

Abstract
This prospective longitudinal study evaluated multiple maternal biomarkers from the preconception and prenatal periods as time-sensitive predictors of child executive functioning (EF) in 100 mother-child dyads. Maternal glycated hemoglobin (HbA1C), C-reactive protein (CRP), and blood pressure (BP) were assayed before pregnancy and during the second and third trimesters. Subsequently, children were followed from birth and assessed for EF (i.e., cognitive flexibility, response inhibition) at ages 4-6 years. Perinatal data were also extracted from neonatal records. Higher maternal CRP, but not maternal HbA1C or BP, uniquely predicted poorer child cognitive flexibility, even with control of maternal HbA1C and BP, relevant demographic factors, and multiple prenatal/perinatal covariates (i.e., preconception maternal body mass index, maternal depression, maternal age at birth, child birth weight, child birth order, child gestational age, and child birth/neonatal complications). Predictions from maternal CRP were specific to the third trimester, and third trimester maternal CRP robustly predicted child cognitive flexibility independently of preconception and second trimester CRP. Child response inhibition was unrelated to maternal biomarkers from all timepoints. These findings provide novel, prospective evidence that maternal inflammation uniquely predicts child cognitive flexibility deficits, and that these associations depend on the timing of exposure before or during pregnancy.
Keywords: pregnancy, preconception, C-reactive protein, glycated hemoglobin, blood pressure, executive function, cognitive flexibility
Executive functioning (EF) domains are separable but related higher-order cognitive processes involved in the control of goal-directed behavior (Pennington & Ozonoff, 1996) and regulated by fronto-striato-parietal networks (e.g., Hart, Radua, Nakao, Mataix-Cols, & Rubia, 2013). Major domains of EF, which include cognitive flexibility and response inhibition (Miyake et al., 2000), reliably predict individual differences in child socioemotional, behavioral, and academic development (e.g., Clark, Prior, & Kinsella, 2002; Diamantopoulou, Rydell, Thorell, & Bohlin, 2007). Moreover, EF deficits are implicated in the etiology of multiple neurodevelopmental disorders including attention-deficit/hyperactivity disorder (ADHD), autism spectrum disorder, and schizophrenia (McGrath et al., 2015; Willcutt, Doyle, Nigg, Faraone, & Pennington, 2005). Given the broad role of EF in child outcomes, EF may be a critical target for prevention studies to promote healthy child development. Additionally, various programs and activities (e.g., cognitive training, school-based curricula, exercise) may improve EF development in young children who are already exhibiting early deficits (Diamond, 2012; Diamond & Lee, 2011), suggesting that EF may be a modifiable risk factor. Thus, improved understanding of well-defined predictors of individual differences in EF would critically inform prevention efforts across major domains of psychopathology and psychosocial functioning.
Maternal physical health during pregnancy is crucial to offspring neurodevelopment. In particular, there is growing evidence that prenatal exposure to maternal inflammation is a biologically plausible risk factor for EF deficits. In addition to predicting offspring cognitive impairments more broadly (Jonakait, 2007; van der Burg et al., 2016), prenatal maternal inflammation is associated with specific EF dimensions (Brown et al., 2009; Graham et al., 2018; Rudolph et al., 2018). For example, higher maternal concentrations of the pro-inflammatory cytokine interleukin-6 during pregnancy prospectively predicted poorer child impulse control at age 24 months (Graham et al., 2018). Additionally, exposure to maternal infections during pregnancy predicted poorer cognitive flexibility in adults with schizophrenia (Brown et al., 2009). Prenatal maternal inflammation is also associated with neurodevelopmental disorders that are characterized by EF deficits, such as autism and schizophrenia (Brown et al., 2014; Canetta et al., 2014; van der Burg et al., 2016). Notably, the association of prenatal maternal inflammation and offspring EF includes exposure to both acute inflammation (e.g., maternal infections; e.g., Brown et al., 2009; Meyer, 2014) and chronic, low-grade inflammation such as persistently elevated pro-inflammatory cytokine and C-reactive protein (CRP) levels associated with maternal obesity (e.g., Brown et al., 2014). Finally, experimental evidence suggests that causal effects of prenatal inflammation on child EF are biologically plausible. For example, in non-human primates, in utero exposure to maternal pro-inflammatory response induced postnatal structural abnormalities in brain regions that modulate EF (i.e., prefrontal cortex; Short et al., 2010). Thus, given growing evidence for biologically plausible and potentially causal associations between prenatal exposure to maternal inflammation and child EF, maternal inflammation is a likely precursor to individual differences in child EF.
Prenatal exposure to adverse maternal metabolic conditions including gestational diabetes and hypertension is also associated with broad cognitive deficits in children (e.g., lower IQ; e.g., Adane et al., 2016; Krakowiak et al., 2012; Tuovinen, Eriksson, Kajantie, & Räikkönen, 2014). Moreover, there is preliminary evidence that maternal gestational diabetes and hypertension specifically predict child EF deficits (Bolanos, Matute, Ramirez-Duenas Mde, & Zarabozo, 2015; Wade & Jenkins, 2016). For example, in a community sample recruited immediately after the birth of the child, retrospectively reported prenatal hypertension negatively predicted an EF composite calculated from cognitive flexibility and response inhibition in preschool-aged children (Wade & Jenkins, 2016). Although not fully understood, maternal hyperglycemia and inflammation as well as fetal hypoxia and oxidative stress, among other factors, are plausible mechanisms underlying the effects of gestational diabetes and hypertension on child outcomes (Adane et al., 2016; Ornoy, Reece, Pavlinkova, Kappen, & Miller, 2015; Tuovinen et al., 2014). Thus, in addition to maternal inflammation, maternal diabetes and hypertension during pregnancy are potential precursors to individual differences in child EF.
Despite growing evidence for prenatal metabolic conditions and inflammation as precursors to child EF development, critical aspects of these associations require clarification. First, because gestational diabetes, hypertension, and inflammation may be intercorrelated (e.g., Hedderson & Ferrara, 2008; Qiu, Sorensen, Luthy, & Williams, 2004; Smith et al., 2005), it is unclear which maternal physiological factors most affect child neurodevelopment. It is also unclear if these maternal factors predict child cognitive deficits specifically or are sensitive to child cognitive deficits via shared variance with potential confounds or correlates including pre-pregnancy maternal obesity (Adane, Mishra, & Tooth, 2016; Christian & Porter, 2014; Mina et al., 2016; van der Burg et al., 2016), prenatal maternal depression (e.g., Deave, Heron, Evans, & Emond, 2008; Kozhimannil, Pereira, & Harlow, 2014), maternal demographic factors (e.g., socioeconomic status; e.g., Hackman, Farah, & Meaney, 2010), parity (e.g., Baker et al., 2008), preterm birth (e.g., Aarnoudse-Moens, Weisglas-Kuperus, van Goudoever, & Oosterlaan, 2009; Sibai et al., 2000), low birth weight (e.g., Burnett et al., 2015; Camerota et al., 2015; Valero De Bernabé et al., 2004), and birth and neonatal complications (e.g., emergency cesarean sections; Scholl, Sowers, Chen, & Lenders, 2001; Wiggs et al., 2016). Second, rather than employing multiple assays of metabolic or pro-inflammatory biomarkers across pregnancy, prior studies typically relied on retrospective report or medical record review of specific maternal diagnoses, which obscures inferences about when during pregnancy particular fetal exposures are most detrimental. Moreover, pre-pregnancy maternal health is also associated with offspring cognitive outcomes (e.g., preconception diabetes; Adane et al., 2016; Adane et al., 2016), yet no studies have directly compared preconception vs. prenatal maternal health factors in prediction of child cognitive functioning. Identifying potential “sensitive periods” could critically inform the timing of interventions to promote maternal health directly and indirectly improve child neurodevelopment. Thus, to meaningfully clarify the specificity of maternal metabolic conditions and inflammation to child neurodevelopment, predictive models must simultaneously evaluate multiple maternal biomarkers with stringent control of prenatal/perinatal risk factors, and directly compare the relative influence of maternal biomarkers from preconception and across pregnancy.
