

Abstract
Influenza A viruses (IAVs) use diverse mechanisms to interfere with cellular gene expression. Although many RNA-seq studies have documented IAV-induced changes in host mRNA abundance, few were designed to allow an accurate quantification of changes in host mRNA splicing. Here, we show that IAV infection of human lung cells induces widespread alterations of cellular splicing, with an overall increase in exon inclusion and decrease in intron retention. Over half of the mRNAs that show differential splicing undergo no significant changes in abundance or in their 3′ end termination site, suggesting that IAVs can specifically manipulate cellular splicing. Among a randomly selected subset of 21 IAV-sensitive alternative splicing events, most are specific to IAV infection as they are not observed upon infection with VSV, induction of interferon expression or induction of an osmotic stress. Finally, the analysis of splicing changes in RED-depleted cells reveals a limited but significant overlap with the splicing changes in IAV-infected cells. This observation suggests that hijacking of RED by IAVs to promote splicing of the abundant viral NS1 mRNAs could partially divert RED from its target mRNAs. All our RNA-seq datasets and analyses are made accessible for browsing through a user-friendly Shiny interface (http://virhostnet.prabi.fr:3838/shinyapps/flu-splicing or https://github.com/cbenoitp/flu-splicing).
INTRODUCTION
Influenza A viruses (IAVs) cause annual epidemics and occasional pandemics with major consequences in terms of mortality and economical loss and are a perennial threat to worldwide public health (1). Their genome consists of eight single-stranded RNA segments of negative polarity, and the virally encoded RNA-dependent RNA polymerase (FluPol) ensures transcription and replication of the viral genome in the nucleus of infected cells. However, viral transcription is also critically dependent on the cellular machinery of transcription. Notably, initiation of viral mRNA synthesis occurs through a unique mechanism known as cap snatching, whereby the FluPol uses short primers derived from capped host RNA polymerase II (PolII) transcripts to prime transcription. Cap snatching is underpinned by a physical association between FluPol and PolII (2). Moreover, some viral mRNAs undergo a tightly regulated splicing, which involves the host splicing machinery. Many splicing factors were identified in proteomic studies or genome-wide loss-of-function genetic screens as being potentially involved in IAV life cycle [e.g. (3,4)]. The RED–SMU1 splicing complex was shown to bind FluPol and to promote splicing of the viral NS1 mRNA (5), whereas hnRNPK and NS1-BP are associated with the NS1 viral protein and promote splicing of the viral M1 mRNA (6).
IAVs not only exploit cellular factors to enable the expression of their own genome, but also interfere with the expression of cellular genes in a way that restricts the cellular response to viral infection and facilitates viral replication (7). One of the mechanisms involved is the disruption of PolII transcription. IAV-infected cells show a genome-wide reduction of PolII occupancy into gene bodies downstream of the transcription start site, suggesting that cap snatching by FluPol interferes with PolII transcriptional elongation (8). IAV infection was also shown to induce a massive failure of PolII termination at poly(A) sites, leading to termination readthrough and continued transcription in the intragenic regions up to several hundreds of bases downstream the gene termini (8–10). Other mechanisms of host-cell shut-off involve the viral NS1 protein, which inhibits the post-transcriptional maturation and nuclear export of cellular mRNAs, and the PA-X protein, which causes their degradation in the cytoplasm [reviewed in (7)].
Until recently, there was no evidence for alterations of host splicing in IAV-infected cells. Alternative splicing (AS) expands the diversity of proteins that can be expressed from a given pre-mRNA and can modulate the stability and translation of mRNAs. Although AS is an essential mechanism for the regulation of gene expression in response to external stimuli, the role of AS in host–pathogen interactions has long been underappreciated. This is likely due to the high complexity of AS regulation and the methodological difficulties of transcriptome-wide analysis of AS. In the recent years, the Illumina RNA-seq technology revealed that up to several hundreds of host genes can show altered mRNA splicing upon infection with herpesviruses (11,12), reoviruses (13,14), flaviviruses (15–17) or IAVs (18,19). However, most studies do not use the sequencing depth that is required for accurate quantification of AS isoform abundance (20), and they provide no or limited validation by an orthogonal methodology.
Here, we aimed at providing a comprehensive analysis of AS alterations induced upon IAV infection of the human alveolar A549 cells. Unlike previously published studies (18,19), we analyzed RNA-seq data using a software that does not rely on existing splice site annotations and therefore can identify novel splicing events. Our data demonstrate widespread changes in AS events (ASEs) upon viral infection with a trend toward increased splicing of cellular pre-mRNAs. Over half of the mRNAs that show differential splicing undergo no significant changes in abundance or in their 3′ end termination site, suggesting that IAVs can specifically manipulate cellular splicing independently of other transcriptional changes. We provide evidence that a substantial proportion of IAV-sensitive ASEs are specific to IAV infection and are conserved across distinct cell lines and viral subtypes. Furthermore, we investigate to what extent hijacking of the RED splicing factor by IAVs to promote splicing of their own mRNAs (5) could account for some of the observed AS changes in infected cells. All our RNA-seq datasets are available in GEO (accession number GSE154596) and our analyses can be explored through a Shiny user-friendly interface (http://virhostnet.prabi.fr:3838/shinyapps/flu-splicing).
MATERIALS AND METHODS
Cells and viruses
A549 (provided by Prof. M. Schwemmle, Freiburg, Germany) and HEK-293T cells (provided by Dr M. Perricaudet, Paris, France) were grown in complete Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (PS). Calu-3 cells (provided by Dr F. Schwalm, Marburg, Germany) were grown in DMEM/F-12 GlutaMax, supplemented with 10% fetal calf serum, 1% PS, 1% sodium pyruvate, 2% sodium bicarbonate and 1% non-essential amino acids. The recombinant A/WSN/33 virus was produced as described in (5). The human seasonal IAV A/Paris/1154/2014 (H3N2) was provided by the National Influenza Center at the Institut Pasteur (Paris, France).
