the growth of tumoural cells in Hodgkin’s lymphoma and B-cell chronic lymphatic leukaemia, and promote resistance to chemotherapy in patients with acute leukaemia, suggesting that patients would likely benefit from IL-17-directed therapies [19–21]. Studies in this direction provide an exciting opportunity also for dermatologists, as they pave the way for using a therapeutic approach that, at the same time, may target the autoimmune disease and interfere with the local environment that promotes tumour expansion.
We would like to highlight that, besides the diseases discussed herein, patients with HM treated with haematopoietic allogenic stem cell transplantation may experience a plethora of skin diseases with an immune-mediate pathogenesis, which, however, do not depend on the HM but rather on the immune system of the donor that reacts against the antigens of the recipient. These manifestations are collectively referred to as ‘graft vs host disease’. Skin involvement is often the earliest sign of graft vs host disease. Lesions vary from maculopapular or morbilliform rush and severe toxic epidermolysis-like (Fig. 9a) mucocutaneous eruptions in acute graft vs host disease, and from lichen-planus-like (Fig. 9b) to scleroderma-like eruptions in chronic graft vs host disease [23, 244, 245]. We believe that extensive discussion of this condition is beyond the scope of the present work, and may be addressed in a separate review.
Fig. 9.

Cutaneous graft-vs-host disease a with extensive multiform erythematous plaques and palmar hyperkeratosis, b lichenoid plaques on the lower lip, and c erosions and small bullae on the base of the tongue
To conclude, exploring the molecular mechanisms behind the interaction between neoplastic cells and the immune system is paramount to develop a targeted approach to the management of immune-mediated diseases of the skin associated with HMs. We also encourage epidemiological studies aimed at assessing the impact of these complications in patients with haematological disorders to identify underlying risk factors and eventually evaluate their possible prognostic significance.
Acknowledgements
Open access funding provided by Università degli Studi di Firenze within the CRUI-CARE Agreement.
Declarations
Funding
No funding was received for the preparation of this article.
Conflict of interest
Roberto Maglie, Giovanni Genovese, Farzan Solimani, Alba Guglielmo, Alessandro Pileri, Francesca Portelli, Michael Hertl, Angelo Valerio Marzano and Emiliano Antiga have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
References
- 1.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–2390. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 3.Moia R, Patriarca A, Schipani M, Ferri V, Favini C, Sagiraju S, et al. Precision medicine management of chronic lymphocytic leukemia. Cancers (Basel) 2020;12(3):642. doi: 10.3390/cancers12030642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zappasodi P, Forno C, Corso A, Lazzarino M. Mucocutaneous paraneoplastic syndromes in hematologic malignancies. Int J Dermatol. 2006;45:14–22. doi: 10.1111/j.1365-4632.2005.02668.x. [DOI] [PubMed] [Google Scholar]
- 5.Li AW, Yin ES, Stahl M, Kim TK, Panse G, Zeidan AM, et al. The skin as a window to the blood: cutaneous manifestations of myeloid malignancies. Blood Rev. 2017;31:370–388. doi: 10.1016/j.blre.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Schmid-Wendtner M, Hornung T, Meurer M, Wendtner C-M. Skin disorders in malignant hematologic diseases. Hautarzt. 2015;66:625–633. doi: 10.1007/s00105-015-3650-9. [DOI] [PubMed] [Google Scholar]
- 7.Maverakis E, Goodarzi H, Wehrli LN, Ono Y, Garcia MS. The etiology of paraneoplastic autoimmunity. Clin Rev Allergy Immunol. 2012;42:135–144. doi: 10.1007/s12016-010-8248-5. [DOI] [PubMed] [Google Scholar]
- 8.Thompson ED, Enriquez HL, Fu Y-X, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med. 2010;207:1791–1804. doi: 10.1084/jem.20092454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Matteis S, Molinari C, Abbati G, Rossi T, Napolitano R, Ghetti M, et al. Immunosuppressive Treg cells acquire the phenotype of effector-T cells in chronic lymphocytic leukemia patients. J Transl Med. 2018;16(1):172. doi: 10.1186/s12967-018-1545-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avalos AM, Meyer-Wentrup F, Ploegh HL. B-cell receptor signaling in lymphoid malignancies and autoimmunity. Adv Immunol. 2014;123:1–49. doi: 10.1016/B978-0-12-800266-7.00004-2. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Bu D, Yang Y, Chen X, Zhu X. Castleman’s tumours and production of autoantibody in paraneoplastic pemphigus. Lancet. 2004;363:525–531. doi: 10.1016/S0140-6736(04)15539-6. [DOI] [PubMed] [Google Scholar]
- 12.Zhu G, Pan D, Zheng T, Lan Q, Chen X, Chen Y, et al. Polymorphisms in Th1/Th2 cytokine genes, hormone replacement therapy, and risk of non-Hodgkin lymphoma. Front Oncol. 2011;1(21):21. doi: 10.3389/fonc.2011.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reuben JM, Lee BN, Johnson H, Fritsche H, Kantarjian HM, Talpaz M. Restoration of Th1 cytokine synthesis by T cells of patients with chronic myelogenous leukemia in cytogenetic and hematologic remission with interferon-alpha. Clin Cancer Res. 2000;6:1671–1677. [PubMed] [Google Scholar]
- 14.Podhorecka M, Dmoszynska A, Rolinski J, Wasik E. T type 1/type 2 subsets balance in B-cell chronic lymphocytic leukemia: the three-color flow cytometry analysis. Leuk Res. 2002;26:657–660. doi: 10.1016/s0145-2126(01)00194-1. [DOI] [PubMed] [Google Scholar]
- 15.Tian T, Yu S, Liu L, Xue F, Yuan C, Wang M, et al. The profile of T helper subsets in bone marrow microenvironment is distinct for different stages of acute myeloid leukemia patients and chemotherapy partly ameliorates these variations. PLoS ONE. 2015;10:e0131761. doi: 10.1371/journal.pone.0131761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lucey DR, Clerici M, Shearer GM. Type 1 and type 2 cytokine dysregulation in human infectious, neoplastic, and inflammatory diseases. Clin Microbiol Rev. 1996;9:532–562. doi: 10.1128/CMR.9.4.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang B, Chu S, Agarwal P, Campbell VL, Hopcroft L, Jørgensen HG, et al. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood. 2016;128:2671–2682. doi: 10.1182/blood-2015-11-679928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuepper MK, Bütow M, Herrmann O, Ziemons J, Chatain N, Maurer A, et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia. 2019;33:1964–1977. doi: 10.1038/s41375-019-0427-7. [DOI] [PubMed] [Google Scholar]
- 19.Wu C, Wang S, Wang F, Chen Q, Peng S, Zhang Y, et al. Increased frequencies of T helper type 17 cells in the peripheral blood of patients with acute myeloid leukaemia. Clin Exp Immunol. 2009;158:199–204. doi: 10.1111/j.1365-2249.2009.04011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bi L, Wu J, Ye A, Wu J, Yu K, Zhang S, et al. Increased Th17 cells and IL-17A exist in patients with B cell acute lymphoblastic leukemia and promote proliferation and resistance to daunorubicin through activation of Akt signaling. J Transl Med. 2016;14:132. doi: 10.1186/s12967-016-0894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrarini I, Rigo A, Zamò A, Vinante F. Classical Hodgkin lymphoma cells may promote an IL-17-enriched microenvironment. Leuk Lymphoma. 2019;60:3395–3405. doi: 10.1080/10428194.2019.1636983. [DOI] [PubMed] [Google Scholar]
