

Abstract
Background & Aims:
It is not clear how pancreatic cancer stem cells (CSCs) are regulated, resulting in ineffective treatments for pancreatic cancer. PAF1, a RNA polymerase II-associated factor 1 complex (PAF1C) component, maintains pluripotency of stem cells, by unclear mechanisms, and is a marker of CSCs. We investigated mechanisms by which PAF1 maintains CSCs and contributes to development of pancreatic tumors.
Methods:
Pancreatic cancer cell lines were engineered to knockdown PAF1 using inducible small hairpin RNAs. These cells were grown as orthotopic tumors in athymic nude mice and PAF1 knockdown was induced by administration of doxycycline in drinking water. Tumor growth and metastasis were monitored via IVIS imaging. CSCs were isolated from pancreatic cancer cell populations using flow cytometry and characterized by tumor sphere formation, tumor formation in nude mice, and expression of CSC markers. Isolated CSCs were depleted of PAF1 using the CRISPR/Cas9 system. PAF1-regulated genes in CSCs were identified via RNA-seq and PCR array analyses of cells with PAF1 knockdown. Proteins that interact with PAF1 in CSCs were identified by immunoprecipitations and mass spectrometry. We performed chromatin immunoprecipitation sequencing of CSCs to confirm the binding of the PAF1 sub-complex to target genes.
Results:
Pancreatic cancer cells depleted of PAF1 formed smaller and fewer tumor spheres in culture and orthotopic tumors and metastases in mice. Isolated CSCs depleted of PAF1 downregulated markers of self-renewal (NANOG, SOX9, and BETA-CATENIN), of CSCs (CD44v6, and ALDH1), and the metastasis-associated gene signature, compared to CSCs without knockdown of PAF1. The role of PAF1 in CSC maintenance was independent of its RNA polymerase II-associated factor 1 complex component identity. We identified DDX3 and PHF5A as proteins that interact with PAF1 in CSCs and demonstrated that the PAF1-PHF5A-DDX3 sub-complex bound to the promoter region of Nanog, whose product regulates genes that control stemness. Levels of the PAF1-DDX3 and PAF1-PHF5A were increased and co-localized in human pancreatic tumor specimens, human pancreatic tumor-derived organoids, and organoids derived from tumors of KPC mice, compared with controls. Binding of DDX3 and PAF1 to the Nanog promoter, and the self-renewal capacity of CSCs, were decreased in cells incubated with the DDX3 inhibitor RK-33. CSCs depleted of PAF1 downregulated genes that regulate stem cell features (Flot2, Taz, Epcam, Erbb2, Foxp1, Abcc5, Ddr1, Muc1, Pecam1, Notch3, Aldh1a3, Foxa2, Plat, and Lif).
Conclusions:
In pancreatic CSCs, PAF1 interacts with DDX3 and PHF5A to regulate expression of NANOG and other genes that regulate stemness. Knockdown of PAF1 reduces the ability of orthotopic pancreatic tumors to develop and progress in mice and their numbers of CSCs. Strategies to target the PAF1-PHF5A-DDX3 complex might be developed to slow or inhibit progression of pancreatic cancer.
Keywords: mouse model, pancreatic ductal adenocarcinoma, side population, transcription
Graphical Abstract
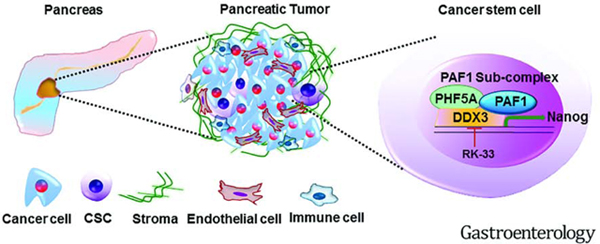
Introduction
Pancreatic cancer (PC) remains a highly aggressive disease with the lowest 5-year survival rate among cancers at only 9%1. The current standard treatment options are FOLFIRINOX and nab-paclitaxel-gemcitabine, which is effective in about 10–30% of cases2. The vast majority of cancer recurrence is believed to be driven by sub-populations of tumor cells with ‘stem-like’ properties and potential for tumor re-growth, which are left unharmed by multidrug chemotherapy regimens. These cancer cells with ‘stem-like’ properties are known as cancer stem cells (CSCs). The aggressiveness of PC, its resistance to standard chemotherapy, and its ability to metastasize have been attributed in part to a lack of effective strategies to target pancreatic CSCs3, 4. Several subpopulations of PC cells have been shown to be responsible for driving tumorigenesis and mediating drug resistance, and are characterized by the expression of CD24+/CD44+/ESA+, CD133, cMet, ALDH, DCLK1, Musashi, PAF1, and RORγ 5, 6. CSCs reactivate embryonic features and overexpress genes associated with self-renewal and pluripotency, such as Oct4, Sox2, Nanog, and C-Myc, particularly in more aggressive, poorly differentiated tumors7, 8. Despite past efforts, effective strategies of targeting CSCs are still lacking due to inadequate understanding of how these CSCs are maintained within the tumor9. Our previous work demonstrated that RNA polymerase II-associated factor 1 (PAF1), or pancreatic differentiation 2 (PD2), is a novel CSC marker that has been attributed to drug resistance and metastasis in PC6, 10. PAF1 is a core component of human RNA polymerase II-associated factor 1 complex (PAF1C), which along with 4 other components (LEO1, CTR9, CDC73, and SKI8) recruits RNA polymerase II for transcriptional elongation11, 12. Previous evidence suggests it plays a role in the regulation of cell cycle progression, modulation of chromatin architecture, and pancreatic acinar to ductal metaplasia13–15. The first indication of a function for PAF1 in stem cells was from studies in mouse embryonic stem cells (ESCs), showing PAF1 interacts with pluripotency master regulator Oct3/4, thereby facilitating self-renewal potential16, 17. The recent identification of functions of PAF1 in CSCs, including pancreatic and ovarian CSCs6, 18, further supports this notion. In addition, PAF1 also regulates RNA polymerase II pause release from stem cell loci19–21. Because maintenance mechanisms for CSCs remain obscure, it is important to understand the factors that support these cells, especially in PC.
We sought to decipher the mechanism by which PAF1 mediates pancreatic CSC maintenance with the hope of uncovering strategies to target CSCs. In this study, we established that PAF1 is critical for pancreatic tumorigenesis and identified a novel PAF1-PHF5A-DDX3 sub-complex, which could be targeted using a specific DDX3 inhibitor, RK-33, with minimal toxicity to normal pancreatic cells.
