

Abstract
OBJECTIVES:
To determine the relationship between LQTS subtype and postnatal cardiac events (CEs).
BACKGROUND:
LQTS presenting with 2:1 atrioventricular (AV) block or torsades de pointes (TdP) in the fetus/neonate has been associated with risk for major CEs, but overall outcomes and predictors remain unknown.
METHODS:
A retrospective study involving 25 international centers evaluated the course of fetuses/newborns diagnosed with congenital LQTS and either 2:1 AV block or TdP. The primary outcomes were age at first CE after dismissal from the newborn hospitalization and death/cardiac transplantation during follow-up. CE was defined as aborted cardiac arrest (ACA), appropriate ICD shock (AS), or sudden cardiac death (SCD).
RESULTS:
Eighty-four fetuses/neonates were identified (12 LQT1, 35 LQT2, 37 LQT3). Median gestational age at delivery was 37 weeks (IQR 35 – 39) and age at hospital discharge was 3 weeks (IQR 2 – 5). Fetal demise occurred in 2 and pre-discharge death in 1. Over a median of 5.2 years, there were 1 LQT1, 3 LQT2, and 23 LQT3 CEs (13 ACA, 5 SCD, and 9 AS). One LQT1 patient and 11 LQT3 patients died or received cardiac transplant during follow-up. The only multivariate predictor of post-discharge CEs was LQT3 status (LQT3 vs LQT2, HR 8.4 [CI 2.6 – 38.9], p<0.001) and LQT3 genotype predicted death/cardiac transplant relative to LQT2 (p<0.001).
CONCLUSIONS:
In this large multicenter study, LQT3 but not LQT1 or LQT2 fetuses/neonates presenting with severe arrhythmias were at high risk of not only frequent, but lethal CEs.
Keywords: atrioventricular block, cardiac sympathetic denervation, genetic testing, implantable cardioverter-defibrillator, long QT syndrome, sudden cardiac death, torsades de pointes, fetus, magnetocardiography, fetal arrhythmia
CONDENSED ABSTRACT
Outcomes of fetuses and neonates with long QT syndrome (LQTS) presenting with 2:1 atrioventricular (AV) block or torsades de pointes (TdP) are unknown. Eighty-four affected fetuses/neonates were identified from 25 international centers. Over a median of 5.2 years, there were 27 post-discharge CEs. The only multivariate predictor of CEs was LQT3 status (p<0.001). Moreover, LQT3 genotype predicted death/cardiac transplant at last follow-up (p<0.001). These data suggest LQT3 fetuses/neonates with 2:1 AV block or TdP are at the highest risk for future CEs and experience not only frequent, but lethal CEs.
INTRODUCTION
Long QT syndrome (LQTS) is a genetic condition characterized by abnormal cardiac repolarization that in severe cases may lead to torsades de pointes (TdP) and sudden cardiac death (SCD). LQTS is estimated to occur in approximately 1 in 2,000 individuals but the true prevalence before birth is unknown.(1) Although usually diagnosed in childhood, cases with severe QTc prolongation may be detected in utero or in infancy and lead to fetal demise or sudden infant death syndrome.(2–5)
First described in 1965,(6) the severe infantile form of LQTS with the signature LQTS-associated rhythms of 2:1 atrioventricular block (AVB) or TdP manifests in approximately 5% of patients(7) and is associated with increased risk for recurrent malignant ventricular arrhythmia. Early studies suggested dismal outcomes with this presentation,(8–10) whereas more recent reports have been mixed with some studies suggesting a more favorable prognosis.(11,12) To date, the optimal management strategy for such patients is largely unknown, given ambiguity with respect to clinical outcomes. The present multicenter study was designed to clarify the postnatal course of fetuses and newborns who manifest severe LQTS rhythms in the perinatal period. It was hypothesized that LQTS genotype would be informative for post-discharge risk stratification.
METHODS
STUDY DESIGN AND POPULATION
Following ethics approval, the medical records at the 25 participating international centers over a 30-year period (June 1989 through June 2019), were queried for unrelated fetuses and newborns (≤30 days of life) presenting with severe LQTS. Severe LQTS was defined as clinical presentation with either TdP or 2:1 AV block as recorded by fetal magnetocardiography (fMCG) from 20 to 40 weeks’ gestation or electrocardiographically after birth in the setting of clinically confirmed LQTS. Only patients with major LQTS genotypes (LQT1, LQT2, and LQT3) as confirmed by genetic analysis, were included in this study.(13) Patients were excluded if genetic testing was not performed or if another genotype or a non-pathogenic variant in LQT1, LQT2, or LQT3 was discovered.(14) Patients with presumptive echocardiographically-diagnosed TdP or 2:1 AV block in utero who did not undergo fMCG or survive to birth to receive a definitive LQTS diagnosis were also excluded. All patient data were securely transmitted to study investigators at the UCLA Medical Center.