Aims
To review, whereas exposure to maternal metabolic conditions and inflammation are biologically plausible risk factors for child EF deficits, their unique associations with child EF are unknown. Moreover, it is also unclear if the timing of these risk factors (i.e., before, early, or later in pregnancy) differentially affect offspring development. The current study combined intensive prospective measurement of maternal health before and during pregnancy with longitudinal follow-up of offspring from birth through early childhood. Metabolic and pro-inflammatory indicators were assayed before and during pregnancy, including maternal glycated hemoglobin (HbA1C), CRP, and blood pressure (BP). Child EF was assessed at ages 4-6 years, when major EF domains may begin to differentiate as well as advance rapidly across childhood and adolescence (for review see Best & Miller, 2010; Zelazo et al., 2013). To improve knowledge on the development of EF deficits from maternal metabolic conditions and inflammation, we evaluated multiple metabolic and pro-inflammatory indicators (i.e., HbA1C, CRP, BP) in prediction of major domains of child EF with rigorous control of potential confounds. We also compared these factors prior to pregnancy and across multiple prenatal time points to ascertain if their associations with EF were temporally specific.
Methods
Participants
Participants were 100 children aged 4 to 6 years (M age = 4.61, SD = 0.65; 59% female; 52% Latino or Hispanic White, 26% non-Hispanic White, 18% African-American/Black, and 4% Multiracial) whose mothers were followed prospectively before and during pregnancy as part of the Community Child Health Network (CCHN), a multi-site research network funded by the Eunice Kennedy Shriver National Institute of Child Health and Human Development to investigate disparities in maternal and child health and improve the health of families (Ramey et al., 2015). Recruitment procedures and criteria as well as maternal demographics are described in detail elsewhere (Dunkel Schetter et al., 2013; Ramey et al., 2015). Briefly, mothers were recruited across five study sites with predominantly low-income recruitment areas in Washington, DC, Baltimore, MD, Los Angeles County, CA, Lake County, IL, and eastern North Carolina immediately after the birth of an index child (i.e., the older siblings of the children included in the present study). CCHN mothers completed up to five study visits between 6 months and 2 years after the birth of the index child (n = 2,089). At three of the study sites (i.e., North Carolina, Washington, DC, and Lake County, IL), mothers who reported they were pregnant with a subsequent child during this 2-year follow-up period (n = 416) were invited to participate in additional study visits. Three hundred and forty-three mothers consented to continued follow-up and completed at least one study visit during or shortly after the subsequent pregnancy. Next, these mothers were invited to participate with their subsequent child in a longitudinal child development study. One hundred and twenty-five children were enrolled and completed a study visit at ages 3-5 years. Of these, 100 children completed a second study visit at ages 4-6 years that included evaluation of EF. Complete demographic data and descriptive statistics for the current sample of 100 children are presented in Table 1. The Institutional Review Boards of all collaborating study sites approved all study procedures.
Table 1.
Sample demographics and descriptive statistics (N = 100)
| % of sample | M (SD), range | ||
|---|---|---|---|
| Child sex (female) | 59 | Child birth order | 1.62 (0.76), 1-4 |
| Child race-ethnicity: | Maternal age at child birth | 28.67 (5.66), 19-43 | |
| African-American/Black | 18 | Maternal education, years | 12.67, (3.40), 6-20 |
| Non-Hispanic White | 26 | Preconception maternal BMI | 29.76 (7.58), 16.66-56.22 |
| Latino or Hispanic White | 52 | Preconception HbA1C, % | 5.36 (0.49), 4.10-6.20 |
| Multiracial | 4 | 2nd trimester HbA1C, % | 4.83 (0.54), 3.80-6.50 |
| Child language (Spanish) | 21 | 3rd trimester HbA1C, % | 5.06 (0.68), 3.60-6.60 |
| Study Site: | Preconception CRP, mg/L | 4.48 (4.06), 0.20-14.80 | |
| North Carolina | 8 | 2nd trimester CRP, mg/L | 8.01 (4.83), 0.70-24.40 |
| Washington, DC | 17 | 3rd trimester CRP, mg/L | 6.94 (4.25), 0.10-20.10 |
| Lake County, IL | 75 | Preconception systolic BP, mmHg | 110.19 (9.67), 79-142 |
| Marital status (married) | 48 | 2nd trimester systolic BP, mmHg | 107.39 (10.13), 87-135 |
| Birth/neonatal complications |
17 |
3rd trimester systolic BP, mmHg | 110.64 (9.89), 89-133 |
| |
M (SD), range |
Preconception diastolic BP, mmHg | 69.69 (8.47), 52-89 |
| Child age, years | 5.15 (0.48), 4.31-6.26 | 2nd trimester diastolic BP, mmHg | 65.30 (8.46), 48-83 |
| Household income, $ | 66,363 (63,027), 265-350,000 | 3rd trimester diastolic BP, mmHg | 66.40 (8.29), 49-87 |
| Child birth weight, grams | 3249.06 (533.94), 1247-4564 | Preconception maternal EPDS | 4.75 (4.32), 0-18 |
| Child gestational age, weeks | 38.73 (1.99), 28-42 | 2nd trimester maternal CESD | 16.63 (4.55), 10-22 |
| Cognitive Flexibility T-score | 50.14 (10.55), 15-71 | 3rd trimester maternal CESD | 16.85 (5.36), 10-35 |
| Response Inhibition T-score | 51.52 (9.52), 14-74 |
Note: Median household income = $41,600; BMI = body mass index; HbA1C = glycated hemoglobin; CRP = C-reactive protein, BP = blood pressure; EPDS = total score on the Edinburgh Postnatal Depression Scale; CESD = total score on the Center for Epidemiological Studies Depression Inventory
Procedures
Complete CCHN study procedures are described in detail elsewhere (e.g., Ramey et al., 2015; Shalowitz et al., 2019). The present study used maternal health data collected during three CCHN study visits: (1) prior to maternal pregnancy with the study child (i.e., preconception), (2) during approximately the second trimester of prenatal development, and (3) during approximately the third trimester of prenatal development. Perinatal data were extracted from neonatal records, and child EF data were collected at the age 4-6 year study visit. See Figure 1 for an outline of the data collection time points used in the present study and the key variables assessed at each of these visits. All study visits were conducted in participants’ homes by community research staff trained in the study protocol, with attempts to match interviewer and participant ethnicity.
Figure 1.
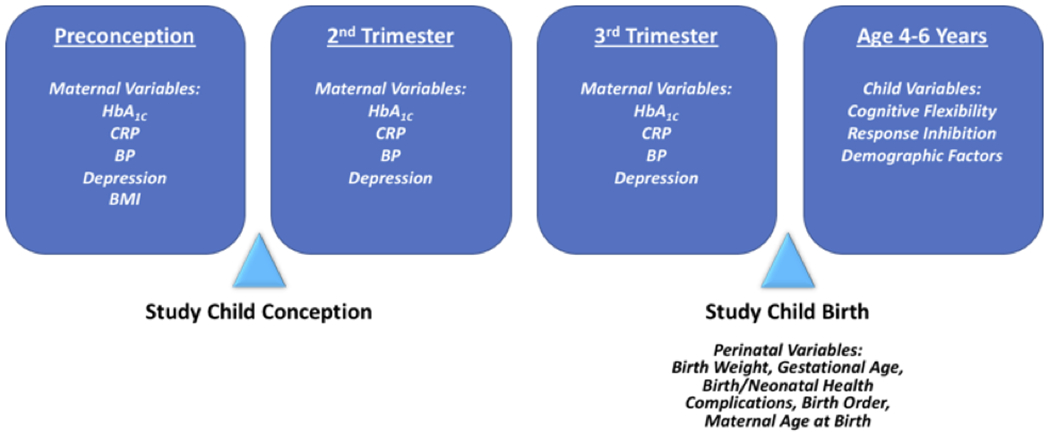
Outline of key study variables and data collection time points used in the present study
Note: HbA1C = glycated hemoglobin; BP = blood pressure; CRP = C-reactive protein; BMI = body mass index
Because mothers became pregnant with the study children at different times during the 2-year CCHN follow-up phase, each individual mother’s most recent CCHN visit prior to conception of the study child was designated as the preconception visit for the current study. The mean length of time in months between the identified preconception visit and the date of study child conception was 5.93 months (SD = 6.07, range = 0-29.54). The first prenatal study visit occurred primarily during the second trimester (M weeks gestation = 20.25, SD = 4.54, range = 6.71-26.57), although due to participant availability, study visits occurred during weeks 6-13 of the pregnancy for a small number of mothers (n = 3). The second prenatal study visit occurred largely during the third trimester (M weeks gestation = 32.85, SD = 3.26, range = 26.71-40.28), with three mothers completing the second prenatal study visit during weeks 26-27. Importantly, although there was variation in the length of time between the preconception visit and conception of the study child, results of the current analyses were unchanged when preconception data collected more than 12 months before the date of conception were excluded (results available upon request). Similarly, the results of the current analyses were unchanged when prenatal data collected outside of strict, non-overlapping trimester cutoffs were excluded (results available upon request). Thus, all analyses described hereafter used all available data from the identified preconception visit, first prenatal visit, and second prenatal visit, and the respective results are interpreted as reflecting the preconception period, second trimester, and third trimester.
