

Abstract
Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) is an evolutionarily conserved essential enzyme in the glycolytic pathway. GAPDH is also involved in a wide spectrum of non‐catalytic cellular ‘moonlighting’ functions. Bacterial surface‐associated GAPDHs engage in many host interactions that aid in colonization, pathogenesis, and virulence. We have structurally and functionally characterized the recombinant GAPDH of the obligate intracellular bacteria Chlamydia trachomatis, the leading cause of sexually transmitted bacterial and ocular infections. Contrary to earlier speculations, recent data confirm the presence of glucose‐catabolizing enzymes including GAPDH in both stages of the biphasic life cycle of the bacterium. The high‐resolution crystal structure described here provides a close‐up view of the enzyme's active site and surface topology and reveals two chemically modified cysteine residues. Moreover, we show for the first time that purified C. trachomatis GAPDH binds to human plasminogen and plasmin. Based on the versatility of GAPDH's functions, data presented here emphasize the need for investigating the Chlamydiae GAPDH's involvement in biological functions beyond energy metabolism.
Keywords: Chlamydia, crystal structure, enzyme kinetics, GAPDH, glycolysis, plasmin binding, plasminogen binding, protein–protein interaction, reactive cysteine, STD/STI
1. INTRODUCTION
Chlamydia trachomatis (Ct) is an obligate intracellular pathogen of eukaryotic cells. Ct is a major cause of sexually transmitted bacterial infections worldwide and a leading cause of preventable blindness in the developed world. 1 , 2 Chronic Chlamydiae infection in women can lead to serious long‐term diseases including cervical and uterine cancers. 2 , 3 Although treatable by antibiotics, treatment failure and reinfections hamper Chlamydiae control in developed countries. 4 , 5 New targets for developing drugs and a vaccine are urgently needed. However, there is a critical gap in our understanding of the biochemical pathways and potential therapeutic targets of Ct.
Chlamydiae have a unique biphasic life cycle alternating between infectious extracellular elementary bodies (EBs) and intracellular replicative reticulate bodies (RBs). Although historically Chlamydiae are considered ‘energy parasites’, bacterial enzymes for glycolysis (except hexokinase), the tricarboxylic acid cycle and pentose phosphate pathway have been identified in both life stages. 1 , 6 Among the most expressed glycolytic enzymes is Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). 1 This tetrameric essential enzyme catalyzes the reversible oxidative phosphorylation of D‐glyceraldehyde 3‐phosphate (D‐G3P) into 1,3‐diphosphoglycerate using NAD+ as cofactor and plays a key role in cellular metabolism by generating NADH. Due to its indispensable role in glycolysis GAPDH is a potential drug target. 7
Additionally, GAPDHs participate in a wide range of catalysis‐independent cellular functions that are coined ‘moonlighting activities’. 8 Although predominantly a cytoplasmic protein GAPDH associated with microbial surfaces facilitates host‐pathogen interactions, colonization, pathogenesis and immunomodulation. 9 , 10 , 11 , 12 , 13 Bacterial surface resident GAPDHs bind to various host macromolecules including fibronectin, plasminogen (Plg) and complement proteins, and are implicated in host cell invasion and immune evasion. 14 , 15 , 16 , 17 , 18 The moonlighting functions may also be potentially useful therapeutic targets. 19 Moreover, Group B Streptococcus GAPDH (GBSGAPDH) is proposed as a potential vaccine candidate. 20 Interestingly, a GAPDH‐derived peptide was found among the T‐cell antigens purified from mice infected with C. muridarum (Cm). 21
The presence of GAPDH in both EBs and RBs underscores its importance throughout the bacterial life cycle. Active GAPDH enzyme was found in the RB extract but the kinetic parameters have not been determined. As part of our studies on microbial GAPDHs, we have expressed, purified and characterized recombinant Ct GAPDH (CtGAPDH) structurally and functionally. We describe here the high‐resolution crystal structure of the holo enzyme refined at 1.5 Å resolution. The structure displays two chemically modified cysteine residues. Furthermore, we report for the first time that CtGAPDH binds to human Plg (hPlg) and human plasmin (hPln) in vitro.
2. RESULTS AND DISCUSSION
2.1. Enzyme activity optimization and kinetics
The optimum pH for CtGAPDH activity was 9.0 (Figure 1a). The optimum pH of GAPDH enzymes from many organisms is between 7.5 and 9.5 with the majority exhibiting highest activity in the pH range of 8.5–9.5. 9 , 22 , 23 , 24 , 25 , 26 Values of K M for NAD+ and D‐G3P calculated from the Michaelis Menten (M‐M) plots were 0.139 (0.016) mM and 0.611 (0.089) mM, respectively, and the value of V max was 49.6 (1.84) μM NADH min−1 mg−1 (Figure 1b, c). In Table 1, we compare the M‐M parameters for CtGAPDH with those of other GAPDH enzymes. These parameters for GAPDHs from different organisms vary widely. It should be noted that the reported values are often based on experiments performed at pHs different from the optimum pH. The kinetics parameters (K M for NAD+ and D‐G3P, and the V max) of CtGAPDH are very similar to those of Mycobacterium tuberculosis GAPDH. 25 The K M for NAD+ is also similar to the values reported for GAPDHs of Group A Streptococcus 9 and B. stearothermophilus (measured at optimum pH). 26 The K M value for D‐G3P of CtGAPDH is closer to those of the bacterial counterparts than enzymes from eukaryotes.
FIGURE 1.
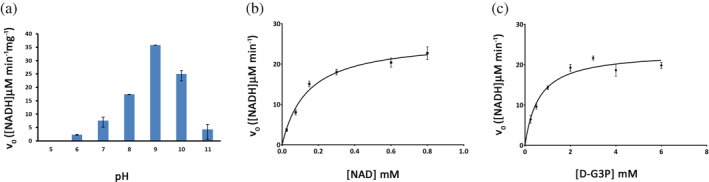
Determination of pH optimum and kinetic parameters of CtGAPDH enzyme activity. (a) CtGAPDH activity is shown at different pH. Initial reaction velocity (v0) was measured. (b) K M and V max for NAD+ were determined from M‐M plot. Assays were done in triplicate. (c) K M and V max for D‐G3P were determined from M‐M plot. Assays were done in triplicate
TABLE 1.