Longitudinal data were collected beginning at the time of the in utero or postnatal presentation until death or the last available follow-up. Baseline characteristics included demographics, presenting rhythm, LQTS genotype, results of obstetrical or fetal imaging and fMCG studies, timing and circumstances of delivery, and QTc from the first post-natal ECG. LQTS rhythms from both the fetal and neonatal period prior to discharge from the hospital were classified as TdP, 2:1 AV block, or both. The details and timing of all interventions including transplacental and postnatal medical therapy, pacemaker or implantable cardioverter-defibrillator (ICD) placement, left (LCSD) or bilateral (BCSD) cardiac sympathetic denervation and cardiac transplantation were also recorded.
The primary outcomes were 1) age at first CE following initial discharge from the hospital, defined as aborted cardiac arrest (ACA), appropriate ICD shock (AS), or SCD as determined by the treating physician and 2) age at death or cardiac transplantation. Secondary outcomes included a diagnosis of epilepsy (defined as ≥2 unprovoked seizures >24 hours apart),(15) developmental delay (further codified as language, cognitive, gross or fine motor as determined by routine clinical assessment), ICD complications, and ventricular dysfunction (defined as left ventricular ejection fraction ≤50% by Simpson’s biplane method).
STATISTICAL ANALYSIS
Data are presented as mean ± standard deviation or median (interquartile range; IQR) for continuous variables as appropriate, and as frequencies and percentages for dichotomous variables. Baseline characteristics among LQTS genotypes were compared with the Fisher’s exact test or Wilcoxon rank sums test for dichotomous and continuous variables, respectively. Cox proportional hazards modeling was used to assess univariate associations between baseline characteristics and age at first post-discharge CE. Covariates were chosen from previously published risk factors and on the basis of biologically-plausible associations. Covariates with a p-value <0.3 were considered as candidates for the multivariable analysis and were entered in the final multivariable Cox proportional hazards model to achieve a minimum Akaike information criterion (AIC) statistic. Freedom from the two main study outcomes were presented using the Kaplan-Meier method. Statistical differences across LQTS genotypes were tested with the overall log-rank test after which pairwise comparisons are reported. Two-tailed p-values <0.05 are considered to be statistically significant. All statistical analysis was performed using JMP Pro version 14.0 (SAS corporation, Cary, NC).
RESULTS
BASELINE POPULATION CHARACTERISTICS
A total of 84 LQTS patients were identified from clinical databases of the 25 participating centers (12 LQT1, 35 LQT2, 37 LQT3). Of these, 38 (45%) presented with a severe LQTS rhythm (2:1 AVB and/or TdP) in utero and 46 (55%) presented for the first time in the early (≤30 days) postnatal period. Baseline characteristics are summarized in Table 1.
Table 1.
Baseline LQTS population characteristics
| Patient Characteristic | n = 84 |
|---|---|
| Sex, female | 51 (61) |
| Prenatal diagnosis | 36 (43) |
| Prenatal documented rhythm | |
| TdP | 12 (14) |
| 2:1 AVB | 18 (21) |
| TdP + 2:1 AVB | 7 (8) |
| fMCG performed | 8 (10) |
| Transplacental therapy | |
| Beta-blocker | 10 (12) |
| Propranolol | 9 (11) |
| Metoprolol | 1 (1) |
| Lidocaine | 1 (1) |
| Mexiletine | 5 (6) |
| Flecainide | 1 (1) |
| Magnesium | 9 (11) |
| Cesarean section | 40 (48) |
| Delivery room intervention | 8 (10) |
| Postnatal documented rhythm | |
| TdP | 19 (23) |
| 2:1 AVB | 27 (32) |
| TdP + 2:1 AVB | 28 (33) |
| Discharge medications | |
| Beta-blocker | 71 (85) |
| Propranolol | 65 (77) |
| Carteolol | 3 (4) |
| Nadolol | 2 (2) |
| Metoprolol | 1 (1) |
| Mexiletine | 42 (50) |
| Flecainide | 4 (5) |
| Initial LV ejection fraction | 60 (49 – 68) |
Abbreviations: TdP, torsades de pointes; AVB, atrioventricular block; fMCG, fetal magnetocardiography; EF, ejection fraction.
Numbers in parentheses represent percentage or interquartile range (IQR) as appropriate.
FETAL COURSE
Two patients died in utero, both with documented TdP and 2:1 AV block in the setting of marked QT prolongation by fMCG (supplemental Figure 1). A genetic diagnosis of LQT3 was confirmed in both patients by fetal tissue analysis or cord blood sampling.
