

Abstract
Photosynthetic electron transport occurs on the thylakoid membrane of chloroplasts. Ferredoxin (Fd), the final acceptor in the electron transport chain, distributes electrons to several Fd‐dependent enzymes including Fd‐thioredoxin reductase (FTR). A cascade from Fd to FTR further reduces Thioredoxin (Trx), which tunes the activity of target metabolic enzymes eventually in a light‐dependent manner. We previously reported that 10 Trx isoforms in Arabidopsis thaliana can be clustered into three classes based on the kinetics of the FTR‐dependent reduction (high‐, middle‐, and low‐efficiency classes). In this study, we determined the X‐ray structure of three electron transfer complexes of FTR and Trx isoform, Trx‐y1, Trx‐f2, and Trx‐m2, as representative examples of each class. Superposition of the FTR structure with/without Trx showed no main chain structural changes upon complex formation. There was no significant conformational change for single and complexed Trx‐m structures. Nonetheless, the interface of FTR:Trx complexes displayed significant variation. Comparative analysis of the three structures showed two types of intermolecular interactions; (i) common interactions shared by all three complexes and (ii) isoform‐specific interactions, which might be important for fine‐tuning FTR:Trx activity. Differential electrostatic potentials of Trx isoforms may be key to isoform‐specific interactions.
Keywords: electron transport, ferredoxin‐reductase, thioredoxin, thioredoxin, x‐ray crystal structure
Short abstract
Abbreviations
- Fd
ferredoxin
- FTR
ferredoxin‐thioredoxin reductase
- PDB
protein data bank
- Trx
thioredoxin
1. INTRODUCTION
Sunlight is the ultimate energy source for plants and algae. Fluctuations in the level of sunlight has led to the evolution of regulatory systems to protect against photodamage and modulate metabolic reactions, such as the Calvin‐Benson cycle. Thiol‐based redox regulation is a mechanism for controlling metabolic pathways in chloroplasts. 1 , 2 Thioredoxin (Trx) possesses a pair of redox‐active cysteines that activates specific enzymes in a light‐dependent manner. The redox level of Trx is maintained by Ferredoxin‐Trx Reductase (FTR) and Ferredoxin (Fd), the latter being the final electron acceptor in the photosynthetic electron transport chain. In this Fd/Trx cascade, electrons are sequentially transmitted from photosystem I, via Fd, FTR, and Trx, to target enzymes such as fructose‐1,6‐bisphosphatase, sedoheptulose‐bisphosphatase, NADP‐malate dehydrogenase, or 2‐Cys peroxiredoxins. 3 , 4 , 5 , 6
Trxs are found in bacteria, plants, and mammals and are thought to share a common fold. 7 , 8 , 9 In green algae and plants, Trx exists in several isoforms. Each Trx displays a different protein surface charge and midpoint redox potential (E m), suggesting that Trx isoforms have specific activation targets. 8 , 10 , 11 , 12 , 13 , 14 Completion of the Arabidopsis thaliana genome sequence revealed 10 chloroplast Trx isoforms, classified into five subtypes (two Trx‐f, four Trx‐m, one Trx‐x, two Trx‐y, and one Trx‐z). 15 , 16 By contrast, A. thaliana has only one gene for the catalytic domain of FTR with a [4Fe‐4S] cluster and a redox active cysteines and two genes for the variable subunit. FTR plays a pivotal role in transferring light‐activated electrons from Fd to each Trx isoform. 17 , 18
Based on FTR‐dependent kinetic parameters, chloroplast Trxs from A. thaliana can be clustered into three classes; high, middle, and low efficiency. 14 These previous observations raise the question of how FTR (E m = −356 ± 7 mV) can distinguish between 10 homologous Trx isoforms. The E m value of each Trx has the correlation with the kinetic parameters in part (high: −296 mV < E m < −276 mV, middle: −321 mV < E m < −310 mV, low: −335 mV < E m < −316 mV), but target selectivity of Trxs is not fully controlled by the E m of Trx. 14 The crystal structures of the complex between Synechocystis FTR and spinach Trx‐f (PDB ID: 2PU9) or ‐m (PDB ID: 2PUK) have been published. 19 Surprisingly, the interactions of Trx‐f and ‐m with FTR were found to be highly similar. Nonetheless, the available structures were from two different organisms and therefore difficult to explain the specific interactions of these proteins in vivo.
