

Abstract
Depsipeptides are compounds that contain both ester bonds and amide bonds. Important natural product depsipeptides include the piscicide antimycin, the K+ ionophores cereulide and valinomycin, the anticancer agent cryptophycin, and the antimicrobial kutzneride. Furthermore, database searches return hundreds of uncharacterized systems likely to produce novel depsipeptides. These compounds are made by specialized nonribosomal peptide synthetases (NRPSs). NRPSs are biosynthetic megaenzymes that use a module architecture and multi‐step catalytic cycle to assemble monomer substrates into peptides, or in the case of specialized depsipeptide synthetases, depsipeptides. Two NRPS domains, the condensation domain and the thioesterase domain, catalyze ester bond formation, and ester bonds are introduced into depsipeptides in several different ways. The two most common occur during cyclization, in a reaction between a hydroxy‐containing side chain and the C‐terminal amino acid residue in a peptide intermediate, and during incorporation into the growing peptide chain of an α‐hydroxy acyl moiety, recruited either by direct selection of an α‐hydroxy acid substrate or by selection of an α‐keto acid substrate that is reduced in situ. In this article, we discuss how and when these esters are introduced during depsipeptide synthesis, survey notable depsipeptide synthetases, and review insight into bacterial depsipeptide synthetases recently gained from structural studies.
Keywords: biosynthesis, depsipeptide, enzyme, ester bond, nonribosomal peptide, synthetase
1. INTRODUCTION TO NONRIBOSOMAL PEPTIDE SYNTHETASES AND THEIR PRODUCTS
Nonribosomal peptide synthetases (NRPSs) are multi‐domain microbial enzymes that synthesize natural products of medical and industrial importance. 1 , 2 Well‐known examples of nonribosomal peptides include the antibiotics penicillin, 3 gramicidins S and D 4 , 5 , 6 , 7 and daptomycin, 8 the antifungal caspofungin, 9 the immunosuppressor cyclosporin, 10 , 11 and the emulsifier surfactin 12 (Figure 1a). Nonribosomal peptides are considered secondary metabolites, compounds that are not essential for organism survival in permissive growth conditions, but confer advantages. 13 They are known to function naturally in growth and reproduction in restrictive environments, 14 , 15 inhibiting, or killing competitor microbes 2 , 13 and modulating symbiont interactions. 16 , 17 , 18 Nonribosomal peptide biosynthesis is common in microbial organisms, with the most prolific producers being members of the bacterial phyla Proteobacteria, Actinobacteria, Firmicutes, and Cyanobacteria, and the fungal phylum Ascomycota. 19
FIGURE 1.
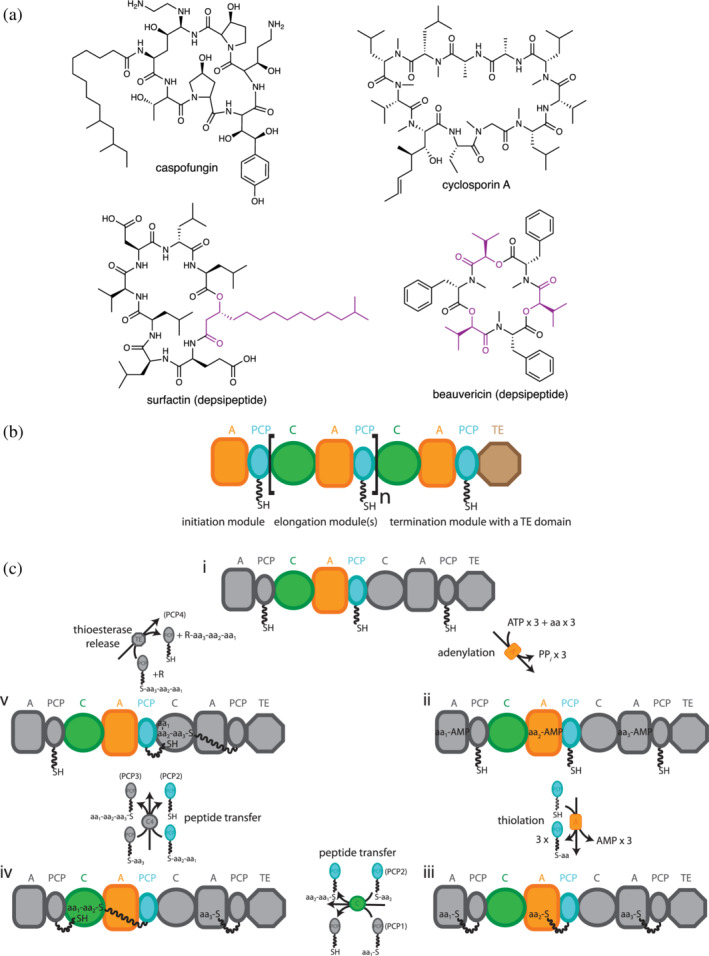
(a) Some nonribosomal peptides (top) and depsipeptides (bottom). Depsipeptides contain amide and ester bonds. Monomers from which the bridging ester oxygen originates are highlighted in purple. (b) The modular organization of nonribosomal peptide synthetases (NRPSs). Brackets highlight an elongation module. NRPS domains are: A, adenylation; C, condensation; PCP, peptidyl carrier protein; TE, thioesterase. (c) A typical NRPS synthetic cycle. The cycle starts with A domains activating monomers by adenylation, followed by their attachment to PCP domains in a phosphopantetheinyl moiety (ppant) as a thioester. PCP domains transport intermediates throughout the cycle. C domains catalyze amide (or ester, see Section 3.1) bond formation between adjacent PCP‐bound intermediates, elongating the peptide chain. Chain release occurs in termination modules, with TE domains the most common termination domains. Terminal reductase domains and terminal condensation domains also occur frequently. aa, amino acid; PPi, inorganic pyrophosphate. Adapted from Huguenin‐Dezot et al. 153
Nonribosomal peptides are small; typically containing ~2–20 residues. They often do not resemble ribosomally‐synthesized peptides, as they can be cyclic, branched or linear, and contain many nonproteinaceous moieties (Figure 1a). More than 500 monomer substrates, including l‐ and d‐ amino acids, hydroxy acids, keto acids, fatty acids, and aryl acids, are known to be incorporated into these peptides. 20 Also, many co‐synthetic modifications, 21 , 22 including methylation, 23 , 24 formylation, 7 , 25 epimerization, 26 , 27 residue cyclization, 28 , 29 , 30 oxidation, 31 , 32 reduction, 33 , 34 and hydroxylation, 35 , 36 are known to occur during peptide synthesis. This large substrate pool and set of tailoring modifications allow nonribosomal peptides to occupy an impressive volume of chemical space and achieve greatly diverse bioactivities. Furthermore, nonribosomal peptides can be postsynthetically modified, 22 , 35 , 37 and in nature NRPS systems are very commonly combined with polyketide synthases (PKSs) to make hybrid peptide‐ketide products. 12 , 38 Incorporation of other metabolites such as lipids 39 , 40 and carbohydrates 41 , 42 , 43 increases chemical diversity even more.
NRPSs synthesize peptides using a modular synthetic scheme that resembles an assembly line. An NRPS module is defined as the set of domains that together possess all the activities required to add one acyl residue to the nascent nonribosomal peptide chain. 44 , 45 , 46 , 47 , 48 A standard elongation module includes a condensation (C) domain, an adenylation (A) domain, and a peptidyl carrier protein (PCP) domain arranged as C‐A‐PCP (Figure 1b). (Note that, the PCP domain is equally commonly called the T domain. In that abbreviation, T stands for thiolation, which is misleading because all other domains perform the reaction after which they are named, while the holo form of this domain is a substrate for, not the catalyst of, thioester formation (“thiolation”). We advocate that T should instead stand for transfer, since the role it plays is analogous to that of the transfer RNA (tRNA) in the ribosomal system.)
A minimal elongation cycle (Figure 1c) starts when the A domain selects the cognate monomer substrate, for example an amino acid, from the pool of cellular metabolites. Selectivity is hard‐wired into each A domain by the set of residues that make up the substrate binding pocket of the A domain, known as “specificity code” residues. 49 , 50 , 51 , 52 The A domain then binds ATP and catalyzes the adenylation of the amino acid to make an activated aminoacyl‐adenylate. 4 , 53 Next, the C‐terminal subdomain of the A domain (Asub) rearranges to enable the PCP domain to bind and insert its 4′‐phosphopantheteinyl (ppant) moiety into the A domain active site. 54 , 55 , 56 , 57 The A domain catalyzes attack of the aminoacyl‐adenylate by the ppant thiol, covalently linking the aminoacyl moiety to the PCP domain as a thioester (aminoacyl‐S‐ppant‐PCP). The acyl thioester is then transported by the PCP domain to the C domain, which has binding sites for this acceptor aminoacyl‐S‐ppant‐PCP and a donor peptidyl‐S‐ppant‐PCP from the upstream module. The C domain will catalyze the condensation reaction, with the amino group from aminoacyl‐S‐ppant‐PCP nucleophilically attacking the thioester carbonyl, which transfers and elongates the nascent peptidyl intermediate. The elongated peptidyl‐S‐ppant‐PCP then moves to the C domain of the downstream module, where it acts as the donor substrate, passing off and further elongating the peptide and liberating the PCP to participate in the synthesis of the next peptide in the next catalytic cycle of its module.
The first and last modules have specialized domain composition and synthetic cycles. Initiation modules do not possess C domains, but only A‐PCP, and after thiolation their aminoacyl‐S‐ppant‐PCP intermediates are transported directly downstream to act as the donor substrate for the second module's C domain. Termination modules possess C, A, and PCP domains plus a specialized termination domain to which the PCP delivers the late‐stage peptidyl intermediate for final processing and release. The most common termination domain, the thioesterase (TE) domain, typically releases the peptide through hydrolysis or cyclization, 58 while reductase (R) domains catalyze release by converting the thioester link to an aldehyde 59 or alcohol 60 and specialized C domains 61 catalyze release by cyclization 62 and amide bond formation with small‐molecule acceptor. 28 , 61 , 63 , 64
There is typically a straightforward correspondence between the composition of a nonribosomal peptide and the NRPS which synthesizes that peptide 47 : Typically, the number of residues in the nonribosomal peptide corresponds to the number of modules in the NRPS, and the order and identity of the residues in the peptide corresponds to the order of the modules in the NRPS and each module's A domain substrate specificity. However, many exceptions to this correspondence are known, which include iterative 5 and module skipping NRPSs. 65 An NRPS system can consist of a single protein or can be split between multiple subunits which interact noncovalently through small docking or communication domains. 66 , 67 , 68 The genes for all the NRPS subunits, the in‐trans tailoring or postsynthetic modifying proteins, and other accessory proteins 69 , 70 , 71 are often found together in the microbial genome, in a “biosynthetic gene cluster” (BGC). This clustering, combined with predictability of A domain substrates and activity of each domain, means that increasingly sophisticated algorithms 51 , 52 , 72 , 73 , 74 , 75 , 76 , 77 , 78 are becoming more and more accurate at predicting the final product of a BGC from gene sequence alone.
All of the common NRPS domains have been well characterized both biochemically and structurally. A domains are ~60 kDa Rossmann‐like fold proteins that belong to the superfamily of adenylating enzymes. 4 , 55 , 79 They consist of a large subdomain of ~50 kDa (Acore, elsewhere called N‐terminal subdomain) which contains binding sites for an acyl monomer and ATP, and a C‐terminal small subdomain of ~10 kDa (Asub, elsewhere called C‐terminal subdomain) which contains a catalytic lysine for adenylation reaction 4 , 53 and which changes position radically during the synthetic cycle. 44 , 79 PCP domains are structured as 4‐helix bundles, homologous to acyl carrier protein (ACP) domains of PKSs and fatty acid synthases. 80 PCP domains require phosphopantetheinylation on the serine of their GGHS motif in PCP helix 2 for the NRPS to be active. This posttranslational modification is imparted by a phosphopantetheinyl transferase (PPTase) using coenzyme A as substrate. 69 , 81 As described in detail in Section 3.1, C domains are bi‐lobed pseudo‐dimers with two binding sites for PCP domains, connected by a tunnel that accesses the active site. 61 , 62 , 82 , 83 , 84 , 85 , 86 TE domains, also described more extensively in Section 4.2, are α/β hydrolase superfamily proteins. 58 , 87 , 88 , 89 , 90 , 91 , 92 They catalyze two half reactions: acyl transfer onto their catalytic Ser, followed by nucleophilic off‐loading, by promoting attack by water (leading to hydrolysis) or by a nucleophile in the peptide intermediate (leading to cyclization). 58 , 87 Another release strategy is reductive release, where R domains are termination domains that release the thioester peptide as an aldehyde or an alcohol using NADH or NADPH as a cofactor. 59 , 93 , 94 Furthermore, the broader structural view of NRPSs is starting to take shape with several recent studies of didomains, modules, or supra‐modular NRPSs showing how domains and modules coordinate in peptide synthesis. 7 , 34 , 44 , 45 , 56 , 57 , 83 , 85 , 86 , 90 , 95 , 96 , 97 , 98 , 99 , 100 , 101 , 102
2. DEPSIPEPTIDES
Nonribosomal depsipeptides are compounds with both ester and amide bonds in their structures 103 (Figure 1a). They are common natural products with a vast array of biological activities of medical and industrial relevance. 104 , 105 , 106 , 107 For example, daptomycin is used as an antibiotic for severe infections, 8 antimycin is used as a piscicide in industrial fish farming, 108 valinomycin has anti‐cancer and anti‐coronavirus activity 109 and surfactin has potential as an industrial tenside. 12 Some depsipeptides are involved in microbial pathogenesis, like the emetic toxin cereulide. 110
Depsipeptides have a variety of natural functions, from increasing organism fitness in K+ deprived environments 14 to participating in symbiotic relationships both in marine 111 and terrestrial 18 , 112 organisms. Among the most interesting examples in symbiosis is the role of depsipeptides in the relationships between microorganisms and ants: Actinomycin, valinomycin, and antimycin participate in a tripartite symbiotic relationship between Streptomyces sp., a fungus, and leaf‐cutter ants, 18 , 112 while dentigerumycin made in Pseudonocardia actinobacteria helps control pathogenic fungal growth in fungal gardens cultivated by ants. 113
As with other NRPS products, the chemical diversity of depsipeptides is outstanding. 105 , 106 , 107 In addition to the occurrence of at least one ester and one amide bond in their structures, most depsipeptides contain a wide variety of acyl groups and other moieties added through the assembly line logic of NRPSs. Depsipeptides may be postsynthetically modified after release from the NRPS, 37 although it should be noted that this review concentrates on nonribosomal synthesis, rather than on downstream tailoring.
Intriguingly, with one exception (asperphenamate, 114 see Section 4.1.1.2), all nonribosomal depsipeptide synthetases characterized to date produce cyclic depsipeptides. Linear products are sometimes observed from cyclic depsipeptide synthetase systems, such as ring‐opened antimycins, 115 but cyclic products predominate even in these systems. Cyclic natural products are very common, and their cyclic nature imparts advantages including reduced entropy costs in binding protein targets, 116 preorganization for binding ions, 15 , 117 and protease resistance. 118 , 119 However, linear nonribosomal peptides are not uncommon, so it is somewhat surprising how few synthetases are known to produce linear nonribosomal depsipeptides.
2.1. Classification systems for depsipeptides
More than 1,300 naturally occurring cyclic depsipeptides are known. 104 De Spiegeleer and coworkers 104 have made a thorough classification and a curated database of these from their chemical features. This classification separates depsipeptides based on whether they have one or more ester bonds, whether multiple ester bonds occur regularly or irregularly, and whether the ester oxygen is in the α, β, or more distal position. It is extremely detailed and very useful. For this review, as a parallel and complementary system to Taevernier et al., we classify the biosynthetic systems based on the characterized or putative mode of introduction of the ester bond(s). Our system of classification is based on the timing of introduction of the ester, which NRPS domain catalyzes ester bond formation, whether a single‐pass synthetic scheme or an iterative synthetic scheme was used by the NRPS to produce the compound, and the origin of the bridging ester hydroxy group (Figure 2). This classification system can be easily expanded if/when novel biosynthetic pathways are described. It is used to structure this review into sections, building from conceptually simpler depsipeptide pathways to more complex ones.
FIGURE 2.
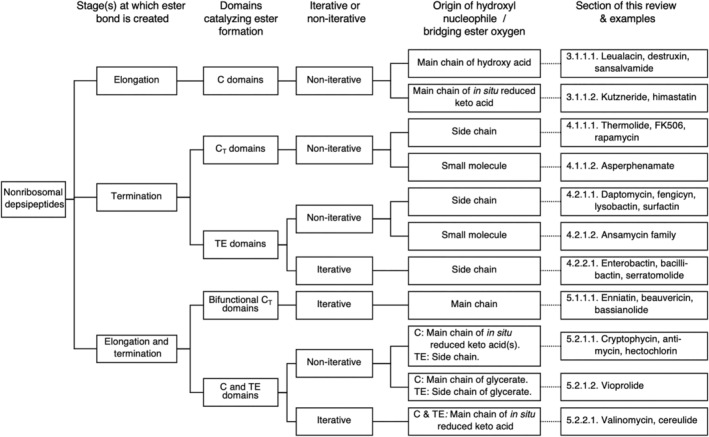
Nonribosomal depsipeptide classification by biosynthetic strategies of ester bond formation. Examples are not comprehensive
3. DEPSIPEPTIDES WITH ESTER BONDS FORMED DURING ELONGATION
3.1. The C domain catalyzes amide and ester bond formation
The condensation domain Cn (i.e., in module n) typically catalyzes peptide bond formation between a peptidyl‐S‐PCPn − 1 donor substrate and an aminoacyl‐S‐PCPn acceptor substrate (Figure 3a). In this condensation reaction, the α‐NH2 of the aminoacyl‐S‐PCPn nucleophilically attacks the thioester carbon of the peptidyl‐S‐PCPn − 1 donor substrate, which transfers the peptidyl moiety to the aminoacyl‐S‐PCPn, and elongates the nascent chain by one amino acid residue.
FIGURE 3.
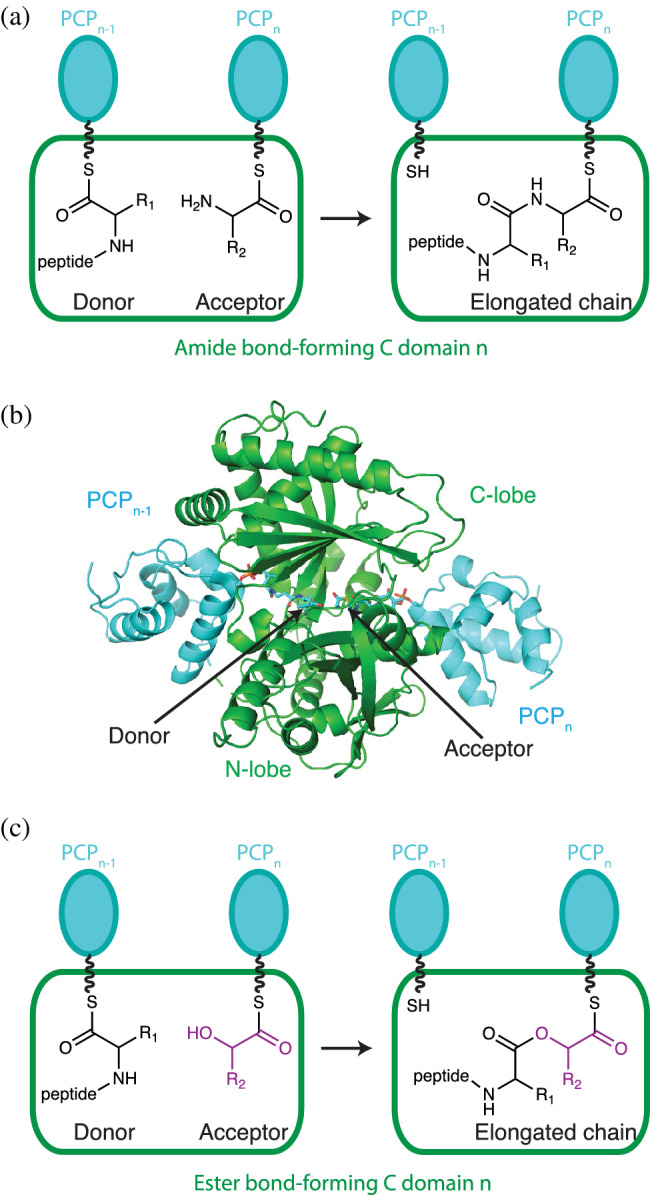
The condensation domain catalyzes amide and/or ester bond formation (a) Amide bond formation occurs in the active site of a condensation domain (green rectangle). Substrates are presented as thioesters attached to phosphopantetheine (wavy line) in PCP domains (cyan). The PCP in the preceding module (n − 1) relays the donor substrate, while the acceptor substrate resides in the current module (n). (b) Structure of condensation conformation of the C domain with acceptor and donor PCP domains, extracted from the dimodular NRPS LgrA. 97 The domain has N‐terminal and C‐terminal lobes which each have chloramphenicol acetyl transferase‐like folds. Donor peptidyl‐ppant and acceptor aminoacyl‐ppant are attached to PCP domains (cyan). (c) Some condensation domains can form ester bonds using OH groups as nucleophiles
Crystal structures reveal C domains to be pseudo‐dimeric domains formed by C‐terminal and N‐terminal lobes 82 (Figure 3b). The active site, identifiable by the conserved motif HHxxxDG, lies in a tunnel that runs through the interface between the lobes. The second His in the motif (His157 based on numbering of first condensation domain of CDA, PDB ID: 4jn3 120 ) is most important for reaction. It has been proposed to be a strong general base, deprotonating the α‐amino group of the acceptor substrate in preparation for nucleophilic attack, or part of an electronic transition state stabilization, 121 or (by us) to principally act in substrate positioning. 84 Mutation of His157 leads to loss of condensation in some systems 122 but not others, 61 , 82 , 123 and heterocyclization domains, which are homologous and related to C domains and also catalyze the same condensation reaction, have a hydrophobic residue in place of His157. 28 The acceptor and donor PCP domain binding sites on the C domain are on the two ends of the active site tunnel. 62 , 83 , 84 , 85 , 86 , 97 Both PCP domain binding sites are made of portions of both N‐terminal and C‐terminal lobes, a reflection of the pseudosymmetry of those lobes 61 (Figure 3b).