NEWBORN HOSPITAL COURSE
Eighty-two live newborns were delivered at a median gestational age of 37 weeks (IQR 35 – 39 weeks). One patient presented with functional 3:1 and 4:1 block and died prior to discharge from the hospital with arrhythmia-related cardiomyopathy. This patient experienced worsened ventricular arrhythmia following LCSD and succumbed to cardiogenic shock with an ICD in place. Genetic testing revealed a previously undescribed pathologic variant in LQT3 (F1760C). Major hospital interventions included pacemaker placement in 28 and ICD placement in 5 (primary in 3 and following pacemaker in 2). LCSD was performed in 5 patients and BCSD in 1, resulting in improved arrhythmia burden in 1 LCSD and 1 BCSD. The most commonly prescribed discharge medications were propranolol in 65 (median 3 mg/kg/day [IQR 2 – 4]) and mexiletine in 42 (median 12 mg/kg/day [IQR 6 – 16]).
POST-DISCHARGE COURSE AND OUTCOMES (Figure 1 – central illustration)
Figure 1 (central illustration).
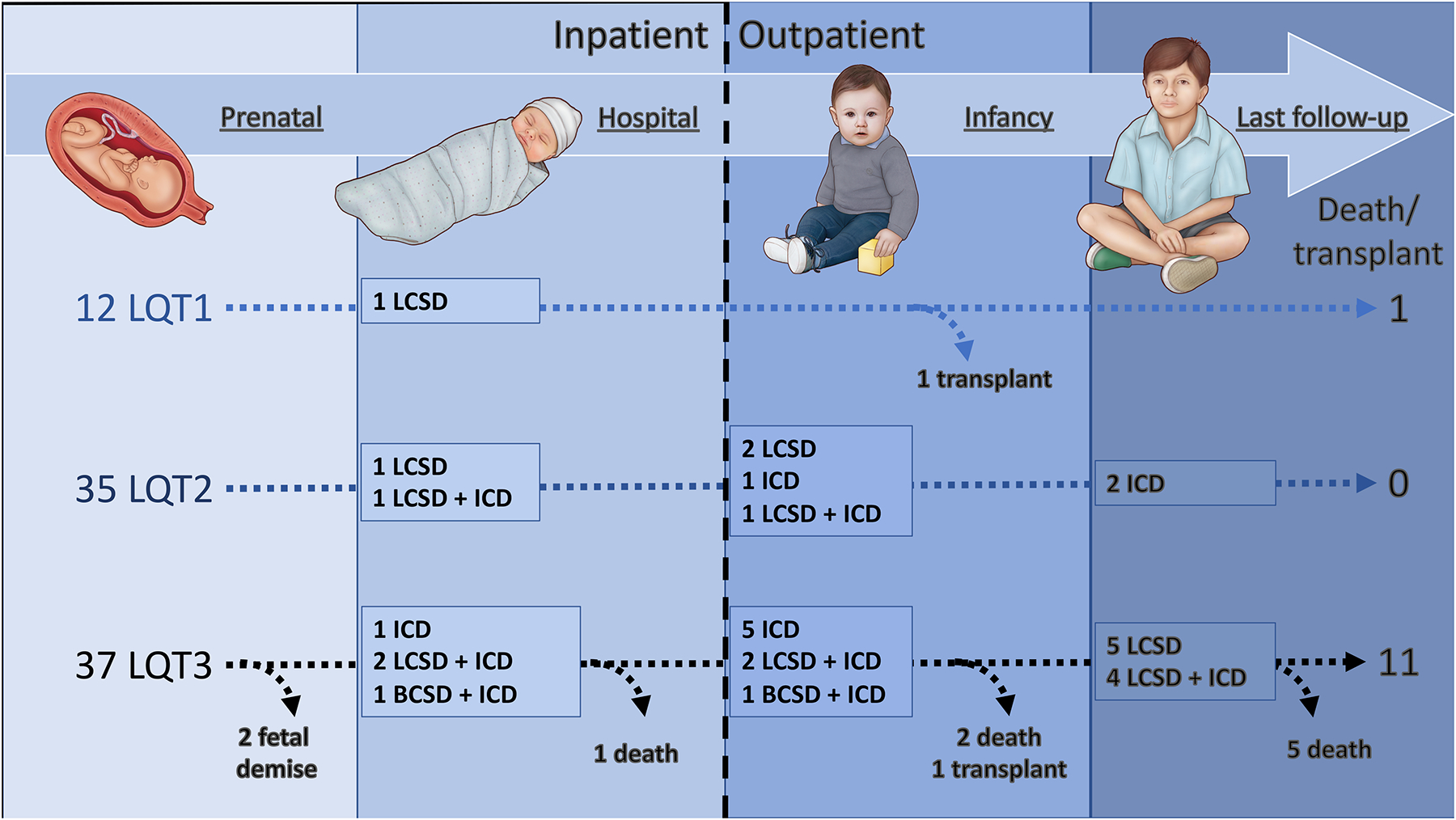
Time-line demonstrating major interventions and outcomes for the study cohort.
Abbreviations: BCSD, bilateral cardiac sympathetic denervation; LCSD, left cardiac sympathetic denervation; ICD, implantable cardioverter-defibrillator; OHT, orthotopic heart transplantation.
