

Abstract
Currently, the world is suffering from the pandemic of coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that uses angiotensin-converting enzyme 2 (ACE2) as a receptor to enter the host cells. So far, 60 million people have been infected with SARS-CoV-2, and 1.4 million people have died because of COVID-19 worldwide, causing serious health, economical, and sociological problems. However, the mechanism of the effect of SARS-CoV-2 on human host cells has not been defined. The present study reports that the SARS-CoV-2 spike protein alone without the rest of the viral components is sufficient to elicit cell signaling in lung vascular cells. The treatment of human pulmonary artery smooth muscle cells or human pulmonary artery endothelial cells with recombinant SARS-CoV-2 spike protein S1 subunit (Val16 – Gln690) at 10 ng/ml (0.13 nM) caused an activation of MEK phosphorylation. The activation kinetics was transient with a peak at 10 min. The recombinant protein that contains only the ACE2 receptor-binding domain of the SARS-CoV-2 spike protein S1 subunit (Arg319 – Phe541), on the other hand, did not cause this activation. Consistent with the activation of cell growth signaling in lung vascular cells by the SARS-CoV-2 spike protein, pulmonary vascular walls were found to be thickened in COVID-19 patients. Thus, SARS-CoV-2 spike protein-mediated cell growth signaling may participate in adverse cardiovascular/pulmonary outcomes, and this mechanism may provide new therapeutic targets to combat COVID-19.
Keywords: Cell signaling, Coronavirus, COVID-19, SARS-CoV-2, Vascular
Abbreviations: ACE2, angiotensin-converting enzyme 2; ARDS, acute respiratory distress syndrome; COVID-19, coronavirus disease 2019; RBD, receptor-binding domain; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Graphical abstract
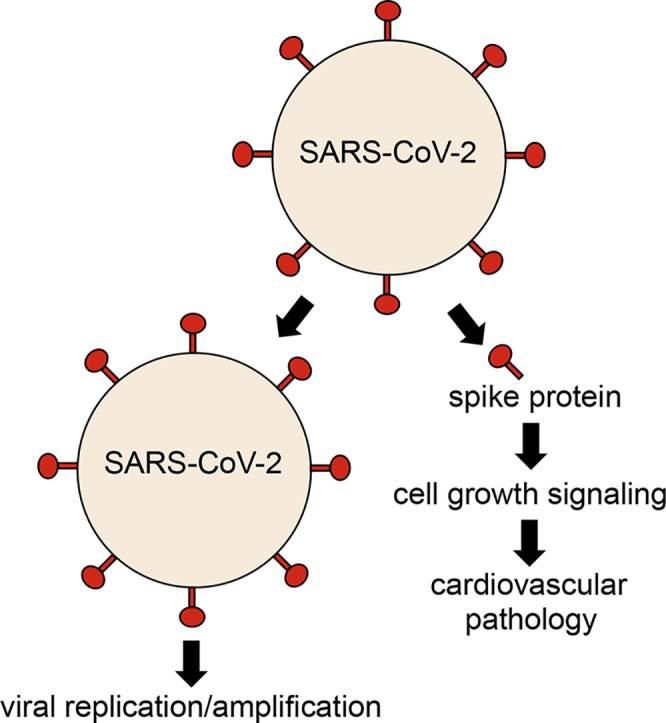
1. Introduction
Coronaviruses are positive sense single stranded RNA viruses that often cause the common cold [1,2]. Some coronaviruses can, however, be lethal. Currently, the world suffer from the pandemic of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [3,4]. So far, 60 million people have been infected with SARS-CoV-2 worldwide, causing serious health, economical, and sociological problems. SARS-CoV-2 uses angiotensin converting enzyme 2 (ACE2) as a receptor to enter the host cells [5,6]. Lung cells are the primary targets of SARS-CoV-2, resulting in severe pneumonia and acute respiratory distress syndrome (ARDS) [7,8]. So far, nearly 1.4 million people have died of COVID-19.