Despite more commonly catalyzing amide bond formation, the ester forming activity of C domains has been confirmed directly in several systems. This activity was first verified biochemically for the biosynthesis of two molecules that happen not to have amide bonds in them and are thus not depsipeptides, the fungal metabolite fumonisin 124 and the antitumor antibiotic C‐1027. 64 , 125 In fumonisin biosynthesis, the C domain Fum14p catalyzes ester bond formation between a donor polyketide thioester and an acceptor tricarballylic thioester. 124 In C‐1027, the C domain SgC5 catalyzes ester bond formation between a donor enediyne core and the acceptor (S)‐3‐chloro‐5‐hydroxy‐β‐tyrosine thioester. 64 , 125 SgC5 is also capable of amide bond formation, suggesting C domains catalyze ester and amide bond formation with analogous catalytic mechanisms. 125 This suggestion is further supported by the conservation of either the full canonical HHXXXD motif. 64 or at least the important second histidine within the motif. 124 Experimental evidence of C domain installation of ester bonds into depsipeptides comes from domain deletion and substitution experiments 126 , 127 and in vitro biochemical assays of an ester bond forming module. 128
3.1.1. Ester bond formation during elongation by a C domain in noniterative synthesis
3.1.1.1 | Ester formed with main chain hydroxyl originating from hydroxy acid substrate
NRPS biosynthetic pathways which introduce hydroxy acid residues during elongation can closely resemble the typical NRPS that introduce amino acid residues. In the conceptually simplest case, the A domain of an elongation module recognizes a hydroxy acid and this hydroxy acid is activated and transferred to the PCP domain. The resulting hydroxyacyl‐S‐PCPn is then used as the acceptor substrate in C domain catalyzed ester bond formation (Figure 3c). Systems that use this conceptually simple case are almost always fungal. 129
To use a hydroxy acid as a building block substrate, both (1) availability and (2) selection of such substrates must be addressed by the BGC. (1) α‐hydroxy acids are not common in primary metabolism, so in fungal systems that use α‐hydroxy acids as NRPS substrates, the BGC often contains standalone ketoreductase (KR) proteins which convert α‐keto acids to α‐hydroxy acids in an NADPH‐dependent reduction reaction 130 , 131 , 132 , 133 , 134 (Figure 4), with keto acids arising from branched‐chain amino acid biosynthesis pathways 135 or from glycolysis. These KR proteins are not closely related to PKS KR domains, 128 , 136 , 137 but belong to the ApbA_C superfamily, which includes ketopantoate reductases. 130 , 133 , 138 Deletion of the KR protein abolishes production of the depsipeptide, which can be rescued by supplementing the media with α‐hydroxy acids. 131 (2) A domain in these systems can discriminate between α‐amino and α‐hydroxy groups through a yet unknown mechanism (Figure 4a). In these A domains, the position of a highly conserved Asp (Asp235 in GrsA‐PheA numbering used here for A domains 4 ) is occupied by a Gly. 130 Asp235 makes key hydrogen bonds with the substrate's α‐amino group in amino acid selecting A domains, and its mutation leads to loss of adenylation cognate amino acids. 139 It is unknown whether this substitution is enough to confer α‐hydroxy acid selectivity, and without a substrate‐bound structure of an α‐hydroxy acid selecting A domain, the mode of α‐hydroxy acid selection remains an open question.
FIGURE 4.
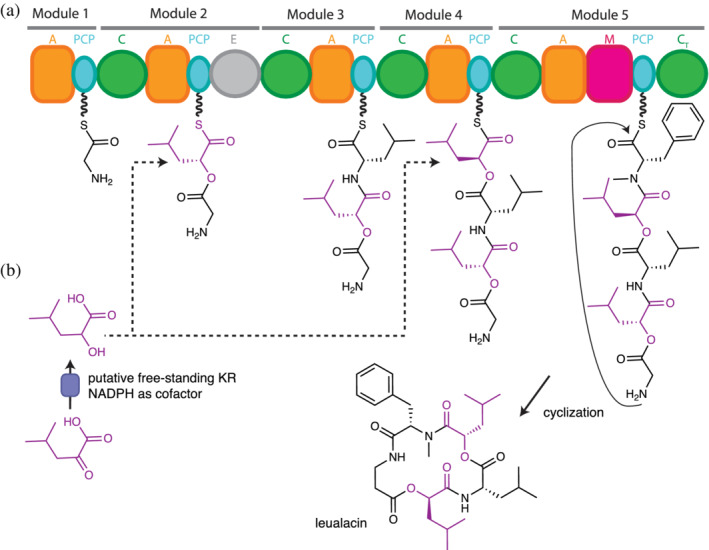
Proposed biosynthesis of leualacin. (a) Specialized C domains in Modules 2 and 4 catalyze ester bond formation using hydroxyacyl thioesters as acceptor substrates. Specialized A domains in the same modules activate the hydroxy acids. (b) Stand‐alone ketoreductases (KR) produce hydroxy acids from keto acids in fungal systems. Figure based on Zhang et al. 140
Ester bond insertion by direct selection and incorporation of α‐hydroxy acid occurs twice leualacin biosynthesis 140 (Figure 4): Leualacin is a cyclic depsipeptide which has two α‐hydroxyacyl residues 140 and acts as a potent calcium channel blocker. 141 The five‐module NRPS responsible for its biosynthesis was identified by genome sequencing and gene disruption. 140 Based on the collinearity rule, it is assumed that the A domains in Modules 2 and 4 select α‐hydroxy‐isocaproic acid (leucic acid), and the C domains of these modules almost certainly perform ester bond formation.
Other depsipeptides for which ester bond formation occurs during elongation and is catalyzed by a C domain include the destruxins 131 , 132 , 142 , 143 and sansalvamide. 132 Destruxins are cyclic insecticidal molecules synthesized in fungal entomopathogens by a 6‐module NRPS. 142 Module 2 is likely responsible for inserting the ester bond, starting with direct selection of the α‐hydroxy‐isocaproic acid by the A domain. 132 , 143 , 144 Timing of the ester bond formation step was controversial, with elongation 143 and terminal cyclization 131 both proposed. Cluster analysis of a BGC for a similar molecule, sansalvamide 132 lent support to the elongation proposal, which is now broadly accepted. 144
3.1.1.2 | Ester formed with main chain hydroxyl originating from keto acid substrate
Bacterial depsipeptide synthetases also form ester bonds during elongation. In contrast to the case in fungi, these hydroxy acids usually arise from specialized modules that select and reduce keto acids. As is clear from this review, the use of these specialized modules is not the only pathway NRPSs employ to insert esters, but their domain composition is dedicated for incorporation of hydroxy acid residues into dipeptides and are thus called “depsipeptide modules”. 34
Discovery, characterization, and an example of the “depsipeptide module” (C)‐A‐KR‐PCP
Though the genes responsible for the production of the bacterial depsipeptide valinomycin were identified as early as 1990, 145 sequencing and annotation of the BGCs for valinomycin 109 and the similar compound cereulide 146 occurred ~15 years later. This sequencing suggested that a KR domain, related to KR domains of PKSs, was inserted into A domains in two modules of these depsipeptide synthetases. The KR insertion site in A domains of depsipeptide modules for cereulide 33 and other bacterial depsipeptides 38 , 147 , 148 was proposed to be between motifs A8 and A9 of the Asub domain. This would create an interrupted A domain arrangement similar to that in the recently determined structure of an A‐methyltransferase didomain. 23
BGCs for both valinomycin and cereulide have two NRPS genes that encode 4‐module, 2‐subunit NRPSs. 109 , 146 The initiation module has domains A‐KR‐PCP, and the third module has domains C–A‐KR‐PCP. The domains in third modules are split between the two subunits, with the C domains the last domains of the first subunit (CesA/Vlm1) and A‐KR‐PCP the first three domains of the second subunit (CesB/Vlm2), meaning each subunit starts with domains A‐KR‐PCP. Magarvey et al. sub‐cloned the two A‐KR‐PCP constructs from cereulide synthetase (Ces) and performed functional assays which revealed crucial information about depsipeptide synthetases. 33 It was shown that the A domains displayed high selectivity for α‐keto acids over α‐hydroxy acids and α‐amino acids of either chirality, 33 , 147 and that the A domain also had side chain specificity. 34 , 147 , 149 In addition, keto reduction was shown to be NADPH‐dependent and highly stereospecific. 33 The substrate for ketoreduction was shown to be the tethered ketoacyl‐S‐PCP, delineating the sequence of reactions in these modules (Figure 5b and 6a).
FIGURE 5.
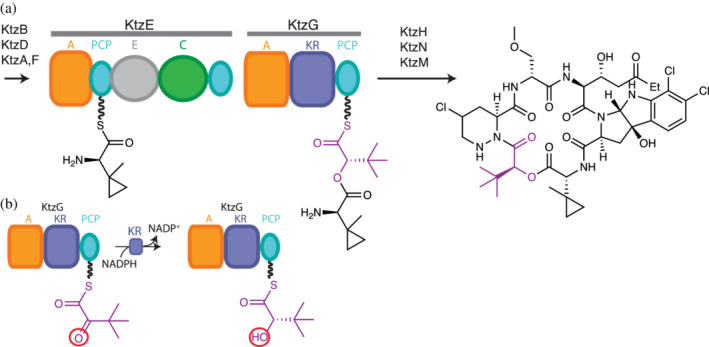
Proposed kutzneride biosynthesis. 147 (a) The C domain in KtzE catalyzes ester bond formation using an α‐hydroxyacyl thioester (magenta) in KtzG as acceptor substrate. (b) The A‐KR‐PCP module KtzG generates α‐hydroxy acids from α‐keto acids. See also Figure 6
FIGURE 6.
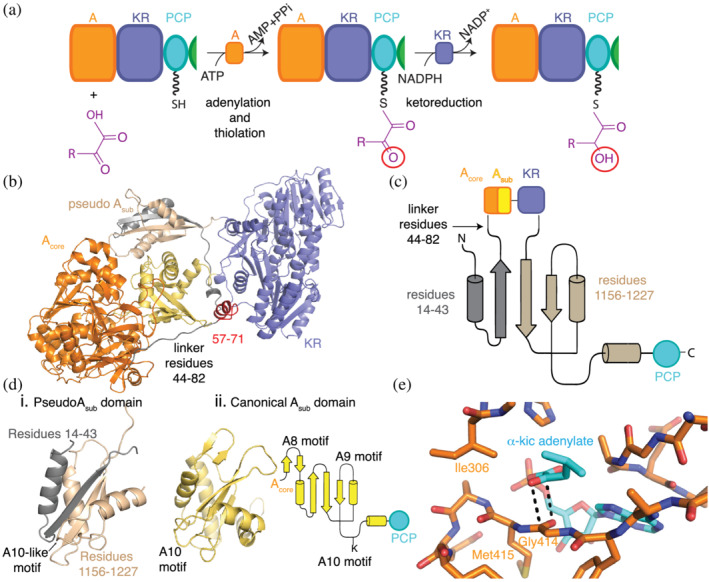
An initiation depsipeptide (A‐KR‐PCP) module. Note that elongation depsipeptide modules include the C domain (C‐A‐KR‐PCP). (a) Specialized A domains activate α‐keto acids by adenylation and attach the keto residue to the PCP domains for transport to ketoreductase (KR) domains within the same module. KR domains reduce the α‐keto group, generating α‐hydroxy acyl thioesters, which can be used in downstream reactions. Cartoon (b) and topology (c) representations show that the A domain has integral Acore (orange) and Asub (yellow) domains in the monomer structural model of the A‐KR‐PCP module of StrA from Bacillus stratosphericus LAMA 585 (based on dimeric structure PDB ID: 6ULW 34 ). A small linker connects the Asub and KR (purple) domains, followed by a small domain (wheat and gray) with a tertiary structure (d) that resembles a canonical Asub domain. (e) α‐ketoacids bind to A domains through an antiparallel carbonyl‐carbonyl interaction (PDB ID: 6ULX, 6ULY). Figure adapted from Alonzo et al. 34
Since this initial characterization, scores of BGCs have been shown to include (C)‐A‐KR‐PCP depsipeptide modules. These modules have an easily recognizable indicator of hydroxy acid incorporation in NRPSs. Among the bacterial depsipeptide synthetases with (C)‐A‐KR‐PCP depsipeptide modules are kutzneride, 147 , 150 antimycin, 18 didemnin, 151 cryptophycin, 38 , 128 cereulide, 33 and valinomycin. 152 , 153 Most substrate α‐keto acids originate in bacteria from branched‐chain amino acid biosynthesis, while pyruvic acid originates from glycolysis. 154 Kutzneride synthetase 147 has a single depsipeptide module as its Module 2, and except for having a bypassed PCP domain situated after the C domain in KtzE (Figure 5a) is a straightforward example for hydroxy acid residue addition into bacterial depsipeptides:
The kutznerides are a family of cyclic hexadepsipeptides with antimicrobial and antifungal properties 147 , 150 (Figure 5a). Kutzneride synthetase has six modules in its four subunits, KtzE‐G‐H‐N. The substrate for Module 1 is the unusual amino acid (1‐methylcyclopropyl)‐d‐glycine (MecPGly; made by KtzB, D, A, F). Module 2, spread across KtzE and D, has the domain architecture C2‐PCP2a–A2‐KR2‐PCP2b (Figure 5b). A2 selects α‐ketoisovaleric acid (or 2‐keto‐3,3‐dimethylbutanoic acid), activates it by adenylation, and transfers the ketoacyl moiety to PCP2b. The α‐ketoisovalyl‐S‐PCP2b is reduced by the imbedded KR2 domain using the co‐substrate NADPH, producing α‐hydroxyisovalyl‐S‐PCP2 (HIV‐S‐PCP2). This will act as the acceptor substrate for C domain‐mediated ester condensation with donor MecPGly‐S‐PCP1, giving MecPGly‐HIV‐S‐PCP2. MecPGly‐HIV‐S‐PCP2 will be donor substrate for C3‐mediated amide bond formation, liberating PCP2 for the next round of depsipeptide synthesis 33 , 34 , 38 , 128 , 147 , 150 , 152 , 153 (Figure 5b). Notably, the only other characterized depsipeptide synthetase to insert a single hydroxy acid residue during NRPS elongation is involved in the biosynthesis of the antibiotic himastatin, which is shares >60% sequence identity and also features a C‐PCP–A‐KR‐PCP module. 155 , 156 Mature himastatin has a peculiar dimeric structure introduced by postsynthetic tailoring.
Structural studies of a depsipeptide module
Until this year, no structural information was available for depsipeptide modules. That changed with our report of the crystallization of the initiation A‐KR‐PCP module from Bacillus stratosphericus LAMA 585 stratospherulide synthetase subunit A (StrA) and its structure determination 34 (Figure 6). This structure reveals a very surprising architecture for depsipeptide modules. Contrary to the predictions of an interrupted A domain, 33 , 38 , 147 , 148 StrA A1 is completely intact, with full‐length Acore and Asub domains (Figure 6b). The KR domain is directly downstream of the intact A1 and connected by a short linker. Even more surprisingly, there was clear density that allowed visualization of another small domain, located in space next to Asub and between the Acore and KR domains (Figure 6b). This domain has the same fold as an Asub domain and was termed a pseudo Asub domain (Figure 6b–d). The pseudo Asub is a dramatically split domain. Around two thirds of this domain comes from a sequence C‐terminal of the KR domain, and approximately one third comes from the very N‐terminus of the protein, meaning that residues over 1,100 amino acids apart in sequence assemble into a folded unit (Figure 6b,c). The N‐terminal portion of the pseudo Asub is connected to the intact A domain by a long linker. Deletion of the pseudo Asub or shortening of the linker gives constructs that are unable to perform depsipeptide synthesis and have reduced stability. Bioinformatics strongly suggests that known depsipeptide modules feature an intact A domain and a split pseudo‐Asub. 34
The revelation that the A domain is intact allowed us to interrogate the mechanism by which depsipeptide synthetase A domains are selective for α‐keto acids. 34 The specificity‐determining residues for keto acid‐selecting A domains had been noted to contain a substitution of residue Asp235 (PheA numbering 4 ) by an aliphatic residue, 147 but it was not clear if/how that allowed α‐keto acid selection. Studies of the promiscuous A domain McyG, which selects unusual phenylpropanoids and which also has an aliphatic residue in place of Asp235, showed that Asp235 enhances binding of the unusual hydrophobic monomer, but the accompanying co‐crystal structure had phenylalanine bound. 99 To elucidate the mechanism of keto acid selection, we excised the intact A domain and determined high resolution structures of it bound to its in situ formed α‐keto adenylate (Figure 6e). 34 These structures revealed that the isoleucine (Ile306) in the analogous position to PheA Asp235 4 was not involved in α‐keto acid binding. Moreover, there is no hydrogen bonding at all between the substrate α‐keto acid and the A domain. Instead, the α‐keto moiety interacts through an antiparallel carbonyl‐carbonyl interaction with the backbone of Gly414. Antiparallel carbonyl‐carbonyl interactions have been previously characterized in small molecules 157 and are involved in the stabilization of unfavorable protein tertiary structure. 158 We observed that the interaction in StrA is facilitated by the presence of an active site methionine that substitutes for a highly conserved proline found in amino acid‐selecting A domains. The substitution effectively creates a flat conformation of Gly414 and Met415 (Figure 6e) that favors the stacking interaction. Mutation of the methionine to proline abolished keto acid selectivity, indicating that the disruption of the flat surface disrupts the carbonyl‐carbonyl interaction. A domains selective for aryl acids (which lack and α‐amino/hydroxy/keto motiety) also harbor the proline to methionine substitution. 57 , 159 , 160
The larger structure from the crystals of the entire A‐KR‐PCP module also enabled the first visualization of an NRPS KR domain. 34 As predicted, the overall fold of the KR domain is similar to that of PKS KR domains and most closely resembles β‐ketoreducing PKS KR domains. 33 These KR domains are pseudo‐dimeric with the N‐terminal Rossmann‐like fold having a structural role and the C‐terminal one containing the active site with its Tyr‐Ser‐Lys catalytic triad. 161 , 162 , 163 They can be classified according to the stereochemistry of their product, A‐type producing l‐β‐hydroxy acids and B‐type producing d‐β‐hydroxy acids. 136 , 137 The type is distinguishable from sequence in PKSs, with a highly conserved Trp in type A and a Leu‐Asp‐Asp in type B KR. 164 In both types, the 4′‐pro‐S hydrogen faces the active site Tyr. The chirality of the product is determined by the approach of the β‐ketoacyl‐S‐ACP, with the conserved motifs directing the β‐ketoacyl‐ACP to one or another ACP binding site with grooves reaching the active site from two opposite directions. 137 , 165 Depsipeptide KS domains have no identifiable sequence motifs 34 , 128 analogous to those found in PKS KR domains, 137 and both putative PCP binding sites and grooves to the active site appear feasible in the KR domain of StrA. 34 Despite being present in the crystals of StrA A‐KR‐PCP, the PCP was disordered. The mechanism of stereospecificity in NRPS KS domains awaits biochemical interrogation and structure determination of depsipeptide modules with each stereospecificity in their KR conformations, such as those underway in our lab. Nonetheless, the study revealed how keto acids can be used for ester bond formation in depsipeptide modules.