RNA-seq
A549 cells plated in 12-well plates (2 × 105 cells/well) were infected with the WSN virus at a multiplicity of infection (MOI) of 5 PFU/cell, or mock infected. Alternatively, they were transfected with 25 nM of anti-RED (5′-GUGAUGAGGAGGUGGAUUA-3′) or control siRNA (Dharmacon) using the Dharmafect-1 reagent, according to the manufacturer’s recommendations. At 6 h post-infection or 48 h post-transfection, total RNA was extracted and treated with DNase using the RNeasy Mini Kit (QIAGEN), according to the manufacturer’s instructions. All samples checked on Bioanalyzer RNA6000 Nano Chip (Agilent Technologies, Santa Clara, CA) had an RNA integrity score >9. Starting from 800 ng of DNA-free total RNA from each sample, poly(A)+ RNA purification and library preparation were performed using the TruSeq Stranded mRNA library preparation kit (Illumina, Inc., San Diego, CA), following the manufacturer’s instructions. Libraries were checked for quality on Bioanalyzer DNA chips (Agilent Technologies, Santa Clara, CA). Accurate quantification was performed using the fluorescence-based quantitation Qubit dsDNA HS Assay Kit (Thermo Fisher). Based on a pilot experiment showing that in infected cell samples >50% of the reads aligned with viral sequences, the eight sample libraries derived from four biological replicates of mock- and virus-infected cells were randomly distributed into four lanes of a HiSeq2500 sequencer flow cell using a non-equimolar ratio of 1 (mock sample) to 2.3 (infected sample) in each lane, and were sequenced in a paired mode (2 × 120 bases). Raw reads were quality checked using FastQC and mapped to the human genome (hg38, Gencode v27 annotation) and to the A/WSN/33 virus genome (accession numbers CY034132–CY034139) using STAR (v2.5.3a) (21).
Identification of changes in gene expression and termination readthrough
Focusing on the reads that mapped to the human genome, we used HTSeq-count (v0.6.1) (22) to count the number of reads per gene and per intergenic region (up to 5000 nt downstream the annotated transcript end sites of genes). The differential expression analysis as well as the estimation of FPKM of genes and their downstream intergenic regions was done with the DESeq2 R package (23) using default parameters. A gene was considered as markedly and significantly differentially expressed if it passed the following thresholds: gene baseMean 10, gene FPKMIAV-infected 1 and/or gene FPKMmock-infected 1, adjusted P-value ≤0.05 and |log2 FC| ≥ 1. The same thresholds were applied to compare the control siRNA and anti-RED siRNA conditions.
The ratio of downstream intergenic FPKM over gene FPKM was computed to estimate the percentage of readthrough (PRT) transcription for each gene in mock and infected samples:
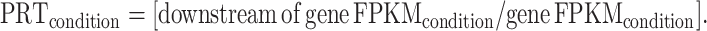 |
ΔPRT represents the magnitude of the change of readthrough transcription between the two conditions and was computed as follows: ΔPRT = [PRTIAV-infected − PRTmock-infected].
A downstream intergenic region was considered as markedly and significantly differentially expressed if it passed the following thresholds: baseMean 10, gene FPKMIAV-infected 1 or gene FPKMmock-infected 1, adjusted P-value ≤0.05 and 2 ≥ ΔPRT ≥ 0.025.
Identification of changes in alternative splicing
All raw reads were assembled using the KisSplice (v2.4.1) (24) local transcriptome assembler. This tool allows to extract splicing events that correspond to specific patterns in the De Bruijn graph, which we call bubbles. KisSplice outputs the sequences and quantification of ASEs. The following parameters were used to run KisSplice: --stranded --strandedAbsoluteThreshold 0 --mismatches 2 --counts 2 --min_overlap 5 --experimental. The sequences of the ASEs were then mapped to the human genome (hg38 using annotation Gencode v27) using STAR, with default settings. Each event was classified in a type of splicing event [alternative acceptor (altA), alternative donor (altD), exon skipping (ES), multiple exon skipping (ES-M) and intron retention (IR)] and assigned to a gene using KisSplice2RefGenome (v1.2.3) (25). Finally, the differential analysis was done with the kissDE R package (v1.1, doi: 10.18129/B9.bioc.kissDE). KissDE outputs three important measures: percent spliced in (PSI), ΔPSI and adjusted P-value. PSI is a measure representing the percentage of inclusion of an exonic or intronic sequence:
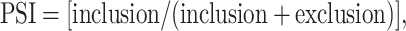 |
where ‘inclusion’ corresponds to the number of reads supporting the inclusion isoform and ‘exclusion’ corresponds to the number of reads supporting the exclusion isoform. A PSI of 1 indicates that the exonic or intronic region is always included, while a PSI of 0 indicates that it is always spliced out in the mature RNA. ΔPSI represents the magnitude of the change of inclusion of the ASE between the two conditions and is computed as follows: ΔPSI = [PSIIAV-infected − PSImock-infected], where PSIIAV-infected and PSImock-infected are the mean PSI values of the four biological replicates. A splicing event was considered as markedly and significantly differentially regulated if it passed the following threshold: |ΔPSI| ≥ 0.10 and adjusted P-value ≤0.05.
To filter out minor isoforms, we used two additional criteria. First, we used a threshold on the level of expression of the gene (FPKMIAV-infected ≥ 1 and/or FPKMmock-infected ≥ 1). Second, because this cutoff turned out to be insufficient to filter out splicing variations among minor isoforms of highly expressed genes, we computed a new measure, which we called local event expression (LEE). This measure gives an estimate of the proportion of the isoforms of the gene containing the splicing event. It is computed as follows:
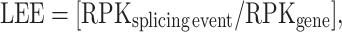 |
where 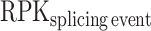 corresponds to the number of reads per kilobase for the splicing event and
corresponds to the number of reads per kilobase for the splicing event and 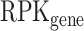 corresponds to the number of reads per kilobase for the full gene. To compute the RPK of the splicing event, we used the number of reads corresponding to the splicing event given by KisSplice and divided it by the effective size of the event (in bp). The effective size corresponds to the number of unique positions where reads, used for the isoform abundance estimation, can be aligned. The RPK of the full gene was computed using the quantification given by HTSeq-count divided by the gene length (in bp). Only the events with LEE ≥ 0.5 in one of the IAV-infected or mock-infected conditions were taken into consideration. The same thresholds were applied to compare the control siRNA and anti-RED siRNA conditions.
corresponds to the number of reads per kilobase for the full gene. To compute the RPK of the splicing event, we used the number of reads corresponding to the splicing event given by KisSplice and divided it by the effective size of the event (in bp). The effective size corresponds to the number of unique positions where reads, used for the isoform abundance estimation, can be aligned. The RPK of the full gene was computed using the quantification given by HTSeq-count divided by the gene length (in bp). Only the events with LEE ≥ 0.5 in one of the IAV-infected or mock-infected conditions were taken into consideration. The same thresholds were applied to compare the control siRNA and anti-RED siRNA conditions.
Principal component and GO term enrichment analyses
Principal component analysis (PCA) was performed using ade4 R package (26) on the normalized gene count values, on the normalized intergenic count values or on the PSI values. We plotted coordinates of the eight samples on the first two principal components using the ggplot2 R package (27). We searched for gene ontology (GO) terms enriched in the differentially expressed genes and in the genes containing a differentially spliced event using the topGO R package (doi: 10.18129/B9.bioc.topGO). We used the elim algorithm with a user-defined score and tested only the GO terms containing at least 50 genes. For the analysis of the differentially expressed genes, the score was defined as the log2 FC value if the adjusted P-value was lower than the threshold of 0.05, and 0 if not. This score gave more importance to genes with a large difference of expression between the two conditions. The same principle was used to define the score for the enrichment analysis on the differentially regulated splicing events. The score was defined as |ΔPSI| if the adjusted P-value was lower than the threshold of 0.05, and 0 if not.
