

Abstract
Decline in insulin sensitivity due to dysfunction of adipose tissue (AT) is one of the earliest pathogenic events in type 2 diabetes. We hypothesize that differential DNA methylation (DNAm) controls insulin sensitivity and obesity by modulating transcript expression in AT. Integrating AT DNAm profiles with transcript profile data measured in a cohort of 230 African Americans (AAs) from the African American Genetics of Metabolism and Expression cohort, we performed cis-expression quantitative trait methylation (cis-eQTM) analysis to identify epigenetic regulatory loci for glucometabolic trait–associated transcripts. We identified significantly associated cytosine-guanine dinucleotide regions for 82 transcripts (false discovery rate [FDR]-P < 0.05). The strongest eQTM locus was observed for the proopiomelanocortin (POMC; ρ = −0.632, P = 4.70 × 10−27) gene. Epigenome-wide association studies (EWAS) further identified 155, 46, and 168 cytosine-guanine dinucleotide regions associated (FDR-P < 0.05) with the Matsuda index, SI, and BMI, respectively. Intersection of EWAS, transcript level to trait association, and eQTM results, followed by causal inference test identified significant eQTM loci for 23 genes that were also associated with Matsuda index, SI, and/or BMI in EWAS. These associated genes include FERMT3, ITGAM, ITGAX, and POMC. In summary, applying an integrative multiomics approach, our study provides evidence for DNAm-mediated regulation of gene expression at both previously identified and novel loci for many key AT transcripts influencing insulin resistance and obesity.
Introduction
Reduced insulin sensitivity, or insulin resistance (IR), is an early marker of type 2 diabetes (T2D) risk, and understanding the biological processes that underlie IR is crucial to the development of new approaches to prevent and treat T2D (1,2). Transcriptional dysregulation of genes in tissues involved in glucose homeostasis is a key molecular mechanism associated with IR and obesity (3–6). As a second dimension to the genome, the epigenome contains important information for shaping the transcriptional landscape of cells (7). DNA methylation (DNAm) takes place most often at cytosines proximal to guanine nucleotides (cytosine-guanine dinucleotide [CpG] sites) (8,9), and it is a major epigenetic modification with a well-documented mechanistic role in gene expression (9,10). However, the contribution of DNAm to variation in glucometabolic traits, including IR and obesity, is poorly understood (11).
Most large-scale methylation studies on glucometabolic traits use DNA from blood leukocytes (12–18). However, because of the tissue-specific nature of DNAm, studies using blood cells may miss important epigenetic regulatory mechanisms associated with IR (8,9). Adipose tissue (AT) shows characteristics of an endocrine organ, affects many metabolic pathways, and has a key role in glucose homeostasis (19). In contrast to blood expression profiles, gene expression in AT strongly correlates with glucose homeostasis and obesity-related traits (20). Thus, analyzing AT may reveal the broader role of DNAm in determining insulin sensitivity and susceptibility to T2D by regulating the transcriptome. Data from epigenetic studies in European ancestry cohorts suggest that differential DNAm in AT may play an important role in glucose homeostasis, obesity, T2D, and related metabolic traits (21–26). U.S. minority populations, including African Americans (AAs), are at high risk for IR, obesity, and T2D. DNAm is highly divergent between populations, and this divergence can be largely attributed to differences in allele frequencies (genetic) and environmental exposures (27). However, studies evaluating CpG methylation in glucose homeostasis–relevant tissues and its role in determining IR and obesity in African ancestry individuals are lacking. Thus, building on the unique resources from our African American Genetics of Metabolism and Expression (AAGMEx) cohort (5,6), we have generated high-density AT DNA-methylation profiles using reduced representation bisulfite sequencing (RRBS).
We hypothesize that differential DNAm in regulatory regions of a subset of genes controls insulin sensitivity and obesity by modulating transcript expression in AT of African ancestry individuals. Integrating DNAm with glucometabolic phenotypes and AT transcript profile data, we first performed cis-expression quantitative trait methylation (cis-eQTM) analysis focused on glucometabolic trait–associated transcripts. eQTM analysis identified local CpGs (within ±100 kilobase pairs [kb] of the respective transcript) whose DNAm levels were correlated with expression levels of glucometabolic trait–associated transcripts. We further completed epigenome-wide association studies (EWAS) to test for the association of DNAm levels of CpGs with insulin sensitivity (Matsuda index and SI derived from oral glucose tolerance test [OGTT] and frequently sampled intravenous glucose tolerance test [FSIGT], respectively) and BMI in the AAGMEx cohort. We integrated results from the EWAS and eQTM analyses to determine if the trait-associated CpGs are cis-eQTMs for glucometabolic trait–associated transcripts in AT. Thus, our study combined available physiological evaluation and gene expression data with AT DNAm profiles in an integrative functional omics paradigm to trace molecular changes through layers of biological information, and it defined epigenetic regulatory mechanisms involved in modulating insulin sensitivity and obesity in AAs.
Research Design and Methods
Human Subjects
Subcutaneous AT biopsy samples collected from 230 individuals without diabetes from the AAGMEx cohort (5,6) were used for epigenome-wide DNAm profiling. The clinical and anthropometric characteristics of the AAGMEx cohort have been previously described (5,6,28). Briefly, cohort participants were healthy, self-reported AA men and women residing in North Carolina, aged 18–60 years, with a BMI between 18 and 42 kg/m2 (Supplementary Table 1). All participants provided written informed consent under protocols approved by the institutional review board at Wake Forest School of Medicine.
Laboratory Measures and Physiological Phenotypes
Details of clinical laboratory measures have been previously described (5,6,28). Among the participants in the AAGMEx cohort, AT transcript profiling data were available for 256 individuals. However, high-quality insulin sensitivity data from both OGTT and FSIGT was available for a subset of 230 individuals (male/female = 124/106), which were selected for this study. Individuals in the AAGMEx cohort had a broad range of insulin sensitivity as measured by the OGTT-derived Matsuda index (mean ± SD 6.2 ± 6.7) and FSIGT-derived SI (mean ± SD 4.0 ± 3.3).
AT DNAm Profiling and Bioinformatic Analysis
Genomic DNA from ∼100 mg frozen subcutaneous AT biopsies was isolated with the Qiagen DNeasy tissue kit (cat. no. 69504). Epigenome-wide profiling of DNAm levels was performed by RRBS. The DNAm profiling and bioinformatics analysis of RRBS data were conducted by Diagenode RRBS service (Diagenode, Liege, Belgium). RRBS libraries were prepared using the Premium RRBS Kit (cat. no. C02030033; Diagenode). RRBS library pools were sequenced on a HiSeq3000 (Illumina, San Diego, CA) using 50 base pair (bp) single-read sequencing (SR50) to obtain at least 30 million reads/sample. The adapter-trimmed sequence reads were aligned to the Homo sapiens reference genome (Genome Reference Consortium 37, hg19), and cytosine methylation values were called using Bismark v0.20.0 (29) with default parameters. Further, MethylKit (30) was used to filter the CpG data set for low coverage and for extremely high coverage to discard reads with possible PCR bias. The data were then normalized for read coverage distribution between samples with the MethylKit default settings (median normalization). Similar to a published study (25), we filtered our data set for CpGs with at least 10× coverage and that are present in at least 75% of the samples, corresponding to 1,073,614 CpGs. Coverage tables and normalized methylation values for the CpG sites covered more than 10× in at least 75% of the samples were generated with MethylKit. Previous studies have shown that methylation levels of neighboring CpGs tend be correlated over a distance of a few 100 bases to 1 kb (31,32), and in contrast to individual CpGs, the methylation level of a methylation region (average methylation of CpG sites in the region) is likely to be robust to measurement errors (25). Additionally, several closely located CpG sites may serve as a more biologically relevant genomic unit in epigenetic regulation of transcript expression. Thus, normalized methylation values summarizing methylation information over tiling windows of 1,000 bp across the genome and covering more than 10× in at least 75% of the samples were extracted using MethylKit (30), and this was considered as a CpG methylation region. These analyses generated methylation levels for 205,566 CpG regions of 1,000-bp windows. We filtered out nonvariable and nonautosomal CpG sites and regions and our analyses focusing on individual CpG sites used data for 1,054,719 CpG sites, while the analyses focusing on CpG regions used data for 200,800 CpG regions. Further detail is included in Supplementary Materials.
Genome-Wide Transcript Profiling
Gene expression and DNAm data were obtained from separate tissue portions from the same subcutaneous AT biopsy site. Genome-wide expression data were generated with HumanHT-12 V4 Expression BeadChips (Illumina) using Illumina GenomeStudio V2011.1. Expression levels were log2 transformed, robust multiarray average normalized (includes quantile normalization), and batch corrected using ComBat (https://www.bu.edu/jlab/wp-assets/ComBat/). Further details of AT gene expression analyses and data quality control methods have previously been published (5,6).
Statistical Analysis
Our previous transcriptome-wide association study (TWAS) analyses identified 7,464 glucometabolic trait–associated transcripts significantly associated with at least 1 of the 23 individual traits and six composite glucometabolic phenotypes tested at Benjamini-Hochberg false discovery rate (FDR)-corrected P value <0.01 in AT of AAGMEx participants (5,6,28,33). The eQTM analysis used linear regression, as implemented in MatrixEQTL to test whether DNAm levels were associated with transcript expression levels of these glucometabolic trait–associated transcripts. Here, the transcript expression level (log2) was the outcome and the methylation level of each CpG site or region was the predictor, with age, sex, and admixture (estimates of African genetic ancestry proportion computed using the ADMIXTURE program; https://dalexander.github.io/admixture/index.html) as covariates. We searched for cis-eQTMs in and around ±100 kb of each transcript. As a measure of the cis-eQTMs effect size and to ensure reporting of reproducible findings independent of statistical methods, we estimated the partial correlation coefficients. These partial correlation coefficients for methylation and transcript expression levels were also adjusted for age, sex, and admixture, and the adjusted residuals for both methylation and transcript levels were used to compute the Spearman correlation coefficient. The P values from the linear regression analysis were adjusted for multiple comparisons using the Benjamini-Hochberg FDR procedure (FDR-adjusted P value). After removing probes with poor expression level (expression P < 0.05 in <25% of the individuals) and probes encompassing common single nucleotide polymorphisms (SNPs), the analysis focused on the results for 6,774 high-quality transcripts.