4. DEPSIPEPTIDES WITH ESTER BONDS FORMED DURING TERMINATION
4.1. The C domain can catalyze termination in depsipeptide synthesis
NRPSs employ a series of termination mechanisms, several of which can result in ester bond formation during peptide release. In fungal NRPSs, including peptide and depsipeptide synthetases, a common pathway of termination uses C‐terminal, cyclizing C domains (CT domains). 62 , 166 These CT domains catalyze nucleophilic attack on the ester carbon of the terminal peptidyl‐S‐PCP by a nucleophile within the peptidyl intermediate, which cyclizes and releases the peptide. Cyclizing CT domains cluster together phylogenetically, 166 , 167 suggesting one or few founding CT domains may have first evolved this specialization. The best known nonribosomal peptide whose biosynthesis terminates in this way is cyclosporin A 10 , 168 , 169 , 170 (Figure 1) and the best characterized CT domain responsible for terminal cyclization is that in the biosynthesis of fumiquinazoline F. 62 , 166 , 171 Structures including CT domains show them to be very similar to elongating C domains, but with adaptations for an active site dedicated to cyclization instead of elongation, such as a narrower solvent channel, a blocked acceptor site and an overall more compact conformation, as observed in TqaA. 62 Neither cyclosporin nor fumiquinazoline F is a depsipeptide, but the knowledge of amide forming CT domains should be relevant for ester forming CT domains.
4.1.1. Ester bond formation during termination by a C domain in noniterative synthesis
4.1.1.1 | Ester formed during cyclization with side chain hydroxyl
Several CT domains which introduce ester bonds are found in hybrid PKS‐NRPSs. Thermolides are nematocidal polyketide macrolactones which include a single amino acid that are produced by Talaromyces thermophilus strains (Figure 7). 172 The polyketide portion is generated by the highly reducing PKS ThmA, while the single amino acid in the structure is added by ThmB, an NRPS subunit with domains C‐A‐PCP‐CT. After amide bond formation between the amino acid and the polyketide intermediate in the first C domain, the CT domain catalyzes cyclization by ester bond formation using a hydroxy group in the polyketide as a nucleophile. The cyclization activity of CT was confirmed by site‐directed mutagenesis of key residues in its active site. 172
FIGURE 7.
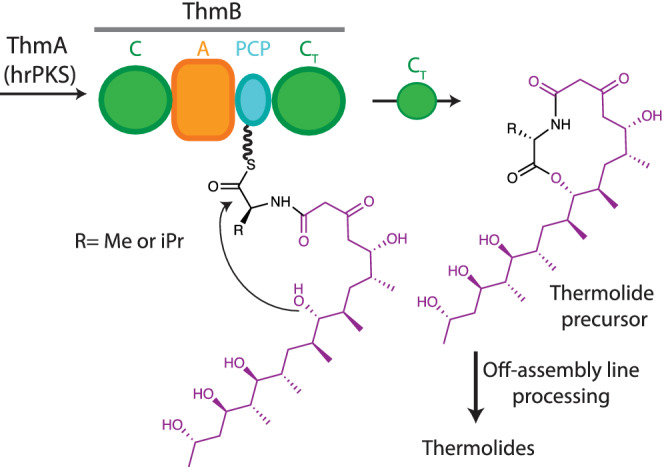
Thermolide biosynthesis. The CT domain catalyzes cyclization using a hydroxy group from the polyketide moiety (magenta) as a nucleophile, forming the lactone portion of the thermolide precursor (right)
Other depsipeptides that are synthesized through ester bond forming cyclization are antalid, 167 the potent macrolide immunosuppressants rapamycin (made famous by its target, mammalian target of rapamycin [mTOR]), and the similar compounds FK506 and FK520. The latter molecules are pipecolate‐containing polyketides 173 , 174 made by BGCs, 173 with three PKS genes and one four‐domain NRPS with the domain arrangement C‐A‐PCP‐C, named RapP or FkbP. Nielson et al. showed ATP‐dependent pipecolate adenylating activity as early as 1991 for FkbP, 175 and sequencing, gene disruption and heterologous expression experiments characterized the analogous role for RapP in rapamycin biosynthesis a few years later. 174 The A domain activates pipecolate, the first C domain condenses the aminoacyl thioester to the polyketide core, 173 and it is believed that the second C domain catalyzes hydroxyl group 26 attack for ester bond forming cyclizing release.
4.1.1.2 | Ester formed during cyclization with a hydroxyl from a small molecule acceptor
Asperphenamate is a fungal linear depsipeptide 114 , 176 , 177 made of two phenylalanine and two benzoic acid residues assembled in an interesting way (Figure 8). 114 Through gene deletion, in vitro work, HPLC, NMR, and MS identification, Li and colleagues 114 showed that two dimodular NRPS, ApmA, and ApmB, make asperphenamate and proposed a biosynthesis pathway. ApmA has domains A‐PCP‐C‐A‐PCP‐R, and ApmB, domains A‐PCP‐C‐A‐PCP‐CT. ApmA assembles N‐benzoylphenylalaninyl‐PCP2‐ApmA, which its terminal R domain, via four electron reductive release, turns into free N‐benzoylphenylalaninol. This is proposed to be the small molecule used by the CT domain of ApmB as the acceptor molecule in reaction with the N‐benzoylphenylalaninyl‐PCP2‐ApmB donor, which couples the two dipeptide intermediates to the final linear depsipeptide asperphenamate. Although this role for the CT domain of ApmB has not been formally shown, it would not be unexpected, as terminal C domains which catalyze release though condensation with small molecule acceptors are known in bacillamide, 28 , 63 wortmanamide B 178 and C‐1027 64 , 125 biosynthetic pathways.
FIGURE 8.
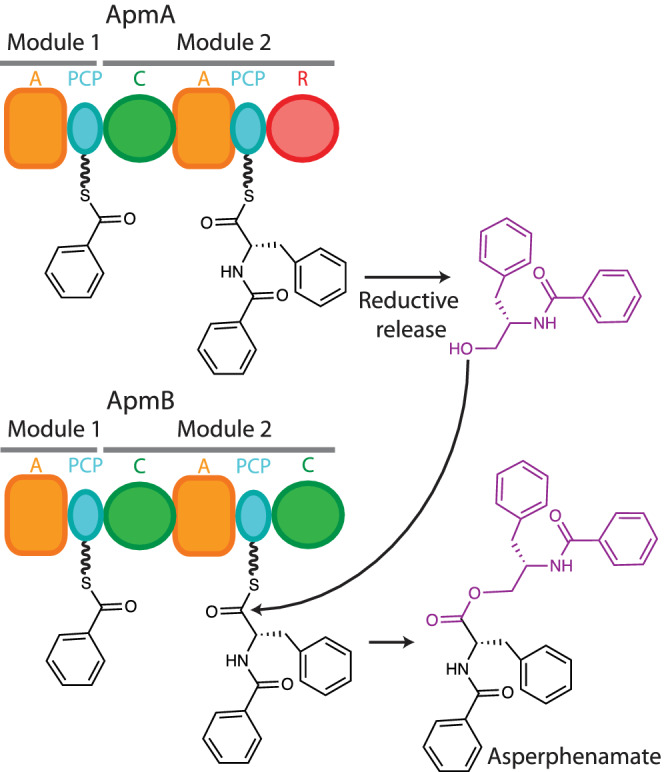
Proposed asperphenamate biosynthesis based on Li et al. 114 The R domain in ApmA releases N‐benzoylphenylalaninol, which acts as a nucleophile in the ester bond forming reaction in the C‐terminal C domain of ApmB, releasing asperphenamate. Note that the famous compound taxol has a depsipeptide moiety that resembles asperphenamate, but taxol is synthesized by yew trees in a completely different manner 251
There are sure to be many other depsipeptides and hybrid‐PKS‐depsipeptides for which C domains install ester bonds. Other review articles 104 , 106 highlight a variety of compounds which include single α‐hydroxy acyl residues, but most of their BGCs have not been identified or characterized. Biochemical and biophysical characterization of these biosynthetic enzymes will shed more light on these processes in the years to come.
4.2. The TE domain can catalyze ester bond formation in depsipeptide synthesis
One of most straightforward routes to produce a cyclic nonribosomal depsipeptide is to introduce the ester bond in the release step, exemplified in Section 4.1 by fungal CT domains. In bacteria, this is very commonly catalyzed by the terminal TE domains. Since the TE domain is so central to depsipeptide synthesis (as well as other nonribosomal peptide and polyketide synthesis), a short general summary of TE structure and function is warranted here (Figure 9).
FIGURE 9.
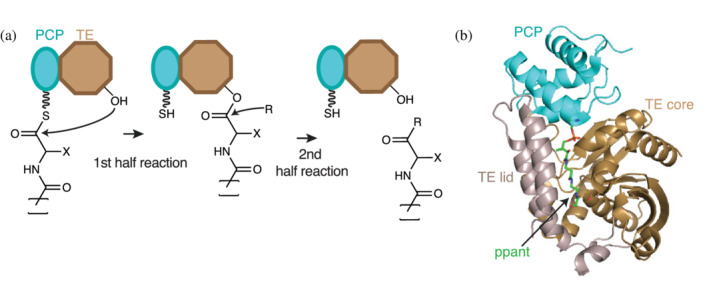
The thioesterase (TE) domain. (a) TE domains (brown) use an active site Ser (or, in rare instances, a Cys) residue to attack the elongated acyl thioester chain in the first half reaction. The resulting ester (thioester in case of Cys) is released by nucleophilic attack in the second half reaction. The nature of the incoming nucleophile determines the chemistry of the final product. (b) Cartoon representation of the holo‐PCP‐TE structure of entF (PDB ID: 3tej). 95 The TE domain has an α/β hydrolase fold formed by core (brown) and lid (grey) regions. TE lids are variable from protein to protein, and can be mobile
TE domains are α/β hydrolase proteins related by evolution and mechanism to serine proteases. 58 , 87 TE domains catalyze chain release through two half reactions using a catalytic Ser‐His‐Asp triad (Figure 9a). 58 , 87 , 88 , 89 , 90 , 91 , 92 In the first half reaction, the His residue increases nucleophilicity of the Ser hydroxy by proton abstraction, allowing attack on the thioester of the peptidyl‐S‐PCP, forming a covalent acyl‐O‐TE intermediate. In the second half reaction, a nucleophile (R in Figure 9a) attacks the acyl‐O‐TE intermediate, displacing the Ser oxygen and releasing the chain from the TE domain. The nucleophile can be water or a hydroxy or amine group, leading to carboxylic acid, ester, or amide formation. 94 If the nucleophile is water, linear peptide is released. 179 If the nucleophile is from within the peptide intermediate (side chain or first residue) a cyclic (depsi)peptide is released (as a branched or unbranched macrocycle, respectively). 180 , 181 If the nucleophile is contained within the next peptide intermediate (peptidyln+1‐S‐PCP), oligomerization occurs (peptidyln‐peptidyln+1‐S‐PCP).
Several TE domains from NRPS and PKS systems have been structurally characterized 58 , 88 , 89 , 90 , 91 , 92 , 153 , 182 , 183 (Figure 9b). They show the α/β hydrolase fold, the active site triad and a putative oxyanion hole to stabilize the negatively charged tetrahedral transition state. The structures also reveal an element inserted into the α/β hydrolase fold between strands 6 and 7, which is positioned on top of the active site, and was thus called “the lid” (Figure 9b). This element has high sequence and structural variability within TE domains, from a simple two‐helix structure (e.g., DEBS‐TE 88 ), to subdomains of >100 residues (e.g., AB3403‐TE 85 ). The lid has been proposed to act as a solvent shield, to interact with the PCP domain, and/or interact with the peptide intermediate, 95 but correlation of lid character with substrate length, complexity or release mechanisms has not been forthcoming. 58 Lids can be mobile, having been observed in different positions or refolded in structures of the same TE domain 153 , 184 or be fully or partially disordered in structural studies. 89 , 185
TE domains must interact with the PCP domain for their first half reaction. The best first views of this interaction come from apo PCP‐TE NMR 90 and holo PCP‐TE crystal 95 structures from enterobactin synthetase (Figure 9b). They show PCP‐TE contacts and show a crevice for ppant binding formed by one of the helices of the lid and the active site canyon. The crystal structure shows putatively important contacts of the ppant arm with core and lid in the TE domain.
Attempts to generate intermediate‐bound acyl‐O‐TE structures, representative of the product of the first half reaction and the substrate of the second half reaction, are hampered by the relative short lifetime of the acyl ester intermediates. 185 , 186 A few structures of TE‐substrate complexes exist, using phosphonate analogues 182 , 183 , 187 or employing an expanded genetic code strategy to generate stable amide intermediates 153 (discussed in Section 5.2.2.1). In the analog‐bound structures of the pikromycin TE bound to a pentaketide phosphonate, 187 the linear pikromycin intermediate curls back in the cavity between the lid and active site toward the catalytic Ser. It was proposed that a “hydrophilic barrier” formed by the lid and ordered water favors the cyclization conformation. There is very little residue‐level conservation between lids of TE domains.
4.2.1. Ester bond formation during termination by cyclization by TE domains in noniterative synthesis
4.2.1.1 | Ester formed during cyclization with side chain hydroxyl
TE‐mediated cyclization commonly uses hydroxy groups from side chains of upstream residues in the peptide intermediate as final acceptor nucleophiles (Figure 10). In a very common case, the TE domain acts on a fully elongated linear peptidyl‐PCP. In the first half reaction, the peptidyl moiety is transferred to the active site Ser of the TE domain forming a peptidyl‐O‐TE intermediate. In the second half reaction, the TE domain catalyzes ester bond formation by nucleophilic attack of the side chain OH on the peptidyl ester, which is followed by cyclization and release of the cyclic nonribosomal depsipeptide. 185 Two examples are in synthesis of daptomycin, where the nucleophile is a threonine side chain hydroxyl 188 (Figure 10a) and fengycin, where the nucleophile is a tyrosine side chain hydroxyl. 189
FIGURE 10.
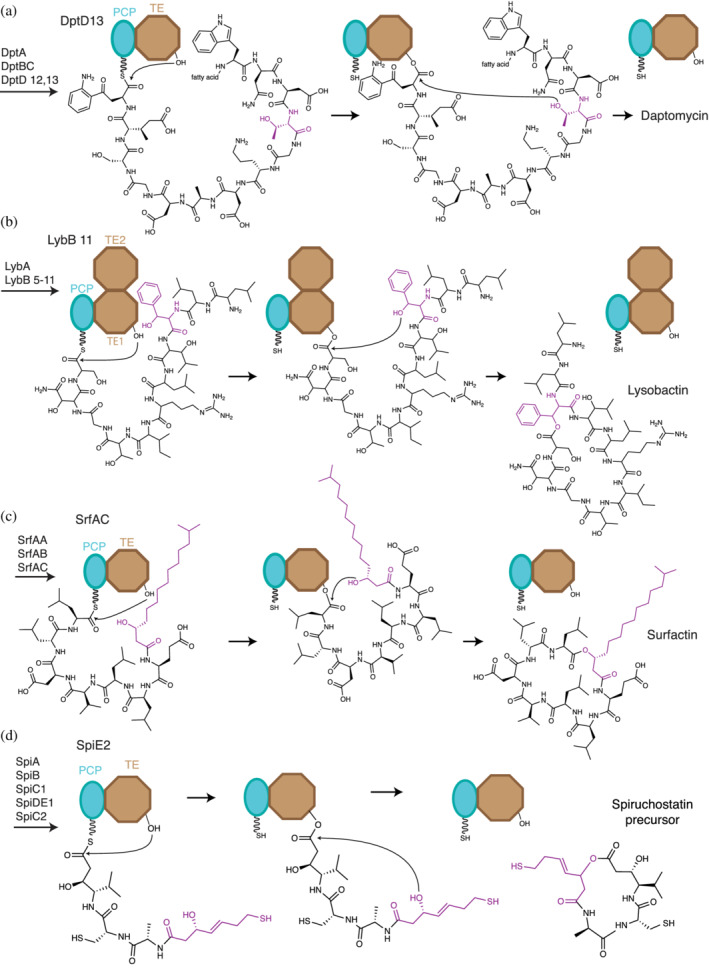
Ester bond formation by cyclization in TE domains. Final steps of (a) daptomycin, (b) lysobactin, (c) surfactin, and (d) spiruchostatin biosynthesis. For simplicity, only the last two domains of the NRPS systems are represented. Cyclization occurs by nucleophilic attack of a hydroxy group within the acyl‐ester intermediate on its own ester carbon linked to the active site serine. The hydroxy can be located in (a) the side chain of a proteogenic amino acid residue, (b), a β‐hydroxylated amino acid residue (c) a fatty acyl moiety or a (d) polyketide moiety
Daptomycin is of high clinical relevance. Sold under the brand name Cubicin, it is a half‐billion dollar cyclic lipodepsipeptide antibiotic used to treat severe skin infections. 8 Daptomycin synthetase is a 13‐module, 3‐subunit NRPS 190 whose TE domain can also accept small molecule thiophenol analogues of peptidyl‐S‐PCP substrates 40 for chemoenzymatic synthesis. Teixobactin, another potent antibiotic against Gram‐positive bacteria, 191 is also released from its assembly line by cyclization with a Thr sidechain hydroxyl acceptor in a way very similar to daptomycin. Fengycin is a decadepsipeptide with antifungal activity that is cyclized through a mechanism similar to daptomycin, using the sidechain OH in residue Tyr4 as a nucleophile. 189 Structures of its TE domain show a single active site lid conformation and allowed the authors to unambiguously identify its oxyanion hole. 185
The hydroxyl nucleophiles in the above two examples derive from proteinaceous amino acids, but other systems use moieties installed during co‐synthetic hydroxylation tailoring. Cyclization through hydroxylated amino acids occurs in polyoxypeptin A, lysobactin and obafluorin synthesis. 102 , 192 , 193 In polyoxypeptin A biosynthesis, a P450 enzyme is proposed to catalyze the hydroxylation of leucine to produce a β‐OH‐Leu moiety containing the nucleophile for ester bond formation. 193 The nucleophilic hydroxyl in lysobactin biosynthesis 192 is installed in a similar way by a tailoring enzyme which hydroxylates Phe‐S‐PCP to β‐OH‐Phe‐S‐PCP (Figure 10b). A peculiarity of the lysobactin biosynthesis system is that the gene for the termination module has two TE domains in tandem. The first TE domain is the normal “type I” TE, catalyzing cyclization and release, while the second is likely proteolyzed to a bona fide free‐standing type II TE domain in vivo. (Type II TE domains that hydrolyze misprimed PCP domains to rescue stalled NRPSs). In obafluorin biosynthesis, the TE domain has an altered triad composed of Cys‐His‐Asp, with a Cys in place of the normal Ser, and where the Asp is in a different topological position from Asps of other TE domains. This positioning is suggested to help in the transthioesterification reaction from PCP to TE thioester. 102
The hydroxyl which is used in ester bond formation in depsipeptides need not come from an aminoacyl side chain. Cyclic lipo‐depsipeptides and polyketide‐depsipeptides are common, and cyclization often occurs through a hydroxy nucleophile from a hydroxylated acyl/ketide chain. Around 20% of depsipeptides in the De Spiegeleer chemical classification are hybrids. 104 For example, romidepsin, or FK228, is an FDA approved anti‐cancer made in Chromobacterium violaceum as a cyclic polyketide‐depsipeptide prodrug containing a disulfide, which is reduced to form the active drug upon cell entry. 194 Romidepsin acts as a histone deacetylase inhibitor and shows promise for treatment of schistosomiasis, a widespread parasite disease with few treatment options. 195 , 196
Another well‐known example is surfactin, a 7‐member lipodepsipeptide with tenside, antitumoral, and antibiotic properties, which is produced by one of the best studied BGCs. 184 , 197 , 198 , 199 Surfactin is generated by the NRPS subunits SrfAA, SrfAB, and SrfAC working in conjunction with fatty acid biogenesis 12 (Figure 10c). The fatty acid substrate 3‐hydroxy myristic acid is activated to a CoA thioester by a fatty acid CoA ligase to be used in surfactin biosynthesis. 12 Then, the N‐terminal C domain in SrfAA catalyzes amide bond formation between the fatty acid and the glutamatyl‐PCP. The synthesis proceeds through normal elongation steps in SrfAB and SrfAC until linear lipopeptidyl‐O‐TE is formed. Finally, the TE domain catalyzes cycle formation using the OH group in the distal hydroxylated lipid for cyclization and surfactin release (Figure 10c). 186 , 199
The structure of SrfAC‐TE was determined in 2002 by Bruner et al. 186 One of the most interesting findings in that study was that the active site lid was in two different conformations in the structure. This was the first structural evidence that the TE domain lid can be very mobile. The authors suggested that it could act as a solvent shield during the cyclization reaction. SrfAC was also a subject of the landmark study, which included the first full (termination) module structure of an NRPS. 86 Among many other vital insights, this and subsequent structures of termination modules, 85 , 96 showed that the TE domain does not have a fixed position relative to the rest of the termination module. Although tethered to the C‐A domain platform through the PCP domain, the TE domain operates independently of the other catalytic domains in the module.