RT-PCR and qRT-PCR
A549 and Calu-3 cells were infected at a MOI of 3–5 PFU/cell with the indicated viruses, or mock infected. Alternatively, A549 cells were treated with 200 mM KCl or mock treated for 2 h, or transfected with 10 ng of total RNA prepared from WSN-infected or mock-infected MDCK cells at 6 hpi, using the transfection reagent Lipofectamine 3000 (Thermo Fisher). Total RNA was extracted using the RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. RT-PCR was performed on 100 ng of total RNA using the forward and reverse primers listed in Supplementary Table S1, and the Superscript III One-Step RT-PCR Kit (Invitrogen) following the manufacturer’s protocol. Amplicons were loaded on a 2% agarose gel. When required, the ImageJ software was used to measure the intensity of the bands and calculate a ΔPSI value. The criterion used for validation was |ΔPSI| ≥ 10%. The ES events subjected to validation were randomly selected using the ‘Random’ function in Excel. In the few cases when the design of adequate validation primers turned out to be unfeasible because of the presence of ≥2 events sharing the same genomic position, or the presence of a very short (<20 bp) or GC-rich flanking exon, the corresponding ES events were skipped and replaced with another randomly selected ES event.
For IFNβ, uc.145 and GAPDH qRT-PCR, reverse transcription was performed on 500 ng of total RNA, using the Maxima First Strand cDNA Synthesis Kit, which includes a mixture of oligo-dT and random hexamer primers (Thermo Scientific), in a final volume of 20 μl. Real-time PCR was performed on 2 μl of a 1:10 dilution of the reverse-transcription reaction, using the Solaris qPCR Master Mix (Thermo Scientific), sets of primers and probe as provided in the Solaris qPCR Gene Expression Assays (Thermo Scientific) or, in the case of uc.145, the forward 5′-GCAGCGAACCCTGCTAAATA-3′ and reverse 5′-AGCCGGCACTAATAGTCCAA-3′, primers and the SYBR Green Master Mix (Roche Life Science), with a Light Cycler 480 (Roche).
FACS
A549 (6 × 105) or Calu-3 (24 × 105) cells seeded in six-well plates were mock infected or infected with the indicated viruses. Cells were harvested with trypsin, followed by fixation (4% paraformaldehyde) and permeabilization (0.1% Triton X). Cells were then stained with a primary mouse monoclonal anti-influenza virus NP antibody (Abcam; ab20343) and a secondary anti-mouse DyLight633 (red) antibody (Thermo Fisher; 35512). The stained cells were subjected to FACS analysis (Attune NxT Flow Cytometer). Data were analyzed and processed using the FlowJo software.
Shiny interface
We used RStudio’s Shiny framework to develop a web-based interface that allows to browse the AS, gene expression and readthrough results generated in this study, as well as the multivariate analysis (PCA) of these three transcriptional processes. This Shiny interface is available online (http://virhostnet.prabi.fr:3838/shinyapps/flu-splicing) and is also available for download on GitHub to install locally (https://github.com/cbenoitp/flu-splicing).
The users can explore and filter the genes or ASEs of interest according to several metrics and can download the list of selected genes or ASEs as an excel file. The interface also gives the possibility to plot some metrics (like ΔPSI or log2 FC) and to download the resulting plot. Finally, users can browse the intersection of different ASE analyses.
RESULTS
Influenza A/WSN/33 infection induces broad changes in cellular splicing
To assess the impact of IAV infection on the AS of host genes, human alveolar A549 cells were mock infected or infected with the IAV strain A/WSN/33 (WSN) at a MOI of 5 PFU/cell. The high infection rate in these conditions was assessed by indirect immunofluorescence and FACS analysis (∼99% cells positively stained for the viral nucleoprotein; Supplementary Figure S1A). An overview of downstream analyses is provided in Figure 1A. Briefly, poly(A)-tailed RNAs were extracted and subjected to HiSeq2500 Illumina sequencing, and the sequencing reads were mapped to the human genome and the WSN genome using the STAR algorithm (21), as detailed in the ‘Materials and Methods’ section. RNA sequencing of control and IAV-infected samples (four independent biological replicates for each condition) yielded 35–80 million paired-end reads (2 × 120 nt) mapping to the host genome (Figure 1B, blue bars, and Supplementary Table S2), allowing for a robust analysis of the transcriptional response to infection. Reads mapping to the viral genome showed the expected distribution across viral open reading frames (Supplementary Figure S1B). The cellular ASEs were analyzed using the KisSplice pipeline (24,25), which enables de novo calling of ASEs and therefore can identify so far non-annotated ASEs. Statistical comparison between mock and infected cell samples from the four independent replicates was carried out using the kissDE algorithm (25) and the magnitude of the perturbation was quantified using the PSI metric, as described in the ‘Materials and Methods’ section. In addition, the DESeq2 algorithm (23) was used for differential gene expression and termination readthrough analysis. PCA indicated that viral infection accounts for 36%, 52% and 54% of the total variance observed in splicing, gene expression and termination readthrough, respectively (Figure 1C).
Figure 1.

Dual RNA-seq analysis of IAV-infected cells. (A) Schematic representation of the dual RNA-seq analysis pipeline. Illumina reads corresponding to viral and cellular mRNAs are represented in red and blue, respectively. (B) Mapping of Illumina sequencing reads. The left panel shows the number of reads mapped to the A/WSN/33 virus genome (red), the human genome (blue) or unmapped (gray) for each technical replicate. In the right panel, the same data are shown as percentages of the total number of reads. (C) PCA on PSI values (left panel), normalized gene counts (middle panel) and normalized intergenic counts (right panel). The samples corresponding to each experimental condition (four biological replicates per condition) were plotted on the first two principal components.
