

Abstract
Corticosteroid‐binding globulin (CBG) transports cortisol and other steroids. High‐affinity CBG (haCBG) undergoes proteolysis of the reactive center loop (RCL) by neutrophil elastase (NE) altering conformation to low‐affinity CBG (laCBG). Elevated temperature reduces CBG:cortisol binding affinity. Surface plasmon resonance was used to determine binding profiles of 19 steroids to haCBG and laCBG at 25, 37, and 39°C mimicking pyrexia and pH 7.4 and 7.0 mimicking acidosis, pathophysiological conditions relevant to sepsis. An expected 4–8‐fold reduction in affinity for cortisol, cortisone, corticosterone, 11‐deoxycortisol, progesterone, 17‐hydroxyprogesterone, and prednisolone occurred with NE‐mediated haCBG‐to‐laCBG conversion. CBG:cortisol binding affinity was further reduced 3.5‐fold at 39°C relative to 37°C, binding affinity was also reduced by acidosis for both haCBG and laCBG. Using a conformational antibody generated against the RCL, we confirmed RCL antibody binding was eliminated by NE cleavage, but preserved in pyrexia and acidosis. Molecular modeling studies performed at 40°C confirmed a critical role for Trp371, positioned within the steroid‐binding pocket, in ligand binding. These studies demonstrated CBG binding affinity to range of steroids is ligand specific and is reduced with NE‐mediated haCBG‐to‐laCBG transition. Reduced CBG:cortisol binding occurs with increased temperature and in acidosis. Increased flexibility of the Trp371 side chain is proposed in the thermo‐coupling mechanism of cortisol release. The synergy of NE cleavage, pyrexia, and acidosis on CBG:cortisol binding may serve to enhance cortisol delivery to the interstitial space in inflammation.
Keywords: acidemia, corticosteroid‐binding globulin, cortisol, ligand binding affinity, pyrexia
1. INTRODUCTION
Corticosteroid‐binding globulin (CBG), a serine protease inhibitor (serpin) family member, 1 is the principal transporter of circulating cortisol. Approximately 80% of blood cortisol is transported by CBG under basal conditions, while the remaining ~15% is bound to albumin and ~ 5% circulates as free hormone. CBG, a 383 amino acid glycoprotein, transports cortisol in a 1:1 stoichiometric ratio increasing the solubility of cortisol in serum, buffering fluctuating cortisol levels and, critically, allowing tissue‐specific delivery. 2 , 3 One well characterized mechanism that facilitates the delivery of cortisol at target sites involves site‐specific proteolysis of CBG by neutrophil elastase (NE). Infection and inflammation generates activated neutrophils that secrete the NE 4 , 5 that cleaves an exposed reactive center loop (RCL) of intact, high affinity CBG (haCBG). The liberated N‐terminal fragment of the RCL then fully and irreversibly inserts itself into the central β‐sheet of CBG as an additional β‐strand. 6 , 7 Subtle conformational changes also accompany this large rearrangement, including unwinding of a connecting loop between α‐helix D and β‐strand 2 in the central β‐sheet, which allosterically alters the structure of ligand pocket. Together these structural rearrangements generate low affinity CBG (laCBG), which has nine‐fold reduced affinity for cortisol. 4 Structural studies have also revealed the plasticity of the ligand binding pocket governs CBG function. The binding pocket itself is dominated by the indole side chain of Trp371 which is essential for a stacking interaction with the hydrophobic rings present in the chemical structure of cortisol. 6 , 8 This side chain is mobile and can rotate to accommodate cortisol with optimal geometry observed when the indole moiety is in the same plane as cortisol. 6 CBG also binds other endogenous steroids of similar chemical structures including progesterone, 17‐hydroxyprogesterone and cortisone, albeit with lower affinity, and is likely responsible for their transport and biodelivery to target tissues. 9 , 10 , 11 Thus NE‐mediated cleavage of CBG leads to a three‐ to four‐fold increase in interstitial fluid free cortisol concentrations 2 , 7 , 12 and represents a prereceptor mechanism for tissue physiologic‐condition dependent steroid delivery, relevant to inflammation. Systemically, we have shown that falling serum concentrations of haCBG correlate to increasing illness severity in sepsis/septic shock and with the risk of mortality due to dysregulated delivery of sterols. 13 , 14
In addition to this NE‐mediated delivery mechanism cortisol is liberated from CBG by increased temperature and possibly also by acidemia, conditions typical of sepsis. 7 , 15 Hydrocortisone binding studies using glycosylated human CBG pooled from plasma revealed that the equilibrium binding constants (K D) increased four‐fold when the temperature was elevated from 37 to 42°C. 7 , 16 CBG:hydrocortisone binding affinity was not affected by pH but technical limitations were reported by the authors. 15 A molecular mechanism to explain the thermal effects on CBG:cortisol binding has been proposed based upon studies performed upon other serpin proteins. The uncleaved RCLs of structurally analogous antithrombin and thyroxine binding globulin partially and reversibly insert into the central β‐sheet, inducing allosteric conformational changes that impact the ligand binding pocket. 6 , 17 This subtle “nudging” conformational change is sensitive to temperature, thereby providing the proteins with a thermocouple mechanism. 17 CBG is proposed to function in a similar manner, although direct evidence for the reversible insertion of the uncleaved RCL has not yet been verified by structural biology. 6 , 7 , 18 Thus, CBG represents a complex biodelivery system for maintaining circulating cortisol levels and mediating tissue specific delivery of steroid hormones.
In this study, surface plasmon resonance (SPR) technology was employed to determine the equilibrium binding constants of a library of steroids to haCBG and laCBG. Purified recombinant human CBG, shown to possess many of the same glycosylation modifications as native protein was utilized. CBG:steroid binding analyses were performed under physiological and pyrexial temperature and acidosis range pH conditions. The immunoreactivity of the RCL on the protein surface was also assessed by SPR using a conformational antibody generated against the RCL epitope. This is the first report to directly probe the proposed “in‐and‐out” submolecular switch movement of the RCL of CBG under various conditions. Finally, in silico molecular dynamic (MD) simulations were performed to delineate the effect of temperature on CBG structure and propose an additional mechanism of altered steroid binding. The in silico findings reinforce a critical role of Trp371 in the thermo‐coupled release of steroid from CBG.
2. MATERIALS
Glycosylated recombinant human CBG, expressed in HEK293 cells and tagged with biotin was purchased from Sino Biological Inc. (Beijing, China, Cat no. 10998‐H27H‐B). Human CBG (UniProtKB, P08185) purified from pooled sera of donors to greater than 90% purity, as assessed by SDS‐PAGE was purchased from Affiland, Belgium. Pure steroid hormones at analytical grade or higher were purchased from Sigma Aldrich, with the exception of allopregnanolone (Med Chem Express, NJ).
3. METHODS
3.1. SDS‐PAGE and western blot analysis of biotinylated recombinant human CBG
Gel electrophoresis of CBG was carried out using a 4–12% acrylamide gel. Following transfer to a nitrocellulose membrane, the membrane was blocked using 5% (w/v) skim milk powder in PBS‐Tween for 48 hr. The membrane was then incubated with either 9G12 (RRID:AB_26632405) (1:50 dilution) or CBG polyclonal antibody (1:1,000 dilution) for 2 hr at room temperature, followed by anti‐mouse IgG2a‐HRP (1:500 dilution) then anti‐rabbit IgG‐HRP (1:2,500 dilution). All dilutions were made in 5% (w/v) skim milk powder PBS‐Tween. Immunoconjugates were developed with BioRad Clarity ECL, containing a mixture of luminal and peroxide, and imaged using a ChemiDoc® Imager (Bio‐Rad Laboratories, CA). A duplicate membrane was also prepared and probed with a Streptavidin‐Alexia Fluor 488 conjugate (Thermo Fisher Scientific, Cat no. S11223) (1:100 dilution) to detect biotinylated protein. After incubating with streptavidin for 1 hr at room temperature, the membrane was washed three times for 5 min in PBS‐Tween and then visualized using a ChemiDoc® Imager.