Eighty-one newborns survived to hospital discharge at a median of 3 weeks (IQR 2 – 5 weeks) and were eligible for the primary outcome analyses. Following discharge, 9 additional patients underwent pacemaker placement and a further 20 underwent ICD placement (8 primary and 12 following prior pacemaker). In addition, LCSD was performed in 15 additional patients (1 followed by BCSD) and resulted in clinical improvement in 6 LCSD and 1 BCSD.
Over a median of 5.2 years (IQR 1.8 – 9.1 years), post-discharge CEs occurred in 1 LQT1, 3 LQT2, and 23 LQT3 patients (initial events were ACA in 13, AS in 9, and SCD in 5). All 5 patients presenting with a first post-discharge event of SCD were in the LQT3 cohort. Significant univariate predictors of post-discharge CEs included LQT3 genotype (LQT3 vs LQT1, HR 6.2 [IQR 1.3–111.5], p=0.02; LQT3 vs LQT2, HR 10.4 [IQR 3.6–43.9], p<0.001), negative family history of LQTS, and first postnatal QTc. When adjusted for other covariates, only LQT3 genotype remained predictive of CEs when compared to LQT2 (Table 2 and Figure 2). LQTS genotype was relatively specific for 1-year cardiac events, with events occurring exclusively in LQT3 and LQT1 infants. The single LQT1 patient who underwent transplant presented at 3 months of age with suspected frequent episodes of ventricular arrhythmia. The patient was admitted to the hospital and 3 days later experienced recurrent TdP, progressing to mechanical circulatory support and eventual cardiac transplantation. Explanted histology data for this patient were not available.
Table 2.
Univariable and multivariable predictors of age at first post-discharge cardiac event
| Univariable predictor | Hazard ratio | 95%CI | p-value | Mutivariable predictor | Hazard ratio | 95%CI | p-value |
|---|---|---|---|---|---|---|---|
| Treatment era a. 2010 | 0.7 | (0.6 – 3.4) | 0.4 | ||||
| Signature rhythm | |||||||
| TdP + AV block (vs isolated TdP) | 2 | (0.8 – 6.1) | 0.17 | ||||
| TdP + AV block (vs isolated AV block) | 1.8 | (0.8 – 4.8) | 0.18 | ||||
| Isolated TdP (vs isolated AV block) | 0.8 | (0.2, 2.5) | 0.7 | ||||
| Prenatal diagnosis | 0.9 | (0.4–1.9) | 0.67 | ||||
| Prenatal characteristic | |||||||
| Female gender | 0.7 | (0.6, 3.0) | 0.39 | ||||
| Family history | 0.4 | (0.1, 1.0) | 0.04 | Family history | 1.2 | (0.2, 3.8) | 0.84 |
| Hydrops fetalis | 1.9 | 0.3, 6.8) | 0.65 | ||||
| Postnatal characteristic | |||||||
| Postnatal QTc per 10 ms | 1.03 | (1.0, 1.1) | 0.22 | Postnatal QTc per 10 ms | 2 | (0.3, 13.8) | 0.26 |
| Genotype, LQT3 (vs LQT1) | 6.2 | (1.3 – 111.5) | 0.02 | Genotype, LQT3 (vs LQT1) | 6.2 | (0.7, 130.0) | 0.1 |
| Genotype, LQT3 (vs LQT2) | 10.4 | (3.6 – 43.9) | <0.001 | Genotype, LQT3 (vs LQT2) | 8.4 | (2.6, 38.9) | <0.001 |
| Genotype, LQT2 (vs LQT1) | 0.3 | (0.03 – 7.4) | 0.38 |
Abbreviations. AVB, atrioventricular block; CI, confidence interval; LQT, long QT; TdP, torsades de pointes
Figure 2.