Here, we determined the crystal structures of three electron transfer complexes between FTR and Trx from A. thaliana; FTR:Trx‐y1, FTR:Trx‐f2 and FTR:Trx‐m2 representing high (S 0.5 = 18 ± 8 nM, t 1/2 = 5.7 ± 1.7 s, and E m = −296 mV), middle (S 0.5 = 157 ± 54 nM, t 1/2 = 20.6 ± 5.2 s, and E m = −321 mV) and low (S 0.5 = 427 ± 83 nM, t 1/2 = 110.9 ± 49.9 s, and E m = −335 mV) FTR:Trx activity class, respectively. These physiological complex structures provide direct information on the protein–protein interaction of FTR and Trx isoforms. Our findings reveal small but significant differences between the three structures, which offer a plausible explanation for Trx isoform‐based fine tuning of FTR activity.
2. RESULTS AND DISCUSSION
2.1. Crystal structure of FTR:Trxs complexes
The crystal structure of FTR:Trx‐y1 was solved using the molecular replacement method at 1.59 Å resolution without any ambiguity. Only the catalytic subunit of FTR was present in the structure (Figure 1a). Crystal packing analysis of FTR:Trx‐y1 implied the supposed position of the FTR variable subunit in the crystal was occupied by the next Trx‐y1 molecule (noted as Trx‐y1' in Figure 1b). It is likely that the chemical composition of the precipitant weakened intrinsic intersubunit interactions and enhanced extrinsic intermolecular interactions between the catalytic subunit and the next Trx‐y1' molecule (Figure 1b). The high concentration of protein in the crystallization droplet may have accelerated this nonphysiological swapping of interacting subunit. The structure of the FTR:Trx‐f2 complex was determined at 1.8 Å resolution by molecular replacement using Synechosystis FTR and spinach Trx‐f structure (PDB ID: 2PU9) as a search model. The crystallographic asymmetric unit comprised one molecule each of FTR and Trx‐f2 (Figure 1c). By contrast, crystals of the FTR:Trx‐m2 complex comprised seven complexes in the asymmetric unit with a high solvent content of 71.9%, giving a resolution of only 2.4 Å. Among the seven FTR:Trx‐m2 molecules related by noncrystallographic symmetry, we selected one complex (Chain A, B for FTR and C for Trx‐m2) as representative (Figure 1d) because its clearer e‐density.
FIGURE 1.
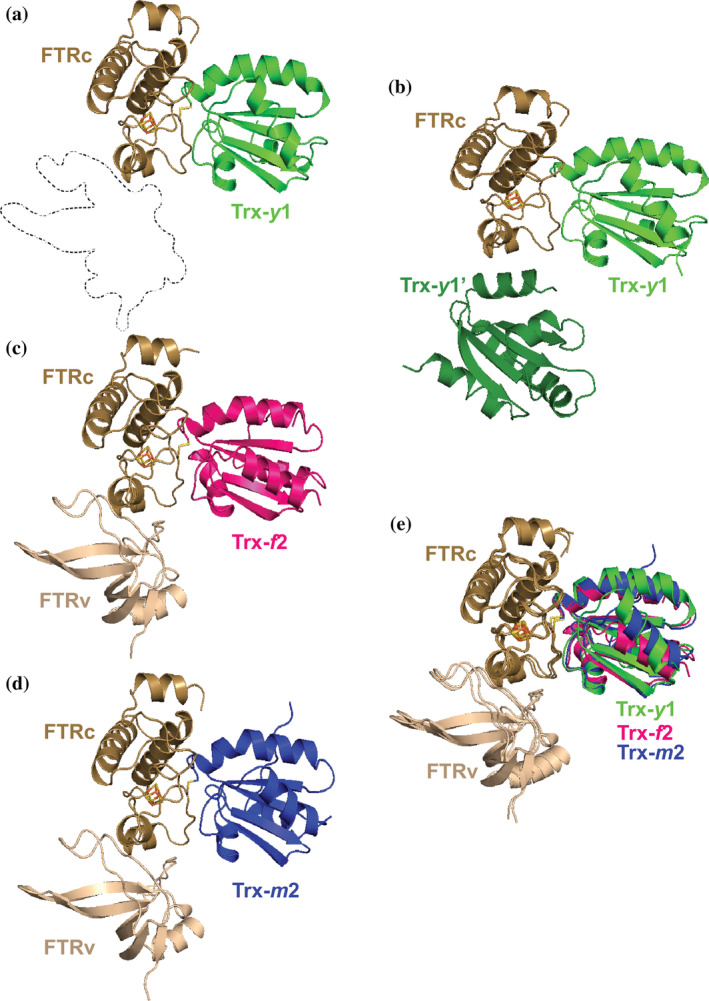
Structures of the three FTR:Trx isoform complexes. All FTR molecules consisting of catalytic and variable subunit (FTRc and FTRv, respectively) are colored in light brown. (a). FTR:Trx‐y1 complex, Trx‐y1 is colored in green. (b). FTR:Trx‐y1 complex shown alongside the crystal structure of next Trx‐y1 molecule labeled Trx‐y1' and colored dark green, (c). FTR:Trx‐f2 complex, Trx‐f2 is colored in magenta, (d). FTR:Trx‐m2 complex, Trx‐m2 is colored in blue, (e). Superimposed model of Trx isoforms based on the Cα atoms of FTRc