Materials and Methods
Cell culture
Human PC cell lines SW1990, CD18, Capan1, and the human pancreatic ductal cell line HPDE were obtained from American type culture collection (Manassas, VA, USA). SW1990, CD18, and Capan1 cells were cultured in DMEM media (HyClone Laboratories, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St Louis, MO, USA) and 1% penicillin-streptomycin solution (Sigma). HPDE was cultured in human keratinocyte serum-free media supplemented with epidermal growth factor 1–53 (EGF 1–53) and bovine pituitary extract (BPE) (Gibco). Normal human fibroblast cell line 9–26 NP was derived from surgically obtained cancer-associated normal tissue. Briefly, the tissue was finely chopped and digested with Liberase (Roche), and cultured in RPMI media (HyClone Laboratories, Logan, UT, USA) supplemented with 1% HEPES, 1% sodium pyruvate, 1% L-glutamine, 1% sodium bicarbonate, 1% non-essential amino acids, and 1% penicillin-streptomycin solution. These cells were subjected to differential trypsinization up to 7 passages, immortalized using hTERT, and then selected using puromycin to generate a stable fibroblast cell line. Cells were incubated in a humidified incubator at 37 °C and supplied with 5% CO2. Cells were subcultured by trypsin-EDTA treatment, and complete medium was changed every other day.
Human PC and normal pancreatic tissues
Paraffin-embedded human PC tissues were obtained from the UNMC tissue bank. Samples were obtained following approval of the protocol by the Institutional Review Board (IRB) at the University of Nebraska Medical Center in Omaha, Nebraska.
Isolation and culture of side population (SP) and non-side population (NSP) from cancer cell lines
Side population (SP) cells or putative CSCs were isolated using flow sorting following Hoechst 33342 (AnaSpec Inc., Fremont, CA, USA) staining as described previously6. Verapamil (Sigma), a calcium channel blocker that reverses the drug resistance phenotype, was used as a control to gate the characteristic SP cells. The non-CSCs exhibit a much higher intensity of Hoechst staining and fall outside the gate and are therefore designated as the ‘non-side population’ (NSP) cells. Isolated SP and NSP fractions from SW1990 and Capan1 were grown in DMEM with 10% FBS for a day to allow the cells to acclimatise. The SP cells were then transferred to a stem cell-specific medium and were cultured in gelatin-coated (MilliporeSigma, Burlington, MA, USA) culture dishes as described previously6. SP cells were treated with 0.2 μM gemcitabine (IC20) for enriching the CSC population.
Depletion of PAF1 using CRISPR/Cas9 and inducible shRNA
PAF1 knockdown in SW1990 SP cells was performed using the CRISPR/Cas9 system. Briefly, cells were transfected with a PAF1 guide RNA (5’-ACCTACCGCATCGACCCCAA-3’) containing a CRISPR/Cas9 vector (pSpCas9 BB-2A-GFP PX458) (Genescript, Piscataway, NJ, USA). GFP positive cells were isolated 72 h after transfection, and the pooled population was collected in a 12-well plate by flow sorting. Cells were allowed to grow into colonies, which were then analyzed for expression of PAF1 by immunoblot analysis.
For inducible knockdown, TRIPZ inducible lentiviral human PAF1 shRNAs from Dharmacon (Lafayette, CO, U.S.A.) were used. HEK293T cells were cultured and transduced with sh23 and sh25 plasmids to generate viral supernatant according to manufacturer’s instructions. Viral supernant was used to infect SW1990, CD18, Capan1, SW1990 SP, and Capan1 SP cells, following which the cells were selected using puromycin (4 μg/ml). Selected cells were subjected to doxycycline (Dox) treatment (2 mg/ml) for 72 h and then sorted for RFP expression. RFP+ cells were maintained under two conditions, with and without Dox treatment, and were cultured under puromycin selection pressure (4 μg/ml). Dox-induced shRNA-mediated knockdown of PAF1 expression was analyzed by immunoblot assay.
SiRNA-mediated transient knockdown
The transient knockdown of PAF1 was performed using PAF1 siRNA (Origene), which is a pool of three target-specific 19–25 nt siRNAs. Capan1 SP cells were plated in a 6-well plate at a concentration of 0.6 million/well. On the following day, the cells were serum-starved for 4 h, and then transfected with PAF1 siRNA or non-targeting control siRNA (scramble siRNA) at a concentration of 80 pmol/well using TurboFect reagent (ThermoFischer Scientific). The serum containing medium was added to the cells 4 h after transfection. The medium was changed every 24 h and lysates were collected 72 h after transfection. Knockdown of DDX3, PHF5A, LEO1, CDC73, and CTR9 was performed using gene-specific siRNA (Origene) as described above.
Additional methods can be found in the Supplementary Materials.
Results
PAF1 promotes pancreatic tumorigenesis
To determine whether PAF1 affects on pancreatic tumorigenesis, we generated a doxycycline-inducible shRNA based-KD system for PAF1 in PC cell lines SW1990, HPAF/CD18, and Capan1 (Fig.1A, Fig.S1A). PAF1 KD significantly impaired pancreatic tumor growth in an orthotopic mouse model (Fig.1B, Fig.1C). Moreover, the metastatic burdens to the liver, stomach, and intestine were reduced upon PAF1 depletion in Capan1 xenograft tumors (Fig.1D, Fig.S1B). Furthermore, loss of PAF1 also impaired transwell migration and wound healing properties of PC cells (Fig.S1C, S1D). The effect of the loss of PAF1 on tumor propagation was validated using orthotopic and subcutaneous implantation from another PC cell line, HPAF/CD18, engineered with PAF1 shRNA (Fig.S1E, S1F). The tumors derived from the PAF1 KD group exhibited much lower proliferative capacity (Fig.S1G) and PAF1 expression (Fig.S1H). Importantly, loss of PAF1 resulted in the downregulation of the self-renewal markers NANOG and SOX9 (Fig.S1I). Based on our previous observations6, 18, 22, we evaluated the effect of PAF1 depletion on CSC maintenance through analysis of CSC markers and the proportion of CSC populations. We found that PAF1 depletion led to a significant downregulation of established CSC markers, such as CD24, CD44v6, and ALDH1, and the self-renewal markers SOX2, OCT3/4, β-CATENIN, and SOX9 (Fig.1E, Fig.S1J). Additionally, PAF1 depletion caused a significant reduction in the percentage of high CD44+ CSCs (Fig.1F, Fig.S1K), indicating that the effect of PAF1 abrogation on tumorigenesis could involve its role in CSC maintenance. Furthermore, we also observed a significant decrease in percentage of ALDH+ CSCs (Fig.S1L) and side population (SP) cells (Fig.S1M). Given that CSCs are capable of forming tumors upon extreme limiting dilution in vivo, we investigated the capacity of control and PAF1-depleted PC cells to initiate tumors in immunocompromised mice using different cell concentrations. At each cell concentration tested (from 100,000 cells to as low as 100 cells), PAF1-depleted PC cells had a significantly impaired capacity for tumor initiation and maintenance (Fig.1G, Fig.S1N). These results collectively indicate that PAF1 plays a cardinal role in pancreatic tumorigenesis, possibly via affecting CSC maintenance.