- 22.Zhu F, McCaw L, Spaner DE, Gorczynski RM. Targeting the IL-17/IL-6 axis can alter growth of chronic lymphocytic leukemia in vivo/in vitro. Leuk Res. 2018;66:28–38. doi: 10.1016/j.leukres.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Ramachandran V, Kolli SS, Strowd LC. Review of graft-versus-host disease. Dermatol Clin. 2019;37:569–582. doi: 10.1016/j.det.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 24.Lopez AT, Geskin L. A case of nivolumab-induced bullous pemphigoid: review of dermatologic toxicity associated with programmed cell death protein-1/programmed death ligand-1 inhibitors and recommendations for diagnosis and management. Oncologist. 2018;23:1119–1126. doi: 10.1634/theoncologist.2018-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Javierre BM, Esteller M, Ballestar E. Epigenetic connections between autoimmune disorders and haematological malignancies. Trends Immunol. 2008;29:616–623. doi: 10.1016/j.it.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Marzano AV, Borghi A, Wallach D, Cugno M. A comprehensive review of neutrophilic diseases. Clin Rev Allergy Immunol. 2018;54:114–130. doi: 10.1007/s12016-017-8621-8. [DOI] [PubMed] [Google Scholar]
- 27.Maalouf D, Battistella M, Bouaziz J-D. Neutrophilic dermatosis: disease mechanism and treatment. Curr Opin Hematol. 2015;22:23–29. doi: 10.1097/MOH.0000000000000100. [DOI] [PubMed] [Google Scholar]
- 28.Marzano AV, Ortega-Loayza AG, Heath M, Morse D, Genovese G, Cugno M. Mechanisms of inflammation in neutrophil-mediated skin diseases. Front Immunol. 2019;10:1059. doi: 10.3389/fimmu.2019.01059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis) Br J Dermatol. 2017;176:1588–1598. doi: 10.1111/bjd.15226. [DOI] [PubMed] [Google Scholar]
- 30.Marzano AV, Fanoni D, Antiga E, Quaglino P, Caproni M, Crosti C, et al. Expression of cytokines, chemokines and other effector molecules in two prototypic autoinflammatory skin diseases, pyoderma gangrenosum and Sweet’s syndrome. Clin Exp Immunol. 2014;178:48–56. doi: 10.1111/cei.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140:784–790. doi: 10.1016/j.cell.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nesterovitch AB, Gyorfy Z, Hoffman MD, Moore EC, Elbuluk N, Tryniszewska B, et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am J Pathol. 2011;178:1434–1441. doi: 10.1016/j.ajpath.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nesterovitch AB, Szanto S, Gonda A, Bardos T, Kis-Toth K, Adarichev VA, et al. Spontaneous insertion of a b2 element in the ptpn6 gene drives a systemic autoinflammatory disease in mice resembling neutrophilic dermatosis in humans. Am J Pathol. 2011;178:1701–1714. doi: 10.1016/j.ajpath.2010.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukens JR, Vogel P, Johnson GR, Kelliher MA, Iwakura Y, Lamkanfi M, et al. RIP1-driven autoinflammation targets IL-1α independently of inflammasomes and RIP3. Nature. 2013;498:224–227. doi: 10.1038/nature12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tartey S, Gurung P, Dasari TK, Burton A, Kanneganti T-D. ASK1/2 signaling promotes inflammation in a mouse model of neutrophilic dermatosis. J Clin Invest. 2018;128:2042–2047. doi: 10.1172/JCI98446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tartey S, Gurung P, Samir P, Burton A, Kanneganti T-D. Cutting edge: dysregulated CARD9 signaling in neutrophils drives inflammation in a mouse model of neutrophilic dermatoses. J Immunol. 2018;201:1639–1644. doi: 10.4049/jimmunol.1800760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marzano AV, Cugno M, Trevisan V, Fanoni D, Venegoni L, Berti E, et al. Role of inflammatory cells, cytokines and matrix metalloproteinases in neutrophil-mediated skin diseases. Clin Exp Immunol. 2010;162:100–107. doi: 10.1111/j.1365-2249.2010.04201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang EA, Steel A, Luxardi G, Mitra A, Patel F, Cheng MY, et al. Classic ulcerative pyoderma gangrenosum is a T cell-mediated disease targeting follicular adnexal structures: a hypothesis based on molecular and clinicopathologic studies. Front Immunol. 2017;8:1980. doi: 10.3389/fimmu.2017.01980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antiga E, Maglie R, Volpi W, Bianchi B, Berti E, Marzano AV, et al. T helper type 1-related molecules as well as interleukin-15 are hyperexpressed in the skin lesions of patients with pyoderma gangrenosum. Clin Exp Immunol. 2017;189:383–391. doi: 10.1111/cei.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prat L, Bouaziz J-D, Wallach D, Vignon-Pennamen M-D, Bagot M. Neutrophilic dermatoses as systemic diseases. Clin Dermatol. 2014;32:376–388. doi: 10.1016/j.clindermatol.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 41.Wallach D, Vignon-Pennamen M-D, Marzano AV, editors. Neutrophilic dermatoses. Heidelberg: Springer International Publishing AG; 2018. p. 55–83. 10.1007/978-3-319-72649-6.
- 42.Uhara H, Saida T, Nakazawa H, Ito T. Neutrophilic dermatoses with acute myeloid leukemia associated with an increase of serum colony-stimulating factor. J Am Acad Dermatol. 2008;59:S10–12. doi: 10.1016/j.jaad.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 43.Asahina A, Koga H, Suzuki Y, Hashimoto T. IgA pemphigus associated with diffuse large B-cell lymphoma showing unique reactivity with desmocollins: unusual clinical and histopathological features. Br J Dermatol. 2013;168:224–226. doi: 10.1111/j.1365-2133.2012.11127.x. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Yang B, Fan J, Yang K, Liu H, Wu G. Peripheral T-cell lymphoma complicated by immunoglobulin A pemphigus: a case report and literature review. Oncol Lett. 2014;8:62–66. doi: 10.3892/ol.2014.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallach D, Vignon-Pennamen M-D. From acute febrile neutrophilic dermatosis to neutrophilic disease: forty years of clinical research. J Am Acad Dermatol. 2006;55:1066–1071. doi: 10.1016/j.jaad.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 46.Lepelletier C, Bouaziz J-D, Rybojad M, Bagot M, Georgin-Lavialle S, Vignon-Pennamen M-D. Neutrophilic dermatoses associated with myeloid malignancies. Am J Clin Dermatol. 2019;20:325–333. doi: 10.1007/s40257-018-00418-2. [DOI] [PubMed] [Google Scholar]
- 47.Szalat R, Monsel G, Le Goff W, Battistella M, Bengouffa D, Schlageter M-H, et al. The spectrum of neutrophilic dermatoses associated with monoclonal gammopathy: association with IgA isotype and inflammatory profile. J Am Acad Dermatol. 2015;73:809–820. doi: 10.1016/j.jaad.2015.07.031. [DOI] [PubMed] [Google Scholar]
- 48.Fauconnier S-A, Leclercq A, Thomas T, Labeille B, Marotte H. Neutrophil dermatosis of the dorsal hands with multiple myeloma. Jt Bone Spine. 2016;83:227. doi: 10.1016/j.jbspin.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 49.Tuncer AM. Acute lymphoblastic leukemia and Sweet’s syndrome. Acta Haematol. 1988;80:224. doi: 10.1159/000205644. [DOI] [PubMed] [Google Scholar]
- 50.Woodrow SL, Munn SE, Basarab T, Russel JR. Sweet’s syndrome in association with non-Hodgkin’s lymphoma. Clin Exp Dermatol. 1996;21:357–359. doi: 10.1111/j.1365-2230.1996.tb00123.x. [DOI] [PubMed] [Google Scholar]
- 51.Sujobert P, Cuccuini W, Vignon-Pennamen D, Martin-Garcia N, Albertini AF, Uzunov M, et al. Evidence of differentiation in myeloid malignancies associated neutrophilic dermatosis: a fluorescent in situ hybridization study of 14 patients. J Invest Dermatol. 2013;133:1111–1114. doi: 10.1038/jid.2012.408. [DOI] [PubMed] [Google Scholar]
- 52.Van Loon K, Gill RM, McMahon P, Chigurupati R, Siddiqi I, Fox L, et al. 20q- clonality in a case of oral sweet syndrome and myelodysplasia. Am J Clin Pathol. 2012;137:310–315. doi: 10.1309/AJCP9I7NRWYLTJHV. [DOI] [PubMed] [Google Scholar]
- 53.Haga N, Iwata H, Yamaguchi Y, Shirato T, Nishimura K, Yamane N, et al. Mucocutaneous pyoderma gangrenosum due to trisomy 8 neutrophilic infiltrates in a patient with myelodysplastic syndrome. Br J Dermatol. 2016;174:239–241. doi: 10.1111/bjd.14102. [DOI] [PubMed] [Google Scholar]
- 54.Paydas S, Sahin B, Zorludemir S. Sweet’s syndrome accompanying leukaemia: seven cases and review of the literature. Leuk Res. 2000;24:83–86. doi: 10.1016/s0145-2126(99)00140-x. [DOI] [PubMed] [Google Scholar]
