

Abstract
Background & Aims:
Intestinal epithelial cells (IECs) regulate intestinal immune cells, particularly development of T-helper 17 (Th17) cells. Deregulation of this process leads to intestinal inflammation and tumorigenesis, via unknown mechanisms. TANK-binding kinase 1 (TBK1) is expressed by IECs and cells in the innate immune system. We studied the functions of TBK1 in the intestinal immune response and tumorigenesis in mice.
Methods:
We performed studies of wild-type mice, mice with conditional disruption of Tbk1 (Tbk1IEC-KO), Tbk1IEC-KO mice crossed with ApcMin/+ mice, and Mt−/− mice crossed with ApcMin/+ mice. Some mice were given intraperitoneal injections of a neutralizing antibody against interleukin 17 (IL17) or IL1B. Intestine tissues were collected from mice and analyzed by histology, for numbers of adenomas and Th17 cells, and expression of inflammatory cytokines by real-time PCR. IECs were isolated from wild-type and Tbk1IEC-KO mice, stimulated with lipopolysaccharide, co-cultured for with bone marrow-derived macrophages, and analyzed by RNA sequencing and biochemical analyses.
Results:
Compared to ApcMin/+Tbk1WT mice, ApcMin/+Tbk1IEC-KO mice had significant increases in number and size of intestinal polyps, and significantly more Th17 cells in lamina propria. Administration of an antibody against IL17 reduced the number of intestinal polyps in ApcMin/+Tbk1IEC-KO mice to that observed in ApcMin/+Tbk1WT mice. In culture, TBK1-deficient IECs promoted expression of IL1B by macrophages, which induced differentiation of naïve CD4+ T cells into Th17 cells. RNA sequencing analysis revealed that the TBK1-deficient IECs had increased expression of metallothionein 1 (MT1), an immune regulator that promotes intestinal inflammation. Intestine tissues from ApcMin/+Mt−/− mice had significant fewer Th17 cells than ApcMin/+Mt+/+ mice, and a significantly lower number of polyps. Analyses of colorectal tumors in the Cancer Genome Atlas found colorectal tumors with high levels of MT1 and IL17 mRNAs to be associated with reduced survival times of patients.
Conclusions:
Expression of TBK1 by IECs suppresses expression of MT1 and prevents expression of IL1B by macrophages and differentiation of Th17 cells, to prevent inflammation and tumorigenesis. Strategies to block this pathway might be developed for colorectal tumorigenesis.
Keywords: tumor suppressor, immune regulation, colon cancer, adenoma
Graphical Abstract
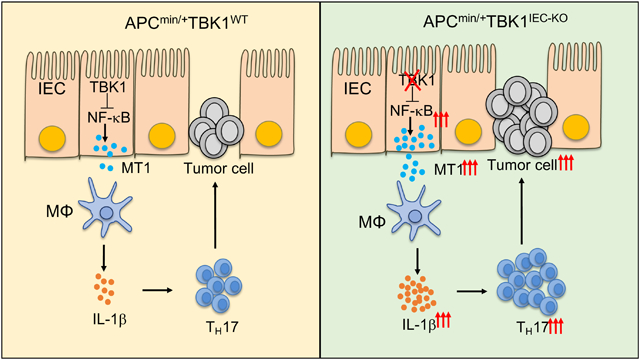
Introduction
Intestine is the largest immune organ of the body, in which the immune system maintains a dynamic interplay with resident microorganisms or microbiota1. A balanced host-microbiota interaction is crucial for intestinal homeostasis, whereby dysregulated mucosal immune responses contribute to the development of inflammatory bowel diseases (IBD) and intestinal tumorigenesis2. The intestinal immune system is composed of a large variety of innate and adaptive immune cells. In particular, T helper 17 (TH17) cells play a crucial role in regulating host defenses against infections in the intestine and also contribute to inflammatory disorders3. The differentiation and maintenance of intestinal TH17 cells require signals from microbiota and are tightly controlled by negative regulatory mechanisms4–6. Dysregulated TH17 cell responses contribute to the pathogenesis of IBD3. Furthermore, TH17 cells and their signature cytokine IL-17 promote the development and progression of intestinal tumorigenesis7–9. The mechanisms that regulate TH17 cell responses in the intestine are incompletely understood, although cytokines produced by dendritic cells (DCs) and macrophages, including IL-1β, IL-23, and TGFβ, contribute to the induction of TH17 cell differentiation and expansion10.
Intestinal epithelial cells (IECs) are vital for the maintenance of intestinal immune homeostasis and host defense11. In addition to forming a physical barrier that segregates host tissue and microbiota, IECs sense microbial components and produce immunomodulatory factors that regulate the development and expansion of immune cells, including TH17 cells. IECs transduce signals from commensal bacteria to induce the differentiation of TH17 cells12, 13. Moreover, IECs also play a critical role in preventing abnormal immune responses to commensal bacteria and limiting steady-state inflammation in the intestine10. The signaling network that mediates the immunoregulatory functions of IECs is incompletely understood.
TANK-binding kinase 1 (TBK1) is an innate immune kinase that mediates induction of type I interferons (IFNs) by various pattern-recognition receptors14. Recent studies suggest that TBK1 responds to additional immune stimuli, some of which do not induce type I IFN expression, implicating novel functions of TBK1 in the immune system15–18. Indeed, emerging evidence suggests that TBK1 regulates immune functions that are beyond IFN induction, including lymphocyte activation and the DC functions17, 19, 20. However, the role of TBK1 in the regulation of intestinal immunity and homeostasis is undefined. In the present study, we demonstrate that TBK1 functions in IECs to control intestinal TH17 cell generation, thereby regulating intestinal tumorigenesis. Our data suggest that TBK1 regulates the crosstalk between IECs and innate immune cells to prevent aberrant induction of proinflammatory cytokines and, thereby, maintain the homeostasis of TH17 cells.
Materials and Methods
Mice
Tbk1-flox mice (on C57BL/6–129Sv/Ev background) was as described16. The Tbk1-flox mice were crossed with Villin-Cre transgenic mice (C57BL/6 genetic background, Jackson Laboratories) to generate age-matched Tbk1 IEC-conditional KO or Tbk1IEC-KO (Tbk1fl/flVillin-Cre) and WT (Tbk1+/+Villin-Cre) control mice. Tbk1IEC-KO mice were further crossed with ApcMin/+ mice (Jackson Laboratory) to generate ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice. B6.SJL mice, expressing the CD45.1 congenic marker, were obtained from the Jackson Lab (C57BL/6 background). Mice with simultaneous knockout of the closely linked Mt1 and Mt2 genes (Mt−/−; Jackson Lab) were crossed with ApcMin/+Tbk1IEC-KO mice to produce Mt−/−ApcMin/+Tbk1IEC-KO and control Mt+/+ApcMin/+Tbk1IEC-KO mice. The genotyping primer sets are listed in Supplementary Table 1. Mice were maintained in a specific pathogen-free facility, and all animal experiments were in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center.
Colon cancer survival analysis
Kaplan–Meier survival analyses for clinical outcomes of colon cancer patients (cohort: GDC TCGA Colon Cancer, COAD) were performed using a web tool, UCSC Xena21. The percentiles of the patients between the upper and lower quartile were auto-selected based on the best performing thresholds as cutoffs. The raw data were from National Cancer Institute Genomic Data Commons (https://docs.gdc.cancer.gov/Data/Release_Notes/Data_Release_Notes/#data-release-180).
Histology
The entire intestine was opened longitudinally, rinsed to clean internal contents with phosphate buffered saline (PBS), and rolled over to obtain a ‘Swiss roll’, which was then fixed in 4% formaldehyde in PBS for 1 hour at room temperature. Fixed tissues were dehydrated by gradually soaking in ethanol (70%, 80%, 90%, 95%, and absolute) and xylene and then embedded in paraffin. The paraffin-embedded specimens were cut into 5-μm sections, stained with hematoxylin-eosin (H&E), and viewed with a digital inverted light microscope (EVOS, Thermo Fisher Scientific, Waltham, MA).
Adenoma quantification
The intestine was opened longitudinally and fixed with 10% formaldehyde in PBS for overnight at 4°C. The adenomas were counted, and their sizes (diameters) were measured after staining with 0.2% methylene blue and decolorizing with ethanol using Olympus SZX16 stereomicroscope attached Olympus SC180 camera and Olympus CellSens Standard software (Olympus co., Center Valley, PA). H&E stained slides were used to count the number of microadenomas, defined as histologically dysplastic glands22.
Real-time quantitative RT-PCR
Total RNA was isolated from sorted macrophages or liquid nitrogen-frozen intestinal tissues using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA) and subjected to real-time quantitative PCR (qRT-PCR) using iCycler Sequence Detection System (Bio-Rad, Hercules, CA) and iQ SYBR Green Supermix (Bio-Rad). All reactions were performed in the same manner: 95°C for 3 min, followed by 45 cycles of 95°C for 10 sec and 60°C for 30 sec. Individual gene expression was calculated by a standard curve method and normalized to the level of Actb. The specific primer sets are listed in Supplementary Table 2.
Cell preparation from intestine
Small intestines were cut into 1 cm pieces and treated with RPMI 1640 containing 0.5 mM EDTA for 20 min at 37°C to remove epithelial cells and mucus. After washing 4–5 times with pre-warmed PBS, tissues were minced and dissociated in RPMI containing 10% FBS, 0.5 mg/ml collagenase D (Sigma-Aldrich, St. Louis, MO) and 100 μg/ml DNase I (Roche Applied Science) twice for 30 min at 37°C with shaking. Large intestines were prepared using a similar procedure, except for the use of a higher concentration (2 mM) of EDTA in the RPMI 1640 incubation medium. Lamina propria lymphocytes were enriched by using 40% and 75% Percoll gradients (Amersham Biosciences) with no brake centrifugation (2,000 rpm, 20 min, room temperature). For IEC isolation, the filtered liquid in the previous washing step was centrifuged using 3 different Percoll gradients (25% / 40% / 75%) to enrich IECs in the boundary between 25% and 40%.