The spiruchostatins are well‐characterized examples of polyketide‐depsipeptides (Figure 10d). Spiruchostatins are antineoplastics, originating from a hybrid PKS‐NRPS biosynthetic cluster. 200 After condensation of the PKS and NRPS portions and chain elongation, the mixed linear ketide‐peptidyl intermediate is released by the TE domain through ester bond formation with a hydroxyl in the ketide portion. The released polyketide‐depsipeptide undergoes uncatalyzed disulfide bond formation to make mature spiruchostatin. Other polyketide‐depsipeptides that very likely have analogous release steps include FK228 (romidepsin), 201 largazole, 202 and virginiamycin. 203
4.2.1.2 | Ester formed using hydroxyl from a small molecule acceptor
Like terminal C domains, TE domains can employ small molecule acceptors to produce linear depsipeptide products, for example in ansatrienin biosynthesis. Ansatrienin is a depsipeptide antibiotic of the ansamycin family, 204 , 205 , 206 with a cyclic polyketide core attached through an ester bond to a cyclohexanoyl‐d‐alanine branch (Figure 11). The Streptomyces collinus ansatrienin BGC was identified as containing PKS and NRPS genes in 1999. 206 Through experiments including gene disruption of astC (which encodes an A‐PCP‐TE tri‐domain NRPS), Shi and colleagues showed conclusively that AstC is responsible for the activation and addition of d‐alanine to the cyclic polyketide core. 205 The TE domain would thus release a d‐alanyl‐polyketide, to which the cyclohexanoyl moiety is later attached, through the alaninyl amino group, by a standalone acyltransferase.
FIGURE 11.
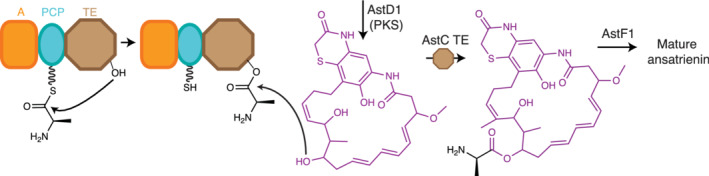
Proposed TE action in the Ast cluster in Streptomyces sp. XZQH13. 205 AstD1 generates a hydroxylated polyketide which acts as a small molecule acceptor, accepting D‐Ala from the NRPS AstC. Mature ansatrienin requires downstream processing steps
The TE‐catalyzed release via a small molecule acceptor mirrors that in mycolic acid biosynthesis where trehalose is the acceptor. 207
4.2.2. Ester bond formation during termination by oligomerization and cyclization in iterative TE domains
4.2.2.1 | Ester formed with side chain hydroxyl
Another well‐known mode of action for certain NRPS TE domains is multi‐functional. Rather than acting only once per biosynthetic cycle, TE domains such as those in NRPSs which synthesize gramicidin S in Bacillus brevis, 5 enterobactin in Escherichia coli 208 , 209 , 210 and bacillibactin in Bacillus subtilis 98 , 211 introduce multiple bonds: They first oligomerize linear peptidyl intermediates, then cyclize the resulting multi‐peptidyl intermediate to release it. In gramicidin S synthesis, the TE domain oligomerizes a pentapeptidyl intermediate via its N‐terminal main chain amine, and then cyclizes the resulting decapeptidyl intermediate to give the macrocyclic decapeptide gramicidin S. By contrast, for depsipeptides enterobactin and bacillibactin, oligomerization and cyclization occur through a side chain hydroxyl, resulting in depsipeptide compounds.
Enterobactin is a siderophore with extreme affinity for Fe (III), 15 which is a trimer of the dipeptide 2,3‐dihydroxybenzoate‐Ser (DHB‐Ser) linked through ester bonds between serine side chain hydroxyls to the serine carbonyl of the adjacent dipeptide 210 (Figure 12). The general synthetic pathway has been clearly delineated. 210 , 212 , 213 The NRPS machinery, including EntE, EntB, and EntF (C‐A‐PCP‐TE), proceeds with standard synthesis, leading to an EntF holding a DHB‐Ser‐S‐PCP intermediate. This is passed to the catalytic serine of the TE domain (DHB‐Ser‐O‐TE). The upstream machinery then assembles a second DHB‐Ser dipeptide on the PCP. The TE domain catalyzes dimerization, and through one or more steps, the dimer is placed on the TE domain ((DHB‐Ser)2‐O‐TE), and a third DHB‐Ser‐S‐PCP is formed on the PCP. The TE domain catalyzes trimerization, and through one or more steps the trimer is placed on the TE domain ((DHB‐Ser)3‐O‐TE). This intermediate is the substrate of the cyclization half reaction by the TE, and in an analogous manner to other cyclization half reactions described above, the TE domain catalyzes attack of the free Ser side chain hydroxyl on the ester bond linking the intermediate to the TE domain, and mature, cyclic enterobactin is released.
FIGURE 12.
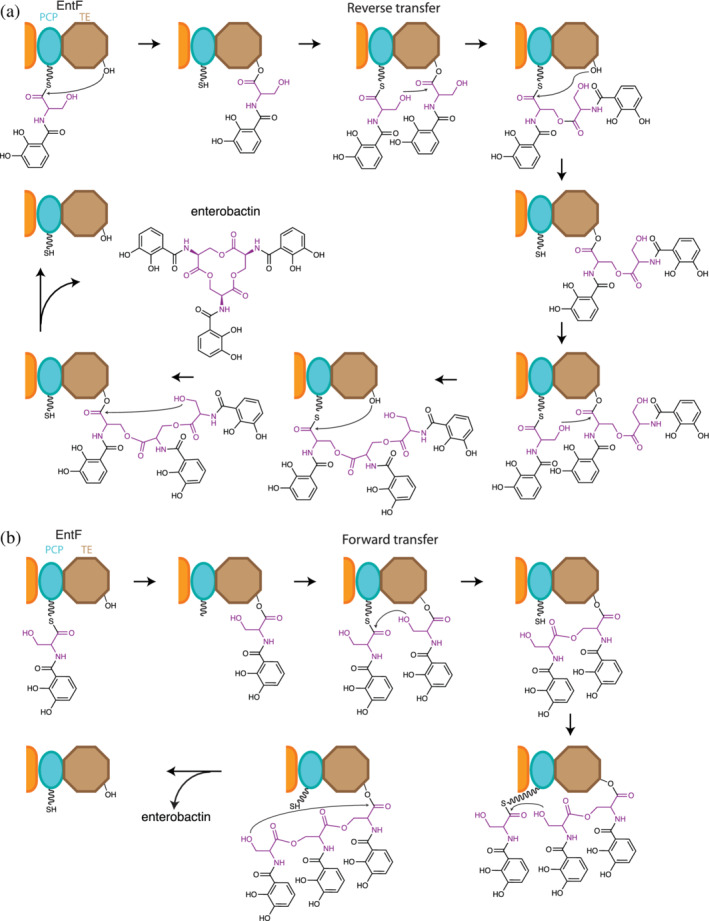
Two proposed oligomerization mechanisms for EntF TE. In both scenarios DHB‐Ser‐S‐PCP is formed on EntF and is transferred to its TE domain, generating DHB‐Ser‐O‐TE. From there, the pathways differ. (a) Reverse transfer oligomerization pathway: A newly made DHB‐Ser‐S‐PCP attacks DHB‐Ser‐O‐TE, forming (DHB‐Ser)2‐S‐PCP. This is transferred to the serine of the TE domain. When a third DHB‐Ser‐S‐PCP is made, another multistep cycle of this oligomerization mechanism takes place to make (DHB‐Ser)3‐S‐PCP and transfer it to the TE domain, giving (DHB‐Ser)3‐O‐TE. (b) Forward transfer oligomerization pathway: DHB‐Ser‐O‐TE attacks the incoming DHB‐Ser‐S‐PCP to make (DHB‐Ser)2‐O‐TE directly. When a third DHB‐Ser‐S‐PCP is made, another iteration of the single step mechanism occurs to make (DHB‐Ser)3‐O‐TE. In both pathways the TE domain releases enterobactin by cyclization of the (DHB‐Ser)3 of (DHB‐Ser)3‐O‐TE
There are two possible pathways for the oligomerization for EntF generally described above 210 (Figure 12). Because the TE domain performs oligomerization, the substrates of the first oligomerization reaction are identical, differing only by their acyl‐enzyme attachment to the NRPS. Therefore, both DHB‐Ser‐S‐PCP and DHB‐Ser‐O‐TE contain a potential hydroxyl side chain nucleophile, and both have an activated (thio)ester linkage to the NRPS. Formally, either dipeptide could provide the nucleophile and either the electrophile. Should the hydroxyl of the DHB‐Ser‐O‐TE attack the DHB‐Ser‐S‐PCP thioester, the dimer is formed on the TE domain, and this dimer could simply wait for the next DHB‐Ser‐S‐PCP to form to attack and make the trimer. This is referred to as forward transfer. Conversely, should the hydroxyl of the DHB‐Ser‐S‐PCP attack the DHB‐Ser‐O‐TE ester, the dimer is formed on the PCP domain and must be transferred to the TE domain to liberate the PCP to receive the next DHB‐Ser dipeptide. Likewise, the dimer would be passed onto the new DHB‐Ser‐S‐PCP, forming the trimer (DHB‐Ser)3 on the PCP. Again, this would be passed to the TE domain for cyclization and release. This pathway is referred to as reverse transfer because intermediates are passed during oligomerization from the TE domain back to the PCP domain.
The forward transfer pathway (Figure 12b) was suggested by Walsh and colleagues 210 because they did not detect (DHB‐Ser)2‐S‐PCP or (DHB‐Ser)3‐S‐PCP intermediates in mass spectrometry of LysC digested EntF, but did detect DHB‐Ser‐O‐TE and (DHB‐Ser)2‐O‐TE. (DHB‐Ser)n‐O‐TE are intermediates in both the reverse and forward mechanisms, and thioesters are more labile than esters, so the forward transfer pathway is not proven. In contrast, the reverse pathway has been definitively demonstrated for biosynthesis of valinomycin (see Section 5.2.2.1), 153 elaiophylin, 214 a macrodiolide side product from the erythromycin system, 215 conglobatin, 216 and gramicidin. 5 This precedence makes it more likely that biosynthesis of enterobactin and the similar compound bacillibactin employs reverse transfer.
As mentioned above, the EntF TE has been fundamentally important for structural understanding of PCP‐TE interactions, 90 , 95 but the oligomerization pathway and the structural determinants of oligomer length are still not clear. Variations on the enterobactin biosynthetic theme include those for depsipeptides corynebactin, which is a trimer of tripeptides linked by the serine side chain hydroxyl, 15 , 217 and bacillibactin, a trimer of tripeptides linked by threonine side chain hydroxyls. 98 , 211 These are likely oligomerized and cyclized using the same iterative path as for enterobactin.
Somewhat further afield is serrawettin W1 (serratamolide), a surfactant lipodepsipeptide 218 of two symmetric fatty acyl‐serinyl (FA‐Ser) moieties joined through an ester bond between a ß‐hydroxyl moiety in the fatty acid and the carboxyl group in Ser. As in enterobactin biosynthesis, its BGC includes an NRPS with domains C‐A‐PCP‐TE. 219 It is likely that the A domain selects Ser and the C domain catalyzes amide bond formation with the activated fatty acyl to give FA‐Ser‐S‐PCP, which is transferred to the TE domain, oligomerized by a forward or reverse pathway to (FA‐Ser)2‐O‐TE and cyclized.
5. DEPSIPEPTIDES WITH ESTER BONDS FORMED DURING BOTH ELONGATION AND TERMINATION
Thus far, we have described BGCs for depsipeptides in which the ester bond is introduced in elongation (by C domains) or in termination (by C and by TE domains). Not surprisingly, these events can occur within the same biosynthetic cluster, and can be steps in either simple or complicated NRPS synthetic cycles. Bacterial NRPSs in which a C domain and the TE domain both install esters exist. These systems can be iterative or noniterative (see Section 5.2). It is likely that a simple, noniterative NRPS which has an ester installing elongating C domain and an ester installing terminating C domain, where each acts only once, also exists, but we are not aware of any.
5.1. The same C domain can catalyze ester bond formation in elongation and termination
5.1.1. Ester bond formation during elongation and termination by bifunctional iterative CT domains
5.1.1.1 | Esters formed with main chain hydroxyls originating from hydroxy acid substrates
Enniatin, beauvericin, 129 and bassianolide 130 are structurally related entomopathogenic fungal toxins. They are cyclized oligomers of dipeptidols of an α‐hydroxy acid and an amino acid 220 (Figure 13). Beauvericin and the enniatins consist of cycles of three dipeptidol residues, while bassianolide consists of a cycle of four dipeptidol residues. The biosynthetic clusters for enniatin, beauvericin, and bassionalide have been characterized. 126 , 127 , 129 , 130 , 221 , 222 Intriguingly, the three synthetases all have the same di‐modular organization: C1‐A1‐PCP1‐C2‐A2‐MT‐PCP2a‐PCP2b‐C3 126 , 220 (Figure 13a), and a similar biosynthetic scheme, delineated independently by two groups 126 , 127 (Figure 13b): C1 appears to be an inactive domain. A1 adenylates its cognate α‐hydroxy acid (D‐Hiv for each synthetase) and transfers it to PCP1. A2 adenylates and transfers a cognate hydrophobic amino acid to either PCP2a or PCP2b. PCP2a and PCP2b are functionally redundant. N‐methylation is likely installed at this point. Next, condensation by C2 generates a dipeptidoyl intermediate on PCP2a/b. Thereafter, an unusual elongation cycle is proposed, whereby PCP1 and PCP2a/b alternate roles as acceptors and donor in condensation. First, A1 produces a new hydroxyisovalyl‐PCP1, which acts as acceptor substrate in CT‐catalyzed condensation with dipeptidoyl‐PCP2a/b, installing the ester bond and making tridepsipeptidyl‐PCP1. Then, A2 produces a new aminoacyl‐PCP2a/b, which is methylated and then acts as acceptor substrate in C2‐catalyzed condensation with tridepsipeptidyl‐PCP1 and making tetradepsipeptidyl‐PCP2a2b (i.e., an intermediate with two dipeptidol residues). One more analogous cycle of elongation occurs in biosynthesis of beauvericin and the enniatins, and two more in bassianolide, to produce the final linear hexadepsipeptidyl‐PCP2a2b (with three dipeptidol residues) or octadepsipeptidyl‐PCP2a2b (with four dipeptidol residues), respectively. These are substrates for CT‐catalyzed cyclization, with the terminal hydroxyl attacking the thioester and releasing the mature natural product. 223 , 224
FIGURE 13.
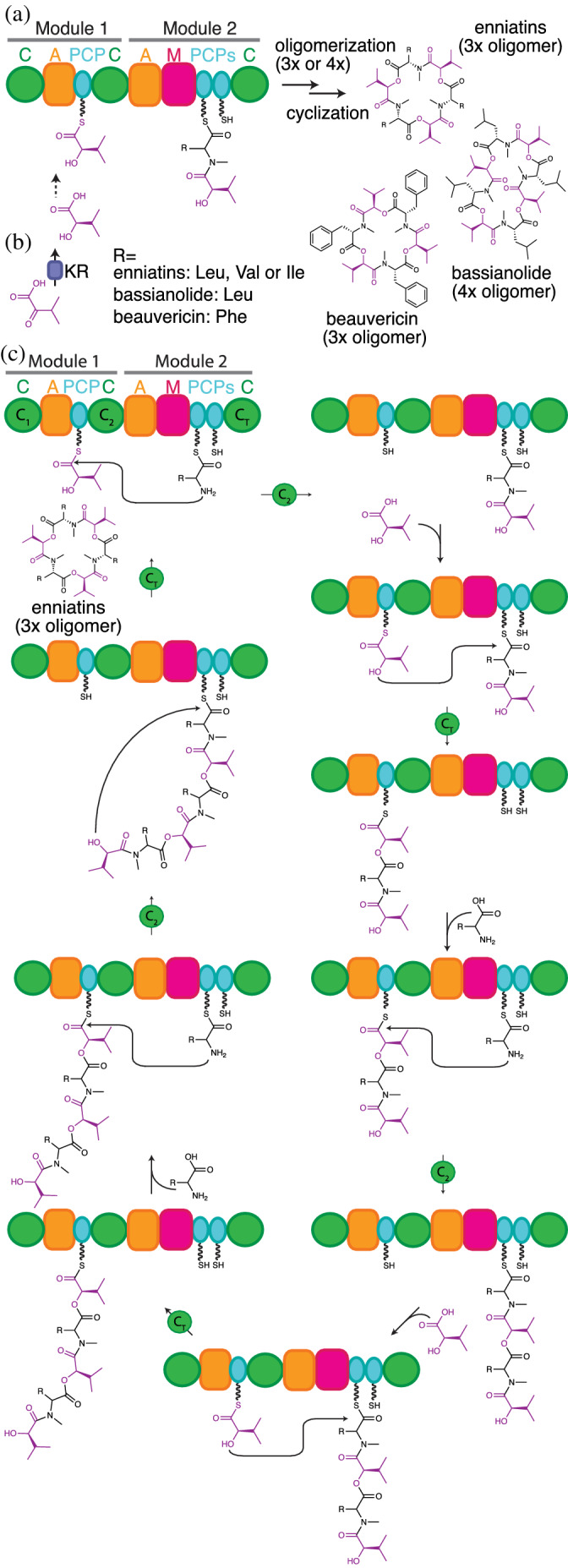
The proposed biosynthesis of enniatin, beauvericin and bassianolide. (a) Enniatin, bassianolide and beauvericin are made by di‐modular NRPSs. The C1 domain is inactive, and the adjacent PCP2a and PCP2b domains have redundant functions (b) Free‐standing ketoreductases (KR), unrelated to PKS KRs, generate α‐hydroxy acids from α‐keto acids. (c) Chain expansion proceeds through alternating condensation reactions catalyzed by the C2 (amide forming) and CT (ester forming) domains, where all PCP domains can alternate between donor or acceptor roles depending on the context of the catalyzed reaction and the nature of the attached substrate. Amino acid acceptor monomers are attached to PCP2a/b during C2 amide bond formation with elongated intermediate donors attached to PCP1, while hydroxy acid acceptor monomers are always attached to PCP1 in CT‐mediated ester bond formation with elongated intermediate donors attached to PCP2a/b. When the elongated chain reaches a specific length, the CT domain switches from elongation to cyclization activity through a yet unknown switching mechanism
The fungal NRPS SidD also uses an unusual iterative pathway. 223 , 224 Fusarinine C is the precursor of the Aspergillus fumigatus virulence factor and siderophore triacetylfusarinine C. SidD has the architecture A1‐PCP1‐C2‐PCP2‐CT. Notably, this includes only one A domain, which is specific for a very large, isopeptide bond‐containing substrate, N5‐hydroxy‐N5‐cis‐anhydromevalonyl‐ornithine. Fusarinine C is a cyclic trimer of this substrate. It will be assembled in an as yet unconfirmed pathway where A1 performs all adenylation and thiolation, and the two elongation steps and final cyclization steps are performed by a combination of C2 and CT acting on PCP1 and PCP2 bound intermediates.