It has been noted that certain populations of infected individuals are severely affected by and die of COVID-19. Elderly patients with systemic hypertension and other cardiovascular diseases are particularly susceptible to developing severe and possibly fatal conditions [4,9,10]. Thus, managing the pulmonary and cardiovascular aspects of COVID-19 is considered to be the key for reducing the severity of the disease and the associated mortality. However, it is unclear exactly how SARS-CoV-2 affects humans. Thus, understanding the mechanism of SARS-CoV-2 actions should help develop therapeutic strategies to reduce the mortality and morbidity associated with COVID-19.
The SARS-CoV-2 spike protein plays a critical role in initiating the interactions between the virus and the host cell surface receptor, facilitating the viral entry into the host cell by assisting in the fusion of the viral and host cell membranes [11,12]. This protein consists of two subunits: Subunit 1 (S1) contains the ACE2 receptor-binding domain (RBD) and Subunit 2 (S2) is responsible for fusion.
The current dogma of the SARS-CoV-2 mechanism is that the binding of the viral spike protein to the host ACE2 receptor results in the entry of the virus into host cells, with the cellular response being the result of the viral infection [5,6,11,12]. However, we herein show that SARS-CoV-2 spike protein alone without the rest of the viral components is sufficient to elicit cell signaling in human host cells, suggesting a novel biological mechanism of SARS-CoV-2 actions.
2. Materials and methods
2.1. Cell culture
Human pulmonary artery smooth muscle cells and human pulmonary artery endothelial cells were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA), and rat pulmonary artery smooth muscle cells were purchased from Cell Applications (San Diego, CA, USA). Cells were cultured in accordance with the manufacturers' instructions in 5% CO2 at 37 °C. Cells at passages 3–6 were maintained in low fetal bovine serum (0.4%)-containing medium overnight before the treatment as routinely performed in experiments on cell signaling and protein phosphorylation [13].
Cells were treated with either the recombinant SARS-CoV-2 spike protein full length S1 subunit, which contains most of the S1 subunit (Val16 - Gln690) with a molecular weight of ~75 kDa (RayBiotech, Peachtree Corners, GA, USA), or the recombinant SARS-CoV-2 spike protein RBD (RayBiotech), which only contains the RBD region (Arg319 - Phe541) with a molecular weight of ~25 kDa. Some cells were pretreated for 1 hour with the rabbit anti-ACE2 antibody (Catalog # 4355; Cell Signaling Technology, Danvers, MA, USA).
2.2. Western blotting
To prepare cell lysates, cells were washed in phosphate buffered saline and solubilized with lysis buffer containing 50 mM Hepes (pH 7.4), 1% (v/v) Triton X-100, 4 mM EDTA, 1 mM sodium fluoride, 0.1 mM sodium orthovanadate, 1 mM tetrasodium pyrophosphate, 2 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Samples were then centrifuged at 16,000g for 10 min at 4 °C, supernatants were collected, and protein concentrations were determined [13].
Equal amounts of protein samples were electrophoresed through a reducing sodium dodecyl sulfate polyacrylamide gel. Proteins were then electro-transferred to the Immobilon-FL Transfer Membrane (MilliporeSigma, Burlington, MA, USA). The membrane was blocked with Odyssey blocking buffer (LI-COR, Lincoln, NE, USA) for 1 h at 25 °C and incubated overnight with the rabbit anti-phospho-MEK1/2 (Ser217/221) antibody (Catalog #9154; Cell Signaling), the mouse anti-MEK1/2 antibody (Catalog #4694; Cell Signaling), the rabbit anti-phospho-Akt (Ser473) antibody (Catalog #9271; Cell Signaling), or the rabbit anti-phospho-Stat3 (Tyr705) antibody (Catalog #9131; Cell Signaling) at 4 °C. Washed membranes were then incubated with IRDye 680RD or IRDye 800CW (LI-COR) for 1 h. Signals were obtained by using the Odyssey Infrared Imaging System (LI-COR).
2.3. Histological examinations of patient samples
Postmortem lung tissues were collected from 10 patients who died of COVID-19 in Ukraine during the period of March–July 2020. For comparison, we used the archival materials of lung tissues from 10 patients who died of influenza A (H1N1) during the epidemic in November and December of 2009. Clinical studies were approved by the regional committee for medical research ethics in Kiev, Ukraine and performed under the Helsinki Declaration of 1975 revised in 2013 or comparable ethical standards.