Measures
Maternal metabolic and pro-inflammatory factors.
Biomarkers of maternal metabolic conditions and inflammation were collected during the preconception, second trimester, and third trimester visits, and included: (1) HbA1C (%) with a clinical cutoff of 5.7% reflecting pre-diabetes (American Diabetes Association, 2017); (2) high-sensitivity CRP (hsCRP, referred to hereafter as CRP; mg/L), with a pro-inflammatory state defined as > 3.0 mg/L (Pearson et al., 2003); and (3) systolic and diastolic BP (mmHg), with clinical cutoffs of 120 for systolic BP and 80 for diastolic BP reflecting prehypertension (WHO criteria). Systolic and diastolic blood pressures were each recorded three times while mothers were seated during the home visit using an OMRON HEM-711DLX or HEM-907XL Pro standardized digital sphygmomanometer (OMRON Global, Osaka, Japan); the three readings of each type of blood pressure were averaged to create composite measures of systolic and diastolic blood pressure. Whole blood spots on Guthrie paper were collected by finger prick with a 14-gauge spring-loaded lancet and dried. Blood specimens were analyzed by ZRT laboratory (Portland, OR). See Shalowitz et al. (2019) for additional details regarding biomarker collection and processing procedures.
HbA1c is a diagnostic indictor of diabetes that reflects long-term glucose concentrations over the prior 60-90 days, and is therefore a highly reliable marker of glycemic control (Goldstein et al., 2003). CRP is a well-characterized marker of inflammation in the body, and is the only pro-inflammatory marker with established clinical cutoffs (Pearson et al., 2003); its production in the liver is stimulated by pro-inflammatory cytokines (i.e., tumor necrosis factor, interleukin-1, interleukin-6) in response to infection, tissue damage, and other harmful stimuli. Blood pressure is a diagnostic indicator of hypertension. Although cutoffs are provided above to aide interpretation, maternal biomarkers were evaluated as continuous variables in all analyses for the current study. Maternal biomarkers were modestly to moderately correlated across the preconception, second trimester, and third trimester visits: HbA1C (rs = .43-55, p < .05), CRP (rs = .39-51, p < .05), and BP (rs = .48-58, p < .01). Pre-pregnancy maternal body mass index (BMI) was also extracted from the preconception visit data and used as covariate in the present analyses. BMI was calculated as weight (kg) divided by height squared (meters), with BMI of 30.0 or greater reflecting obesity.
Epidemiological studies of systemic inflammation in non-pregnant individuals have typically excluded those with CRP values greater than 10 mg/L because higher values may reflect acute inflammation secondary to infection or injury (Ridker, 2003). However, based on the substantial evidence for the negative impact of both chronic low-grade and acute maternal inflammation on child neurodevelopment (e.g., Brown et al., 2014; Brown et al., 2009; Meyer et al., 2011; van der Burg et al., 2016), and because CRP levels may increase during pregnancy (Hwang, Kwon, Kim, Park, & Kim, 2007), excluding participants with CRP values greater than 10 mg/L would likely diminish meaningful variance in prediction of child EF. Therefore, we used sample-specific criteria to classify and exclude outliers, whereby CRP values greater than three standard deviations from the sample mean were excluded. This resulted in exclusion of second trimester CRP data for only one mother with a value of 24.4 mg/L, whereas all CRP values from the preconception and third trimester time points were within three standard deviations of the mean for those time points.
Maternal depression.
Maternal depression was assessed at the preconception, second trimester, and third trimester visits and included as a covariate in tests of the biomarkers from each of the respective time points. Because mothers were recruited immediately after the birth of a child, the 10-item Edinburgh Postnatal Depression Scale (EPDS; Cox, Holden, & Sagovsky, 1987) was used to measure maternal depression during these initial study visits (i.e., during the preconception period for the purposes of the present study). Mothers rated the severity of their symptoms experienced in the past 7 days on a 4-point scale, and a total score was summed (α = .83). At the two prenatal visits, maternal depression was assessed with the short form of the Center for Epidemiological Studies Depression Inventory (CES-D; Santor & Coyne, 1997), a 9-item measure of depression with excellent psychometric properties that has been validated specifically in pregnant women (Marcus, Flynn, Blow, & Barry, 2005). Mothers rated the severity of their symptoms in terms of days per week on a 4-point scale, and a total score was summed (second trimester α = .80; third trimester α = .76). In the present sample, the preconception EPDS scores were correlated as expected with the second and third trimester CES-D scores (respectively, rs = .43, p = .001; rs = .48, p < .001).
Perinatal factors.
Factors relevant to child cognitive functioning were extracted from medical records and included as covariates in analyses: birth weight (grams), gestational age (weeks), and birth or neonatal health complications (combined into a single variable coded yes/no). Examples of birth/neonatal health complications in the current sample included emergency cesarean section, jaundice, respiratory problems, and hypoplastic left heart syndrome.
Child EF.
Child EF domains were assessed with the Early Childhood version of the NIH Toolbox Cognition Battery (NIHTB-CB; Gershon et al., 2013). The NIHTB-CB was developed through a large multi-site initiative to design state-of-the-art, standardized, and easily-administered measures of cognitive functioning across the lifespan, in addition to other health domains, with funding from the NIH Blueprint for Neuroscience Research (Gershon et al., 2013). The Early Childhood version of the NIHTB-CB was specifically designed for children aged 3-6 years, and included age-appropriate computerized measures of cognitive flexibility and inhibitory control (Zelazo et al., 2013). Cognitive flexibility, which refers to the ability to switch fluidly between two separate tasks or mental sets (Miyake et al., 2000), was assessed using the Dimensional Change Card Sort Task (DCCS). The DCCS required children to sort a series of pictorial stimuli according to one dimension (i.e., shape or color) and then according to the other dimension across four different test condition blocks. Response inhibition, or the ability to inhibit inappropriate or automatic responses (Miyake et al., 2000), was assessed via the Flanker Inhibitory Control and Attention Test. For the Flanker, children indicated the orientation of a centrally presented stimulus while inhibiting their attention to other surrounding stimuli (i.e., the flankers) across three test blocks. Both tasks have administration times of approximately 3-4 minutes. See Zelazo et al. (2013) for additional details regarding task administration. As described in Zelazo et al. (2013), these specific measures of cognitive flexibility and response inhibition were selected for the NIHTB-CB based on their availability in the public domain and ability to be modified to meet key NIH Toolbox usability objectives (e.g., brief, computer-administered, suitable for participants aged 3-85 years). The Early Childhood NIHTB-CB has extensively-validated English- and Spanish-language versions with excellent psychometrics (Akshoomoff et al., 2014; Casaletto et al., 2015, 2016; Mungas et al., 2013; Zelazo et al., 2013).
The NIHTB-CB was administered to the study children in their primary language, English (n = 79; 79%) or Spanish (n = 21; 21%). Because DCCS and Flanker scores did not differ between English- and Spanish-speaking children in the current sample (respectively, t(94) = −0.28, p = .71; t(94) = −0.25, p = .81), EF data were collapsed across languages. As recommended by the NIHTB-CB developers, we used T-scores for each EF domain that were adjusted for child age, sex, race-ethnicity, and maternal education level (Casaletto et al., 2015, 2016). Children with DCCS and Flanker T-scores more than three standard deviations from the mean (n = 1 for both measures) were designated missing. DCCS and Flanker T-scores were moderately correlated (r = .31, p < .01).
Statistical Analysis
Missing data.
All 100 children had maternal biomarker data from at least one of the three time points (i.e., preconception, second trimester, third trimester), 74 (74%) had biomarker data at two time points, and 36 (36%) had biomarker data at all three time points. The number of children with available maternal biomarker data at each time point was as follows: preconception (n = 75; 75%), second trimester (n = 56; 56%), third trimester (n = 75; 75%). Additionally, 97 children (97%) had usable EF data. Given the missing data secondary to the longitudinal follow-up, we used full information maximum likelihood (FIML) estimation to maximize sample size for all analyses. FIML optimally remediates missing data when the amount of missingness per variable is up to 50% and data are missing at random or missing completely at random (MCAR; Schlomer, Bauman, & Card, 2010). Little’s MCAR Test (Little, 1988) indicated that the study data were indeed MCAR (χ2(1188) = 603.05, p = .99). Thus, all analyses described below were conducted on the full sample of 100 children using FIML estimation.