- 55.Arbetter KR, Hubbard KW, Markovic SN, Gibson LE, Phyliky RL. Case of granulocyte colony-stimulating factor-induced Sweet’s syndrome. Am J Hematol. 1999;61:126–129. doi: 10.1002/(sici)1096-8652(199906)61:2<126::aid-ajh9>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 56.Giasuddin AS, El-Orfi AH, Ziu MM, El-Barnawi NY. Sweet’s syndrome: is the pathogenesis mediated by helper T cell type 1 cytokines? J Am Acad Dermatol. 1998;39:940–943. doi: 10.1016/s0190-9622(98)70266-x. [DOI] [PubMed] [Google Scholar]
- 57.Shinojima Y, Toma Y, Terui T. Sweet syndrome associated with intrahepatic cholangiocarcinoma producing granulocyte colony-stimulating factor. Br J Dermatol. 2006;155:1103–1104. doi: 10.1111/j.1365-2133.2006.07521.x. [DOI] [PubMed] [Google Scholar]
- 58.Paydas S. Sweet’s syndrome: a revisit for hematologists and oncologists. Crit Rev Oncol Hematol. 2013;86:85–95. doi: 10.1016/j.critrevonc.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 59.Tseng E, Alhusayen R, Sade S, Buckstein R, Prica A. Pyoderma gangrenosum secondary to azacitidine in myelodysplastic syndrome. Br J Haematol. 2015;169:461. doi: 10.1111/bjh.13341. [DOI] [PubMed] [Google Scholar]
- 60.Ayirookuzhi SJ, Ma L, Ramshesh P, Mills G. Imatinib-induced sweet syndrome in a patient with chronic myeloid leukemia. Arch Dermatol. 2005;141:368–370. doi: 10.1001/archderm.141.3.368. [DOI] [PubMed] [Google Scholar]
- 61.Dasanu CA, Bockorny B, Alexandrescu DT. Pyoderma gangrenosum due to lenalidomide use for multiple myeloma. J Oncol Pharm Pract. 2015;21:471–473. doi: 10.1177/1078155214541975. [DOI] [PubMed] [Google Scholar]
- 62.Paydaş S, Sahin B, Seyrek E, Soylu M, Gonlusen G, Acar A, et al. Sweet’s syndrome associated with G-CSF. Br J Haematol. 1993;85:191–192. doi: 10.1111/j.1365-2141.1993.tb08668.x. [DOI] [PubMed] [Google Scholar]
- 63.Wang JY, French LE, Shear NH, Amiri A, Alavi A. Drug-induced pyoderma gangrenosum: a review. Am J Clin Dermatol. 2018;19:67–77. doi: 10.1007/s40257-017-0308-7. [DOI] [PubMed] [Google Scholar]
- 64.Nelson CA, Noe MH, McMahon CM, Gowda A, Wu B, Ashchyan HJ, et al. Sweet syndrome in patients with and without malignancy: a retrospective analysis of 83 patients from a tertiary academic referral center. J Am Acad Dermatol. 2018;78:303–309.e4. doi: 10.1016/j.jaad.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 65.Langan SM, Groves RW, Card TR, Gulliford MC. Incidence, mortality, and disease associations of pyoderma gangrenosum in the United Kingdom: a retrospective cohort study. J Invest Dermatol. 2012;132:2166–2170. doi: 10.1038/jid.2012.130. [DOI] [PubMed] [Google Scholar]
- 66.DeFilippis EM, Feldman SR, Huang WW. The genetics of pyoderma gangrenosum and implications for treatment: a systematic review. Br J Dermatol. 2015;172:1487–1497. doi: 10.1111/bjd.13493. [DOI] [PubMed] [Google Scholar]
- 67.Alavi A, French LE, Davis MD, Brassard A, Kirsner RS. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18:355–372. doi: 10.1007/s40257-017-0251-7. [DOI] [PubMed] [Google Scholar]
- 68.Jacobson S, Martin DB, Deng A, Cooper JZ. Pyoderma gangrenosum following tattoo placement in a patient with acute myelogenous leukemia. J Dermatol Treat. 2008;19:58–60. doi: 10.1080/09546630701713501. [DOI] [PubMed] [Google Scholar]
- 69.Vacas AS, Bollea-Garlatti ML, Torre AC, Galimberti RL. Bullous pyoderma gangrenosum as a predictor of hematological malignancies. An Bras Dermatol. 2018;93:133–134. doi: 10.1590/abd1806-4841.20187031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nieto D, Sendagorta E, Rueda JM, Herranz P. Successful treatment with ustekinumab and vacuum-assisted closure therapy in recalcitrant myelodysplastic syndrome-associated pyoderma gangrenosum: case report and literature review. Clin Exp Dermatol. 2019;44:116–119. doi: 10.1111/ced.13679. [DOI] [PubMed] [Google Scholar]
- 71.Razmi TM, Muhammed H, Handa S, Malhotra P, Sharma S, Saikia UN, et al. Bullous pyoderma gangrenosum secondary to underlying multiple myeloma: treated with ciclosporin. Clin Exp Dermatol. 2019;44:e13–e15. doi: 10.1111/ced.13804. [DOI] [PubMed] [Google Scholar]
- 72.Velasco-Tamariz V, Carreño-Tarragona G, Tous-Romero F, Gil-de la-Cruz E, Martín-Clavero E, Rivera-Díaz R. Dramatic resolution of disseminated pyoderma gangrenosum associated with monoclonal gammopathy after therapy with bortezomib and dexamethasone. Int Wound J. 2017;14:1382–1384. doi: 10.1111/iwj.12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Padhi T, Pradhan S, Pradhan K, Kumar SK. Pyoderma gangrenosum associated with mantle cell lymphoma. Indian Dermatol Online J. 2016;7:332–334. doi: 10.4103/2229-5178.185467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Montagnon CM, Fracica EA, Patel AA, Camilleri MJ, Murad MH, Dingli D, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82(6):1346–1359. doi: 10.1016/j.jaad.2019.09.032. [DOI] [PubMed] [Google Scholar]
- 75.Bennett ML, Jackson JM, Jorizzo JL, Fleischer AB, White WL, Callen JP. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore). 2000;79:37–46. doi: 10.1097/00005792-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 76.Kazmi SM, Pemmaraju N, Patel KP, Cohen PR, Daver N, Tran KM, et al. Characteristics of Sweet syndrome in patients with acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015;15:358–363. doi: 10.1016/j.clml.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.El-Khalawany M, Aboeldahab S, Mosbeh A-S, Thabet A. Clinicopathologic, immunophenotyping and cytogenetic analysis of Sweet syndrome in Egyptian patients with acute myeloid leukemia. Pathol Res Pract. 2017;213:143–153. doi: 10.1016/j.prp.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 78.Kulasekararaj AG, Kordasti S, Basu T, Salisbury JR, Mufti GJ, du Vivier AWP. Chronic relapsing remitting Sweet syndrome: a harbinger of myelodysplastic syndrome. Br J Haematol. 2015;170:649–656. doi: 10.1111/bjh.13485. [DOI] [PubMed] [Google Scholar]
- 79.Rochet NM, Chavan RN, Cappel MA, Wada DA, Gibson LE. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557–564. doi: 10.1016/j.jaad.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 80.Cohen PR, Talpaz M, Kurzrock R. Malignancy-associated Sweet’s syndrome: review of the world literature. J Clin Oncol. 1988;6:1887–1897. doi: 10.1200/JCO.1988.6.12.1887. [DOI] [PubMed] [Google Scholar]
- 81.Neoh CY, Tan AWH, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentations and associations. Br J Dermatol. 2007;156:480–485. doi: 10.1111/j.1365-2133.2006.07677.x. [DOI] [PubMed] [Google Scholar]
- 82.So JK, Carlos CA, Frucht CS, Cohen PR. Histiocytoid giant cellulitis-like Sweet’s syndrome: case report and review of the literature. Dermatol Online J. 2015;21:13030/qt0682f5wp. [PubMed] [Google Scholar]
- 83.Surovy AM, Pelivani N, Hegyi I, Buettiker U, Beltraminelli H, Borradori L. Giant cellulitis-like Sweet syndrome, a new variant of neutrophilic dermatosis. JAMA Dermatol. 2013;149:79–83. doi: 10.1001/2013.jamadermatol.548. [DOI] [PubMed] [Google Scholar]
- 84.Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, Rodríguez-Peralto JL, Alegre V, Cerroni L, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651–659. doi: 10.1001/jamadermatol.2016.6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ghoufi L, Ortonne N, Ingen-Housz-Oro S, Barhoumi W, Begon E, Haioun C, et al. Histiocytoid Sweet syndrome is more frequently associated with myelodysplastic syndromes than the classical neutrophilic variant: a comparative series of 62 patients. Medicine (Baltimore) 2016;95:e3033. doi: 10.1097/MD.0000000000003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raza S, Kirkland RS, Patel AA, Shortridge JR, Freter C. Insight into Sweet’s syndrome and associated-malignancy: a review of the current literature. Int J Oncol. 2013;42:1516–1522. doi: 10.3892/ijo.2013.1874. [DOI] [PubMed] [Google Scholar]