An EWAS was performed to identify the associations between methylation levels of all measured CpG sites/regions with Matsuda index, SI, and BMI. For these EWAS analyses, we computed a linear regression model with the glucometabolic phenotypes as outcome, methylation level as predictor and age, sex, and admixture as covariates. FDR-adjusted P values are reported.
To explore potential mechanistic links, we integrated both layers of omic (methylome and transcriptome) data with phenotypic data. We intersected results from EWAS (CpG methylation to trait) and TWAS (transcript level to trait) analyses for each trait (Matsuda index, SI, and BMI) separately with results from eQTM (CpG methylation to transcript level) analysis. Intersecting results from these three analyses help to identify the CpG sites/regions that may determine IR and obesity by regulating expression of glucometabolic trait–associated transcripts. CpG sites/regions that are eQTMs for a glucometabolic trait–associated transcript and are associated with a glucometabolic trait (Matsuda index, SI, or BMI) are filtered through the intersection of three analyses (eQTM, TWAS, and EWAS) and further evaluated by using a causal inference test (CIT) in which CpG is the instrument variable. CpG sites/regions that are eQTMs (FDR-P < 0.05) for a transcript (FDR-P < 0.01 in TWAS) and are also associated with the specific trait (EWAS P < 0.01) were selected for the CIT, a formal test of causality that simultaneously evaluates multiple conditions known to be consistent with causal mediation. Since EWAS of three glucometabolic traits yielded few significant signals at FDR-P < 0.05, a less stringent P value (P < 0.01) in EWAS was considered for this intersection. The CIT (34) implemented in the R package CIT (https://cran.r-project.org/web/packages/cit/index.html) was used to evaluate potential causal relationships between selected CpGs, transcript expression, and clinical traits (Matsuda index, SI, and BMI). In this test, CpG is an instrument variable (L), trait is the outcome (T), and transcript level is the potential causal mediator (G). The CIT performs statistical significance testing for four conditions: 1) L and T are associated, 2) L is associated with G|T, 3) G is associated with T|L, and 4) L is independent of T|G. The largest of the four P values (PCIT) is the primary inferential metric in CIT. In this exploratory analysis, PCIT <0.05 for a SNP loci was considered significant and implied putative causation (L→G→T). The study design and analysis strategy are shown in Fig. 1.
Figure 1.
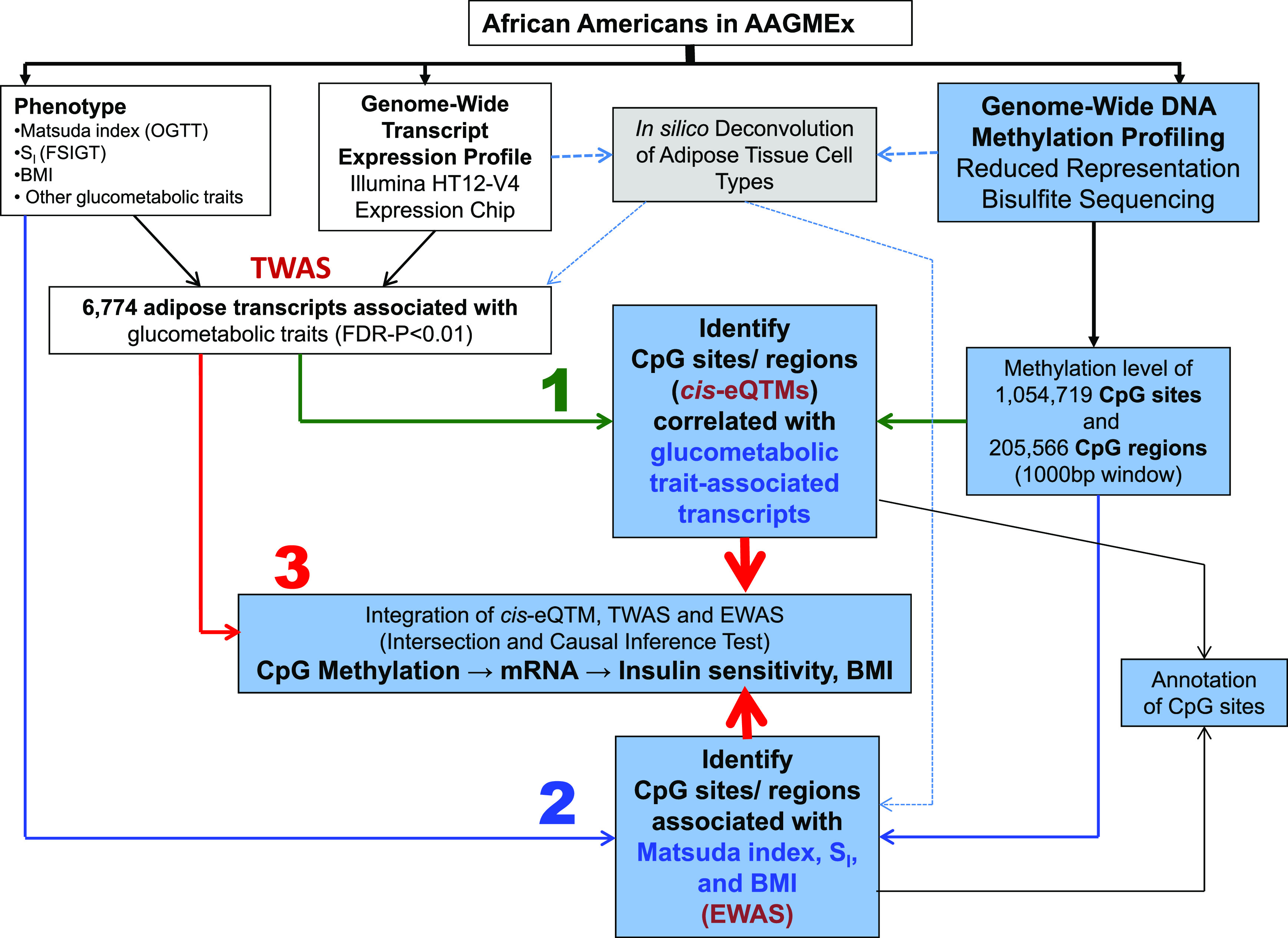
A schematic diagram of the study design and analyses. We measured the methylation levels of CpG sites (DNAm) across the genome in subcutaneous AT of 230 deeply phenotyped individuals of African ancestry from the AAGMEx cohort to evaluate their role in modulating insulin sensitivity and BMI. To explore potential mechanistic links and determine if the altered methylation levels in CpG sites or regions may increase the susceptibility to IR and obesity via epigenetic regulation of transcript levels, we implemented a three-step analysis strategy focusing on glucometabolic trait–associated transcripts identified in our previous TWAS. 1) The first step of our study involved integration of two molecular traits, namely, DNAm and transcript expression profile of AT in eQTM analyses; 2) the second step of our study involved integration of DNAm and glucometabolic phenotypes (Matsuda index, SI, and BMI) in EWAS; and 3) to explore potential mechanistic links (CpG → mRNA → glucometabolic trait) in the final step, we integrated both layers of omic (methylome and transcriptome) data with phenotypic data by intersecting TWAS, eQTM, and EWAS results and by formal CITs on selected loci.
Annotation of CpG Sites and Regions
Selected CpG sites and regions were annotated with the R/Bioconductor package “annotatr” (https://bioconductor.org/packages/release/bioc/html/annotatr.html) (35). Annotation of 1,054,719 CpG sites based on the distance to a CpG island identified 44.9% CpG sites in CpG islands, 12.8% in CpG shores (2,000 bp of the flanking regions of the CpG islands), 3.7% in CpG shelves (2,000 bp of the flanking regions of the shores), and 38.6% in open sea (outside CpG regions or cpg_inter). The gene region annotation analysis classified methylation sites/regions in the context of genes. Among CpG sites in this analysis, only 6.9% were intergenic, while 93.1% were annotated within or very close (±5 kb) to 19,821 genes. These CpG sites were located in exons (15.5%), introns (35.8%), intron-exon boundary (13.2%), untranslated regions (UTRs) (7.1%), promoters (13%), or close (1–5 kb) to an annotated gene (8.6%). To obtain additional evidence on the regulatory potential of significant CpG sites identified in cis-eQTM and EWAS analyses, each site was categorized based on chromatin state annotation data (ChromeHMM, Core 15-state model) downloaded from the Roadmap Epigenomics Project (https://www.roadmapepigenomics.org/). ChromeHMM data for the three cell types/adult tissue sources most relevant to AT were used for this functional annotation (see Supplementary Materials).
In Silico Deconvolution of AT Cell Types
Direct assessment of cell type composition in AT is challenging (36), and estimates that better reflect an individual’s AT are warranted (37). We utilized in silico deconvolution to estimate the relative proportions of cell types in AT, using both transcriptome and methylome data and two different reference-based methods. First, we used the “Adipose Signature Matrix” recently developed by Glastonbury et al. (36) in the CIBERSORTx analytical tool (https://cibersortx.stanford.edu/) to deconvolute AT transcriptome data and estimate the relative proportions of cell types in AT of individuals in the AAGMEx cohort. Second, a reference-based deconvolution algorithm was utilized as detailed in a previous study (25) to deconvolute AT methylome data of 230 individuals. In this reference-based method, references were collected from the methylation profiles of purified cell types from the Blueprint Epigenome Project and the International Human Epigenome Consortium data portal (see Supplementary Materials). We used these predicted proportions of cell types in secondary exploratory statistical analyses as described below.
Partial correlation coefficients were computed adjusting for age and sex to test for correlations between cell type composition and the glucometabolic phenotypes. A principal component (PC) analysis with all of the cell type proportions derived from CIBERSORTx was performed. The first PC, PC1, was the contrast adipocytes minus M2 macrophage. We performed secondary TWAS (transcript to glucometabolic trait association) and EWAS (CpG to glucometabolic trait association) analyses adjusting for cell type (using PC1) in addition to age, sex, and admixture.
Data and Resource Availability
The transcript expression data set used in this study was deposited in the Gene Expression Omnibus (identification no. GSE95674). DNAm profile data generated for the current study will be deposited to the Gene Expression Omnibus Sequence Read Archive.