In the FTR:Trx‐y1, ‐f2, and ‐m2 complex structures, Trxs interact specifically with the catalytic subunit of FTR, whereas the variable subunit of FTR does not participate in the intermolecular interaction. Residues 1–23 in the N‐terminal region of the FTR variable subunit were invisible in both FTR:Trx‐f2 and FTR:Trx‐m2 complexes. These regions are rich in serine residues, which can cause structural instability. 20 In the case of spinach FTR, removal of up to 24 N‐terminal residues of the variable subunit stabilizes FTR and does not affect the catalytic properties. 21 The binding modes of Trx‐y1, ‐f2, and ‐m2 to the FTR catalytic subunit are highly similar. 19 The distances between [4Fe‐4S] cluster (FE2 atom of the cluster) of FTR and the disulfide bond (SG atom of Cys39) of Trx‐y1, ‐f2, and ‐m2 are 8.31, 8.01, and 8.51 Å, respectively. To analyze detailed structural differences among the three complexes, we superimposed the structures based on the common FTR catalytic subunit (Figure 1e). Superposition of the three complexes shows an almost identical structure of the FTR catalytic subunit (aveage rmsd = 0.588 Å) but some differences in the rotational orientation of the Trx isoforms. The differences between the three superimposed Trx isoform structures were 1.2–1.5 Å in rmsd and 7.9–16.1° in the polar angle of kappa.
2.2. Interaction between FTR and Trxs in the complex
The FTR:Trx complexes were stabilized by an intermolecular disulfide bridge and additional interactions at the interface. Common or isoform‐specific amino acids for the protein–protein interactions are listed in Figure 2a. Figure 2b presents the molecular surfaces of the interface between FTR (left) and Trx isoform (right), showing the interacting residues from each counterpart. A conserved sequence of W30, C31, G32, and P33 (Trx‐y1 numbering) was located in the middle of the interface (Figure 2a,b). C31 made an intermolecular disulfide bridge with C56 of FTR. These residues in the WCGP consensus sequence of Trx made common interaction with V37, V38, K40, G41, P55, C56, H85, and C86 of FTR (Figure S1). In addition, structurally conserved interactions were found at I71, E72, A73, and L74 (Trx‐y1 numbering, Figure 2a). One charged residue (E72 of Trx‐y1, R80 of Trx‐f2, and R79 of Trx‐m2) interacted with R57 and H58 of FTR. The other three uncharged residues (I71, A73, and L74 in Trx‐y1 numbering) were involved in hydrophobic interactions with C56, R57, and H58 of FTR (Figure S1). V34 of FTR was involved in complex formation with all three Trx isoforms (Figure 2a), but the counterparts were unique and not part of the two consensus regions.
FIGURE 2.