Kinetic parameters for CtGAPDH and GAPDHs of some other organisms
| Organism | KM NAD+mM | KM G3P mM | Vmax μM NADH min−1 mg−1 | Ref. |
|---|---|---|---|---|
| T. brucei (glycosomal) | 0.45 (0.18) a | 0.15 (0.06) | 294 (59) | 22 |
| T. brucei (cytosolic) | 0.04 (0.02) | 0.17 (0.01) | 270 (60) | 22 |
| Human erythrocyte | 0.05 (0.01) | 0.07 (0.01) | 69 (5) | 22 |
| Rabbit muscle | 0.06 (0.01) | 0.082 (0.009) | 73 (2) | 22 |
| Yeast | 0.090 (0.009) | 0.21 (0.03) | 99 (18) | 22 |
| B. stearothermophilus | 0.039 (0.006) | 0.09 (0.01) | 225 (6) | 22 |
| B. stearothermophilus b | 0.15 (0.03) | 0.80 (0.09) | 70 (6) | 27 |
| C. trachomatis | 0.139 (0.016) | 0.611 (0.089) | 49.6 (1.84) | This work |
| P. falciparum | 0.57 (0.06) | 0.9 (0.1) | 3.1 (0.2) | 23 |
| C. parvum | 0.032 (0.002) | 0.763 (0.043) | 72 (2) | 25 |
| Group A Strep | 0.156 | 1.33 | 19.48 | 9 |
| M. tuberculosis | 0.135 (0.05) | 0.58 (0.1) | 55 (5) | 26 |
Note: T. brucei: Trypanosoma brucei. B. stearothermophilus: Bacillus stearothermophilus (Geobacillus stearothermophilus). C. trachomatis: Chlamydia trachomatis. P. falciparum: Plasmodium falciparum. C. parvum: Cryptosporidium parvum. Group A Strep: Group A Streptococcus. M. tuberculosis: Mycobacterium tuberculosis.
SDs are shown in parenthesis.
Assays were performed at the optimum pH (8.9) for B. stearothermophilus GAPDH. The study in Reference 22 was conducted at pH 7.6.
2.2. Overall structure quality
CtGAPDH crystallized in space group P21 as a tetramer in the asymmetric unit with subunits (labelled as A, B, C, and D) related by 222 non‐crystallographic symmetry (Figure 2). The crystal structure was refined to 1.5 Å resolution. Due to insufficient electron density the C‐terminal residue Lys334 (all subunits) and Val100 (in subunits B and C) were not modeled. The overall quality of the structures is excellent (Table 2) with only one outlier, Val238. Val238 is in a well‐defined loop linking adjacent antiparallel β‐strands. The atypical backbone geometry is stabilized by a hydrogen bond between the peptide nitrogen atom of Val238 and Asn314 as observed in other GAPDH structures. 24 , 27 The root mean square deviation (rmsd) of bond distances (0.013 Å) and bond angles (1.69°) indicate excellent geometry. Recently, the 2.4 Å resolution structure of CtGAPDH in space group P212121 was reported. 28 Our high‐resolution structure is similar to the reported structure (PDBID: 6OK4) with an rmsd of 0.29 Å for superposition of the tetramers but reveals additional features not noted in 6OK4.
FIGURE 2.

Structure of CtGAPDH monomer and the assembly into dimers and tetramer. (a) Cartoon drawing showing the tetrameric assembly of CtGAPDH. NAD+ is shown in stick model. Major interface axes P and Q are labeled. S‐loop is colored fire brick. (b) Subunits A, B and C, D form dimers. The AB dimer is shown with a close‐up view of the major interface highlighted in green color on subunit A. (c) Subunits A and C assemble across the Q‐axis. Interface area on subunit A is shown in magenta color. The S‐loop in subunit C is shown in fire brick color. (d) The catalytic domain and the NAD+‐binding domain in subunit A are colored yellow and light orange, respectively. Dimer interface area is shown in green. The S‐loop is in fire brick color. The S‐loop is also part of the second largest interface. Areas with strictly conserved residues (7–13 and 145–154) near the NAD+‐binding pocket are shown in magenta color. NAD+ is shown in stick model
TABLE 2.
Data collection and refinement statistics for CtGAPDH (6WYC)
| Data collection | |
|---|---|
| Space group | P21 |
| Unit cell parameters (Å; °) | a = 66.60, b = 104.05, c = 86.47; β = 97.4 |
| Resolution (Å) | 66.17–1.50 (1.53–1.50) a |
| Unique reflections | 173,217 (8596) |
| Completeness (%) | 93.2 (93.0) |
| Redundancy | 3.3 (3.3) |
| R merge (%) | 7.6 (62.6) |
| R meas (%) | 8.9 (74.2) |
| R pim (%) | 4.5 (38.8) |
| I/σ(I) | 10.2 (1.9) |
| CC1/2 | 0.997 (0.681) |
| Refinement | |
| Resolution (Å) | 66.26–1.50 (1.54–1.50) |
| No. of unique reflections | 173,177 (12748) |
| Completeness (%) | 92.9 (92.8) |
| R work (%) | 17.0 (27.7) |
| R free (%) | 18.9 (29.1) |
| Atoms (protein; ligands; waters) | 10,251;176; 1,230 |
| Wilson B (Å2) | 12.3 |
| Average B‐factors (Å2) | |
| Overall, protein, ligands, waters | 17.1, 15.7, 10.0, 29.3 |
| Rmsd bonds (Å) | 0.013 |
| Rmsd angles (°) | 1.69 |
| CC (Fo‐Fc) | 0.96 |
| Ramachandran | |
| Favored, allowed, outlier (%) | 97, 2.7, 0.3 |
| Clash score b | 2.50 |
| Molprobity score b | 1.21 |
Numbers in the parenthesis are for the highest resolution shell.
Clash score and Molprobity score are calculated by using Molprobity (4.2). 52
2.3. CtGAPDH tetramer assembly
The CtGAPDH tetramer consists of dimers AB and CD, and exhibits two major and one minor interfaces (Figure 2). The major interface P is contributed by subunits A, B and C, D with the dimer interfaces formed by 108 residues spanning across 3,741 Å2 and 3,767 Å2 of buried surface area, respectively. The second largest interface Q is generated by subunit pairs A, C and B, D with 72 and 74 residues contributing to 2,805 and 2,846 Å2 of interface area (buried surface area), respectively. GAPDHs display an evolutionarily conserved three‐dimensional structure and topology. Figure S1 shows a structure‐based alignment of GAPDH sequences from selected organisms. Each GAPDH subunit folds into two domains: the cofactor‐binding domain (residues 1–150, 314–334) and the catalytic domain (151–313).
2.4. Cofactor‐binding domain
The cofactor‐binding domain exhibits the typical Rossmann fold (β‐α‐β fold). 29 In all subunits, the electron density for NAD+ was excellent (Figure S2) and their average B values were lower than the B average for the protein chains (Table 2). The ADP moiety of NAD+ binds to the N‐terminal β‐α‐β motif. Active site residues Gly7‐Phe‐Gly‐Arg‐Ile‐Gly‐Arg13 and Ile145/Val‐Ser‐Asn/Gly‐Ala‐Ser‐Cys‐Thr‐Thr‐Asn‐Cys154/Ser are conserved in GAPDHs of various organisms (Figures S1 and S3). In CtGAPDH, six residues (Arg10, Ile11, Asp33, Lys77, Thr119, and Asn314) directly contact NAD+ (Figure 3a). Main chain nitrogen atoms of Arg10 and Ile11 form hydrogen bonds to the pyrophosphate while side chain oxygen atoms of the conserved Asp33 interact with hydroxyl oxygen atoms of the adenosine ribose. The peptide oxygen atom of Lys77 forms a hydrogen bond with the adenine amino group. The corresponding residue Arg80 in human GAPDH (hGAPDH, 1U8F) also exhibits a similar hydrogen bond. 30 Notably, this hydrogen bond was absent from the 6OK4 model. At the nicotinamide end of the dinucleotide, Thr119 is hydrogen bonded to the ribose and Asn314 forms a hydrogen bond with the amide oxygen of nicotinamide. Table S1 lists the hydrogen bonds between the protein and the cofactor.