Results
Expression Levels of Glucometabolic Trait–Correlated Transcripts Are Regulated by eQTMs
Our previous TWAS in AT of AAGMEx participants identified 6,774 high-quality transcripts that were significantly correlated (FDR-P < 0.01) with glucometabolic phenotypes (5,6,28,33). Expression levels of these transcripts were associated with key glucose homeostasis (e.g., fasting glucose, fasting insulin, HbA1c, HOMA-IR, Matsuda index, and SI), anthropometric (e.g., BMI, waist-to-hip ratio, and % of fat mass), and serum lipid (e.g., triglyceride and HDL cholesterol) traits and are of high physiological relevance. A total of 627,021 CpGs were mapped within and around ±100 kb of these 6,774 selected transcripts. We tested for an association between methylation levels of each local CpG site with the expression levels for each of these transcripts (cis-eQTMs), with, on average, 207 ± 201 cis-CpG sites tested for each of these transcripts (Fig. 1, step 1). The cis-eQTM analysis identified 268 CpG-transcript pairs that met both linear regression FDR-P value < 0.05 and partial correlation coefficient of ≥0.2 or less than or equal to −0.2, comprising 107 transcripts and 200 CpG sites (Supplementary Table 2A). Methylation levels of CpG sites were both positively (31%) and negatively (69%) correlated with expression levels (Fig. 2). These 200 significant cis-CpG sites did not exhibit enrichment for any particular CpG geography (Fisher exact test [FET] P = 0.44), with 55.6%, 10.1%, and 3.0% located in CpG islands, CpG shores, and CpG shelves, respectively. There was an enrichment of intergenic (>5 kb away from a RefSeq annotated gene) sites (37.1% vs. 6.9%; enrichment FET-P = 0.001) among the significant cis-CpGs. On the basis of hg19 gene annotations, “annotatr” determined that 62.9% of these significant CpG sites were located in (exons, introns, UTRs) or near (<5 kb) a gene. However, of these gene-annotated sites, 38.1% were not located in or near the target gene determined by the eQTM analyses. The cis-eQTM analyses directly determined the target gene and suggested functional importance for the CpG sites. Gleaning AT/adipocyte-relevant chromatin state annotation data suggested that 30.4%, 34.6%, 9.6%, and 16.7% of these significant CpG sites in cis-eQTM loci are in or flanking transcription start sites, in actively transcribed (weak or strong) regions, in enhancers, and in repressors (polycomb), respectively, and supports a putative functional role of these CpG sites.
Figure 2.
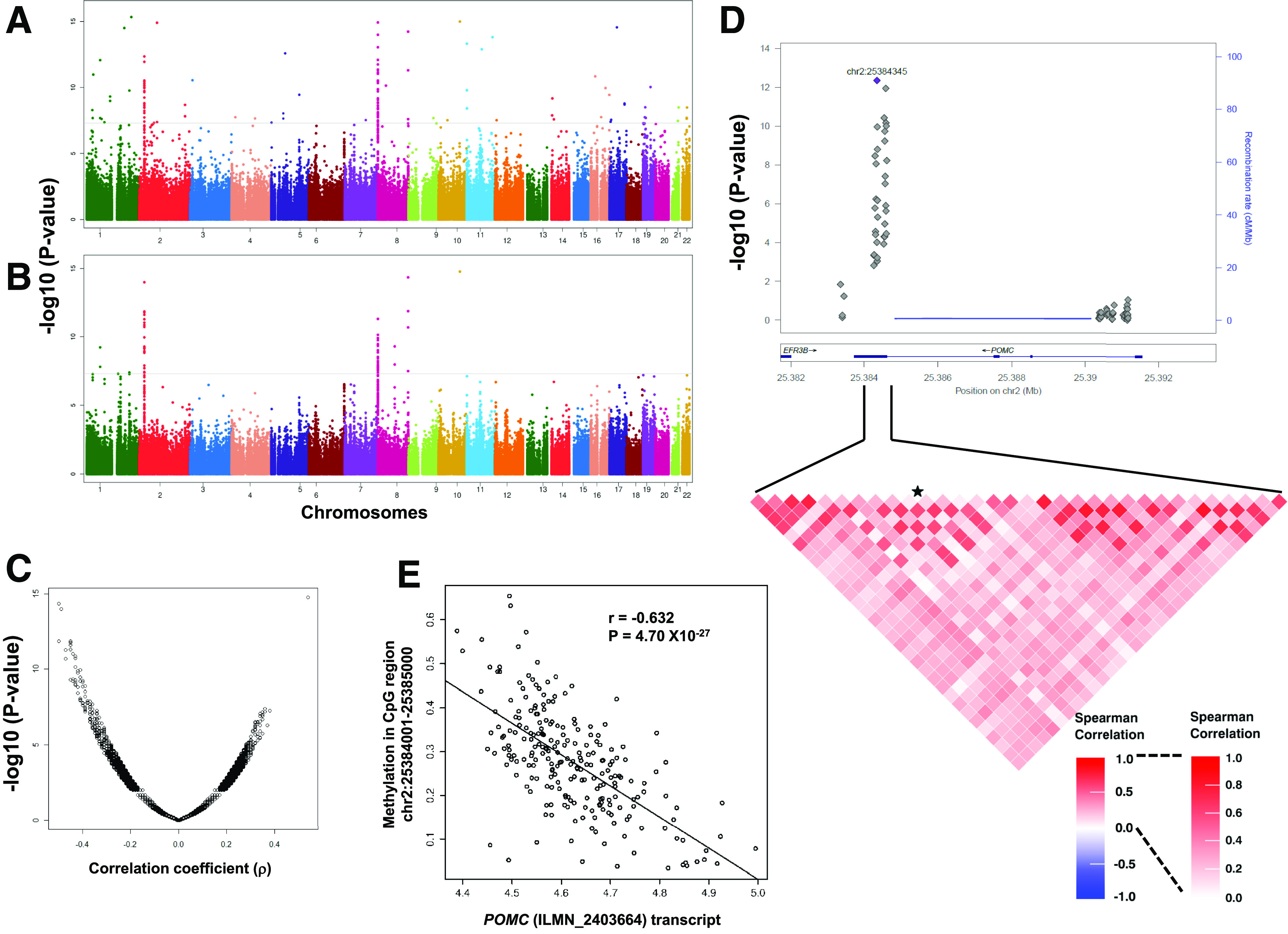
The eQTM analysis identified association of DNAm levels in CpGs with expression of AT transcripts in AAs. Manhattan plots showing chromosomal distribution of −log10 (P values) for linear regression (A) and partial correlation (B), and a scatterplot (C) showing direction of effect (effect size, ρ) and distribution of −log10 (P values) for correlation of all cis-CpG sites (±100 kb of the transcript start and end) tested for 6,774 glucometabolic trait–associated transcripts in adipose. Locus zoom plot shows association of methylation levels of CpG sites with POMC transcript (ILMN_2403664) expression (D). Data for CpG sites around ±2 kb of POMC gene is shown. Heat map show correlation among CpG sites in the marked genomic region. The CpG site chr2:25384345 is marked with a star. Scatterplots show correlation of DNAm levels at chr2:25,384,001–25,385,000 CpG region with POMC transcript (ILMN_2403664) levels (E).
Compared with individual CpG sites, average methylation levels of CpG sites in a genomic region are more robust (less likely to be affected by stochastic variation or measurement errors) and may reduce spurious association signals (25). The cis-eQTM analysis using methylation levels of CpG regions (1,000-bp window) identified significant cis-eQTMs for 82 transcripts (representing 71 Entrez Gene identification numbers) (Supplementary Table 2B). A cis-eQTM for 53 of these 82 transcripts was also identified at the same statistical threshold in the analyses using CpG sites, underscoring the complementary value of considering CpG regions. The best eQTM/CpG regions (i.e., most strongly correlated CpG region-transcript pair) for the 20 most significant genes are shown in Table 1. Top eQTM loci were identified for genes involved in mitochondrial function (e.g., ATPIF1, CYBA, MRPL21, MRPL43, and SDHC), integrin-mediated signaling pathway (e.g., ITGAX), genes recently identified to be involved in IR (ECHDC3), and genes with unknown functional connection with glucometabolic phenotypes (ERICH1, MACROD1). Important corroborating evidence comes from the Multi Tissue Human Expression Resource (MuTHER) cohort, where using the Illumina Infinium HumanMethylation450K BeadChips and Human HT-12 V3 Expression BeadChips, they reported cis-eQTMs for 43 of these 82 transcripts in AT of European ancestry female twins (FDR 1%; P = 2.6 × 10−4) (32). The strongest eQTM locus in AT of AAGMEx individuals is within the proopiomelanocortin (POMC; r = −0.632, P = 4.70 × 10−27) gene at the chr2:25,384,001–25,385,000 region (CpG island in the exon 3 and the intron 2/exon 3 boundary) (Fig. 2). A significant eQTM for POMC was also identified in AT of Europeans in the MuTHER cohort (32), and blood samples of European ancestry subjects suggested that hypermethylation in this region reduces expression of POMC transcripts (38). Therefore, in addition to identifying eQTMs for novel genes, our study validated known eQTM loci for important genes involved in glucometabolic phenotypes.
Table 1.