Microbiome analysis
16S rRNA gene sequencing was performed at the Alkek Center for Metagenomics and Microbiome Research (CMMR), Baylor College of Medicine, as previously reported23. Briefly, bacterial genomic DNA was extracted from feces using MO BIO PowerSoil DNA Isolation Kit (MO BIO Laboratories). The 16S rDNA V4 region was amplified by PCR and sequenced in the MiSeq platform (Illumina) using the 2 × 250 bp paired-end protocol yielding pair-end reads that overlap almost completely. The primers used for amplification contain adapters for MiSeq sequencing and single-index barcodes so that the PCR products may be pooled and sequenced directly24, targeting at least 10,000 reads per sample. 16S rRNA sequencing data analysis was performed by ATIMA software developed at the CMMR, Baylor College of Medicine23.
Cytokine neutralization by antibodies.
For IL-17 neutralization, mice were injected intraperitoneally (i.p.) with a neutralizing antibody for mouse IL-17 (Clone 17F3) or isotype control IgG1 (Clone MOPC-21) (10 μg/g mouse body weight) every other day for 2 months. A similar approach was used for IL-1β neutralization, in which mice were injected i.p. with anti-IL-1β (clone B122) or isotype IgG1 control (100 μg/mouse) every other day for 2 months. For IL-1β neutralization in vitro, naïve CD4+ T cells were co-cultured with intestinal macrophages or supernatants of small intestinal tissue culture in the presence of anti-mouse IL-1β (clone B122) or PBS as control.
Cell sorting strategy
For TH17 cell differentiation experiments, lymphocytes were isolated from spleen and lymph nodes. The enriched CD4+ T cells were further stained with FITC-conjugated anti-mouse CD44, Pacific Blue-conjugated anti-mouse CD4 and PE-conjugated anti-mouse CD25, and sorted as CD4+CD25−CD44− naïve CD4+ T cells using FACSJazz™ (BD Biosciences, San Jose, CA). For sorting intestinal macrophages, DCs, and IECs, lamina propria cells were isolated and stained with FITC-conjugated anti-mouse CD45, PE-conjugated anti-mouse CD64, Pacific Blue-conjugated anti-mouse CD11c and PE-Cy7-conjugated anti-mouse MHCII. Intestinal resident macrophages and DCs were sorted as CD45+MHCII+CD64+CD11c+ cells and CD45+MHCII+CD64− CD11c+ using FACSJazz™, respectively. IECs isolated as described above were stained with FITC-conjugated anti-mouse CD45, Pacific Blue-conjugated anti-mouse EpCAM, and sorted as CD45−EpCAM+ population using FACSJazz™.
In vitro TH17 cell differentiation
TH17 cell differentiation was done as previously described25. In brief, purified naïve CD4+ T cells (CD44loCD62Lhi) were activated with plate-coated anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml) under TH17 condition (5 μg/ml anti-IL-4, 5 μg/ml anti-IFN-γ, 20 ng/ml IL-6, and 2.5 ng/ml TGF-β). After 5 days, harvested cells were re-stimulated for 4–5 hours with PMA (50 ng/ml) and ionomycin (750 ng/ml) in the presence of a protein transport inhibitor, monensin (Invitrogen), followed by intracellular staining of IL-17A.
Crypts isolation
Crypts were isolated as described elsewhere26. In brief, cleaned small intestines were cut into 1 cm pieces and incubated in 1 mM EDTA in PBS for 30 minutes at 4°C. Tissues were minced, transferred into 5 mM EDTA in PBS and further incubated for 1 hour at 4°C with gentle shaking. Crypts were dissociated by pipetting with pre-cooled PBS and filtered suspended PBS out using a 70-μm cell strainer (Corning, NY).
Organoid culture
For construction of organoids, 200–500 crypts per well were suspended in Matrigel (Corning). We used complete ENR medium of advanced DMEM/F12 (Gibco by Thermo Fisher Scientific, Waltham, MA) containing penicillin/streptomycin (Gibco), 1 mM N-acetyl cysteine (Sigma Aldrich), B27 supplement (Invitrogen), N2 supplement (Invitrogen), EGF (R&D Systems, Minneapolis, MN), Noggin (R&D Systems), and R-spondin-1 (R&D Systems). ENR medium was replaced every 2 days and organoids were passaged around 7 days of culture by mechanical disruption with a pipette. Detached organoid fragments were washed several times by centrifugation (1,200 rpm, 5 min, 4°C) and replaced with new Matrigel. To isolate single cells from organoids, the mechanically dissociated organoid pellet was further incubated in TrypLE Express (Gibco) for 5 min at 37°C.
For tumor organoid culture, mouse intestinal adenomas were cut into 5 mm pieces using a razor blade, followed by incubation in a digestion buffer containing collagenase IV and dispase II for 30 min at 37°C. The supernatant was filtered using a 100-μm cell strainer and centrifuged for 5 min at 300 × g. Adenoma cells were resuspended in Matrigel (Corning) and seeded in 24-well plates (500–1000 cells per well). To maintain tumoroid culture, ENR medium was replaced every 2 days and tumoroid culture were monitored daily.
Preparation of bone marrow-derived macrophages (BMDMs)
BMDMs were generated by cultivating bone marrow cells in growth medium supplemented with recombinant M-CSF (30 ng/ml)25. Fresh M-CSF-containing medium was added on day 3, and the fully differentiated macrophages were harvested on day 8 for further studies.
In vitro cocultivation of macrophages and IECs
IECs isolated from Tbk1WT or Tbk1IEC-KO mice were stimulated with LPS (1 μg/ml) for 1 hour. The cells were washed twice with PBS and then co-cultured for 24 hours with BMDMs with 10:1 ratio (1 × 105 IECs per each well). Supernatants were collected for ELISA analysis of the indicated cytokines.
Statistical analysis.
GraphPad Prism software (GraphPad, La Jolla, CA) was adopted for statistical analysis. Significant change between two groups were analyzed with two-tailed unpaired t-test. Data are presented as means ± s.e.m. A p-value less than 0.05 is considered statistically significant.
Results
IEC-specific TBK1 deficiency promotes tumorigenesis in ApcMin/+ mice
TBK1 is known to mediate survival and proliferation of Kras-dependent lung cancer cells27–29. Interestingly, our analysis of the TCGA database revealed the association of TBK1 high expression with improved patient survival in colon cancer (Fig. 1A). To directly examine the role of TBK1 in regulating intestinal tumorigenesis, we generated Tbk1IEC-KO mice (Figure 1B and C) and crossed them with ApcMin/+ mice, a frequently used intestinal tumorigenesis model carrying a heterozygous mutation in the Apc tumor suppressor30, 31. Genotyping PCR and immunoblot confirmed the generation of ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO progeny mice and IEC-specific TBK1 deletion (Figure 1D and E). As expected, ApcMin/+Tbk1WT mice spontaneously developed multiple adenoma or polyps in the small intestine at the age of five months (Fig. 1F and G). Importantly, the ApcMin/+Tbk1IEC-KO mice had drastically increased numbers of polyps compared to the ApcMin/+Tbk1WT control mice (Fig. 1F and G). The number of polyps smaller than 0.5 mm was not significantly influenced by TBK1 deficiency, suggesting a role for TBK1 in regulating polyp growth (Fig. 1G). However, since the total number of polyps were drastically increased in the ApcMin/+Tbk1IEC-KO mice, a role for TBK1 in regulating tumor initiation could not be ruled out. Although the number of polyps in large intestine was extremely low, it was also significantly increased in the ApcMin/+Tbk1IEC-KO mice. These results suggest that IEC-specific deletion of TBK1 promotes intestinal tumorigenesis in both the small and large intestines.
Figure 1.