3.2. N‐glycan release and handling
The cysteine residues of human CBG isolated from donor blood (hereafter “Native CBG”) and biotinylated recombinant human CBG (hereafter “Biotin‐CBG”) were reduced using 10 mM aqueous dithiothreitol (DTT) (Sigma‐Aldrich, MO), 45 min at 56°C and carbamidomethylated using 40 mM aqueous iodoacetamide (Sigma‐Aldrich, MO), 30 min in the dark at 20°C (all stated as final concentrations). Native and Biotin‐CBG were each immobilized in three discrete spots (5 μg/spot) on a primed 0.45 μm polyvinylidene fluoride (PVDF) membrane (Merck‐Millipore, MA) and processed essentially as described previously. 19 In brief, the dried protein spots were stained with Direct Blue (Sigma‐Aldrich, MO), excised, transferred to separate wells in a flat bottom polypropylene 96‐well plate (Corning Life Sciences, NY), blocked with 1% (w/v) polyvinylpyrrolidone in 50% (v/v) aqueous methanol and washed with water. The N‐glycans were exhaustively released from the immobilised proteins using 10 U peptide: N‐glycosidase F (PNGase F) (Elizabethkingia miricola, Promega, WI) in 10 μl water/well, 16 hr at 37°C. The released N‐glycans were transferred into fresh tubes and deaminated by the addition of 10 μl 100 mM aqueous ammonium acetate, pH 5, 1 hr at 20°C. The N‐glycans were then dried and subsequently reduced to alditols using 20 μl 1 M sodium borohydride in 50 mM aqueous potassium hydroxide, 3 hr at 50°C. The reactions were quenched by the addition of 2 μl glacial acetic acid to each sample. Dual desalting of the reduced N‐glycans was performed using a strong cation exchange column (AG 50 W X8, Bio‐Rad, Hercules, CA) and then porous graphitised carbon (PGC) packed as micro‐columns on top of C18 discs in P10 solid‐phase extraction (SPE) formats. The N‐glycans were eluted from the PGC micro‐columns using 0.05% trifluoroacetic acid:40% acetonitrile (ACN):59.95% water (v/v/v), dried and redissolved in 10 μl water for LC–MS/MS‐based N‐glycan profiling.
3.3. N‐glycan profiling
The liberated N‐glycans were profiled using LC–MS/MS performed on LTQ Velos Pro ion trap mass spectrometer (Thermo Scientific, CA) coupled to a Dionex Ultimate‐3000 HPLC (Dionex, CA). The N‐glycans were separated on a PGC LC capillary column (Hypercarb KAPPA, 5 μm particle size, 250 Å pore size, 0.18 × 100 mm, Thermo Fisher Scientific, CA) using a linearly increasing concentration of solvent B (2.6–64% (v/v) 10 mM NH4HCO3 in ACN over 86 min) in solvent A (10 mM aqueous NH4HCO3) at a constant flow rate of 3 μl/min. The setup employed a postcolumn make‐up flow configuration by supplying 100% isopropanol at a constant 4 μl/min flow rate throughout the LC–MS/MS run for enhanced sensitivity. 20 The acquisition MS1 scan range was set to m/z 570–2,000 with a zoom scan resolution of m/z 0.25 at full width at half maximum (FWHM) and a source voltage of +2.7 kV. Detection was performed in negative ion polarity mode with data‐dependent MS/MS acquisition. The automatic gain control (AGC) for the MS1 scans was set to 5 × 104 with a maximum accumulation time of 50 ms. For the MS/MS events, the resolution was set to m/z 0.35 FWHM, the AGC was 2 × 104 and the maximum accumulation time was 300 ms. The top‐five most abundant precursors in each MS1 scan were selected for MS/MS using resonance‐activation (ion trap) based collision‐induced dissociation (CID) at a fixed 35% normalized collision energy (NCE). Dynamic exclusion was disabled. The raw LC–MS/MS data was browsed and interrogated using Xcalibur v2.2 (Thermo Fisher Scientific, CA). The glycan structures were characterized using relative and absolute PGC‐LC retention time, monoisotopic mass, and CID‐MS/MS fragmentation pattern as previously described. 21
3.4. Preparation of sensor chips for SPR assays
SPR binding assays were conducted on a Biacore S200 (GE Healthcare, Uppsala, Sweden) using SPR running buffer containing 10 mM HEPES pH 7.4, 150 mM NaCl and 0.05% (w/v) Tween 20. Streptavidin‐coated CM5 sensor chips were prepared as previously described. 22 Briefly, all four flow cells (hereafter called Flow cell 1–4) of a CM5 sensor chip (GE Healthcare) were activated using NHS‐EDC chemistry (GE Healthcare) for 900 s at a constant 10 μl/min flow rate. Flow cell 1, which served as a no‐protein control, was then treated with blocking buffer (50 mM Tris pH 8.0) for 420 s at 10 μl/min to block unreacted sites. Streptavidin (100 μg/ml, pH 4.5 in sodium acetate) was immobilized onto the three remaining flow cells by injecting the protein solution at 2 μl/min until ~10,000 resonance units (RU) had been deposited. The activated surface was then washed with blocking buffer to any remove uncoupled streptavidin. Flow cell 2 remained a streptavidin only control and was not treated with biotinylated‐CBG. Biotinylated‐CBG (25 μg/ml in SPR running buffer) was then injected across Flow cells 3 and 4 until ~6,000 RU was captured. Human NE (Sigma Aldrich, Cat no. E8140) was reconstituted to 19.5 U/ml in a 50% glycerol solution (Sigma Aldrich Cat no. 56–81‐5) then further diluted in SPR running buffer to a final concentration of 0.5 U/ml. NE was applied only to Flow cell 4, containing immobilized CBG, for 1,800 s at 25°C, at a flow rate of 10 μl/min to digest CBG in situ and generate laCBG, followed by equilibration in SPR running buffer for 900 s at 10 μl/min.
3.5. Ligand binding assays using SPR
A library of 19 endogenous and exogenous steroid hormones was selected for binding analysis based on their known or predicted binding to CBG. 9 , 10 , 11 All steroid hormones were dissolved in dimethyl sulfoxide (DMSO), and diluted in SPR running buffer to a final concentration of 5% (v/v) DMSO. For each CBG‐ligand interaction, a serial dilution of analyte covering a concentration range spanning 6,000–8.2 nM was prepared in SPR running buffer pH 7.4 containing 5% (v/v) DMSO. Binding curves were obtained by injecting varying hormone concentrations across the sensor chip for 40 s followed by a 100 s dissociation phase at a constant flow rate of 30 μl/min. Initial validation binding studies to both haCBG and laCBG were performed at room temperature 25°C. Ligands that demonstrated binding at 25°C were subsequently analyzed at 37 and 39°C, and pH 7.0 designed to mimic the pathophysiologic conditions of sepsis, while those which did not were not further analyzed at increased temperature as binding was not expected. Change in pH was achieved by adjusting the SPR running buffer from pH 7.4 to pH 7.0 by the addition of 0.1 M HCl. As all hormones completely dissociated from the surface during the acquisition period, no regeneration steps were required between samples. To correct for variations in DMSO concentration during the preparation of the analytes a solvent correction curve was included in the analysis by preparing a series of test solutions between 4.5 and 5.8% (v/v) DMSO. The increase in SPR signal observed with CBG:steroid binding interaction is expressed in response units (RU) and is calculated by subtracting the reference RU from the CBG:steroid RU. All RUs were referenced against the blank immobilised reference surface in Flow cell 1 and five buffer only injections to minimize system and injection artifacts. Each binding experiment was technically repeated 2–28 times. The double referenced data was globally fitted using a simple 1:1 L model using the Biacore T200 V3.0 evaluation software (GE Healthcare, Uppsala, Sweden).
3.6. Antibody binding assays by SPR
Concentrations of total CBG and haCBG (and arithmetically inferred laCBG) were measured using an in‐house polyclonal rabbit antibody and a monoclonal mouse antibody, 9G12 (RRID:AB_26632405), respectively. The 9G12 antibody recognizes the well‐defined linear epitope of the RCL comprising the 341STGVTLNL348 sequence of human CBG that encompasses the NE cleavage site at Val344‐Thr345. NE‐based cleavage of CBG reportedly disrupts of this epitope causing a loss of 9G12 binding. 23 , 24 Hence, 9G12, by implication recognizes only intact haCBG. The CBG polyclonal antibody recognizes multiple epitopes on CBG thus measuring total CBG regardless of conformation and structural integrity. Antibody binding was performed at the end of each binding series to confirm the presence of total CBG and laCBG on the respective flow cells. The polyclonal and 9G12 antibodies (1 mg/ml) were diluted in SPR running buffer to final concentrations of 6,000 and 4,000 ng/ml, respectively, run over the flow cells for a contact time of 30 s and dissociation time of 90 s at a flow rate of 10 μl/min. Regeneration was performed using 0.1 M glycine at pH 2.5 with a contact time of 60 s at 10 μl/min. The antibody binding experiments were performed upon completion of the ligand binding studies as the regeneration step was observed to denature CBG with concomitant loss of ligand binding.
3.7. Equilibrium MD simulations
The structure of human cleaved CBG in complex with progesterone (PDB 4BB2, 2.48 Å) 25 was downloaded from the RCSB protein databank and missing residues between Ly96 and Ser100 were added using the molecular modeling program MODELLER. 26 The ligand‐receptor complex was parameterized using the classical Chemistry at Harvard Macromolecular Mechanics 36 (CHARMM36) 27 force field with the aid of the CHARMM‐GUI. 28 The system was solvated in a rectangular TIP3 water box fitted to the size of the protein with padding of 10.0 Å. KCl was added to the system for neutralizing, randomly replacing water molecules to simulate a concentration of 0.15 M.