The 20 most significant eQTMs for glucometabolic trait–associated AT transcripts in AAGMEx
| CpG Region_1,000 bp (hg19) | Methylation level | Transcript probe identification | Symbol | Partial correlation | Linear regression | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Mean | SD | Spearman ρ | P | FDR-P | β | SE | P | FDR-P | |||
| chr2:25384001–25385000 | 230 | 0.278 | 0.119 | ILMN_2403664 | POMC | −0.632 | 4.70E−27 | 1.17E−21 | −0.584 | 0.047 | 3.30E−27 | 8.18E−22 |
| chr8:600001–601000 | 230 | 0.822 | 0.162 | ILMN_2104696 | ERICH1 | −0.521 | 1.95E−17 | 1.61E−12 | −0.418 | 0.045 | 1.82E−17 | 1.41E−12 |
| chr1:28573001–28574000 | 230 | 0.164 | 0.111 | ILMN_1685978 | ATPIF1 | −0.392 | 7.55E−10 | 3.75E−05 | −0.263 | 0.045 | 2.02E−08 | 4.18E−04 |
| chr7:45068001–45069000 | 230 | 0.692 | 0.119 | ILMN_1692295 | MYO1G | −0.386 | 1.42E−09 | 5.89E−05 | −0.500 | 0.080 | 1.66E−09 | 4.59E−05 |
| chr19:53119001–53120000 | 200 | 0.782 | 0.201 | ILMN_2190414 | ZNF83 | −0.372 | 5.92E−08 | 1.13E−03 | −0.356 | 0.069 | 5.17E−07 | 4.14E−03 |
| chr11:47399001–47400000 | 230 | 0.618 | 0.136 | ILMN_1696463 | SPI1 | −0.366 | 1.04E−08 | 3.22E−04 | −0.748 | 0.137 | 1.28E−07 | 1.49E−03 |
| chr6:168443001–168444000 | 229 | 0.807 | 0.143 | ILMN_1676679 | KIF25 | −0.365 | 1.22E−08 | 3.36E−04 | −1.175 | 0.185 | 1.26E−09 | 3.91E−05 |
| chr10:11784001–11785000 | 230 | 0.088 | 0.057 | ILMN_2072178 | ECHDC3 | −0.359 | 2.09E−08 | 4.72E−04 | −2.368 | 0.384 | 3.18E−09 | 7.44E−05 |
| chr22:39531001–39532000 | 218 | 0.336 | 0.188 | ILMN_2412172 | APOBEC3F | 0.356 | 6.61E−08 | 1.17E−03 | 0.293 | 0.047 | 3.30E−09 | 7.44E−05 |
| chr14:31909001–31910000 | 219 | 0.793 | 0.183 | ILMN_1704238 | C14orf126 | −0.351 | 9.61E-08 | 1.49E−03 | −0.308 | 0.053 | 2.49E−08 | 4.75E−04 |
| chr16:88677001–88678000 | 230 | 0.902 | 0.030 | ILMN_1744604 | CYBA | 0.348 | 5.92E−08 | 1.13E−03 | 3.695 | 0.686 | 1.84E−07 | 1.82E−03 |
| chr10:102714001–102715000 | 229 | 0.931 | 0.058 | ILMN_2258774 | MRPL43 | 0.345 | 8.46E−08 | 1.40E−03 | 0.975 | 0.171 | 3.78E−08 | 6.70E−04 |
| chr11:63974001–63975000 | 228 | 0.339 | 0.184 | ILMN_1740960 | MACROD1 | 0.341 | 1.25E−07 | 1.82E−03 | 0.189 | 0.037 | 4.96E−07 | 4.10E−03 |
| chr5:134364001–134365000 | 230 | 0.250 | 0.071 | ILMN_1674386 | PITX1 | 0.335 | 1.96E−07 | 2.43E−03 | 0.429 | 0.077 | 6.73E−08 | 1.11E−03 |
| chr11:68611001–68612000 | 230 | 0.236 | 0.087 | ILMN_1744835 | MRPL21 | 0.334 | 2.21E−07 | 2.55E−03 | 0.573 | 0.105 | 1.38E−07 | 1.49E−03 |
| chr16:31488001–31489000 | 230 | 0.592 | 0.125 | ILMN_2254635 | ITGAX | −0.333 | 2.26E−07 | 2.55E−03 | −0.757 | 0.169 | 1.20E−05 | 3.92E−02 |
| chr19:54677001–54678000 | 229 | 0.529 | 0.123 | ILMN_1784884 | LILRB3 | 0.329 | 3.39E−07 | 3.50E−03 | 0.868 | 0.185 | 4.76E−06 | 1.89E−02 |
| chr1:161349001–161350000 | 230 | 0.596 | 0.223 | ILMN_2323366 | SDHC | 0.328 | 3.53E−07 | 3.50E−03 | 0.164 | 0.030 | 1.38E−07 | 1.49E−03 |
| chr3:44349001–44350000 | 176 | 0.893 | 0.114 | ILMN_1754423 | C3orf23 | 0.328 | 9.11E−06 | 3.28E−02 | 0.256 | 0.054 | 4.21E−06 | 1.81E−02 |
| chr9:107547001–107548000 | 228 | 0.570 | 0.162 | ILMN_1786308 | NIPSNAP3B | −0.325 | 5.42E−07 | 5.12E−03 | −0.395 | 0.071 | 8.99E−08 | 1.31E−03 |
The most significantly correlated CpG region (1,000 bp)-transcript pair for each gene is shown.
AT CpG Methylation Is Associated With Insulin Sensitivity and Obesity
The above results suggest that there are strong detectable effects of eQTMs on transcript expression. However, previous studies in non-African ancestry populations found a modest effect of specific CpG sites on glucometabolic phenotypes (23). AAGMEx participants were evaluated by OGTT and FSIGT, and individuals in the AAGMEx cohort had a broad range of insulin sensitivity and BMI values. Thus, to define the role of DNAm in determining IR and obesity, we performed EWAS to examine the association of DNAm level of CpGs with Matsuda index, SI, and BMI in the AAGMEx cohort (Fig. 1, step 2). The genomic inflation factor λ was 1.13, 1.14, and 1.12 for the Matsuda index, SI, and BMI EWAS, respectively (Fig. 3). We recognize that genomic inflation factors (originally designed for genome-wide association studies) commonly overestimate the true level of inflation in EWAS and TWAS (39). Among the 1,054,719 CpG sites analyzed for EWAS, 26 and 15 CpG sites were significantly associated (FDR-P < 0.05) with Matsuda index and BMI, respectively. No CpG site was associated with SI at FDR-P < 0.05 (Supplementary Tables 3–5 show the association of CpG sites with traits up to FDR-P < 0.1).
Figure 3.
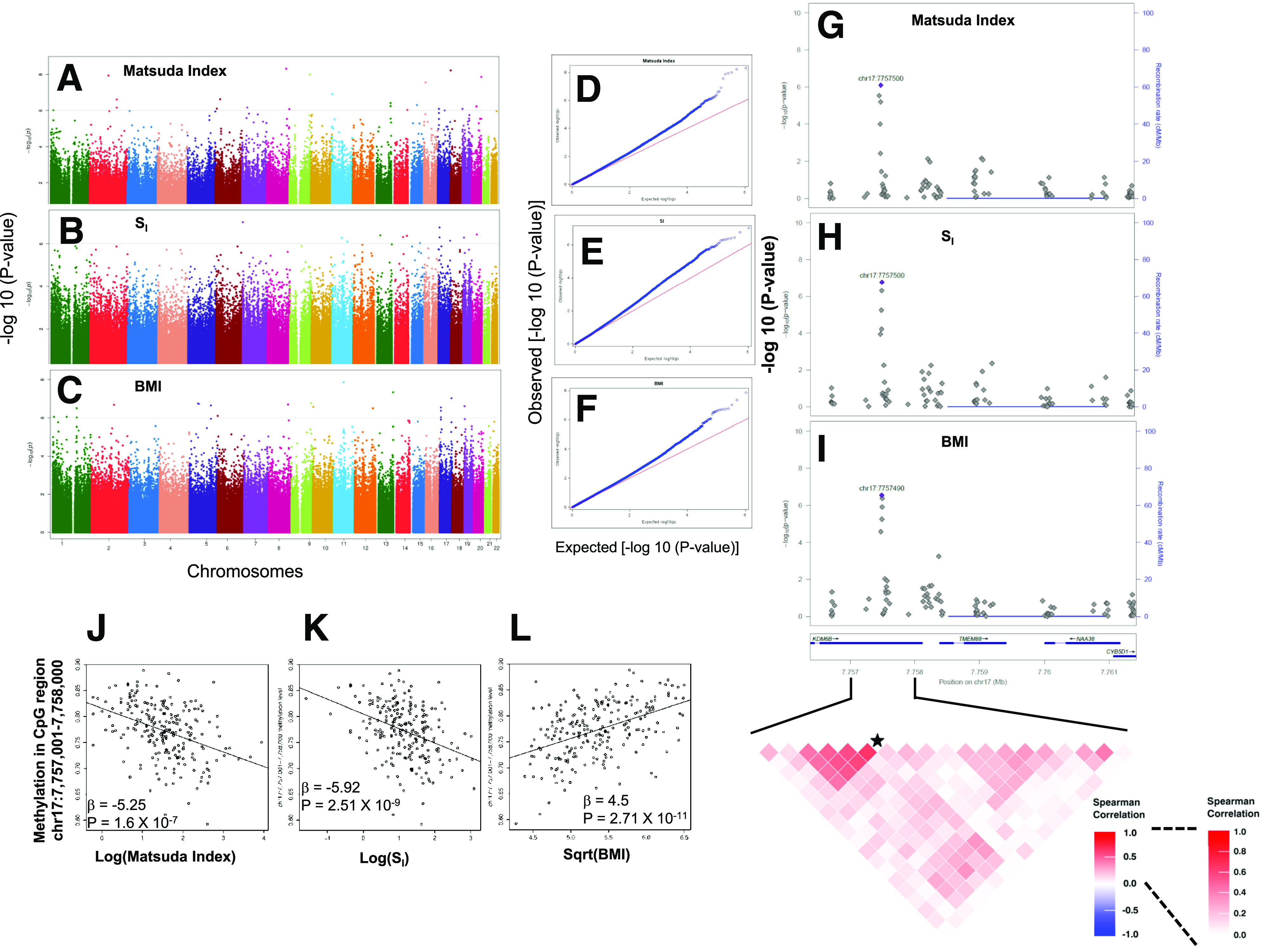
EWAS identified association of AT CpG methylation levels with insulin sensitivity and obesity. Genome-wide Manhattan plots showing −log10 (P values) for association of 1,054,719 CpG sites with Matsuda index (A), SI (B), and BMI (C). Q-Q plots showing distribution of observed and expected –log10 (P values) in EWAS for Matsuda index (D), SI (E), and BMI (F). DNAm levels of CpG sites in the promoter region of the TMEM88 gene are associated with glucometabolic phenotypes in AAs. Locus zoom plots showing association of methylation levels of CpG sites with Matsuda index (G), SI (H), and BMI (I). Data for CpG sites around ±2 kb of TMEM88 (which also includes 3′UTR of KDM6B) is shown. A heat map shows a correlation among CpG sites in the marked genomic region. The CpG site chr17:7757500 is marked with a star. Scatterplots show correlation of DNAm levels at the chr17:7757001–7758000 CpG region with Matsuda index (J), SI (K), and BMI (L).