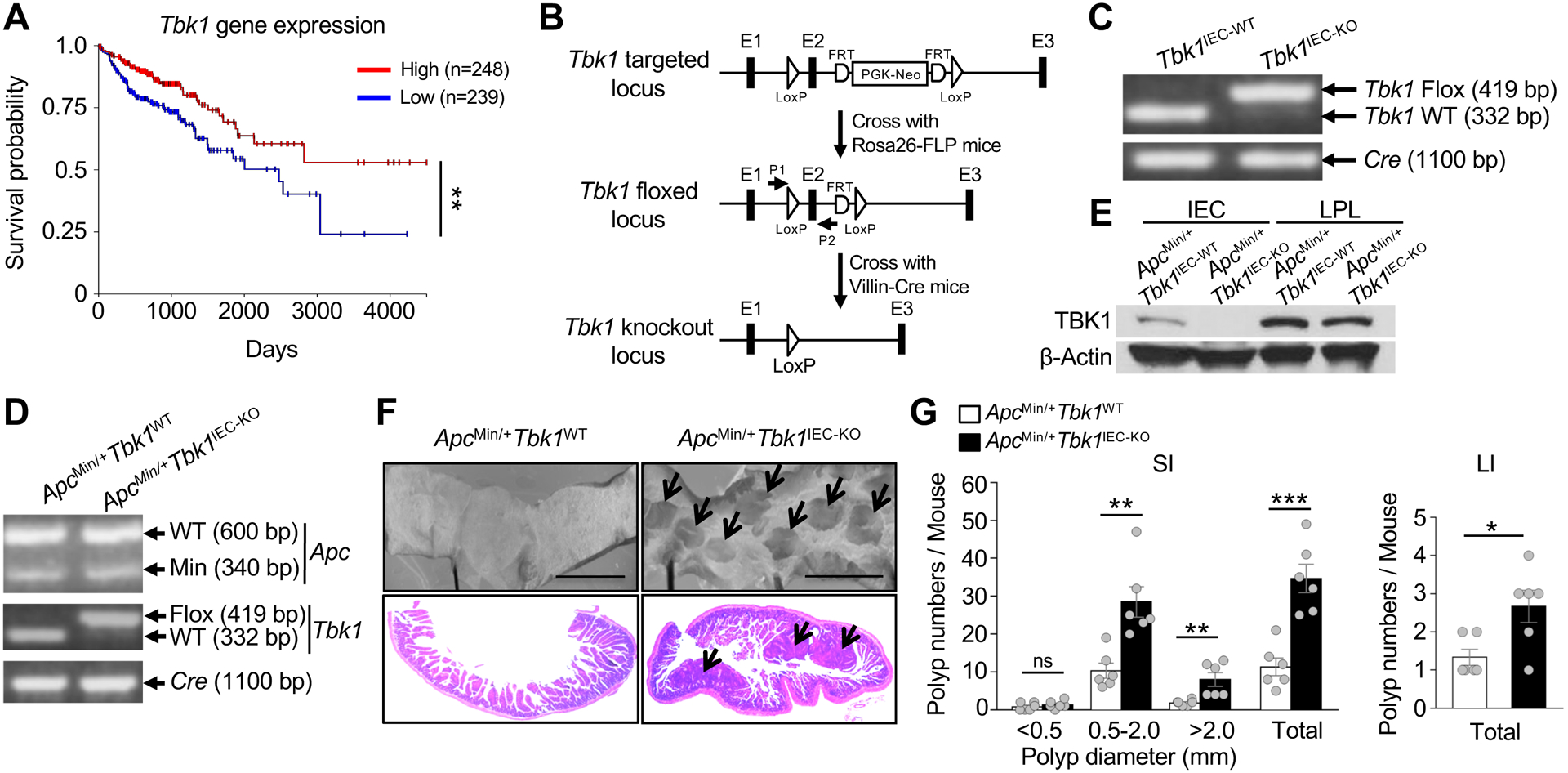
IEC-specific TBK1 ablation promotes adenomas formation in ApcMin/+ mice. (A) Association of Tbk1 high expression with improved patient survival in colorectal cancer revealed through analysis of TCGA database using the UCSC Xena software (n = 571). (B) Schematic of strategies for generating Tbk1 flox and Tbk1IEC-KO mice. (C) Genotyping PCR analysis using tail DNAs from Tbk1WT (Tbk1+/+Villin-Cre) and Tbk1IEC-KO (Tbk1fl/flVillin-Cre) mice showing the floxed and WT alleles of Tbk1 and the Cre transgene. (D) Genotyping PCR analysis using tail DNAs from ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice, showing the WT and Min alleles of Apc, WT and flox alleles of Tbk1, and the Cre transgene. (E) Immunoblot analysis of TBK1 and β-Actin in primary IECs and lamina propria lymphocytes (LPL) isolated from the small intestine of the indicated mice. (F) Representative images of tumor development in small intestines of ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice (5-month old) analyzed using stereomicroscope (top) and H&E staining (bottom). (G) Polyp numbers were counted in the small intestine (SI) and large intestine (LI) of ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice (5-month old; n = 6 per genotype). Data are representative of 3 independent experiments. Summary data are mean ± s.e.m. with P values being determined by Kaplan-Meier analysis by the log rank test (A) or unpaired two-tailed Student’s t-test (G). **P < 0.01, ***P < 0.001; ns, not significant.
IEC-specific deletion of TBK1 increases the frequency of TH17 cells in lamina propria
TBK1 deficiency did not alter the growth of intestinal stem cells in organoid, arguing against a cell-intrinsic function of TBK1 in controlling stem cell propagation (Supplementary Fig. 1A). TBK1 deficiency also did not alter polyp growth in tumoroid culture (Supplementary Fig. 1B). These findings suggest an indirect role for TBK1 in regulating intestinal tumorigenesis. Immune and inflammatory factors, including TH17 cells and the TH17 signature cytokine IL-17, contribute to intestinal tumorigenesis and progression7–9,32. Although Tbk1IEC-KO mice did not display overt inflammation in the intestine (Data not shown; also see Figure 3C), their small intestine displayed a markedly higher level of IL-17 expression than that of wildtype mice (Fig. 2A). Consistently, the lamina propria of the Tbk1IEC-KO mice had an increased frequency and absolute number of TH17 cells, but not TH1 or regulatory T (TReg) cells (Fig. 2B–D). The frequency of IFNγ/IL-17 double positive cells was very low in both the wildtype and Tbk1IEC-KO mice (Fig. 2B). The Tbk1IEC-KO mice also had a moderately increased frequency of group 3 innate lymphoid cells (ILC3s) known to produce IL-1733 (Supplementary Fig. 2). TBK1 deletion did not significantly alter the frequency of total CD4 or CD8 T cells in the lamina propria of small intestine and several other lymphoid organs (Supplementary Fig. 3A–D). Thus, the IEC-specific TBK1 has a specific role in regulating TH17 cell abundance in the intestine.
Figure 3.
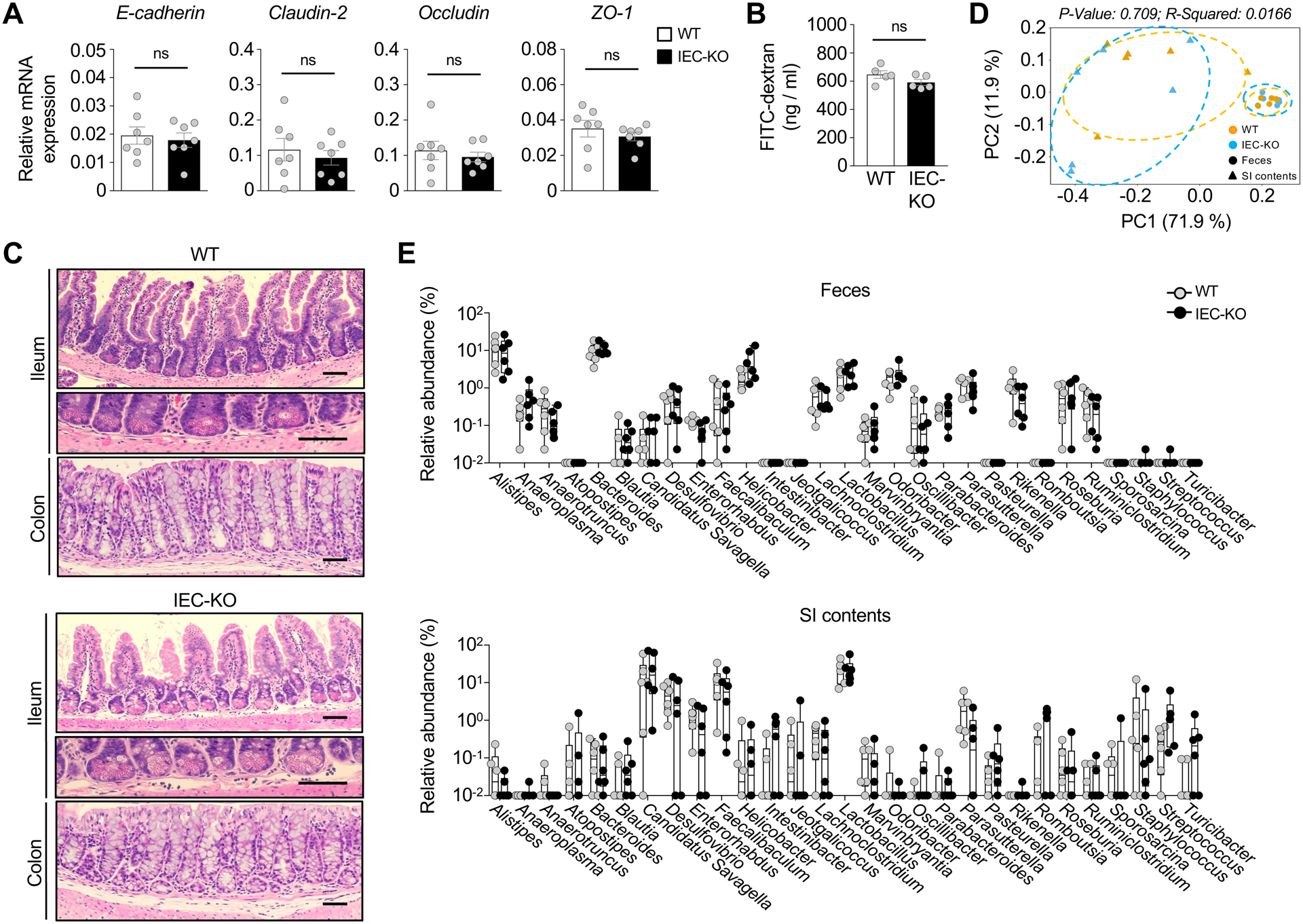
IEC-specific TBK1 ablation does not affect the integrity and barrier function of intestinal epithelia. (A) qRT-PCR analysis of the expression of tight junction-related genes in IECs of Tbk1WT and Tbk1IEC-KO mice (n = 7 per genotype). (B) Intestinal integrity measured based on FITC-dextran levels in serum (n = 5 per genotype). (C) H&E histology analysis of the ileum of small intestine or colon of Tbk1WT and Tbk1IEC-KO mice. Scale bar = 100 μm. (D and E) Beta diversity were visualized by Principal Coordinates Analysis (PCoA; D), and the microbiota composition at genus level (E) were analyzed in feces and small intestine contents of Tbk1WT and Tbk1IEC-KO mice (n = 6 each) by 16S rRNA sequencing analysis. Data are representative of 3 independent experiments, and summary data are mean ± s.e.m. with P values being determined by unpaired two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant.