All MD simulations were performed in GPU‐accelerated nodes of the National Computing Infrastructure (NCI) Gadi supercomputer using the NAMD 2.13 package. 29 All simulations were performed with periodic boundary conditions in the NpT ensemble. The temperature of the simulation system was set to either 310 K or 313 K using the Langevin dynamics for temperature coupling. The system pressure was set to 1 atm using a Langevin piston. A distance cut‐off of 11.0 Å was applied to short‐range nonbonded interactions, whereas long‐range electrostatic interactions were treated using the particle‐mesh Ewald. Lennard‐Jones interactions were updated at every step and electrostatic interactions at every two steps. The time step of integration was chosen to be 2 fs for all simulations performed. Before the production MD simulations, systems were minimized for 10,000 steps and equilibrated for 125,000 steps. Finally, production simulations were carried out for 60 ns with a 20 ps output timing.
3.8. Simulation data analysis
Visualization was carried out using the VMD package. 30 The trajectories analysis, including root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were carried out using TCL scripts developed in‐house. Data plotting was carried out using the plotting tool Grace (https://plasma-gate.weizmann.ac.il/Grace/).
3.9. Statistical analysis
SPR data was analyzed using GraphPad Prism version 8 for Mac OS X (GraphPad Software, Inc., CA). Two‐way analysis of variance (ANOVA) was used to compare variables between the four temperature and pH conditions for both haCBG and laCBG. Results are expressed as mean ± standard error mean (SEM) unless otherwise stated. p < 0.05 was considered statistically significant.
4. RESULTS
4.1. Production and characterization of recombinant mammalian human CBG
To facilitate the development of a direct ligand binding assay using SPR, an engineered variant of recombinant human CBG was used for these studies. Specifically, two additional sequences were fused onto the C‐terminus of the protein: a hexahistidine sequence to enable protein purification and a 14 amino acid avi‐tag that allowed site‐specific biotinylation of the protein for the SPR assay. Inspection of available X‐ray structures of both haCBG and laCBG revealed that the C‐terminus was optimal for the tags as it was distal to both the ligand binding pocket and the RCL containing the NE scissile site. 7 Therefore, we predicted that fusion of the additional amino acid residues onto the C‐terminus was unlikely to disrupt ligand binding nor NE‐proteolysis of the RCL. The attachment of biotin onto a single lysine residue present in the avi‐tag permitted the immobilization of recombinant CBG onto streptavidin coated SPR sensor surfaces under mild binding conditions that maintained protein structure and biological activity. The commercially sourced CBG was expressed recombinantly in mammalian HEK293 cells to allow for human‐type protein glycosylation and purified using immobilized metal ion chromatography. SDS‐PAGE analysis revealed that, while CBG has a calculated molecular mass of 42 kDa, the apparent molecular weight was 50–60 kDa due to known N‐glycosylation heterogeneity 31 , 32 (Figure 1). Western blot analysis showed that the purified protein was immunoreactive toward a polyclonal anti‐CBG antibody as well as the conformational 9G12 monoclonal antibody that recognized a specific epitope within the intact RCL (Figure 1). A similar outcome was also observed on a Streptavidin blot, thus confirming the recombinant CBG was both biotinylated and uncleaved (Figure 1).
FIGURE 1.
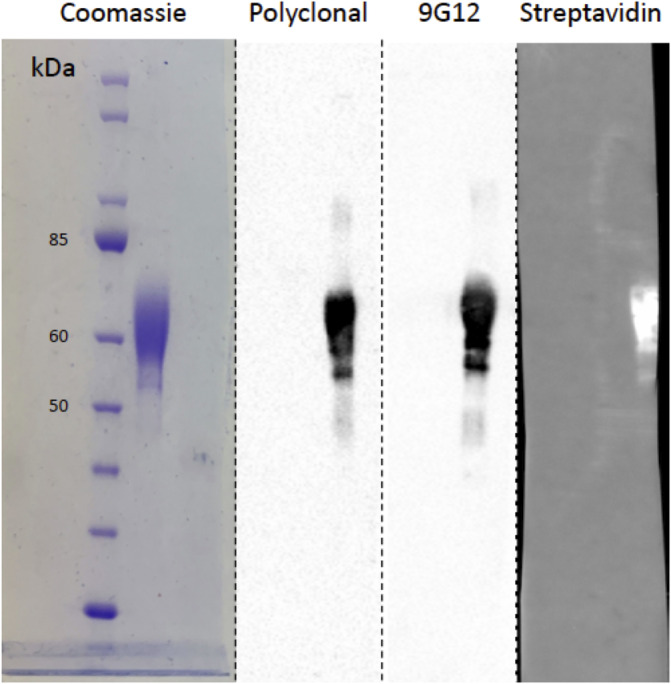
Analysis of recombinant human CBG. Coomassie‐blue stained SDS‐PAGE (left lane) and the corresponding Western blot analysis (three right lanes) of biotinylated recombinant human CBG probed with polyclonal anti‐CBG antibody, anti‐RCL monoclonal antibody 9G12 and streptavidin to detect biotinylated protein. This validation step demonstrates that biotinylated recombinant human CBG is uncleaved. Migration of molecular mass markers are also shown
Glycemic analysis further confirmed that the recombinant human CBG contained many of the same N‐glycosylation features as native human CBG isolated from healthy donor blood, albeit at different levels between the two variants of CBG obtained from different cellular sources (Figure 2). These included highly branched bi‐, tri‐, and possibly even higher branched complex sialoglycans variably modified with core fucosylation. 34 Our glycomics data revealed mostly quantitative differences between the N‐glycans of HEK293‐expressed biotinylated human CBG and native CBG, but also a few structures uniquely carried by recombinant CBG 34 including LacdiNAc and sialyl‐LacdiNAc and oligomannosidic‐type N‐glycans. Together these data suggested that the recombinant protein employed in this study possessed similar glycosylation properties as the native protein and was a suitable surrogate for in vitro ligand binding studies.
FIGURE 2.
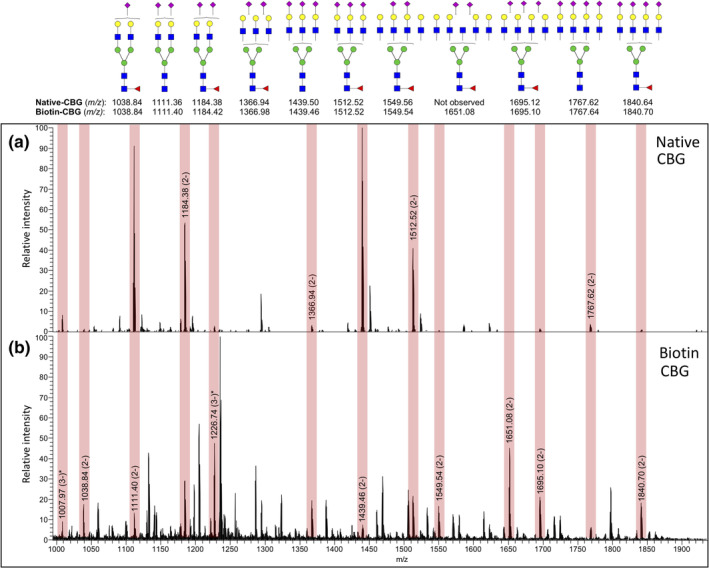
Comparative glycomic analysis of native and recombinant human CBG. Native human CBG isolated from donor blood (Native CBG, upper panel) and biotinylated recombinant human CBG expressed in HEK293 cells (Biotin‐CBG, lower panel) are glycosylated with broadly similar N‐glycosylation profiles, as documented using comparative PGC‐LC–MS/MS N‐glycome profiling. Vertical boxes in light red shading on the summed mass spectra highlight the N‐glycans that are shared between the two CBG forms, albeit at different relative abundances. The N‐glycan structures corresponding to the observed m/z values as supported by CID‐MS/MS data are shown above using the conventional symbol nomenclature for glycans. 33 “*” denotes an already depicted N‐glycan structure also present in another charge state
4.2. Development and validation of SPR binding assay
Using the characterized recombinant protein we developed a direct ligand binding assay using SPR technology which allowed us to systematically investigate ligand binding to haCBG and laCBG under a variety of conditions that mimic those encountered under normal physiology, septic shock and other pathophysiological states. Streptavidin was first immobilized on to three SPR sensor surfaces then biotinylated human CBG was captured onto two of the SPR flow cells, while a third flow cell was left as a streptavidin only control. A polyclonal anti‐CBG antibody was employed to demonstrate successful immobilization of CBG onto the sensor chip (Figure 3). One of the flow cells containing immobilised CBG then underwent treatment with NE for 30 min to facilitate the conversion of haCBG to laCBG in situ. Proteolysis was verified using the 9G12 monoclonal antibody with immunoreactivity specifically against the intact RCL (Figure 3). Complete conversion of haCBG‐to‐laCBG by NE treatment was confirmed by the loss of binding by 9G12 monoclonal antibody while maintaining polyclonal antibody binding. The known ligand hydrocortisone was then used to functionally validate the binding assay (Figure 4, for an example of the SPR sensograms for hydrocortisone binding to haCBG and laCBG, as well as the corresponding binding isotherms that underpinned the K D determination). The measured equilibrium binding constants at 25°C for hydrocortisone (haCBG, K D 42 nM; laCBG K D 217 nM) were in excellent agreement with previous reports (haCBG, K D 45 nM; laCBG K D 422 nM). 25 Hydrocortisone is the name used for synthetic cortisol employed therapeutically.