We focused our splicing analysis on the five major types of ASEs, i.e. altA and altD sites, ES, ES-M and IR. To filter out minor isoforms, including minor isoforms derived from highly expressed genes, we computed for each of the >66 000 ASEs that were identified by kissDE an LEE value that provides an estimation of the proportion of isoforms showing the ASE (see the ‘Materials and Methods’ section). Only the ASEs showing FPKM ≥ 1 and LEE ≥ 0.5 in at least one of the two conditions (IAV-infected and/or mock-infected) were taken into consideration. The resulting reference splicing dataset comprised 25 037 ASEs corresponding to 7043 distinct genes (Figure 2A, outer circle). Upon additional filtering for ASEs showing a marked and significant change in ΔPSI values (|ΔPSI| ≥ 10% and P ≤ 0.05), we identified 3969 ASEs, corresponding to 2076 genes, that are IAV sensitive (Figure 2A, inner circle, and Supplementary Table S3). A substantial proportion of these shows at least one non-annotated splice site (Figure 2B), therefore highlighting the added value of performing de novo calling of ASEs with KisSplice. A trend for increased exon inclusion and intron removal in IAV-infected cells, i.e. overall increased splicing, was observed. Indeed, the median ΔPSI (PSIIAV-infected − PSImock-infected) value was positive for ES (+11%) and ES-M events (+10.3%) and negative for IR events (−17.5%) (Figure 2C). The distribution of the lengths of IAV-sensitive ASEs is shown in Figure 2D and Supplementary Figure S2. Within the IAV-sensitive dataset, the median length of skipped exons was close to the median length of all exons in the human genome (127 nt versus 149 nt), whereas the median length of retained introns was smaller than the median length of all introns in the human genome (368 nt versus 2036 nt). This observation could partly be due to the limited capacity of KisSplice to assemble long introns, since it requires that they are fully covered by reads to be correctly assembled. However, we obtained similar findings using IRFinder, a pipeline dedicated to the analysis of IR (28): the overall decrease in IR was clearly confirmed (median dPSI = −15.4%), and the median length of retained introns in the IAV-sensitive dataset (707 nt) was higher compared with that when the KisSplice pipeline was used but clearly smaller than the median length of all introns (Supplementary Figure S3).
Figure 2.

Global alterations of the cellular splicing landscape upon IAV infection. (A) Filtering of the ASE dataset. Out of the >66 000 ASEs that were analyzed by kissDE, the 25 037 ASEs showing FPKM ≥ 1 and LEE ≥ 0.5 in the mock-infected and/or IAV-infected condition were considered as the reference splicing dataset (outer circle). Upon additional filtering for |ΔPSI| ≥ 10% and P ≤ 0.05, 3969 differentially regulated ASEs were identified (IAV-sensitive splicing dataset, inner circle). (B) Number of annotated (plain bars) and non-annotated (hatched bars) events in the IAV-sensitive splicing dataset. The percentage of non-annotated events for each of the indicated types of ASE is indicated above. (C-D) Box plots showing the distribution of ΔPSI values (PSIIAV-infected − PSImock-infected) (C) or the distribution of the lengths in nucleotides of the variable part (D), for each of the indicated type of ASE within the IAV-sensitive splicing dataset. The median values are shown as a line in the center of the boxes.
IAV-induced changes in cellular splicing are specific and not merely a consequence of interferon induction or cellular stress
To assess the specificity of influenza-induced changes in splicing, A549 cells were subjected in parallel to infection with the WSN strain or three distinct treatments: (i) infection with the VSV virus, which unlike IAV does not replicate in the nucleus of infected cells; (ii) transfection with RNA extracted from WSN-infected cells, to induce interferon expression; and (iii) incubation with 200 mM KCl to induce an osmotic stress. The efficacy of each treatment was controlled by quantifying the IFNβ mRNA and the uc.145 long noncoding RNA as a marker of the osmotic stress response (29) (Supplementary Figure S4). We focused on ES events because they represent almost 50% of the influenza-sensitive splicing events (Figure 2B) and are the most amenable to RT-PCR validation using gene-specific primers. A subset of 21 ES events, randomly selected among the IAV-sensitive events identified by RNA-seq, was characterized in the four above-described experimental conditions using RT-PCR. The increase or decrease in exon inclusion expected from the RNA-seq data was confirmed for all 21 ES events; in 15 cases, it was detected exclusively in A549 cells infected with the WSN virus and not in the other experimental conditions (Figure 3, left panels, Supplementary Figure S5 and Supplementary Table S4). These results indicate that influenza infection induces specific changes in the host AS program that are independent from the interferon response and distinct from a general stress response.
Figure 3.

RT-PCR validation and further characterization of IAV-sensitive splicing events detected by RNA-seq. (Left) A549 cells were subjected to viral infection at a high MOI (WSN versus VSV or mock infection), RNA transfection (RNA extracted from WSN-infected versus mock-infected cells) or osmotic stress (200 mM KCl versus mock treatment). (Right) A549 or Calu-3 cells were infected at a high MOI with the WSN virus or a seasonal H3N2 IAV, or mock infected. Total RNA was extracted and RT-PCR was performed using primers flanking the exon of interest. The amplification products were loaded on a 2% agarose gel. The expected size in case of exon inclusion or exclusion is indicated, as well as the ΔPSI value as determined from RNA-seq data. MW: molecular weight. The 300-bp band of the 100-bp DNA Ladder (NEB) is indicated by a star.
To evaluate to what extent the observed changes in splicing were dependent on the experimental virus–cell system, a subset of eight IAV-sensitive ES events identified by RNA-seq was characterized in parallel in A549 or Calu-3 cells infected at a high MOI with the WSN strain (H1N1 subtype) or with a virus representative of the circulating human seasonal IAVs (H3N2 subtype). Both cell lines were infected at high rates, as assessed by FACS analysis (∼99% and 85% NP-positive A549 cells and ∼90% and 80% NP-positive Calu-3 cells upon infection with the WSN and H3N2 viruses, respectively) (Supplementary Figure S1A). The expected increase or decrease in exon inclusion was detected systematically with the WSN and H3N2 viruses in both A549 and Calu-3 cells (Figure 3, right panels, Supplementary Figure S6 and Supplementary Table S4). Our findings suggest that a substantial proportion of the IAV-induced changes in host splicing detected by RNA-seq are largely conserved across cell lines and shared between different influenza viruses.
Overall, RT-PCR validation on RNAs extracted from new batches of mock- or WSN-infected cells was performed on a total of 46 randomly selected ES events showing IAV sensitivity in RNA-seq data. All (including 13 for which at least one splice site was non-annotated) showed the same pattern upon RT-PCR as expected from the RNA-seq data (Supplementary Figures S5–S7 and Supplementary Table S4). Our careful filtering out of minor isoforms likely contributes to this high validation rate, which was obtained irrespective of whether |ΔPSI| was in the 10–20%, 20–30% or >30% range (17/17, 12/12 and 17/17 validated events, respectively; Supplementary Table S4), therefore establishing the robustness of our experimental setting and bioinformatics pipeline.