Figure 1: Loss of PAF1 reduces pancreatic tumorigenesis and proportions of CSCs.

A. Immunoblot depicting the efficiency of PAF1 KD using a doxycycline-based inducible shRNA system. B. Representative IVIS image of mice from control and PAF1 KD group. The right panel represents a quantification of total photon counts at indicated time points. Data represented as mean ± SD (n = 10 mice per group). C. Representative images and quantification of tumor weights generated with orthotopic implantation of engineered SW1990 and Capan1 cells in pancreas of athymic nude mice. D. Graphical representation of metastatic incidence in xenografts generated from orthotopic implantation of control and PAF1-depleted Capan1 cells. E. Immunoblot analysis of PAF1 and CSC markers with and without doxycycline treatment in engineered SW1990 and Capan1 cells. F. Percentage of high CD44+ cells in control and PAF1 KD SW1990 and Capan1 cells. G. Graphical representation of tumor weights of control and PAF1 KD cohorts versus the number of cells injected per tumor subcutaneously.*P <0.05, **P <0.01, ***P <0.001.
PAF1 is overexpressed in CSCs and maintains pancreatic CSCs
CSCs isolated from PC cells SW1990 and Capan1 using flow sorting (Fig.S2A) showed higher expression of established CSC (Fig.2A) and self-renewal markers in SP compared to NSP cells (Fig.2B). PAF1 was significantly overexpressed in CSCs compared to non-CSCs (Fig.2A). Purity of isolated CSCs and non-CSCs was confirmed using in vitro tumor sphere assay (Fig.S2B) and in vivo tumorigenicity assay (Fig.S2C). Depletion of PAF1 caused a significant downregulation of CSC markers ALDH1 and CD44v6, and of self-renewal markers SOX9 and β-CATENIN at the RNA (Fig.S2D) and protein levels (Fig.2C, Fig.S2E). Depletion of PAF1 also abrogated the functional CSC traits, such as proportions of ALDH+ cells (Fig.2D), the self-renewal capacity (Fig.2E, Fig.S2F), and the proliferation of CSCs (Fig.S2G). Since tumor recurrence is phenotypic trait of CSCs3, 5, we investigated the propensity of control and PAF1-depleted PC cells to cause recurrence following the primary tumor’s surgical resection. Our results indicate that loss of PAF1 impairs the ability of CSCs to cause tumor recurrence (Fig.S2H, S2I, S2J, S2K, S2L). These results collectively indicate that PAF1 is required for self-renewal and maintenance of CSCs.
Figure 2: PAF1 is upregulated in pancreatic CSCs and is required for sustenance of CSCs.

A. Immunoblot analysis depicting the expression of PAF1 and CSC markers in isolated CSCs (SP cells) from SW1990 and Capan1 compared to non-CSCs (NSP cells). B. Immunoblot analysis representing variation in expression of self-renewal markers between NSP and SP cells. C. Immunoblot analysis depicting the effect of depletion of PAF1 on CSC and self-renewal markers. D. Representative scatter plots and quantification of percentage of ALDH+ cells in control versus PAF1-depleted CSCs. E. Representative images of tumor spheres and quantification of the number of tumor spheres generated by control versus PAF1 KD CSCs. Scale bars are 1000 μm. Data is represented as mean ± SEM.
Role of PAF1 in pancreatic CSC maintenance independent from PAF1C function
To determine whether PAF1-mediated CSC maintenance involves PAF1C, we assessed the contribution of each subunit of PAF1C to maintain the CSC state. PAF1 and other PAF1C subunits (LEO1, CTR9, and CDC73) exhibited significantly higher expression in human PC tissues than normal pancreas tissues, as analyzed using the GEPIA database (Fig.S3A). Intriguingly, the expression of CTR9, LEO1, and CDC73 did not vary between SP and NSP cells from SW1990 and Capan1 (Fig.3A). Furthermore, we found that individual depletion of each of these PAF1C subunits did not affect the expression of CSC markers (Fig.3B), and the ability of CSCs to form tumor spheres (Fig.3C), indicating that these PAF1C members likely do not affect pancreatic CSC maintenance individually. Comparing expression of different PAF1C subunits in high purity tumor tissues from The Cancer Genome Atlas, PAF1 displayed the highest expression compared to other PAF1C subunits (Fig.S3B). In line with our observations in ESCs17, the depletion of PAF1 from SP cells did not affect the expression of other PAF1C components (Fig.3D). However, downregulation of PAF1 in NSP cells decreased the expression of other PAF1C subunits (Fig.3D), indicating differential regulation of PAF1C in CSCs versus non-CSCs. Interestingly, individual knockdown of LEO1, CTR9, and CDC73 significantly downregulated other PAF1C subunits, including PAF1 (Fig.S3C). Further, depletion of PAF1 in CSCs elicited extensive apoptosis, whereas in non-CSCs minimal cell death was seen (Fig.3E), suggesting that the loss of PAF1 doesn’t lead to general cell death.
Figure 3: LEO1, CTR9, and CDC73 are not major contributors to CSC maintenance.

A. Immunoblot analysis of PAF1C subunits in SP and NSP cells. Equal amounts of protein were loaded in each well. B. Immunoblot analysis of CSC and self-renewal markers with individual KD of CDC73, CTR9, and LEO1. C. Average number of tumor spheres formed with individual knockdown of LEO1, CDC73, and CTR9 at 0 h and 96 h. Data representative of two independent experiments and three replicates per condition. D. Immunoblot analysis of PAF1C subunits (CTR9, LEO1, and CDC73) with PAF1 depletion in SP and NSP cells. E. Scatter plots demonstrating apoptotic cells following 72 h treatment with Scr or PAF1 si in SW1990 SP and SW1990 NSP cells. Quantification of the percentage of late apoptotic cells is shown on the right. n.s. = non-significant.