The EWAS analysis using methylation levels of CpG regions (1,000-bp window) identified 155, 46, and 168 CpG regions associated with Matsuda index, SI, and BMI, respectively (FDR-P < 0.05) (Supplementary Tables 6–8). The “annotatr” analysis suggested that CpG regions associated with Matsuda index, SI, and BMI (FDR-P ≤ 0.05) were linked to 100, 31, and 113 genes, respectively. The CpG regions annotated to seven genes (TMEM88, AGAP2, PTPRN2, SYTL1, GTPBP3, RAP1GAP2, and LINC00982) were associated with all three traits. The CpG region spanning chr17:7757001–7758000 is located in the putative promoter region of transmembrane protein 88 (TMEM88) and is within the 3′UTR of lysine demethylase 6B (KDM6B). This was the most strongly associated CpG region for SI (β = −5.92, adj-R2 = 0.147, P = 2.51 × 10−9) and BMI (β = 4.5, adj-R2 = 0.228, P = 2.71 × 10−11) (Fig. 3). This region was also strongly associated with the Matsuda index (β = −5.25, adj-R2 = 0.154, P = 1.6 × 10−7).
An RRBS analysis of AT of Finnish individuals identified associations of four CpG regions in FASN with multiple glucometabolic traits including BMI (chr17:80,051,500–80,053,080; β = 9.44, P = 1.31 × 10−11) and Matsuda index (25). Despite the differences in RRBS coverage for the FASN gene region, we observed a significant association of methylation levels at the chr17:80,052,001–80,053,000 CpG region with BMI in AAs (β = 1.91, P = 9.04 × 10−6). Also, consistent with published studies (38,40), the strongest eQTM locus identified in our study at chr2:25,384,001–25,385,000 for the POMC gene was associated with BMI (β = 0.77, P = 0.007) but did not meet FDR significance in the EWAS.
AT Cell Type Composition and Association of CpG Methylation With Glucometabolic Phenotypes
In silico deconvolution of AT cell types based on transcriptome data estimated that the adipocytes were the most dominant relative cell type (proportion: mean ± SD 0.78 ± 0.11, median 0.80), followed by M2 macrophages (0.20 ± 0.10) in AT of AAGMEx participants (Supplementary Fig. 1). Similarly, in silico deconvolution of AT cell types based on CpG DNAm profile data also suggested that adipocytes were the most common cell type (0.799 ± 0.104, median 0.818) (Supplementary Fig. 2), followed by endothelial cells (0.067 ± 0.31), macrophages (0.042 ± 0.056), and neutrophils (0.038 ± 0.059) in the AT from the AAGMEx cohort. The AT macrophage (M1 and M2 combined) proportion estimated by the transcriptome-based CIBERSORTx was positively correlated with BMI (r = 0.49, P = 1.8 × 10−16) and waist-to-hip ratio (r = 0.46, P = 4.72 × 10−14), but it was inversely correlated with Matsuda index (r = −0.46, P = 5.32 × 10−14) (Supplementary Fig. 1). Similarly, the AT macrophage proportion estimated by the methylome-based analysis was positively correlated with BMI (r = 0.21, P = 1.46 × 10−3) but was inversely correlated with Matsuda index (r = −0.20, P = 3.33 × 10−3) (Supplementary Fig. 2). Thus, both in silico analyses suggested that ∼80% of the cells in AT of our cohort were adipocytes and were a major contributor in the AT transcriptome and methylome data. However, other cell types contributed to expression and methylation levels in bulk tissue data. Adjustment for cell type proportions in the analysis models is a statistical approach to remove the confounding effects related to cell type heterogeneity. However, accuracy of the cell type estimates obtained through in silico deconvolution methods in our study could not be validated by empirical measurements of cell type composition in our frozen tissue samples. Thus, we used these predicted proportions of cell types in secondary exploratory EWAS and TWAS analysis adjusted for age, sex, admixture, and cell type estimates. In general, we observed an overall decrease in significance levels in EWAS (Supplementary Fig. 3) and TWAS (Supplementary Fig. 4) after adjusting for cell types. Among the 155, 46, and 168 CpG regions that were significantly associated (FDR-P ≤ 0.05) with Matsuda index, SI, and BMI, respectively, in the cell type–unadjusted EWAS analysis, all except one BMI-associated region remained nominally associated (P < 0.05) in cell type–adjusted EWAS analysis (Supplementary Tables 6–8). Similarly, among the 4,441, 2,251, and 4,664 transcripts selected for their significant association (FDR-P ≤ 0.01) with Matsuda index, SI, and BMI, respectively, based on our cell type–unadjusted TWAS analysis, 3,705 (83.4%), 1,838 (81.7%), and 3,476 (74.5%) remained nominally associated (P < 0.05) with Matsuda index, SI, and BMI, respectively, in the cell type–adjusted TWAS analysis. Our exploratory secondary analysis with an additional adjustment for estimated cell type proportions suggested a minor impact of AT cell type heterogeneity on our top ranked findings. Thus, in the absence of empirical cell counts, we considered our results from the cell type–unadjusted primary analysis for the final interpretation of our findings.
Insulin Sensitivity– and Obesity-Associated CpGs Regulate Levels of Transcript Expression in AT
To explore potential mechanistic links and determine if altered methylation levels may increase the susceptibility to IR and obesity via epigenetic regulation of transcript levels, we jointly analyzed the EWAS, TWAS, and eQTM data (Fig. 1, step 3). Intersection of cis-eQTM, EWAS, and TWAS results identified CpGs that were eQTM for glucometabolic trait–associated transcripts and were also associated with Matsuda index, SI, and BMI. We restricted this search to CpGs that were significant cis-eQTM (FDR-P < 0.05 and |ρ| ≥ 0.2) for a transcript (TWAS FDR-P < 0.01), associated with the same glucometabolic trait (EWAS P < 0.01), and directionally consistent. We identified 11, 12, and 14 cis-CpG sites that were associated with Matsuda index, SI, and BMI, respectively, and were eQTMs for 10, 11, and 10 genes associated with the respective glucometabolic phenotypes (Supplementary Tables 9–11). Intersection of results for CpG regions (1,000 bp) using the same statistical thresholds identified 18, 12, and 16 cis-CpG regions that were associated with Matsuda index, SI, and BMI, respectively, and were eQTMs for 19, 13, and 18 genes associated with the respective glucometabolic phenotypes (Supplementary Tables 12–14). Among the loci identified by this intersection analysis, methylation levels of CpG regions associated with transcript expression levels for nine genes (CYBA, MAP4K1, ITGAM, ITGAX, MACROD1, FERMT3, MYO1G, FGR, and SLC16A3) were associated with Matsuda index, SI, and BMI. CpG sites and regions selected by the intersection method above were further evaluated by CIT to identify potential mechanistic relationships between CpGs and glucometabolic traits mediated by transcript expression (CpG → mRNA → trait) (Supplementary Tables 9–14). A subset of significant CpG regions based on CIT are shown in Table 2. Significant CpG regions identified by CIT (PCIT <0.05) are putative epigenetic regulators of 23 genes, including genes involved in the integrin-mediated signaling pathway (FCER1G, ITGAM, ITGAX, FERMT3, and FGR; enrichment P = 1.83 × 10−4). Methylation levels at the chr11:63974001–63975000 CpG region covering the 5′UTR, first intron and exon of FERMT3 were associated with Matsuda index (β = 0.69, P = 9.78 × 10−3) and BMI (β = −0.95, P = 1.03 × 10−7). Interestingly, this locus is an eQTM with methylation levels inversely correlated with expression of FERMT3 (ILMN_2366330, ρ = −0.32, P = 5.57 × 10−7) and directly correlated with expression of MACROD1 (ILMN_1740960, ρ = 0.34, P = 1.25 × 10−7) (Fig. 4). Fermitin family member 3 (FERMT3) is involved in integrin activation, while the MACRO domain containing 1 (MACROD1, also known as Leukemia-related protein 16/LRP16) enhances inflammatory responses by upregulating a Rac1-dependent pathway, and reduces insulin-stimulated glucose uptake in adipocytes (41). In our study, FERMT3 expression was inversely correlated (ILMN_2366330, β = −1.32, P = 6.0 × 10−14), while MACROD1 expression was positively correlated (ILMN_1740960, β = 3.10, P = 3.44 × 10−11) with Matsuda index. The CIT suggested (PCIT <0.01) the chr11:63974001–63975000 CpG region as a putative causal determinant of insulin sensitivity and obesity via its role in epigenetic regulation of the expression of the FERMT3 and MACROD1 transcripts.
Table 2.