IAV-induced changes in cellular splicing are largely independent from the other cellular transcriptional responses to infection
The observed cellular splicing changes in IAV-infected cells (Figure 2) could possibly result from other transcriptional changes. Therefore, we examined whether the 2076 genes that show altered splicing upon IAV infection were exhibiting changes in the level of expression and/or defects in transcription termination. To this end, we performed DESeq2 analysis on both genic and intergenic regions. Upon DESeq2 analysis on genic regions, 5527 genes were found to be differentially expressed (log2 FC ≥ 1, P ≤ 0.05), among which 2481 and 3046 genes were significantly up- and downregulated in infected cells, respectively (Supplementary Table S5). When DESeq2 analysis was performed on intergenic regions that are in a 5-kb window upstream and downstream genic regions, it revealed an overall 2-fold increase in intergenic transcription in WSN-infected cells compared with mock-infected cells (Supplementary Figure S8) in agreement with recently published studies (8–10). Using a ΔPRT metric analogous to ΔPSI (see the ‘Materials and Methods’ section), a marked and significant change in transcriptional termination (ΔPRT ≥ 0.025, P ≤ 0.05) was observed for 2012 genes, among which a vast majority of 1777 genes showed a positive ΔPRT value, i.e. an increased termination readthrough in IAV-infected cells compared with mock-infected cells (Supplementary Table S6).
Slightly over 50% of the genes showing differential splicing upon IAV infection showed no differential expression or termination readthrough (Figure 4A). Besides, GO analysis using the topGO bioinformatics resource (30) revealed different enrichment patterns for the genes showing differential splicing, expression or termination readthrough (Figure 4B and Supplementary Figure S8B). Therefore, a large proportion of viral-induced alterations of cellular splicing are not merely a side effect of other transcriptional dysregulations, and possibly reflect a direct manipulation of the splicing machinery in infected cells. To more precisely assess the level of interdependence between splicing and expression changes, we plotted the mean ΔPSI values of IAV-sensitive ES and IR splicing events as a function of the log2 FC value of the corresponding gene (Figure 4C, left and right panels, respectively). A slightly positive correlation (R2 = 5%, P = 7 × 10−11) was observed for IR events only (Figure 4C, black curves). When this analysis was restricted to genes involved in the regulation of transcription by PolII, which are significantly enriched among both differentially spliced and differentially expressed genes (Figure 4B, indicated by red stars), the observed correlation coefficient was still not significant for ES (R2 = 0.3%, P = 0.26) but higher for IR (R2 = 9%, P = 2.2 × 10−3) (Figure 4C, red curves). Overall, our data establish that splicing and expression changes induced by IAV expression are generally independent from each other. However, they demonstrate a low level of correlation between increased splicing and decreased expression, which is more pronounced for IR than for ES events.
Figure 4.

Cross-analysis of splicing alterations and other transcriptional changes induced by IAV infection. (A) Venn diagram representing the sets of genes showing differential splicing, expression and/or readthrough upon IAV infection. (B) GO analysis. The top 10 GO terms most enriched among the genes differentially spliced in the CDS (upper panel) or the genes differentially expressed (lower panel) are indicated. The dot size is proportional to the number of genes annotated with the GO term in the full genome, as indicated. (C) Plot representing ΔPSI as a function of the log2 FC value, for differentially spliced ES events (left panel) and IR events (right panel). Each dot represents a distinct splicing event. Regression curves are shown. Black curve: all splicing events. Red dots and curve: splicing events related to genes annotated with the GO term ‘Regulation of transcription by RNA polymerase II’ (GO0006357).
IAV-sensitive and RED protein-controlled splicing events show a limited but significant overlap
As a substantial proportion of IAV-induced changes in splicing appeared to be independent from other global host responses to infection, we hypothesized that some of them could result from a direct interplay between IAV and the splicing machinery. We focused on the RED splicing factor because we previously showed that it is recruited by the influenza polymerase, regulates splicing of the abundant viral NS1 mRNAs and is essential for efficient viral replication (5). Although RED showed no dramatic changes of its accumulation level or subcellular localization in infected cells (5), its splicing function could possibly be impacted by viral-induced changes, e.g. the nuclear accumulation of viral polymerase and NS1 pre-mRNAs. To investigate this possibility, we performed a global profiling of ASEs controlled by RED in A549 cells. Cells were treated with an siRNA targeting RED or a control siRNA (Supplementary Figure S9A). Efficient depletion of RED was achieved at 48 h post-treatment as shown at the RNA (Supplementary Figure S9B) and protein (Supplementary Figure S9C) levels. Poly(A)-tailed RNAs were extracted and subjected to HiSeq2500 Illumina sequencing, and splicing changes induced by RED depletion were analyzed using the same pipeline as described in Figure 1A. PCA indicated that the control siRNA had no effect on splicing and that RED depletion accounted for 45% of the total variance observed in splicing and up to 63% of the total variance observed in the subset of IR events (Supplementary Figure S10A), in agreement with the specific requirement of RED–SMU1 for the splicing of short introns (31). IAV infection accounted for a lower percentage of the variance (16%) along a clearly separate axis. We identified 11 571 ASEs corresponding to 4570 genes, 14% of which are non-annotated splicing events, that undergo marked (|ΔPSI| ≥ 10%) and significant (P ≤ 0.05) differential regulation upon depletion of RED (Supplementary Figure S10B and Supplementary Table S7). A clear trend for decreased exon inclusion and increased IR was observed in RED-depleted cells (Supplementary Figure S10C), in agreement with previously published studies (31,32).
The similarity between the two sets of IAV-sensitive and RED protein-controlled splicing events was limited, and more so for IR than for ES events as expected from the opposite trends observed earlier (i.e. increased versus decreased intron removal upon IAV infection and RED depletion, respectively; Supplementary Figure S11). We compared the observed numbers of ES and IR splicing events that were dysregulated upon both IAV infection and RED depletion, with ΔPSI of same or opposite sign, with the numbers expected under the null hypothesis that the two variables are independent. Interestingly, the observed numbers were significantly higher than expected for events with ΔPSI of same sign (Figure 5, blue color) and lower than expected for events with ΔPSI of opposite sign (Figure 5, orange color). A Fisher exact test confirmed that the trend was significant for both ES (P < 2 × 10−16) and IR (P = 1.4 × 10−3). These findings are indicative of some level of relatedness between IAV-sensitive and RED protein-controlled splicing events (further discussed below).
Figure 5.

Overlap of IAV-sensitive and RED protein-controlled splicing events. The numbers of ES (A) and IR (B) events that are dysregulated upon both IAV infection and RED depletion are indicated in a chart, and compared with those expected under the null hypothesis that the two variables are independent, according to the sign of ΔPSI in each condition. Blue color: ΔPSI of same sign; orange color: ΔPSI of opposite sign; gray color: expected numbers.