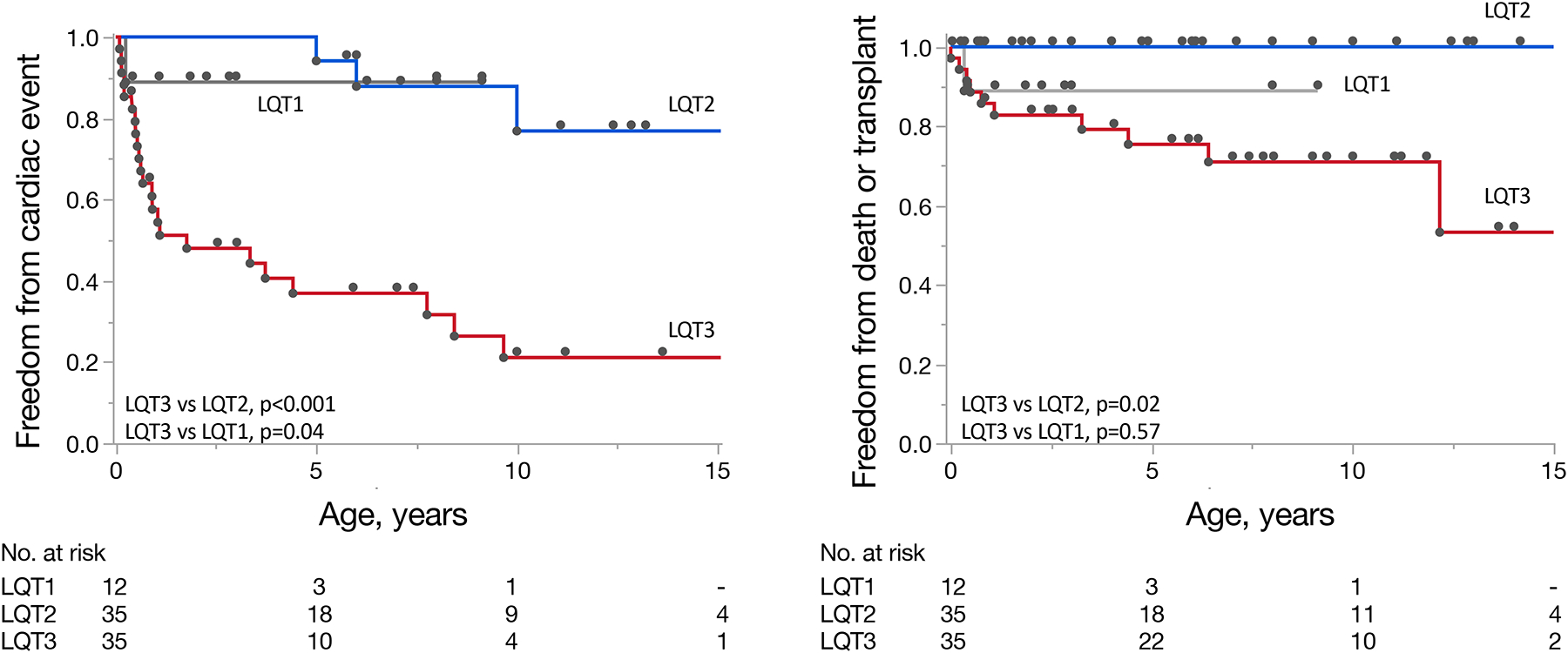
A) Kaplan-Meier curves demonstrating time to major cardiac events and B) time to death or heart transplantation for patients with LQT1, LQT2, and LQT3 genotypes. P-values are calculated using the log-rank test.
Abbreviations. LQT, long QT
At last follow-up, death or transplantation occurred in 1 LQT1 (8%), no LQT2 (0%), and 9 LQT3 (24%) patients (not considering 2 LQT3 patients with fetal demise) and worse transplant-free survival was apparent for LQT3 vs LQT2 (p<0.001) (Figure 2). Four LQT3 patients died or received cardiac transplantation despite both prior ICD placement and either LCSD or BCSD, and all 9 had been treated with sodium channel blocker (7 mexiletine [median 10, IQR 5 – 26 mg/kg/day] or 2 flecainide [6 mg/kg/day and 100 mg/m2]) and 9 propranolol (median 4, IQR 4 – 5 mg/kg/day). Three of these LQT3 patients experienced uncontrolled arrhythmia and one died from status epilepticus. All 3 patients undergoing cardiac transplantation were alive at last follow-up (1.6, 4.5 and 14.9 years of age). Although similar at baseline, LQTS survivors showed a significant reduction in QTc from baseline to last follow-up (571 vs 484 ms, p<0.001) whereas a similar reduction in QTc was not observed for those experiencing death or transplantation (601 ms vs 594 ms, p=0.54).
COMPLICATIONS RELATED TO CARDIAC INTERVENTIONS
There were 8 patients with cardiac implantable device-related complications. Three experienced lead fracture, 3 infection and 2 device migration. Acute complications of LCSD occurred in 2 cases, with 1 Horner’s syndrome and 1 inadvertent pleural puncture.
NEURO-DEVELOPMENTAL OUTCOMES
Overall, 10 (12%) patients were diagnosed with ≥1 form of developmental delay. The delay was classified as language in 7, cognitive in 7, gross motor in 4, and fine motor in 4. Ten patients (12%) were diagnosed with epilepsy. LQT3 was common among patients with poor neuro-developmental outcomes, comprising 8 (80%) of the patients with developmental delay and all of the patients with epilepsy. A numerically greater proportion of those with non-fatal initial CEs were diagnosed with developmental delay than those without CEs (6 [22%] vs 5 [10%], p=0.14). There was no apparent association between the severity of CEs and neurodevelopmental outcome.
GENETIC ASSOCIATIONS
LQT3 genotype was associated with negative family history of LQTS, postnatal interventions of LCSD/BSCD and ICD placement, left ventricular systolic dysfunction by echocardiography, and adverse developmental and neurologic outcomes (Table 3). See Figure 3 for a complete description of the genetic variants identified during clinical evaluation and final outcomes.