DNAm levels in CpG regions determine insulin sensitivity and obesity by modulating transcript expression in AT of African ancestry individuals
| CpG_region (L) | Transcript (G) | Gene Symbol | Trait (T) | Intersection | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EWAS | eQTM | TWAS | CIT | ||||||||
| β | P value | β | P value | ρ | β | P value | PCIT | ||||
| chr1:153486001–153487000 | ILMN_1684306 | S100A4 | Matsuda index | 1.882 | 5.77E−05 | −0.967 | 1.24E−05 | −0.283 | −0.942 | 5.11E−12 | 3.53E−03 |
| chr1:161256001–161257000 | ILMN_2123743 | FCER1G | Matsuda index | 1.584 | 7.92E−04 | −1.291 | 1.99E−06 | −0.300 | −0.917 | 2.13E−19 | 7.92E−04 |
| chr1:161256001–161257000 | ILMN_2123743 | FCER1G | BMI | −1.158 | 3.63E−04 | −1.291 | 1.99E−06 | −0.300 | 0.602 | 1.59E−19 | 3.40E−04 |
| chr1:27941001–27942000 | ILMN_2368318 | FGR | BMI | 0.901 | 9.05E−04 | 1.028 | 2.75E−06 | 0.293 | 0.456 | 5.80E−10 | 1.87E−03 |
| chr7:1163001–1164000 | ILMN_1684227 | GPR146 | Matsuda index | −1.091 | 8.98E−04 | −0.325 | 1.27E−06 | −0.317 | 2.225 | 7.63E−13 | 8.98E−04 |
| chr9:107547001–107548000 | ILMN_3236656 | LOC286367 | Matsuda index | −0.909 | 2.63E−03 | −0.560 | 2.78E−06 | −0.293 | 1.261 | 1.75E−16 | 2.63E−03 |
| chr9:107547001–107548000 | ILMN_3236656 | LOC286367 | BMI | 0.726 | 4.51E−04 | −0.560 | 2.78E−06 | −0.293 | −0.881 | 5.27E−19 | 4.70E−04 |
| chr9:107547001–107548000 | ILMN_1786308 | NIPSNAP3B | Matsuda index | −0.909 | 2.63E−03 | −0.395 | 8.99E−08 | −0.325 | 2.210 | 2.46E−13 | 2.63E−03 |
| chr9:107547001–107548000 | ILMN_1786308 | NIPSNAP3B | BMI | 0.726 | 4.51E−04 | −0.395 | 8.99E−08 | −0.325 | −1.023 | 2.09E−09 | 1.48E−03 |
| chr10:129949001–129950000 | ILMN_1734543 | PTPRE | BMI | 1.556 | 2.77E−04 | 0.700 | 1.42E−05 | 0.287 | 1.097 | 7.07E−12 | 9.36E−04 |
| chr11:47399001–47400000 | ILMN_1696463 | SPI1 | BMI | −0.737 | 2.13E−03 | −0.748 | 1.28E−07 | −0.366 | 0.797 | 3.11E−16 | 2.98E−03 |
| chr11:63974001–63975000 | ILMN_2366330 | FERMT3 | Matsuda index | 0.686 | 9.78E−03 | −0.396 | 1.25E−05 | −0.324 | −1.319 | 6.00E−14 | 9.78E−03 |
| chr11:63974001–63975000 | ILMN_2366330 | FERMT3 | BMI | −0.949 | 1.03E−07 | −0.396 | 1.25E−05 | −0.324 | 0.957 | 1.21E−17 | 3.64E−03 |
| chr11:63974001–63975000 | ILMN_1740960 | MACROD1 | Matsuda index | 0.686 | 9.78E−03 | 0.189 | 4.96E−07 | 0.341 | 3.098 | 3.44E−11 | 9.78E−03 |
| chr11:63974001–63975000 | ILMN_1740960 | MACROD1 | SI | 0.847 | 1.29E−03 | 0.189 | 4.96E−07 | 0.341 | 3.057 | 4.76E−10 | 5.21E−03 |
| chr11:63974001–63975000 | ILMN_1740960 | MACROD1 | BMI | −0.949 | 1.03E−07 | 0.189 | 4.96E−07 | 0.341 | −2.421 | 9.21E−19 | 1.94E−03 |
| chr16:31363001–31364000 | ILMN_1685009 | ITGAM | Matsuda index | 1.170 | 3.49E−04 | −0.483 | 7.55E−06 | −0.308 | −1.430 | 1.84E−15 | 1.00E−03 |
| chr16:31363001–31364000 | ILMN_1685009 | ITGAM | BMI | −0.836 | 1.37E−04 | −0.483 | 7.55E−06 | −0.308 | 1.034 | 2.30E−19 | 3.29E−03 |
| chr16:31488001–31489000 | ILMN_2254635 | ITGAX | Matsuda index | 1.431 | 2.24E−04 | −0.757 | 1.20E−05 | −0.333 | −0.805 | 9.02E−09 | 9.04E−03 |
| chr16:31488001–31489000 | ILMN_2254635 | ITGAX | BMI | −0.729 | 6.48E−03 | −0.757 | 1.20E−05 | −0.333 | 0.592 | 7.58E−11 | 6.72E−03 |
| chr16:88677001–88678000 | ILMN_1744604 | CYBA | Matsuda index | −8.028 | 6.43E−06 | 3.695 | 1.84E−07 | 0.348 | −1.335 | 1.13E−18 | 2.62E−04 |
| chr16:88677001–88678000 | ILMN_1744604 | CYBA | SI | −6.090 | 6.61E−04 | 3.695 | 1.84E−07 | 0.348 | −0.898 | 3.80E−08 | 7.89E−03 |
| chr16:88677001–88678000 | ILMN_1744604 | CYBA | BMI | 5.148 | 2.43E−05 | 3.695 | 1.84E−07 | 0.348 | 0.807 | 3.43E−16 | 1.98E−04 |
| chr17:72528001–72529000 | ILMN_1693552 | CD300A | Matsuda index | 0.818 | 6.08E−03 | −0.339 | 1.74E−05 | −0.313 | −1.403 | 6.95E−09 | 6.08E−03 |
| chr17:80262001–80263000 | ILMN_2364022 | SLC16A3 | BMI | 1.376 | 8.88E−05 | 0.985 | 7.22E−08 | 0.306 | 0.697 | 7.16E−10 | 2.98E−03 |
| chr18:74818001–74819000 | ILMN_1672660 | MBP | Matsuda index | 1.014 | 7.68E−04 | −0.466 | 9.83E−07 | −0.311 | −1.259 | 1.07E−10 | 2.57E−03 |
| chr19:39086001–39087000 | ILMN_1665943 | MAP4K1 | Matsuda index | −1.600 | 3.19E−04 | 0.463 | 1.02E−07 | 0.300 | −1.933 | 2.78E−09 | 5.45E−04 |
| chr19:39086001–39087000 | ILMN_1665943 | MAP4K1 | BMI | 1.069 | 4.66E−04 | 0.463 | 1.02E−07 | 0.300 | 1.371 | 6.08E−11 | 1.74E−03 |
Results from intersection of eQTM, TWAS, and EWAS results followed by CIT for selected loci (PCIT < 0.01) are shown. Best CpG region for a gene with PCIT < 0.01 are shown. Complete results are presented in Supplementary Tables 12–14.
Figure 4.
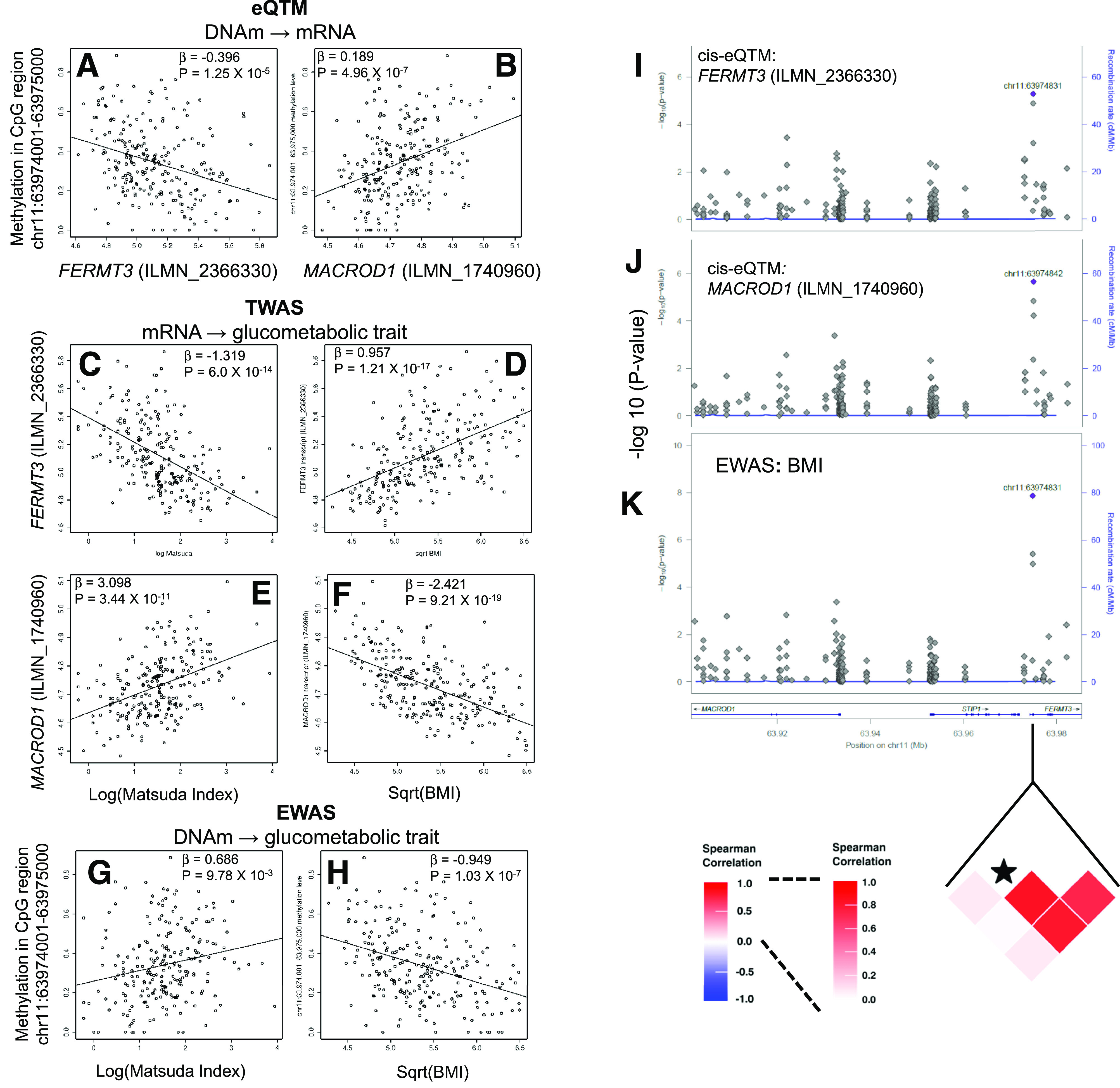
Integration of the eQTM analysis, EWAS, and TWAS results suggests that the DNAm level at the chr11:63974001–63975000 CpG region determines insulin sensitivity and obesity by regulating expression of FERMT3 and MACROD1 in AT of AAs. Scatterplot showing correlation between methylation level of the CpG region chr11:63974001–63975000 with FERMT3 (ILMN_2366330) transcript expression (A), methylation level of CpG region chr11:63974001–63975000 with MACROD1 (ILMN_1740960) transcript expression (B), FERMT3 (ILMN_2366330) transcript expression and Matsuda index (C), FERMT3 (ILMN_2366330) transcript expression and BMI (D), MACROD1 (ILMN_1740960) transcript expression and Matsuda index (E), MACROD1 (ILMN_1740960) transcript expression and BMI (F), methylation level of the CpG region chr11:63974001–63975000 with Matsuda index (G), and methylation level of the CpG region chr11:63974001–63975000 with BMI (H). Locus zoom plots showing (CpG sites in chr11:63,901,609–63,985,308) eQTM: association of methylation level of CpG sites with FERMT3 (ILMN_2366330) transcript expression (I); eQTM: association of methylation level of CpG sites with MACROD1 (ILMN_1740960) transcript expression (J); and EWAS: association of methylation level of CpG sites with BMI (K). A heat map shows a correlation among CpG sites in the marked genomic region. The CpG site chr11:63974831 is marked with a star.