Figure 2.
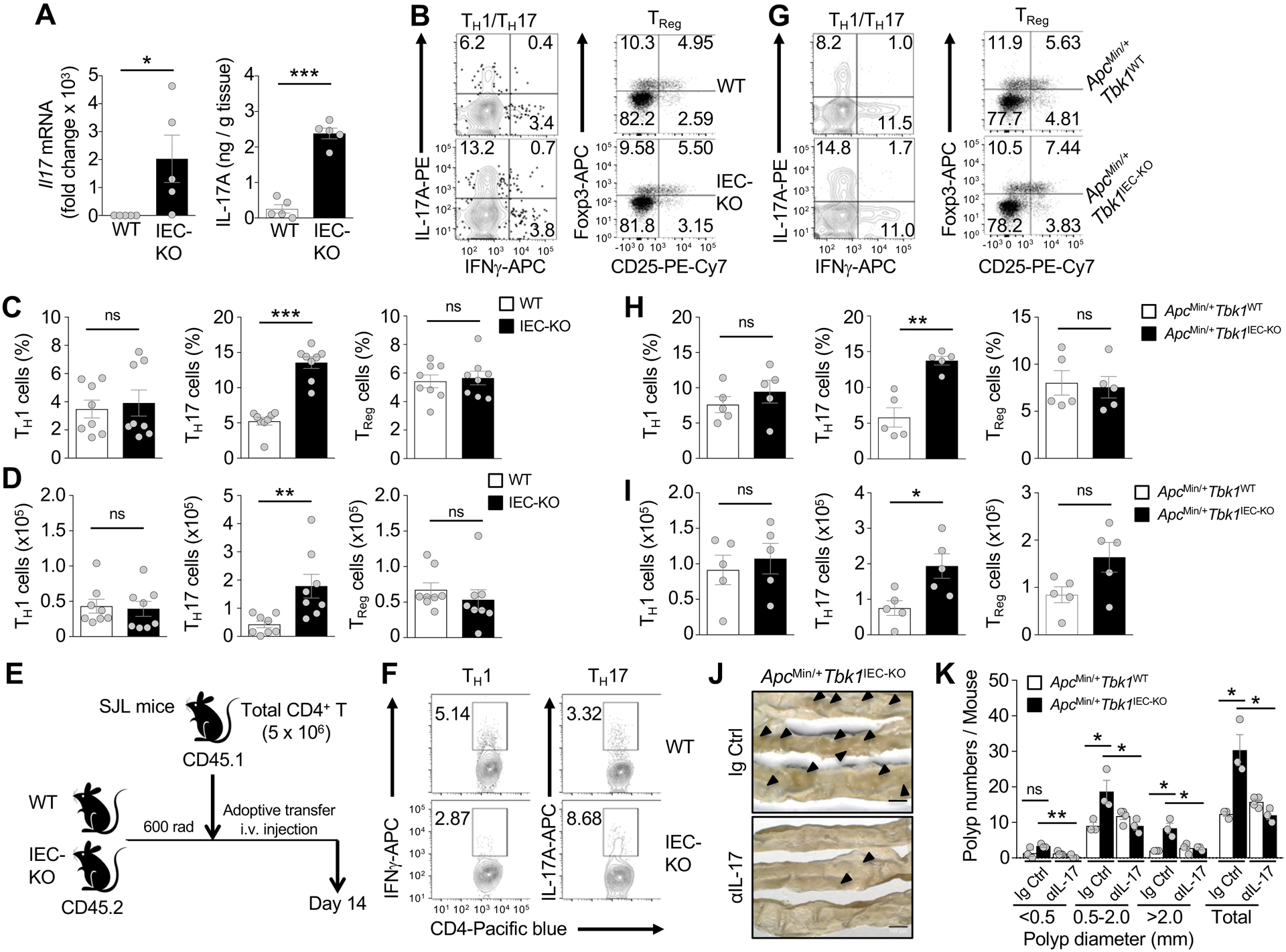
IECs-specific deletion of TBK1 enhances the frequency of TH17 cells in lamina propria. (A) qRT-PCR analysis of Il17 mRNA (left) and ELISA of IL-17A protein (right) levels in whole tissue homogenates of Tbk1WT and Tbk1IEC-KO mice (n = 5 per genotype). (B-D). Flow cytometric analysis of IFNγ-producing TH1 cells, IL-17-producing TH17 cells, and CD25+Foxp3+ T + Reg cells in lamina propria CD4 lymphocyte population of 6-week-old Tbk1WT and Tbk1IEC-KO mice (n = 8 per genotype). Data are presented as a representative flow cytometry images (B) and summary graphs (C, frequency; D, absolute cell numbers; each circle represents a mouse). (E) Schematic of experimental design. WT or Tbk1IEC-KO mice (CD45.2+) were irradiated, and after 3 h, transferred with CD4+ T cells from SJL mice (CD45.1+). (F) Flow cytometric analysis of TH1 and TH17 cells within the CD45.1+ donor CD4+ T cell population of the Tbk1WT and Tbk1IEC-KO recipient mice depicted in E. (G-I) Flow cytometric analysis of TH1 cells, TH17 cells, and TReg cells in the lamina propria CD4 T cell population of 5-month-old ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice (n = 5 per genotype). Data are presented as representative FACS plots (G) and summary graphs (H, frequency; I, absolute cell numbers; each circle represents a mouse). (J and K) Representative intestinal polyp images (J) and quantification of intestinal polyp numbers (K) in 5-month-old ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice following injection of an IL-17 neutralizing antibody or IgG control (n = 3 or more; each circle represents a mouse). Data are representative of 2 (F and J-K) or 3 (A-D and G-I) independent experiments. Summary data are mean ± s.e.m. with P values being determined by unpaired two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant.
To confirm that IEC-specific TBK1 deletion had no effect on T cell development, we adoptively transferred wildtype CD4 T cells derived from B6.SJL mice (CD45.1+) to sublethally irradiated wildtype or Tbk1IEC-KO mice (CD45.2+) (Fig. 2E). Compared to the wildtype recipient mice, the Tbk1IEC-KO recipient mice had a profoundly higher frequency of transferred TH17 cells (CD45.1+) in lamina propria, emphasizing the role of IEC-specific TBK1 in regulating intestinal TH17 cell abundance (Fig. 2F). The Tbk1IEC-KO recipient mice produced a lower frequency of TH1 cells (Fig. 2F), probably due to increased production of TH17 cells.
Analysis of ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice revealed a comparable frequency and absolute number of TH1 cells, but a significant higher frequency and absolute number of TH17 cells in the ApcMin/+ Tbk1IEC-KO mice (Fig. 2G–I). This phenotype was specific for TH17 cells, since the total populations of CD4 or CD8 T cells in lamina propria and other peripheral lymphoid organs were comparable between ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice (Supplementary Fig. 3E–H). Importantly, antibody-mediated IL-17 neutralization significantly reduced the polyp number in ApcMin/+Tbk1WT mice, erasing the differences between the ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice (Fig. 2J and K). These results suggest that TBK1 functions in IECs to control the abundance of TH17 cells, thereby inhibiting tumorigenesis.
TBK1 is dispensable for epithelial barrier function and microbiota regulation
Epithelial barrier function of IECs plays a crucial role in regulating the intestinal homeostasis and inflammation34. However, the TBK1-deficient and wildtype control IECs had comparable level of expression of several major tight junction proteins, including E-cadherin, Claudin-2, Occludin, and Zonula occludens-1 (ZO-1) (Fig. 3A). Intestinal barrier permeability assay also did not reveal a significant difference between the wildtype and TBK1-deficient mice (Fig. 3B). Consistently, histological analysis did not show obvious abnormalities of the Tbk1IEC-KO mice in the morphology of the villi and crypts of the small intestine (Fig. 3C).
Because of the role of microbiota in TH17 cell regulation6, we next examined the effect of TBK1 deficiency on microbiota compositions by performing 16S rRNA sequencing analysis. Principal coordinate analysis (PCoA) did not reveal significant differences in microbial communities between Tbk1IEC-KO and WT control mice, although paired samples of feces and small intestinal contents were distant from each other (Fig. 3D). The wildtype and Tbk1IEC-KO mice also did not show significant differences in any of the genera, including the segmented filamentous bacteria (SFB) Candidatus savagella known to stimulate TH17 cell differentiation (Fig. 3E). These results suggest a dispensable role for TBK1 in regulating epithelial barrier function and microbiota composition.