PAF1-PHF5A sub-complex regulates the stem cell state in pancreatic CSCs
Based on our findings of differential regulation of PAF1 in CSCs and non-CSCs, we hypothesized that PAF1’s role in CSC maintenance requires interaction with other proteins. The first candidate we explored was PHD finger protein 5a (PHF5A), a nuclear protein that regulates pluripotency in ESCs21. PHF5A exhibited significant upregulation in PC patient tissues compared to normal pancreas tissues (Fig.S4A). Given the similarities between ESCs and CSCs, we investigated the status of PHF5A in SP and NSP cells and found that PHF5A is overexpressed in SP compared to NSP cells isolated from SW1990 and Capan1 (Fig.4A). Further, PAF1 and PHF5A co-localized in the nucleus of CSCs, as observed using confocal microscopy, whereas minimal expression was detected in SW1990 NSP (non-CSCs) cells (Fig.4B, Fig.S4B). Using reciprocal co-immunoprecipitation, we observed that PAF1 interacts with PHF5A (Fig.4C). Of interest, the PAF1C subunits LEO1, CTR9, and CDC73 interacted with PAF1 in CSCs, but not with PHF5A (Fig.4C). We observed that PAF1 co-localized with PHF5A in distinct cells from human PC tumor tissues, alluding to the clinical significance of their interaction (Fig.S4C). These results suggest that PAF1 possibly forms a sub-complex with PHF5A and other proteins in CSCs, which does not include LEO1, CTR9, and CDC73.
Figure 4: PAF1 interacts with PHF5A and jointly regulates Nanog.

A. Immunoblot analysis of PHF5A in SW1990 and Capan1 SP and NSP. B. Representative immunofluorescence image of PAF1 and PHF5A representing co-localization in nuclei of isolated CSCs. Data representative of three independent experiments and quantification is shown in Figure S4B. C. Immunoprecipitation was performed with PAF1 and PHF5A antibodies from SW1990 SP, and immunoblotting was performed for PAF1, PHF5A, CTR9, LEO1, and CDC73. D. Immunoblot analysis of CSC and self-renewal markers with depletion of PHF5A in SW1990 SP cells. E. Heatmap of mean binding enrichment of PAF1 and PHF5A on promoters of stem cell genes. F. Representative examples of UCSC genome browser tracks of PAF1 and PHF5A ChIP-Seq in SW1990 SP cells. G. Immunoblot analysis of Nanog with PAF1 and PHF5A depletion. H. Immunofluorescence analysis of PHF5A and NANOG in SW1990 SP cells with depleted PHF5A. Quantification of mean fluorescence intensity (MFI) is shown on the right. Lower panel shows expression of PAF1 and NANOG in control and PAF1-depleted SW1990 SP cells. Data representative of two independent experiments and is presented as a mean of five individual images per condition.
To determine the functional relevance of PHF5A for the maintenance of pancreatic CSCs, we investigated the effect of PHF5A loss on the expression of CSC markers. Surprisingly, most established CSC markers, including CD133, ESA, SOX9, OCT3/4, β-CATENIN, and PAF1, were not affected (Fig.4D). Thus, we used a global approach to identify the genes that are jointly regulated by PAF1 and PHF5A by performing ChIP-Seq with PAF1 and PHF5A-pulldown in CSCs. Chromosome-wide PAF1 peaks were more frequent than PHF5A peaks in SW1990 SP cells, at a false discovery rate of less than 0.05 (Fig.S4D), potentially explaining why PAF1 depletion had a greater effect on CSC markers compared with PHF5A depletion. Promoters of several stemness regulators, including Nanog, SOX9, ABCB5, ERBB2, LIF, POU5F1 and others, were occupied by PAF1 and PHF5A in SW1990 SP cells (Fig.4E). Nanog was amongst the top genes whose promoters were jointly occupied by PAF1 and PHF5A in SW1990 SP cells. Furthermore, PAF1 and PHF5A occupied the Nanog promoter at identical loci (Fig.4F). For subsequent analyses, we focused on Nanog, as it is the master regulator for pluripotency and stem cell state23. Knockdown of PAF1 and PHF5A led to a significant downregulation of Nanog, as observed by immunoblot and qRT-PCR analysis (Fig.4G, Fig.S4E). Importantly, knockdown of PAF1 and PHF5A also reduced nuclear localization of NANOG in CSCs, observed by confocal microscopy (Fig.4H).
PAF1 sub-complex interacts with DDX3 to regulate Nanog transcriptionally
To identify additional interacting partners of PAF1 in pancreatic CSCs, we performed mass spectrometry using immunoprecipitation of PAF1 from pancreatic CSCs (SW1990 SP), mouse embryonic carcinoma cells (pluripotent stem cell line F9), and a normal pancreatic ductal cell line (HPDE). Several unique interaction partners for PAF1 were identified in addition to the canonical PAF1C members. Proteins that served as the common interactors of PAF1 in pancreatic CSCs and ESCs were identified (Fig.5A). These proteins might be a part of the PAF1 sub-complex and facilitate effects on pancreatic CSC stemness. Of the 19 proteins identified, an RNA helicase, DDX3 showed the highest probability of interaction with PAF1 in SW1990 SP and F9 cells, validated using co-immunoprecipitation (Fig.5B, Fig.S5A). Emerging evidence indicates that DDX3 can participate in the epithelial-to-mesenchymal transition (EMT), apoptosis, cell cycle regulation, and tumorigenesis24. We found DDX3 was overexpressed in human PC tumors compared to normal tissues (Fig.S5B), in oncogene-transduced HPNE cells (E6/E7/Htert/KRAS HPNE versus HPNE control), and CSCs compared to non-CSCs (Fig.5C). We found co-localization of PAF1 and DDX3 in specific cells within human PC tissues, PC patient tissue-derived organoids, and KPC (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre) mouse organoids, while PAF1 and DDX3 showed basal expression in respective normal tissues and did not co-localize (Fig.5D, 5E, Fig.S5C). Moreover, levels of DDX3 and PAF1 were increased and co-localized in SW1990 SP cells compared to NSP cells (Fig.5F, Fig.S5D). To determine whether DDX3 was a part of PAF1-PHF5A sub-complex, we investigated if DDX3 and PHF5A interact. We found an interaction of DDX3 and PHF5A in SW1990 SP cells (Fig.5G), indicating a ternary complex composed of PAF1, PHF5A, and DDX3. Importantly, DDX3 was found to bind to a specific region of the Nanog promoter (Fig.5H), indicating that the PAF1-PHF5A-DDX3 sub-complex regulates Nanog. Consistent with roles of Nanog and PAF1 in CSC maintenance, we observed co-overexpression of PAF1 and NANOG in human PC tissues compared to the normal pancreas (Fig.S5E) and poorer survival in patients with higher expression of NANOG (Fig.S5F). These results collectively demonstrate that the PAF1-PHF5A-DDX3 sub-complex regulates Nanog in pancreatic CSCs, with potential clinical relevance.