FIGURE 3.
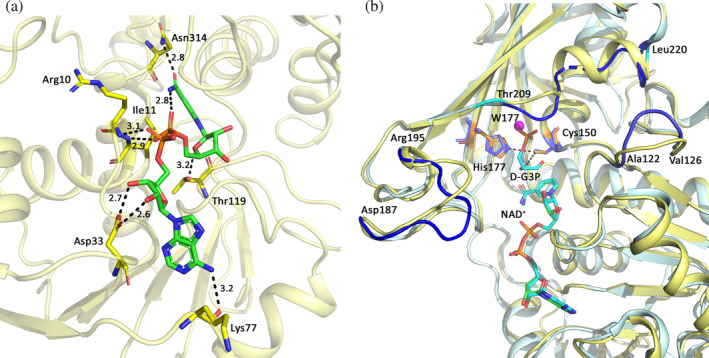
Interactions between NAD+ and CtGAPDH, and comparison of active sites in the binary and ternary complex. (a) Interactions between CtGAPDH (subunit A) residues and NAD+. CtGAPDH residues (C: yellow) and NAD+ (C: green) are shown in stick models. Hydrogen bonding distances (Å) are shown. (b) Superposition of subunits A of CtGAPDH and GBSGAPDH ternary complex (5JYA). NAD+ and the substrate in 5JYA, and the catalytic residues Cys150 and His177 in CtGAPDH are in stick model (C: wheat color). The distance between Cys150Sγ and the C1 atom of the substrate is 3.6 Å, and between Cys150Sγ and His177Nε it is 3.3 Å. The catalytic residues of 5JYA are also shown in stick model (C: slate color). The catalytic cysteine was mutated to serine in 5JYA. Areas where the conformations differ significantly are shown in dark blue on the GBSGAPDH structures and the CtGAPDH residues bordering the divergent areas are labeled. One water molecule (W177 shown as magenta sphere) is found at or near the site for the O3P atom of the substrate
2.5. Active site
The GAPDH catalytic domain is composed of a six‐stranded β‐sheet, three α‐helices and two additional short helices, with catalytic cysteine (Cys150) and histidine (His177) residues positioned for reaction with the substrate. The overall structures of CtGAPDH holo‐form and GBSGAPDH ternary complex (5JYA) are similar (rmsd 0.95 Å for tetramers), and the catalytic residues in these structures superpose well (Figure 3b). In CtGAPDH the Cys150 sulfhydryl atom is at a distance of 3.5–3.6 Å from the carbonyl carbon (C1) atom of D‐G3P (based on its position in 5JYA subunit A). Residues that interact with the C3‐phosphate are also conserved in CtGAPDH (Ser149, Thr151, and Gly210). One water molecule is found at or near the predicted location for the O3P atom of the C3‐phosphate group (Figure 3b). When compared with 5JYA, the CtGAPDH structure deviates in the helix–loop segment (residues 209–220). In 5JYA, two conserved residues, Thr212 and Gly213, within this loop interact with the phosphate of the substrate. Divergence between these structures is also observed in a loop consisting of residues 122–126 and in another loop known as the S‐loop.
2.6. The T‐cell antigenic peptide is located within the S‐loop
GAPDH structures feature a long loop called the S‐loop. In the CtGAPDH structure, the electron density for the S‐loop (residues 179–200) is excellent. The average B value for these residues (12.96 Å2) in each subunit is lower than the B‐average for the protein (15.7 Å2). Several residues in the S‐loop are involved in inter‐subunit interactions across the P and Q interfaces and thus in the assembly of the tetramer. Arg195 and Arg198 of subunit A form salt bridges with Asp294 and Asp283 of subunit B, respectively. Subunit A residues Asp187, Arg191 and Arg198 form hydrogen bonds with Arg13, Leu34, Tyr42 and Ser48 of subunit C, respectively (Figure S4). The peptide nitrogen atom of Ala181 forms a water‐mediated interaction with NAD+ in the same subunit.
The MHC class II bound peptides purified from Cm‐infected mice included a 15 residue peptide corresponding to Cm GAPDH residues 173–187 (MTTVHAATATQ SVVD, where the nine core residues are shown in bold). 21 The equivalent peptide in CtGAPDH is located at the major interface on a long β‐strand and extends into the S‐loop. Three peptide residues (Ala179, Ser184 and Val185, underscored) belonging to the S‐loop are conserved in GAPDHs of Ct and Cm but different in other organisms (Figure S5). Markos et al. noted that the sequence of the S‐loop carries the distinct signature for enzymes of eubacterial and eukaryotic origin. 31 Selection of the T‐cell antigenic peptide within the S‐loop is therefore highly interesting. The S‐loop sequences of GAPDHs of Ct and Cm exhibit some differences from the prokaryotic and eukaryotic proteins but cluster more closely with E. coli and eukaryotic GAPDHs (Table 3).
TABLE 3.