FIGURE 3.
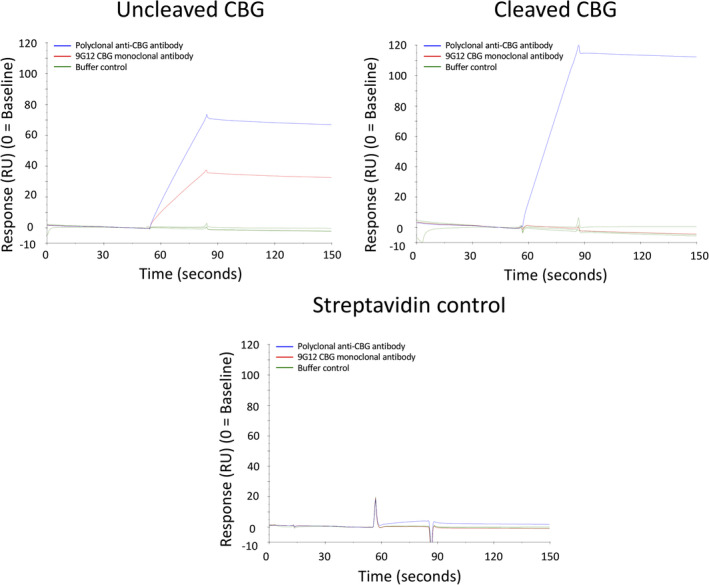
SPR analysis of immobilised CBG. Streptavidin was immobilized onto three separate flow cells on a CM5 sensorchip. The sensorchip was then treated with either biotinylated recombinant CBG (uncleaved CBG), biotinylated recombinant CBG followed by proteolysis with NE (cleaved CBG) or no protein (streptavidin control). Sensorgrams are shown for the binding of polyclonal anti‐CBG antibody (blue trace), 9G12 monoclonal antibody recognizing only uncleaved haCBG (red trace) and a buffer control (green trace). The streptavidin only flow cell, devoid of CBG, served as a negative control to address nonspecific antibody binding
FIGURE 4.
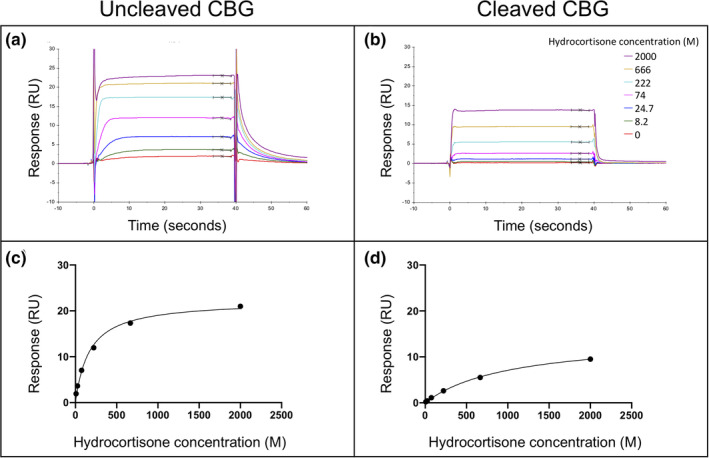
Hydrocortisone binding to CBG. Representative SPR sensorgrams are shown for hydrocortisone binding to (a) uncleaved CBG and (b) cleaved CBG at 37°C and pH 7.4. The corresponding binding isotherms used for determining equilibrium binding constants (K D) are also shown (c) and (d). Each colored band indicates a different hydrocortisone concentration, ranging from 0 to 2000 nM
4.3. Steroid ligand binding studies
The validated SPR assay was then used to systematically determine the affinities of an extended library of 19 steroid ligands. Binding to both haCBG and laCBG were initially assayed at 25°C for validation with subsequent analysis at temperature and pH designed to mimic the pathophysiologic conditions of sepsis. Table 1 shows the equilibrium binding constants of all steroid hormones to haCBG and laCBG at both 25 and 37°C at pH 7.4. Affinities were measured for hydrocortisone, cortisone, 11‐deoxycortisol, corticosterone, prednisolone, progesterone, 17‐OH progesterone, aldosterone, and testosterone. The most potent binding was recorded for haCBG binding to hydrocortisone (K D 42 nM) and corticosterone (K D 45 nM). The data demonstrated the expected four‐ to five‐fold decrease in affinity of the tested hormone upon haCBG‐to‐laCBG conversion. The greatest change, a seven‐fold reduction in hormone binding affinity, was observed for cortisone (haCBG K D 400 nM; la CBG K D 2957 nM; p <.0001). Glucocorticoids and progestagens were the only hormones that displayed measurable binding to laCBG (i.e., K D <6 μM). No binding to either ha‐CBG or la‐CBG (K D >6,000 nM) was detected for 17‐OH pregnenolone, β‐estradiol, dexamethasone, dehydroepiandrosterone (DHEA), estrone, 5α‐androstan‐17β‐ol‐3‐one, adrenosterone, dehydroisoandrosterone 3‐sulphate, pregnenolone, or allopregnanolone.
TABLE 1.
Equilibrium binding constants (K D, nM) of uncleaved and cleaved CBG determined at a constant condition of 25°C, 37°C, and pH 7.4
| Steroid ligand | Uncleaved CBG (K D, nM) 25°C | Cleaved CBG (K D, nM) 25°C | Uncleaved CBG (K D, nM) 37°C | Cleaved CBG (K D, nM) 37°C |
|---|---|---|---|---|
| Hydrocortisone (cortisol) | 42.0 (3.8) | 217.5 (8.7) | 214.4 (18.4) | 1,064.5 (139.5) |
| Corticosterone | 45.2 (6.4) | 170.8 (6.6) | 181.3 (2.3) | 898.0 (29.3) |
| Prednisolone | 77.9 (14.7) | 458 (48.5) | 329.8 (53.2) | 2,265.5 (345.5) |
| 11‐deoxycortisol | 85.3 (25.5) | 348.7 (42.7) | 249.8 (22.6) | 1931 (201) |
| 17‐OH progesterone | 118.4 (10.2) | 516.4 (53.9) | 889.1 (373.9) | 2,837.5 (257.5) |
| Progesterone | 318.2 (67.6) | 1,570.3 (214.9) | 1,680.5 (101.5) | 6,143.0 (1266) |
| Cortisone | 400.5 (97.8) | 2,957.5 (243.5) | 1,338 (281) | 5,242 |
| Testosterone | 609.5 (22.7) | 3,102.3 (564.5) | N/D | N/D |
| Aldosterone | 1,008.8 (236.3) | 4,634.5 (846.4) | N/D | N/D |
| 17‐OH pregnenolone | >6,000 | >6,000 | N/D | N/D |
| B‐estradiol | >6,000 | >6,000 | N/D | N/D |
| Dexamethaxone | >6,000 | >6,000 | N/D | N/D |
| Dehydroepiandrosterone (DHEA) | >6,000 | >6,000 | N/D | N/D |
| Estrone | >6,000 | >6,000 | N/D | N/D |
| 5a‐Androstan‐17B‐ol‐3‐one | >6,000 | >6,000 | N/D | N/D |
| Adrenosterone | >6,000 | >6,000 | N/D | N/D |
| Dehydroisoandrosterone 3‐sulphate | >6,000 | >6,000 | N/D | N/D |
| Pregnenolone | >6,000 | >6,000 | N/D | N/D |
| Allopregnanolone | >6,000 | >6,000 | N/D | N/D |
Note: All data points are represented as the mean values and their variance indicated in brackets as SEM as determined from at least three technical replicates.
Abbreviation: N/D, not determined.
The effects of temperature and pH on hydrocortisone (cortisol) binding to CBG were subsequently addressed (Table 2). Increasing temperature from 37 to 39°C, as observed in inflammation, caused a significant decrease in hydrocortisone binding to haCBG (37°C K D 214 nM; 39°C K D 507 nM; p = .0042). Likewise, acidification from the physiological pH 7.4 to pH 7.0 at 39°C caused further loss in binding activity (pH 7.4 K D 507 nM; pH K D 740 nM, p = .0308).
TABLE 2.
Hydrocortisone (cortisol) equilibrium binding constants (K D, nM) of uncleaved and cleaved CBG at different temperatures and pH
| Hydrocortisone (cortisol) | Uncleaved CBG (K D, nM) | Cleaved CBG (K D, nM) | ΔK D (fold change) |
|---|---|---|---|
|
25°C, pH 7.4 N = 10 |
42.0 (3.8) |
217.5 (8.7) |
5.2 |
|
37°C, pH 7.4 N = 26 |
214.4 a , b (18.4) | 1,064.5 d , e (139.5) | 5.0 |
|
39°C, pH 7.4 N = 28 |
507 a , c (19.0) | 1,398.9 d (37.7) | 2.8 |
|
39°C, pH 7.0 N = 26 |
740.1 b , c (24.8) | 1,505.1 e (44.1) | 2.0 |
Note: All data points are represented as mean values and their variance indicated in brackets as SEM. Two‐way ANOVA overall interaction p = .0002, row factor p = <.0001, column factor p <.0001.