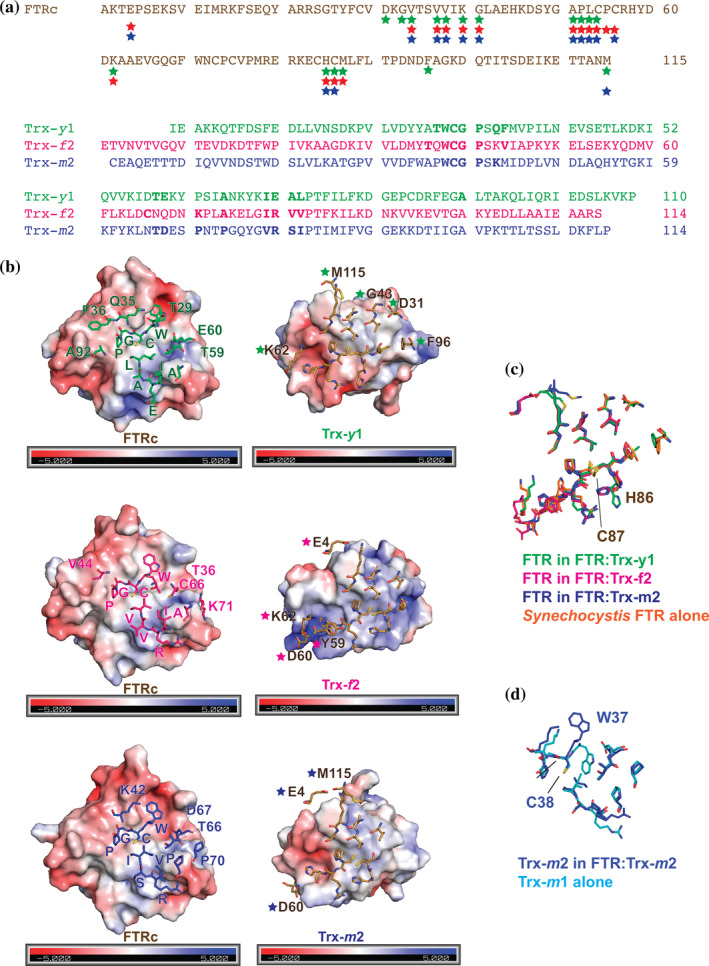
Structural comparison of the three FTR:Trx complexes. (a). Amino acid sequence alignment of the catalytic subunit of FTR (upper) and the three Trx isoforms (bottom). A colored star below the FTR sequence indicates that the corresponding residue interacts with the Trx molecule. Color code for the stars are as follows: green for Trx‐y1, magenta for Trx‐f2, or blue for Trx‐m2. A bold letter in the Trx sequences indicates that the residue is involved in interaction with FTR. (b). The molecular surfaces of FTR and Trx colored according to the electrostatic potentials are shown in open‐book style with interacting residues shown as stick models. A one letter code with a sequence number correspond to isoform‐specific residues. (c). Superimposed models of FTR in the three complexes and Synechocystis FNR alone (1DJ7). (d). Superimposed models of Trx‐m
In addition to the above core interactions, several isoform‐specific interactions were found at the periphery of each interface, which may contribute to fine tuning the efficiency of electron transfer from FTR to Trxs. In the FTR:Trx‐y1 complex, six additional residues of Trx‐y1, namely A92, F36, Q35, T29, E60, and T59, were involved in forming a complex with FTR whose counterparts were D31, G43, K62, F96, and M115, respectively (Figure 2b, upper panel). Similarly, in FTR:Trx‐f2 four additional residues, namely V44, T36, C66, K71 of Trxs‐f2, interacted with four residues of FTR, namely E4, K62, Y59, and D60 (Figure 2b, middle panel). In FTR:Trx‐m2 residues K42, D67, T66, P60 of Trx‐m2 interacted with three specific residues of FTR, namely E4, D60, and M115 (Figure 2b, lower panel). The accessible buried surface area between FTR and Trxs was calculated. The corresponding area for the FTR:Trx‐y1 complex was 1,622 Å2, whereas the area for FTR:Trx‐f2 and FTR:Trx‐m2 was 1,470 and 1,509 Å2, respectively. Based on our reported kinetic parameter of S 0.5 which would correlate with the affinity of each FTR:Trx complex, 14 S 0.5 of Trx‐y1 is 10‐fold smaller than that of Trx‐f2, whereas S 0.5 of Trx‐m2 is not so different from that of Trx‐f2. The differences in buried surface area may explain why Trx‐y1 has more isoform‐specific peripheral interactions than Trx‐f2 or ‐m2.