Hypothesis testing.
We first constructed separate regression models predicting child cognitive flexibility (i.e., DCCS T-scores) as follows: (1) simultaneously evaluating preconception maternal HbA1C, CRP, and BP; (2) simultaneously evaluating second trimester maternal HbA1C, CRP, and BP; and (3) simultaneously evaluating third trimester maternal HbA1C, CRP, and BP. Each model employed robust standard errors and controlled for both the study site where the child was assessed and the language used to administer the cognitive battery; these covariates were selected because DCCS data from both English- and Spanish-language versions were included in analyses and because DCCS scores from the Lake County, IL study site were lower than those from the two other sites (t(95) = 2.11, p = .04). The DCCS T-scores were also already adjusted for child age, sex, race-ethnicity, and maternal education level.1 To rigorously control for potential prenatal/perinatal correlates associated with maternal metabolic and pro-inflammatory factors and/or child cognitive functioning outcomes in prior studies, the following covariates were added at Step 2: maternal depression from the respective measurement time point, maternal preconception BMI, maternal age, maternal marital status, household income, child birth order, child birth weight, child gestational age, and child birth/neonatal complications. Next, for any biomarker that predicted child cognitive flexibility, we analyzed whether the association was temporally specific. That is, we constructed an additional model that simultaneously evaluated measures of that biomarker from preconception, second trimester, and third trimester as predictors of child cognitive flexibility. The same analytic strategy was then repeated but in prediction of child response inhibition (i.e., Flanker T-scores).
Results
Prediction of Child Cognitive Flexibility from Maternal Biomarkers
Bivariate correlations among key study variables are presented in Table 2. We first evaluated whether preconception (i.e., M = 5.93 months prior to the date of conception) maternal HbA1C, CRP, and BP uniquely predicted child cognitive flexibility (i.e., DCCS T-scores). To facilitate interpretation, standardized regression coefficient values (β) are reported after the unstandardized regression parameters values (B and SE). Covarying for study site and cognitive battery language (T-scores were also adjusted for child age, sex, race-ethnicity, and maternal education level), none of the preconception biomarkers predicted child cognitive flexibility: CRP (B = −0.20, SE = 0.32, p = .52; β = −0.08), HbA1C (B = −2.65, SE = 2.26, p = .24, β = −0.13), BP (B = 1.17, SE = 8.47, p = .89, β = 0.02). Thus, no further preconception analyses were conducted in prediction of child cognitive flexibility.
Table 2.
Bivariate associations of child executive functions and maternal biomarkers with all study variables
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. Cognitive flexibility | - | ||||||||||
| 2. Response Inhibition | .30* | - | |||||||||
| 3. Preconception HbA1C | −.05 | .05 | - | ||||||||
| 4. 2nd trimester HbA1C | −.24 | −.16 | .47* | - | |||||||
| 5. 3rd trimester HbA1C | −.01 | .06 | .43** | .55** | - | ||||||
| 6. Preconception CRP | −.07 | .09 | −.01 | .42* | .14 | - | |||||
| 7. 2nd trimester CRP | −.14 | .01 | .13 | .35* | .35* | .51** | - | ||||
| 8. 3rd trimester CRP | −.28* | .08 | −.24 | .15 | .04 | .41* | .39* | - | |||
| 9. Preconception systolic BP | −.01 | −.09 | .09 | −.10 | .09 | −.43* | −.30* | −.11 | - | ||
| 10. 2nd trimester systolic BP | −.08 | −.01 | .25 | .17 | .17 | −.16 | .17 | .02 | .48** | - | |
| 11. 3rd trimester systolic BP | −.11 | .06 | −.03 | −.11 | −.02 | −.11 | .16 | .11 | .58** | .52** | - |
| 12. Preconception maternal EPDS | −.07 | −.10 | .13 | .18 | −.01 | −.20 | .02 | .09 | .24* | .30* | .06 |
| 13. 2nd trimester maternal CESD | .06 | −.01 | .29 | .19 | .15 | −.01 | .02 | .05 | .19 | .07 | .07 |
| 14. 3rd trimester maternal CESD | .12 | −.04 | −.09 | −.26 | −.17 | −.24 | −.16 | −.08 | .06 | −.06 | .01 |
| 15. Preconception maternal BMI | −.10 | .03 | −.03 | .45* | .20 | .63** | .47* | .22 | −.37* | −.24 | −.10 |
| 16. Maternal age at child birth | −.04 | .14 | −.05 | .16 | .10 | −.05 | .04 | −.05 | .15* | −.06 | −.01 |
| 17. Marital status (married) | −.03 | .02 | −.27* | −.25 | −.01 | .10 | −.10 | −.17 | −.09 | −.03 | −.08 |
| 18. Family income | .11 | .04 | −.19 | −.08 | −.04 | −.06 | −.07 | −.01 | .03 | .07 | .08 |
| 19. Child birth order | −.10 | −.14 | .04 | −.01 | −.02 | .26* | .35* | .08 | .01 | .31* | .17 |
| 20. Child birth weight | .03 | −.16 | −.06 | .03 | .08 | .05 | .01 | .20 | −.28* | −.08 | −.03 |
| 21. Child gestational age | .21 | −.14 | −.02 | −.11 | −.06 | −.10 | −.39* | −.03 | −.12 | −.20 | −.21 |
| 22. Birth/neonatal complications | −.04 | −.08 | .04 | .14 | −.10 | .21 | .21 | −.10 | −.03 | .22 | −.01 |
p < .001
p < .01
Note: Spearman and point biserial correlation coefficients were calculated for continuous and dichotomous variable, respectively; HbA1C = glycated hemoglobin; CRP = C-reactive protein, BP = blood pressure; EPDS = total score on the Edinburgh Postnatal Depression Scale; CESD = total score on the Center for Epidemiological Studies Depression Inventory; BMI = body mass index
Second, we evaluated whether second trimester maternal HbA1C, CRP, and BP uniquely predicted child cognitive flexibility. Covarying for study site and cognitive battery language, none of the second trimester biomarkers predicted child cognitive flexibility: CRP (B = −0.10, SE = 0.34, p = .76; β = −0.04), HbA1C (B = −4.59, SE = 3.00, p = .13, β = −0.25), BP (B = 3.85, SE = 7.47, p = .61, β = 0.07). Thus, no further second trimester analyses were conducted in prediction of child cognitive flexibility.
Third, we evaluated whether third trimester maternal HbA1C, CRP, and BP uniquely predicted child cognitive flexibility. Controlling for study site and cognitive battery language, third trimester maternal CRP inversely predicted child cognitive flexibility (B = −0.67, SE = 0.27, p = .01; β = −0.29); neither third trimester maternal HbA1C (B = −0.16, SE = 1.42, p = .99, β = −0.001) nor third trimester maternal BP (B = −0.72, SE = 6.68, p = .92, β = −0.01) was associated with child cognitive flexibility. When the prenatal/perinatal covariates were added to the model (i.e., third trimester maternal depression, preconception maternal BMI, maternal age, maternal marital status, household income, child birth order, child birth weight, child gestational age, and child birth/neonatal complications), third trimester maternal CRP continued to predict child cognitive flexibility (B = −0.92, SE = 0.35, p = .01, β = −0.39; standardized regression coefficients for the fully saturated model are presented in Table 3). Thus, higher maternal CRP during the third trimester of pregnancy uniquely and robustly predicted poorer child cognitive flexibility at ages 4-6 years.
Table 3.