Abbreviation: N, number of technical replicates.
Comparison of haCBG:cortisol at 37°C pH 7.4 versus 39°C pH 7.4 p = .0042.
Comparison of haCBG:cortisol at 37°C pH 7.4 versus 39°C pH 7.0 p <.0001.
Comparison of haCBG:cortisol at 39°C pH 7.4 versus 39°C pH 7.0 p = .0308.
Comparison of laCBG:cortisol at 37°C pH 7.4 versus 39°C pH 7.4 p = .0010.
Comparison of laCBG:cortisol at 37°C pH 7.4 versus 39°C pH 7.0 p <.0001.
There was an additive effect of NE treatment, temperature elevation and acidification on CBG:hydrocortisone binding affinity. Hydrocortisone showed 3.5‐fold reduced binding from haCBG 37°C, pH 7.4 (K D 214 nM) to 39°C, pH 7.0 (K D 740 nM; p = <.0001). NE‐treatment further reduced binding with seven‐fold reduced affinity observed between haCBG at 37°C, pH 7.4 (K D 214 nM) and laCBG at 39°C at pH 7.0 (K D 1,505 nM; p = <.0001). Similarly, a significant reduction in cortisol binding to laCBG was observed with increased temperature (Table 2). The data clearly demonstrate that temperature, pH, and NE‐mediated proteolysis all contribute to the release of CBG bound hormone under the pathophysiological conditions that mimic septic shock.
4.4. Antibody binding studies to investigate movement of the RCL
The effect of temperature and pH upon CBG structure was next investigated using SPR. Specifically, allosteric structural changes involving the reversible insertion of the RCL into the central β‐sheet of haCBG were probed using the 9G12 conformational monoclonal antibody. Binding studies were performed at 25 and 37°C at pH 7.4, and 39°C at pH 7.4 and 7.0 (Table 3). No changes in binding response were observed across the three temperatures tested, consistent with the RCL epitope being solvent exposed and immunoreactive despite the varying conditions. These results support the observation that the RCL remains intact and available for synergistic NE proteolysis, even at the elevated temperature of 39°C. Likewise, changes in pH did not alter 9G12 antibody binding at 25°C. However, reduced binding was observed at 39°C which may reflect synergistic effects of temperature and acidic pH on the solvent‐accessibility of the RCL, as reflected by CBG:hydrocortisone binding studies performed under these conditions (Table 3).
TABLE 3.
RCL remains available over different temperature and pH ranges as demonstrated by antibody binding to uncleaved and cleaved CBG
| Temperature | Uncleaved CBG | Cleaved CBG | ||
|---|---|---|---|---|
| Polyclonal (RU) | 9G12 (RU) | Polyclonal (RU) | 9G12 (RU) | |
| 25°C, pH 7.4 | 51.7 (3.5) | 34.2 (7.2) | 85.0 (3.9) | 1.1 (0.4) |
| 37°C, pH 7.4 | 63.9 (4.1) | 32.2 (1.0) | 87.6 (1.6) | −0.6 (0.3) |
| 39°C, pH 7.4 | 84.5 | 31.9 | 126.8 | 0.7 |
| 39°C, pH 7.0 | 97.5 | 15.9 | 172.2 | −2.5 |
Note: The binding response was measured in response units (RU). RU, increase in SPR signal observed with CBG:steroid binding interaction—reference RU. All data points are represented as mean values and their variance indicated in brackets as SEM.
4.5. Equilibrium MD simulations
Finally, MD simulations were performed to further understand the impact of temperature upon CBG structure. Here the high‐resolution crystal structure of human cleaved CBG in complex with progesterone (PDB ID 4BB2, 2.48 Å) was subjected to MD simulations after parameterization with the CHARMM36 all tom force field. This structure was selected as the crystal structure of human uncleaved CBG has not been reported, and the homologous rat CBG structures have regions unresolved including the RCL. During these initial equilibrium simulations at 310 and 313 K, no large changes in CBG structure were observed. The protein backbone reached equilibrium dynamics of only ~1.6 Å deviated from the original conformation. Similarly, the ligand also reached an equilibrium structure ~0.3 Å from the original crystallographic position. Therefore, temperature‐induced changes in CBG structure were most likely due to localized movement of amino acid side chains.
The relationship between ligand binding and temperature (37 and 40°C) was further probed by studying the dynamics of the CBG‐ligand complex at 310 and 313 K for a period of 60 ns. Those amino acid residues located within 8 Å from the ligand were interrogated in further detail (Figure 5a). A comparison of the RMSF values between the two conditions showed that the indole side chain of Trp371 experienced the largest and most significant movement of any amino acid in the binding site (Figure 5b; Movies A https://figshare.com/s/5739e72874f70476ba2c and B https://figshare.com/s/fd5769ab24bbed371fe2). Visual inspection of the structure at 310 K revealed that the indole sidechain adopted the stable conformation required for a direct stacking interaction with the hydrophobic rings of the steroid. In sharp contrast, the indole heterocycle experienced much greater mobility at 313 K and was observed to rotate outside of the plane necessary for this binding interaction. Given the critical role of Trp371 in ligand binding, this increased rotation and mobility would conceivably impact upon a thermo‐coupled loss of ligand binding.
FIGURE 5.
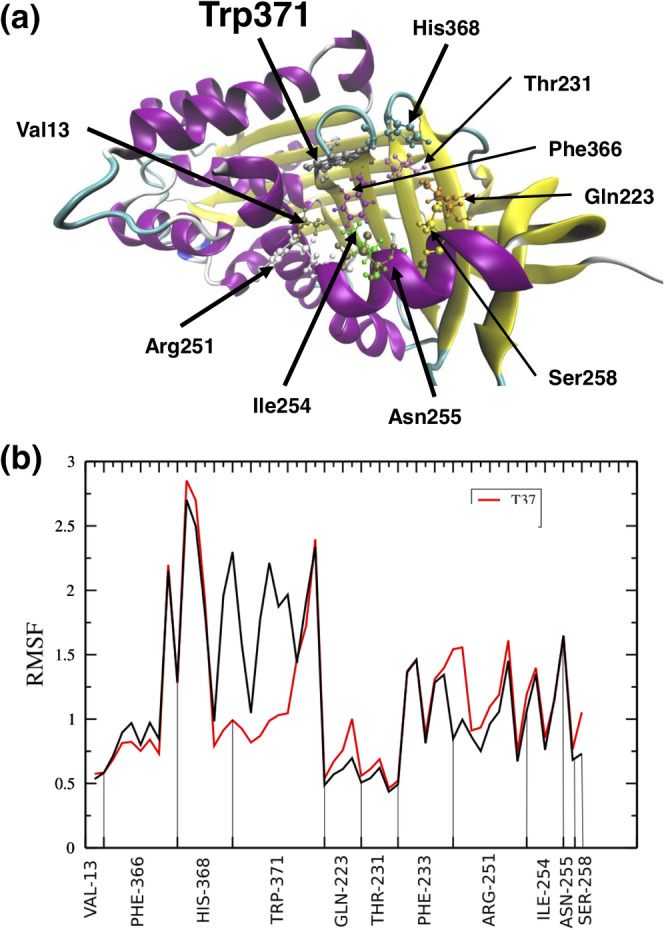
Molecular dynamic simulations. (a). The tertiary structure of the human CBG with key amino acids required for ligand binding highlighted. The human CBG (PBD ID 4BB2, 2.48 Å) structure was used. (b). The root mean squire fluctuation (RMSF) values for select amino acids in the ligand binding site during the simulation at 310 K (red curve) and 313 K (black curve). A comparison of curves show that the indole side chain of Trp371 experienced the largest and most significant movement of any amino acid within the ligand binding site. The naturally occurring Trp371 to Ser mutation (CBG Athens variant) resulting in the complete loss of CBG:cortisol binding activity in a Greek patient 35 further supports the critical role of Trp371 in the thermo‐coupled release of steroid from CBG
5. DISCUSSION
This study confirms that the steroid binding to CBG is ligand specific and is reduced upon NE‐mediated conversion of haCBG‐to‐laCBG. Importantly, this study has shown that CBG:cortisol binding is significantly reduced with increases in temperature and in acidosis for both haCBG and laCBG, representative of that which is experienced in systemic inflammatory states such as in sepsis and septic shock. Furthermore, the immunoreactivity of the RCL of haCBG remains largely preserved at 39°C and in acidosis. Molecular modeling studies of CBG performed at 40°C revealed the critical role of Trp371 in ligand binding. Taken together we propose the thermo‐coupling mechanism of cortisol release involves Trp371 and occurs while maintaining RCL integrity. Thus, the synergy of NE cleavage, pyrexia and acidosis on CBG structure may serve to enhance cortisol delivery to the interstitium at sites of inflammation.