For histological examinations, samples of autopsy materials approximately 10 mm thick were fixed overnight in 10% buffered formalin at room temperature. Fixed tissues were embedded in paraffin. From paraffin blocks, 5 μm thick sections were made using a microtome. The sections were stained with hematoxylin and eosin. Histological specimens were examined using a Leica BX 51 microscope, a Leica MC 190 digital camera, and the Leica LAS software at magnifications of 100× – 200×. Morphometric investigations included the assessment of pulmonary arterial wall thickness and pulmonary arterial lumen index (the ratio of internal vessel area to external vessel area) using the ImageJ software. Seven vessels were analyzed for each patient.
2.4. Statistical analysis
For Western blotting data, means and standard errors of mean (SEM) were computed. Two groups were compared using a two-tailed Student's t-test, and differences between more than two groups were determined by the analysis of variance (ANOVA). p < 0.05 was defined to be statistically significant. For the morphometric analysis, IBM SPSS Statistics software version 22.0 was used for the statistical calculations. The Mann-Whitney U Test was used to define the statistical significance at p < 0.05.
3. Results
3.1. The SARS-CoV-2 spike protein alone can activate cell signaling in human cells
To test the hypothesis that the SARS-CoV-2 spike protein can elicit cell signaling on its own, human pulmonary artery smooth muscle cells were treated with the full length S1 subunit (Val16 - Gln690) of the SARS-CoV-2 spike protein for 0, 10, and 30 min. As shown in Fig. 1A, the spike protein at a concentration of 10 ng/ml (0.133 nM) strongly activated the phosphorylation of 45 kDa MEK at Ser217 and Ser221 residues. The kinetics of MEK phosphorylation promoted by the full-length S1 subunit (Val16 - Gln690) of the SARS-CoV-2 spike protein was consistently found to be transient with a peak at 10 min. This fast activation suggests that this may be a receptor-mediated cell-signaling event. Similarly, the full-length S1 subunit SARS-CoV-2 spike protein promoted the phosphorylation of MEK in human pulmonary artery endothelial cells (Fig. 1B). The SARS-CoV-2 spike protein, however, did not activate other signaling events such as Akt (Fig. 1A) and Stat3 (Fig. 1B) pathways.
Fig. 1.

The SARS-CoV-2 spike protein S1 promotes the MEK phosphorylation in human cells. (A) Human pulmonary artery smooth cells (N = 5) and (B) human pulmonary artery endothelial cells (N = 3) were treated with recombinant full length S1 subunit of the SARS-CoV-2 spike protein (Val16 - Gln690) at 10 ng/ml for the durations indicated. Cell lysates were prepared and subjected to Western blotting using antibodies against phosphorylated MEK (p-MEK), MEK protein, phosphorylated Akt (p-Akt), and phosphorylated Stat3 (p-Stat3). Bar graphs represent means ± SEM. *Significantly different from 0 min at p < 0.05.
As we performed experiments to determine the effects of the full-length S1 subunit of the SARS-CoV-2 spike protein on rat pulmonary artery smooth muscle cells, we surprisingly found that, not only does the SARS-CoV-2 spike protein not promote the MEK phosphorylation, but it even decreases this phosphorylation. As shown in Fig. 2 , the treatment of the full length SARS-CoV-2 spike protein S1 resulted in the dephosphorylation of MEK as early as 10 min after the treatment, and this dephosphorylation event was maintained for at least 60 min. Thus, the SARS-CoV-2 spike protein promotes the MEK phosphorylation in human cells but not in rat cells.
Fig. 2.

In rat cells, the SARS-CoV-2 spike protein S1 does not phosphorylate, but rather dephosphorylates MEK. Rat pulmonary artery smooth muscle cells were treated with recombinant full length S1 subunit of the SARS-CoV-2 spike protein (Val16 - Gln690) at 10 ng/ml for the durations indicated. Cell lysates were prepared and subjected to Western blotting using antibodies against phosphorylated MEK (p-MEK) and MEK protein. The bar graph represents means ± SEM (N = 4). *Significantly different from 0 min at p < 0.05.