What drives these additional peripheral isoform‐specific interactions? The electrostatic potential of each protein may be key to this molecular recognition. Although the FTR interface has the same electrostatic potential in all three complexes, each Trx isoform counterpart has a slightly different charge distribution on the molecular surface (Figure 2b). Trx‐y1 has a more negatively charged surface than Trx‐m2, whereas the interface of Trx‐f2 is mostly positively charged. Small but significant differences in the charged surface area of the Trx isoforms may induce additional isoform‐specific interactions. The resulting variety of buried surface areas of the electron transfer complexes could fine tune FTR:Trx activity.
2.3. Structural changes of FTR and Trx upon complex formation
Sequence alignment of the catalytic subunit of FTR from Arabidopsis thaliana (AtFTR) and Synechocystis (PDB ID: 1DJ7) shows a high sequence identity of 65%. Unfortunately, the structure of AtFTR alone was unavailable. Thus, the structure of a single Synechocystis FTR in the database and AtFTR structures in our complexes were superimposed based on Cα atoms. These models did not suggest any significant structural changes upon complex formation. However, conformation of the side chain of H85 (A. thaliana numbering) did vary. This histidine is located next to C86, which is spatially close to C56 involved in disulfide bond formation with a cysteine from Trx. The side chain of H85 in the FTR:Trx‐y1 complex showed a unique conformer facing differently from the equivalent residue in both of the FTR:Trx‐f2 and FTR:Trx‐m2 complexes (Figure 2c). This conformational difference in the side chain may be caused by the tighter interaction between FTR and Trx‐y1, as discussed above.
Structural comparison of Trx‐m2 in the complex and the single Trx‐m1 isoform, determined independently at 1.1 Å resolution, revealed no significant conformational changes before and after complex formation except for the N‐ or C‐terminal regions. However, significant differences in these two Trx‐m structures were identified in the side chain conformations at the interface (Figure 2d). Specifically, the side chain of conserved residue W37, located next to C38 that is involved in disulfide bonding to FTR (W33 and C34 of Trx‐m1), was flipped to facilitate hydrophobic interactions with FTR. The same flip of the tryptophan side chain was also found in Trx‐f2 when compared to the structure of spinach Trx‐f (PDB ID: 1FAA), which displays 75% sequence identity to A. thaliana Trx‐f2. Unfortunately, we were unable to perform a similar analysis for Trx‐y1 due to the lack of an available structure. We predict isoform‐specific differences in the conformer of this conserved tryptophan in the FTR:Trx‐y1 complex.
In summary, we have determined the crystal structures of three higher plant electron transfer complexes between FTR and Trx isoforms with different enzymatic activities. The three representative classes of FTR:Trx complex with high, middle, and low activity exhibit a similar main chain structure. Small rotational divergence in binding mode of Trxs was detected, but no significant changes of the main chain structure of FTR and Trx upon complex formation were found. We identified two modes of interaction between FTR and Trx isoforms; one is common to all three classes and the other is isoform‐specific. Some peripheral nonconserved residues identified in the isoform‐specific interactions are structurally important and may explain the differences in FTR:Trx activity.
3. MATERIALS AND METHODS
3.1. Protein expression and purification
The 10 isoforms of chloroplast Trx can be grouped into three classes based on their FTR‐dependent kinetic properties. Trx‐y1, Trx‐f2, and Trx‐m2 were used as representative examples of high, middle, and low affinity class, respectively. The variable subunit of FTR A2 was used in this study. FTR and Trxs from A. thaliana were expressed and purified as previously described. 14 Mutation of buried Cys residue in the active‐site of Trx to Ser facilitates stabilization of the intermolecular disulfide bridge between FTR and Trx. A covalent bond between wild‐type FTR and mutant Trx was generated by mixing equimolar amounts of FTR and Trx. First, the mixture of FTR and Trx in 25 mM Tris–HCl buffer (pH 7.5) was reduced by addition of DTT at a final concentration of 10 mM, and then the solution was dialyzed overnight against 25 mM Tris–HCl buffer (pH 7.5) at 4°C. The resulting protein complex was repurified by ion exchange chromatography.