S‐loop sequences in GAPDHs
| Bst | Y | T | N | D | Q | R | I | L | D | L | P | ‐ | H | K | D | L | R | R | A | R | A | A | A | E | ||||
| Bsu | Y | T | N | D | Q | Q | I | L | D | L | P | ‐ | H | K | D | Y | R | R | A | R | A | A | A | E | ||||
| Ta | Y | T | N | D | Q | R | L | L | D | L | P | ‐ | H | K | D | L | R | R | A | R | A | A | A | I | ||||
| Tv | Y | T | N | D | Q | V | V | A | D | T | M | ‐ | H | K | D | L | R | R | A | R | A | A | G | M | ||||
| Cg | Y | T | G | D | Q | R | L | H | D | A | P | ‐ | H | R | D | L | R | R | A | R | A | A | A | V | ||||
| At | Y | T | G | D | Q | R | L | L | D | A | S | ‐ | H | R | D | L | R | R | A | R | A | A | A | L | ||||
| Sp | T | T | G | D | Q | M | I | L | D | G | P | ‐ | H | R | G | G | D | L | R | R | A | R | A | G | A | A | ||
| Sag | Y | T | G | D | Q | M | I | L | D | G | P | ‐ | H | R | G | G | D | L | R | R | A | R | A | G | A | A | ||
| Sa | Y | T | G | D | Q | N | T | Q | D | A | P | ‐ | H | R | K | G | D | K | R | R | A | R | A | A | A | E | ||
| Clp | Y | T | N | D | Q | N | T | L | D | G | P | H | R | K | G | D | F | R | R | A | R | A | A | A | V | |||
| Ct | A | T | A | T | Q | S | V | V | D | G | P | S | R | K | D | W | R | G | G | R | G | A | F | Q | ||||
| Cm | A | T | A | T | Q | S | V | V | D | G | P | S | R | K | D | W | R | G | G | R | G | A | F | Q | ||||
| Ec | T | T | A | T | Q | K | T | V | D | G | P | S | H | K | D | W | R | G | G | R | G | A | S | Q | ||||
| Sc | L | T | A | T | Q | K | T | V | D | G | P | S | H | K | D | W | R | G | G | R | T | A | S | G | ||||
| Tc | T | T | A | T | Q | K | T | V | D | G | P | S | Q | K | D | W | R | G | G | R | G | A | A | Q | ||||
| Tb | T | T | A | T | Q | K | T | V | D | G | P | S | Q | K | D | W | R | G | G | R | G | A | A | Q | ||||
| Eh | T | T | A | T | Q | K | T | V | D | G | P | S | G | K | D | W | R | A | G | R | C | A | C | A | ||||
| Ce | V | T | A | T | Q | K | T | V | D | G | P | S | G | K | L | W | R | D | G | R | G | A | G | Q | ||||
| Hs | I | T | A | T | Q | K | T | V | D | G | P | S | G | K | L | W | R | D | G | R | G | A | L | Q | ||||
| Mm | I | T | A | T | Q | K | T | V | D | G | P | S | G | K | L | W | R | D | G | R | G | A | A | Q | ||||
| Rn | I | T | A | T | Q | K | T | V | D | G | P | S | G | K | L | W | R | D | G | R | G | A | A | Q | ||||
| Pf | S | T | A | N | Q | L | V | V | D | G | P | S | K | G | G | K | D | W | R | A | G | R | C | A | L | S | ||
| Cp | L | T | A | N | Q | L | T | V | D | G | P | S | K | G | G | K | D | W | R | A | G | R | C | A | G | N | ||
| Tg | M | T | A | N | Q | L | T | V | D | G | P | S | K | G | G | K | D | W | R | A | G | R | S | A | G | V | ||
| a | a |
Note: Chlamydiae specific residues Ala179, Ser184, and Val185 in the T‐cell antigenic peptide are underlined. These residues are also underlined in Figure S3.
Abbreviations: At, Arabidopsis thaliana; Bst, Bacillus stearothermophilus (Geobacillus stearothermophilus); Bsu, Bacillus subtilis; Ce, Caenorhabditis elegans; Cg, Corynebacterium glutamicum; Clp, Clostridium pasteurianum; Cm, Chlamydia muridarum; Cp, Cryptosporidium parvum; Ct, Chlamydia trachomatis; Ec, Escherichia coli (Gap A); Eh, Entamoeba histolytica; Hs, Homo sapiens (Human); Mm, Mus musculus (Mouse); Pf, Plasmodium falciparum; Rn, Rattus norvegicus (Rat); Sa, Staphylococcus aureus; Sag, Streptococcus agalactiae (GBS); Sc, Saccharomyces cerevisiae (yeast); Sp, Streptococcus pneumoniae; Ta, Thermus aquaticus; Tb, Trypanosoma brucei (cytoplasmic); Tc, Trypanosoma cruzi; Tg, Toxoplasma gondii; Tv, Trichomonas vaginalis.
Signature residues for eukaryotic GAPDHs are shaded in CtGAPDH and CmGAPDH S‐loop sequences.
2.7. CtGAPDH structure reveals modified cysteine residues
The electron density for Cys63 and Cys287 indicated that these residues were chemically modified (Figure 4a). Based on the shape of the electron density and refinement parameters we predicted that Cys63 was nitrosylated (SNC) presumably within E. coli and/or in the crystals, and Cys287 was oxidized by β‐mercaptoethanol (βME, from the buffers) to S,S‐(2‐hydroxyethyl)thiocysteine (CME). Mass spectrometric analysis of freshly purified CtGAPDH revealed the presence of several modified protein derivatives along with unmodified protein (Figure S6). The doubly modified protein containing SNC63 and CME287 represented a minor peak (~36,380 Da, indicated with * in Figure S6) while the mass for the two most abundant forms matched those of the BME oxidation product (~36,350 Da) and the unmodified protein (~36,274 Da). There were additional peaks of higher molecular masses. Similar modifications were observed in the structure determined from a second crystal (coordinates and structure factors deposited in PDB as 6X2E). Details of crystallization, data collection and refinement parameters for 6X2E are described in the Supplementary Materials and Table S2.
FIGURE 4.
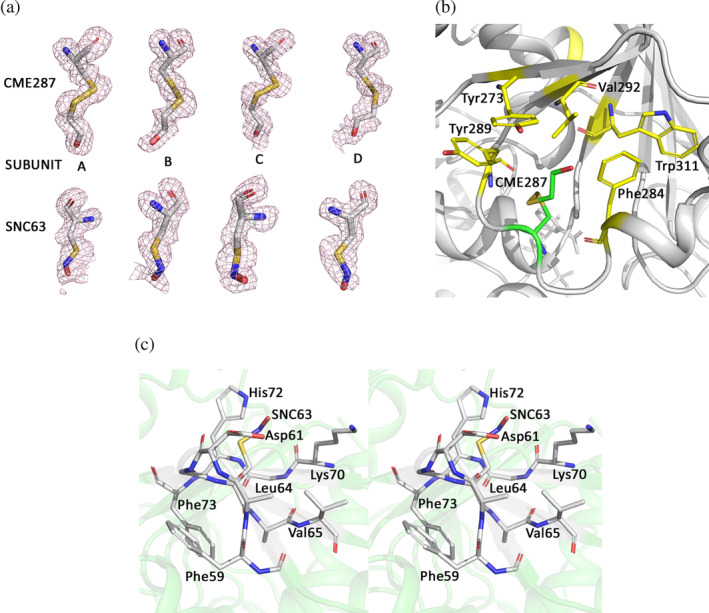
Electron density for the modified residues CME287 and SNC63, and their environment in CtGAPDH crystal structure. (a) Fo‐Fc omit maps (mesh in light pink contoured at 3σ) for CME287 (upper panel) and SNC63 (lower panel). The residues are shown in stick model (C: white, N: blue, O: red, S: yellow). (b) Aromatic and hydrophobic residues surrounding CME287 (C: green) are shown in stick model (C: yellow). (c) Stereo diagram showing aromatic and basic residues within 5 Å of SNC63 (stick model, C: white)
SNC and CME modifications have been observed in protein structures. 32 , 33 In GAPDH sequences a cysteine residue is present at position 287 only in Chlamydiae and certain coccidia (Figure S3). In CtGAPDH residue CME287 is in a hydrophobic pocket formed by Tyr273, Phe284, Tyr289, Val292 and Trp311 (Figure 4b). Notably, the electron density map for the published CtGAPDH structure (6OK4) shows additional density near the Cys287 sulfur atom (Figure S7) even though somewhat different purification and crystallization conditions were used by Barrett et al. 28
The cysteine residue in position 63 is unique in Chlamydiae GAPDH and is located on the protein surface. Regulatory functions of endogenously S‐nitrosylated cysteine residues in GAPDH have been reported. 34 While a consensus linear motif for cysteine nitrosylation has not been identified, interactions with aromatic and basic residues are suggested to render cysteine residues susceptible to S‐nitrosylation. 35 In the CtGAPDH structure, aromatic residues Phe59, Leu64, Val65 and Phe73 and basic residues Lys70 and His72 are within 5 Å of SNC63 (Figure 4c).