Table 3.
Patient characteristics grouped by LQTS genotype
| Characteristic | LQT1 (N=12) | LQT2 (N=35) | LQT3 (N=37) |
|---|---|---|---|
| Sex, female | 6 (50) | 25 (71) | 20 (54) |
| Family history of LQTS* | 11 (92) | 19 (54) | 6 (16) |
| -Number of patients - pre-natal presentation | 8 | 15 | 14 |
| Gestational age at diagnosis, wks | 26 (22 – 30) | 33 (28 – 34) | 31 (26 – 36) |
| LQTS rhythm* | |||
| Isolated TdP | 0 (0) | 8 (53) | 4 (29) |
| Isolated 2:1 AVB | 8 (100) | 3 (20) | 7 (50) |
| TdP +2:1 AVB | 0 (0) | 4 (27) | 2 (14) |
| Hydrops fetalis | 0 (0) | 2 (13) | 3 (21) |
| Gestational age at delivery, wks | 38 (36 – 39) | 37 (35 – 39) | 37 (34 – 39) |
| -Number of patients - postnatal follow-up | 12 | 36 | 35 |
| LQTS rhythm* | |||
| Isolated TdP | 1 (8) | 14 (39) | 5 (14) |
| Isolated 2:1 AVB | 6 (50) | 10 (28) | 12 (38) |
| TdP + 2:1 AVB | 0 (0) | 10 (28) | 18 (49) |
| First post-natal QTc | 558 (531 – 582) | 585 (522 – 640) | 592 (536 – 667) |
| Post-natal intervention | |||
| Cardiac sympathetic denervation* | 1 (8) | 5 (14) | 15 (43) |
| Pacemaker placement | 1 (8) | 17 (47) | 19 (54) |
| Defibrillator placement* | 0 (0) | 5 (14) | 20 (57) |
| Beta-blocker therapy | 9 (75) | 32 (89) | 30 (86) |
| Mexiletine* | 0 (0) | 19 (53) | 23 (66) |
| Age at discharge, wks | 4 (1 – 12) | 2 (1 – 5) | 3 (2 – 6) |
| LVEF <50% during follow-up* | 1 (8) | 0 (0) | 9 (24) |
| -Number of patients - defibrillator outcomes | 0 | 5 | 20 |
| ≥1 appropriate shock* | - | 1 (20) | 16 (80) |
| ≥1 inappropriate shock | - | 0 (0) | 4 (20) |
| VT storm | - | 0 (0) | 5 (25) |
| Device infection | - | 1 (20) | 2 (10) |
| Lead failure | - | 1 (20) | 3 (15) |
| -Number of patients - neurodevelopmental outcomes | 9 | 33 | 33 |
| Developmental delay* | 1 (11) | 1 (3) | 8 (24) |
| Cognitive | 0 | 0 | 7 |
| Fine motor | 0 | 0 | 4 |
| Gross motor | 0 | 1 | 3 |
| Language | 1 | 0 | 6 |
| Epilepsy* | 0 (0) | 0 (0) | 10 (30) |
Abbreviations. AVB, atrioventricular block; CI, confidence interval; LQT, long QT; TdP, torsades de pointes; VT, ventricular tachycardia; wks, weeks
indicates p-value <0.05 for overall group comparisons
Figure 3.

Bar graph demonstrating the genetic variants detected among study patients. Asterisks denote those that succumbed to death/cardiac transplantation during follow-up. For 9 patients, only the LQTS subtype and not the specific genetic variant was contained in the available medical record (4 KCNQ1, 4 KCNH2, 1 SCN5A)
Abbreviations. LQT, long QT
DISCUSSION
KEY FINDINGS
This large multicenter study examined major outcomes of newborns with genetically-confirmed LQTS1, 2, and 3 discharged from the hospital after an initial presentation with either 2:1 AVB or TdP in either fetal or postnatal life. This study convincingly shows for the first time that LQTS genotype accurately predicts post-discharge CEs, with significantly worse outcomes for patients with LQT3 as compared to LQT1 and LQT2. LQT3 patients displayed a rapid trajectory toward CEs, of which 20% were fatal. Conversely, with one exception both LQT1 and LQT2 patients experienced a relatively benign clinical course and outcomes, experiencing no deaths and a single cardiac transplantation. These novel observations are expected to be useful for prenatal counseling and may facilitate postnatal clinical care and implementation of therapeutic strategies for a particularly malignant LQT3 subgroup.