To confirm that the action of the SARS-CoV-2 spike protein is through its well-known receptor ACE2,5,6 human pulmonary artery smooth muscle cells were pretreated with the ACE2 antibody for 1 h before being treated with the full-length S1 subunit of the SARS-CoV-2 spike protein. The ACE2 antibody alone caused the activation of MEK, and the SARS-CoV-2 spike protein did not further increase this MEK phosphorylation signal (Fig. 3 ).
Fig. 3.

The SARS-CoV-2 spike protein S1 does not promote the phosphorylation of MEK in the presence of the neutralizing antibody against ACE2. Human pulmonary artery smooth muscle cells were pre-treated with the ACE2 antibody for 1 h and then treated with recombinant full length S1 subunit of the SARSCoV-2 spike protein (Val16 – Gln690) for 10 min. Cell lysates were prepared and subjected to Western blotting using antibodies against phosphorylated MEK (p-MEK) and MEK protein. The bar graph represents means ± SEM (N = 4).*Significantly different from untreated control at p < 0.05. NS denotes that the two values are not significantly different from each other at p < 0.05.
3.2. RBD domain alone is not sufficient to activate cell signaling
To test whether the RBD binding to ACE2 is sufficient to stimulate cell signaling for MEK phosphorylation, human cells were treated with the recombinant S1 RBD of the SARS-CoV-2 spike protein containing only the RBD (Arg319 - Phe541). In contrast to the full-length S1 subunit (Val16 - Gln690) that strongly phosphorylated MEK, S1 RBD (Arg319 - Phe541) did not activate the MEK phosphorylation in human pulmonary artery smooth muscle cells (Fig. 4A) or in human pulmonary artery endothelial cells (Fig. 4B). Thus, other regions of the spike protein in addition to the RBD may be required for eliciting cell signaling for MEK phosphorylation.
Fig. 4.

The RBD only containing SARS-CoV-2 spike protein S1 does not activate the MEK phosphorylation. (A) Human pulmonary artery smooth muscle cells and (B) human pulmonary artery endothelial cells were treated with recombinant full length S1 subunit (Val16 - Gln690) or the RBD region of S1 subunit (Arg319 - Phe541) at 100 ng/ml for the durations indicated. Cell lysates were prepared and subjected to Western blotting using antibodies against phosphorylated MEK (p-MEK) and the MEK protein. Bar graphs represent means ± SEM (N = 3). *Significantly different from 0 min control at p < 0.05.
3.3. Pulmonary vascular walls are thickened in COVID-19 patients
Our results show that the SARS-CoV-2 spike protein is capable of activating the MEK/ERK pathway in pulmonary artery smooth muscle and endothelial cells, which suggests that cell growth signaling may be triggered in the pulmonary vascular walls in response to SARS-CoV-2. To test this, we examined the lung histology results of patients who died of COVID-19.
Fig. 5A shows the representative pulmonary vessels of patients who died of COVID-19. These pulmonary arteries consistently exhibit histological characteristics of vascular wall thickening. The walls of the pulmonary arteries are thickened mainly due to the hypertrophy of the tunica media. The vessels have lost the clarity of the boundaries with the surrounding lung parenchyma. Enlargement of smooth muscle cells of the middle lining of the arteries is observed, and the nuclei of smooth muscle cells also are swollen, increased in size, and have become rounded, while vacuoles are identified in the cytoplasm.
Fig. 5.

Pulmonary arterial walls are thickened in COVID-19 patients. (A) Representative hematoxylin and eosin staining images of the pulmonary arteries of patients who died of ARDS due to COVID-19 and H1N1 influenza. In COVID-19 patients, but not in H1N1 influenza patients, thickening of the pulmonary arterial walls was observed. Scale bars indicate 100 μm. (B) Box plots represent quantifications of pulmonary arterial wall thicknesses and lumen areas in COVID-19 and H1N1 influenza patients using H&E stained slides. The number of patients used for these analyses was 10 for COVID-19 and 10 for H1N1. *The Mann-Whitney U Test indicated that the two values are significantly different at p = 0.001.
By contrast, pulmonary vessels of patients who died of H1N1 influenza infection showed no thickened pulmonary arteries (Fig. 5A). Unlike in COVID-19, smooth muscle cells form a compact layer of tunica media in H1N1 patients, there are no signs of the enlargement of smooth muscle cells.