The glucocorticoid cortisol is important in the regulation and modulation of the immune system in response to inflammation and essential for survival. In acute physiological stress, such as that experienced in sepsis, glucocorticoids work through secondary messenger pathways on pro‐inflammatory transcription factors and at genomic sites to reduce inflammation. 36 Initially, minor insults elevate cortisol with pro‐inflammatory actions enhancing the innate immune response, 37 after which cortisol exerts anti‐inflammatory effects through glucocorticoid receptor (GR)‐α mediated suppression of intracellular pathways induced by the pro‐inflammatory transcription factors NF‐κB and AP‐1 and direct GR transcriptional actions at genomic glucocorticoid response elements. 38 , 39 Glucocorticoids target inflammatory cytokine synthesis, neutrophil migration, T cell apoptosis and macrophage inhibition. Cortisol has pleiotropic effects increasing hepatic glucose output and blood pressure through synergistic action with catecholamines on peripheral vasculature and the heart, immunomodulation, increased cognition and arousal. 36 , 40 Up to 80% of the body's cortisol is bound to CBG. CBG optimizes the human body's response to inflammation by facilitating cortisol delivery via four key mechanisms: pyrexia, reduced hepatic synthesis of CBG, NE cleavage, and tissue acidosis. These mechanisms feature in each pathogenic step in the cascade from localized inflammation to overwhelming septic shock. Our previous human studies have demonstrated that a lack of targeted delivery of natural cortisol and/or administered hydrocortisone to damaged tissue is related to deficient serum concentrations of CBG, which perpetuates organ failure and death in sepsis. 13 , 14
To further probe the action of CBG in the finely tuned delivery of cortisol and other steroids, we developed a novel ligand binding assay. The chosen SPR technology provided a reproducible and readily applicable, direct ligand binding assay to quantitate the bimolecular interaction between recombinant CBG and a library of 19 steroids. Our ligand panel consisted of both hormones with reported binding to CBG alongside previously untested hormones displaying similar chemical structures to the bona fide ligands that play possible neuro‐modulating roles in the brain. 41 , 42 Sex steroids were also profiled, in part due to the reported association of the CBG A51V polymorphism with higher female‐to‐male live births in Han Chinese. 43 Moreover, the assay was developed such that both uncleaved (haCBG) and cleaved (laCBG) isoforms could be tested in parallel under varying temperatures and pH. This is the first study to extensively investigate CBG binding interactions using SPR. Equilibrium binding constants for cortisol to haCBG (K D 42 nM) and laCBG (K D 217 nM) at 25°C pH 7.4 were in excellent agreement with those reported in the literature (i.e., Qi et al. 18 ; K D 26.9 nM for uncleaved recombinant CBG at 22°C, Gardill et al. 25 K D 45 and 422 nM for uncleaved and cleaved recombinant CBG, respectively, at 25°C; and Chan et al 7 K D 42.8 nM and 165 nM for uncleaved and cleaved recombinant CBG, respectively, at 25°C).
The SPR binding assay demonstrated a 3‐to‐8‐fold decrease in binding affinity between haCBG and laCBG for cortisol, cortisone, corticosterone, 11‐deoxycortisol, progesterone, 17‐hydroxyprogesterone and prednisolone at physiological conditions (37°C, pH 7.4). Cortisol binding affinity also significantly decreased at higher temperatures and acidic pH for both haCBG and laCBG. With respect to other steroids, progesterone bound to haCBG with an eight‐fold lower affinity than cortisol (K D 1680.5 vs. 214.4 nM, respectively). During pregnancy progesterone concentrations at the intervillous space are 15‐fold higher than in the peripheral maternal circulation, and CBG is largely progesterone‐laden at this site. 44 The high circulating progesterone concentrations compete with cortisol for binding, contributing to the increase in free cortisol required for organ maturation neurodevelopment and neonatal transition. 45 17‐OH progesterone binds to CBG with four‐fold lower affinity than cortisol (K D 889.1 vs. 214.4 nM, respectively). In untreated and undertreated congenital adrenal hyperplasia, elevated 17‐OH progesterone levels would preferentially bind CBG, increasing free cortisol.
Unique to our assay was the ability to directly study the RCL under temperature and pH conditions which would only be achievable using alternative methodologies such as mass spectrometry or X‐ray crystallography. The 9G12 antibody recognition was preserved for haCBG across the tested temperature and pH range confirming that the RCL remains solvent exposed and available for NE proteolysis in these conditions. Presumably, CBG loses ligand affinity during pyrexia due to temperature‐dependent flexibility of the hormone binding pocket independent of RCL modification (discussed below). Previous studies have proposed cortisol release in pyrexia may be driven by a reversible conformational change of haCBG due to a temperature‐dependent insertion of the RCL into the CBG protein core, 6 , 7 , 18 described as the “flip‐flop” or “nudging” submolecular switch mechanism as observed for other serpins. 17 , 18 Here, we demonstrated intact thermo‐coupling occurs even after NE cleavage for laCBG, corroborating that the temperature‐dependent mechanism of steroid release from CBG persists despite the full insertion of the RCL. Thus, these findings support a synergistic effect of both NE cleavage of haCBG and temperature‐dependent mechanisms in the release of bound cortisol at sites of inflammation.
Serpins have the capacity to form a series of conformers due to RCL flexibility and A‐sheet configurations resulting in graded ligand affinity. This equilibrium phenomenon is observed only for intact serpins which have not undergone proteolytic cleavage of the RCL. 6 CBG is proposed to exist within this dynamic landscape, but appears to have a more limited range of RCL flexibility than that observed for other serpins due to its higher ligand binding affinity and slightly shorter RCL by four residues. 6 With respect to the equilibrium of model of serpins, we acknowledge the potential limitation using antibody binding studies in this work to investigate RCL movement. Use of the 9G12 monoclonal antibody directed against the NE‐cleavage site of the RCL will differentiate between a fully incorporated or exposed RCL, however, it may lack sensitivity to detect subtle micro movements of the RCL. Furthermore, the equilibrium of RCL movement under study conditions may be influenced by the virtue of antibody binding.
Our MD simulation experiments highlighted an important role for Trp371 in the thermo‐coupled release of bound steroid. Previous studies have, similarly, implicated this amino acid residue in ligand binding. Substitution of Trp371 with threonine or lysine generated a mutant that was unable to bind cortisol in assays using radiolabelled ligand. 46 Likewise, a naturally occurring Trp371 to Ser mutation in a Greek patient with low morning cortisol levels (the CBG Athens variant) was characterized and shown to be devoid of cortisol binding activity. 35 Structural biology has shed further insights into the molecular basis of cortisol binding and mechanism of release. Comparative analysis of the available crystal structures of human and rat CBG in the unliganded state as well as both haCBG and laCBG in complex with steroids reveal that the ligand binding sites are all highly conserved. 6 , 8 , 25 , 47 The primary point of structural difference is the orientation of the indole sidechain of Trp371. 6 In co‐complexes with cortisol or progesterone, the indole ring of Trp371 is positioned parallel to ligand necessary for stacking interactions with the common A and B rings of these steroids. This is in stark contrast to the unliganded structure where the same side chain is rotated 90° and adopts a conformation suggestive that it is unreceptive to ligand binding. 6 In the current study, we measured significantly greater mobility of the same amino acid side chain essential for ligand binding when temperature was elevated above those expected under normal physiology. Our MD studies, in concert with biophysical binding assays and structural biology, provide an alternative mechanism for the thermo‐coupled dissociation of ligand during pyrexia involving movement of the Trp371 side chain.
CBG glycosylation is known to be essential for cortisol binding through its influence on CBG protein folding and steroid binding at glycosylation site Asn238, one of five glycosylation sites positioned in proximity of the ligand binding site, 48 and influence RCL cleavability given there is glycosylation at Asn347, within the RCL, modulating the rate at which cortisol is released. 49 In the current study we used recombinant human CBG that was characterized for glycosylation modifications and demonstrated 9G12 immunoreactivity to untreated protein and the expected complete loss of 9G12 reactivity upon NE‐mediated haCBG‐to‐laCBG conversion. Our previous research has revealed falling serum haCBG concentrations over time in proportion to septic shock severity and lower haCBG levels is associated with septic shock mortality. 13 , 14 In sepsis and septic shock with attendant multi‐organ failure and hepatic dysfunction, protein glycosylation does not increase, thus it is unlikely that the reduction in 9G12 reactive CBG is due to an increase in CBG glycosylation of the RCL in these states. Furthermore, a loss in 9G12 reactive CBG correlates with rising free cortisol concentrations in sepsis and septic shock. 13 , 14 Data from the current study shows a clear temporal connection between in vitro NE cleavage under controlled conditions, reduction of CBG:cortisol binding and loss of 9G12 immunoreactivity. Thus, a reduction in 9G12 reactive CBG, as observed in human sepsis and septic shock, likely reflects a loss of circulating haCBG due to NE cleavage in vivo. The HEK293‐expressed human CBG used in this study shared many glycosylation features with native human CBG. While some measurable differences may contribute to different 9G12 reactivity profiles, it is highly unlikely these minor changes would contribute to the results reported here further supporting the use of 9G12 antibodies to quantify haCBG in our human clinical studies. 14 , 45 , 50 , 51 , 52 , 53 , 54
This study (1) systematically determines the binding affinity of glycosylated haCBG and laCBG to a large panel of steroid ligands, (2) demonstrates reduced CBG:cortisol binding affinity in response to acidosis, and (3) reveals CBG:cortisol binding affinity is reduced upon increases in temperature with the preservation of the thermo‐coupling mechanism even after NE‐cleavage. We demonstrate through molecular modeling the importance of Trp371 critically located within the binding pocket in the thermo‐coupling mechanism of cortisol release and through RCL antibody binding studies that the NE‐cleavage site remains largely intact at increased temperature. This has implications for the study of cortisol delivery in human sepsis and suggests that the conditions associated with sepsis separately contribute to tissue cortisol delivery via discrete structural effects on the CBG molecule. In sepsis, a sequential four‐part process may operate to optimize free cortisol delivery, with pyrexia, 53 cytokine‐based inhibition of hepatic CBG synthesis, 55 NE‐mediated cleavage and acidosis acting in concert to produce immune activation, metabolic and neurocognitive effects, direct delivery of immunomodulatory cortisol at inflammatory sites and cortisol protection to healthy tissues affected by the inflammatory process. These findings demonstrate the modifiable hormone binding and delivery characteristics of CBG in both physiologic and pathologic conditions, supporting its significance in cortisol delivery to obviate systemic inflammation 4 , 5 and multi‐organ failure in patients with septic shock and its association with mortality. 13 , 14
AUTHOR CONTRIBUTIONS
Emily Meyer: Conceptualization; data curation; formal analysis; investigation; methodology; writing‐original draft. David Torpy: Funding acquisition; supervision; writing‐review and editing. Anastasia Chernykh: Investigation; writing‐review and editing. Morten Thaysen‐Andersen: Investigation; supervision; writing‐review and editing. Marni Nenke: Writing‐review and editing. John Lewis: Writing‐review and editing. Harinda Rajapaksha: Investigation; writing‐review and editing. Wayne Rankin: Conceptualization; funding acquisition; supervision; writing‐review and editing. Steven Polyak: Conceptualization; methodology; supervision; writing‐review and editing.