Chemical modifications of cysteine residues of GAPDH influence various cellular processes. 34 , 36 Although modifications in native CtGAPDH has not been reported, our results suggest the presence of one or more reactive cysteine residues in the protein.
2.8. GAPDH structures feature regions of low sequence identity
Alignment of CtGAPDH and human GAPDH (hGAPDH) sequences exhibits ~55% identity and 69% similarity. In two areas within the cofactor‐binding domain (Region 1: residues 18–41 and Region 2: 52–91), and in one area (Region 3: 248–273) in the catalytic domain, CtGAPDH and hGAPDH are only 21–31% identical (Figure 5a). Among the residues that directly interact with NAD+, Lys77 and Thr119 are replaced by an arginine and a serine residue, respectively, in hGAPDH. Differences are also apparent near the adenine‐binding pocket (within Region 1). In this area, the hGAPDH sequence has one amino acid insertion (Pro36), and Phe37 in hGAPDH replaces Leu34 (Figure 5b). Similarly, despite the high overall sequence identity between CtGAPDH and GBSGAPDH (~45%) in Region 2 they share only ~18% identity (Figure 5c). Pairwise sequence alignment using Needle/EMBOSS (https://www.ebi.ac.uk/Tools/psa/) reveals that compared to GBSGAPDH there is an insertion (270Asn‐Ile‐Met‐Tyr273) in Region 3 of CtGAPDH. Moreover, the characteristic extension of a two‐stranded β‐sheet exposed on some bacterial GAPDHs is absent from the CtGAPDH structure. 37 , 38 The regions of low sequence identity are mainly distributed on the GAPDH protein surface and may be involved in specific intermolecular interactions.
FIGURE 5.
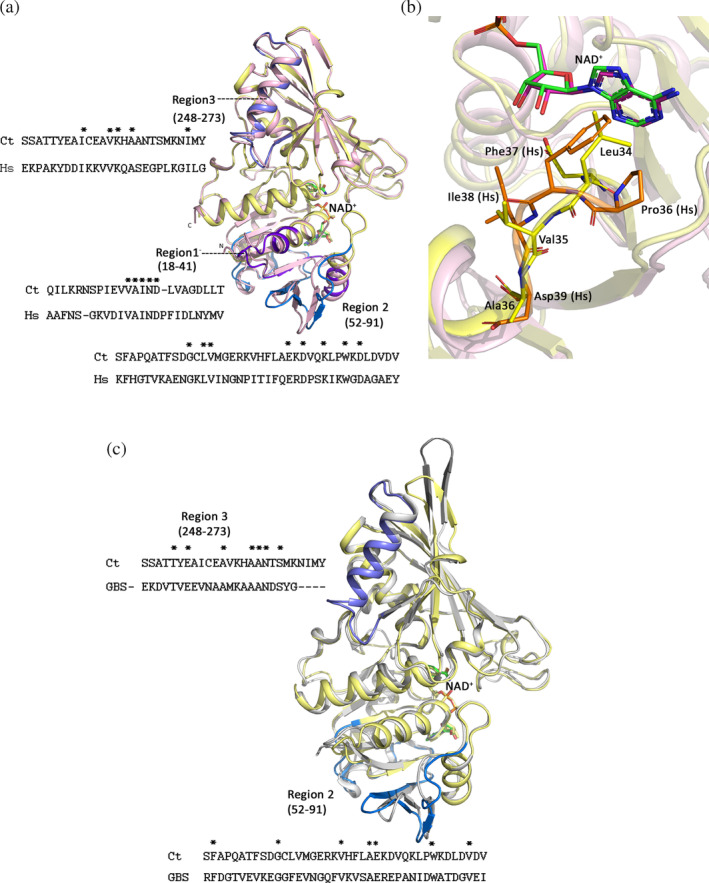
Comparison of different areas where CtGAPDH sequence diverges from human and GBS GAPDH. (a) Cartoon drawing showing superposition of subunit A of CtGAPDH (light yellow) and hGAPDH (1U8F subunit O, light pink). Regions where the sequence identities are low are marked and shown in purple blue, slate and marine color. Comparison of these regions in the sequences for Ct and human (Hs) GAPDHs are shown. NAD+ is shown in stick model (C: green). (b) Close‐up view of the adenine binding pocket. Residues in hGAPDH (C: orange), CtGAPDH (C: yellow) and NAD+ (C: magenta and green in hGAPDH and CtGAPDH respectively) are shown in stick models. (c) Superposition of subunit A of CtGAPDH (light yellow) and GBSGAPDH (5JYA subunit A, grey). Regions 2 and 3 where the sequence identities are low are marked and shown in slate and marine color. Comparison of these regions in the sequences for CtGAPDH and GBSGAPDH are shown. NAD+ is shown in stick model (C: green)
2.9. CtGAPDH binds to plasminogen and plasmin
Some bacterial GAPDHs are known to bind host extracellular matrix proteins but such interactions have not been reported for CtGAPDH. We examined the binding of CtGAPDH with hPlg and hPln using biolayer interferometry (BLI). Our results show that CtGAPDH binds to both proteins with similar affinity (Figure S8). The values for the dissociation constant (K D) measured in our experiments were 0.33 μM (R 2 = 0.988; X 2 = 1.388) for hPlg and 0.14 μM (R 2 = 0.9988; X 2 = 1.505) for hPln, respectively. The association and dissociation rate constants for hPlg interactions were 3.25 x 105 (0.28 x 105) M−1 s−1 and 1.083 x 10−1 (0.036 x 10−1) s−1, respectively. The association and dissociation rate constants for hPln interactions were 3.673 x 103 (0.056 x 103) M−1 s−1 and 5.294 x 10−4 (0.277 x 10−4) s−1, respectively.