We performed a morphometric analysis of the pulmonary vessels of patients who died of SARS-CoV-2 and H1N1 influenza virus. The median wall thickness value of the pulmonary arteries of COVID-19 patients was calculated to be 15.4 μm, while that of patients with H1N1 influenza was 6.7 μm (Fig. 5B, left), indicating that pulmonary arterial walls are more than two-fold thicker in COVID-19 patients compared with H1N1 patients. These two values are significantly different from each other, with p = 0.001. The pulmonary arterial lumen area was found to be significantly smaller in COVID-19 patients compared with H1N1 patients, with p = 0.001 (Fig. 5B, right).
4. Discussion
The major finding of this study is that even without the rest of the virus, the SARS-CoV-2 spike protein can elicit cell signaling, specifically the activation of the MEK/ERK pathway, in human host lung vascular smooth muscle and endothelial cells. MEK is a mitogen-activated protein kinase kinase (MAPKK) that phosphorylates and activates extracellular-regulated kinase (ERK), one type of mitogen-activated protein kinases (MAPK). In this MEK/ERK pathway of cell signaling, MEK is activated by the phosphorylation by Raf1 kinase [14]. The MEK/ERK pathway is a well-known cell growth mechanism [15] and has also been shown to facilitate viral replication cycles [16].
In contrast to the full-length S1 subunit of the SARS-CoV-2 spike protein (Val16 - Gln690), which strongly activated MEK, the RBD only containing protein (Arg319 - Phe541) did not activate MEK in human pulmonary artery smooth muscle or endothelial cells. These results suggest that the protein regions other than the RBD (i.e. Val16 - Arg319 and/or Phe541 - Gln690) are required for eliciting cell signaling for the MEK phosphorylation. Thus, we propose that SARS-CoV-2 spike protein-mediated cell signaling is not merely cells responding to anything binding to the membrane surface protein, but rather is a growth factor/hormone-like specific cell signal transduction event that is well coordinated by the RBD as well as other protein regions that facilitate cell signaling. Our ACE2 antibody experiments, as described in Fig. 3, support the involvement of ACE2. However, the binding of the ACE2 antibody to ACE2 seems sufficient to cause the activation of MEK. While we interpreted that the ACE2 antibody blocked and interfered with the binding of the RBD to ACE2, it is possible that ACE2 antibody-mediated cell signaling may have desensitized the subsequent spike protein-mediated effects to activate MEK. Thus, SARS-CoV-2 spike protein-mediated cell signaling may occur through a receptor other than ACE2.
In contrast to the effects of the SARS-CoV-2 spike protein on human cells to activate the MEK phosphorylation, this protein promoted the dephosphorylation of MEK in rat cells. This species difference in the actions of the SARS-CoV-2 spike protein may be important to understand how this virus severely affects humans. We previously reported a novel ligand-mediated dephosphorylation mechanism of MEK induced by neurotensin and neuromedin N [17]. The SARS-CoV-2 spike protein signaling in rat pulmonary artery smooth muscle cells represents another example of the MEK dephosphorylation mechanism. These results also suggest that the species-specific actions of SARS-CoV-2 may depend on how spike protein-mediated cell signaling mechanisms differ among species.
It has been noted that elderly patients with systemic hypertension and other cardiovascular diseases are particularly susceptible to developing severe and possibly fatal conditions of COVID-19 [4,9,10]. Thus, the pathology of the disease does not seem to be explained merely by the action of SARS-CoV-2 to enter host cells for destruction. The present study shows that cell growth signaling may be triggered by this virus in both cultured cells and in COVID-19 patient samples. We propose that SARS-CoV-2 spike protein-mediated cell signaling promotes the hyperplasia and/or hypertrophy of vascular smooth muscle and endothelial cells, contributing to the complex cardiovascular outcomes in COVID-19. Further studies investigating this concept are warranted in order to develop effective therapeutic strategies to reduce the mortality associated with COVID-19.