Both our biosynthetic classification and Taevernier's depsipeptide chemical classification 104 clusters enniatin, bassianolide and beauvericin together. However, it is notable that Taevernier includes bacterial depsipeptides such as antimycin, cereulide and valinomycin in the same class. This is appropriate chemically, but comparing the description of biosynthesis above to that for valinomycin in Section 5.2.2.1 illustrates how the biosynthetic pathways of fungal and bacterial depsipeptide synthetases can be strikingly different.
5.2. The C domain and TE domain can both install esters into the same depsipeptide
5.2.1. Ester bond formation during elongation by C domains and termination by TE domains in noniterative synthesis
5.2.1.1 | Ester formed with main chain hydroxyl originating from keto acid substrate (elongation) and from side chain hydroxyl (termination)
Cryptophycins are cyanobacterial depsipeptides with tubulin‐depolymerizing properties, 225 whose synthetic analogues have been tested in phase II clinical trials for their anticancer properties. 38 , 226 There are more than 25 naturally occurring cryptophycin variants. 38 , 227 The two ester bonds in the structure are formed using an α‐hydroxy acid and side chain hydroxyl, inserted during elongation and termination respectively. Subunits CrpA, CrpB, and CrpC synthesize a hybrid polyketide/peptide moiety, which is passed as a linear intermediate to CrpD (Figure 14), a di‐modular NRPS subunit with a C‐A‐PCP‐C‐A‐KR‐PCP‐TE architecture. The first module (CrpD1: C‐A‐PCP) adds β‐alanine to the growing PKS‐peptidyl chain, while the second module (CrpD2: C‐A‐KR‐PCP‐TE) adds α‐hydroxyisocaproic acid (α‐HIC) through α‐keto acid activation and reduction in the A‐KR domains 38 , 128 , 147 (see below). The CrpD2 C domain uses the α‐hydroxyacyl‐S‐PCP intermediate as an acceptor substrate in the ester bond forming condensation reaction. The ester bond forming activity of CrpD2 C domain was verified by heterologous expression and biochemical assays, making it the first characterized depsipeptide ester bond forming C domain. 128 The second ester bond in the cryptophycin structure arises from macrocyclization in a TE domain‐catalyzed reaction, using the hydroxy group in the polyketide as a nucleophile. 128 , 227
FIGURE 14.
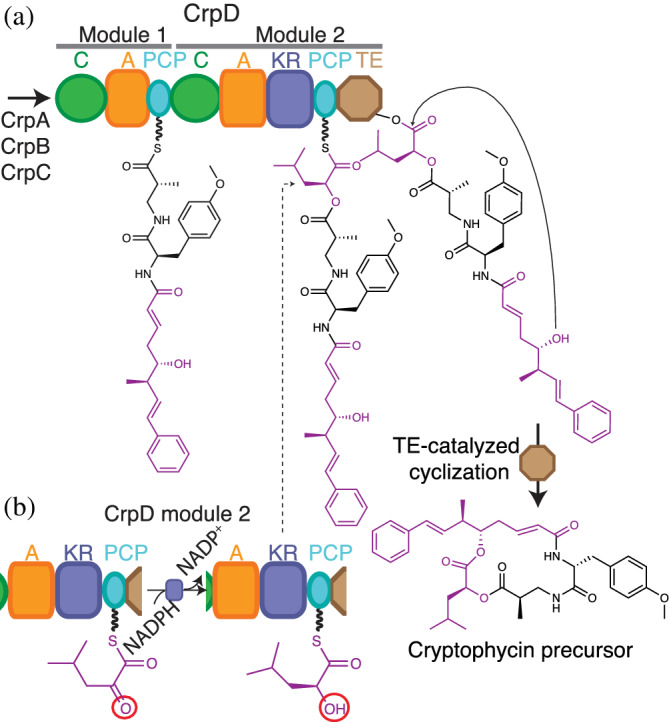
CrpD inserts ester bonds during elongation and termination steps in cryptophycin biosynthesis. (a) The C domain in CrpD Module 2 catalyzes ester bond formation between the growing polyketide‐peptide chain and an α‐hydroxy acid. The TE domain catalyzes cyclization using the OH group in the polyketide portion as nucleophile, releasing a cryptophycin precursor. The cycle proceeds in off‐assembly line reactions. (b) The A‐KR‐PCP module in CrpD activates and reduces an α‐ketoacid to generate an α‐hydroxy acyl thioester. Figure based on Magarvey et al. 38
The antimycin‐like family is a group of depsipeptides where ester bonds are also inserted during elongation and termination in an analogous manner to cryptophycin. 115 , 228 , 229 They possess an impressive diversity of bioactivities, including piscicidal, 108 insecticidal, nematocidal action 115 , 230 as well as symbiosis with ants. 18 , 231 Antimycin‐like peptides are synthesized by mixed PKS‐NRPS systems. 228 , 229 They all have at least one C‐A‐KR‐T depsipeptide module for incorporation of keto acids into depsipeptides in elongation but can have multiple extras which increase the lactone ring size. 232 , 233 , 234 Awakawa and coworkers were able to generate novel antimycin‐like compounds by employing a shuffling and deletions strategy between the JBIR‐06 and neoantimycin NRPSs. 235 This led to functional chimeric depsipeptide synthetases, an elegant proof of concept of the bioengineering potential of these megaenzymes.
Didemnins are produced by sea bacteria from the Tristrella genus, and have antineoplastic, anti‐viral and immunosuppressive activities. 151 , 236 , 237 Dehydrodidemnin B (plitidepsin or Aplidin®) 238 , 239 is approved for the treatment of multiple myeloma in Australia (Australian Register of Therapeutic Goods ID 291661) and has been reported by the company PhamaMar to show nanomolar in vitro inhibitory activity against coronavirus HCoV‐229E, a similar virus to the one causing the current COVID‐19 pandemic. Didemnins have two ester bonds formed by α‐hydroxy acyl residues reduced in situ from keto acyl residues by depsipeptide modules in addition to an ester bond formed by a threonine side chain in termination. 151 , 236 , 238
Another example in this depsipeptide classification, hectochlorin, is an antifungal compound isolated from the cyanobacterium Lyngbya majuscula. 240 The biosynthetic pathway also employs two elongating C‐A‐KR‐PCP modules for ester bond formation during elongation. 241 The cyclizing TE domain catalyzes ester bond formation using a hydroxy group from the ketide portion of the molecule as a nucleophile. 241
5.2.1.2 | Esters formed using main chain hydroxyl (elongation) and side chain hydroxyl (termination), both originating from bisphosphoglycerate
Vioprolide is a cyclic lipodepsipeptide (Figure 15) with an unusual origin for its bridging ester oxygens. 242 The 10‐module NRPS system which produces the depsipeptide precursor of valpromide contains several tailoring domains and incorporates several nonproteinogenic amino acids. Of relevance for this review are the first elongation step and the termination step, which Mueller and coworkers described. 242 , 243 In Module 2, in place of an A domain there is a domain with homology to FkbH proteins of the haloacid dehalogenase superfamily. The FkbH domain in VioA, like the FkbH domain in the PKS OzmB, 244 has affinity for 1,3 bisphosphoglycerate. The phosphorylated carboxylate in 1,3 bisphosphoglycerate is energetically equal to the acyl‐adenylates which A domains generate, so activation of 1,3 bisphosphoglycerate is not required for transfer to the PCP domain. The three‐step reaction catalyzed by FkbH proceeds first with acyl‐enzyme intermediate formation on an active site Cys in the FkbH domain (3‐phosphoglyceryl‐S‐FkbG) displacing carboxylate phosphate, then dephosphorylation of the β‐hydroxyl (to glyceryl‐S‐FkbG) and finally transthioesterification to glyceryl‐S‐PCP2. This glyceryl moiety contains both the hydroxyls that will provide the bridging oxygens in the two ester bonds of vioprolide: the α‐hydroxyl is the nucleophile in C2‐mediated condensation with an acyl‐PCP1 in the first elongation step, and the β‐hydroxyl is the nucleophile in TE10‐mediated cyclization in termination (Figure 15).
FIGURE 15.
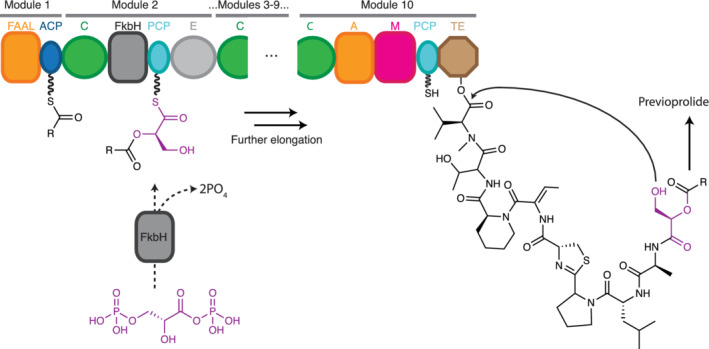
Proposed vioprolide biosynthesis. A decamodular NRPS generates previoprolide. It is suggested that the FkbH domain in Module 2 catalyzes stepwise covalent binding of D‐1,3‐biphosphoglycerate, dephosphorylation and transfer of glycerate to PCP2, generating D‐glyceryl‐S‐PCP2. The α‐hydroxy group serves as a nucleophile in the condensation reaction in the C2, and the β‐hydroxy group in the glyceryl side chain serves as the nucleophile in cyclic termination. This releases a previoprolide intermediate that undergoes further modification to generate mature vioprolide
5.2.2. Ester bond formation during elongation by C domains and termination by TE domains in iterative synthesis
5.2.2.1 | Ester formed with main chain hydroxyls originating from keto acid substrates
Cereulide and valinomycin are dodecadepsipeptides formed by alternating arrays of α‐amino and α‐hydroxy acids. 145 , 146 , 245 , 246 They can also be described as trimers of a tetradepsipeptide unit. In spite of their strong structural resemblance to the fungal products enniatin, beauvericin, and bassianolide, their biosynthesis is very different (Figure 16).
FIGURE 16.
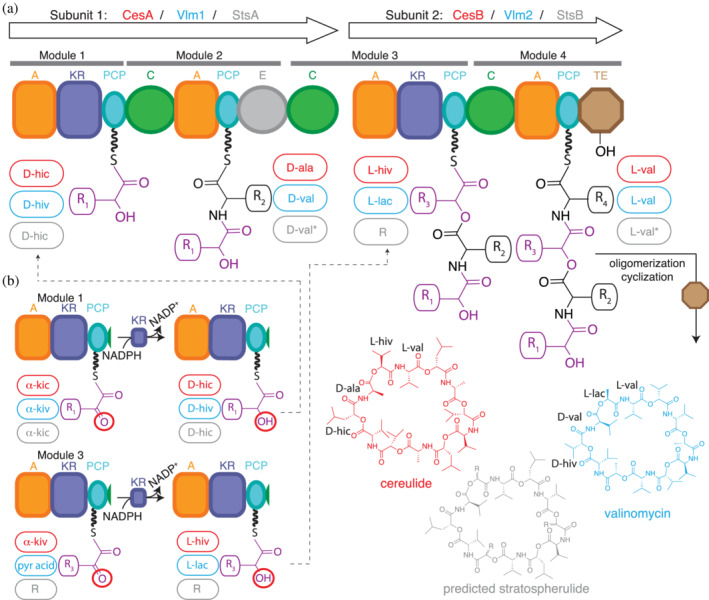
Proposed biosynthesis of cereulide, valinomycin and a hypothetical product from Bacillus stratosphericus LAMA 585. (a) During elongation ester bonds are formed by C3 domains. During termination, the TE domains catalyzes oligomerization of tetradepsipeptidyl units, as well as cyclic release, using the distal α‐hydroxy groups as nucleophiles (see also Figure 17). (b) Depsipeptide Modules 1 and 3 activate, thiolate and reduce α‐keto acids to generate α‐hydroxy acyl thioesters. Adapted from Alonzo et al. 34
The cereulide and valinomycin biosynthetic clusters are 2‐subunit, 4‐module NRPSs 33 , 109 , 152 , 247 (Figure 16a). CesA and Vlm1 have the architecture A1‐KR1‐PCP1‐C2‐A2‐PCP2‐E2‐C3, while CesB and Vlm2 have A3‐KR3‐PCP3‐C4‐A4‐KR4‐PCP4‐TE4. Depsipeptide initiation modules CesA1 and Vlm11 and elongation modules CesB3 and Vlm23 activate and reduce α‐ketoacids through the action of their A‐KR domains (Figure 16b). 33 However, the fate of each generated acyl thioester depends on whether the module is in an initiation or elongation position. The reduced hydroxyl in Residue 1 is not used immediately, but is first a passive part of the donor substrate (e.g., α‐D‐HIC‐S‐PCP1 in cereulide biosynthesis) for condensation in C2 with L‐Ala‐S‐PCP2 as acceptor substrate. In contrast, the reduced hydroxyl in Residue 3 is used immediately after reduction to α‐D‐HIV‐S‐PCP3, as it is the acceptor substrate for C3 in that ester forming condensation reaction. The tridepsipeptidyl chain will then be elongated in the C4 to a tetradepsipeptide intermediate with a free distal α‐hydroxyl group and one internal ester bond. This tetradepsipeptide intermediate is then oligomerized and cyclized by the terminal TE domain, 5 , 248 but until recently, the details of this step were opaque.
Several outstanding questions pertaining to the oligomerization and termination cycles of these systems were addressed in a collaborative paper from our group, the Chin group and the Boddy group 153 (Figure 17). First, we addressed the question of whether the oligomerization mechanism proceeded through a forward 210 or reverse 5 transfer mechanism (Figure 17a; see also enterobactin Figure 12). Using Vlm2 TE heterologously expressed in E. coli and reacting it with a tetradepsipeptidyl‐SNAC substrate, we confirmed that the reverse transfer mechanism is the most likely due to the detection of reaction intermediates of only part of the reverse transfer pathway. Then, we used a novel chemical biology approach to obtain co‐complex structural information. It is classically challenging to obtain structural information on TE‐intermediate complexes because of the instability of the ester bond in the acyl‐enzyme intermediates. 185 , 186 Other groups have recently used phosphonate analogues to obtain analog co‐complexes of the bifunctional nocardicin TE domain. 183 In the case of depsipeptide synthetases, we employed an expanded genetic code strategy 249 , 250 to replace the active site serine with the amine‐containing amino acid 2,3‐diaminopropionic acid (DAP) 153 (Figure 17b). This strategy effectively produced recombinant DAP‐containing protein that, when incubated with SNAC analogues or valinomycin, formed stable amide TE‐intermediate complexes analogous to the first and last steps of the oligomerization/cyclization reactions, whose structures we determined by X‐ray crystallography.
FIGURE 17.
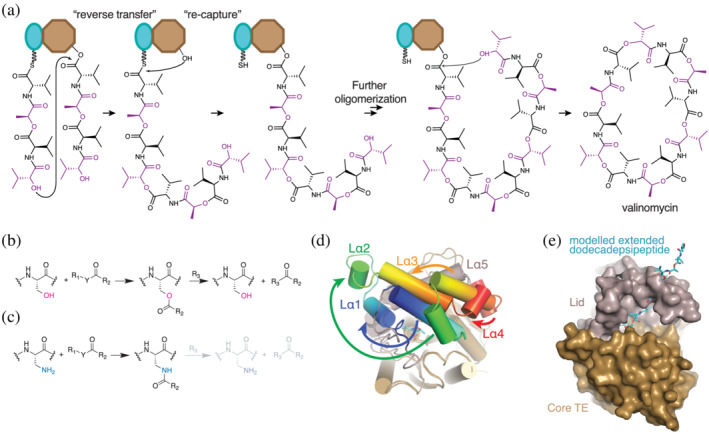
Valinomycin oligomerization and cyclization. (a) Oligomerization of tetradepsipeptidyl units proceeds through a reverse transfer pathway, where the distal α‐hydroxyl in tetradepsipeptidyl‐S‐PCP4 attacks the ester carbon in tetradepsipeptidyl‐O‐TE4, generating (tetradepsipeptidyl)2‐S‐PCP4 after one oligomerization. This is transferred to TE4 and the next tetradepsipeptidyl‐S‐PCP4 is used to make (tetradepsipeptidyl)3‐S‐PCP4 in an analogous way. Cyclization of the (tetradepsipeptidyl)3‐S‐PCP4 via its α‐hydroxyl releases mature valinomycin. (b) Vlm 2 TE forms ester intermediates during its reaction cycle. (c) Replacing the active site serine in Vlm2 TE by DAP generates a protein that can form amide bonds instead of ester bonds, making the intermediates more stable and amenable for structural and biophysical analysis. (d) The active site lid of Vlm2 TE undergoes massive conformational arrangements to favor the cyclization conformation of the dodecadepsipeptide in the active site. (e) A model of a fully elongated dodecadepsipeptide (cyan) shows that such conformation would clash with the active site lid (grey) observed in the X‐ray structure of the dodecadepsipeptidyl‐NH‐TE complex, thus supporting the hypothesis that lid remodeling favors the cyclization reaction. Figure based on Huguenin‐Dezot et al. 153
The TE domain co‐complex structures showed remarkable placidity in the lid. The Vlm TE lid is 88 residues, folded into an extended loop: bundled helices α1–3, short helix α4, long helix α5, and short helix α6. In the absence of substrates, one structure showed disordered lid and the other showed an ordered lid. The ordered position of the lid in the apo state was similar to that when bound with one tetradepsipeptidyl moiety (tetradepsipeptidyl‐NH‐TE). However, when the TE domain was bound with last‐stage (tetradepsipeptidyl)3‐NH‐TE the lid was not only in a different position, but also the secondary structure elements had markedly rearranged relative to each other (Figure 17d). This rearranged lid formed a hemisphere‐like pocket, which likely helps curl the dodecadepsipeptide back toward the active site serine for cyclization (Figure 17e). Only a few of the substrate depsipeptidyl residues were visible in the density, which is perhaps not surprising considering the TE domain must accommodate a range of length of substrates during its cycle. Indeed, specific and strong binding interactions could slow the synthetic cycle, as the tetradepsipeptide must transition back and forth between being ligated to the PCP domain and ligated to the TE domain, and the same tetradepsipeptide must assume multiple different positions in the course of a cycle.
6. CONCLUDING WORDS
The outstanding chemical diversity of depsipeptides and the array of ester bond forming strategies in depsipeptide synthetases create scientific and practical interest in the activities and biosynthesis of these compounds. Although the field has gained good knowledge of the biosynthetic pathways processes, there is still much to be discovered.
There are many orphan clusters and compounds throughout nature. Of note are the numerous uncharacterized fungal depsipeptide synthetases. If these act, as we believe, through a cycle that includes C domain‐catalyzed ester bond formation, it will mean that ester‐bond‐forming‐C domains are more widespread than once thought. Furthermore, if bifunctional CT domains such as those in enniatin biosynthesis require promiscuity to catalyze both elongation and cyclization, they may be extremely promising targets for bioengineering efforts. 127 More structural, biophysical, and biochemical studies of the domain will increasingly shed light into the mechanistical details that separate the elongation and the cyclization activities of these domains, and whether they could eventually be exploited for combinatorial biosynthetic efforts.
Another open question in fungal systems concerns the production of α‐hydroxyl acids in standalone KR domains. Do these KR domains transiently associate with the NRPSs to ensure a high local concentration of NRPS substrate, or is there another means of substrate channeling? It would seem wasteful to generate millimolar concentrations of hydroxy acids throughout the fungal cytoplasm. Answering this question could have relevance for many biosynthetic clusters.
An active topic of research in our laboratory pertains to the stereospecificity of KR domains in depsipeptide (C)‐A‐KR‐T module. Because the motifs that identify stereochemistry outcomes in PKS KR domains are absent or degenerated in NRPS A‐KR‐PCP modules, additional information is required to predict depsipeptide stereochemistry. It is likely that stereochemistry motifs are different and that bioinformatic analysis and co‐complex structure determination of these systems is necessary to elucidate the stereospecificity determinants.