3.2. Crystallization
Crystals of FTR:Trx‐y1, FTR:Trx‐f2, and FTR:Trx‐m2 were obtained by the hanging‐drop vapor diffusion method. Crystals of FTR:Trx‐y1 were obtained from droplets comprising 1 μl of protein sample (10 mg ml−1) and 1 μl of reservoir solution containing 100 mM magnesium citric acid (pH 3.5), 5% (v/v) 2‐propanol, and 5–9% (w/v) PEG 3,350, at 293 K. Crystals of FTR:Trx‐f2 were obtained from droplets comprising 1 μl of protein sample (10 mg ml−1) and 1 μl of reservoir solution containing 200 mM magnesium nitrate and 16–20% (w/v) PEG 3,350, at 293 K. Crystals of FTR:Trx‐m2 were obtained from droplets comprising 1 μl of protein sample (10 mg ml−1) and 1 μl of reservoir solution containing 100 mM sodium citrate (pH 6.2) and 18% (w/v) PEG 3,350, at 277 K. Crystals of Trx‐m1 were obtained by the sitting‐drop vapor diffusion method from droplets comprising 200 nl of protein sample (10 mg ml−1) and 200 nl of reservoir solution containing in 0.1 M HEPES‐NaOH (pH 7.5), 10% (v/v) 2‐propanol, and 20% (w/v) PEG 4,000, at 293 K.
3.3. Data collection and structure determination
X‐ray diffraction data of FTR:Trx‐y1, FTR:Trx‐f2, and Trx‐m1 were collected on beamline BL44XU at SPring‐8 (Harima, Japan) using EIGER X16M (Dectris, Baden, Switzerland), and those of FTR:Trx‐m2 was collected on beamline TPS 05A at NSRRC (Hsinchu, Taiwan) using CCD detector MX‐300HE (Rayonix, Evanston, IL). All diffraction data were collected at 100 K and processed and scaled using XDS. 22 The structures were determined by molecular replacement with program PHASER in the CCP4 program package 23 using Synechocystis FTR and mutant spinach Trxs complex (PDB ID: 2PU9 and 2PUK) as search models. The structure models of FTR:Trx‐y1, FTR:Trx‐f2, and FTR:Trx‐m2 were refined with phenix refine 24 while the Trx‐m1 structure was refined using SHELXL. 25 Data collection and refinement statistics are given in Table 1. Figures were prepared using PyMol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC) and protein–protein interactions were checked by Dimplot in LigPlot. 26
TABLE 1.
Data collection and refinement statistics
| FTR:Trx‐y1 | FTR:Trx‐f2 | FTR:Trx‐m2 | Trx‐m1 | |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength (Å) | 0.90000 | 0.90000 | 1.00000 | 0.90000 |
| Resolution range (Å) | 43.07–1.59 (1.69–1.59) | 48.71–1.79 (1.90–1.79) | 42.52–2.4 (2.54–2.4) | 26.68–1.10 (1.13–1.10) |
| Space group | P212121 | P212121 | C2 | P21 |
| Unit cell | ||||
| a, b, c (Å) | 54.96, 60.76, 61.06 | 68.74, 83.94, 69.04 | 174.26, 137.21, 192.46 | 24.01, 64.33, 29.49 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90.21, 90 | 90, 96.01, 90 |
| Total reflections | 184,414 (27,906) | 257,225 (42,160) | 663,199 (105,966) | 238,324 (38,607) |
| Unique reflections | 27,938 (4,419) | 37,812 (5,988) | 175,399 (27,733) | 35,880 (5,726) |
| Multiplicity | 6.6 (6.3) | 6.1 (6.3) | 6.2 (6.3) | 6.2 (6.3) |
| Completeness (%) | 99.8 (99.1) | 99.4 (98.6) | 99.14 (96.86) | 99.69 (98.70) |
| Mean I/sigma(I) | 10.40 (3.03) | 13.16 (2.53) | 8.82 (1.31) | 20.98 (9.8) |
| R‐merge | 0.12 (0.736) | 0.105 (0.943) | 0.123 (1.285) | 0.058 (0.252) |
| R‐means | 0.13 (0.754) | 0.114 (0.965) | 0.144 (1.494) | 0.063 (0.274) |
| CC1/2 | 0.995 (0.903) | 0.998 (0.833) | 0.995 (0.539) | 0.998 (0.98) |
| Refinement | ||||
| Reflections used in refinement | 27,910 | 37,802 | 175,276 | 35,799 |
| Reflections used for R‐free | 1,397 | 1891 | 8,764 | 1,668 |
| R‐work | 0.1699 | 0.1793 | 0.2095 | 0.1437 |
| R‐free | 0.1929 | 0.2105 | 0.2445 | 0.1877 |
| Number of nonhydrogen atoms | 1841 | 2,773 | 18,993 | 1,062 |