- 87.Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594–1602. doi: 10.1111/jdv.12566. [DOI] [PubMed] [Google Scholar]
- 88.Thomas CL, Ffolkes L, Akhras V. A case of mistaken identity: unilateral erythema elevatum diutinum associated with IgA paraproteinaemia. Clin Exp Dermatol. 2015;40:761–764. doi: 10.1111/ced.12622. [DOI] [PubMed] [Google Scholar]
- 89.Yiannias JA, El-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38–44. doi: 10.1016/0190-9622(92)70003-x. [DOI] [PubMed] [Google Scholar]
- 90.Queipo de Llano M, Yebra M, Cabrera R, Suarez E. Myelodysplastic syndrome in association with erythema elevatum diutinum. J Rheumatol. 1992;19:1005–1006. [PubMed] [Google Scholar]
- 91.Archimandritis AJ, Fertakis A, Alegakis G, Bartsokas S, Melissinos K. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613–614. doi: 10.1136/bmj.2.6087.613-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Delaporte E, Alfandari S, Fenaux P, Piette F, Bergoend H. Erythema elevatum diutinum and chronic lymphocytic leukemia. Clin Exp Dermatol. 1994;19:188. doi: 10.1111/j.1365-2230.1994.tb01160.x. [DOI] [PubMed] [Google Scholar]
- 93.Atallah J, Garces JC, Loayza E, Carlson JA. Chronic localized fibrosing leukocytoclastic vasculitis associated with lymphedema, intralymphatic and intravascular lymphocytosis, and chronic myelogenous leukemia: a case report of unilateral erythema elevatum diutinum. Am J Dermatopathol. 2017;39:479–484. doi: 10.1097/DAD.0000000000000802. [DOI] [PubMed] [Google Scholar]
- 94.Hatzitolios A, Tzellos TG, Savopoulos C, Tzalokostas V, Kaiafa G, Psomas E, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297–300. doi: 10.1111/j.1346-8138.2008.00470.x. [DOI] [PubMed] [Google Scholar]
- 95.Watts PJ, Khachemoune A. Subcorneal pustular dermatosis: a review of 30 years of progress. Am J Clin Dermatol. 2016;17:653–671. doi: 10.1007/s40257-016-0202-8. [DOI] [PubMed] [Google Scholar]
- 96.Kasha EE, Epinette WW. Subcorneal pustular dermatosis (Sneddon-Wilkinson disease) in association with a monoclonal IgA gammopathy: a report and review of the literature. J Am Acad Dermatol. 1988;19:854–858. doi: 10.1016/s0190-9622(88)70245-5. [DOI] [PubMed] [Google Scholar]
- 97.Kavala M, Karadag AS, Zindancı I, Turkoglu Z, Ozturk E, Zemheri E, et al. A case of subcorneal pustular dermatosis with IgG monoclonal gammopathy of undetermined significance: a rare association. Int J Dermatol. 2015;54:e551–e553. doi: 10.1111/ijd.12956. [DOI] [PubMed] [Google Scholar]
- 98.Atukorala DN, Joshi RK, Abanmi A, Jeha MT. Subcorneal pustular dermatosis and IgA myeloma. Dermatology. 1993;187:124–126. doi: 10.1159/000247221. [DOI] [PubMed] [Google Scholar]
- 99.Park BS, Cho KH, Eun HC, Youn JI. Subcorneal pustular dermatosis in a patient with aplastic anemia. J Am Acad Dermatol. 1998;39:287–289. doi: 10.1016/s0190-9622(98)70093-3. [DOI] [PubMed] [Google Scholar]
- 100.Ratnarathorn M, Newman J. Subcorneal pustular dermatosis (Sneddon-Wilkinson disease) occurring in association with nodal marginal zone lymphoma: a case report. Dermatol Online J. 2008;14:6. [PubMed] [Google Scholar]
- 101.Brown SJ, Barrett PD, Hendrick A, Langtry JAA. Subcorneal pustular dermatosis in association with chronic lymphocytic leukaemia. Acta Derm Venereol. 2003;83:306–307. doi: 10.1080/00015550310016661. [DOI] [PubMed] [Google Scholar]
- 102.Bachmeyer C, Aractingi S. Neutrophilic eccrine hidradenitis. Clin Dermatol. 2000;18:319–330. doi: 10.1016/s0738-081x(99)00123-6. [DOI] [PubMed] [Google Scholar]
- 103.Grandi V, Maglie R, Antiga E, Vannucchi M, Delfino C, Lastrucci I, et al. Eosinophilic dermatosis of hematologic malignancy: a retrospective cohort of 37 patients from an Italian center. J Am Acad Dermatol. 2019;81:246–249. doi: 10.1016/j.jaad.2018.11.048. [DOI] [PubMed] [Google Scholar]
- 104.Weed RI. Exaggerated delayed hypersensitivity to mosquito bites in chronic lymphocytic leukemia. Blood. 1965;26:257–268. doi: 10.1182/blood.V26.3.257.257. [DOI] [PubMed] [Google Scholar]
- 105.Bairey O, Goldschmidt N, Ruchlemer R, Tadmor T, Rahimi-Levene N, Yuklea M, et al. Insect-bite-like reaction in patients with chronic lymphocytic leukemia: a study from the Israeli Chronic Lymphocytic Leukemia Study Group. Eur J Haematol. 2012;89:491–496. doi: 10.1111/ejh.12015. [DOI] [PubMed] [Google Scholar]
- 106.Butzmann CM, Kern JS, Stanislawski G, Meiss F. Insect bite-like reaction in a patient with chronic lymphocytic leukemia. J Dtsch Dermatol Ges. 2014;12:734–737. doi: 10.1111/ddg.12326. [DOI] [PubMed] [Google Scholar]
- 107.Farber MJ, La Forgia S, Sahu J, Lee JB. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690–695. doi: 10.1111/j.1600-0560.2012.01906.x. [DOI] [PubMed] [Google Scholar]
- 108.Byrd JA, Scherschun L, Chaffins ML, Fivenson DP. Eosinophilic dermatosis of myeloproliferative disease: characterization of a unique eruption in patients with hematologic disorders. Arch Dermatol. 2001;137:1378–1380. [PubMed] [Google Scholar]
- 109.Jencks A, Kiavash K, Daveluy S, Thompson AD. Eosinophilic dermatosis of myeloproliferative disease in a young adult with diffuse large B-cell lymphoma. Am J Dermatopathol. 2019;41:303–308. doi: 10.1097/DAD.0000000000001256. [DOI] [PubMed] [Google Scholar]
- 110.Visseaux L, Durlach A, Barete S, Beylot-Barry M, Bonnet N, Chassine A, et al. T-cell papulosis associated with B-cell malignancy: a distinctive clinicopathologic entity. J Eur Acad Dermatol Venereol. 2018;32:1469–1475. doi: 10.1111/jdv.14805. [DOI] [PubMed] [Google Scholar]
- 111.Jayasekera PS, Bakshi A, Al-Sharqi A. Eosinophilic dermatosis of haematological malignancy. Clin Exp Dermatol. 2016;41:692–695. doi: 10.1111/ced.12846. [DOI] [PubMed] [Google Scholar]
- 112.Qiao J, Sun C-E, Zhu W, Zhu D, Fang H. Flame figures associated with eosinophilic dermatosis of hematologic malignancy: is it possible to distinguish the condition from eosinophilic cellulitis in patients with hematoproliferative disease? Int J Clin Exp Pathol. 2013;6:1683–1687. [PMC free article] [PubMed] [Google Scholar]
- 113.Meiss F, Technau-Hafsi K, Kern JS, May AM. Eosinophilic dermatosis of hematologic malignancy: correlation of molecular characteristics of skin lesions and extracutaneous manifestations of hematologic malignancy. J Cutan Pathol. 2019;46:175–181. doi: 10.1111/cup.13389. [DOI] [PubMed] [Google Scholar]
- 114.Maglie R, Antiga E, Vannucchi M, Del Bianco E, Bianchi B, Massi D, et al. Bullous eruption in a patient with B-cell chronic lymphocytic leukemia: a diagnostic challenge. Int J Dermatol. 2017;56:1445–1447. doi: 10.1111/ijd.13807. [DOI] [PubMed] [Google Scholar]
- 115.Takamura S, Teraki Y. Eosinophilic pustular folliculitis associated with hematological disorders: a report of two cases and review of Japanese literature. J Dermatol. 2016;43:432–435. doi: 10.1111/1346-8138.13088. [DOI] [PubMed] [Google Scholar]
- 116.Owen RL, Shah KK, Sosis AC, Purcell SM. Eosinophilic pustular folliculitis with underlying mantle cell lymphoma. Cutis. 2018;101:454–457. [PubMed] [Google Scholar]
- 117.Bari O, Cohen PR. Eosinophilic dermatosis of hematologic malignancy mimicking varicella zoster infection: report in a woman with chronic lymphocytic leukemia and review of the literature. Dermatol Pract Concept. 2017;7:6–15. doi: 10.5826/dpc.0703a02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mashima E, Sawada Y, Yamaguchi T, Ohmori S, Haruyama S, Yoshioka M, et al. Eosinophilic cellulitis possibly due to mosquito bite with high IL-5 production. J Investig Allergol Clin Immunol. 2017;27:149–150. doi: 10.18176/jiaci.0142. [DOI] [PubMed] [Google Scholar]