In addition, the mechanisms by which the TE domains, with their very varied lid composition, direct the proper nucleophile for iterative oligomerization and/or cyclization requires further study. Only with a substantial number of co‐crystal structures can excellent generalizable insight be obtained. We are hopeful that the DAP expanded genetic code strategy employed for valinomycin TE will facilitate these studies. Controlling TE domain function is an important frontier for most comprehensive bioengineering efforts.
As much as fundamental insight into known pathways is needed, perhaps the most exciting question is that of what novel modes of depsipeptide biosynthesis are yet to be discovered. There are clear positions in the schematic (Figure 2) that could be filled in with yet‐to‐be discovered pathways (e.g., by a system that iteratively oligomerizes and cyclizes through the distal α‐hydroxyl acid residue, similar to how the nondepsipeptide cyclic gramicidin is made). The filling in of those positions is probably merely a matter of time. However, these authors look most forward to the description of completely novel depsipeptide NRPS synthetic logic, awaiting discovery in earth's huge microbial diversity.
AUTHOR CONTRIBUTIONS
Diego Alonzo: Visualization; writing‐original draft; writing‐review and editing. T Schmeing: Conceptualization; funding acquisition; project administration; resources; supervision; visualization; writing‐original draft; writing‐review and editing.
ACKNOWLEDGMENTS
The authors thank Elias Kalthoff for insightful comments on the vioprolide and fusarinin sections, Clarisse Chiche‐Lapierre, Michael Tarry, and members of the Schmeing lab for many interesting discussions about depsipeptides and their biosyntheses, Nathan Magarvey for initially suggesting depsipeptide synthetases as a research topic, Nicolas Huguenin‐Dezot, Jason Chin, Jimin Wang, Graham Heberlig, and Chris Boddy for fruitful collaborations on depsipeptide synthetases, and Nancy Rogerson for editorial help. This work was supported by a Canada Research Chair to T. Martin Schmeing.
Alonzo DA, Schmeing TM. Biosynthesis of depsipeptides, or Depsi: The peptides with varied generations. Protein Science. 2020;29:2316–2347. 10.1002/pro.3979
Funding information Canada Research Chairs; Canadian Institutes of Health Research, Grant/Award Number: FDN‐148472
REFERENCES
- 1. Felnagle EA, Jackson EE, Chan YA, et al. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol Pharm. 2008;5:191–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Challinor VL, Bode HB. Bioactive natural products from novel microbial sources. Ann N Y Acad Sci. 2015;1354:82–97. [DOI] [PubMed] [Google Scholar]
- 3. van Liempt H, von Döhren H, Kleinkauf H. Delta‐(L‐alpha‐aminoadipyl)‐L‐cysteinyl‐D‐valine synthetase from Aspergillus nidulans. The first enzyme in penicillin biosynthesis is a multifunctional peptide synthetase. J Biol Chem. 1989;264:3680–3684. [PubMed] [Google Scholar]
- 4. Conti E, Stachelhaus T, Marahiel MA, Brick P. Structural basis for the activation of phenylalanine in the non‐ribosomal biosynthesis of gramicidin S. EMBO J. 1997;16:4174–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoyer KM, Mahlert C, Marahiel MA. The iterative gramicidin s thioesterase catalyzes peptide ligation and cyclization. Chem Biol. 2007;14:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stein T, Kluge B, Vater J, Franke P, Otto A, Wittmann‐Liebold B. Gramicidin S synthetase 1 (phenylalanine racemase), a prototype of amino acid racemases containing the cofactor 4′‐phosphopantetheine. Biochemistry. 1995;34:4633–4642. [DOI] [PubMed] [Google Scholar]
- 7. Reimer JM, Aloise MN, Harrison PM, Schmeing TM. Synthetic cycle of the initiation module of a formylating nonribosomal peptide synthetase. Nature. 2016;529:239–242. [DOI] [PubMed] [Google Scholar]
- 8. Robbel L, Marahiel MA. Daptomycin, a bacterial lipopeptide synthesized by a nonribosomal machinery. J Biol Chem. 2010;285:27501–27508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bills G, Li Y, Chen L, Yue Q, Niu XM, An Z. New insights into the echinocandins and other fungal non‐ribosomal peptides and peptaibiotics. Nat Prod Rep. 2014;31:1348–1375. [DOI] [PubMed] [Google Scholar]
- 10. Hoppert M, Gentzsch C, Schorgendorfer K. Structure and localization of cyclosporin synthetase, the key enzyme of cyclosporin biosynthesis in Tolypocladium inflatum. Arch Microbiol. 2001;176:285–293. [DOI] [PubMed] [Google Scholar]
- 11. Lawen A. Biosynthesis and mechanism of action of cyclosporins. Prog Med Chem. 1996;33:53–97. [DOI] [PubMed] [Google Scholar]
- 12. Kraas FI, Helmetag V, Wittmann M, Strieker M, Marahiel MA. Functional dissection of surfactin synthetase initiation module reveals insights into the mechanism of lipoinitiation. Chem Biol. 2010;17:872–880. [DOI] [PubMed] [Google Scholar]
- 13. O'Brien J, Wright GD. An ecological perspective of microbial secondary metabolism. Curr Opin Biotechnol. 2011;22:552–558. [DOI] [PubMed] [Google Scholar]
- 14. Ekman JV, Kruglov A, Andersson MA, Mikkola R, Raulio M, Salkinoja‐Salonen M. Cereulide produced by Bacillus cereus increases the fitness of the producer organism in low‐potassium environments. Microbiology. 2012;158:1106–1116. [DOI] [PubMed] [Google Scholar]
- 15. Raymond KN, Dertz EA, Kim SS. Enterobactin: An archetype for microbial iron transport. Proc Natl Acad Sci USA. 2003;100:3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van der Meij A, Worsley SF, Hutchings MI, van Wezel GP. Chemical ecology of antibiotic production by actinomycetes. FEMS Microbiol Rev. 2017;41:392–416. [DOI] [PubMed] [Google Scholar]
- 17. Demain AL, Fang A. The natural functions of secondary metabolites. Adv Biochem Eng Biotechnol. 2000;69:1–39. [DOI] [PubMed] [Google Scholar]
- 18. Seipke RF, Barke J, Brearley C, et al. A single Streptomyces symbiont makes multiple antifungals to support the fungus farming ant Acromyrmex octospinosus . PLoS One. 2011;6:e22028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang H, Fewer DP, Holm L, Rouhiainen L, Sivonen K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc Natl Acad Sci USA. 2014;111:9259–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caboche S, Leclere V, Pupin M, Kucherov G, Jacques P. Diversity of monomers in nonribosomal peptides: Towards the prediction of origin and biological activity. J Bacteriol. 2010;192:5143–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Konz D, Marahiel MA. How do peptide synthetases generate structural diversity? Chem Biol. 1999;6:R39–R48. [DOI] [PubMed] [Google Scholar]
- 22. Walsh CT, Chen H, Keating TA, et al. Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines. Curr Opin Chem Biol. 2001;5:525–534. [DOI] [PubMed] [Google Scholar]
- 23. Mori S, Pang AH, Lundy TA, Garzan A, Tsodikov OV, Garneau‐Tsodikova S. Structural basis for backbone N‐methylation by an interrupted adenylation domain. Nat Chem Biol. 2018;14:428–430. [DOI] [PubMed] [Google Scholar]
- 24. Al‐Mestarihi AH, Villamizar G, Fernández J, Zolova OE, Lombó F, Garneau‐Tsodikova S. Adenylation and S‐methylation of cysteine by the bifunctional enzyme TioN in thiocoraline biosynthesis. J Am Chem Soc. 2014;136:17350–17354. [DOI] [PubMed] [Google Scholar]
- 25. Schoenafinger G, Schracke N, Linne U, Marahiel MA. Formylation domain: An essential modifying enzyme for the nonribosomal biosynthesis of linear gramicidin. J Am Chem Soc. 2006;128:7406–7407. [DOI] [PubMed] [Google Scholar]
- 26. Samel SA, Czodrowski P, Essen LO. Structure of the epimerization domain of tyrocidine synthetase a. Acta Cryst D. 2014;70:1442–1452. [DOI] [PubMed] [Google Scholar]
- 27. Stein DB, Linne U, Hahn M, Marahiel MA. Impact of epimerization domains on the intermodular transfer of enzyme‐bound intermediates in nonribosomal peptide synthesis. Chembiochem. 2006;7:1807–1814. [DOI] [PubMed] [Google Scholar]
- 28. Bloudoff K, Fage CD, Marahiel MA, Schmeing TM. Structural and mutational analysis of the nonribosomal peptide synthetase heterocyclization domain provides insight into catalysis. Proc Natl Acad Sci USA. 2017;114:95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duerfahrt T, Eppelmann K, Muller R, Marahiel MA. Rational design of a bimodular model system for the investigation of heterocyclization in nonribosomal peptide biosynthesis. Chem Biol. 2004;11:261–271. [DOI] [PubMed] [Google Scholar]
- 30. Gehring AM, Mori II, Perry RD, Walsh CT. The nonribosomal peptide synthetase HMWP2 forms a thiazoline ring during biogenesis of yersiniabactin, an iron‐chelating virulence factor of yersinia pestis. Biochemistry. 1998;37:17104. [DOI] [PubMed] [Google Scholar]
- 31. Du L, Chen M, Sanchez C, Shen B. An oxidation domain in the BlmIII non‐ribosomal peptide synthetase probably catalyzing thiazole formation in the biosynthesis of the anti‐tumor drug bleomycin in Streptomyces verticillus ATCC15003. FEMS Microbiol Lett. 2000;189:171–175. [DOI] [PubMed] [Google Scholar]
- 32. Schneider TL, Shen B, Walsh CT. Oxidase domains in epothilone and bleomycin biosynthesis: Thiazoline to thiazole oxidation during chain elongation. Biochemistry. 2003;42:9722–9730. [DOI] [PubMed] [Google Scholar]
- 33. Magarvey NA, Ehling‐Schulz M, Walsh CT. Characterization of the cereulide NRPS alpha‐hydroxy acid specifying modules: Activation of alpha‐keto acids and chiral reduction on the assembly line. J Am Chem Soc. 2006;128:10698–10699. [DOI] [PubMed] [Google Scholar]
- 34. Alonzo DA, Chiche‐Lapierre C, Tarry MJ, Wang J, Schmeing TM. Structural basis of keto acid utilization in nonribosomal depsipeptide synthesis. Nat Chem Biol. 2020;16:493–496. [DOI] [PubMed] [Google Scholar]
- 35. van Wageningen AM, Kirkpatrick PN, Williams DH, et al. Sequencing and analysis of genes involved in the biosynthesis of a vancomycin group antibiotic. Chem Biol. 1998;5:155–162. [DOI] [PubMed] [Google Scholar]
- 36. Uhlmann S, Sussmuth RD, Cryle MJ. Cytochrome p450sky interacts directly with the nonribosomal peptide synthetase to generate three amino acid precursors in skyllamycin biosynthesis. ACS Chem Biol. 2013;8:2586–2596. [DOI] [PubMed] [Google Scholar]
- 37. Ding Y, Seufert WH, Beck ZQ, Sherman DH. Analysis of the cryptophycin P450 epoxidase reveals substrate tolerance and cooperativity. J Am Chem Soc. 2008;130:5492–5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Magarvey NA, Beck ZQ, Golakoti T, et al. Biosynthetic characterization and chemoenzymatic assembly of the cryptophycins. Potent anticancer agents from cyanobionts. ACS Chem Biol. 2006;1:766–779. [DOI] [PubMed] [Google Scholar]
- 39. Hojati Z, Milne C, Harvey B, et al. Structure, biosynthetic origin, and engineered biosynthesis of calcium‐dependent antibiotics from Streptomyces coelicolor. Chem Biol. 2002;9:1175–1187. [DOI] [PubMed] [Google Scholar]
- 40. Kopp F, Grunewald J, Mahlert C, Marahiel MA. Chemoenzymatic design of acidic lipopeptide hybrids: New insights into the structure‐activity relationship of daptomycin and A54145. Biochemistry. 2006;45:10474–10481. [DOI] [PubMed] [Google Scholar]
- 41. Losey HC, Peczuh MW, Chen Z, et al. Tandem action of glycosyltransferases in the maturation of vancomycin and teicoplanin aglycones: Novel glycopeptides. Biochemistry. 2001;40:4745–4755. [DOI] [PubMed] [Google Scholar]
- 42. Peschke M, Haslinger K, Brieke C, Reinstein J, Cryle MJ. Regulation of the P450 oxygenation cascade involved in glycopeptide antibiotic biosynthesis. J Am Chem Soc. 2016;138:6746–6753. [DOI] [PubMed] [Google Scholar]
- 43. Haslinger K, Peschke M, Brieke C, Maximowitsch E, Cryle MJ. X‐domain of peptide synthetases recruits oxygenases crucial for glycopeptide biosynthesis. Nature. 2015;521:105–109. [DOI] [PubMed] [Google Scholar]
- 44. Reimer JM, Haque AS, Tarry MJ, Schmeing TM. Piecing together nonribosomal peptide synthesis. Curr Opin Struct Biol. 2018;49:104–113. [DOI] [PubMed] [Google Scholar]
- 45. Gulick AM. Structural insight into the necessary conformational changes of modular nonribosomal peptide synthetases. Curr Opin Chem Biol. 2016;35:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schwarzer D, Finking R, Marahiel MA. Nonribosomal peptides: From genes to products. Nat Prod Rep. 2003;20:275–287. [DOI] [PubMed] [Google Scholar]
- 47. Marahiel MA, Stachelhaus T, Mootz HD. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem Rev. 1997;97:2651–2674. [DOI] [PubMed] [Google Scholar]
- 48. Cane DE, Walsh CT, Khosla C. Harnessing the biosynthetic code: Combinations, permutations, and mutations. Science. 1998;282:63–68. [DOI] [PubMed] [Google Scholar]
- 49. Stachelhaus T, Mootz HD, Marahiel MA. The specificity‐conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505. [DOI] [PubMed] [Google Scholar]
- 50. Challis GL, Ravel J, Townsend CA. Predictive, structure‐based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem Biol. 2000;7:211–224. [DOI] [PubMed] [Google Scholar]
- 51. Bachmann BO, Ravel J. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217. [DOI] [PubMed] [Google Scholar]
- 52. Prieto C, Garcia‐Estrada C, Lorenzana D, Martin JF. NRPSsp: Non‐ribosomal peptide synthase substrate predictor. Bioinformatics. 2012;28:426–427. [DOI] [PubMed] [Google Scholar]
- 53. Conti E, Franks NP, Brick P. Crystal structure of firefly luciferase throws light on a superfamily of adenylate‐forming enzymes. Structure. 1996;4:287–298. [DOI] [PubMed] [Google Scholar]
- 54. Gulick AM, Starai VJ, Horswill AR, Homick KM, Escalante‐Semerena JC. The 1.75 A crystal structure of acetyl‐CoA synthetase bound to adenosine‐5′‐propylphosphate and coenzyme a. Biochemistry. 2003;42:2866–2873. [DOI] [PubMed] [Google Scholar]
- 55. Yonus H, Neumann P, Zimmermann S, May JJ, Marahiel MA, Stubbs MT. Crystal structure of DltA. Implications for the reaction mechanism of non‐ribosomal peptide synthetase adenylation domains. J Biol Chem. 2008;283:32484–32491. [DOI] [PubMed] [Google Scholar]
- 56. Mitchell CA, Shi C, Aldrich CC, Gulick AM. Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry. 2012;51:3252–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sundlov JA, Shi C, Wilson DJ, Aldrich CC, Gulick AM. Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem Biol. 2012;19:188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Horsman ME, Hari TPA, Boddy CN. Polyketide synthase and non‐ribosomal peptide synthetase thioesterase selectivity: Logic gate or a victim of fate? Nat Prod Rep. 2015;33:183–202. [DOI] [PubMed] [Google Scholar]
- 59. Wilson DJ, Shi C, Teitelbaum AM, Gulick AM, Aldrich CC. Characterization of AusA: A dimodular nonribosomal peptide synthetase responsible for the production of aureusimine pyrazinones. Biochemistry. 2013;52:926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Manavalan B, Murugapiran S, Lee G, Choi S. Molecular modeling of the reductase domain to elucidate the reaction mechanism of reduction of peptidyl thioester into its corresponding alcohol in non‐ribosomal peptide synthetases. BMC Struct Biol. 2010;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bloudoff K, Schmeing TM. Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: Discovery, dissection and diversity. Biochim Biophys Acta. 1865;2017:1587–1604. [DOI] [PubMed] [Google Scholar]
- 62. Zhang J, Liu N, Cacho RA, et al. Structural basis of nonribosomal peptide macrocyclization in fungi. Nat Chem Biol. 2016;12:1001–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yuwen L, Zhang FL, Chen QH, Lin SJ, Zhao YL, Li ZY. The role of aromatic L‐amino acid decarboxylase in bacillamide C biosynthesis by Bacillus atrophaeus C89. Sci Rep. 2013;3:1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chang CY, Lohman JR, Huang T, et al. Structural insights into the free‐standing condensation enzyme SgcC5 catalyzing ester‐bond formation in the biosynthesis of the enediyne antitumor antibiotic C‐1027. Biochemistry. 2018;57:3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wenzel SC, Kunze B, Hofle G, et al. Structure and biosynthesis of myxochromides S1‐3 in Stigmatella aurantiaca: Evidence for an iterative bacterial type I polyketide synthase and for module skipping in nonribosomal peptide biosynthesis. Chembiochem. 2005;6:375–385. [DOI] [PubMed] [Google Scholar]
- 66. Hacker C, Cai X, Kegler C, et al. Structure‐based redesign of docking domain interactions modulates the product spectrum of a rhabdopeptide‐synthesizing NRPS. Nat Commun. 2018;9:4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chiocchini C, Linne U, Stachelhaus T. In vivo biocombinatorial synthesis of lipopeptides by COM domain‐mediated reprogramming of the surfactin biosynthetic complex. Chem Biol. 2006;13:899–908. [DOI] [PubMed] [Google Scholar]
- 68. Dehling E, Volkmann G, Matern JC, et al. Mapping of the communication‐mediating interface in nonribosomal peptide synthetases using a genetically encoded photocrosslinker supports an upside‐down helix‐hand motif. J Mol Biol. 2016;428:4345–4360. [DOI] [PubMed] [Google Scholar]
- 69. Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. [DOI] [PubMed] [Google Scholar]
- 70. Yeh E, Kohli RM, Bruner SD, Walsh CT. Type II thioesterase restores activity of a NRPS module stalled with an aminoacyl‐S‐enzyme that cannot be elongated. Chembiochem. 2004;5:1290–1293. [DOI] [PubMed] [Google Scholar]
- 71. Felnagle EA, Barkei JJ, Park H, et al. MbtH‐like proteins as integral components of bacterial nonribosomal peptide synthetases. Biochemistry. 2010;49:8815–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Weber T, Blin K, Duddela S, et al. antiSMASH 3.0‐a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–W243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Blin K, Shaw S, Steinke K, et al. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47:W81–W87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dejong CA, Chen GM, Li H, et al. Polyketide and nonribosomal peptide retro‐biosynthesis and global gene cluster matching. Nat Chem Biol. 2016;12:1007–1014. [DOI] [PubMed] [Google Scholar]
- 75. Skinnider MA, Dejong CA, Franczak BC, McNicholas PD, Magarvey NA. Comparative analysis of chemical similarity methods for modular natural products with a hypothetical structure enumeration algorithm. J Cheminform. 2017;9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Skinnider MA, Dejong CA, Rees PN, et al. Genomes to natural products PRediction informatics for secondary Metabolomes (PRISM). Nucleic Acids Res. 2015;43:9645–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Khater S, Gupta M, Agrawal P, et al. SBSPKSv2: Structure‐based sequence analysis of polyketide synthases and non‐ribosomal peptide synthetases. Nucleic Acids Res. 2017;45:W72–W79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS One. 2012;7:e34064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gulick AM. Conformational dynamics in the acyl‐CoA synthetases, adenylation domains of non‐ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lohman JR, Ma M, Cuff ME, et al. The crystal structure of BlmI as a model for nonribosomal peptide synthetase peptidyl carrier proteins. Proteins. 2014;82:1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lambalot RH, Gehring AM, Flugel RS, et al. A new enzyme superfamily ‐ the phosphopantetheinyl transferases. Chem Biol. 1996;3:923–936. [DOI] [PubMed] [Google Scholar]