GENOTYPE AND POSTNATAL OUTCOMES
Despite a clear association with lethal CEs,(10) the clinical course of LQTS newborns presenting with TdP or 2:1 AV block has remained incompletely characterized. Of the LQTS subtypes, LQT2 and LQT3 have generally been associated with the most severe arrhythmias.(11,12,16,17) While favorable outcomes after this presentation have been observed in some descriptive studies,(11,12) others have reported mixed or even dismal outcomes.(16,17) There have been preliminary data that LQT3 by itself could portend a particularly severe prognosis, but definitive studies have been lacking. One report of 33 LQT3 patients provided the first evidence that those with cardiac events in the first year of life were at higher risk than asymptomatic patients during the first year of life,(18) and another demonstrated that many LQTS stillbirth victims host rare nonsynonymous SCN5A variants.(2) Recently, review of comprehensive fMCG data suggested a severe phenotype and high mortality for fetuses with LQT3.(19) Nevertheless, a comparative study exploring the relationship between genotype and clinical outcome in the fetal/neonatal population has not previously been performed. The present results confirm the predominance of LQT2 and LQT3 among the population of fetuses and neonates presenting with severe LQTS arrhythmias, but more importantly, conclusively show that the risk chiefly resides among those affected by LQT3. Such patients faced CEs significantly more frequently during follow-up and experienced a high proportion of lethal events. These data also indicate the utility of early genetic diagnosis for fetuses and neonates presenting with 2:1 AV block or TdP, so that the physicians may implement aggressive medical and interventional strategies prior to birth and prior to dismissal from the newborn hospitalization.
TREATMENT MODALITIES AND EFFICACY
Treatment strategies for infants and children with severe LQTS phenotype consisted primarily antiarrhythmic drugs, cardiac devices (pacemakers or defibrillators), and LCSD. All of these treatment modalities were extensively utilized in this high-risk population, consistent with prior experience.(12) Overall, medication failure was observed and resulted in either cardiac device placement or LCSD. As these latter treatment strategies were more invasive, treatment-related comorbidities were not infrequent and were observed after 8 cardiac implantable electronic device and 2 LCSD procedures. In addition, even these more aggressive approaches were inconsistently successful. In the present study, LCSD resulted in decreased arrhythmia burden in fewer than half of cases. Likewise, deaths or cardiac transplantation still occurred in 4 LQT3 patients despite both LCSD and ICD placement as well as standard sodium channel and beta-blocker administration.
Given the higher arrhythmia burden, greater requirement for invasive procedures, and frequent treatment failure for LQT3, these patients experienced the greatest consequences of their disease. The risks associated with this genotype should therefore be disclosed to families as early as possible with careful counseling and provision of tailored therapeutic strategies. Interestingly, the survival among LQT3 cases in this large multicenter cohort was similar to the estimated 5-year survival (approximately 70%) associated with primary transplantation in infancy.(20) This last observation begets the question of whether severe arrhythmias (e.g. VT storm despite optimal medical management) in a subset of LQT3 neonates should prompt early referral for cardiac transplantation. Further study is required to address this issue.
STUDY LIMITATIONS
The study was retrospective and spanned a 30-year period. As such, some data were occasionally unavailable. For instance, familial cascade screening to adjudicate de novo LQTS variants were inconsistently available and reliance was predominantly on the family history. Moreover, patients were excluded from the study if the results of previously performed genetic tests were negative. Given the importance of the recently-described CALM variants and the associated severe LQTS-like presentation, it is possible that some of the excluded patients harbored one of these more elusive genetic variants. Therefore, future studies should evaluate the influence of these other LQTS genotypes on the outcomes of fetuses and neonates presenting with severe arrhythmia. Finally, although the study outcomes were identified a priori, the analysis was partially exploratory in nature. It is therefore expected that these findings be validated in the context of larger prospective investigations.
CONCLUSION
Fetuses/infants with LQTS, who present with either 2:1 AVB or TdP, are at increased risk for adverse CEs following hospital discharge. LQT3 predicts both frequent and lethal post-discharge events and should be considered in pre-discharge management, with early consideration for cardiac transplant evaluation in select neonates.
Supplementary Material
Supplemental figure 1. Demonstration of severe LQTS rhythms in separate patients by fetal magnetocardiography (fMCG). A) FMCG taken at a gestational age of 33 weeks and 4 days demonstrating 2:1 atrioventricular block, and B) tracing taken at 34 weeks and 6 days showing torsade de pointes.
Social media summary:
This was a multicenter study to evaluate the outcomes in a large cohort of severe LQTS fetuses and neonates
Cardiac events and mortality were frequent among LQT3 neonates and were rare among those with LQT1 and LQT2
Early genotyping may be useful for clinical management of severe neonatal LQTS
#JACCCEP #cvEP #cvGenetics #LQTS #cvPEd #SuddenCardiacArrest #fetus, #magnetocardiography
ACKNOWLEDGEMENTS
The authors wish to thank Danielle Harake, MD for her contribution to data collection.