Our histological finding that COVID-19 patients have thickened pulmonary arterial walls also has clinical significance. These changes may reflect the tissue responses of the lungs to hypoxia and may play a vital role in the development of acute respiratory failure in COVID-19 patients. These results also indicate the possibility that patients who recovered from COVID-19 may be predisposed to developing pulmonary arterial hypertension and right-sided heart failure. We also noticed that published lung histology images of patients who died of ARDS during the 2002–2004 SARS outbreak due to the infection with SARS-CoV-1 did not exhibit the signs of thickened pulmonary vascular walls [18,19]. Thus, pulmonary vascular wall thickening is a unique feature of the SARS-CoV-2 infection and COVID-19
5. Conclusions
Our results in human pulmonary artery smooth muscle and endothelial cells revealed that the SARS-CoV-2 spike protein S1 subunit is sufficient to trigger biological responses in human host cells in the absence of the participation of the rest of the SARS-CoV-2 viral particle. We also found that the pulmonary vascular walls are thickened in COVID-19 patients. We propose that SARS-CoV-2 spike protein-mediated cell growth signaling participates in adverse cardiovascular/pulmonary outcomes seen in COVID-19. This mechanism may provide new therapeutic targets to combat the SARS-CoV-2 infection and COVID-19.
Funding
This work was supported in part by the National Institutes of Health [grant numbers R21AI142649, R03AG059554, R03AA026516] to Y.J.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Declaration of Competing Interest
None.
References
- 1.Su S., Wong G., Shi W., Liu J., Lai A.C.K., Zhou J., Liu W., Bi Y., Gao G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satija N., Lal S.K. The molecular biology of SARS coronavirus. Ann. N. Y. Acad. Sci. 2007;1102:26–38. doi: 10.1196/annals.1408.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., Yuan M.L., Zhang Y.L., Dai F.H., Liu Y., Wang Q.M., Zheng J.J., Xu L., Holmes E.C., Zhang Y.Z. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tai W., He L., Zhang X., Pu J., Voronin D., Jiang S., Zhou Y., Du L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020;17:613–620. doi: 10.1038/s41423-020-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L., Tai Y., Bai C., Gao T., Song J., Xia P., Dong J., Zhao J., Wang F.S. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu C., Chen X., Cai Y., Xia J., Zhou X., Xu S., Huang H., Zhang L., Zhou X., Du C., Zhang Y., Song J., Wang S., Chao Y., Yang Z., Xu J., Zhou X., Chen D., Xiong W., Xu L., Zhou F., Jiang J., Bai C., Zheng J., Song Y. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020;180:1–11. doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B., Yang J., Zhao F., Zhi L., Wang X., Liu L., Bi Z., Zhao Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020;109:531–538. doi: 10.1007/s00392-020-01626-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J., Zheng Y., Gou X., Pu K., Chen Z., Guo Q., Ji R., Wang H., Wang Y., Zhou Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int. J. Infect. Dis. 2020;94:91–95. doi: 10.1016/j.ijid.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong C.M., Cheema A.K., Zhang L., Suzuki Y.J. Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 2008;102:310–318. doi: 10.1161/CIRCRESAHA.107.159814. [DOI] [PubMed] [Google Scholar]
- 14.Wortzel I., Seger R. The ERK cascade. Distinct functions within various subcellular organelles. Genes Cancer. 2011;2:195–209. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W., Liu H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 16.Bonjardim C.A. Viral exploitation of the MEK/ERK pathway - a tale of vaccinia virus and other viruses. Virology. 2017;507:267–275. doi: 10.1016/j.virol.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Shults N.V., Almansour F.S., Rybka V., Suzuki D.I., Suzuki Y.J. Ligand-mediated dephosphorylation signaling for MAP kinase. Cell. Signal. 2018;52:147–154. doi: 10.1016/j.cellsig.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding Y., Wang H., Shen H., Li Z., Geng J., Han H., Cai J., Li X., Kang W., Weng D., Lu Y., Wu D., He L., Yao K. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J. Pathol. 2003;200:282–289. doi: 10.1002/path.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang D.M., Chamberlain D.W., Poutanen S.M., Low D.E., Asa S.L., Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod. Pathol. 2005;18:1–10. doi: 10.1038/modpathol.3800247. [DOI] [PMC free article] [PubMed] [Google Scholar]