Figure 5: Mass spectrometric analysis identified RNA helicase DDX3 as a novel interacting partner of PAF1.

A. Venn diagram depicting percentage of interacting proteins of PAF1 in F9, HPDE, and SW1990 SP cells. Common interacting proteins are listed. B. Reciprocal immunoprecipitation analysis of PAF1 and DDX3 in SW1990 SP and HPDE cells. C. Immunoblotting analysis of DDX3 in NSP and SP cells isolated from SW1990 and Capan1 and in control versus E6/E7/Htert/KRAS HPNE cells. D. Immunofluorescence analysis of PAF1 and DDX3 in human PC tissues and normal pancreas. White arrows indicate individual cells with co-localized PAF1 and DDX3. E. Immunofluorescence images showing co-localization of PAF1 and DDX3 in human PC organoids and mouse KPC organoids. Representative images depicting PAF1 and DDX3 expression in human and mouse normal organoids are shown in lower panel. F. Representative immunofluorescence images depicting expression of PAF1 and DDX3 in SW1990 SP cells and SW1990 NSP cells. Quantification is shown in Figure S5C. G. Immunoprecipitation analysis of PHF5A and DDX3 in SW1990 SP followed by immunoblotting for PHF5A. H. Representative ChIP-PCR gel images demonstrating DDX3 binding on binding site 4 (B.S. 4) on Nanog promoter. Primers specific for B.S. 1 and B.S. 8 did not amplify. Primer sequences are shown in Supplementary Table 1.
Abrogation of DDX3 activity impairs cancer stem cell maintenance
Since we found a role of the PAF1-PHF5A-DDX3 sub-complex in pancreatic CSC maintenance, we next sought to determine a means of disrupting this sub-complex to target CSCs. To this end, we investigated the effect of RK-33, a specific small molecule inhibitor of DDX3 helicase activity25. Of importance, CSCs (SW1990 SP and Capan1 SP) were more sensitive to RK-33 than normal human fibroblast cells (IC50 in 9–26 NP fibroblasts was 1.5- to 2-fold higher than that of SW1990 SP and Capan1 SP; SW1990 SP: 4.32 μM; Capan1 SP: 5.53 μM; 9–26 NP: 9.23 μM) (Fig.S6A). Treatment of CSCs with RK-33 for 48 h led to a significant downregulation of CSC markers (β-CATENIN, CD44v6, SOX9, and NANOG) (Fig.6A). Surprisingly, we found a robust downregulation of PAF1 with RK-33 treatment (Fig.6A). Further, the effect of RK-33 on the expression of CSC markers in NSP cells was not as profound as that seen with SP cells (Fig.S6B). Functionally, DDX3 inhibition impaired the tumor sphere formation capacity (Fig.6B) and colony formation ability (Fig.S6C) of CSCs. Additionally, treatment with RK-33 resulted in apoptosis of CSCs (Fig.S6D, S6E), while minimal cell death was seen in normal human fibroblasts (9–26 NP), supporting the clinical applicability of RK-33. It is important to note that both PAF1 and DDX3 were found to bind on the Nanog promoter, and treatment with RK-33 decreased their binding significantly (Fig.6C). Furthermore, we validated downregulation of PAF1 and Nanog expression following RK-33 treatment using qPCR (Fig.6D). Knockdown of DDX3 in CSCs phenocopied the effects observed with RK-33 treatment, causing downregulation of CSC markers CD44v6, β-CATENIN, SOX9, and NANOG (Fig.6E).
Figure 6: DDX3 inhibition downregulates CSC markers and decreases binding on Nanog promoter.

A. Immunoblotting analysis for DDX3, PAF1, CD44v6, NANOG, SOX9, and β-CATENIN following RK-33 treatment for 48 h. B. Representative images of tumor spheres in control and RK-33 (2.5 μM)-treated SW1990 SP cells. Quantification of the average number of tumor spheres is shown on the right and is represented as mean ± SEM. Data representative of two independent experiments and six replicates per condition. Scale bars are 1000 μm. C. ChIP-PCR gel images for PAF1 and DDX3 using primers specific for B.S. 4, B.S. 1, and B.S. 8 in DMSO- and RK-33 (5 μM)-treated SW1990 SP cells. ChIP qRT PCR analysis of PAF1 and DDX3 using B.S. 4 primers in DMSO- and RK-33 (5 μM)-treated SW1990 SP cells is represented on right. D. Quantification of PAF1 and Nanog transcripts in DMSO- and RK-33-treated CSCs. Error bars indicate SEM values. E. Immunoblotting analysis for DDX3, PAF1, CD44v6, NANOG, and β-CATENIN in control and DDX3 KD CSCs.
Stemness and metastasis-promoting genes are targets of PAF1
PAF1 depletion in CSCs led to downregulation of stemness genes (Gata3, Elk1, Stat3, Stat5b, Stat6, Jun-B, Cebpb, Cebpa, Ctnnb1, Crebbp, and Foxa2) (Fig.7A) and tumor-promoting pathways (JAK/STAT signaling, IL-6 signaling, ERK/MAPK signaling, and CXCR4 signaling) (Fig.7B). Of importance, genes with important roles in stemness maintenance (Flot2, Taz, Epcam, Erbb2, Foxp1, Abcc5, Ddr1, Muc1, Pecam1, Notch3, Aldh1a3, Foxa2, Plat, and Lif) and metastasis (Itga3, St6galnac4, Flot1, S100a14, Mmp14, Flot2, Itgb2, S100a9, St6galnac2, S100a11, Muc1, Itgb7, and St6galnac1) were significantly downregulated via RNA-Seq (Fig.7C, Fig.S7A). Given that CSCs mediate tumor relapse, we evaluated the expression of genes associated with relapse in control and PAF1-depleted CSCs and found significant downregulation of relapse genes, such as Sema3b, Abcc5, Fgfr3, Tspan1, Muc1, Tff1, Ephb2, and Alcam (Fig.S7B). Comparing PCR array and ChIP-sequencing data, we found a greater enrichment of PAF1 on promoters of those genes that were downregulated on PAF1 KD (Fig.S7C). Overall, these data indicate that PAF1 functions as the master regulator for stem cell maintenance by regulating the transcription of several stem cell-related genes.