- 119.Tatsuno K, Fujiyama T, Matsuoka H, Shimauchi T, Ito T, Tokura Y. Clinical categories of exaggerated skin reactions to mosquito bites and their pathophysiology. J Dermatol Sci. 2016;82:145–152. doi: 10.1016/j.jdermsci.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 120.Rossini MS, de Souza EM, Cintra ML, Pagnano KB, Chiari AC, Lorand-Metze I. Cutaneous adverse reaction to 2-chlorodeoxyadenosine with histological flame figures in patients with chronic lymphocytic leukaemia. J Eur Acad Dermatol Venereol. 2004;18:538–542. doi: 10.1111/j.1468-3083.2004.00969.x. [DOI] [PubMed] [Google Scholar]
- 121.Mitteldorf C, Tronnier M, Merz H, Haenssle HA, Bertsch HP, Schön MP, et al. Insect bite-like reactions in a patient with B-cell chronic lymphocytic leukaemia: fluorescence in situ hybridization analysis revealed neoplastic B cells within the skin infiltrate. Br J Dermatol. 2012;167:944–946. doi: 10.1111/j.1365-2133.2012.10966.x. [DOI] [PubMed] [Google Scholar]
- 122.Thiesen I, Wehkamp U, Brüggemann M, Ritgen M, Murga Penas EM, Klapper W, et al. Skin involvement by chronic lymphocytic leukaemia is frequently associated with unrelated neoplastic or inflammatory cutaneous disease and is not indicative of general disease progression. Br J Dermatol. 2019;180:227–228. doi: 10.1111/bjd.17135. [DOI] [PubMed] [Google Scholar]
- 123.Li J, Best OG, Mulligan SP, Fernando SL. Serum from a subset of patients with chronic lymphocytic leukemia and large local reactions to mosquito bites induces upregulation of CD63 surface expression on basophils in atopic donors. Leuk Lymphoma. 2016;57:2417–2420. doi: 10.3109/10428194.2016.1173208. [DOI] [PubMed] [Google Scholar]
- 124.Maglie R, Grandi V, Massi D, Caproni M, Pimpinelli N, Antiga E. Cutaneous eruptions associated with haematologic malignancies: the need for a unifying nomenclature. J Eur Acad Dermatol Venereol. 2019;33:e191–e192. doi: 10.1111/jdv.15452. [DOI] [PubMed] [Google Scholar]
- 125.Maglie R, Grandi V, Antiga E. Response to: “Hematologic-Related Malignancy-Induced Eosinophilic Dermatosis (He Remained): an eosinophilic dermatosis predominantly associated with chronic lymphocytic leukemia”. J Am Acad Dermatol. 2020;82:e15–e16. doi: 10.1016/j.jaad.2019.08.061. [DOI] [PubMed] [Google Scholar]
- 126.Sato-Sano M, Teixeira SP, Vargas JC, Baiocchi OCCG, Enokihara MMSES, Gomes EE, et al. Lenalidomide in the management of eosinophilic dermatosis of hematological malignancy. J Dermatol. 2019;46:618–621. doi: 10.1111/1346-8138.14916. [DOI] [PubMed] [Google Scholar]
- 127.Jin A, Pousti BT, Savage KT, Mollanazar NK, Lee JB, Hsu S. Eosinophilic dermatosis of hematologic malignancy responding to dupilumab in a patient with chronic lymphocytic leukemia. JAAD Case Rep. 2019;5:815–817. doi: 10.1016/j.jdcr.2019.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cohen PR. Hematologic-related malignancy-induced eosinophilic dermatosis (he eemained): an eosinophilic dermatosis predominantly associated with chronic lymphocytic leukemia. J Am Acad Dermatol. 2020;82:e13–e14. doi: 10.1016/j.jaad.2019.08.062. [DOI] [PubMed] [Google Scholar]
- 129.Kim JH, Kim S-C. Paraneoplastic pemphigus: paraneoplastic autoimmune dsease of the skin and mucosa. Front Immunol. 2019;10:1259. doi: 10.3389/fimmu.2019.01259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Park GT, Lee JH, Yun SJ, Lee SC, Lee JB. Paraneoplastic pemphigus without an underlying neoplasm. Br J Dermatol. 2007;156:563–566. doi: 10.1111/j.1365-2133.2006.07605.x. [DOI] [PubMed] [Google Scholar]
- 131.Lee J, Bloom R, Amber KT. A systematic review of patients with mucocutaneous and respiratory complications in paraneoplastic autoimmune multiorgan syndrome: Castleman’s disease is the predominant malignancy. Lung. 2015;193:593–596. doi: 10.1007/s00408-015-9732-8. [DOI] [PubMed] [Google Scholar]
- 132.Ohzono A, Sogame R, Li X, Teye K, Tsuchisaka A, Numata S, et al. Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br J Dermatol. 2015;173:1447–1452. doi: 10.1111/bjd.14162. [DOI] [PubMed] [Google Scholar]
- 133.Choi Y, Nam K-H, Lee J-B, Lee JY, Ihm C-W, Lee SE, et al. Retrospective analysis of 12 Korean patients with paraneoplastic pemphigus. J Dermatol. 2012;39:973–981. doi: 10.1111/j.1346-8138.2012.01655.x. [DOI] [PubMed] [Google Scholar]
- 134.Didona D, Maglie R, Eming R, Hertl M. Pemphigus: current and future therapeutic strategies. Front Immunol. 2019;10:1418. doi: 10.3389/fimmu.2019.01418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primer. 2017;3:17026. doi: 10.1038/nrdp.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Frew JW, Murrell DF. Paraneoplastic pemphigus (paraneoplastic autoimmune multiorgan syndrome): clinical presentations and pathogenesis. Dermatol Clin. 2011;29:419–25, viii. doi: 10.1016/j.det.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 137.Solimani F, Maglie R, Pollmann R, Schmidt T, Schmidt A, Ishii N, et al. Thymoma-associated paraneoplastic autoimmune multiorgan syndrome: from pemphigus to lichenoid dermatitis. Front Immunol. 2019;10:1413. doi: 10.3389/fimmu.2019.01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Passeron T, Bahadoran P, Lacour JP, Perrin C, Gilbert D, Benzaken S, et al. Paraneoplastic pemphigus presenting as erosive lichen planus. Br J Dermatol. 1999;140:552–553. doi: 10.1046/j.1365-2133.1999.02737.x. [DOI] [PubMed] [Google Scholar]
- 139.Mahoney MG, Aho S, Uitto J, Stanley JR. The members of the plakin family of proteins recognized by paraneoplastic pemphigus antibodies include periplakin. J Invest Dermatol. 1998;111:308–313. doi: 10.1046/j.1523-1747.1998.00279.x. [DOI] [PubMed] [Google Scholar]
- 140.Numata S, Teye K, Tsuruta D, Sogame R, Ishii N, Koga H, et al. Anti-α-2-macroglobulin-like-1 autoantibodies are detected frequently and may be pathogenic in paraneoplastic pemphigus. J Invest Dermatol. 2013;133:1785–1793. doi: 10.1038/jid.2013.65. [DOI] [PubMed] [Google Scholar]
- 141.Poot AM, Diercks GFH, Kramer D, Schepens I, Klunder G, Hashimoto T, et al. Laboratory diagnosis of paraneoplastic pemphigus. Br J Dermatol. 2013;169:1016–1024. doi: 10.1111/bjd.12479. [DOI] [PubMed] [Google Scholar]
- 142.Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T. Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest. 1998;102:775–782. doi: 10.1172/JCI3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Solimani F, Pollmann R, Ishii N, Eming R, Hashimoto T, Schmidt T, et al. Diagnosis of anti-laminin γ-1 pemphigoid by immunoblot analysis. J Eur Acad Dermatol Venereol. 2019;33:735–741. doi: 10.1111/jdv.15170. [DOI] [PubMed] [Google Scholar]
- 144.Poot AM, Siland J, Jonkman MF, Pas HH, Diercks GFH. Direct and indirect immunofluorescence staining patterns in the diagnosis of paraneoplastic pemphigus. Br J Dermatol. 2016;174:912–915. doi: 10.1111/bjd.14282. [DOI] [PubMed] [Google Scholar]
- 145.Witte M, Zillikens D, Schmidt E. Diagnosis of autoimmune blistering diseases. Front Med. 2018;5:296. doi: 10.3389/fmed.2018.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Amber KT, Valdebran M, Grando SA. Paraneoplastic autoimmune multiorgan syndrome (PAMS): beyond the single phenotype of paraneoplastic pemphigus. Autoimmun Rev. 2018;17:1002–1010. doi: 10.1016/j.autrev.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 147.Daneshpazhooh M, Moeineddin F, Kiani A, Naraghi ZS, Firooz A, Akhyani M, et al. Fatal paraneoplastic pemphigus after removal of Castleman’s disease in a child. Pediatr Dermatol. 2012;29:656–657. doi: 10.1111/j.1525-1470.2011.01670.x. [DOI] [PubMed] [Google Scholar]
- 148.Lane JE, Woody C, Davis LS, Guill MF, Jerath RS. Paraneoplastic autoimmune multiorgan syndrome (paraneoplastic pemphigus) in a child: case report and review of the literature. Pediatrics. 2004;114:e513–e516. doi: 10.1542/peds.2004-0436. [DOI] [PubMed] [Google Scholar]