The Plg binding sites on different GAPDHs are not well characterized. Plg contains five “Kringle” domains (K1, K2, K3, K4, and K5) that are lysine‐binding modules known to be important for receptor binding. 39 Moreau et al. identified three lysine residues (Lys304, Lys115, and Lys116) of S. pneumoniae GAPDH (SpGAPDH) critical for binding to hPlg. 38 Among these, Lys304 is located at the edge of the extended β‐sheet structure seen in some bacterial GAPDHs. The structurally equivalent residue in CtGAPDH is Asp303. SpGAPDH residues Lys115 and Lys116 are replaced by Lys114 and Arg115 in CtGAPDH, which are located on the protein surface. CtGAPDH has 76 lysine residues (19 per subunit) exposed on the tetramer surface.
To predict the possible binding modes, we performed protein–protein docking using two different programs, ClusPro 40 and Gramm‐X 41 with CtGAPDH tetramer (6WYC) as receptor and hPlg monomer (4DUR) 42 as ligand. From the top 10 docking models predicted by each program we selected a total of three based on (a) stable docked assembly (positive ΔG diss value calculated by PISA 43 ) and (b) utilization of Kringle domain for binding: model 1 (GrammX), and models 2 and 3 (ClusPro) with ΔG diss values of 9.3, 7.2, and 6.2 kcal/mol, respectively (Table S3). In models 1 and 2, hPlg domains K3 (residues 255–340) and K4 (355–440) dock onto adjacent subunits of the CtGAPDH tetramer. In model 1, the K3 domain interacts with three CtGAPDH subunits. The hPlg K3 inserts into the inter‐subunit space near Lys192 of subunit C (Figure S9). The K4 docking surface shows interactions with Lys225 (subunit C), which forms a salt bridge/hydrogen bond with Glu395 of hPlg. In addition, Lys223 of subunit C is also located near the K4 domain. In model 2, the K4 of hPlg docks on to a similar surface on CtGAPDH as in model 1 but the K3 domain does not show interaction with any lysine residue (Figure S10). In model 3 all hPlg residues except Arg312 (K3) that are involved in CtGAPDH binding are located outside any Kringle domain. In this pose, Lys85 and Lys114 are ~7–8 Å away from the nearest residue in the K3 domain. However, Plg also binds to arginine residues. 44 In all three binding poses discussed above the CtGAPDH active site remains accessible. Hydrogen bonds and salt bridges between the receptor and ligand in the three models are listed in Table S3.
3. CONCLUSIONS
The presence of GAPDH in both life stages of Ct is intriguing considering the versatile functions of GAPDHs. To establish and survive its intracellular life Ct interacts with many host molecules. It is not known if CtGAPDH is present on the bacterial surface. However, its ability to bind to hPlg and hPln suggests that if attached to the outer surface CtGAPDH can serve as a ligand for interaction with host extracellular matrix proteins. High‐resolution crystal structure reveals structural differences in the active sites of CtGAPDH and hGAPDH, and shows areas of low sequence identity on the tetramer surface. These regions may be involved in specific functions. Although GAPDH is an evolutionarily conserved protein with considerable homology across the species, antibodies raised against bacterial GAPDHs can be highly specific. 45 Our data indicate the presence of reactive cysteine residues that may modulate non‐enzymatic cellular functions of CtGAPDH.
4. MATERIALS AND METHODS
4.1. Expression and purification
Ct gapdh sequence was cloned into Escherichia coli expression vector pJ411 (DNA2.0). CtGAPDH was expressed in E. coli BL21(DE3) cells in LB medium containing 100 μg/ml ampicillin. At mid‐log phase recombinant protein expression was induced by adding 0.4 mM isopropyl‐thio‐β‐D‐galactoside and the culture was grown ~20 hrs at 22°C. All subsequent steps were performed at 4°C. CtGAPDH was isolated from the bacterial extract using ammonium sulfate precipitation. The majority of CtGAPDH was obtained in the pellet at 50–60% ammonium sulfate saturation. The pellet was suspended in a minimum volume of 20 mM HEPES, pH 7.5, 0.1 mM NaCl, 5 mM βME and fractionated on a Superdex 200 column (GE Healthcare) in the same buffer. Pooled fractions containing tetrameric CtGAPDH were further purified on a DEAE Sephacel column (GE Healthcare) and eluted between 250–300 mM NaCl concentration in elution buffer (20 mM Tris HCl, pH 8.0, 5 mM βME).
4.2. Enzyme activity and kinetics
The optimum pH for CtGAPDH activity was determined by measuring initial reaction velocities in the pH range of 5–11 in a reaction buffer (50 mM triethanolamine, 50 mM sodium bisphosphate, and 0.2 mM EDTA) using 0.5 μg/ml CtGAPDH, 0.6 mM NAD+ and 2 mM D‐G3P. For each reaction the pH was adjusted after the addition of D‐G3P and NAD+ prior to the addition of enzyme. Kinetic experiments were performed in a total volume of 1 ml with either saturating D‐G3P (3 mM) and varying NAD+ (0.025–0.8 mM) or constant NAD+ (0.6 mM) and varying D‐G3P (0.25–6 mM) in the reaction buffer (pH 9.0). Reactions (in triplicate) were initiated with the addition of 0.5 μg CtGAPDH. Reaction velocities were measured by monitoring the increase in absorbance (340 nM) for 1.3 min in a 1 cm path‐length quartz cuvette in an HP 8542A Diode Array UV–Vis spectrophotometer. The K M and V max parameters were determined by non‐linear least square fitting of the (v o) versus (S) curve using GraphPad Prism (v7.0).
4.3. Crystallization and data collection
CtGAPDH (30 mg/ml) was incubated at 4°C with 1 mM NAD+ for 30 min and used for crystallization trials at 21°C using commercial screens. Crystals were obtained in PEG Suite I (Qiagen) condition #24 (25% PEG 2000 MME, 0.1 M Tris, pH 8.5) and flash frozen in the reservoir solution supplemented with 20% glycerol. X‐ray diffraction data were collected on a Pilatus 6 M hybrid pixel detector at the NE‐CAT 24‐ID‐C beamline (Advanced Photon Source). Intensity data were integrated, merged and scaled with XDS 46 followed by Aimless in CCP4. 47 Data collection parameters are listed in Table 2.
4.4. Crystal structure determination and refinement
The structure of CtGAPDH was solved by molecular replacement using Phaser 48 with a monomer of the GBSGAPDH (4QX6) as search model. 37 After refinement of the protein chains, one NAD+ molecule was unambiguously placed in each subunit. Refinement was carried out by Refmac 49 and Phenix, 50 and Coot 51 was used for model building. Figures were created with PyMOL (Version 2.2.0; Schrödinger LLC). Refinement parameters are listed in Table 2. Clashscore and Molprobity score reported in Table 2 were calculated by using Molprobity (4.2). 52 The coordinates and structure factors have been deposited in the Protein Data Bank under the code 6WYC.