| Macromolecules | 1725 | 2,470 | 18,397 | 931 |
| Ligands | 8 | 65 | 66 | 1 |
| Solvent | 108 | 238 | 530 | 130 |
| RMS (bonds; Å) | 0.018 | 0.007 | 0.013 | 0.0135 |
| RMS (angles; °) | 1.41 | 0.81 | 1.04 | 2.21 |
| Ramachandran favored (%) | 98.10 | 97.37 | 98.76 | 100.00 |
| Ramachandran allowed (%) | 1.90 | 2.63 | 1.06 | 0.00 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.18 | 0.00 |
| Clashscore | 2.63 | 1.39 | 7.78 | 4.79 |
| Average B‐factor (Å2) | 22.42 | 28.38 | 67.22 | 18.28 |
| Macromolecules | 22.00 | 27.67 | 67.64 | 16.32 |
| Ligands | 12.70 | 35.04 | 46.26 | 27.40 |
| Solvent | 29.77 | 33.94 | 55.32 | 32.20 |
| PDB ID | 7BZK | 7C2B | 7C3F | 7C65 |
Note: Values for the highest resolution shell are shown in parentheses.
AUTHOR CONTRIBUTIONS
Linda Juniar: Data curation; investigation; methodology; writing‐original draft. Hideaki Tanaka: Data curation; formal analysis; methodology; validation. Keisuke Yoshida: Conceptualization; investigation; methodology. Toru Hisabori: Conceptualization; funding acquisition; investigation; supervision. Genji Kurisu: Conceptualization; funding acquisition; supervision; validation; writing‐review and editing.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank Dr. Ju Yaen Kim for her contribution of initial sample preparation, and the beamline staff of BL44XU, at SPring‐8, and TPS 05A, at NSRRC for their kind support during data collection under the proposal numbers of 2019A6500 and 2019‐1‐348‐1, respectively. This research was supported by a Grants‐in‐Aid for Scientific Research under grant number 16H06560 (Genji Kurisu), 16H06556 (Toru Hisabori), and 19H03241 (Keisuke Yoshida) from MEXT‐KAKENHI, and in part by Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials.
Juniar L, Tanaka H, Yoshida K, Hisabori T, Kurisu G. Structural basis for thioredoxin isoform‐based fine‐tuning of ferredoxin‐thioredoxin reductase activity. Protein Science. 2020;29:2538–2545. 10.1002/pro.3964
Funding information Japan Society for the Promotion of Science, Grant/Award Numbers: Grants‐in‐Aid for Scientific Research/ 16H06556, Grants‐in‐Aid for Scientific Research/ 16H06560, Grants‐in‐Aid for Scientific Research/ 19H03241; Ministry of Education, Culture, Sports, Science and Technology, Grant/Award Number: Dynamic Alliance for Open Innovation Bridging Huma
REFERENCES
- 1. Michelet L, Zaffagnini M, Morisse S, et al. Redox regulation of the Calvin‐Benson cycle: Something old, something new. Front Plant Sci. 2013;4:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dietz KJ, Hell R. Thiol switches in redox regulation of chloroplasts: Balancing redox state, metabolism and oxidative stress. Biol Chem. 2015;396:483–494. [DOI] [PubMed] [Google Scholar]
- 3. Schürmann P, Wolosiuk RA. Studies on the regulatory properties of chloroplast fructose‐1,6‐bisphosphatase. Biochim Biophys Acta. 1978;522:130–138. [DOI] [PubMed] [Google Scholar]
- 4. Queiroz‐Claret C, Meunier JC. Light activation of sedoheptulose‐1,7‐bisphosphatase by a reconstituted thylakoid system compared with DTT activation. Evidence for protein‐enzyme interactions. J Plant Physiol. 1995;145:45–49. [Google Scholar]
- 5. Buchanan BB. Role of light in the regulation of chloroplast enzymes. Annu Rev Plant Physiol. 1980;31:341–374. [Google Scholar]
- 6. Collin V, Issakidis‐Bourguet E, Marchand C, et al. The Arabidopsis plastidial thioredoxins. New functions and new insights into specificity. J Biol Chem. 2003;278:23747–23752. [DOI] [PubMed] [Google Scholar]