Regression model predicting child cognitive flexibility from third trimester maternal HbA1C, CRP, and BP
| Child Cognitive Flexibility |
||||
|---|---|---|---|---|
| Independent Variables | β | SE | p | 95% CI |
| Cognitive battery language (Spanish) | .14 | .11 | .19 | - |
| Study Site (North Carolina) | .13 | .14 | .34 | - |
| Study Site (Washington, DC) | .09 | .14 | .51 | - |
| Maternal age at child birth | −.06 | .16 | .71 | - |
| Marital status (married) | −.19 | .17 | .26 | - |
| Family income | .47 | .20 | .02* | .08, .86 |
| Child birth order | .13 | .10 | .20 | - |
| Child birth weight | .13 | .15 | .40 | - |
| Child gestational age at birth | .10 | .12 | .45 | - |
| Child birth or neonatal complications | −.11 | .13 | .40 | - |
| Preconception maternal BMI | .02 | .15 | .91 | - |
| Third trimester maternal depression | .05 | .12 | .69 | - |
| Third trimester maternal HbA1C | −.02 | .11 | .88 | - |
| Third trimester maternal BP | −.08 | .12 | .54 | - |
| Third trimester maternal CRP | −.39 | .14 | .01** | −.68, −.11 |
p < .05
p < .01
Note: β = standardized coefficient; reference group for Study Site = “Lake Country, IL”; BMI = body mass index; HbA1C = glycated hemoglobin; BP = blood pressure (systolic/diastolic); CRP = C-reactive protein
Temporal Specificity of CRP to Child Cognitive Flexibility
To further clarify that the observed association between maternal CRP and child cognitive flexibility was specific to exposure during the third trimester only, we simultaneously evaluated preconception, second trimester, and third trimester maternal CRP in prediction of child cognitive flexibility. Consistent with the model comparing all biomarkers from the third trimester above, third trimester maternal CRP also inversely predicted child cognitive flexibility over and above preconception and second trimester maternal CRP (B = −1.22, SE = 0.40, p < .01; β = −0.54). Consistent with the prior models testing all preconception and second trimester biomarkers, neither preconception CRP (B = 0.13, SE = 0.28, p = .65, β = 0.05) nor second trimester CRP (B = 0.52, SE = 0.46, p = .26, β = 0.23) was associated with child cognitive flexibility over and above third trimester CRP. Thus, higher maternal CRP specifically during the third trimester predicted poorer child cognitive flexibility at ages 4-6 years.
Prediction of Child Response Inhibition from Maternal Biomarkers
We then repeated the regression models for each time point (i.e., maternal biomarkers from preconception, second trimester, and third trimester) but in prediction of child response inhibition (i.e., Flanker T-scores). Covarying for study site and cognitive battery language, maternal HbA1C, CRP, and BP from all time points were unrelated to child response inhibition. Thus, no further analyses were conducted in prediction of child response inhibition.
Discussion
We evaluated multiple maternal metabolic and pro-inflammatory factors (i.e., HbA1C, CRP, and BP) as unique predictors of child EF (i.e., cognitive flexibility, response inhibition) in an intensive, prospective longitudinal study of prenatal health and child development. Multiple maternal biomarkers were assayed at preconception, second trimester, and third trimester time points, allowing for temporally specific comparisons in prediction of child EF. Higher maternal CRP during the third trimester of pregnancy, but not third trimester HbA1C and BP, uniquely predicted poorer child cognitive flexibility at ages 4-6 years, even with stringent control of relevant demographic factors, concurrent third trimester maternal HbA1C and BP, and multiple prenatal/perinatal covariates (i.e., preconception maternal BMI, third trimester maternal depression, maternal age, maternal marital status, household income, child birth order, child birth weight, child gestational age, and child birth/neonatal complications). Predictions from maternal CRP were specific to the third trimester only, and third trimester CRP robustly predicted child cognitive flexibility over and above preconception and second trimester CRP. None of the preconception or second trimester maternal biomarkers predicted child cognitive flexibility, and child response inhibition was unrelated to maternal biomarkers from all time points. These findings reflect prospective evidence that exposure to maternal inflammation uniquely predicts cognitive flexibility deficits in children, and that this association is dependent on the timing of the exposure before or during pregnancy.
That maternal CRP specifically from the third trimester uniquely and robustly predicted child cognitive flexibility converges with a large literature implicating maternal inflammation in offspring neurodevelopment and individual differences in offspring EF (e.g., Brown et al., 2009; Jonakait, 2007; Ross, Graham, Money, & Stanwood, 2015; Rudolph et al., 2018; van der Burg et al., 2016). However, to our knowledge, this is the first study to observe an association of maternal CRP with child EF. Because this association was adjusted for maternal BMI, HbA1C, and BP, maternal CRP likely reflected inflammation secondary to infection and/or other non-metabolic triggers of pro-inflammatory response. Likewise, the effect size for the observed association was relatively small, suggesting that clinically meaningful decreases in child cognitive flexibility required substantial elevations in maternal CRP, such as those resulting from infectious processes. Although there is a substantial human and non-human animal literature supporting the role of prenatal maternal infection in offspring neurodevelopment (Brown et al., 2009; Kundakovic, 2017; Madore et al., 2016; Meyer et al., 2006; Meyer, 2014; Short et al., 2010), there is also growing evidence that inflammation may mediate associations of prenatal maternal metabolic conditions (i.e., diabetes, hypertension, obesity) with subsequent child cognitive outcomes (e.g., van der Burg et al., 2016). In fact, based on reliable associations between maternal CRP, maternal metabolic conditions, and adverse birth outcomes (e.g., prematurity and delivery complications) in prior studies (e.g., Christian & Porter, 2014; Elovitz et al., 2011; Qiu et al., 2004; Smith et al., 2005), child EF predictions from CRP may either mediate or be mediated by these correlated factors. Notably, however, in the present study, third trimester maternal CRP uniquely predicted child cognitive flexibility, even with control of concurrent HbA1C and BP as well as other prenatal/perinatal correlates (e.g., preconception maternal BMI, child gestational age, birth/neonatal complications). Moreover, none of these covariates significantly predicted cognitive flexibility over and above third trimester CRP. Although this does not rule out mediated effects of maternal CRP from metabolic conditions or through perinatal complications per se, if replicated, these findings may complement experimental evidence in non-human animals suggesting that maternal inflammation is highly proximal to offspring neurodevelopment. For example, maternal pro-inflammatory factors not only altered placenta functioning, but also transfer to amniotic fluid and enter fetal circulation (Urakubo, Jarskog, Lieberman, & Gilmore, 2001). In turn, this can trigger a pro-inflammatory response in the fetus that may permeate the blood-brain barrier (Meyer et al., 2006). Thus, maternal inflammation may rapidly influence fetal brain development through various biologically plausible mechanisms, such as inhibition of fetal neurotrophic factors (Golan, Lev, Hallak, Sorokin, & Huleihel, 2005) and neurotransmitter levels (Vuillermot, Weber, Feldon, & Meyer, 2010).
Because child cognitive flexibility was predicted specifically from third trimester maternal CRP, and not preconception or second trimester CRP, it is important to note that the prefrontal cortex continues to develop substantially during the third trimester of pregnancy (as well as postnatally; Monk, Webb, & Nelson, 2001); it therefore remains susceptible to adverse prenatal environments like maternal pro-inflammatory state. For example, in rhesus monkeys, maternal infection during the third trimester elicited postnatal structural abnormalities in offspring prefrontal cortex and other brain regions relevant to EF (Short et al., 2010). The third trimester may in fact distinctively reflect a time of particular neurodevelopmental susceptibility to maternal inflammation, as exposure to maternal infection during late pregnancy, but not mid-pregnancy, induced elevations in cytokine gene expression in fetal mouse brain (Meyer et al., 2006). Collectively, therefore, the non-human animal literature suggests that exposure to maternal inflammation (as reflected by elevated maternal CRP) during the third trimester might initiate a causal chain of events that hinders healthy development of brain regions regulating EF. Although our correlational human findings complement these non-human animal studies, further research is needed to identify proximal mechanisms that may mediate third trimester maternal inflammation and child EF specifically in humans. If the third trimester further proves to be a sensitive period for maternal inflammation and offspring cognitive flexibility (or EF more generally), this would inform the timing of prenatal interventions and preventive strategies to promote maternal and fetal health.