- 82. Keating TA, Marshall CG, Walsh CT, Keating AE. The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains. Nat Struct Biol. 2002;9:522–526. [DOI] [PubMed] [Google Scholar]
- 83. Chen W‐H, Li K, Guntaka NS, Bruner SD. Interdomain and intermodule organization in epimerization domain containing nonribosomal peptide synthetases. ACS Chem Biol. 2016;11:2293–2303. [DOI] [PubMed] [Google Scholar]
- 84. Bloudoff K, Alonzo DA, Schmeing TM. Chemical probes allow structural insight into the condensation reaction of nonribosomal peptide synthetases. Cell Chem Biol. 2016;23:331–339. [DOI] [PubMed] [Google Scholar]
- 85. Drake EJ, Miller BR, Shi C, et al. Structures of two distinct conformations of holo‐non‐ribosomal peptide synthetases. Nature. 2016;529:235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tanovic A, Samel SA, Essen LO, Marahiel MA. Crystal structure of the termination module of a nonribosomal peptide synthetase. Science. 2008;321:659–663. [DOI] [PubMed] [Google Scholar]
- 87. Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. [DOI] [PubMed] [Google Scholar]
- 88. Tsai SC, Miercke LJ, Krucinski J, et al. Crystal structure of the macrocycle‐forming thioesterase domain of the erythromycin polyketide synthase: Versatility from a unique substrate channel. Proc Natl Acad Sci USA. 2001;98:14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tsai SC, Lu H, Cane DE, Khosla C, Stroud RM. Insights into channel architecture and substrate specificity from crystal structures of two macrocycle‐forming thioesterases of modular polyketide synthases. Biochemistry. 2002;41:12598–12606. [DOI] [PubMed] [Google Scholar]
- 90. Frueh DP, Arthanari H, Koglin A, et al. Dynamic thiolation‐thioesterase structure of a non‐ribosomal peptide synthetase. Nature. 2008;454:903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Whicher JR, Florova G, Sydor PK, et al. Structure and function of the RedJ protein, a thioesterase from the prodiginine biosynthetic pathway in Streptomyces coelicolor. J Biol Chem. 2011;286:22558–22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Guntaka NS, Healy AR, Crawford JM, Herzon SB, Bruner SD. Structure and functional analysis of ClbQ, an unusual intermediate‐releasing thioesterase from the colibactin biosynthetic pathway. ACS Chem Biol. 2017;12:2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chhabra A, Haque AS, Pal RK, et al. Nonprocessive [2 + 2]e‐ off‐loading reductase domains from mycobacterial nonribosomal peptide synthetases. Proc Natl Acad Sci USA. 2012;109:5681–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Du L, Lou L. PKS and NRPS release mechanisms. Nat Prod Rep. 2010;27:255–278. [DOI] [PubMed] [Google Scholar]
- 95. Liu Y, Zheng T, Bruner SD. Structural basis for phosphopantetheinyl carrier domain interactions in the terminal module of nonribosomal peptide synthetases. Chem Biol. 2011;18:1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Miller BR, Drake EJ, Shi C, Aldrich CC, Gulick AM. Structures of a nonribosomal peptide synthetase module bound to MbtH‐like proteins support a highly dynamic domain architecture. J Biol Chem. 2016;291:22559–22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Reimer JM, Eivaskhani M, Harb I, Guarne A, Weigt M, Schmeing TM. Structures of a dimodular nonribosomal peptide synthetase reveal conformational flexibility. Science. 2019;366:eaaw4388. [DOI] [PubMed] [Google Scholar]
- 98. Tarry MJ, Haque AS, Bui KH, Schmeing TM. X‐ray crystallography and electron microscopy of cross‐ and multi‐module nonribosomal peptide synthetase proteins reveal a flexible architecture. Structure. 2017;25:783–793. [DOI] [PubMed] [Google Scholar]
- 99. Tan XF, Dai YN, Zhou K, et al. Structure of the adenylation‐peptidyl carrier protein didomain of the Microcystis aeruginosa microcystin synthetase McyG. Acta Cryst D. 2015;71:873–881. [DOI] [PubMed] [Google Scholar]
- 100. Weissman KJ. The structural biology of biosynthetic megaenzymes. Nat Chem Biol. 2015;11:660–670. [DOI] [PubMed] [Google Scholar]
- 101. Gahloth D, Dunstan MS, Quaglia D, et al. Structures of carboxylic acid reductase reveal domain dynamics underlying catalysis. Nat Chem Biol. 2017;13:975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kreitler DF, Gemmell EM, Schaffer JE, Wencewicz TA, Gulick AM. The structural basis of N‐acyl‐alpha‐amino‐beta‐lactone formation catalyzed by a nonribosomal peptide synthetase. Nat Commun. 2019;10:3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ballard CE, Yu H, Wang B. Recent developments in depsipeptide research. Curr Med Chem. 2002;9:471–498. [DOI] [PubMed] [Google Scholar]
- 104. Taevernier L, Wynendaele E, Gevaert B, Spiegeleer B. Chemical classification of cyclic depsipeptides. Curr Prot Peptide Sci. 2017;18:425–452. [DOI] [PubMed] [Google Scholar]
- 105. Wang X, Gong X, Li P, Lai D, Zhou L. Structural diversity and biological activities of cyclic depsipeptides from fungi. Molecules. 2018;23:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sivanathan S, Scherkenbeck J. Cyclodepsipeptides: A rich source of biologically active compounds for drug research. Molecules. 2014;19:12368–12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Moore RE. Cyclic peptides and depsipeptides from cyanobacteria: A review. J Ind Microbiol. 1996;16:134–143. [DOI] [PubMed] [Google Scholar]
- 108. Derse PH, Strong FM. Toxicity of antimycin to fish. Nature. 1963;200:600–601. [DOI] [PubMed] [Google Scholar]
- 109. Cheng YQ. Deciphering the biosynthetic codes for the potent anti‐SARS‐CoV cyclodepsipeptide valinomycin in Streptomyces tsusimaensis ATCC 15141. Chembiochem. 2006;7:471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Agata N, Ohta M, Mori M, Isobe M. A novel dodecadepsipeptide, cereulide, is an emetic toxin of Bacillus‐Cereus. FEMS Microbiol Lett. 1995;129:17–19. [DOI] [PubMed] [Google Scholar]
- 111. Zan J, Li Z, Tianero MD, Davis J, Hill RT, Donia MS. A microbial factory for defensive kahalalides in a tripartite marine symbiosis. Science. 2019;364:eaaw6732. [DOI] [PubMed] [Google Scholar]
- 112. Schoenian I, Spiteller M, Ghaste M, Wirth R, Herz H, Spiteller D. Chemical basis of the synergism and antagonism in microbial communities in the nests of leaf‐cutting ants. Proc Natl Acad Sci USA. 2011;108:1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Oh DC, Poulsen M, Currie CR, Clardy J. Dentigerumycin: A bacterial mediator of an ant‐fungus symbiosis. Nat Chem Biol. 2009;5:391–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Li W, Fan A, Wang L, et al. Asperphenamate biosynthesis reveals a novel two‐module NRPS system to synthesize amino acid esters in fungi. Chem Sci. 2018;9:2589–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu J, Zhu X, Kim SJ, Zhang W. Antimycin‐type depsipeptides: Discovery, biosynthesis, chemical synthesis, and bioactivities. Nat Prod Rep. 2016;33:1146–1165. [DOI] [PubMed] [Google Scholar]
- 116. Clardy J, Walsh C. Lessons from natural molecules. Nature. 2004;432:829–837. [DOI] [PubMed] [Google Scholar]
- 117. Makarasen A, Yoza K, Isobe M. Higher structure of cereulide, an emetic toxin from Bacillus cereus, and special comparison with valinomycin, an antibiotic from Streptomyces fulvissimus. Chemistry. 2009;4:688–698. [DOI] [PubMed] [Google Scholar]
- 118. Sakurai N, Koike KA, Irie Y, Hayashi H. The rice culture filtrate of Bacillus cereus isolated from emetic‐type food poisoning causes mitochondrial swelling in a HEp‐2 cell. Microbiol Immunol. 1994;38:337–343. [DOI] [PubMed] [Google Scholar]
- 119. Mikami T, Horikawa T, Murakami T, et al. An improved method for detecting cytostatic toxin (emetic toxin) of Bacillus cereus and its application to food samples. FEMS Microbiol Lett. 1994;119:53–57. [DOI] [PubMed] [Google Scholar]
- 120. Bloudoff K, Rodionov D, Schmeing TM. Crystal structures of the first condensation domain of CDA synthetase suggest conformational changes during the synthetic cycle of nonribosomal peptide synthetases. J Mol Biol. 2013;425:3137–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. Structural and functional insights into a peptide bond‐forming bidomain from a nonribosomal peptide synthetase. Structure. 2007;15:781–792. [DOI] [PubMed] [Google Scholar]
- 122. Bergendahl V, Linne U, Marahiel MA. Mutational analysis of the C‐domain in nonribosomal peptide synthesis. Eur J Biochem. 2002;269:620–629. [DOI] [PubMed] [Google Scholar]
- 123. Marshall CG, Hillson NJ, Walsh CT. Catalytic mapping of the vibriobactin biosynthetic enzyme VibF. Biochemistry. 2002;41:244–250. [DOI] [PubMed] [Google Scholar]
- 124. Zaleta‐Rivera K, Xu C, Yu F, et al. A bidomain nonribosomal peptide synthetase encoded by FUM14 catalyzes the formation of tricarballylic esters in the biosynthesis of fumonisins. Biochemistry. 2006;45:2561–2569. [DOI] [PubMed] [Google Scholar]
- 125. Lin S, Van Lanen SG, Shen B. A free‐standing condensation enzyme catalyzing ester bond formation in C‐1027 biosynthesis. Proc Natl Acad Sci USA. 2009;106:4183–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yu D, Xu F, Zhang S, Zhan J. Decoding and reprogramming fungal iterative nonribosomal peptide synthetases. Nat Commun. 2017;8:15349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Steiniger C, Hoffmann S, Mainz A, et al. Harnessing fungal nonribosomal cyclodepsipeptide synthetases for mechanistic insights and tailored engineering. Chem Sci. 2017;8:7834–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ding Y, Rath CM, Bolduc KL, Hakansson K, Sherman DH. Chemoenzymatic synthesis of cryptophycin anticancer agents by an ester bond‐forming non‐ribosomal peptide synthetase module. J Am Chem Soc. 2011;133:14492–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Liuzzi VC, Mirabelli V, Cimmarusti MT, et al. Enniatin and beauvericin biosynthesis in fusarium species: Production profiles and structural determinant prediction. Toxins. 2017;9:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Xu Y, Orozco R, Kithsiri Wijeratne EM, et al. Biosynthesis of the cyclooligomer depsipeptide bassianolide, an insecticidal virulence factor of Beauveria bassiana. Fungal Genet Biol. 2009;46:353–364. [DOI] [PubMed] [Google Scholar]
- 131. Wang B, Kang Q, Lu Y, Bai L, Wang C. Unveiling the biosynthetic puzzle of destruxins in Metarhizium species. Proc Natl Acad Sci USA. 2012;109:1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Romans‐Fuertes P, Sondergaard TE, Sandmann MI, et al. Identification of the non‐ribosomal peptide synthetase responsible for biosynthesis of the potential anti‐cancer drug sansalvamide in Fusarium solani . Curr Genet. 2016;62:799–807. [DOI] [PubMed] [Google Scholar]
- 133. Zhang T, Jia X, Zhuo Y, et al. Cloning and characterization of a novel 2‐ketoisovalerate reductase from the beauvericin producer Fusarium proliferatum LF061. BMC Biotechnol. 2012;12:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lee C, Gorisch H, Kleinkauf H, Zocher R. A highly specific D‐hydroxyisovalerate dehydrogenase from the enniatin producer Fusarium sambucinum. J Biol Chem. 1992;267:11741–11744. [PubMed] [Google Scholar]
- 135. Kohlhaw GB. Leucine biosynthesis in fungi: Entering metabolism through the back door. Microbiol Mol Biol Rev. 2003;67:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zheng J, Taylor CA, Piasecki SK, Keatinge‐Clay AT. Structural and functional analysis of A‐type ketoreductases from the amphotericin modular polyketide synthase. Structure. 2010;18:913–922. [DOI] [PubMed] [Google Scholar]
- 137. Liu C, Yuan M, Xu X, et al. Substrate‐bound structures of a ketoreductase from amphotericin modular polyketide synthase. J Struct Biol. 2018;203:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Xu Y, Wijeratne EM, Espinosa‐Artiles P, Gunatilaka AA, Molnar I. Combinatorial mutasynthesis of scrambled beauvericins, cyclooligomer depsipeptide cell migration inhibitors from Beauveria bassiana. Chembiochem. 2009;10:345–354. [DOI] [PubMed] [Google Scholar]
- 139. Hamoen LW, Eshuis H, Jongbloed J, Venema G, van Sinderen D. A small gene, designated comS, located within the coding region of the fourth amino acid‐activation domain of srfA, is required for competence development in Bacillus subtilis. Mol Microbiol. 1995;15:55–63. [DOI] [PubMed] [Google Scholar]
- 140. Zhang S, Qiu Y, Kakule TB, et al. Identification of cyclic depsipeptides and their dedicated synthetase from Hapsidospora irregularis. J Nat Prod. 2017;80:363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hamano K, Kinoshita M, Tanzawa K, et al. Leualacin, a novel calcium blocker from Hapsidospora irregularis II. Structure determination. J Antibiot. 1992;45:906–913. [DOI] [PubMed] [Google Scholar]
- 142. Pedras MS, Irina Zaharia L, Ward DE. The destruxins: Synthesis, biosynthesis, biotransformation, and biological activity. Phytochemistry. 2002;59:579–596. [DOI] [PubMed] [Google Scholar]
- 143. Giuliano Garisto Donzelli B, Krasnoff SB, Moon YS, Churchill AC, Gibson DM. Genetic basis of destruxin production in the entomopathogen Metarhizium robertsii. Curr Genet. 2012;58:105–116. [DOI] [PubMed] [Google Scholar]
- 144. Wang B, Kang Q, Lu Y, Bai L, Wang C. Correction for Wang et al., unveiling the biosynthetic puzzle of destruxins in Metarhizium species. Proc Natl Acad Sci USA. 2016;113:E4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Perkins JB, Guterman SK, Howitt CL, Williams VE 2nd, Pero J. Streptomyces genes involved in biosynthesis of the peptide antibiotic valinomycin. J Bacteriol. 1990;172:3108–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ehling‐Schulz M, Vukov N, Schulz A, et al. Identification and partial characterization of the nonribosomal peptide synthetase gene responsible for cereulide production in emetic Bacillus cereus . Appl Environ Microbiol. 2005;71:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT. Cloning and characterization of the biosynthetic gene cluster for kutznerides. Proc Natl Acad Sci USA. 2007;104:16498–16503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Labby KJ, Watsula SG, Garneau‐Tsodikova S. Interrupted adenylation domains: Unique bifunctional enzymes involved in nonribosomal peptide biosynthesis. Nat Prod Rep. 2015;32:641–653. [DOI] [PubMed] [Google Scholar]
- 149. Alonzo DA, Magarvey NA, Schmeing TM. Characterization of cereulide synthetase, a toxin‐producing macromolecular machine. PLoS One. 2015;10:e0128569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Broberg A, Menkis A, Vasiliauskas R. Kutznerides 1‐4, depsipeptides from the actinomycete Kutzneria sp. 744 inhabiting mycorrhizal roots of Picea abies seedlings. J Nat Prod. 2006;69:97–102. [DOI] [PubMed] [Google Scholar]
- 151. Xu Y, Kersten RD, Nam S‐J, et al. Bacterial biosynthesis and maturation of the didemnin anti‐cancer agents. J Am Chem Soc. 2012;134:8625–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Jaitzig J, Li J, Sussmuth RD, Neubauer P. Reconstituted biosynthesis of the nonribosomal macrolactone antibiotic valinomycin in Escherichia coli . ACS Synthet Biol. 2014;3:432–438. [DOI] [PubMed] [Google Scholar]
- 153. Huguenin‐Dezot N, Alonzo DA, Heberlig GW, et al. Trapping biosynthetic acyl‐enzyme intermediates with encoded 2,3‐diaminopropionic acid. Nature. 2019;565:112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Amorim Franco TM, Blanchard JS. Bacterial branched‐chain amino acid biosynthesis: Structures, mechanisms, and drugability. Biochemistry. 2017;56:5849–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Zhang H, Chen J, Wang H, et al. Structural analysis of HmtT and HmtN involved in the tailoring steps of himastatin biosynthesis. FEBS Lett. 2013;587:1675–1680. [DOI] [PubMed] [Google Scholar]
- 156. Ma J, Wang Z, Huang H, et al. Biosynthesis of himastatin: Assembly line and characterization of three cytochrome P450 enzymes involved in the post‐tailoring oxidative steps. Angew Chem Int Ed Engl. 2011;50:7797–7802. [DOI] [PubMed] [Google Scholar]
- 157. Allen FH, Baalham CA, Lommerse JPM, Raithby PR. Carbonyl‐carbonyl interactions can be competitive with hydrogen bonds. Acta Cryst B. 1998;54:320–329. [Google Scholar]
- 158. Deane CM, Allen FH, Taylor R, Blundell TL. Carbonyl‐carbonyl interactions stabilize the partially allowed Ramachandran conformations of asparagine and aspartic acid. Protein Eng. 1999;12:1025–1028. [DOI] [PubMed] [Google Scholar]
- 159. May JJ, Kessler N, Marahiel MA, Stubbs MT. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc Natl Acad Sci USA. 2002;99:12120–12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ishikawa F, Miyanaga A, Kitayama H, et al. An engineered aryl acid adenylation domain with an enlarged substrate binding pocket. Angew Chem Int Ed Engl. 2019;58:6906–6910. [DOI] [PubMed] [Google Scholar]
- 161. Reid R, Piagentini M, Rodriguez E, et al. A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry. 2003;42:72–79. [DOI] [PubMed] [Google Scholar]
- 162. Keatinge‐Clay AT, Stroud RM. The structure of a ketoreductase determines the organization of the beta‐carbon processing enzymes of modular polyketide synthases. Structure. 2006;14:737–748. [DOI] [PubMed] [Google Scholar]
- 163. Keatinge‐Clay AT. A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem Biol. 2007;14:898–908. [DOI] [PubMed] [Google Scholar]
- 164. Caffrey P. Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. Chembiochem. 2003;4:654–657. [DOI] [PubMed] [Google Scholar]
- 165. Yin Y, Gokhale R, Khosla C, Cane DE. Erythromycin biosynthesis. The 4‐pro‐S hydride of NADPH is utilized for ketoreduction by both module 5 and module 6 of the 6‐deoxyerythronolide B synthase. Bioorg Med Chem Lett. 2001;11:1477–1479. [DOI] [PubMed] [Google Scholar]
- 166. Gao X, Haynes SW, Ames BD, et al. Cyclization of fungal nonribosomal peptides by a terminal condensation‐like domain. Nat Chem Biol. 2012;8:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Tautz T, Hoffmann J, Hoffmann T, et al. Isolation, structure elucidation, biosynthesis, and synthesis of antalid, a secondary metabolite from polyangium species. Org Lett. 2016;18:2560–2563. [DOI] [PubMed] [Google Scholar]