Figure 7: PAF1 behaves as the master regulator of stem cell maintenance.

A. RT2 profiler PCR array analysis of human transcription factors in SW1990 SP control and PAF1-depleted cells. Upregulated genes are indicated in red, and genes significantly downregulated upon PAF1 depletion are indicated in green. Genes encircled in red were validated using qRT PCR. B. Representative network of significantly downregulated pathways upon PAF1 loss in SW1990 SP cells, derived from ingenuity pathway analysis of downregulated genes from PCR array and RNA-Seq. C. Heatmap representing genes significantly downregulated upon PAF1 depletion in CSCs, and expression pattern of stemness genes in control and PAF1-depleted CSCs. D. Schematic illustration of the mechanism of PAF1-mediated maintenance of pancreatic CSCs. PAF1, PHF5A, and DDX3 are upregulated in PC and form a sub-complex that supports pancreatic CSCs. Depletion of PAF1 results in a decreased percentage of CSCs, reduced tumorigenesis and impaired tumor sphere formation.
Discussion
Previous work from our laboratory and others has demonstrated that PAF1 is essential in normal stem cell maintenance16, 17. In addition, a role for PAF1 in the self-renewal of CSCs in pancreatic and ovarian cancer has recently been established6, 18, 22. While these studies point to the importance of PAF1 in stem cells, little was understood about how PAF1 mediates these CSC functions. In this study, we mechanistically established PAF1 as an essential regulator of CSC maintenance in PC and demonstrated the existence of a unique PAF1-PHF5A-DDX3 sub-complex in CSCs. Using different strategies to downregulate PAF1 in several PC cell lines, we demonstrated that PAF1 depletion led to decreases in CSC markers, CSC frequency, stemness features, and tumor burden and recurrence. PAF1 ablation downregulated CSC gene expression signatures, and PAF1 was found to bind on several stem cell gene promoters in pancreatic CSCs. PAF1C has been connected to both activating and repressing effects on gene transcription, depending on the cellular context19, 26. In our system, we have focused on activating functions, as our ChIP-seq analysis revealed a strong binding of PAF1 at CSC genes that were significantly downregulated upon PAF1 depletion. Nevertheless, it would be interesting to address the repressive functions of PAF1 in pancreatic CSCs.
We showed that PAF1 downregulation significantly impaired pancreatic tumorigenesis. Similar to previous reports6, 22, we found PAF1 to be important for regulating the expression of CSC markers and phenotypic traits, suggesting that the decreased tumorigenesis on PAF1 depletion is, in part, due to the role of PAF1 in CSC maintenance. Importantly, PAF1 loss adversely affected the capacity to initiate tumors upon limiting dilution in vivo. Although we found an upregulation of LEO1, CTR9, and CDC73 transcripts in human PC tissues compared to the normal pancreas, these other PAF1C subunits were not overexpressed in isolated CSCs. Interestingly, the other PAF1C subunits, LEO1, CTR9, and CDC73, appear dispensable for the maintenance of stem cell state, as their downregulation affected neither the expression of CSC markers nor the formation of tumor spheres. This reiterates the idea that PAF1C subunits may play PAF1C-independent roles. For instance, the PAF1C subunit CTR9 has been shown to regulate ERα+ breast tumorigenesis and control morphology, proliferative capacity, and tamoxifen sensitivity of luminal breast cancer cells, while no other PAF1C subunit except PAF1 could modulate these features27. Similarly, CDC73, another PAF1C subunit, was recently shown to play an indispensable role in maintaining mouse hematopoietic stem cells and in regulating leukemia-specific gene programs28. Our observation of differential expression of PAF1C subunits in CSCs (SP) versus non-CSCs (NSP) following PAF1 loss alludes to the concept that PAF1 directs unique gene programs in undifferentiated versus differentiated cells. We postulated that PAF1 achieves cell-type-specific functions based on the availability of its interacting partners. In line with this, PAF1 interacts with another pluripotency regulator, PHF5A, in ESCs and in turn, regulates RNA polymerase II pause-release of pluripotency genes in ESCs21. Strikoudis et al. demonstrated that the interaction of PAF1 with PHF5A maintained the stability of PAF1C in ESCs21. Although PHF5A was found to interact with other PAF1C members in ESCs; it only interacts with PAF1 and not LEO1, CTR9, or CDC73 in pancreatic CSCs. Furthermore, the treatment of NSP cells with DDX3 inhibitor had limited effects on the expression of CSC markers, in contrast to the effects observed in SP cells. This reiterates that the PAF1 sub-complex with DDX3 and PHF5A is unique to CSCs.
A previous report showed that MED12, a member of a transcriptional complex, maintained stem/progenitor cells of the hematopoietic system by regulating hematopoietic stem cell-specific enhancers, independently of its kinase role29. By contrast, we found that PAF1, along with its binding partners, regulates the promoters of stem cell genes. PAF1 has been suggested to modulate metazoan gene expression by several means, including modulation of the RNA polymerase II paused state and enhancer activation, in addition to its direct roles in transcription elongation19, 20, 26. For instance, PAF1 has been implicated in restraining full activation of a subset of enhancers and consequently impairs the release of paused RNA polymerase II from nearby target genes in colorectal carcinoma HCT116 cells20. However, it would be interesting to investigate if PAF1 is required for hyperactivation of stem cell enhancers in pancreatic CSCs.
In contrast to PAF1, which had a profound effect on stem cell gene regulation, PHF5A depletion altered the expression of NANOG, but not other known CSC markers. This observation was supported by our ChIP-Seq analysis, wherein chromosome-wide binding for PAF1 was more frequent compared to PHF5A. However, Nanog emerged as the common target of PAF1 and PHF5A in pancreatic CSCs. Although Nanog is considered a stem cell master regulator, it is now known to be de-repressed in several cancers, including breast, ovarian, lung, colorectal, pancreatic, and others30, 31. A body of literature suggests that the drug-resistant cancer cells responsible for tumor recurrence express Nanog following conventional treatment32, 33. CSCs expressing Nanog have been targeted with inhibitors and shRNA; more recently, the use of dendritic cell vaccination against NANOG has been suggested34–36. These studies emphasize the clinical and functional significance of Nanog for CSC maintenance and underscore the relevance of our findings. Of importance, we demonstrated co-localization of PAF1 and NANOG in human pancreatic tumor tissues and loss of NANOG expression upon PHF5A and PAF1 depletion in pancreatic CSCs. Our ChIP-Seq results also identified LIF, an important self-renewal regulator, as a common target for PAF1 and PHF5A. Shi et al. showed that LIF receptor signaling promotes PC via STAT3 activation and mediates intrinsic chemoresistance of CSCs, making it an attractive therapeutic target in PC37. Possible regulation of LIF via PAF1 sub-complex alludes to the therapeutic potential of PAF1 sub-complex targeted approaches.