Whereas maternal CRP robustly predicted child cognitive flexibility, we did not observe predictions of child cognitive flexibility from other maternal biomarkers (i.e., HbA1C and BP), or predictions of child response inhibition from any biomarker (i.e., CRP, HbA1C, and BP). These null findings diverge somewhat from prior research suggesting that preconception maternal diabetes, gestational diabetes, and gestational hypertension are associated with broad cognitive deficits in children such as lower IQ (e.g., Adane et al., 2016; Krakowiak et al., 2012; Tuovinen, Eriksson, Kajantie, & Räikkönen, 2014), and one study showing that prenatal maternal inflammation predicted poorer child impulse control (Graham et al., 2018). However, the current findings are consistent with a prior study in which gestational diabetes was unrelated to child cognitive flexibility (Bolanos et al., 2015).2 Given the stringent control of multiple potential confounds in the present study, including concurrent HbA1C and BP, one plausible interpretation of the present findings is that maternal inflammation is more detrimental to child neurodevelopment than maternal hyperglycemia or hypertension. Alternatively, methodological differences may also contribute to differing results. For example, whereas Graham et al. (2018) found an association between maternal inflammation and child impulse control, their measure of impulse control did not differentiate between child response inhibition (the EF domain assessed in the present study) and other related processes (i.e., motivation and reactivity). Additionally, whereas previous studies relied on retrospective reports of specific diagnoses to assess maternal metabolic conditions, the intensive and prospective nature of the current study, including direct assay of maternal biomarkers across multiple time points, may have yielded more accurate measurements of maternal health before and across pregnancy.
Several key limitations should be noted. First, despite the use of reliable measures and an intensive, prospective longitudinal research design to maximize statistical power, the analyses were limited by the modest sample size. Second, although we controlled for a range of pertinent perinatal factors, which was a unique strength of the present study, other potential confounds beyond the scope of the present study are plausible. For example, we did not account for postnatal factors (e.g., postnatal depression, maternal caregiving style), maternal cognitive functioning, or other domains of child cognitive functioning (e.g., IQ), all of which are correlated with child EF development (e.g., Bernier, Carlson, & Whipple, 2010; Friedman et al., 2008). In particular, it is plausible that the observed association of third trimester maternal inflammation and child cognitive flexibility also reflects associations with children’s general cognitive abilities. Third, because prenatal data from the first trimester were not available for the current study, we were unable to examine first trimester maternal biomarkers as predictors of child EF. Fourth, there was a higher percentage of missing data from the 2nd trimester compared to data from the preconception and 3rd trimester periods. Thus, although power was maximized in all analyses using FIML estimation to address missing data, we cannot rule out that missing data impacted the present second trimester results. Fifth, although response inhibition and cognitive flexibility begin to develop early in life and are definitely present at ages 4-6 years, they advance substantially across middle childhood (e.g., Best & Miller, 2010). Thus, it will be important to replicate the present findings not only in larger samples but also in prospective longitudinal studies of youth across development. For example, predictions of child response inhibition from maternal biomarkers, although not observed in the present study, may be evident in older children once response inhibition is more fully developed. Similarly, although maternal HbA1C and BP at preconception and prenatally were unrelated to child cognitive flexibility here, these factors may predict later child EF outcomes. It is also important to consider why maternal CRP was specifically associated with child cognitive flexibility, but not response inhibition. Notably, there is replicated evidence from rodent models that in utero exposure to maternal immune activation results in poorer offspring cognitive flexibility (Meyer, 2014), but these effects have not been examined with respect to response inhibition. Thus, further research is needed to clarify if maternal inflammation predicts cognitive flexibility specifically, or EF more generally. Another logical extension of the present findings is to examine whether the observed effect of maternal CRP on child cognitive flexibility also extends to neurodevelopmental disorders that are associated with poor cognitive flexibility (e.g., ADHD).
We observed individual differences in third trimester maternal CRP as a temporally-specific and unique predictor of child cognitive flexibility. To our knowledge, this study was the first to employ multiple assays of maternal metabolic and pro-inflammatory factors over time, directly compare preconception and prenatal exposures, and control for numerous potential confounds in prediction of child EF. Future studies must aim to characterize the proximal mechanisms that mediate third trimester maternal CRP and child cognitive flexibility. Identification of biologically plausible mechanisms underlying EF development will be critical to informing prevention and intervention efforts across major domains of child psychopathology and psychosocial functioning.
Acknowledgments
This work was supported by National Institute of Health grant HD072021.
Footnotes
Data Availability Statement: Data sharing is not applicable to this article as no new data were created or analyzed in this study.
The pattern of results was unchanged when models were conducted without the initial study site and battery language covariates and when age-adjusted (vs. fully-adjusted) DCCS and Flanker T-scores were used (results available upon request), alleviating concerns that the present results were hindered by Type II error secondary to overly stringent models.
Gestational diabetes did predict child working memory, a dimension of child EF not assessed in the present study, in Bolanos et al. (2015).
References
- Aarnoudse-Moens CSH, Weisglas-Kuperus N, van Goudoever JB, & Oosterlaan J (2009). Meta-analysis of neurobehavioral outcomes in very preterm and/or very low birth weight children. Pediatrics, 124(2), 717–728. doi: 10.1542/peds.2008-2816 [DOI] [PubMed] [Google Scholar]
- Adane AA, Mishra GD, & Tooth LR (2016). Maternal pre-pregnancy obesity and childhood physical and cognitive development of children: A systematic review. International Journal of Obesity, 40(11), 1608–1618. doi: 10.1038/ijo.2016.140 [DOI] [PubMed] [Google Scholar]
- Adane Akilew Awoke, Mishra GD, Tooth LR, Bhattarai M, Dunstan D, Zimmet P, … Kirkham J (2016). Diabetes in pregnancy and childhood cognitive development: A systematic review. Pediatrics, 137(5), 173–179. doi: 10.1542/peds.2015-4234 [DOI] [PubMed] [Google Scholar]
- Akshoomoff N, Newman E, Thompson WK, McCabe C, Bloss CS, Chang L, … Jernigan TL (2014). The NIH toolbox cognition battery: Results from a large normative developmental sample (PING). Neuropsychology, 28(1), 1–10. doi: 10.1037/neu0000001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association. (2017). Classification and diagnosis of diabetes. Diabetes Care, 40(Supplement 1), S11–S24. doi: 10.2337/dc17-S005 [DOI] [PubMed] [Google Scholar]
- Baker AM, Braun JM, Salafia CM, Herring AH, Daniels J, Rankins N, & Thorp JM (2008). Risk factors for uteroplacental vascular compromise and inflammation. American Journal of Obstetrics and Gynecology, 199(3), 256.e1–256.e9. doi: 10.1016/j.ajog.2008.06.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier A, Carlson SM, & Whipple N (2010). From external regulation to self-regulation: Early parenting precursors of young children’s executive functioning. Child Development, 81(1), 326–39. doi: 10.1111/j.1467-8624.2009.01397.x [DOI] [PubMed] [Google Scholar]
- Best JR, & Miller PH (2010). A developmental perspective on executive function. Child Development, 81(6), 1641–1660. doi: 10.1111/j.1467-8624.2010.01499.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos L, Matute E, Ramirez-Duenas Mde L, & Zarabozo D (2015). Neuropsychological impairment in school-aged children born to mothers with gestational diabetes. Journal of Child Neurology, 30(12), 1616–1624. doi: 10.1177/0883073815575574 [DOI] [PubMed] [Google Scholar]