DISCUSSION
Here, we provide an integrated view of changes in the host transcriptome that occur in response to IAV infection, with a focus on IAV-induced changes in splicing that have been documented in only a few studies so far (18,19). Upon RNA-seq analysis of A549 cells infected with the A/WSN/33 virus, we found that >2000 genes show a significant dysregulation of one or several ASEs at 6 h post-infection. RT-PCR yielded a high validation rate of IAV-sensitive splicing events identified through the KisSplice pipeline (all of the 46 ES events tested). Our findings are consistent with the previous observation of viral-induced alterations of host splicing by Fabozzi et al. (18). The sequencing depth (30M reads per replicate) and number of replicates (two) are lower than in our study, therefore hindering a thorough comparison. When our pipeline for differential splicing was applied to the RNA-seq dataset of Fabozzi et al., only 95 cellular genes were found to exhibit significant splicing changes. Out of these 95 genes, only 27 (28%) also showed splicing changes in our dataset, which most likely relates to differences in the experimental protocol and to some degree of dependence on the virus–cell system used (A/Udorn/307/72-Beas2B versus A/WSN/33-A549). Yet, a subset of eight IAV-induced splicing changes from our dataset, which were not present in the dataset of Fabozzi et al., was consistently observed with two distinct cell lines and two distinct viruses, suggesting that the overall degree of conservation of IAV-induced splicing changes is actually higher.
Our data reveal an increase in exon inclusion and intron removal in IAV-infected cells. We asked whether this is a global trend for increased splicing activity. Such an increased splicing could potentially be related to the fact that IAV infection strongly interferes with PolII transcription at the initiation, elongation and termination stages, in a way that contributes to the shut-off of host gene expression (2,8–10). Indeed, pre-mRNA splicing is tightly coupled to PolII transcription. A slowdown of PolII elongation is thought to increase the accessibility of splice sites and therefore to enhance co-transcriptional assembly of the spliceosome (33,34). The splicing and 3′ end processing of pre-mRNAs are functionally interconnected (34,35). We found only a low level of correlation between increased splicing and decreased expression, which was more pronounced for IR than for ES events. This, taken together with the little overlap between the differentially spliced genes and those showing a defect in PolII termination upon IAV infection, suggests that PolII targeting by the virus is not a major causative mechanism for the observed splicing changes. Similar to our findings, little crossover between the set of genes showing differential splicing and differential expression was also observed in other systems [e.g. (36,37)].
We investigated to what extent IAV-induced splicing changes might indirectly result from the cellular sensing of virus-derived nucleic acids and downstream stimulation of innate immune and inflammatory signaling pathways. It should be noted that A549 cells infected with A/WSN/33 showed no transcriptomic signatures of the interferon response at 6 h post-infection, in agreement with others’ findings and with the strong interferon antagonistic activity of the viral NS1 protein (38). In addition, we assessed whether the osmotic stress response, previously shown to induce PolII termination defects similar to IAV infection (8), also induced similar splicing changes. Among a randomly selected subset of 21 IAV-induced splicing changes, only a very minor proportion was triggered in cells subjected to an osmotic shock or infected with the VSV virus. These results suggest that a majority of the observed IAV-induced splicing changes are not merely a secondary consequence of a general stress response or the innate immune/inflammatory response caused by infection.
Other mechanisms of viral manipulation of the splicing machinery include the inhibition, relocalization and/or post-translational modification of splicing factors, mediated by direct or indirect interactions with viral proteins or RNAs [reviewed in (20,39)]. We showed previously that the RED splicing factor, which promotes splicing of the viral NS1 mRNA, is bound by the IAV polymerase (5). Here, we show that the vast majority of IAV-sensitive splicing events are not regulated by RED. Conversely, the silencing of RED induces a wide array of splicing changes, only a fraction of which are recapitulated by IAV infection. Although the overlap between IAV-sensitive and RED-dependent splicing events is limited, it is significantly higher than expected under the null hypothesis of full independence, therefore suggesting that IAV-induced changes are partially mediated by RED. A number of factors may explain why IAV infection only partially phenocopies RNA-mediated depletion of RED. On the one hand, a 90% knockdown of RED is likely to have a more drastic effect on RED function, as the levels and nuclear localization of RED remain unchanged in IAV-infected cells (5). Viral-induced changes in the nuclear environment of RED may affect its function in a more subtle way than depletion, e.g. only a minor fraction of RED may be bound to the viral polymerase and/or the abundant NS1 mRNA. On the other hand, the transcription slowdown in IAV-infected cells (40) may contribute to the global increase in intron removal we observed and may overcome more subtle effects such as increased IR in a specific subset of mRNAs due to altered RED function. Interestingly, in a recent preprint by Thompson et al., an approach similar to ours is used to assess to what extent hnRNP K could be mediating IAV-induced changes in ES events (37). The findings are similar to ours, i.e. the overlap is in the same range whether the IAV infection dataset is compared with the hnRNP K depletion dataset in the study by Thompson et al. (21% of ES events are found in common, among which 63% show a concordant ΔPSI) or with the RED depletion dataset in our study (35% of ES events are found in common, among which 72% show a concordant ΔPSI; Supplementary Figure S11A). These observations support the hypothesis that multiple splicing factors could be involved in the reprogramming of the splicing landscape in IAV-infected cells: RED, hnRNP K and potentially other non-core splicing factors that have been proposed to regulate the splicing of IAV mRNAs, such as SF2 (41) and TRA2A (42). There is evidence that the NS1 protein of IAVs can modulate host splicing through binding to the U6 snRNA (43) or by inducing a relocalization of the SRSF2 factor (44). The recent finding that NS1 primarily binds intronic sequences (45) might contribute to the marked decrease in IR we observed upon IAV infection, also observed by Rotival et al. in IAV-infected human macrophages (19). Finally, our RNA-seq data reveal that 74 and 34 genes corresponding to splicing factors show differential expression or splicing, respectively (Supplementary Table S8), which could in turn be the cause of other splicing changes.
Given the magnitude of splicing changes observed at 6 h post-infection, analyses performed at earlier time points could help elucidate the key mechanisms involved. Separate analysis of poly(A)+ and poly(A)− mRNAs from the cytoplasmic and nuclear fractions would provide a more accurate picture of the splicing landscape and changes induced by IAV infection. From a methodological perspective, our RNA-seq datasets provide a valuable basis to train and improve bioinformatic pipelines for the analysis of AS. Indeed, our high-depth sequencing uncovers a large fraction of unannotated splice sites, IRs and complex splicing events, whose identification and quantification remain challenging with the currently available softwares. Our datasets also offer opportunities for further investigations aimed at uncovering the functional significance of the splicing alterations induced by IAV infection. Notably, genes involved in the regulation of transcription by the cellular PolII were the most enriched among the differentially spliced gene list (Figure 4B), which likely points to so far unexplored mechanisms for viral-induced host shut-off.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Leandro Lima for his help in developing a stranded version of KisSplice, and Tim Krischuns for helpful discussions. This work was performed using the computing facilities of the CC LBBE/PRABI.