We identified DDX3 as a novel binding partner of PAF1 in pancreatic CSCs. It belongs to the DEAD-box family of RNA helicases, which have primary roles in unwinding complex RNA secondary and tertiary structures. However, several members of this family additionally function in transcriptional regulation38. RNA helicase DDX3 is involved in transcription, RNA splicing, nuclear export of mRNA, and translation initiation24, 39. We showed that PAF1 and DDX3 play a role in the maintenance of pancreatic CSCs. Inhibition of DDX3 activity by RK-33 downregulated CSC markers and PAF1, while a loss of DDX3 alone had a similar effect on expression of CSC markers. This indicates that DDX3 alone may have important roles in CSC biology. This notion is supported by a recent publication that showed DDX3 is overexpressed in undifferentiated human ESCs compared to differentiated cells, and that perturbation of DDX3 activity reduced expression of the pluripotency regulators OCT4, SOX2, and NANOG and impaired teratoma formation40. Importantly, we demonstrated that RK-33 treatment reduced the localization of DDX3 and PAF1 on the Nanog promoter.
We finally show that PAF1 depletion downregulated stem cell-related transcription factors and genes, including JAK/STAT, ERK/MAPK, and CXCR4 signaling pathways. The role of PAF1 in regulating these stem cell genes is indicated by its localization on promoters of these genes. Several of these pathways are also drivers of tumorigenesis. For instance, several studies have shown the importance of the JAK/STAT pathway, particularly STAT3 activation in mediating tumorigenic ability of CSCs in breast41, colon cancer42, and several other cancers43. STAT3 is constitutively active in PC44 and plays a role in proliferation, survival, and metastasis of pancreatic tumor cells45, 46. Additionally, the STAT3/Sox2 axis has been documented to play an important role in maintaining stem cell phenotypes in PC cells47. Similarly, PAF1 KD downregulated pathways involved in CSC-mediated cancer pathogenesis, including CXCR44 and Wnt/β-catenin signaling48. Together, these observations underscore the pivotal role of PAF1 in mediating stemness and indicate that PAF1 may serve as a common upstream regulator of these pathways. Given that PAF1 has multiple functions and serves as a converging point for several co-transcriptional processes, including transcription elongation, termination, and mRNA processing, its basic functions in different cell types and disease settings merit further study. Nonetheless, this study serves as a stepping-stone for targeting PAF1-dependent pancreatic CSC maintenance through DDX3. Taken together, these data reveal that the PAF1-PHF5A-DDX3 sub-complex is required for sustenance of pancreatic CSCs (Fig.7D) and present a therapeutic window to target this sub-complex through DDX3 inhibition.
Supplementary Material
What you need to know:
Background and Context:
It is not clear how pancreatic cancer stem cells (CSCs) are regulated, resulting in ineffective treatments for pancreatic cancer. PAF1, a RNA polymerase II-associated factor 1 complex component, maintains stem cells and might be important for the function of cancer stem cells.
New Findings:
In pancreatic CSCs, PAF1 forms a sub-complex that regulates expression of genes that control stem cell features. Knockout of PAF1 reduced the ability of pancreatic tumors to develop and progress in mice and numbers of CSCs.
Limitations:
This study was performed in cell lines and mice with orthotopic tumors; further studies are needed in other mouse models and humans.
Impact:
Strategies to target the PAF1-PHF5A-DDX3 complex might be developed to slow or inhibit progression of pancreatic cancer.
Lay Summary:
This study identified a protein that is upregulated in pancreatic cancer cells that allows them to proliferate indefinitely and promote tumor development.
Acknowledgements and writing assistance
The authors acknowledge the invaluable technical support of Ms. Kavita Mallya. We thank the advanced microscopy core, flow cytometry core, sequencing core, and mass spectrometry and proteomics core at UNMC for their help and support. We also thank Dr. Jessica Mercer for editing this manuscript. Questions associated with RK-33 should be addressed to vraman2@jhmi.edu.
Funding
The authors/work in this article were supported, in part, by the following grants from the National Institutes of Health (F99 CA234962, R01 CA210637, R01 CA206444, P01 CA217798, U01 CA200466 and U01 CA210240).
Abbreviations
- (CSCs)
Cancer stem cells
- (PC)
pancreatic cancer
- (PAF1C)
RNA polymerase II-associated factor 1 complex
- (PAF1)
RNA polymerase II-associated factor 1
- (PD2)
pancreatic differentiation 2
- (ESCs)
embryonic stem cells
- (IVIS)
in vivo imaging systems
- (SP)
side population
- (NSP)
non-side population
- (ChIP-Seq)
chromatin immunoprecipitation sequencing
- (RNA-Seq)
RNA sequencing
- (EMT)
epithelial-to-mesenchymal transition
- (KD)
knockdown
- (SEM)
standard error of mean
- (n.s.)
non-significant
- (B.S.)
binding site
- (IP)
immunoprecipitation
- (MS)
mass spectrometry
- (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre)
KPC
Footnotes
Data Resources
RNA sequencing and ChIP sequencing data: GSE144371 (https://www.ncbi.nlm.nih.gov/geo/).
Author names in bold designate shared co-first authorship.