- 168. Dittmann J, Wenger RM, Kleinkauf H, Lawen A. Mechanism of cyclosporin A biosynthesis. Evidence for synthesis via a single linear undecapeptide precursor. J Biol Chem. 1994;269:2841–2846. [PubMed] [Google Scholar]
- 169. Weber G, Schörgendorfer K, Schneider‐Scherzer E, Leitner E. The peptide synthetase catalyzing cyclosporine production in Tolypocladium niveum is encoded by a giant 45.8‐kilobase open reading frame. Curr Genet. 1994;26:120–125. [DOI] [PubMed] [Google Scholar]
- 170. Tedesco D, Haragsim L. Cyclosporine: A review. J Transplant. 2012;2012:230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Haynes SW, Ames BD, Gao X, Tang Y, Walsh CT. Unraveling terminal C‐domain‐mediated condensation in fungal biosynthesis of imidazoindolone metabolites. Biochemistry. 2011;50:5668–5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zhang J‐M, Wang H‐H, Liu X, Hu C‐H, Zou Y. Heterologous and engineered biosynthesis of nematocidal polyketide‐nonribosomal peptide hybrid macrolactone from extreme thermophilic fungi. J Am Chem Soc. 2020;142:1957–1965. [DOI] [PubMed] [Google Scholar]
- 173. Gatto GJ Jr, McLoughlin SM, Kelleher NL, Walsh CT. Elucidating the substrate specificity and condensation domain activity of FkbP, the FK520 pipecolate‐incorporating enzyme. Biochemistry. 2005;44:5993–6002. [DOI] [PubMed] [Google Scholar]
- 174. Konig A, Schwecke T, Molnar I, et al. The pipecolate‐incorporating enzyme for the biosynthesis of the immunosuppressant rapamycin‐nucleotide sequence analysis, disruption and heterologous expression of rapP from Streptomyces hygroscopicus. Eur J Biochem. 1997;247:526–534. [DOI] [PubMed] [Google Scholar]
- 175. Nielsen JB, Hsu MJ, Byrne KM, Kaplan L. Biosynthesis of the immunosuppressant immunomycin: The enzymology of pipecolate incorporation. Biochemistry. 1991;30:5789–5796. [DOI] [PubMed] [Google Scholar]
- 176. Zheng CJ, Shao CL, Wu LY, et al. Bioactive phenylalanine derivatives and cytochalasins from the soft coral‐derived fungus. Aspergillus elegans. Mar Drugs. 2013;11:2054–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Frisvad JC, Houbraken J, Popma S, Samson RA. Two new Penicillium species Penicillium buchwaldii and Penicillium spathulatum, producing the anticancer compound asperphenamate. FEMS Microbiol Lett. 2013;339:77–92. [DOI] [PubMed] [Google Scholar]
- 178. Hai Y, Tang Y. Biosynthesis of long‐chain N‐acyl amide by a truncated polyketide synthase‐nonribosomal peptide synthetase hybrid megasynthase in fungi. J Am Chem Soc. 2018;140:1271–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Gunsior M, Breazeale SD, Lind AJ, Ravel J, Janc JW, Townsend CA. The biosynthetic gene cluster for a monocyclic beta‐lactam antibiotic, nocardicin A. Chem Biol. 2004;11:927–938. [DOI] [PubMed] [Google Scholar]
- 180. Konz D, Klens A, Schorgendorfer K, Marahiel MA. The bacitracin biosynthesis operon of Bacillus licheniformis ATCC 10716: Molecular characterization of three multi‐modular peptide synthetases. Chem Biol. 1997;4:927–937. [DOI] [PubMed] [Google Scholar]
- 181. Trauger JW, Kohli RM, Mootz HD, Marahiel MA, Walsh CT. Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature. 2000;407:215–218. [DOI] [PubMed] [Google Scholar]
- 182. Argyropoulos P, Bergeret F, Pardin C, et al. Towards a characterization of the structural determinants of specificity in the macrocyclizing thioesterase for deoxyerythronolide B biosynthesis. Biochim Biophys Acta. 1860;2016:486–497. [DOI] [PubMed] [Google Scholar]
- 183. Patel KD, d'Andrea FB, Gaudelli NM, Buller AR, Townsend CA, Gulick AM. Structure of a bound peptide phosphonate reveals the mechanism of nocardicin bifunctional thioesterase epimerase‐hydrolase half‐reactions. Nat Commun. 2019;10:3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Koglin A, Lohr F, Bernhard F, et al. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature. 2008;454:907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Samel SA, Wagner B, Marahiel MA, Essen LO. The thioesterase domain of the fengycin biosynthesis cluster: A structural base for the macrocyclization of a non‐ribosomal lipopeptide. J Mol Biol. 2006;359:876–889. [DOI] [PubMed] [Google Scholar]
- 186. Bruner SD, Weber T, Kohli RM, et al. Structural basis for the cyclization of the lipopeptide antibiotic surfactin by the thioesterase domain SrfTE. Structure. 2002;10:301–310. [DOI] [PubMed] [Google Scholar]
- 187. Akey DL, Kittendorf JD, Giraldes JW, Fecik RA, Sherman DH, Smith JL. Structural basis for macrolactonization by the pikromycin thioesterase. Nat Chem Biol. 2006;2:537–542. [DOI] [PubMed] [Google Scholar]
- 188. Baltz RH. Biosynthesis and genetic engineering of lipopeptide antibiotics related to daptomycin. Curr Top Med Chem. 2008;8:618–638. [DOI] [PubMed] [Google Scholar]
- 189. Steller S, Vollenbroich D, Leenders F, et al. Structural and functional organization of the fengycin synthetase multienzyme system from Bacillus subtilis b213 and A1/3. Chem Biol. 1999;6:31–41. [DOI] [PubMed] [Google Scholar]
- 190. Grunewald J, Sieber SA, Mahlert C, Linne U, Marahiel MA. Synthesis and derivatization of daptomycin: A chemoenzymatic route to acidic lipopeptide antibiotics. J Am Chem Soc. 2004;126:17025–17031. [DOI] [PubMed] [Google Scholar]
- 191. Guo C, Mandalapu D, Ji X, Gao J, Zhang Q. Chemistry and biology of teixobactin. Chemistry. 2018;24:5406–5422. [DOI] [PubMed] [Google Scholar]
- 192. Hou J, Robbel L, Marahiel MA. Identification and characterization of the lysobactin biosynthetic gene cluster reveals mechanistic insights into an unusual termination module architecture. Chem Biol. 2011;18:655–664. [DOI] [PubMed] [Google Scholar]
- 193. Du Y, Wang Y, Huang T, Tao M, Deng Z, Lin S. Identification and characterization of the biosynthetic gene cluster of polyoxypeptin a, a potent apoptosis inducer. BMC Microbiol. 2014;14:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Cheng YQ, Yang M, Matter AM. Characterization of a gene cluster responsible for the biosynthesis of anticancer agent FK228 in Chromobacterium violaceum no. 968. Appl Environ Microbiol. 2007;73:3460–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Chua MJ, Arnold MS, Xu W, et al. Effect of clinically approved HDAC inhibitors on Plasmodium, Leishmania and Schistosoma parasite growth. Int J Parasitol Drugs Drug Resist. 2017;7:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Krauth SJ, Coulibaly JT, Knopp S, Traore M, N'Goran EK, Utzinger J. An in‐depth analysis of a piece of shit: Distribution of Schistosoma mansoni and hookworm eggs in human stool. PLoS Negl Trop Dis. 2012;6:e1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Menkhaus M, Ullrich C, Kluge B, Vater J, Vollenbroich D, Kamp RM. Structural and functional organization of the surfactin synthetase multienzyme system. J Biol Chem. 1993;268:7678–7684. [PubMed] [Google Scholar]
- 198. Vollenbroich D, Mehta N, Zuber P, Vater J, Kamp RM. Analysis of surfactin synthetase subunits in srfA mutants of Bacillus subtilis OKB105. J Bacteriol. 1994;176:395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Tseng CC, Bruner SD, Kohli RM, Marahiel MA, Walsh CT, Sieber SA. Characterization of the surfactin synthetase C‐terminal thioesterase domain as a cyclic depsipeptide synthase. Biochemistry. 2002;41:13350–13359. [DOI] [PubMed] [Google Scholar]
- 200. Potharla VY, Wang C, Cheng YQ. Identification and characterization of the spiruchostatin biosynthetic gene cluster enable yield improvement by overexpressing a transcriptional activator. J Ind Microbiol Biotechnol. 2014;41:1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Narita K, Kikuchi T, Watanabe K, et al. Total synthesis of the bicyclic depsipeptide HDAC inhibitors spiruchostatins A and B, 5″‐epi‐spiruchostatin B, FK228 (FR901228) and preliminary evaluation of their biological activity. Chemistry. 2009;15:11174–11186. [DOI] [PubMed] [Google Scholar]
- 202. Hong J, Luesch H. Largazole: From discovery to broad‐spectrum therapy. Nat Prod Rep. 2012;29:449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Pulsawat N, Kitani S, Nihira T. Characterization of biosynthetic gene cluster for the production of virginiamycin M, a streptogramin type A antibiotic, in Streptomyces virginiae. Gene. 2007;393:31–42. [DOI] [PubMed] [Google Scholar]
- 204. Qu X, Lei C, Liu W. Transcriptome mining of active biosynthetic pathways and their associated products in Streptomyces flaveolus. Angew Chem Intl Ed Engl. 2011;50:9651–9654. [DOI] [PubMed] [Google Scholar]
- 205. Shi G, Shi N, Li Y, et al. D‐Alanylation in the assembly of ansatrienin side chain is catalyzed by a modular NRPS. ACS Chem Biol. 2016;11:876–881. [DOI] [PubMed] [Google Scholar]
- 206. Chen S, von Bamberg D, Hale V, et al. Biosynthesis of ansatrienin (mycotrienin) and naphthomycin. Identification and analysis of two separate biosynthetic gene clusters in Streptomyces collinus Tu 1892. Eur J Biochem. 1999;261:98–107. [DOI] [PubMed] [Google Scholar]
- 207. Gavalda S, Bardou F, Laval F, et al. The polyketide synthase Pks13 catalyzes a novel mechanism of lipid transfer in mycobacteria. Chem Biol. 2014;21:1660–1669. [DOI] [PubMed] [Google Scholar]
- 208. Gehring AM, Mori I, Walsh CT. Reconstitution and characterization of the Escherichia coli enterobactin synthetase from EntB, EntE, and EntF. Biochemistry. 1998;37:2648–2659. [DOI] [PubMed] [Google Scholar]
- 209. Rusnak F, Sakaitani M, Drueckhammer D, Reichert J, Walsh CT. Biosynthesis of the Escherichia coli siderophore enterobactin: Sequence of the entF gene, expression and purification of EntF, and analysis of covalent phosphopantetheine. Biochemistry. 1991;30:2916–2927. [DOI] [PubMed] [Google Scholar]
- 210. Shaw‐Reid CA, Kelleher NL, Losey HC, Gehring AM, Berg C, Walsh CT. Assembly line enzymology by multimodular nonribosomal peptide synthetases: The thioesterase domain of E. coli EntF catalyzes both elongation and cyclolactonization. Chem Biol. 1999;6:385–400. [DOI] [PubMed] [Google Scholar]
- 211. May JJ, Wendrich TM, Marahiel MA. The dhb operon of Bacillus subtilis encodes the biosynthetic template for the catecholic siderophore 2,3‐dihydroxybenzoate‐glycine‐threonine trimeric ester bacillibactin. J Biol Chem. 2001;276:7209–7217. [DOI] [PubMed] [Google Scholar]
- 212. Zhou Z, Lai JR, Walsh CT. Interdomain communication between the thiolation and thioesterase domains of EntF explored by combinatorial mutagenesis and selection. Chem Biol. 2006;13:869–879. [DOI] [PubMed] [Google Scholar]
- 213. Lai JR, Fischbach MA, Liu DR, Walsh CT. Localized protein interaction surfaces on the EntB carrier protein revealed by combinatorial mutagenesis and selection. J Am Chem Soc. 2006;128:11002–11003. [DOI] [PubMed] [Google Scholar]
- 214. Zhou Y, Prediger P, Dias LC, Murphy AC, Leadlay PF. Macrodiolide formation by the thioesterase of a modular polyketide synthase. Angew Chem Int Ed Engl. 2015;54:5232–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Hari TP, Labana P, Boileau M, Boddy CN. An evolutionary model encompassing substrate specificity and reactivity of type I polyketide synthase thioesterases. Chembiochem. 2014;15:2656–2661. [DOI] [PubMed] [Google Scholar]
- 216. Zhou Y, Murphy AC, Samborskyy M, Prediger P, Dias LC, Leadlay PF. Iterative mechanism of macrodiolide formation in the anticancer compound conglobatin. Chem Biol. 2015;22:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Budzikiewicz H, Bössenkamp A, Taraz K, Pandey A, Meyer J. Corynebactin, a cyclic catecholate siderophore from: Corynebacterium Glutamicum Atcc 14067 (brevibacterium Sp. Dsm 20411). ZNC. 1997;52:551–554. [Google Scholar]
- 218. Thies S, Santiago‐Schubel B, Kovacic F, Rosenau F, Hausmann R, Jaeger KE. Heterologous production of the lipopeptide biosurfactant serrawettin W1 in Escherichia coli . J Biotechnol. 2014;181:27–30. [DOI] [PubMed] [Google Scholar]
- 219. Li H, Tanikawa T, Sato Y, Nakagawa Y, Matsuyama T. Serratia marcescens gene required for surfactant serrawettin W1 production encodes putative aminolipid synthetase belonging to nonribosomal peptide synthetase family. Microbiol Immunol. 2005;49:303–310. [DOI] [PubMed] [Google Scholar]
- 220. Sussmuth R, Muller J, von Dohren H, Molnar I. Fungal cyclooligomer depsipeptides: From classical biochemistry to combinatorial biosynthesis. Nat Prod Rep. 2011;28:99–124. [DOI] [PubMed] [Google Scholar]
- 221. Jirakkakul J, Punya J, Pongpattanakitshote S, et al. Identification of the nonribosomal peptide synthetase gene responsible for bassianolide synthesis in wood‐decaying fungus Xylaria sp. BCC1067. Microbiology. 2008;154:995–1006. [DOI] [PubMed] [Google Scholar]
- 222. Xu Y, Orozco R, Wijeratne EM, Gunatilaka AA, Stock SP, Molnar I. Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem Biol. 2008;15:898–907. [DOI] [PubMed] [Google Scholar]
- 223. Schrettl M, Bignell E, Kragl C, et al. Distinct roles for intra‐ and extracellular siderophores during Aspergillus fumigatus infection. PLoS Pathog. 2007;3:1195–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Oide S, Moeder W, Krasnoff S, et al. NPS6, encoding a nonribosomal peptide synthetase involved in siderophore‐mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant Cell. 2006;18:2836–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Smith CD, Zhang X, Mooberry SL, Patterson GM, Moore RE. Cryptophycin: A new antimicrotubule agent active against drug‐resistant cells. Cancer Res. 1994;54:3779–3784. [PubMed] [Google Scholar]
- 226. D'Agostino G, del Campo J, Mellado B, et al. A multicenter phase II study of the cryptophycin analog LY355703 in patients with platinum‐resistant ovarian cancer. Int J Gynecol Cancer. 2006;16:71–76. [DOI] [PubMed] [Google Scholar]
- 227. Schmidt JJ, Khatri Y, Brody SI, et al. A versatile chemoenzymatic synthesis for the discovery of potent cryptophycin analogs. ACS Chem Biol. 2020;15:524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Yan Y, Zhang L, Ito T, et al. Biosynthetic pathway for high structural diversity of a common dilactone core in antimycin production. Org Lett. 2012;14:4142–4145. [DOI] [PubMed] [Google Scholar]
- 229. Sandy M, Rui Z, Gallagher J, Zhang W. Enzymatic synthesis of dilactone scaffold of antimycins. ACS Chem Biol. 2012;7:1956–1961. [DOI] [PubMed] [Google Scholar]
- 230. Shiomi K, Hatae K, Hatano H, et al. A new antibiotic, antimycin Ag, produced by Streptomyces sp. K01‐0031. J Antibiot. 2005;58:74–78. [DOI] [PubMed] [Google Scholar]
- 231. Seipke RF, Crossman L, Drou N, et al. Draft genome sequence of Streptomyces strain S4, a symbiont of the leaf‐cutting ant Acromyrmex octospinosus . J Bacteriol. 2011;193:4270–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Li X, Zvanych R, Torchia J, Magarvey NA. Structures and biosynthesis of 12‐membered macrocyclic depsipeptides from Streptomyces sp. ML55. Bioorg Med Chem Lett. 2013;23:4150–4153. [DOI] [PubMed] [Google Scholar]
- 233. Skyrud W, Liu J, Thankachan D, Cabrera M, Seipke RF, Zhang W. Biosynthesis of the 15‐membered ring depsipeptide neoantimycin. ACS Chem Biol. 2018;13:1398–1406. [DOI] [PubMed] [Google Scholar]
- 234. Li X, Zvanych R, Vanner SA, Wang W, Magarvey NA. Chemical variation from the neoantimycin depsipeptide assembly line. Bioorg Med Chem Lett. 2013;23:5123–5127. [DOI] [PubMed] [Google Scholar]
- 235. Awakawa T, Fujioka T, Zhang L, et al. Reprogramming of the antimycin NRPS‐PKS assembly lines inspired by gene evolution. Nat Commun. 2018;9:3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Lee J, Currano JN, Carroll PJ, Joullie MM. Didemnins, tamandarins and related natural products. Nat Prod Rep. 2012;29:404–424. [DOI] [PubMed] [Google Scholar]
- 237. Sakai R, Rinehart KL, Kishore V, et al. Structure‐activity relationships of the didemnins. J Med Chem. 1996;39:2819–2834. [DOI] [PubMed] [Google Scholar]
- 238. Urdiales JL, Morata P, Nunez De Castro I, Sanchez‐Jimenez F. Antiproliferative effect of dehydrodidemnin B (DDB), a depsipeptide isolated from Mediterranean tunicates. Cancer Lett. 1996;102:31–37. [DOI] [PubMed] [Google Scholar]
- 239. Newman DJ, Cragg GM. Marine natural products and related compounds in clinical and advanced preclinical trials. J Nat Prod. 2004;67:1216–1238. [DOI] [PubMed] [Google Scholar]
- 240. Marquez BL, Watts KS, Yokochi A, et al. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of Actin assembly. J Nat Prod. 2002;65:866–871. [DOI] [PubMed] [Google Scholar]
- 241. Ramaswamy AV, Sorrels CM, Gerwick WH. Cloning and biochemical characterization of the hectochlorin biosynthetic gene cluster from the marine cyanobacterium Lyngbya majuscula . J Nat Prod. 2007;70:1977–1986. [DOI] [PubMed] [Google Scholar]
- 242. Auerbach D, Yan F, Zhang Y, Muller R. Characterization of an unusual glycerate esterification process in vioprolide biosynthesis. ACS Chem Biol. 2018;13:3123–3130. [DOI] [PubMed] [Google Scholar]
- 243. Yan F, Auerbach D, Chai Y, et al. Biosynthesis and heterologous production of vioprolides: Rational biosynthetic engineering and unprecedented 4‐methylazetidinecarboxylic acid formation. Angew Chem Int Ed Engl. 2018;57:8754–8759. [DOI] [PubMed] [Google Scholar]
- 244. Dorrestein PC, Van Lanen SG, Li W, et al. The bifunctional glyceryl transferase/phosphatase OzmB belonging to the HAD superfamily that diverts 1,3‐bisphosphoglycerate into polyketide biosynthesis. J Am Chem Soc. 2006;128:10386–10387. [DOI] [PubMed] [Google Scholar]
- 245. Toh M, Moffitt MC, Henrichsen L, et al. Cereulide, the emetic toxin of Bacillus cereus, is putatively a product of nonribosomal peptide synthesis. J Appl Microbiol. 2004;97:992–1000. [DOI] [PubMed] [Google Scholar]
- 246. Matter AM, Hoot SB, Anderson PD, Neves SS, Cheng YQ. Valinomycin biosynthetic gene cluster in Streptomyces: Conservation, ecology and evolution. PLoS One. 2009;4:e7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Ehling‐Schulz M, Fricker M, Grallert H, Rieck P, Wagner M, Scherer S. Cereulide synthetase gene cluster from emetic Bacillus cereus: Structure and location on a mega virulence plasmid related to Bacillus anthracis toxin plasmid pXO1. BMC Microbiol. 2006;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Heberlig GW, Boddy CN. Thioesterase from cereulide biosynthesis is responsible for oligomerization and macrocyclization of a linear tetradepsipeptide. J Nat Prod. 2020;83:1990–1997. [DOI] [PubMed] [Google Scholar]
- 249. Chin JW. Expanding and reprogramming the genetic code of cells and animals. Annu Rev Biochem. 2014;83:379–408. [DOI] [PubMed] [Google Scholar]
- 250. Chin JW. Expanding and reprogramming the genetic code. Nature. 2017;550:53–60. [DOI] [PubMed] [Google Scholar]
- 251. Jennewein S, Croteau R. Taxol: Biosynthesis, molecular genetics, and biotechnological applications. Appl Microbiol Biotechnol. 2001;57:13–19. [DOI] [PubMed] [Google Scholar]