IEC-specific TBK1 controls IL-1β expression in the intestine
In an in vitro TH17 differentiation assay, we found that the supernatant of Tbk1IEC-KO intestinal culture displayed a much stronger TH17-stimulatory activity than that of the wildtype intestinal culture (Fig. 4A). Interestingly, the intestinal tissue of Tbk1IEC-KO mice had increased expression of IL-1β and IL-23 (Fig. 4B), proinflammatory cytokines known to promote TH17 cell differentiation and expansion10. The expression of intestinal IL-22 was also increased in the Tbk1IEC-KO mice (Fig. 4B), reflecting the enhanced production of TH17 cells known to express IL-2235. The TBK1 deficiency did not significantly alter the expression of two other cytokines, IL-6 and IL-21 (Fig. 4B). Increase IL-1β expression in the small intestine, as well as large intestine, of Tbk1IEC-KO mice was also detected at the protein level by ELISA (Fig. 4C and 4D). Antibody-mediated IL-1β neutralization largely blocked the hyper-induction of TH17 cells by the culture supernatant of the Tbk1IEC-KO small intestine (Fig. 4E and 4F). Furthermore, in vivo blockade of IL-1β using a neutralizing antibody drastically reduced the frequency of TH17 cells in the small intestine of ApcMin/+Tbk1IEC-KO mice, thereby erasing the differences between ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice (Fig. 4G). The antibody-mediated IL-1β blockade also reduced the number of polyps in the ApcMin/+Tbk1IEC-KO mice (Fig. 4H). These results suggest that TBK1-mediated TH17 cell regulation and tumorigenesis involves control of IL-1β expression in the intestine.
Figure 4.
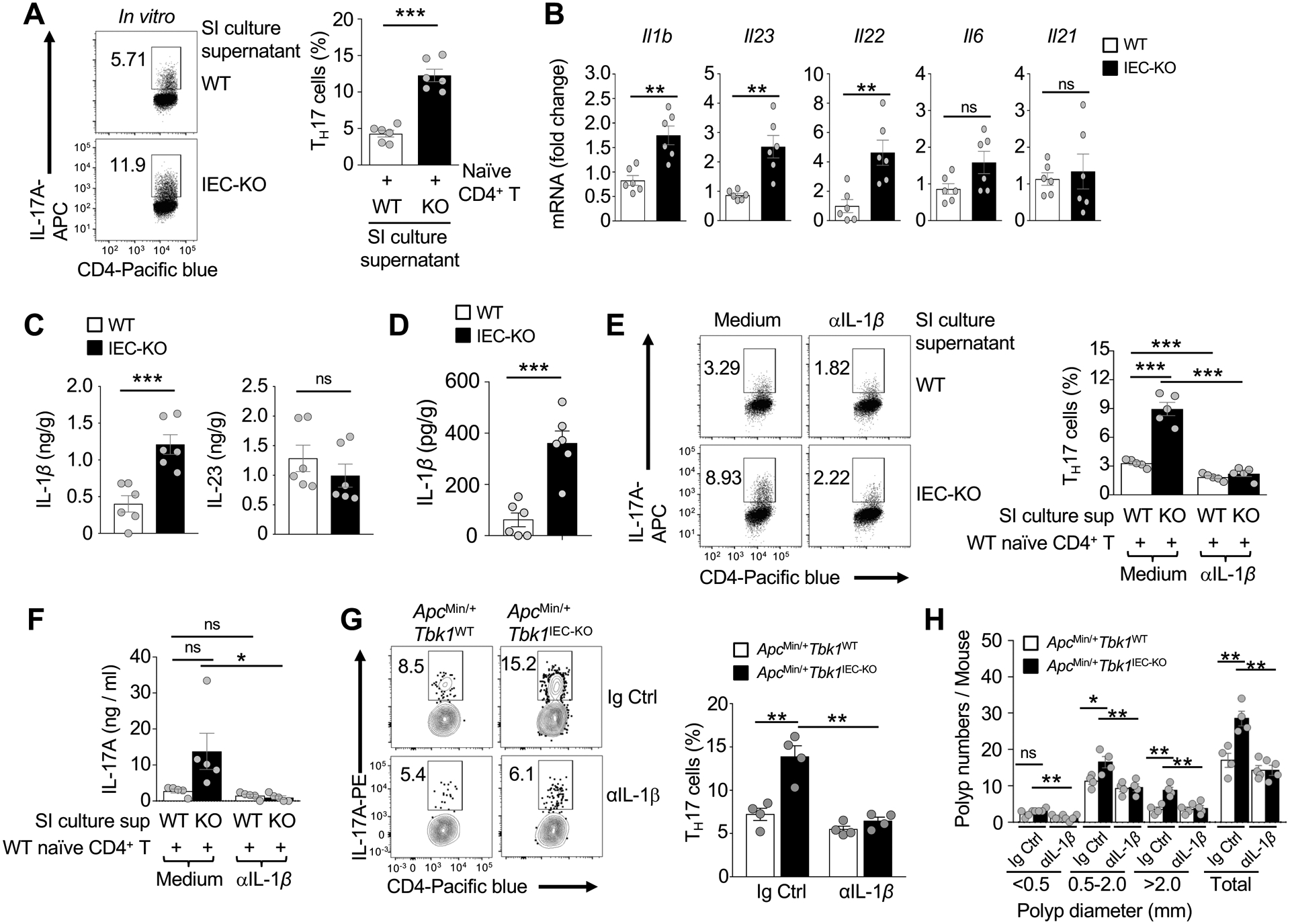
Upregulated expression of IL-1β in the intestine of Tbk1IEC-KO mice contributes to aberrant TH17 cell generation. (A) Flow cytometric analysis of TH17 cells differentiated in vitro by co-cultivating WT splenic naïve CD4+ T cells (for 5 days) with supernatants derived from 24-hour culture of small intestinal tissues of Tbk1WT and Tbk1IEC-KO mice (n = 6 per genotype). Data are presented as a representative FACS plot (left) and summary graph (right; each circle represents a mouse). (B) qRT-PCR analysis of the indicated cytokine mRNAs in the small intestine of Tbk1WT and Tbk1IEC-KO mice (n = 6 per genotype). (C and D) ELISA of IL-1β and IL-23 protein levels in the homogenates of small intestine (C) and large intestine (D) tissues of Tbk1WT and Tbk1IEC-KO mice (n = 6 per genotype). (E) Flow cytometric analysis of TH17 cells differentiated in vitro by co-cultivating WT splenic naïve CD4+ T cells (for 5 days) with supernatants of 24-hour small intestine cultures of Tbk1WT and Tbk1IEC-KO mice (n = 5 per genotype), in the presence of either PBS control or an IL-1β neutralization antibody (5 μg/ml). Data are presented as a representative FACS plot (left) and summary graph (right; each circle represents a mouse). (F) ELISA of IL-17A in supernatants of day 5 co-cultivation of naïve CD4+ T cells and small intestine (SI) tissue culture supernatant, in the presence of PBS or anti-IL-1β. (G and H) Flow cytometric analysis of IL-17A-expressing TH17 cells in the small intestine (G) and quantification of intestinal polyp numbers (H) of 5-month old ApcMin/+Tbk1WT and ApcMin/+Tbk1IEC-KO mice following repeated injection (i.p.) with an IL-1β neutralizing antibody or an IgG isotype control (each circle represents a mouse). Data are representative of 2 (G-H) or 3 (A-F) independent experiments. Summary data are mean ± s.e.m. with P values being determined by unpaired two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant.
TBK1 regulates the crosstalk between IECs and macrophages via promoting IL-1β production
Innate immune cells are a major source of IL-1β production in the intestine36, 37. The lamina propria of Tbk1IEC-KO and wildtype control mice did not have significant differences in the frequency of DC, macrophage, or neutrophil populations (Supplementary Fig. 4). However, the lamina propria macrophages, although not DCs, from Tbk1IEC-KO mice were profoundly more active than those from wildtype mice in promoting TH17 cell differentiation (Fig. 5A and B). In line with these results, the small intestinal lamina propria macrophages, as well as colon macrophages, of Tbk1IEC-KO mice expressed a significantly higher level of IL-1β compared to those of wildtype mice (Fig. 5C and 5D).
Figure 5.