Discussion
Glucose homeostasis and obesity traits are driven by a complex interplay among genetic, epigenetic, and environmental factors (42). Thus, epigenetic modifications, including DNAm, are hypothesized to play a role in the pathogenesis of IR and obesity, likely via regulation of gene expression in tissues involved in glucose homeostasis (23,43). In this study, we measured the methylation levels of CpG sites across the genome in subcutaneous AT of deeply phenotyped individuals of African ancestry from the AAGMEx cohort to evaluate their role in modulating insulin sensitivity and BMI. One cannot determine causality without perturbation experiments, particularly in human studies, and we must rely primarily on observational studies leveraging the natural variation in DNAm levels to develop hypotheses. However, concurrent availability of other molecular phenotypes, especially those falling in the putative causal cascade, strengthen resulting hypotheses that can be tested by experimental approaches. AAGMEx is a unique cohort that has genome-wide multiomic profiles, including DNAm and transcript expression levels, in AT and insulin sensitivity as measured by both FSIGT and OGTT. Our analyses of glucometabolic trait–associated transcripts identified eQTMs, or loci in which CpG methylation levels were correlated with expression levels of a subset of these transcripts, suggesting epigenetic regulation of these pathophysiologically relevant genes in AT of African ancestry individuals. EWAS in the AAGMEx cohort detected associations between methylation levels at many CpG regions with Matsuda index, SI, and BMI. Focusing on epigenetic regulation of glucometabolic trait–associated transcript expression, we identified significant CpG regions or eQTM loci for 19, 13, and 18 genes that were also nominally associated with Matsuda index, SI, and BMI, respectively, in EWAS. CITs further supported the putative regulatory role of a subset of these CpG regions in determining insulin sensitivity or obesity by epigenetic regulation of expression of 23 genes. These associations include integrin α M (ITGAM), integrin α X (ITGAX), FERMT3, and POMC. Previous studies in European ancestry cohorts reported CpG methylation–mediated transcriptional regulation and/or association with glucometabolic phenotypes for some of these loci, while others were novel. Thus, results from this study supported our hypothesis and provided evidence that differential DNAm at putative regulatory regions contributes to insulin sensitivity and obesity by modulating AT transcript expression of a subset of genes in AAs.
Previous studies in AAGMEx identified strong associations between quantitative glucometabolic phenotypes and transcript levels of 6,774 genes in AT (5,6,28,33). In this study, we focused on testing whether and which expression levels of these 6,774 transcripts was modulated by altered DNAm levels in nearby regulatory regions (±100 kb). Our analyses identified cis-eQTMs (FDR-P ≤ 0.05) for 107 and 82 transcripts based on analysis using methylation levels of CpG sites (uncorrected P ≤ 1.94 × 10−5) and CpG regions (uncorrected P ≤ 1.96 × 10−5), respectively. Among this cumulative list of 136 transcripts, cis-eQTM was previously reported for 64 transcripts in AT of European ancestry individuals from the MuTHER cohort (FDR <0.01, uncorrected P = 2.6 × 10−4, N = 662, female twins) (32). Compared with >1 million CpG sites in our RRBS-based methylation profiling, MuTHER used data on 344,303 CpG sites from a fixed panel of sites on the Illumina HumanMethylation450 K BeadChips. Additionally, a cis-eQTM analysis in MuTHER was restricted to an average of 17 CpG sites within or 1.5 kb upstream of each transcript, while in the AAGMEx cohort, we tested an average of 207 CpG sites within or ±100 kb of each transcript. Despite differences in study design and specific CpG sites analyzed, our study in AAs provides gene-level evidence of the epigenetic regulation of a subset of glucometabolic trait–associated transcripts. These and other studies interrogated only a small fraction of the ∼30 million CpG sites in the human genome. Thus, further studies with higher coverage of CpG sites will be required to uncover the full spectrum of epigenetic regulation and to determine ancestry-specific regulation of these glucometabolic trait–associated transcripts.
The strongest eQTM in our study was POMC in the chr2:25384001–25385000 region (P = 4.70 × 10−27), which was nominally associated with BMI (P = 0.007) (Supplementary Fig. 5). CIT suggested (PCIT = 0.015) that the chr2:25384001–25385000 CpG region was a putative causal determinant of BMI via its role in epigenetic regulation of the expression of POMC transcript. The CpG site cg06846259 (chr2:25,384,655) was the strongest cis-eQTM site for POMC transcript expression (ILMN_2403664, β = −0.122, P = 1.51 × 10−29) in AT from the MuTHER cohort (32). POMC is mostly strongly synthesized in the pituitary and arcuate nucleus of the hypothalamus, but the POMC transcript is expressed in other tissues (38,44). POMC encodes a preproprotein that undergoes extensive, tissue-specific, posttranslational processing and may yield as many as 10 biologically active peptide hormones, namely, adrenocorticotrophic hormone, melanocyte-stimulating hormone, and β-lipotropin, which are involved in diverse cellular functions (44). Mutations in this gene have been associated with early-onset obesity. In POMC gene-centric studies, higher DNAm of CpG sites in this region was observed in arcuate nucleus neurons (r = 0.34, P = 0.028, N = 41), as well as in peripheral blood cells (r = 0.18, P = 0.008, N = 228) of obese German individuals compared with normal weight control subjects (38,40), and it was considered a predictor of obesity, independent of tissue. The CpG sites at chr2:25,384,590 and chr2:25,384,569 were the most significantly associated with obesity in a study by Kühnen et al. (40). In our analysis, we had high-quality data for 33 CpG sites in this region, and 21 of them were significantly negatively correlated with POMC expression (ILMN_2403664 at FDR-P ≤ 0.05). Although CpG sites at chr2:25,384,569 and chr2:25,384,590 were among the significant sites, chr2:25,384,585 (r = −0.49, P = 1.06 × 10−14) was the most significant eQTM site and was associated with BMI (β = 0.464, P = 0.0029) in AAGMEx. Thus, replicating previous studies in European ancestry cohorts, our study suggests a role for cis-eQTM loci in the exon 3 and intron 2/exon 3 boundary of the POMC gene in regulating BMI in AAs as well.
The cis-eQTM analysis in this study identified association of 136 AT transcripts with DNAm of individual CpG sites or average methylation levels of CpG sites in CpG regions, which suggests epigenetic regulation of the transcript levels of these genes. A previously completed cis-eQTL analysis on this cohort (45) suggested the association of 42 of these 136 transcripts with genotypes of a local SNP (P ≤ 6.95 × 10−6, FDR = 0.01). For example, we identified the strongest eQTM for POMC expression in AT (ILMN_2403664; chr2:25384001–25385000; β = −0.584, P = 3.30 × 10−27). Our cis-eQTL analysis suggested that rs7591899 (chr2: 25389159) and rs6713396 (chr2: 25384705) were the strongest associated genotyped and imputed cis-expression regulatory single nucleotide polymorphism, respectively. The minor allele C of rs6713396 was associated (minor allele frequency = 0.27, β = 0.12, P = 5.51 × 10−45) with higher expression of POMC in AT of AAGMEx participants (Supplementary Fig. 5). The minor allele C of rs6713396 was also associated (effect = 0.88, P = 2.30 × 10−12) with higher levels of POMC (ENSG00000115138.10) in subcutaneous AT in GTEx project (V8; https://gtexportal.org/) participants. Previous studies suggested an association of CpG DNAm levels in adipose and other tissues with genotypes of nearby SNPs in European ancestry populations (32,46). Interestingly, methylation levels in the chr2:25384001–25385000 region were associated with SNPs rs7591899 and rs6713396. The allele C of rs6713396 was associated (β = −0.1022, P = 5.42 × 10−22; adjusted for age, sex, and admixture) with lower methylation levels of the chr2:25384001–25385000 CpG region in AT of AAGMEx participants (Supplementary Fig. 5). Association between POMC transcript level and methylation level of the chr2:25384001–25385000 CpG region (cis-eQTM) was reduced upon adjustment for genotype of rs6713396 (β = −0.5015, P = 2.09 × 10−8). Thus, genotype-dependent modulation of DNAm at the chr2:25384001–25385000 CpG region may modulate the transcript levels of POMC in AT. Previous studies in blood samples of European ancestry subjects suggested that hypermethylation in this region interferes with the binding of the transcription enhancer P300 and reduces expression of POMC transcripts (38). A complete methylation quantitative trait analysis will be required to determine the role of genetic polymorphisms in epigenetic modulation of the expression of all glucometabolic trait–associated transcripts by differential DNAm in AAs.
Compared with molecular traits, such as DNAm and gene expression, complexity of regulatory mechanisms determining clinical and physiological traits, such as IR and obesity, is expected to be greater (47). IR in a population is a heterogeneous mix of molecular phenotypes, caused by derangements in the expression of multiple genes contributing to obesity and altered insulin sensitivity. Differential DNAm at each CpG locus is likely to have a stronger effect on the transcript level of a nearby gene but a comparatively weaker effect on insulin sensitivity or obesity. Published EWAS in European ancestry case-control cohorts comparing AT DNAm levels in insulin resistance with sensitive participants (48) and patients with and without diabetes (24) identified CpG sites nominally associated with these complex traits, with mean difference in methylation level of only 2–3% between groups. Additionally, EWAS cannot directly implicate the target gene for clinical trait–associated CpG sites. For example, the insulin sensitivity and BMI-associated CpG region chr17:7757001–7758000 spans the promoter of TMEM88 and 3′UTR of KDM6B, but the eQTM analysis suggests TMEM88 as the putative target gene in AT. Also, expression of TMEM88 (ILMN_1757129) but not KDM6B in AT is associated with Matsuda index and BMI. These data suggest the importance of the availability of additional molecular data in determining the target genes for EWAS-implicated trait-associated CpG sites. Thus, to identify epigenetic regulatory loci for glucometabolic traits, integration of two molecular traits, namely, DNAm and transcript expression profile of AT in eQTM analyses was the first step of our study. We identified eQTMs for several genes involved in integrin-mediated signaling pathway (including ITGAX, ITGAM, FERMT3, FCER1G, and FGR), and CITs further provide statistical evidence that altered DNAm levels in a subset of those loci may determine IR and/or obesity by regulating transcript expression in AT of AAs. Thus, our multiomic approach initiated with eQTM analyses was successful in determining epigenetic regulatory mechanisms of insulin sensitivity and obesity in AAs.