4.5. Whole protein mass spectrometry
Purified CtGAPDH was diluted to a final concentration of 0.3 μM in water with 0.1% formic acid (v/v). The sample was chromatographically separated over a Phenomenex SecurityGuard Ultra C4 trap utilizing a gradient composed of SA (water with 0.1% [vol/vol] formic acid) and SB (acetonitrile with 0.1% [vol/vol] formic acid) on a Shimadzu SPD‐20 series pump system. Detection was carried out on a Waters G2‐Si Synapt mass spectrometer. Analysis was performed using the MaxEnt function in Waters MassLynx software.
4.6. Protein–protein interaction: BLI
The BLItz BLI instrument (Pall ForteBio) was used to measure the binding affinities between CtGAPDH and hPln or hPlg. The High Precision Streptavidin (SAX) biosensors were used to immobilize biotin‐labeled hPln (Sigma, P1867) and hPlg (Sigma, P1517) via interaction with the Streptavidin. The amino groups of hPln and hPlg were labeled with biotin using EZ‐Link NHS‐PEG4‐Biotin (Thermo Fisher Scientific) according to manufacturer's instructions. Titrations were performed using varying concentrations of CtGAPDH in the range of 0.15–0.57 μM for hPlg binding and 1.5–3.7 μM for hPln binding. The running buffer consisted of 1X PBS and 1X Kinetic buffer (Pall ForteBio). The composition of the Kinetic buffer was as follows: 10 mM phosphate, 150 mM NaCl, 0.02% Tween 20, 0.05% sodium azide, and 1 mg/ml BSA, pH 7.4.
AUTHOR CONTRIBUTIONS
Norbert Schormann: Formal analysis; investigation; methodology; validation; writing; review and editing. Juan Campos: Investigation; writing; review and editing. Rachael Motamed: Investigation; writing; review and editing. Katherine Hayden: Investigation; methodology; validation; writing; review and editing. Joseph Gould: Investigation; methodology; writing; review and editing. Olga Senkovich: Investigation; methodology; writing; review and editing. Todd Green: Investigation; writing; review and editing. Surajit Banerjee: Investigation; methodology; writing; review and editing. Glen Ulett: Investigation; writing; review and editing. Debasish Chattopadhyay: Conceptualization; investigation; writing; reviewing and editing.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest with the content of this article.
Supporting information
Data S1: Supporting Information
ACKNOWLEDGMENTS
We thank Dr. Peter Edward Prevelige, Jr. (UAB Microbiology Department) for assistance with mass spectroscopy data collection. This work was supported in part with funding from a Project Grant from the National Health and Medical Research Council (NHMRC) Australia (APP1129928) to G. C. U. and D. C. NE‐CAT beamlines and the detector are supported by National Institutes of Health Grant Number: (P30 GM124165) and RR029205 at the APS (DE‐AC02‐06CH11357).
Schormann N, Campos J, Motamed R, et al. Chlamydia trachomatis glyceraldehyde 3‐phosphate dehydrogenase: Enzyme kinetics, high‐resolution crystal structure, and plasminogen binding. Protein Science. 2020;29:2446–2458. 10.1002/pro.3975
Funding information National Institutes of Health, Grant/Award Numbers: DE‐AC02‐06CH11357, P30 GM124165; National Health and Medical Research Council, Grant/Award Number: APP1129928
REFERENCES
- 1. Skipp PJ, Hughes C, McKenna T, et al. Quantitative proteomics of the infectious and replicative forms of Chlamydia trachomatis. PLoS One. 2016;11:e0149011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dolat L, Valdivia RH. A renewed tool kit to explore chlamydia pathogenesis: From molecular genetics to new infection models. F1000Res. 2019;8:F1000 Faculty Rev‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Price MJ, Ades AE, De Angelis D, et al. Risk of pelvic inflammatory disease following chlamydia trachomatis infection: Analysis of prospective studies with a multistate model. Am J Epidemiol. 2013;178:484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kong FY, Hocking JS. Treatment challenges for urogenital and anorectal chlamydia trachomatis. BMC Infect Dis. 2015;15:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Phillips S, Quigley BL, Timms P. Seventy years of chlamydia vaccine research ‐ limitations of the past and directions for the future. Front Microbiol. 2019;10:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iliffe‐Lee ER, McClarty G. Glucose metabolism in chlamydia trachomatis: The 'energy parasite' hypothesis revisited. Mol Microbiol. 1999;33:177–187. [DOI] [PubMed] [Google Scholar]
- 7. Aronov AM, Verlinde CL, Hol WG, Gelb MH. Selective tight binding inhibitors of trypanosomal glyceraldehyde‐3‐phosphate dehydrogenase via structure‐based drug design. J Med Chem. 1998;41:4790–4799. [DOI] [PubMed] [Google Scholar]
- 8. Chauhan AS, Kumar M, Chaudhary S, et al. Moonlighting glycolytic protein glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH): An evolutionarily conserved plasminogen receptor on mammalian cells. FASEB J. 2017;31:2638–2648. [DOI] [PubMed] [Google Scholar]
- 9. Pancholi V, Fischetti VA. A major surface protein on group a streptococci is a glyceraldehyde‐3‐phosphate‐dehydrogenase with multiple binding activity. J Exp Med. 1992;176:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crowe JD, Sievwright IK, Auld GC, Moore NR, Gow NA, Booth NA. Candida albicans binds human plasminogen: Identification of eight plasminogen‐binding proteins. Mol Microbiol. 2003;47:1637–1651. [DOI] [PubMed] [Google Scholar]
- 11. Seifert KN, McArthur WP, Bleiweis AS, Brady LJ. Characterization of group B streptococcal glyceraldehyde‐3‐phosphate dehydrogenase: Surface localization, enzymatic activity, and protein‐protein interactions. Can J Microbiol. 2003;49:350–356. [DOI] [PubMed] [Google Scholar]
- 12. Madureira P, Baptista M, Vieira M, et al. Streptococcus agalactiae GAPDH is a virulence‐associated immunomodulatory protein. J Immunol. 2007;178:1379–1387. [DOI] [PubMed] [Google Scholar]
- 13. Matta SK, Agarwal S, Bhatnagar R. Surface localized and extracellular Glyceraldehyde‐3‐phosphate dehydrogenase of Bacillus anthracis is a plasminogen binding protein. Biochim Biophys Acta. 2010;1804:2111–2120. [DOI] [PubMed] [Google Scholar]
- 14. Magalhaes V, Andrade EB, Alves J, et al. Group B Streptococcus hijacks the host plasminogen system to promote brain endothelial cell invasion. PLoS One. 2013;8:e63244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Terao Y, Yamaguchi M, Hamada S, Kawabata S. Multifunctional glyceraldehyde‐3‐phosphate dehydrogenase of Streptococcus pyogenes is essential for evasion from neutrophils. J Biol Chem. 2006;281:14215–14223. [DOI] [PubMed] [Google Scholar]