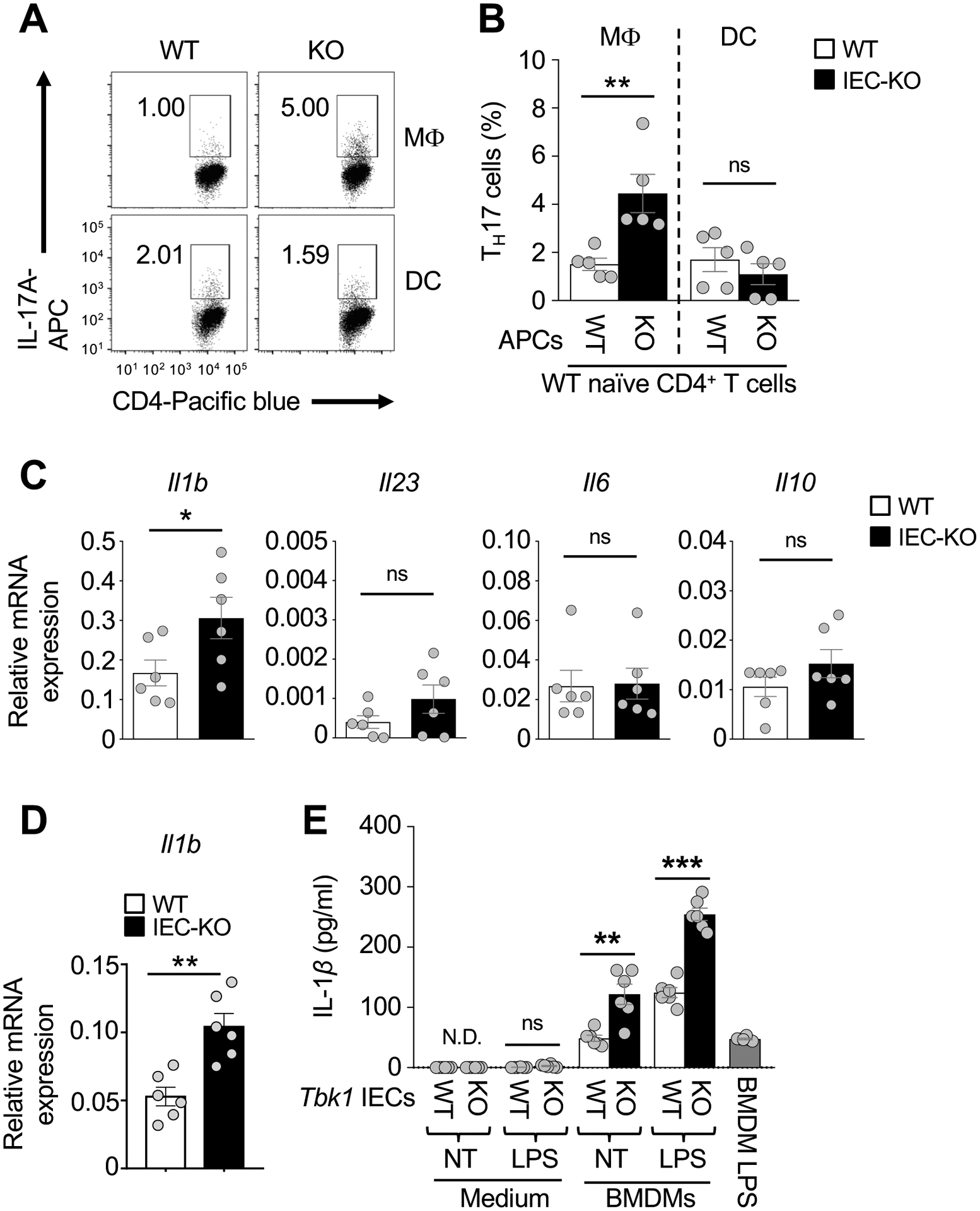
IEC-specific deletion of TBK1 promotes IL-1β expression in macrophages and induction of TH17 cell differentiation. (A and B) Flow cytometric analysis of TH17 cells differentiated (for 5 days) in vitro by stimulating WT splenic naïve CD4+ T cells (CD4+CD25−CD44−) with anti-CD3, anti-CD28 and 10-fold low concentration of IL-6 (2 ng/ml) and TGF-β (250 pg/ml) in the presence of intestinal macrophages (MΦ; CD45+MHCIIhiCD11c+CD64+) or DCs (CD45+MHCIIhiCD11c+CD64−) sorted from Tbk1WT and Tbk1IEC-KO mice (n = 5 each). Data are presented as a FACS plot (A) and summary graph (B, each circle represents a mouse). (C and D) qRT-PCR analysis of Il1b and the other indicated mRNAs in freshly sorted macrophages of the small intestine (C) or large intestine (D) of Tbk1WT and Tbk1IEC-KO mice (n = 6 each). (E) ELISA of IL-1β in the supernatants of wildtype BMDMs co-cultivated for 24 hours with Tbk1WT (WT) or Tbk1IEC-KO (KO) IECs that had been either not treated (NT) or stimulated with LPS (1 μg/ml) for 1 hour. A sample of LPS-stimulated (1h) BMDMs was included as a control. Data are representative of 3 independent experiments. Summary data are mean ± s.e.m. with P values being determined by unpaired two-tailed Student’s t-test. **P < 0.01, ***P < 0.001; ns, not significant.
Based on the results described above, we surmised that TBK1 might function in IECs to regulate the crosstalk between IECs and macrophages. To examine this possibility, we co-cultivated bone marrow derived macrophages (BMDMs) with either wildtype or TBK1-deficient IECs in the absence or presence of TLR stimulations. Compared to the wildtype IECs, the TBK1-deficient IECs displayed a much stronger activity in IL-1β induction (Fig. 5E). Collectively, these results suggest that TBK1 functions in IECs to negatively regulate IL-1β expression in macrophages.
TBK1 regulates MT1 expression in IECs
RNA sequencing analysis using freshly isolated wildtype and TBK1-deficient IECs identified several up- or down-regulated genes in the TBK1-deficient IECs (Fig. 6A and supplementary Fig. 5). qRT-PCR confirmed the significant downregulation of Tetraspanin 32 (Tspan32) and upregulation of Mt1 in the TBK1-deficient IECs, although the differential expression of the other genes did not reach statistical significance (Fig. 6B). MT1 was also the most abundantly expressed and strikingly upregulated gene in TBK1-deficient IECs (Fig. 6B,C and Supplementary Fig. 5). Similar results were obtained with IECs isolated from the large intestine (Fig. 6D and E).
Figure 6.
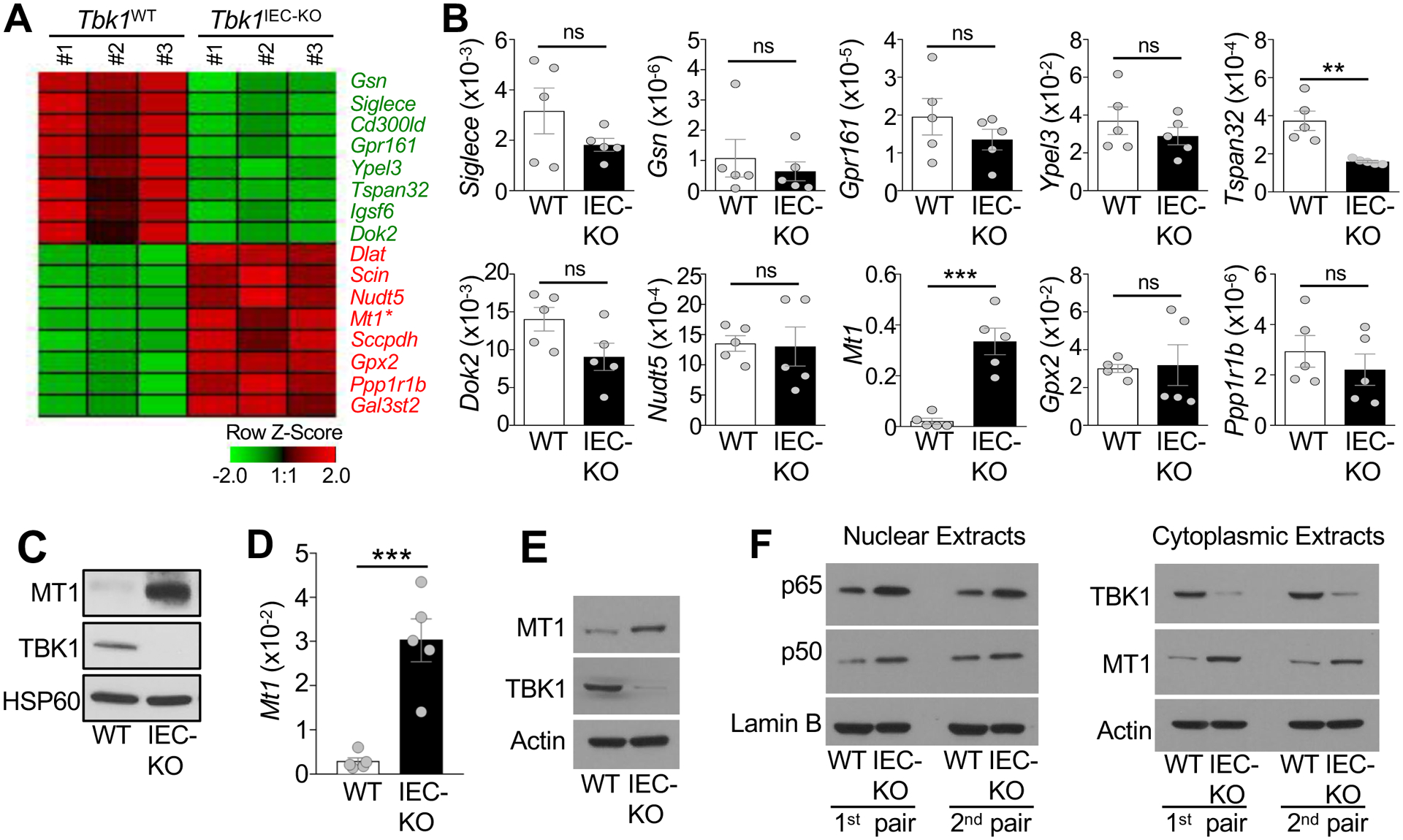
TBK1 controls the expression of Mt1 in IECs. (A) A Heat map of highly variable genes detected in RNA sequencing analysis using primary IECs from Tbk1WT and Tbk1IEC-KO mice (n = 3 each). (B) qRT-PCR analysis of the indicated mRNAs in freshly isolated IECs from Tbk1WT and Tbk1IEC-KO mice (each circle represents a mouse). Data are presented as values relative to Actb. (C) Immunoblot analysis of the indicated proteins in small intestinal IECs of Tbk1WT and Tbk1IEC-KO mice. (D and E) qRT-PCR analysis of Mt1 mRNA expression relative to Actb (D) and immunoblot analysis of MT1 protein expression (E) in freshly isolated IECs derived from the large intestines of Tbk1WT and Tbk1IEC-KO mice. (F) Immunoblot analysis of the indicated proteins in the nuclear or cytoplasmic extracts of freshly isolated small intestine IECs of Tbk1WT and Tbk1IEC-KO mice. Data are representative of 1 (A), or 2 (D-F), or 3 (B and C) independent experiments. Summary data are mean ± s.e.m. with P values being determined by unpaired two-tailed Student’s t-test. **P < 0.01, ***P < 0.001; ns, not significant.