In summary, this study partially delineates the epigenetic architecture of AT in AAs and suggests a role of DNAm-mediated regulation of gene expression at both previously identified and novel loci in determining IR and obesity. Integration of TWAS of glucometabolic traits, eQTM, and EWAS analyses enabled us to determine a plausible causal link between CpG methylation, transcript expression, and glucometabolic traits. However, directionality of the DNAm-gene expression relationships cannot be fully ascertained from our analyses based on a cross-sectional cohort. Further, epigenetic analyses in longitudinal or interventional cohorts and functional studies, including targeted editing of the methylation level (49,50) at those eQTM loci, will be required for direct experimental validation.
Article Information
Acknowledgments. The authors thank Drs. Jorge Calles-Escandon, Jamel Demons, Samantha Rogers, Barry Freedman, and the dedicated staff of the Clinical Research Unit at Wake Forest School of Medicine (WFSM) for support of the clinical studies and assistance with clinical data management for the AAGMEx cohort. The authors thank Lata Menon, Joyce Byers, Ethel Kouba, and Donna Davis of WFSM for support in participant recruitment for AAGMEx cohort. The authors thank the staff in the Genomics Core Laboratory at Center for Genomics and Personalized Medicine Research, WFSM, for their extensive support in gene expression analysis using the Illumina microarray platform for the AAGMEx cohort. The authors thank the laboratory and bioinformatics team including Damien Calay, Paola Genevini, and Sol Schvartzman at Diagenode service laboratory in Belgium for extensive technical support for RRBS analysis. The authors acknowledge the Center for Public Health Genomics at WFSM and Dr. Hannah C. Ainsworth at WFSM for computational resources and software support.
Funding. This work was primarily supported by the American Diabetes Association Innovative Clinical or Translational Science Research Award 1-18-ICTS-113 to S.K.D. and was additionally supported by the National Institute of Diabetes and Digestive and Kidney Diseases grants R01-DK-090111 and R01-DK-118243 to S.K.D.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. N.K.S. performed experiments, analyzed data, and reviewed and edited the manuscript. M.E.C. performed the statistical analysis and reviewed and edited the manuscript. D.M. performed the cell deconvolution analysis, interpreted the data, and reviewed and edited the manuscript. M.P. provided key resources for the deconvolution analysis and reviewed and edited the manuscript. T.D.H. performed the CpG annotation analysis, interpreted the data, and reviewed and edited the manuscript. C.D.L. supervised all statistical analyses, performed the statistical analysis, interpreted the data, and reviewed and edited the manuscript. S.K.D. designed the study, analyzed and interpreted the data, and wrote the manuscript. C.D.L. and S.K.D. are the guarantors of this work, and as such, had full access to all study data and take responsibility for the integrity of the data and accuracy of data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 80th Scientific Sessions of the American Diabetes Association, 12–16 June 2020.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.12896636.
References
- 1.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med 2017;23:804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature 2019;576:51–60 [DOI] [PubMed] [Google Scholar]
- 3.Das SK, Sharma NK, Hasstedt SJ, et al. An integrative genomics approach identifies activation of thioredoxin/thioredoxin reductase-1-mediated oxidative stress defense pathway and inhibition of angiogenesis in obese nondiabetic human subjects. J Clin Endocrinol Metab 2011;96:E1308–E1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sales V, Patti ME. The ups and downs of insulin resistance and type 2 diabetes: lessons from genomic analyses in humans. Curr Cardiovasc Risk Rep 2013;7:46–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma NK, Sajuthi SP, Chou JW, et al. Tissue-specific and genetic regulation of insulin sensitivity-associated transcripts in African Americans. J Clin Endocrinol Metab 2016;101:1455–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma NK, Chuang Key CC, Civelek M, et al. Genetic regulation of enoyl-CoA hydratase domain-containing 3 in adipose tissue determines insulin sensitivity in African Americans and Europeans. Diabetes 2019;68:1508–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rivera CM, Ren B. Mapping human epigenomes. Cell 2013;155:39–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varley KE, Gertz J, Bowling KM, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 2013;23:555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziller MJ, Gu H, Müller F, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013;500:477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heyn H, Moran S, Hernando-Herraez I, et al. DNA methylation contributes to natural human variation. Genome Res 2013;23:1363–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 2009;58:2718–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chambers JC, Loh M, Lehne B, et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol 2015;3:526–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demerath EW, Guan W, Grove ML, et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet 2015;24:4464–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet 2014;383:1990–1998 [DOI] [PubMed] [Google Scholar]
- 15.Hidalgo B, Irvin MR, Sha J, et al. Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network study. Diabetes 2014;63:801–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Ding J, Reynolds LM, et al. Methylomics of gene expression in human monocytes. Hum Mol Genet 2013;22:5065–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toperoff G, Aran D, Kark JD, et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum Mol Genet 2012;21:371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wahl S, Drong A, Lehne B, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017;541:81–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006;444:847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature 2008;452:423–428 [DOI] [PubMed] [Google Scholar]
- 21.Agha G, Houseman EA, Kelsey KT, Eaton CB, Buka SL, Loucks EB. Adiposity is associated with DNA methylation profile in adipose tissue. Int J Epidemiol 2015;44:1277–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benton MC, Johnstone A, Eccles D, et al. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol 2015;16:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab 2019;29:1028–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilsson E, Jansson PA, Perfilyev A, et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 2014;63:2962–2976 [DOI] [PubMed] [Google Scholar]
- 25.Orozco LD, Farrell C, Hale C, et al. Epigenome-wide association in adipose tissue from the METSIM cohort. Hum Mol Genet 2018;27:1830–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allum F, Hedman ÅK, Shao X, et al. Dissecting features of epigenetic variants underlying cardiometabolic risk using full-resolution epigenome profiling in regulatory elements. Nat Commun 2019;10:1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fraser HB, Lam LL, Neumann SM, Kobor MS. Population-specificity of human DNA methylation. Genome Biol 2012;13:R8–R13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langefeld CD, Comeau ME, Sharma NK, Bowden DW, Freedman BI, Das SK. Transcriptional regulatory mechanisms in adipose and muscle tissue associated with composite glucometabolic phenotypes. Obesity (Silver Spring) 2018;26:559–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011;27:1571–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 2012;13:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Spector TD, Deloukas P, Bell JT, Engelhardt BE. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome Biol 2015;16:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grundberg E, Meduri E, Sandling JK, et al.; Multiple Tissue Human Expression Resource Consortium . Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet 2013;93:876–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sajuthi SP, Sharma NK, Comeau ME, et al. Genetic regulation of adipose tissue transcript expression is involved in modulating serum triglyceride and HDL-cholesterol. Gene 2017;632:50–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Millstein J, Chen GK, Breton CV. cit: hypothesis testing software for mediation analysis in genomic applications. Bioinformatics 2016;32:2364–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cavalcante RG, Sartor MA. annotatr: genomic regions in context. Bioinformatics 2017;33:2381–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glastonbury CA, Couto Alves A, El-Sayed Moustafa JS, Small KS. Cell-type heterogeneity in adipose tissue is associated with complex traits and reveals disease-relevant cell-specific eQTLs. Am J Hum Genet 2019;104:1013–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allum F, Grundberg E. Capturing functional epigenomes for insight into metabolic diseases. Mol Metab 2020;38:100936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuehnen P, Mischke M, Wiegand S, et al. An Alu element-associated hypermethylation variant of the POMC gene is associated with childhood obesity. PLoS Genet 2012;8:e1002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Iterson M, van Zwet EW, Heijmans BT; BIOS Consortium . Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol 2017;18:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kühnen P, Handke D, Waterland RA, et al. Interindividual variation in DNA methylation at a putative POMC metastable epiallele is associated with obesity. Cell Metab 2016;24:502–509 [DOI] [PubMed] [Google Scholar]
- 41.Zang L, Hong Q, Yang G, et al. MACROD1/LRP16 enhances LPS-stimulated inflammatory responses by up-regulating a rac1-dependent pathway in adipocytes. Cell Physiol Biochem 2018;51:2591–2603 [DOI] [PubMed] [Google Scholar]
- 42.Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature 2019;571:489–499 [DOI] [PubMed] [Google Scholar]
- 43.Ma X, Kang S. Functional implications of DNA methylation in adipose biology. Diabetes 2019;68:871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cawley NX, Li Z, Loh YP. 60 Years of POMC: biosynthesis, trafficking, and secretion of pro-opiomelanocortin-derived peptides. J Mol Endocrinol 2016;56:T77–T97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sajuthi SP, Sharma NK, Chou JW, et al. Mapping adipose and muscle tissue expression quantitative trait loci in African Americans to identify genes for type 2 diabetes and obesity. Hum Genet 2016;135:869–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schübeler D. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet 2011;43:1091–1097 [DOI] [PubMed] [Google Scholar]
- 47.Claringbould A, de Klein N, Franke L. The genetic architecture of molecular traits. Curr Opin Syst Biol 2017;1:25–31 [Google Scholar]
- 48.Arner P, Sahlqvist AS, Sinha I, et al. The epigenetic signature of systemic insulin resistance in obese women. Diabetologia 2016;59:2393–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu XS, Wu H, Ji X, et al. Editing DNA methylation in the mammalian genome. Cell 2016;167:233–247.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDonald JI, Celik H, Rois LE, et al. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol Open 2016;5:866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]