- Brown AS, Sourander A, Hinkka-Yli-Saloma S, McKeague IW, Sundvall J, Surcel H, & Autism. (2014). Elevated maternal C-reactive protein and autism in a national birth cohort. Molecular Psychiatry, 19, 259–264. doi: 10.1176/appi.ajp.2014.13121579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown Alan S., Vinogradov S, Kremen WS, Poole JH, Deicken RF, Penner JD, … Schaefer CA (2009). Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. American Journal of Psychiatry, 166(6), 683–690. doi: 10.1176/appi.ajp.2008.08010089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett AC, Scratch SE, Lee KJ, Cheong J, Searle K, Hutchinson E, … Anderson PJ (2015). Executive function in adolescents born <1000 g or <28 Weeks: A prospective cohort study. Pediatrics, 135(4), e826–834. doi: 10.1542/peds.2014-3188 [DOI] [PubMed] [Google Scholar]
- Camerota M, Willoughby MT, Cox M, & Greenberg MT (2015). Executive function in low birth weight preschoolers: The moderating effect of parenting. Journal of Abnormal Child Psychology. doi: 10.1007/s10802-015-0032-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canetta S, Sourander A, Surcel H, Hinkka-Yli-Salomäki S, Leiviskä J, Kellendonk C, … Brown AS (2014). Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. American Journal of Psychiatry, 171(9), 960–968. doi: 10.1176/appi.ajp.2014.13121579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaletto KB, Umlauf A, Beaumont J, Gershon R, Slotkin J, Akshoomoff N, & Heaton RK (2015). Demographically corrected normative standards for the English language version of the NIH Toolbox Cognition Battery. Journal of the International Neuropsychological Society, 21(5), 378–391. doi: 10.1017/S135561771500137X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaletto KB, Umlauf A, Marquine M, Beaumont JL, Mungas D, Gershon R, … Heaton RK (2016). Demographically corrected normative standards for the Spanish language version of the NIH Toolbox Cognition Battery, 22(3), 364–374. doi: 10.1017/S1355617715000351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian LM, & Porter K (2014). Longitudinal changes in serum proinflammatory markers across pregnancy and postpartum: Effects of maternal body mass index. Cytokine, 70(2), 134–140. doi: 10.1016/j.cyto.2014.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark C, Prior M, & Kinsella G (2002). The relationship between executive function abilities, adaptive, behaviour, and academic achievement in children with externalising behaviour problems. Journal of Child Psychology and Psychiatry, 43(6), 785–796. doi: 10.1111/1469-7610.00084 [DOI] [PubMed] [Google Scholar]
- Cox JL, Holden JM, & Sagovsky R (1987). Detection of postnatal depression: Development of the 10-item Edinburgh Postnatal Depression Scale. British Journal of Psychiatry, 150(6), 782–786. doi: 10.1007/978-94-007-1694-0_2 [DOI] [PubMed] [Google Scholar]
- Deave T, Heron J, Evans J, & Emond A (2008). The impact of maternal depression in pregnancy on early child development. BJOG:, 115(8), 1043–1051. doi: 10.1111/j.1471-0528.2008.01752.x [DOI] [PubMed] [Google Scholar]
- Diamantopoulou S, Rydell A-M, Thorell LB, & Bohlin G (2007). Impact of executive functioning and symptoms of attention deficit hyperactivity disorder on children’s peer relations and school performance. Developmental Neuropsychology, 32(1), 521–542. doi: 10.1080/87565640701360981 [DOI] [PubMed] [Google Scholar]
- Diamond A (2012). Activities and programs that improve children’s executive functions. Current Directions in Psychological Science, 21(5), 335–341. doi: 10.1177/0963721412453722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond A, & Lee K (2011). Interventions shown to aid executive function development in children 4 to 12 years old. Science, 333(6045), 959–964. doi: 10.1126/science.1204529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkel Schetter C, Schafer P, Lanzi RG, Clark-Kauffman E, Raju TNK, & Hillemeier MM (2013). Shedding Light on the Mechanisms Underlying Health Disparities Through Community Participatory Methods: The Stress Pathway. Perspectives on Psychological Science, 8(6), 613–633. doi: 10.1177/1745691613506016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elovitz MA, Brown AG, Breen K, Anton L, Maubert M, & Burd I (2011). Intrauterine inflammation, insufficient to induce parturition, still evokes fetal and neonatal brain injury. International Journal of Developmental Neuroscience, 29(6), 663–671. doi: 10.1016/j.ijdevneu.2011.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman NP, Miyake A, Young SE, Defries JC, Corley RP, & Hewitt JK (2008). Individual differences in executive functions are almost entirely genetic in origin. Journal of Experimental Psychology: General, 137(2), 201–25. doi: 10.1037/0096-3445.137.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon RC, Wagster MV, Hendrie HC, Fox NA, Cook KF, & Nowinski CJ (2013). NIH Toolbox for assessment of neurological and behavioral function. Neurology, 80(Suppl 3), S2–S6. doi: 10.1212/WNL.0b013e3182872e5f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan HM, Lev V, Hallak M, Sorokin Y, & Huleihel M (2005). Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology, 48(6), 903–917. doi: 10.1016/j.neuropharm.2004.12.023 [DOI] [PubMed] [Google Scholar]
- Goldstein DE, Little RR, Lorenz RA, Malone JI, Nathan D, Peterson CM, & Sacks DB (2003). Tests of glycemia in diabetes. Diabetes Care, 27(7), 1761–1773. doi: 10.2337/diacare.26.2007.S106 [DOI] [PubMed] [Google Scholar]
- Graham AM, Rasmussen JM, Rudolph MD, Heim CM, Gilmore JH, Styner M, … Buss C (2018). Maternal systemic interleukin-6 during pregnancy is associated with newborn amygdala phenotypes and subsequent behavior at 2 years of age. Biological Psychiatry, 83(2), 109–119. doi: 10.1016/j.biopsych.2017.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackman DA, Farah MJ, & Meaney MJ (2010). Socioeconomic status and the brain: Mechanistic insights from human and animal research. Nature Reviews Neuroscience, 11, 651–659. doi: 10.1038/nrn2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart H, Radua J, Nakao T, Mataix-Cols D, & Rubia K (2013). Meta-analysis of functional magnetic resonance imaging studies of inhibition and attention in attention-deficit/hyperactivity disorder. JAMA Psychiatry, 70(2), 185–198. doi: 10.1001/jamapsychiatry.2013.277 [DOI] [PubMed] [Google Scholar]
- Hedderson MM, & Ferrara A (2008). High blood pressure before and during early pregnancy is associated with an increased risk of gestational diabetes. Diabetes Care, 31(12), 2362–2367. doi: 10.2337/dc08-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HS, Kwon JY, Kim MA, Park YW, & Kim YH (2007). Maternal serum highly sensitive C-reactive protein in normal pregnancy and pre-eclampsia. International Journal of Gynecology and Obstetrics, 98(2), 105–109. doi: 10.1016/j.ijgo.2007.03.050 [DOI] [PubMed] [Google Scholar]
- Jonakait GM (2007). The effects of maternal inflammation on neuronal development: Possible mechanisms. International Journal of Developmental Neuroscience, 25(7), 415–425. doi: 10.1016/j.ijdevneu.2007.08.017 [DOI] [PubMed] [Google Scholar]
- Kozhimannil KB, Pereira MA, & Harlow BL (2014). Association between diabetes and perinatal depression among low-income mothers. JAMA, 301(8), 842–847. [DOI] [PubMed] [Google Scholar]
- Krakowiak P, Walker CK, Bremer AA, Baker AS, Ozonoff S, Hansen RL, & Hertz-Picciotto I (2012). Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics, 129(5), e1121–8. doi: 10.1542/peds.2011-2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundakovic M (2017). Fearing the Mother’s Virus: The Lasting Consequences of Prenatal Immune Activation on the Epigenome and Brain Function. Biological Psychiatry, 81(3), e23–e25. doi: 10.1016/j.biopsych.2016.11.005 [DOI] [PubMed] [Google Scholar]
- Little RJA (1988). A test of missing completely at random for multivariate data with missing values. Journal of the American Statistical Association, 83(404), 1198–1202. [Google Scholar]
- Madore C, Leyrolle Q, Lacabanne C, Benmamar-Badel A, Joffre C, Nadjar A, & Lay S (2016). Neuroinflammation in Autism: Plausible Role of Maternal Inflammation, Dietary Omega 3, and Microbiota. Neural Plasticity, 2016. doi: 10.1155/2016/3597209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus SM, Flynn HA, Blow F, & Barry K (2005). A screening study of antidepressant treatment rates and mood symptoms in pregnancy. Archives of Women’s Mental Health, 8(1), 25–27. doi: 10.1007/s00737-005-0072-1 [DOI] [PubMed] [Google Scholar]