- 149.Cervini AB, Tosi V, Kim SH, Bocian M, Chantada G, Nousari C, et al. Paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome: report of 2 cases in children and a review of the literature. Actas Dermosifiliogr. 2010;101:879–886. doi: 10.1016/j.ad.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 150.Mimouni D, Anhalt GJ, Lazarova Z, Aho S, Kazerounian S, Kouba DJ, et al. Paraneoplastic pemphigus in children and adolescents. Br J Dermatol. 2002;147:725–732. doi: 10.1046/j.1365-2133.2002.04992.x. [DOI] [PubMed] [Google Scholar]
- 151.Eming R, Hennerici T, Bäcklund J, Feliciani C, Visconti KC, Willenborg S, et al. Pathogenic IgG antibodies against desmoglein 3 in pemphigus vulgaris are regulated by HLA-DRB1*04:02-restricted T cells. J Immunol. 2014;193:4391–4399. doi: 10.4049/jimmunol.1401081. [DOI] [PubMed] [Google Scholar]
- 152.Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M. Autoreactive T cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol. 2013;22:699–704. doi: 10.1111/exd.12229. [DOI] [PubMed] [Google Scholar]
- 153.Billet SE, Grando SA, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;39:617–630. doi: 10.1080/08916930600972099. [DOI] [PubMed] [Google Scholar]
- 154.Kwatra SG, Boozalis E, Pasieka H, Anhalt GJ. Decreased recognition of paraneoplastic pemphigus in patients previously treated with anti-CD 20 monoclonal antibodies. Br J Dermatol. 2019;180:1238–1239. doi: 10.1111/bjd.17577. [DOI] [PubMed] [Google Scholar]
- 155.Vezzoli P, Berti E, Marzano AV. Rationale and efficacy for the use of rituximab in paraneoplastic pemphigus. Expert Rev Clin Immunol. 2008;4:351–363. doi: 10.1586/1744666X.4.3.351. [DOI] [PubMed] [Google Scholar]
- 156.Menenakos C, Braumann C, Hartmann J, Jacobi CA. Retroperitoneal Castleman’s tumor and paraneoplastic pemphigus: report of a case and review of the literature. World J Surg Oncol. 2007;5:45. doi: 10.1186/1477-7819-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nousari HC, Kimyai-Asadi A, Anhalt GJ. Elevated serum levels of interleukin-6 in paraneoplastic pemphigus. J Invest Dermatol. 1999;112:396–398. doi: 10.1046/j.1523-1747.1999.00520.x. [DOI] [PubMed] [Google Scholar]
- 158.Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135:1353–1364. doi: 10.1182/blood.2019000931. [DOI] [PubMed] [Google Scholar]
- 159.Abid MB, Peck R, Abid MA, Al-Sakkaf W, Zhang Y, Dunnill GS, et al. Is tocilizumab a potential therapeutic option for refractory unicentric Castleman disease? Hematol Oncol. 2018;36:320–323. doi: 10.1002/hon.2420. [DOI] [PubMed] [Google Scholar]
- 160.Gu L, Ye S. Tocilizumab cannot prevent the development of bronchiolitis obliterans in patients with Castleman disease-associated paraneoplastic pemphigus. J Clin Rheumatol. 2019;25:e77–e78. doi: 10.1097/RHU.0000000000000675. [DOI] [PubMed] [Google Scholar]
- 161.Lee S, Yamauchi T, Ishii N, Hashimoto T, Kinoshita K, Imamura S, et al. Achievement of the longest survival of paraneoplastic pemphigus with bronchiolitis obliterans associated with follicular lymphoma using R-CHOP chemotherapy. Int J Hematol. 2017;106:852–859. doi: 10.1007/s12185-017-2305-2. [DOI] [PubMed] [Google Scholar]
- 162.Ouedraogo E, Gottlieb J, de Masson A, Lepelletier C, Jachiet M, Salle de Chou C, et al. Risk factors for death and survival in paraneoplastic pemphigus associated with hematologic malignancies in adults. J Am Acad Dermatol. 2019;80:1544–1549. doi: 10.1016/j.jaad.2018.03.043. [DOI] [PubMed] [Google Scholar]
- 163.Kridin K, Zelber-Sagi S, Comaneshter D, Batat E, Cohen AD. Pemphigus and hematologic malignancies: a population-based study of 11,859 patients. J Am Acad Dermatol. 2018;78(1084–9):e1. doi: 10.1016/j.jaad.2017.11.039. [DOI] [PubMed] [Google Scholar]
- 164.Atzmony L, Mimouni I, Reiter O, Leshem YA, Taha O, Gdalevich M, et al. Association of bullous pemphigoid with malignancy: a systematic review and meta-analysis. J Am Acad Dermatol. 2017;77:691–699. doi: 10.1016/j.jaad.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 165.Roberto M, Emiliano A, Marzia C. Comment on “Association of bullous pemphigoid with malignancy: a systematic review and meta-analysis”. J Am Acad Dermatol. 2018 doi: 10.1016/j.jaad.2017.10.053. [DOI] [PubMed] [Google Scholar]
- 166.Barnadas MA, Moreno A, Brunet S, González MJ, Rodriguez JL, Gelpí C, et al. Linear IgA bullous dermatosis associated with Hodgkin’s disease. J Am Acad Dermatol. 1988;19:1122–1124. doi: 10.1016/s0190-9622(98)80012-1. [DOI] [PubMed] [Google Scholar]
- 167.Kienzler JL, Blanc D, Laurent R, Agache P. Linear IgA bullous dermatosis and Hodgkin’s disease. Ann Dermatol Venereol. 1983;110:727–728. [PubMed] [Google Scholar]
- 168.Tiger JB, Rush JT, Barton DT, Danilov AV, Chapman MS. Urticarial linear IgA bullous dermatosis (LABD) as a presenting sign of chronic lymphocytic leukemia (CLL) JAAD Case Rep. 2015;1:412–414. doi: 10.1016/j.jdcr.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Godfrey K, Wojnarowska F, Leonard J. Linear IgA disease of adults: association with lymphoproliferative malignancy and possible role of other triggering factors. Br J Dermatol. 1990;123:447–452. doi: 10.1111/j.1365-2133.1990.tb01448.x. [DOI] [PubMed] [Google Scholar]
- 170.Shaw M, McKee PH, Gaminara E, Pearson TC, Evans B, McGibbon DH. Epidermolysis bullosa acquisita associated with chronic lymphatic leukaemia. Clin Exp Dermatol. 1985;10:162–168. doi: 10.1111/j.1365-2230.1985.tb00546.x. [DOI] [PubMed] [Google Scholar]
- 171.Shaffer BR, Schleicher SM. Epidermolysis bullosa acquisita in association with mantle cell lymphoma. Cutis. 2018;101:E13–E15. [PubMed] [Google Scholar]
- 172.Baler GR. Epidermolysis bullosa acquisita associated with lymphoma. J Am Acad Dermatol. 1987;17:856–859. doi: 10.1016/s0190-9622(87)80296-7. [DOI] [PubMed] [Google Scholar]
- 173.Trump DL, Allen H, Olson J, Wright J, Humphrey RL. Epidermolysis bullosa acquisita: association with amyloidosis and multiple myeloma. JAMA. 1980;243:1461–1462. doi: 10.1001/jama.243.14.1461. [DOI] [PubMed] [Google Scholar]
- 174.Engineer L, Dow EC, Braverman IM, Ahmed AR. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943–946. doi: 10.1067/mjd.2002.113682. [DOI] [PubMed] [Google Scholar]
- 175.Antiga E, Maglie R, Quintarelli L, Verdelli A, Bonciani D, Bonciolini V, et al. Dermatitis herpetiformis: novel perspectives. Front Immunol. 2019;10:1290. doi: 10.3389/fimmu.2019.01290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sunderkötter CH, Zelger B, Chen K-R, Requena L, Piette W, Carlson JA, et al. Nomenclature of cutaneous vasculitis: dermatologic addendum to the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol. 2018;70:171–184. doi: 10.1002/art.40375. [DOI] [PubMed] [Google Scholar]
- 177.Kurzrock R, Cohen PR. Vasculitis and cancer. Clin Dermatol. 1993;11:175–187. doi: 10.1016/0738-081x(93)90115-s. [DOI] [PubMed] [Google Scholar]
- 178.Loricera J, Calvo-Río V, Ortiz-Sanjuán F, González-López MA, Fernández-Llaca H, Rueda-Gotor J, et al. The spectrum of paraneoplastic cutaneous vasculitis in a defined population: incidence and clinical features. Medicine (Baltimore) 2013;92:331–343. doi: 10.1097/MD.0000000000000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sánchez-Guerrero J, Gutiérrez-Ureña S, Vidaller A, Reyes E, Iglesias A, Alarcón-Segovia D. Vasculitis as a paraneoplastic syndrome: report of 11 cases and review of the literature. J Rheumatol. 1990;17:1458–1462. [PubMed] [Google Scholar]
- 180.Carlson JA, Chen K-R. Cutaneous vasculitis update: small vessel neutrophilic vasculitis syndromes. Am J Dermatopathol. 2006;28:486–506. doi: 10.1097/01.dad.0000246646.45651.a2. [DOI] [PubMed] [Google Scholar]