NF-κB signaling pathway is crucial for maintaining the expression of MT138. Notably, TBK1 has been implicated as a negative regulator of NF-κB signaling16, 39, 40, although the role of TBK1 in NF-κB regulation in primary IECs is unknown. Interestingly, we found that TBK1 deficiency profoundly increased the level of nuclear NF-κB p65 and p50, suggesting hyperactivation of the NF-κB pathway under in vivo conditions (Fig. 6F). These studies suggest that TBK1 controls the expression of MT1 in IECs, which likely involves regulation of NF-κB signaling.
MT1 regulates intestinal TH17 cell abundance and tumorigenesis
MT1 and MT2 are immune modulators implicated in the pathogenesis of IBD41–44. MT1,2 can be released to extracellular compartments and provide a danger signal for stimulating inflammation41, 44. Remarkably, we found that Mt−/− mice had a drastically reduced frequency of TH17 cells in the intestinal lamina propria (Fig. 7A and B). This phenotype was not due to T cell-intrinsic function of MT, since the MT-deficient and wildtype CD4 T cells displayed similar level of TH17 cell differentiation in vitro (Fig. 7C). Contrary to the Tbk1IEC-KO mice, the MT-deficient mice had significantly reduced Il1b expression in the intestine compared to the wildtype control mice (Fig. 7D). Consistently, the MT-deficient IECs had reduced function to stimulate IL-1β expression in macrophages in an in vitro cocultivation assay (Fig. 7E). These results suggest that controlling MT1 expression in the IECs is an important part of the mechanism by which TBK1 regulates intestinal immune homeostasis.
Figure 7.
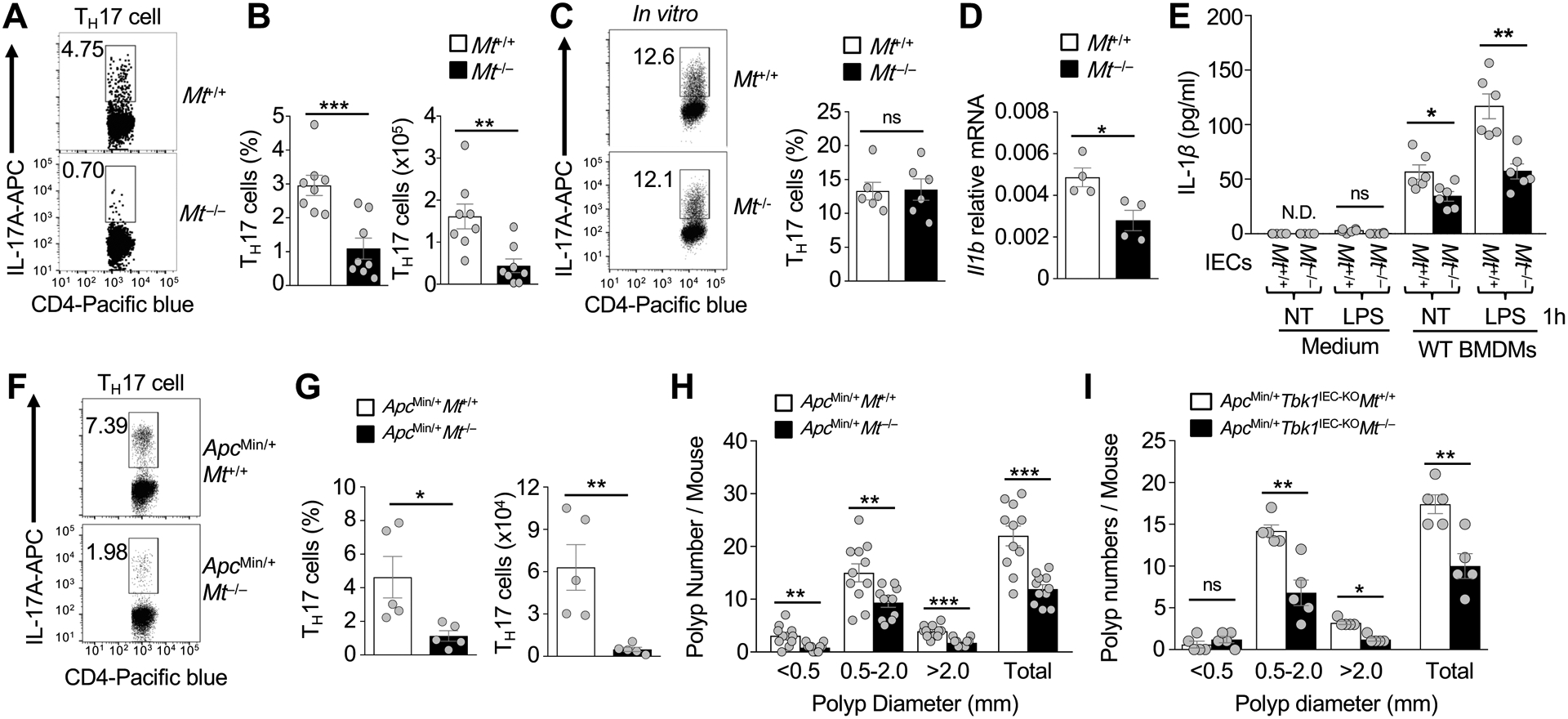
MT1 regulates intestinal TH17 cell abundance and tumorigenesis. (A and B) Flow cytometric analysis of IL-17-producing TH17 cells in the CD3+CD4+ lymphocyte population of lamina propria of 6-week-old Mt+/+ or Mt−/− mice (n = 8 each). Data are presented as a representative FACS plot (A) and summary graph (B, each circle represents a mouse). (C) Flow cytometric analysis of TH17 cells differentiated in vitro from splenic naïve CD4+ T cells of Mt+/+ or Mt−/− mice (n = 5 each). Data are presented as a representative FACS plot (left) and summary graph (right, each circle represents a mouse). (D) qRT-PCR analysis of Il1b expression in the small intestine tissue of Mt+/+ or Mt−/− mice. (E) ELISA of IL-1β in the supernatants of wildtype BMDMs co-cultivated for 24 hours with Mt+/+ or Mt−/− IECs that had been either not treated (NT) or stimulated with LPS (1 μg/ml). (F and G) Flow cytometric analysis of IL-17-producing TH17 cells in the CD3+CD4+ lymphocyte population of lamina propria of 5-month-old ApcMin/+Mt+/+ and ApcMin/+Mt−/− mice (n = 5 each). Data are presented as a representative FACS plot (F) and summary graph (G, each circle represents a mouse). (H and I) Polyp numbers of whole intestine were counted in 5-month-old of ApcMin/+Mt+/+ or ApcMin/+Mt−/− mice (H, n = 11 each) and in ApcMin/+Tbk1IEC-KOMt+/+ or ApcMin/+Tbk1IEC-KOMt−/− mice (I, n = 5 each). Data are representative of 2 (C-E) or 3 (A, B, and F-I) independent experiments. Summary data are mean ± s.e.m. with P values being determined by unpaired two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant.
To examine whether MT1 also regulates intestinal tumorigenesis, we crossed the Mt−/− mice with ApcMin/+ mice. As seen with the Mt−/− mice, the ApcMin/+Mt−/− mice had drastically reduced TH17 cells compared to ApcMin/+Mt+/+ mice (Fig. 7F and G). The ApcMin/+Mt−/− mice also had a significantly lower number of polyps than the ApcMin/+Mt+/+ mice (Fig. 7H). Consistently, TCGA database analysis revealed significant correlation of high expression of Mt1, as well as Il17, with reduced patient survival in colon cancer (Supplementary Fig. 6). Furthermore, MT deletion significantly reduced polyp burden in ApcMin/+Tbk1IEC-KO mice (Fig. 7I). These findings establish MT1,2 as crucial regulators of intestinal TH17 cell abundance and tumorigenesis and suggest an important role for TBK1-mediated MT1 regulation in controlling TH17 cell differentiation and tumorigenesis in the intestine.
Discussion
By generating IEC-conditional TBK1 KO mice, we demonstrated a novel function of TBK1 in the regulation of intestinal immune homeostasis and tumorigenesis. Although the Tbk1IEC-KO mice did not develop histological features of intestinal inflammation, they displayed low-grade inflammation characterized by elevated IL-1β production and had increased adenoma formation when crossed with the ApcMin/+ mice. Our data suggest that TBK1 functions in IECs to regulate the expression of immune and inflammatory modulators, particularly MT1.
IECs form a physical barrier to prevent invasion by microorganisms as well as sense microbial signals to maintain intestinal immune homeostasis11. The signaling network that regulates the dynamic functions of IECs is still poorly understood. Our data suggest that TBK1 is dispensable for epithelial cell survival, epithelial barrier function, or microbiota maintenance. However, TBK1 plays an important role in controlling the crosstalk between IECs and innate immune cells, particularly induction of IL-1β expression in lamina propria macrophages. TBK1 deletion in IECs causes hyperproduction of intestinal IL-1β, which in turn promotes generation or expansion of TH17 subset of proinflammatory T cells.
TBK1 is important for the survival and proliferation of some Kras-dependent cancer cell lines, implicating TBK1 as an oncogenic kinase27–29. Interestingly, our animal model study demonstrated a negative role for TBK1 in regulating intestinal tumorigenesis, and our analysis of TCGA database revealed the association of TBK1 high expression with improved survival of colorectal cancer patients. The tumor-suppressing function of TBK1 does not involve a cell-intrinsic mechanism, since organoid culture experiments did not reveal an important function of TBK1 in regulating the propagation of intestinal stem cells or polyp cells. Instead, TBK1 indirectly regulates intestinal tumorigenesis through controlling the abundance of TH17 cells. We obtained strong evidence that aberrant production of the TH17 cell signature cytokine IL-17 contributes to the elevated adenoma formation in ApcMin/+Tbk1IEC-KO mice, which is in line with previous finding that IL-17 promotes intestinal tumorigenesis7–9. Our data suggest that TBK1 functions in IECs to regulate IL-1β expression in lamina propria macrophages, which in turn facilitates the differentiation of TH17 cells.