Conflicts of interest
SKB is one of co-founders of Sanguine Diagnostics and Therapeutics, Inc. Venu Raman has a patent for RK-33, and he is one of the co-founders and advisory board members of Natsar Pharmaceuticals. The other authors disclose no potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25. [DOI] [PubMed] [Google Scholar]
- 3.Bednar F, Simeone DM. Pancreatic cancer stem cell biology and its therapeutic implications. J Gastroenterol 2011;46:1345–52. [DOI] [PubMed] [Google Scholar]
- 4.Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007;1:313–23. [DOI] [PubMed] [Google Scholar]
- 5.Lytle NK, Ferguson LP, Rajbhandari N, et al. A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 2019;177:572–586 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaz AP, Ponnusamy MP, Rachagani S, et al. Novel role of pancreatic differentiation 2 in facilitating self-renewal and drug resistance of pancreatic cancer stem cells. Br. J Cancer 2014;111:486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008;40:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong DJ, Liu H, Ridky TW, et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008;2:333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005;5:275–84. [DOI] [PubMed] [Google Scholar]
- 10.Vaz AP, Deb S, Rachagani S, et al. Overexpression of PD2 leads to increased tumorigenicity and metastasis in pancreatic ductal adenocarcinoma. Oncotarget 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaudhary K, Deb S, Moniaux N, et al. Human RNA polymerase II-associated factor complex: dysregulation in cancer. Oncogene 2007;26:7499–7507. [DOI] [PubMed] [Google Scholar]
- 12.Moniaux N, Nemos C, Schmied BM, et al. The human homologue of the RNA polymerase II-associated factor 1 (hPaf1), localized on the 19q13 amplicon, is associated with tumorigenesis. Oncogene 2006;25:3247–3257. [DOI] [PubMed] [Google Scholar]
- 13.Dey P, Ponnusamy MP, Deb S, et al. Human RNA polymerase II-association factor 1 (hPaf1/PD2) regulates histone methylation and chromatin remodeling in pancreatic cancer. PLoS. One 2011;6:e26926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dey P, Rachagani S, Vaz AP, et al. PD2/Paf1 depletion in pancreatic acinar cells promotes acinar-to-ductal metaplasia. Oncotarget 2014;5:4480–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moniaux N, Nemos C, Deb S, et al. The human RNA polymerase II-associated factor 1 (hPaf1): a new regulator of cell-cycle progression. PLoS. One 2009;4:e7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding L, Paszkowski-Rogacz M, Nitzsche A, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell 2009;4:403–415. [DOI] [PubMed] [Google Scholar]
- 17.Ponnusamy MP, Deb S, Dey P, et al. RNA polymerase II associated factor 1/PD2 maintains self-renewal by its interaction with Oct3/4 in mouse embryonic stem cells. Stem Cells 2009;27:3001–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karmakar S, Seshacharyulu P, Lakshmanan I, et al. hPaf1/PD2 interacts with OCT3/4 to promote self-renewal of ovarian cancer stem cells. Oncotarget 2017;8:14806–14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen FX, Woodfin AR, Gardini A, et al. PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell 2015;162:1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen FX, Xie P, Collings CK, et al. PAF1 regulation of promoter-proximal pause release via enhancer activation. Science 2017;357:1294–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strikoudis A, Lazaris C, Trimarchi T, et al. Regulation of transcriptional elongation in pluripotency and cell differentiation by the PHD-finger protein Phf5a. Nat Cell Biol 2016;18:1127–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karmakar S, Dey P, Vaz AP, et al. PD2/PAF1 at the Crossroads of the Cancer Network. Cancer Res 2018;78:313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu X, Mazur SJ, Lin T, et al. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene 2014;33:2655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bol GM, Xie M, Raman V. DDX3, a potential target for cancer treatment. Mol Cancer 2015;14:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bol GM, Vesuna F, Xie M, et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol Med 2015;7:648–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu M, Yang W, Ni T, et al. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science 2015;350:1383–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng H, Xu W. Ctr9, a key subunit of PAFc, affects global estrogen signaling and drives ERalpha-positive breast tumorigenesis. Genes Dev 2015;29:2153–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saha N, Ropa J, Chen L, et al. The PAF1c Subunit CDC73 Is Required for Mouse Hematopoietic Stem Cell Maintenance but Displays Leukemia-Specific Gene Regulation. Stem Cell Reports 2019;12:1069–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aranda-Orgilles B, Saldana-Meyer R, Wang E, et al. MED12 Regulates HSC-Specific Enhancers Independently of Mediator Kinase Activity to Control Hematopoiesis. Cell Stem Cell 2016;19:784–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeter CR, Badeaux M, Choy G, et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells 2009;27:993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Y, Zhu H, Shan H, et al. Knockdown of Oct4 and Nanog expression inhibits the stemness of pancreatic cancer cells. Cancer Lett 2013;340:113–23. [DOI] [PubMed] [Google Scholar]
- 32.Noh KH, Kim BW, Song KH, et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J Clin Invest 2012;122:4077–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noh KH, Lee YH, Jeon JH, et al. Cancer vaccination drives Nanog-dependent evolution of tumor cells toward an immune-resistant and stem-like phenotype. Cancer Res 2012;72:1717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding Y, Yu AQ, Li CL, et al. TALEN-mediated Nanog disruption results in less invasiveness, more chemosensitivity and reversal of EMT in Hela cells. Oncotarget 2014;5:8393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han J, Zhang F, Yu M, et al. RNA interference-mediated silencing of NANOG reduces cell proliferation and induces G0/G1 cell cycle arrest in breast cancer cells. Cancer Lett 2012;321:80–8. [DOI] [PubMed] [Google Scholar]
- 36.Wefers C, Schreibelt G, Massuger L, et al. Immune Curbing of Cancer Stem Cells by CTLs Directed to NANOG. Front Immunol 2018;9:1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y, Gao W, Lytle NK, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019;569:131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuller-Pace FV. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res 2006;34:4206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ariumi Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front Genet 2014;5:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerr CL, Bol GM, Vesuna F, et al. Targeting RNA helicase DDX3 in stem cell maintenance and teratoma formation. Genes Cancer 2019;10:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin L, Hutzen B, Lee HF, et al. Evaluation of STAT3 signaling in ALDH+ and ALDH+/CD44+/CD24-subpopulations of breast cancer cells. PLoS One 2013;8:e82821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin L, Fuchs J, Li C, et al. STAT3 signaling pathway is necessary for cell survival and tumorsphere forming capacity in ALDH(+)/CD133(+) stem cell-like human colon cancer cells. Biochem Biophys Res Commun 2011;416:246–51. [DOI] [PubMed] [Google Scholar]
- 43.Yin X, Zhang BH, Zheng SS, et al. Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of Stat3/Snail signaling. J Hematol Oncol 2015;8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scholz A, Heinze S, Detjen KM, et al. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003;125:891–905. [DOI] [PubMed] [Google Scholar]
- 45.Fofaria NM, Srivastava SK. STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis 2015;36:142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei D, Le X, Zheng L, et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003;22:319–29. [DOI] [PubMed] [Google Scholar]
- 47.Jiang J, Li Z, Yu C, et al. MiR-1181 inhibits stem cell-like phenotypes and suppresses SOX2 and STAT3 in human pancreatic cancer. Cancer Lett 2015;356:962–70. [DOI] [PubMed] [Google Scholar]
- 48.Ram Makena M, Gatla H, Verlekar D, et al. Wnt/beta-Catenin Signaling: The Culprit in Pancreatic Carcinogenesis and Therapeutic Resistance. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.