- 7. Katti SK, LeMaster DM, Eklund H. Crystal structure of thioredoxin from Escherichia coli at 1.68 Å resolution. J Mol Biol. 1990;212:167–184. [DOI] [PubMed] [Google Scholar]
- 8. Capitani G, Marković‐Housley Z, DelVal G, Morris M, Jansonius JN, Schürmann P. Crystal structures of two functionally different thioredoxins in spinach chloroplasts. J Mol Biol. 2000;302:135–154. [DOI] [PubMed] [Google Scholar]
- 9. Weichsel A, Gasdaska JR, Powis G, Montfort WR. Crystal structures of reduced, oxidized, and mutated human thioredoxins: Evidence for a regulatory homodimer. Structure. 1996;4:735–751. [DOI] [PubMed] [Google Scholar]
- 10. Hirasawa M, Schürmann P, Jacquot JP, et al. Oxidation‐reduction properties of chloroplast thioredoxins, ferredoxin:Thioredoxin reductase, and thioredoxin f‐regulated enzymes. Biochemistry. 1999;38:5200–5205. [DOI] [PubMed] [Google Scholar]
- 11. Lemaire SD, Miginiac‐Maslow M. The thioredoxin superfamily in Chlamydomonas reinhardtii . Photosynth Res. 2004;82:203–220. [DOI] [PubMed] [Google Scholar]
- 12. Lemaire SD, Tedesco D, Crozet P, et al. Crystal structure of chloroplastic thioredoxin f2 from Chlamydomonas reinhardtii reveals distinct surface properties. Antioxidants. 2018;7:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshida K, Hara S, Hisabori T. Thioredoxin selectivity for thiol‐based redox regulation of target proteins in chloroplasts. J Biol Chem. 2015;290:14278–14288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshida K, Hisabori T. Distinct electron transfer from ferredoxin‐thioredoxin reductase to multiple thioredoxin isoforms in chloroplasts. Biochem J. 2017;474:1347–1360. [DOI] [PubMed] [Google Scholar]
- 15. The Arabidopsis Genome Initiative . Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature. 2000;408:796–815. [DOI] [PubMed] [Google Scholar]
- 16. Meyer Y, Reichheld JP, Vignols F. Thioredoxins in Arabidopsis and other plants. Photosynth Res. 2005;86:419–433. [DOI] [PubMed] [Google Scholar]
- 17. Buchanan BB, Schürmann P, Wolosiuk RA, Jacquot JP. The ferredoxin/thioredoxin system: From discovery to molecular structures and beyond. Photosynth Res. 2002;73:215–222. [DOI] [PubMed] [Google Scholar]
- 18. Glauser DA, Bourquin F, Manieri W, Schürmann P. Characterization of ferredoxin:Thioredoxin reductase modified by site‐directed mutagenesis. J Biol Chem. 2004;279:16662–16669. [DOI] [PubMed] [Google Scholar]
- 19. Dai S, Friemann R, Glauser DA, et al. Structural snapshots along the reaction pathway of ferredoxin‐thioredoxin reductase. Nature. 2007;448:92–96. [DOI] [PubMed] [Google Scholar]
- 20. Gaymard E, Franchini L, Manieri W, Stutz E, Schürmann P. A dicistronic construct for the expression of functional spinach chloroplast ferredoxin: Thioredoxin reductase in Escherichia coli . Plant Sci. 2000;158:107–113. [DOI] [PubMed] [Google Scholar]
- 21. Manieri W, Franchini L, Raeber L, Dai S, Stritt‐Etter AL, Schürmann P. N‐terminal truncation of the variable subunit stabilizes spinach ferredoxin:Thioredoxin reductase. FEBS Lett. 2003;549:167–170. [DOI] [PubMed] [Google Scholar]
- 22. Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liebschner D, Afonine PV, Baker ML, et al. Macromolecular structure determination using X‐rays, neutrons, and electrons: Recent developments in Phenix. Acta Crystallogr. 2019;D75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sheldrick GM. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015;C71:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laskowski RA, Swindells MB. LigPlot+: Multiple ligand‐protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information