CONFLICT OF INTEREST
The authors report no conflict of interest.
Supporting information
Movies A and B Tertiary structure of CBG at 37°C and 40°C. Supplementary movies of the tertiary structure of human CBG (PBD ID 4BB2, 2.48 Å) at 37°C (Movie A) and 40°C (Movie B) with key amino acids located within the steroid binding pocket required for ligand binding highlighted. Comparison shows spatial instability of Trp371 (depicted in silver), experiencing the most amount of movement of any amino acid within the binding site at 40°C compared to 37°C. The naturally occurring Trp371 to Ser mutation (CBG Athens variant) resulting in the complete loss of CBG:cortisol binding activity in a Greek Patient supports the critical role of Trp371 in the thermo‐coupled release of steroid from CBG Reference 46.
ACKNOWLEDGMENT
The work was supported in part by the Health Services Charitable Gifts Board, Royal Adelaide Hospital. Emily J. Meyer was supported by the AR Clarkson Scholarship, Royal Adelaide Hospital Research Fund. Morten Thaysen‐Andersen was supported by a Macquarie University Research Seeding Grant. Steven W. Polyak was supported by the National Health and Medical Research Council of Australia (GN1147538). Marni A. Nenke was supported by the Mary Overton Early Career Research Fellowship, Royal Adelaide Hospital Research Fund. Anastasia Chernykh was supported by International Macquarie University Research Excellence Scholarship.
Meyer EJ, Torpy DJ, Chernykh A, et al. Pyrexia and acidosis act independently of neutrophil elastase reactive center loop cleavage to effect cortisol release from corticosteroid‐binding globulin. Protein Science. 2020;29:2495–2509. 10.1002/pro.3982
Wayne Rankin and Steven W. Polyak senior co‐authors.
Funding information Macquarie University; National Health and Medical Research Council, Grant/Award Number: GN1147538; Royal Adelaide Hospital
REFERENCES
- 1. Seal US, Doe RP. Corticosteroid‐binding globulin. I. Isolation from plasma of diethylstilbestrol‐treated men. J Biol Chem. 1962;237:3136–3140. [PubMed] [Google Scholar]
- 2. Meyer EJ, Nenke MA, Rankin W, Lewis JG, Torpy DJ. Corticosteroid‐binding globulin: A review of basic and clinical advances. Horm Metab Res. 2016;48:359–371. [DOI] [PubMed] [Google Scholar]
- 3. Meyer EJ, Nenke MA, Lewis JG, Torpy DJ. Corticosteroid‐binding globulin: Acute and chronic inflammation. Expert Rev Endocrinol Metab. 2017;2017:1–11. [DOI] [PubMed] [Google Scholar]
- 4. Pemberton PA, Stein PE, Pepys MB, Potter JM, Carrell RW. Hormone binding globulins undergo serpin conformational change in inflammation. Nature. 1988;336:257–258. [DOI] [PubMed] [Google Scholar]
- 5. Loke I, Ostergaard O, Heegaard NHH, Packer NH, Thaysen‐Andersen M. Paucimannose‐rich N‐glycosylation of spatiotemporally regulated human neutrophil elastase modulates its immune functions. Mol Cell Proteomics. 2017;16:1507–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou A, Wei Z, Stanley PL, Read RJ, Stein PE, Carrell RW. The S‐to‐R transition of corticosteroid‐binding globulin and the mechanism of hormone release. J Mol Biol. 2008;380:244–251. [DOI] [PubMed] [Google Scholar]
- 7. Chan WL, Carrell RW, Zhou A, Read RJ. How changes in affinity of corticosteroid‐binding globulin modulate free cortisol concentration. J Clin Endocrinol Metab. 2013;98:3315–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klieber MA, Underhill C, Hammond GL, Muller YA. Corticosteroid‐binding globulin, a structural basis for steroid transport and proteinase‐triggered release. J Biol Chem. 2007;282:29594–29603. [DOI] [PubMed] [Google Scholar]
- 9. Mickelson KE, Forsthoefel J, Westphal U. Steroid‐protein interactions. Human corticosteroid binding globulin: Some physicochemical properties and binding specificity. Biochemistry. 1981;20:6211–6218. [DOI] [PubMed] [Google Scholar]
- 10. Pugeat MM, Dunn JF, Nisula BC. Transport of steroid hormones: Interaction of 70 drugs with testosterone‐binding globulin and corticosteroid‐binding globulin in human plasma. J Clin Endocrinol Metab. 1981;53:69–75. [DOI] [PubMed] [Google Scholar]
- 11. Dunn JF, Nisula BC, Rodbard D. Transport of steroid hormones: Binding of 21 endogenous steroids to both testosterone‐binding globulin and corticosteroid‐binding globulin in human plasma. J Clin Endocrinol Metab. 1981;53:58–68. [DOI] [PubMed] [Google Scholar]
- 12. Nguyen PT, Lewis JG, Sneyd J, Lee RS, Torpy DJ, Shorten PR. Development of a formula for estimating plasma free cortisol concentration from a measured total cortisol concentration when elastase‐cleaved and intact corticosteroid binding globulin coexist. J Steroid Biochem Mol Biol. 2014;141:16–25. [DOI] [PubMed] [Google Scholar]
- 13. Nenke MA, Rankin W, Chapman MJ, et al. Depletion of high‐affinity corticosteroid‐binding globulin corresponds to illness severity in sepsis and septic shock; clinical implications. Clin Endocrinol. 2015;82:801–807. [DOI] [PubMed] [Google Scholar]
- 14. Meyer EJ, Nenke MA, Rankin W, et al. Total and high‐affinity corticosteroid‐binding globulin depletion in septic shock is associated with mortality. Clin Endocrinol. 2019;90:232–240. [DOI] [PubMed] [Google Scholar]
- 15. Cameron A, Henley D, Carrell R, Zhou A, Clarke A, Lightman S. Temperature‐responsive release of cortisol from its binding globulin: A protein thermocouple. J Clin Endocrinol Metab. 2010;95:4689–4695. [DOI] [PubMed] [Google Scholar]
- 16. Henley D, Lightman S, Carrell R. Cortisol and CBG—Getting cortisol to the right place at the right time. Pharmacol Ther. 2016;166:128–135. [DOI] [PubMed] [Google Scholar]
- 17. Beauchamp NJ, Pike RN, Daly M, et al. Antithrombins Wibble and wobble (T85M/K): Archetypal conformational diseases with in vivo latent‐transition, thrombosis, and heparin activation. Blood. 1998;92:2696–2706. [PubMed] [Google Scholar]
- 18. Qi X, Loiseau F, Chan WL, et al. Allosteric modulation of hormone release from thyroxine and corticosteroid‐binding globulins. J Biol Chem. 2011;286:16163–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jensen PH, Karlsson NG, Kolarich D, Packer NH. Structural analysis of N‐ and O‐glycans released from glycoproteins. Nat Protoc. 2012;7:1299–1310. [DOI] [PubMed] [Google Scholar]
- 20. Hinneburg H, Chatterjee S, Schirmeister F, et al. Post‐column make‐up flow (PCMF) enhances the performance of capillary‐flow PGC‐LC‐MS/MS‐based glycomics. Anal Chem. 2019;91:4559–4567. [DOI] [PubMed] [Google Scholar]
- 21. Ashwood C, Lin CH, Thaysen‐Andersen M, Packer NH. Discrimination of isomers of released N‐ and O‐glycans using diagnostic product ions in negative ion PGC‐LC‐ESI‐MS/MS. J Am Soc Mass Spectrom. 2018;29:1194–1209. [DOI] [PubMed] [Google Scholar]
- 22. Thompson AP, Salaemae W, Pederick JL, et al. Mycobacterium tuberculosis dethiobiotin synthetase facilitates nucleoside triphosphate promiscuity through alternate binding modes. ACS Catalysis. 2018;8:10774–10783. [Google Scholar]
- 23. Lewis JG, Elder PA. Corticosteroid‐binding globulin reactive Centre loop antibodies recognise only the intact natured protein: Elastase cleaved and uncleaved CBG may coexist in circulation. J Steroid Biochem Mol Biol. 2011;127:289–294. [DOI] [PubMed] [Google Scholar]
- 24. Lewis JG, Elder PA. Intact or "active" corticosteroid‐binding globulin (CBG) and total CBG in plasma: Determination by parallel ELISAs using monoclonal antibodies. Clin Chim Acta. 2013;416:26–30. [DOI] [PubMed] [Google Scholar]