Notes
Present address: Clara Benoit-Pilven, Institute of Molecular Medicine, Helsinki University, Helsinki, Finland.
Present address: Guillaume Fournier, Laboratoire National de Santé, Dudelange, Luxembourg.
Present address: Nadia Naffakh, Unité Biologie des ARN et Virus Influenza, Institut Pasteur, CNRS UMR3569, Paris, France.
Contributor Information
Usama Ashraf, Unité de Génétique Moléculaire des Virus à ARN, Institut Pasteur, CNRS UMR3569, Université de Paris, 75015 Paris, France; Université de Paris, Sorbonne Paris Cité, 75013 Paris, France.
Clara Benoit-Pilven, Lyon Neuroscience Research Center, INSERM U1028, CNRS UMR5292, 69675 Bron, France; Laboratoire de Biométrie et Biologie Evolutive, CNRS UMR5558, Université Lyon 1, 69622 Villeurbanne, France; EPI ERABLE, INRIA Grenoble Rhône-Alpes, 38330 Montbonnot-Saint-Martin France.
Vincent Navratil, PRABI, Rhône-Alpes Bioinformatics Center, Université Lyon 1, 69622 Villeurbanne, France; European Virus Bioinformatics Center, 07743 Jena, Germany; Institut Français de Bioinformatique, IFB-core, UMS 3601, 91057 Évry, France.
Cécile Ligneau, Unité de Génétique Moléculaire des Virus à ARN, Institut Pasteur, CNRS UMR3569, Université de Paris, 75015 Paris, France.
Guillaume Fournier, Unité de Génétique Moléculaire des Virus à ARN, Institut Pasteur, CNRS UMR3569, Université de Paris, 75015 Paris, France.
Sandie Munier, Unité de Génétique Moléculaire des Virus à ARN, Institut Pasteur, CNRS UMR3569, Université de Paris, 75015 Paris, France.
Odile Sismeiro, Institut Pasteur, Pôle BIOMICS, Plateforme Transcriptome et Epigenome, 75015 Paris, France.
Jean-Yves Coppée, Institut Pasteur, Pôle BIOMICS, Plateforme Transcriptome et Epigenome, 75015 Paris, France.
Vincent Lacroix, Laboratoire de Biométrie et Biologie Evolutive, CNRS UMR5558, Université Lyon 1, 69622 Villeurbanne, France; EPI ERABLE, INRIA Grenoble Rhône-Alpes, 38330 Montbonnot-Saint-Martin France.
Nadia Naffakh, Unité de Génétique Moléculaire des Virus à ARN, Institut Pasteur, CNRS UMR3569, Université de Paris, 75015 Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
French National Research Agency [ANR-16-CE23-0001 to V.L.]; LabEx IBEID [10-LABX-0062 to N.N.]; Horizon 2020—Research and Innovation Framework Programme [665807 to U.A. as a participant in the Pasteur-Paris University International PhD Program]; Institut Carnot Pasteur Microbes & Santé [to U.A. as a participant in the Pasteur-Paris University International PhD Program].
Conflict of interest statement. None declared.
REFERENCES
- 1. Krammer F., Smith G.J.D., Fouchier R.A.M., Peiris M., Kedzierska K., Doherty P.C., Palese P., Shaw M.L., Treanor J., Webster R.G. et al.. Influenza. Nat. Rev. Dis. Primers. 2018; 4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walker A.P., Fodor E.. Interplay between influenza virus and the host RNA polymerase II transcriptional machinery. Trends Microbiol. 2019; 27398–27407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stertz S., Shaw ML.. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 2011; 13:516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tripathi S., Pohl M.O., Zhou Y., Rodriguez-Frandsen A., Wang G., Stein D.A., Moulton H.M., DeJesus P., Che J., Mulder L.C.F. et al.. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe. 2015; 18:723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fournier G., Chiang C., Munier S., Tomoiu A., Demeret C., Vidalain P.O., Jacob Y., Naffakh N.. Recruitment of RED–SMU1 complex by influenza A virus RNA polymerase to control viral mRNA splicing. PLoS Pathog. 2014; 10:e1004164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thompson M.G., Munoz-Moreno R., Bhat P., Roytenberg R., Lindberg J., Gazzara M.R., Mallory M.J., Zhang K., García-Sastre A., Fontoura B.M.A. et al.. Co-regulatory activity of hnRNP K and NS1-BP in influenza and human mRNA splicing. Nat. Commun. 2018; 9:2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levene R.E., Gaglia MM.. Host shutoff in influenza A virus: many means to an end. Viruses. 2018; 10:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bauer D.L.V., Tellier M., Martinez-Alonso M., Nojima T., Proudfoot N.J., Murphy S., Fodor E.. Influenza virus mounts a two-pronged attack on host RNA polymerase II transcription. Cell Rep. 2018; 23:2119–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heinz S., Texari L., Hayes M.G.B., Urbanowski M., Chang M.W., Givarkes N., Rialdi A., White K.M., Albrecht R.A., Pache L. et al.. Transcription elongation can affect genome 3D structure. Cell. 2018; 174:1522–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao N., Sebastiano V., Moshkina N., Mena N., Hultquist J., Jimenez-Morales D., Ma Y., Rialdi A., Albrecht R., Fenouil R. et al.. Influenza virus infection causes global RNAPII termination defects. Nat. Struct. Mol. Biol. 2018; 25:885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Batra R., Stark T.J., Clark E., Belzile J.P., Wheeler E.C., Yee B.A., Huang H., Gelboin-Burkhart C., Huelga S.C., Aigner S. et al.. RNA-binding protein CPEB1 remodels host and viral RNA landscapes. Nat. Struct. Mol. Biol. 2016; 23:1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu B., Li X., Huo Y., Yu Y., Zhang Q., Chen G., Zhang Y., Fraser N.W., Wu D., Zhou J.. Cellular responses to HSV-1 infection are linked to specific types of alterations in the host transcriptome. Sci. Rep. 2016; 6:28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boudreault S., Martenon-Brodeur C., Caron M., Garant J.M., Tremblay M.P., Armero V.E., Durand M., Lapointe E., Thibault P., Tremblay-Létourneau M. et al.. Global profiling of the cellular alternative RNA splicing landscape during virus–host interactions. PLoS One. 2016; 11:e0161914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rivera-Serrano E.E., Fritch E.J., Scholl E.H., Sherry B.. A cytoplasmic RNA virus alters the function of the cell splicing protein SRSF2. J. Virol. 2017; 91:e02488-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Maio F.A., Risso G., Iglesias N.G., Shah P., Pozzi B., Gebhard L.G., Mammi P., Mancini E., Yanovsky M.J., Andino R. et al.. The dengue virus NS5 protein intrudes in the cellular spliceosome and modulates splicing. PLoS Pathog. 