- McGrath LM, Braaten EB, Doty ND, Willoughby BL, Wilson HK, O’Donnell EH, … Doyle AE (2015). Extending the ‘cross-disorder’ relevance of executive functions to dimensional neuropsychiatric traits in youth. Journal of Child Psychology and Psychiatry. doi: 10.1111/jcpp.12463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, … Feldon1 J (2006). The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. Journal of Neuroscience, 26(18), 4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer Urs. (2014). Prenatal Poly(I:C) exposure and other developmental immune activation models in rodent systems. Biological Psychiatry, 75(4), 307–315. doi: 10.1016/j.biopsych.2013.07.011 [DOI] [PubMed] [Google Scholar]
- Meyer Urs, Feldon J, & Dammann O (2011). Schizophrenia and autism: Both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatric Research, 69(5), 26–33. doi: 10.1203/PDR.0b013e318212c196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina TH, Lahti M, Drake AJ, Raikkonen K, Minnis H, Denison FC, … Reynolds RM (2016). Prenatal exposure to very severe maternal obesity is associated with adverse neuropsychiatric outcomes in children. Psychological Medicine, 1–10. doi: 10.1017/S0033291716002452 [DOI] [PubMed] [Google Scholar]
- Miyake A, Friedman NP, Emerson MJ, Witzki AH, Howerter A, & Wager TD (2000). The unity and diversity of executive functions and their contributions to complex “Frontal Lobe” tasks: A latent variable analysis. Cognitive Psychology, 41(1), 49–100. doi: 10.1006/cogp.1999.0734 [DOI] [PubMed] [Google Scholar]
- Monk CS, Webb SJ, & Nelson CA (2001). Prenatal neurobiological development: Molecular mechanisms and anatomical change. Developmental Neuropsychology, 19(2), 147–171. doi: 10.1207/S15326942DN1902 [DOI] [PubMed] [Google Scholar]
- Mungas D, Widaman K, Zelazo PD, Tulsky D, Heaton RK, Slotkin J, … Gershon RC (2013). NIH toolbox cognition battery (CB): Factor structure for 3 to 15 year olds. Monographs of the Society for Research in Child Development, 78(4), 103–118. doi: 10.1111/mono.12037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornoy A, Reece EA, Pavlinkova G, Kappen C, & Miller RK (2015). Effect of maternal diabetes on the embryo, fetus, and children: Congenital anomalies, genetic and epigenetic changes and developmental outcomes. Birth Defects Research Part C - Embryo Today: Reviews, 105(1), 53–72. doi: 10.1002/bdrc.21090 [DOI] [PubMed] [Google Scholar]
- Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, Criqui M, … Vinicor F (2003). Markers of inflammation and cardiovascular disease: Application to clinical and public health practice: A statement for healthcare professionals from the centers for disease control and prevention and the American Heart Association. Circulation, 107(3), 499–511. doi: 10.1161/01.CIR.0000052939.59093.45 [DOI] [PubMed] [Google Scholar]
- Pennington BF, & Ozonoff S (1996). Executive functions and developmental psychopathology. Journal of Child Psychology and Psychiatry, 37(1), 51–87. doi: 10.1111/j.1469-7610.1996.tb01380.x [DOI] [PubMed] [Google Scholar]
- Qiu C, Sorensen TK, Luthy DA, & Williams MA (2004). A prospective study of maternal serum C-reactive protein (CRP) concentrations and risk of gestational diabetes mellitus. Paediatric and Perinatal Epidemiology, 18(5), 377–384. doi: 10.1111/j.1365-3016.2004.00578.x [DOI] [PubMed] [Google Scholar]
- Ramey SL, Schafer P, DeClerque JL, Lanzi RG, Hobel C, Shalowitz M, … Raju TNK (2015). The Preconception Stress and Resiliency Pathways Model: A Multi-Level Framework on Maternal, Paternal, and Child Health Disparities Derived by Community-Based Participatory Research. Maternal and Child Health Journal, 19(4), 707–719. doi: 10.1007/s10995-014-1581-1 [DOI] [PubMed] [Google Scholar]
- Ridker PM (2003). Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation, 107(3), 363–369. doi: 10.1161/01.CIR.0000053730.47739.3C [DOI] [PubMed] [Google Scholar]
- Ross EJ, Graham DL, Money KM, & Stanwood GD (2015). Developmental Consequences of Fetal Exposure to Drugs: What We Know and What We Still Must Learn. Neuropsychopharmacology, 40(1), 61–87. doi: 10.1038/npp.2014.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MD, Graham AM, Feczko E, Miranda-Dominguez O, Rasmussen JM, Nardos R, … Fair DA (2018). Maternal IL-6 during pregnancy can be estimated from newborn brain connectivity and predicts future working memory in offspring. Nature Neuroscience, 21(5), 765–772. doi: 10.1038/s41593-018-0128-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santor DA, & Coyne JC (1997). Shortening the CES-D to improve its ability to detect cases of depression. Psychological Assessment, 9(3), 233–243. doi: 10.1037/1040-3590.9.3.233 [DOI] [Google Scholar]
- Schlomer GL, Bauman S, & Card NA (2010). Best practices for missing data management in counseling psychology. Journal of Counseling Psychology, 57(1), 1–10. doi: 10.1037/a0018082 [DOI] [PubMed] [Google Scholar]
- Scholl TO, Sowers M, Chen X, & Lenders C (2001). Maternal glucose concentration influences fetal growth, gestation, and pregnancy complications. American Journal of Epidemiology, 154(6), 514–520. doi: 10.1093/aje/154.6.514 [DOI] [PubMed] [Google Scholar]
- Shalowitz MU, Schetter CD, Hillemeier MM, Chinchilli VM, Adam EK, Hobel CJ, … Raju TNK (2019). Cardiovascular and Metabolic Risk in Women in the First Year Postpartum: Allostatic Load as a Function of Race, Ethnicity, and Poverty Status. American Journal of Perinatology, 36(10), 1079–1089. doi: 10.1055/s-0038-1675618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SJ, Lubach GR, Karasin AI, Olsen CW, Styner M, Knickmeyer RC, … Coe CL (2010). Maternal influenza infection during pregnancy impacts postnatal brain development in the rhesus monkey. Biological Psychiatry, 67(10), 965–973. doi: 10.1016/j.biopsych.2009.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibai BM, Caritis SN, Hauth JC, MacPherson C, VanDorsten JP, Klebanoff M, … Roberts J (2000). Preterm delivery in women with pregestational diabetes mellitus or chronic hypertension relative to women with uncomplicated pregnancies. American Journal of Obstetrics and Gynecology, 183(6), 1520–1524. doi: 10.1067/mob.2000.107621 [DOI] [PubMed] [Google Scholar]
- Smith GD, Lawlor DA, Harbord R, Timpson N, Rumley A, Lowe GDO, … Ebrahim S (2005). Association of C-reactive protein with blood pressure and hypertension: Life course confounding and Mendelian randomization tests of causality. Arteriosclerosis, Thrombosis, and Vascular Biology, 25(5), 1051–1056. doi: 10.1161/01.ATV.0000160351.95181.d0 [DOI] [PubMed] [Google Scholar]
- Tuovinen S, Eriksson JG, Kajantie E, & Räikkönen K (2014). Maternal hypertensive pregnancy disorders and cognitive functioning of the offspring: A systematic review. Journal of the American Society of Hypertension, 8(11), 832–847. doi: 10.1016/j.jash.2014.09.005 [DOI] [PubMed] [Google Scholar]
- Urakubo A, Jarskog LF, Lieberman JA, & Gilmore JH (2001). Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophrenia Research, 47, 27–36. [DOI] [PubMed] [Google Scholar]
- Valero De Bernabé J, Soriano T, Albaladejo R, Juarranz M, Calle ME, Martínez D, & Domínguez-Rojas V (2004). Risk factors for low birth weight: A review. European Journal of Obstetrics Gynecology and Reproductive Biology, 116, 3–15. doi: 10.1016/j.ejogrb.2004.03.007 [DOI] [PubMed] [Google Scholar]
- van der Burg JW, Sen S, Chomitz VR, Seidell JC, Leviton A, & Dammann O (2016). The role of systemic inflammation linking maternal body mass index to neurodevelopment in children: Inflammation and neurodevelopment. Pediatric Research, 79, 3–12. doi: 10.1002/aur.1474.Replication [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S, Weber L, Feldon J, & Meyer U (2010). A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. Journal of Neuroscience, 30(4), 1270–1287. doi: 10.1523/JNEUROSCI.5408-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M, & Jenkins JM (2016). Pregnancy hypertension and the risk for neuropsychological difficulties across early development: A brief report. Child Neuropsychology, 22(2), 247–254. doi: 10.1080/09297049.2014.958070 [DOI] [PubMed] [Google Scholar]
- Wiggs K, Elmore AL, Nigg JT, & Nikolas MA (2016). Pre- and perinatal risk for attention-deficit hyperactivity disorder: Does neuropsychological weakness explain the link? Journal of Abnormal Child Psychology. doi: 10.1007/s10802-016-0142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcutt EG, Doyle AE, Nigg JT, Faraone SV, & Pennington BF (2005). Validity of the executive function theory of attention-deficit/hyperactivity disorder: A meta-analytic review. Biological Psychiatry, 57(11), 1336–1346. doi: 10.1016/j.biopsych.2005.02.006 [DOI] [PubMed] [Google Scholar]
- Zelazo PD, Anderson JE, Richler J, Wallner-Allen K, Beaumont JL, & Weintraub S (2013). NIH Toolbox Cognition Battery (CB): Measuring Executive Function and Attention. Monographs of the Society for Research in Child Development, 78(4), 16–33. doi: 10.1111/mono.12032 [DOI] [PubMed] [Google Scholar]