By RNA sequencing, we identified Mt1 as a major gene regulated by the TBK1 signaling network in IECs. MT1 and its close homolog, MT2, have been implicated as important regulators of intestinal inflammation, although the underlying mechanism of MT regulation and function is poorly understood41–44. Our data suggest that TBK1 controls the expression of MT1 in IECs. Hyperproduction of MT1 in TBK1-deficient IECs contributes to aberrant expression of IL-1β in macrophages and, thus, increased abundance of intestinal TH17 cells, which in turn promotes intestinal tumorigenesis. Mechanistically, TBK1 controlled the activation of NF-κB, a crucial transcription factor mediating Mt1 gene expression38. We obtained genetic evidence that MT1 and MT2 are required for the maintenance of intestinal TH17 cells as well as for intestinal tumorigenesis in the ApcMin/+ mice. MT deletion potently inhibited the adenoma formation in ApcMin/+Tbk1IEC-KO mice, suggesting that MT1 regulation serves as a crucial mechanism by which TBK1 controls TH17 cell abundance and tumorigenesis. Consistent with these findings with animal models, our analysis of TCGA database revealed the association of high expression of MT1, as well as IL-17, with poor survival of colorectal cancer patients. As such, our work establishes TBK1 as a pivotal signaling factor that functions in IECs to mediate regulation of intestinal immune homeostasis and tumor formation.
Supplementary Material
What you need to know:
Background and Context:
Intestinal epithelial cells (IECs) regulate intestinal immune cells, particularly development of T-helper 17 (Th17) cells. Alterations in this process can lead to intestinal inflammation and tumorigenesis, via unknown mechanisms. TANK-binding kinase 1 (TBK1) is expressed by IECs and cells in the innate immune system.
New Findings:
Expression of TBK1 by IECs suppressed expression of MT1 and prevented expression of IL1B by macrophages and differentiation of Th17 cells, to prevent inflammation and tumorigenesis.
Limitations:
This study was performed in mice and tissues and cells. Further studies are needed in humans.
Impact:
Strategies to block this pathway might be developed for colorectal tumorigenesis.
Lay Summary:
The authors identified a protein produced by intestinal cells that regulates the intestinal immune response to prevent inflammation and cancer.
Acknowledgments
We thank personnel from the flow cytometry, sequencing and microarray, and animal facility of the CCSG-shared resources at The MD Anderson Cancer Center.
Grant Support: This study was supported by a grant from the National Institutes of Health (AI057555) and partially supported by a seed fund from the Center for Inflammation and Cancer at the MD Anderson Cancer Center. A.M. was supported by a Ruth L. Kirschstein National Research Service Award (5 T32 CA009598-24) from the National Institutes of Health.
Abbreviations:
- IEC
intestinal epithelial cell
- MT1
metallothionein 1
- IBD
inflammatory bowel disease
- TH17
T helper 17
- IL-17
interleukin-17
- SFB
segmented filamentous bacteria
- DC
dendritic cell
- TBK1
TANK binding kinase 1
- H&E
hematoxylin-eosin; bone marrow-derived macrophage (BMDM)
- APC
adenomatous polyposis coli
- ZO-1
Zonula occludens-1
- FITC
fluorescein isothiocyanate
- qRT-PCR
quantitative RT-PCR
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors disclose no conflicts of interest relevant to this manuscript.
References
- 1.Chassaing B, Kumar M, Baker MT, et al. Mammalian gut immunity. Biomed J 2014;37:246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Z, Cao AT, Cong Y. Microbiota regulation of inflammatory bowel disease and colorectal cancer. Semin Cancer Biol 2013;23:543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu W, Chen F, Liu Z, et al. Microbiota-specific Th17 Cells: Yin and Yang in Regulation of Inflammatory Bowel Disease. Inflamm Bowel Dis 2016;22:1473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung H, Pamp SJ, Hill JA, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 2012;149:1578–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lecuyer E, Rakotobe S, Lengline-Garnier H, et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 2014;40:608–20. [DOI] [PubMed] [Google Scholar]
- 6.Pandiyan P, Bhaskaran N, Zou M, et al. Microbiome Dependent Regulation of Tregs and Th17 Cells in Mucosa. Front Immunol 2019;10:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chae WJ, Gibson TF, Zelterman D, et al. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A 2010;107:5540–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Kim MK, Di Caro G, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014;41:1052–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurtado CG, Wan F, Housseau F, et al. Roles for Interleukin 17 and Adaptive Immunity in Pathogenesis of Colorectal Cancer. Gastroenterology 2018;155:1706–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chewning JH, Weaver CT. Development and survival of Th17 cells within the intestines: the influence of microbiome- and diet-derived signals. J Immunol 2014;193:4769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 2014;14:141–53. [DOI] [PubMed] [Google Scholar]
- 12.Atarashi K, Tanoue T, Ando M, et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015;163:367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ladinsky MS, Araujo LP, Zhang X, et al. Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science 2019;363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiscott J Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem 2007;282:15325–15329. [DOI] [PubMed] [Google Scholar]
- 15.Clark K, Plater L, Peggie M, et al. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J. Biol. Chem 2009;284:14136–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin J, Xiao Y, Chang JH, et al. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat Immunol. 2012;13:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu J, Zhou X, Chang M, et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat. Commun 2015;6:6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015;347:aaa2630. [DOI] [PubMed] [Google Scholar]
- 19.Xiao Y, Zou Q, Xie X, et al. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J Exp Med 2017;214:1493–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi JH, Xie X, Sun SC. TBK1 as a regulator of autoimmunity and antitumor immunity. Cell Mol Immunol 2018;15:743–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldman M, Craft B, Hastie M, et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 2019:326470. [Google Scholar]
- 22.Boivin GP, Washington K, Yang K, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 2003;124:762–777. [DOI] [PubMed] [Google Scholar]
- 23.Ajami NJ, Cope JL, Wong MC, et al. Impact of Oral Fidaxomicin Administration on the Intestinal Microbiota and Susceptibility to Clostridium difficile Colonization in Mice. Antimicrob Agents Chemother 2018;62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 2012;6:1621–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin J, Xie X, Xiao Y, et al. Epigenetic regulation of the expression of Il12 and Il23 and autoimmune inflammation by the deubiquitinase Trabid. Nat Immunol 2016;17:259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee YS, Kim TY, Kim Y, et al. Microbiota-Derived Lactate Accelerates Intestinal Stem-Cell-Mediated Epithelial Development. Cell Host Microbe 2018;24:833–846 e6. [DOI] [PubMed] [Google Scholar]
- 27.Chien Y, Kim S, Bumeister R, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 2006;127:157–170. [DOI] [PubMed] [Google Scholar]
- 28.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ou YH, Torres M, Ram R, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol. Cell 2011;41:458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moser AR, Luongo C, Gould KA, et al. ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur. J. Cancer 1995;A(7–8):1061–1064. [DOI] [PubMed] [Google Scholar]
- 31.Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology 2009;136:780–798. [DOI] [PubMed] [Google Scholar]
- 32.Terzic J, Grivennikov S, Karin E, et al. Inflammation and colon cancer. Gastroenterology 2010;138:2101–2114 e5. [DOI] [PubMed] [Google Scholar]
- 33.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat Rev Immunol 2013;13:75–87. [DOI] [PubMed] [Google Scholar]
- 34.Schulzke JD, Ploeger S, Amasheh M, et al. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci 2009;1165:294–300. [DOI] [PubMed] [Google Scholar]
- 35.Dong C TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat. Rev. Immunol 2008;8:337–348. [DOI] [PubMed] [Google Scholar]
- 36.McAlindon ME, Hawkey CJ, Mahida YR. Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 1998;42:214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coccia M, Harrison OJ, Schiering C, et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med 2012;209:1595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng Z, Peng L, Fan Y, et al. A critical role for IkappaB kinase beta in metallothionein-1 expression and protection against arsenic toxicity. J Biol Chem 2007;282:21487–96. [DOI] [PubMed] [Google Scholar]
- 39.Clark K, Peggie M, Plater L, et al. Novel cross-talk within the IKK family controls innate immunity. Biochem. J 2011;434:93–104. [DOI] [PubMed] [Google Scholar]
- 40.Zhao P, Wong KI, Sun X, et al. TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell 2018;172:731–743 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devisscher L, Hindryckx P, Lynes MA, et al. Role of metallothioneins as danger signals in the pathogenesis of colitis. J Pathol 2014;233:89–100. [DOI] [PubMed] [Google Scholar]
- 42.Waeytens A, De Vos M, Laukens D. Evidence for a potential role of metallothioneins in inflammatory bowel diseases. Mediators Inflamm 2009;2009:729172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devisscher L, Hindryckx P, Olievier K, et al. Inverse correlation between metallothioneins and hypoxia-inducible factor 1 alpha in colonocytes and experimental colitis. Biochem Biophys Res Commun 2011;416:307–12. [DOI] [PubMed] [Google Scholar]
- 44.Lynes MA, Zaffuto K, Unfricht DW, et al. The physiological roles of extracellular metallothionein. Exp Biol Med (Maywood) 2006;231:1548–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.