- 25. Gardill BR, Vogl MR, Lin HY, Hammond GL, Muller YA. Corticosteroid‐binding globulin: Structure‐function implications from species differences. PloS One. 2012;7:e52759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2017;1654:39–54. [DOI] [PubMed] [Google Scholar]
- 27. Huang J, MacKerell AD Jr. CHARMM36 all‐atom additive protein force field: Validation based on comparison to NMR data. J Comput Chem. 2013;34:2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim S, Lee J, Jo S, Brooks CL 3rd, Lee HS, Im W. CHARMM‐GUI ligand reader and modeler for CHARMM force field generation of small molecules. J Comput Chem. 2017;38:1879–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:27–38. [DOI] [PubMed] [Google Scholar]
- 31. Hammond GL, Smith CL, Goping IS, et al. Primary structure of human corticosteroid binding globulin, deduced from hepatic and pulmonary cDNAs, exhibits homology with serine protease inhibitors. Proc Natl Acad Sci USA. 1987;84:5153–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kato EA, Hsu BR, Kuhn RW. Comparative structural analyses of corticosteroid binding globulin. J Steroid Biochem. 1988;29:213–220. [DOI] [PubMed] [Google Scholar]
- 33. Varki A, Cummings RD, Aebi M, et al. Symbol nomenclature for graphical representations of glycans. Glycobiology. 2015;25:1323–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sumer‐Bayraktar Z, Kolarich D, Campbell MP, Ali S, Packer NH, Thaysen‐Andersen M. N‐glycans modulate the function of human corticosteroid‐binding globulin. Mol Cell Proteomics. 2011;M111(009100):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hill LA, Vassiliadi DA, Simard M, et al. Two different corticosteroid‐binding globulin variants that lack cortisol‐binding activity in a greek woman. J Clin Endocrinol Metab. 2012;97:4260–4267. [DOI] [PubMed] [Google Scholar]
- 36. Cruz‐Topete D, Cidlowski JA. One hormone, two actions: Anti‐ and pro‐inflammatory effects of glucocorticoids. Neuroimmunomodulation. 2015;22:20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dhabhar FS. Stress‐induced augmentation of immune function—the role of stress hormones, leukocyte trafficking, and cytokines. Brain Behav Immun. 2002;16:785–798. [DOI] [PubMed] [Google Scholar]
- 38. De Bosscher K, Vanden Berghe W, Vermeulen L, Plaisance S, Boone E, Haegeman G. Glucocorticoids repress NF‐kappaB‐driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc Natl Acad Sci USA. 2000;97:3919–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. De Bosscher K, Vanden Berghe W, Haegeman G. Mechanisms of anti‐inflammatory action and of immunosuppression by glucocorticoids: Negative interference of activated glucocorticoid receptor with transcription factors. J Neuroimmunol. 2000;109:16–22. [DOI] [PubMed] [Google Scholar]
- 40. Lee SR, Kim HK, Youm JB, et al. Non‐genomic effect of glucocorticoids on cardiovascular system. Pflüg AArch Eur J Phy. 2012;464:549–559. [DOI] [PubMed] [Google Scholar]
- 41. Solomon MB, Herman JP. Sex differences in psychopathology: Of gonads, adrenals and mental illness. Physiol Behav. 2009;97:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moore DE, Kawagoe S, Davajan V, Nakamura RM, Mishell DR. An in vivo system in man for quantitation of estrogenicity II. Pharmacologic changes in binding capacity of serum corticosteroid‐binding globulin induced by conjugated estrogens, mestranol, and ethinyl estradiol. Am J Obstet Gynecol. 1978;130:482–486. [PubMed] [Google Scholar]
- 43. Lei JH, Yang X, Peng S, et al. Impact of corticosteroid‐binding globulin deficiency on pregnancy and neonatal sex. J Clin Endocrinol Metab. 2015;100:1819–1827. [DOI] [PubMed] [Google Scholar]
- 44. Benassayag C, Souski I, Mignot TM, et al. Corticosteroid‐binding globulin status at the fetomaternal interface during human term pregnancy. Biol Reprod. 2001;64:812–821. [DOI] [PubMed] [Google Scholar]
- 45. Hodyl NA, Stark MJ, Meyer EJ, Lewis JG, Torpy DJ, Nenke MA. High binding site occupancy of corticosteroid‐binding globulin by progesterone increases fetal free cortisol concentrations. Eur J Obstet Gynecol Reprod Biol. 2020;251:129–135. [DOI] [PubMed] [Google Scholar]
- 46. Avvakumov GV, Hammond GL. Substitutions of tryptophan residues in human corticosteroid‐binding globulin: Impact on steroid binding and glycosylation. J Steroid Biochem Mol Biol. 1994;49:191–194. [DOI] [PubMed] [Google Scholar]
- 47. Chan WL, Zhou A, Read RJ. Towards engineering hormone‐binding globulins as drug delivery agents. PLoS One. 2014;9:e113402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Avvakumov GV, Warmels‐Rodenhiser S, Hammond GL. Glycosylation of human corticosteroid‐binding globulin at aspargine 238 is necessary for steroid binding. J Biol Chem. 1993;268:862–866. [PubMed] [Google Scholar]
- 49. Sumer‐Bayraktar Z, Grant OC, Venkatakrishnan V, Woods RJ, Packer NH, Thaysen‐Andersen M. Asn347 glycosylation of corticosteroid‐binding globulin fine‐tunes the host immune response by modulating proteolysis by Pseudomonas aeruginosa and neutrophil elastase. J Biol Chem. 2016;291:17727–17742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nenke MA, Holmes M, Rankin W, Lewis JG, Torpy DJ. Corticosteroid‐binding globulin cleavage is paradoxically reduced in alpha‐1 antitrypsin deficiency: Implications for cortisol homeostasis. Clin Chim Acta. 2016;452:27–31. [DOI] [PubMed] [Google Scholar]
- 51. Nenke MA, Lewis JG, Rankin W, Shaw D, Torpy DJ. Corticosteroid‐binding globulin cleavage may be pathogen‐dependent in bloodstream infection. Clin Chim Acta. 2017;464:176–181. [DOI] [PubMed] [Google Scholar]
- 52. Nenke MA, Lewis JG, Rankin W, et al. Reduced corticosteroid‐binding globulin cleavage in active rheumatoid arthritis. Clin Endocrinol. 2016;85:369–377. [DOI] [PubMed] [Google Scholar]
- 53. Nenke MA, Zeng A, Meyer EJ, et al. Differential effects of estrogen on corticosteroid‐binding globulin forms suggests reduced cleavage in pregnancy. J Endocr Soc. 2017;1:202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nenke MA, Lewis JG, Rankin W, Torpy DJ. Evidence of reduced CBG cleavage in abdominal obesity: A potential factor in development of the metabolic syndrome. Horm Metab Res. 2016;48:523–528. [DOI] [PubMed] [Google Scholar]
- 55. Emptoz‐Bonneton A, Crave JC, LeJeune H, Brébant C, Pugeat M. Corticosteroid‐binding globulin synthesis regulation by cytokines and glucocorticoids in human hepatoblastoma‐derived (HepG2) cells. J Clin Endocrinol Metab. 1997;82:3758–3762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movies A and B Tertiary structure of CBG at 37°C and 40°C. Supplementary movies of the tertiary structure of human CBG (PBD ID 4BB2, 2.48 Å) at 37°C (Movie A) and 40°C (Movie B) with key amino acids located within the steroid binding pocket required for ligand binding highlighted. Comparison shows spatial instability of Trp371 (depicted in silver), experiencing the most amount of movement of any amino acid within the binding site at 40°C compared to 37°C. The naturally occurring Trp371 to Ser mutation (CBG Athens variant) resulting in the complete loss of CBG:cortisol binding activity in a Greek Patient supports the critical role of Trp371 in the thermo‐coupled release of steroid from CBG Reference 46.