Abbreviations:
- ACA
Aborted cardiac arrest
- AS
Appropriate shock
- AVB
Atrioventricular block
- BCSD
Bilateral cardiac sympathetic denervation
- CE
Cardiac events
- ICD
Implantable cardioverter-defibrillator
- LCSD
Left cardiac sympathetic denervation
- LQTS
Long QT syndrome
- SCD
Sudden cardiac death
- TdP
Torsades de pointes
Footnotes
PERSPECTIVES: Among fetuses and newborns presenting with severe LQTS rhythms, LQT3 genotype predicts both more frequent post-discharge cardiac events as well as death or cardiac transplantation at last follow-up.
COMPETENCY IN MEDICAL KNOWLEDGE: Knowledge of LQTS genotype may be used to tailor management strategies for affected newborns prior to hospital discharge.
TRANSLATIONAL OUTLOOK: Future studies are needed to clarify the optimal clinical care pathways for affected LQTS patients in light of the increased risk associated with LQT3 fetuses and neonates.
REFERENCES
- 1.Schwartz PJ, Stramba-Badiale M, Crotti L et al. Prevalence of the congenital long-QT syndrome. Circulation 2009;120:1761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crotti L, Tester DJ, White WM et al. Long QT syndrome-associated mutations in intrauterine fetal death. JAMA 2013;309:1473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller TE, Estrella E, Myerburg RJ et al. Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome. Circulation 2004;109:3029–34. [DOI] [PubMed] [Google Scholar]
- 4.Arnestad M, Crotti L, Rognum TO et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation 2007;115:361–7. [DOI] [PubMed] [Google Scholar]
- 5.Ackerman MJ, Siu BL, Sturner WQ et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA 2001;286:2264–9. [DOI] [PubMed] [Google Scholar]
- 6.Romano C. Congenital Cardiac Arrhythmia. Lancet 1965;1:658–9. [DOI] [PubMed] [Google Scholar]
- 7.Garson A Jr., Dick M 2nd, Fournier A et al. The long QT syndrome in children. An international study of 287 patients. Circulation 1993;87:1866–72. [DOI] [PubMed] [Google Scholar]
- 8.Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. The long QT syndrome with impaired atrioventricular conduction: a malignant variant in infants. J Cardiovasc Electrophysiol 1998;9:1225–32. [DOI] [PubMed] [Google Scholar]
- 9.Saoudi N, Bozio A, Kirkorian G, Atallah G, Normand J, Touboul P. Prolonged QT, atrioventricular block, and sudden death in the newborn: an electrophysiologic evaluation. Eur Heart J 1991;12:838–41. [DOI] [PubMed] [Google Scholar]
- 10.Trippel DL, Parsons MK, Gillette PC. Infants with long-QT syndrome and 2:1 atrioventricular block. Am Heart J 1995;130:1130–4. [DOI] [PubMed] [Google Scholar]
- 11.Lupoglazoff JM, Denjoy I, Villain E et al. Long QT syndrome in neonates: conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J Am Coll Cardiol 2004;43:826–30. [DOI] [PubMed] [Google Scholar]
- 12.Aziz PF, Tanel RE, Zelster IJ et al. Congenital long QT syndrome and 2:1 atrioventricular block: an optimistic outcome in the current era. Heart Rhythm 2010;7:781–5. [DOI] [PubMed] [Google Scholar]
- 13.Blais BA, Satou G, Sklansky MS, Madnawat H, Moore JP. The diagnosis and management of long QT syndrome based on fetal echocardiography. HeartRhythm Case Rep 2017;3:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher RS, Acevedo C, Arzimanoglou A et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475–82. [DOI] [PubMed] [Google Scholar]
- 16.Horigome H, Nagashima M, Sumitomo N et al. Clinical characteristics and genetic background of congenital long-QT syndrome diagnosed in fetal, neonatal, and infantile life: a nationwide questionnaire survey in Japan. Circ Arrhythm Electrophysiol 2010;3:10–7. [DOI] [PubMed] [Google Scholar]
- 17.Cuneo BF, Etheridge SP, Horigome H et al. Arrhythmia phenotype during fetal life suggests long-QT syndrome genotype: risk stratification of perinatal long-QT syndrome. Circ Arrhythm Electrophysiol 2013;6:946–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD: true or false? Heart Rhythm 2009;6:113–20. [DOI] [PubMed] [Google Scholar]
- 19.Strand S, Strasburger JF, Cuneo BF, Wakai RT. Complex and Novel Arrhythmias Precede Stillbirth in Fetuses with De Novo Long QT Syndrome. Circulation: Arrhythmia and Electrophysiology 2020;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossano JW, Cherikh WS, Chambers DC et al. The Registry of the International Society for Heart and Lung Transplantation: Twentieth Pediatric Heart Transplantation Report-2017; Focus Theme: Allograft ischemic time. J Heart Lung Transplant 2017;36:1060–1069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. Demonstration of severe LQTS rhythms in separate patients by fetal magnetocardiography (fMCG). A) FMCG taken at a gestational age of 33 weeks and 4 days demonstrating 2:1 atrioventricular block, and B) tracing taken at 34 weeks and 6 days showing torsade de pointes.