- 181.Carlson JA, Cavaliere LF, Grant-Kels JM. Cutaneous vasculitis: diagnosis and management. Clin Dermatol. 2006;24:414–429. doi: 10.1016/j.clindermatol.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 182.Fain O, Hamidou M, Cacoub P, Godeau B, Wechsler B, Pariès J, et al. Vasculitides associated with malignancies: analysis of sixty patients. Arthritis Rheum. 2007;57:1473–1480. doi: 10.1002/art.23085. [DOI] [PubMed] [Google Scholar]
- 183.Pileri A, Baraldi C, Broccoli A, Maglie R, Patrizi A, Zinzani P, et al. Cutaneous leukocytoclastic vasculitis in B-cell chronic lymphocytic leukemia patients. G Ital Dermatol E Venereol. 2019;154:605–606. doi: 10.23736/S0392-0488.18.05916-3. [DOI] [PubMed] [Google Scholar]
- 184.Podjasek JO, Wetter DA, Pittelkow MR, Wada DA. Cutaneous small-vessel vasculitis associated with solid organ malignancies: the Mayo Clinic experience, 1996 to 2009. J Am Acad Dermatol. 2012;66:e55–65. doi: 10.1016/j.jaad.2010.09.732. [DOI] [PubMed] [Google Scholar]
- 185.Curgunlu A, Karter Y, Uyanik O, Tunçkale A, Curgunlu S. Leukocytoclastic vasculitis and renal cell carcinoma. Intern Med Tokyo Jpn. 2004;43:256–257. doi: 10.2169/internalmedicine.43.256. [DOI] [PubMed] [Google Scholar]
- 186.Solans-Laqué R, Bosch-Gil JA, Pérez-Bocanegra C, Selva-O’Callaghan A, Simeón-Aznar CP, Vilardell-Tarres M. Paraneoplastic vasculitis in patients with solid tumors: report of 15 cases. J Rheumatol. 2008;35:294–304. [PubMed] [Google Scholar]
- 187.Sánchez NB, Canedo IF, García-Patos PE, de Unamuno PP, Benito AV, Pascual AM. Paraneoplastic vasculitis associated with multiple myeloma. J Eur Acad Dermatol Venereol. 2004;18:731–735. doi: 10.1111/j.1468-3083.2004.01069.x. [DOI] [PubMed] [Google Scholar]
- 188.Martín Oterino JA, Sánchez Rodríguez AS, Chimpén Ruiz VA, Fidalgo Fernández MA. Hypersensitivity vasculitis as a paraneoplastic manifestation prior to acute monocytic leukemia. Med Clin (Barc) 1997;109:238–239. [PubMed] [Google Scholar]
- 189.Das M, Chhabra R, Hinton SW. Cutaneous leukocytoclastic vasculitis and myelodysplastic syndrome with little or no evidence of associated autoimmune disorders-a case report and a brief review of the literature. Am J Med Sci. 2008;336:368–371. doi: 10.1097/MAJ.0b013e31815e8c0b. [DOI] [PubMed] [Google Scholar]
- 190.Paydaş S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773–779. doi: 10.1046/j.1365-2133.2000.03774.x. [DOI] [PubMed] [Google Scholar]
- 191.Cañueto J, Meseguer-Yebra C, Román-Curto C, Santos-Briz A, Fernández-López E, Fraile C, et al. Leukemic vasculitis: a rare pattern of leukemia cutis. J Cutan Pathol. 2011;38:360–364. doi: 10.1111/j.1600-0560.2010.01623.x. [DOI] [PubMed] [Google Scholar]
- 192.Audemard A, Lamy T, Bareau B, Sicre F, Suarez F, Truquet F, et al. Vasculitis associated with large granular lymphocyte (LGL) leukemia: presentation and treatment outcomes of 11 cases. Semin Arthritis Rheum. 2013;43:362–366. doi: 10.1016/j.semarthrit.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 193.Youinou P, Mackenzie LE, Lamour A, Mageed RA, Lydyard PM. Human CD5-positive B cells in lymphoid malignancy and connective tissue diseases. Eur J Clin Invest. 1993;23:139–150. doi: 10.1111/j.1365-2362.1993.tb00753.x. [DOI] [PubMed] [Google Scholar]
- 194.Robak E, Robak T. Skin lesions in chronic lymphocytic leukemia. Leuk Lymphoma. 2007;48:855–865. doi: 10.1080/10428190601137336. [DOI] [PubMed] [Google Scholar]
- 195.Wick MR, Patterson JW. Cutaneous paraneoplastic syndromes. Semin Diagn Pathol. 2019;36:211–228. doi: 10.1053/j.semdp.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 196.Magro CM, Crowson AN. A clinical and histologic study of 37 cases of immunoglobulin A-associated vasculitis. Am J Dermatopathol. 1999;21:234–240. doi: 10.1097/00000372-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 197.Pamuk GE, Uyanik MS, Demir M, Tekgündüz E, Turgut B, Soy M. Systemic antineutrophil cytoplasmic antibody vasculitis in a patient with chronic lymphocytic leukemia: quite a rare diagnosis. Leuk Res. 2007;31:1149–1151. doi: 10.1016/j.leukres.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 198.Astudillo L, Recher C, Launay F, Lamant L, Brousset P, Arlet-Suau E. Malignant lymphoma presenting as cutaneous granulomatous vasculitis. Br J Dermatol. 2005;152:820–821. doi: 10.1111/j.1365-2133.2005.06516.x. [DOI] [PubMed] [Google Scholar]
- 199.Gibson LE, Winkelmann RK. Cutaneous granulomatous vasculitis: its relationship to systemic disease. J Am Acad Dermatol. 1986;14:492–501. doi: 10.1016/s0190-9622(86)70064-9. [DOI] [PubMed] [Google Scholar]
- 200.Cohen MJ, Shyman A, Klein M, Ben-Yehuda A, Rubinow A, Or R, et al. Large vessel (Takayasu’s) arteritis in a patient with myelodysplastic syndrome: is there a common pathogenesis? Clin Lymphoma Myeloma Leuk. 2011;11:60–63. doi: 10.3816/CLML.2011.n.008. [DOI] [PubMed] [Google Scholar]
- 201.Oksuz MF, Coskun BN, Tufan AN, Orucoglu N, Dalkilic E, Oztürk Nazlıoğlu H, et al. Hairy cell leukemia presenting initially with symptoms of Behçet’s disease. Int J Rheum Dis. 2014;17:689–692. doi: 10.1111/1756-185X.12370. [DOI] [PubMed] [Google Scholar]
- 202.Folci M, Ramponi G, Shiffer D, Zumbo A, Agosti M, Brunetta E. ANCA-associated vasculitides and hematologic malignancies: lessons from the past and future perspectives. J Immunol Res. 2019;2019:1732175. doi: 10.1155/2019/1732175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hemminki K, Liu X, Ji J, Försti A. Origin of B-cell neoplasms in autoimmune disease. PLoS ONE. 2016 doi: 10.1371/journal.pone.0158360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Din L, Sheikh M, Kosaraju N, Smedby KE, Bernatsky S, Berndt SI, et al. Genetic overlap between autoimmune diseases and non-Hodgkin lymphoma subtypes. Genet Epidemiol. 2019;43:844–863. doi: 10.1002/gepi.22242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Onishi A, Sugiyama D, Kumagai S, Morinobu A. Cancer incidence in systemic sclerosis: meta-analysis of population-based cohort studies. Arthritis Rheum. 2013;65:1913–1921. doi: 10.1002/art.37969. [DOI] [PubMed] [Google Scholar]
- 206.Yu K-H, Kuo C-F, Huang LH, Huang W-K, See L-C. Cancer risk in patients with inflammatory systemic autoimmune rheumatic diseases: a nationwide population-based dynamic cohort study in Taiwan. Medicine (Baltimore) 2016;95:e3540. doi: 10.1097/MD.0000000000003540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Klein A, Polliack A, Gafter-Gvili A. Systemic lupus erythematosus and lymphoma: Incidence, pathogenesis and biology. Leuk Res. 2018;75:45–49. doi: 10.1016/j.leukres.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 208.Vasaitis L, Nordmark G, Theander E, Backlin C, Smedby KE, Askling J, et al. Population-based study of patients with primary Sjögren’s syndrome and lymphoma: lymphoma subtypes, clinical characteristics, and gender differences. Scand J Rheumatol. 2020;49:225–232. doi: 10.1080/03009742.2019.1696403. [DOI] [PubMed] [Google Scholar]
- 209.Sebastian A, Madej M, Sebastian M, Butrym A, Woytala P, Hałoń A, et al. Prevalence and clinical presentation of lymphoproliferative disorder in patients with primary Sjögren’s syndrome. Rheumatol Int. 2020;40:399–404. doi: 10.1007/s00296-020-04522-7. [DOI] [PubMed] [Google Scholar]
- 210.Castanet J, Taillan B, Lacour JP, Garnier G, Perrin C, Ortonne JP. Subacute cutaneous lupus erythematosus associated with Hodgkin’s disease. Clin Rheumatol. 1995;14:692–694. doi: 10.1007/bf02207938. [DOI] [PubMed] [Google Scholar]
- 211.Cedeno-Laurent F, Obstfeld AE, Boni A, Lipoff JB. Lupus-like eruption as the presenting sign of acute myeloblastic leukemia. Lupus. 2016;25:312–314. doi: 10.1177/0961203315611497. [DOI] [PubMed] [Google Scholar]
- 212.Queen D, Lopez A, Geskin LJ. Paraneoplastic scleroderma in the setting of CD30+ large cell transformation of mycosis fungoides. JAAD Case Rep. 2019;5:201–204. doi: 10.1016/j.jdcr.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Park JK, Subhawong AP, Ziminski CM, Grader-Beck T. Dermatomyositis as the initial presentation of a large anaplastic T-cell lymphoma. J Clin Oncol. 2011;29:e378–e380. doi: 10.1200/JCO.2010.33.2528. [DOI] [PubMed] [Google Scholar]