- 16. Terrasse R, Tacnet‐Delorme P, Moriscot C, et al. Human and pneumococcal cell surface glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) proteins are both ligands of human C1q protein. J Biol Chem. 2012;287:42620–42633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lama A, Kucknoor A, Mundodi V, Alderete JF. Glyceraldehyde‐3‐phosphate dehydrogenase is a surface‐associated, fibronectin‐binding protein of Trichomonas vaginalis. Infect Immun. 2009;77:2703–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bergmann S, Rohde M, Hammerschmidt S. Glyceraldehyde‐3‐phosphate dehydrogenase of Streptococcus pneumoniae is a surface‐displayed plasminogen‐binding protein. Infect Immun. 2004;72:2416–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jung DW, Kim WH, Seo S, et al. Chemical targeting of GAPDH moonlighting function in cancer cells reveals its role in tubulin regulation. Chem Biol. 2014;21:1533–1545. [DOI] [PubMed] [Google Scholar]
- 20. Alves J, Madureira P, Baltazar MT, et al. A safe and stable neonatal vaccine targeting GAPDH confers protection against group B Streptococcus infections in adult susceptible mice. PLoS One. 2015;10:e0144196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karunakaran KP, Rey‐Ladino J, Stoynov N, et al. Immunoproteomic discovery of novel T cell antigens from the obligate intracellular pathogen chlamydia. J Immunol. 2008;180:2459–2465. [DOI] [PubMed] [Google Scholar]
- 22. Lambeir AM, Loiseau AM, Kuntz DA, Vellieux FM, Michels PA, Opperdoes FR. The cytosolic and glycosomal glyceraldehyde‐3‐phosphate dehydrogenase from Trypanosoma brucei. Kinetic properties and comparison with homologous enzymes. Eur J Biochem. 1991;198:429–435. [DOI] [PubMed] [Google Scholar]
- 23. Penkler G, du Toit F, Adams W, et al. Construction and validation of a detailed kinetic model of glycolysis in Plasmodium falciparum . FEBS J. 2015;282:1481–1511. [DOI] [PubMed] [Google Scholar]
- 24. Cook WJ, Senkovich O, Chattopadhyay D. An unexpected phosphate binding site in glyceraldehyde 3‐phosphate dehydrogenase: Crystal structures of apo, holo and ternary complex of Cryptosporidium parvum enzyme. BMC Struct Biol. 2009;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmalhausen EV, Shumkov MS, Muronetz VI, Svedas VK. Expression of glyceraldehyde‐3‐phosphate dehydrogenase from M. tuberculosis in E. coli. Purification and characteristics of the untagged recombinant enzyme. Protein Expr Purif. 2019;157:28–35. [DOI] [PubMed] [Google Scholar]
- 26. Michels S, Rogalska E, Branlant G. Phosphate‐binding sites in phosphorylating glyceraldehyde‐3‐phosphate dehydrogenase from Bacillus stearothermophilus . Eur J Biochem. 1996;235:641–647. [DOI] [PubMed] [Google Scholar]
- 27. Kim H, Feil IK, Verlinde CL, Petra PH, Hol WG. Crystal structure of glycosomal glyceraldehyde‐3‐phosphate dehydrogenase from Leishmania mexicana: Implications for structure‐based drug design and a new position for the inorganic phosphate binding site. Biochemistry. 1995;34:14975–14986. [DOI] [PubMed] [Google Scholar]
- 28. Barrett KF, Dranow DM, Phan IQ, et al. Structures of glyceraldehyde 3‐phosphate dehydrogenase in Neisseria gonorrhoeae and chlamydia trachomatis. Protein Sci. 2020;29:768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rao ST, Rossmann MG. Comparison of super‐secondary structures in proteins. J Mol Biol. 1973;76:241–256. [DOI] [PubMed] [Google Scholar]
- 30. Jenkins JL, Tanner JJ. High‐resolution structure of human D‐glyceraldehyde‐3‐phosphate dehydrogenase. Acta Cryst. 2006;D62:290–301. [DOI] [PubMed] [Google Scholar]
- 31. Markos A, Miretsky A, Muller M. A glyceraldehyde‐3‐phosphate dehydrogenase with eubacterial features in the amitochondriate eukaryote, Trichomonas vaginalis. J Mol Evol. 1993;37:631–643. [DOI] [PubMed] [Google Scholar]
- 32. Chen YY, Chu HM, Pan KT, et al. Cysteine S‐nitrosylation protects protein‐tyrosine phosphatase 1B against oxidation‐induced permanent inactivation. J Biol Chem. 2008;283:35265–35272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lau K, Podolec R, Chappuis R, Ulm R, Hothorn M. Plant photoreceptors and their signaling components compete for COP1 binding via VP peptide motifs. EMBO J. 2019;38:e102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hara MR, Cascio MB, Sawa A. GAPDH as a sensor of NO stress. Biochim Biophys Acta. 2006;1762:502–509. [DOI] [PubMed] [Google Scholar]
- 35. Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S‐nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci USA. 2006;103:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hildebrandt T, Knuesting J, Berndt C, Morgan B, Scheibe R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol Chem. 2015;396:523–537. [DOI] [PubMed] [Google Scholar]
- 37. Schormann N, Ayres CA, Fry A, et al. Crystal structures of group B Streptococcus glyceraldehyde‐3‐phosphate dehydrogenase: Apo‐form, binary and ternary complexes. PLoS One. 2016;11:e0165917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moreau C, Terrasse R, Thielens NM, Vernet T, Gaboriaud C, Di Guilmi AM. Deciphering key residues involved in the virulence‐promoting interactions between Streptococcus pneumoniae and human plasminogen. J Biol Chem. 2017;292:2217–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miles LA, Lighvani S, Baik N, et al. New insights into the role of Plg‐RKT in macrophage recruitment. Int Rev Cell Mol Biol. 2014;309:259–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein‐protein docking. Nat Protoc. 2017;12:255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tovchigrechko A, Vakser IA. GRAMM‐X public web server for protein‐protein docking. Nucleic Acids Res. 2006;34:W310–W314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Law RH, Caradoc‐Davies T, Cowieson N, et al. The X‐ray crystal structure of full‐length human plasminogen. Cell Rep. 2012;1:185–190. [DOI] [PubMed] [Google Scholar]
- 43. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- 44. Sanderson‐Smith ML, Dowton M, Ranson M, Walker MJ. The plasminogen‐binding group a streptococcal M protein‐related protein Prp binds plasminogen via arginine and histidine residues. J Bacteriol. 2007;189:1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Madureira P, Andrade EB, Gama B, et al. Inhibition of IL‐10 production by maternal antibodies against group B Streptococcus GAPDH confers immunity to offspring by favoring neutrophil recruitment. PLoS Pathog. 2011;7:e1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kabsch W. Xds. Acta Cryst. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Cryst. 2011;D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum‐likelihood method. Acta Cryst. 1997;D53:240–255. [DOI] [PubMed] [Google Scholar]
- 50. Afonine PV, Grosse‐Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.Refine. Acta Cryst. 2012;D68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Cryst. 2010;D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen VB, Arendall WB 3rd, Headd JJ, et al. MolProbity: All‐atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information