2016; 12:e1005841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu B., Huo Y., Yang L., Chen G., Luo M., Yang J., Zhou J.. ZIKV infection effects changes in gene splicing, isoform composition and lncRNA expression in human neural progenitor cells. Virol. J. 2017; 14:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sessions O.M., Tan Y., Goh K.C., Liu Y., Tan P., Rozen S., Ooi E.E.. Host cell transcriptome profile during wild-type and attenuated dengue virus infection. PLoS Negl. Trop. Dis. 2013; 7:e2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fabozzi G., Oler A.J., Liu P., Chen Y., Mindaye S., Dolan M.A., Kenney H., Gucek M., Zhu J., Rabin R.L. et al.. Strand-specific dual RNA sequencing of bronchial epithelial cells infected with influenza A/H3N2 viruses reveals splicing of gene segment 6 and novel host–virus interactions. J. Virol. 2018; 92:e00518-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rotival M., Quach H., Quintana-Murci L.. Defining the genetic and evolutionary architecture of alternative splicing in response to infection. Nat. Commun. 2019; 10:1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ashraf U., Benoit-Pilven C., Lacroix V., Navratil V., Naffakh N.. Advances in analyzing virus-induced alterations of host cell splicing. Trends Microbiol. 2019; 27:268–281. [DOI] [PubMed] [Google Scholar]
- 21. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anders S., Pyl P.T., Huber W.. HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sacomoto G.A., Kielbassa J., Chikhi R., Uricaru R., Antoniou P., Sagot M.F., Peterlongo P., Lacroix V.. KISSPLICE: de-novo calling alternative splicing events from RNA-seq data. BMC Bioinformatics. 2012; 13:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benoit-Pilven C., Marchet C., Chautard E., Lima L., Lambert M.P., Sacomoto G., Rey A., Cologne A., Terrone S., Dulaurier L. et al.. Complementarity of assembly-first and mapping-first approaches for alternative splicing annotation and differential analysis from RNAseq data. Sci. Rep. 2018; 8:4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dray S., Dufour AB.. The ade4 package: implementing the duality diagram for ecologists. J.Stat. Software. 2007; 22:1. [Google Scholar]
- 27. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2016; NY: Springer. [Google Scholar]
- 28. Middleton R., Gao D., Thomas A., Singh B., Au A., Wong J.J., Bomane A., Cosson B., Eyras E., Rasko J.E.J. et al.. IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol. 2017; 18:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vilborg A., Passarelli M.C., Yario T.A., Tycowski K.T., Steitz J.A.. Widespread inducible transcription downstream of human genes. Mol. Cell. 2015; 59:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexa A., Rahnenfuhrer J.. topGO: enrichment analysis for gene ontology. 2019; R package version 2360.
- 31. Keiper S., Papasaikas P., Will C.L., Valcarcel J., Girard C., Luhrmann R.. Smu1 and RED are required for activation of spliceosomal B complexes assembled on short introns. Nat. Commun. 2019; 10:3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Papasaikas P., Tejedor J.R., Vigevani L., Valcarcel J.. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell. 2015; 57:7–22. [DOI] [PubMed] [Google Scholar]
- 33. Dujardin G., Lafaille C., de la Mata M., Marasco L.E., Munoz M.J., Le Jossic-Corcos C., Corcos L., Kornblihtt A.R.. How slow RNA polymerase II elongation favors alternative exon skipping. Mol. Cell. 2014; 54:683–690. [DOI] [PubMed] [Google Scholar]
- 34. Tellier M., Maudlin I., Murphy S.. Transcription and splicing: a two-way street. Wiley Interdiscip. Rev. RNA. 2020; 11:e1593. [DOI] [PubMed] [Google Scholar]
- 35. Misra A., Green MR.. From polyadenylation to splicing: dual role for mRNA 3′ end formation factors. RNA Biol. 2016; 13:259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huntley M.A., Srinivasan K., Friedman B.A., Wang T.M., Yee A.X., Wang Y., Kaminker J.S., Sheng M., Hansen D.V., Hanson J.E.. Genome-wide analysis of differential gene expression and splicing in excitatory neurons and interneuron subtypes. J. Neurosci. 2020; 40:958–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thompson M.G., Dittmar M., Mallory M.J., Bhat P., Ferretti M.B., Fontoura M.A., Cherry S., Lynch K.W.. Viral-induced alternative splicing of host genes promotes influenza replication. 2020; bioRxiv doi:30 May 2020, preprint: not peer reviewed 10.1101/2020.05.28.122044. [DOI] [PMC free article] [PubMed]
- 38. Killip M.J., Fodor E., Randall R.E.. Influenza virus activation of the interferon system. Virus Res. 2015; 209:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boudreault S., Roy P., Lemay G., Bisaillon M.. Viral modulation of cellular RNA alternative splicing: a new key player in virus–host interactions. Wiley Interdiscip. Rev. RNA. 2019; 10:e1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Engelhardt O.G., Fodor E.. Functional association between viral and cellular transcription during influenza virus infection. Rev. Med. Virol. 2006; 16:329–345. [DOI] [PubMed] [Google Scholar]
- 41. Huang X., Zheng M., Wang P., Mok B.W., Liu S., Lau S.Y., Chen P., Liu Y.-.C., Liu H., Chen Y. et al.. An NS-segment exonic splicing enhancer regulates influenza A virus replication in mammalian cells. Nat. Commun. 2017; 8:14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu Y., Wang R., Yu L., Sun H., Tian S., Li P., Jin M., Chen H., Ma W., Zhou H.. Human TRA2A determines influenza A virus host adaptation by regulating viral mRNA splicing. Sci. Adv. 2020; 6:eaaz5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qiu Y., Nemeroff M., Krug R.M.. The influenza virus NS1 protein binds to a specific region in human U6 snRNA and inhibits U6–U2 and U6–U4 snRNA interactions during splicing. RNA. 1995; 1:304–316. [PMC free article] [PubMed] [Google Scholar]
- 44. Fortes P., Lamond A.I., Ortin J.. Influenza virus NS1 protein alters the subnuclear localization of cellular splicing components. J. Gen. Virol. 1995; 76:1001–1007. [DOI] [PubMed] [Google Scholar]
- 45. Zhang L., Wang J., Munoz-Moreno R., Kim M., Sakthivel R., Mo W., Shao D., Anantharaman A., García-Sastre A., Conrad N.K. et al.. Influenza virus NS1 protein–RNA interactome reveals intron targeting. J. Virol. 2018; 92:e01634-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.