- 214.Ledwich LJ, Olenginski TP. A rare lymphoma in a patient with amyopathic dermatomyositis. Am J Clin Dermatol. 2010;11:151–155. doi: 10.2165/11530170-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 215.Marie I, Guillevin L, Menard J-F, Hatron PY, Cherin P, Amoura Z, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615–620. doi: 10.1016/j.autrev.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 216.Stübgen J-P. Juvenile dermatomyositis/polymyositis and lymphoma. J Neurol Sci. 2017;377:19–24. doi: 10.1016/j.jns.2017.03.033. [DOI] [PubMed] [Google Scholar]
- 217.Nan X, Adike A, Rice L, Bunin V. Primary thyroid lymphoma presenting as dermatomyositis: a first case and review of the literature. BMJ Case Rep. 2016 doi: 10.1136/bcr-2016-215057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Veraitch O, Curto-Garcia N, Harrison C, Stefanato CM, McGibbon D. Hydroxyurea-induced dermatomyositis koebnerizing at the site of previous shingles. Clin Exp Dermatol. 2019;44:546–548. doi: 10.1111/ced.13772. [DOI] [PubMed] [Google Scholar]
- 219.Oskay T, Kutluay L, Ozyilkan O. Dermatomyositis-like eruption after long-term hydroxyurea therapy for polycythemia vera. Eur J Dermatol. 2002;12:586–588. [PubMed] [Google Scholar]
- 220.Vennepureddy A, Siddique MN, Odaimi M, Terjanian T. Bleomycin-induced flagellate erythema in a patient with Hodgkin’s lymphoma: a case report and review of literature. J Oncol Pharm Pract. 2016;22:556–560. doi: 10.1177/1078155215580468. [DOI] [PubMed] [Google Scholar]
- 221.Schmitt A, Volz A. Non-infectious granulomatous dermatoses. J Dtsch Dermatol Ges. 2019;17:518–533. doi: 10.1111/ddg.13848. [DOI] [PubMed] [Google Scholar]
- 222.Rosenbach M, English JC. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373–387. doi: 10.1016/j.det.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 223.Tabata MM, Novoa RA, Martires KJ. Paraneoplastic granulomatous dermatitis in a patient with Hodgkin’s disease: a diagnostic pitfall. BMJ Case Rep. 2018 doi: 10.1136/bcr-2018-224961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Pei S, Hinshaw MA. Palisaded neutrophilic granulomatous dermatitis leading to diagnosis of Hodgkin lymphoma: report of rare case and literature review of paraneoplastic granulomatous dermatitides. Am J Dermatopathol. 2019;41:835–845. doi: 10.1097/DAD.0000000000001411. [DOI] [PubMed] [Google Scholar]
- 225.Deen J, Banney L, Perry-Keene J. Palisading neutrophilic and granulomatous dermatitis as a presentation of Hodgkin lymphoma: a case and review. J Cutan Pathol. 2018;45:167–170. doi: 10.1111/cup.13076. [DOI] [PubMed] [Google Scholar]
- 226.Weed J, Ko C, Stahl M, Much M, Witt D, Zeidan AM, et al. Reactive granulomatous dermatitis presenting as subcutaneous nodules and cords in a patient with advanced myelodysplastic syndrome. Ann Hematol. 2017;96:1037–1039. doi: 10.1007/s00277-017-2954-5. [DOI] [PubMed] [Google Scholar]
- 227.Balin SJ, Wetter DA, Kurtin PJ, Letendre L, Pittelkow MR. Myelodysplastic syndrome presenting as generalized granulomatous dermatitis. Arch Dermatol. 2011;147:331–335. doi: 10.1001/archdermatol.2011.39. [DOI] [PubMed] [Google Scholar]
- 228.Maehara E, Wada N, Takamatsu H, Harada K, Imayama S, Okamura S, et al. Generalised granuloma annulare associated with myelodysplastic syndrome. Eur J Dermatol. 2016;26:410–412. doi: 10.1684/ejd.2016.2806. [DOI] [PubMed] [Google Scholar]
- 229.Hinckley MR, Walsh SN, Molnár I, Sheehan DJ, Sangueza OP, Yosipovitch G. Generalized granuloma annulare as an initial manifestation of chronic myelomonocytic leukemia: a report of 2 cases. Am J Dermatopathol. 2008;30:274–277. doi: 10.1097/DAD.0b013e318166ea1a. [DOI] [PubMed] [Google Scholar]
- 230.Pham LV, Pogue E, Ford RJ. The role of macrophage/B-cell interactions in the pathophysiology of B-cell lymphomas. Front Oncol. 2018;8:147. doi: 10.3389/fonc.2018.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Dadban A, Slama B, Azzedine A, Lepeu G. Widespread granuloma annulare and Hodgkin’s disease. Clin Exp Dermatol. 2008;33:465–468. doi: 10.1111/j.1365-2230.2008.02730.x. [DOI] [PubMed] [Google Scholar]
- 232.Cornejo KM, Lum CA, Izumi AK. A cutaneous interstitial granulomatous dermatitis-like eruption arising in myelodysplasia with leukemic progression. Am J Dermatopathol. 2013;35:e26–e29. doi: 10.1097/DAD.0b013e31826ff6a6. [DOI] [PubMed] [Google Scholar]
- 233.Bouchliou I, Miltiades P, Nakou E, Spanoudakis E, Goutzouvelidis A, Vakalopoulou S, et al. Th17 and Foxp3(+) T regulatory cell dynamics and distribution in myelodysplastic syndromes. Clin Immunol. 2011;139:350–359. doi: 10.1016/j.clim.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 234.Hagiwara A, Fujimura T, Furudate S, Kambayashi Y, Numata Y, Haga T, et al. Generalized granulomatous dermatitis accompanied by myelodysplastic syndrome. Acta Derm Venereol. 2014;94:223–224. doi: 10.2340/00015555-1656. [DOI] [PubMed] [Google Scholar]
- 235.Oliwiecki S, Kotecha B, Kingston T, Rothera MP. Sarcoidosis-lymphoma syndrome. J R Soc Med. 1992;85:176–177. [PMC free article] [PubMed] [Google Scholar]
- 236.Cohen PR, Kurzrock R. Sarcoidosis and malignancy. Clin Dermatol. 2007;25:326–333. doi: 10.1016/j.clindermatol.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 237.Cohen PR. Granuloma annulare, relapsing polychondritis, sarcoidosis, and systemic lupus erythematosus: conditions whose dermatologic manifestations may occur as hematologic malignancy-associated mucocutaneous paraneoplastic syndromes. Int J Dermatol. 2006;45:70–80. doi: 10.1111/j.1365-4632.2005.02667.x. [DOI] [PubMed] [Google Scholar]
- 238.Zoroquiain P, González S, Molgó M, Rodríguez A, Valbuena JR. Leukocytoclastic vasculitis as early manifestation of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly. Am J Dermatopathol. 2012;34:330–334. doi: 10.1097/DAD.0b013e3182318dd9. [DOI] [PubMed] [Google Scholar]
- 239.Broccoli A, Gandolfi L, Pellegrini C, Agostinelli C, Argnani L, Zinzani PL. Leukocytoclastic vasculitis associated with hairy cell leukemia at diagnosis: a case report and review of the literature. Tumori. 2016 doi: 10.5301/tj.5000487. [DOI] [PubMed] [Google Scholar]
- 240.Lee A, Sandhu S, Imlay-Gillespie L, Mulligan S, Shumack S. Successful use of Bruton’s kinase inhibitor, ibrutinib, to control paraneoplastic pemphigus in a patient with paraneoplastic autoimmune multiorgan syndrome and chronic lymphocytic leukaemia. Australas J Dermatol. 2017;58:e240–e242. doi: 10.1111/ajd.12615. [DOI] [PubMed] [Google Scholar]
- 241.Forbat E, Ali FR, Al-Niaimi F. Intravenous immunoglobulins in dermatology. Part 2: clinical indications and outcomes. Clin Exp Dermatol. 2018;43:659–666. doi: 10.1111/ced.13552. [DOI] [PubMed] [Google Scholar]
- 242.Quist SR, Kraas L. Treatment options for pyoderma gangrenosum. J Dtsch Dermatol Ges. 2017;15:34–40. doi: 10.1111/ddg.13173. [DOI] [PubMed] [Google Scholar]
- 243.Na I-K, Buckland M, Agostini C, Edgar JDM, Friman V, Michallet M, et al. Current clinical practice and challenges in the management of secondary immunodeficiency in hematological malignancies. Eur J Haematol. 2019;102:447–456. doi: 10.1111/ejh.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Cho A, Paulitschke V, Just U, Knobler R. Cutaneous manifestations of acute and chronic graft-versus-host disease. G Ital Dermatol Venereol. 2020;155:76–87. doi: 10.23736/S0392-0488.19.06535-0. [DOI] [PubMed] [Google Scholar]
- 245.Strong-Rodrigues K, Oliveira-Ribeiro C, de Abreu Fiuza Gomes S, Knobler R. Cutaneous graft-versus-host disease: diagnosis and treatment. Am J Clin Dermatol. 2018;19:33–50. doi: 10.1007/s40257-017-0306-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.