

Abstract
Lipids present in lipoproteins cleared from the circulation are processed sequentially by three major proteins within the late endosomal / lysosomal (E/L) compartment of all cells: lysosomal acid lipase (LAL), Niemann-Pick (NPC) C2 and NPC1. When all three of these proteins are functioning normally, unesterified cholesterol (UC) exits the E/L compartment and is used in plasma membrane maintenance and various pathways in the endoplasmic reticulum including esterification by sterol O-acyltransferase 2 (SOAT2) or SOAT1 depending partly on cell type. Mutations in either NPC2 or NPC1 result in continual entrapment of UC and glycosphingolipids leading to neurodegeneration, pulmonary dysfunction, splenomegaly and liver damage. To date, the most effective agent for promoting release of entrapped UC in nearly all organs of NPC1-deficient mice and cats is 2-hydroxypropyl-β-cyclodextrin (2HPβCD). The cytotoxic nature of the liberated UC triggers various defenses including suppression of sterol synthesis and increased esterification. The present studies, using the Npc1−/− nih mouse model, measured the comparative quantitative importance of these two responses in the liver versus the spleen of Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice in the 24 h following a single acute treatment with 2HPβCD. In the liver but not the spleen of both types of mice suppression of synthesis alone or in combination with increased esterification provided the major defense against the rise in unsequestered cellular UC content. These findings have implications for systemic 2HPβCD treatment in NPC1 patients in view of the purportedly low levels of SOAT2 activity in human liver.
Keywords: Lysosomal storage disease, Unesterified cholesterol sequestration, Cholesterol synthesis and esterification, Liver, Spleen
Graphical Abstract
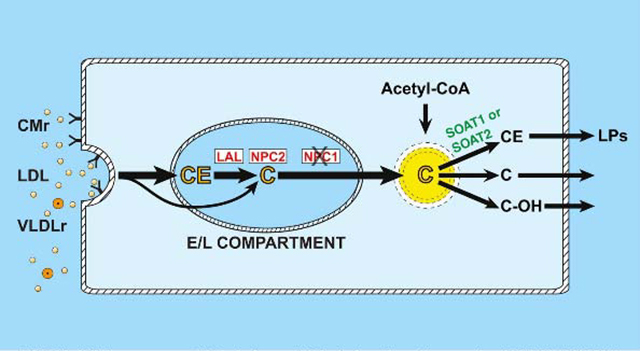
In NPC1 deficiency, unesterified cholesterol (C), derived from lipoproteins (CMr, LDL. VLDLr), becomes trapped in the E/L compartment of cells. 2-Hydroxypropyl-β-cyclodextrin releases sequestered C causing compensatory changes in the rates of cholesterol synthesis (from Acetyl-CoA) and esterification by SOAT1 or 2.
1. Introduction
Three proteins, lysosomal acid lipase (LAL), Niemann-Pick C (NPC) 2, and NPC1 facilitate the sequential trafficking of lipoprotein-derived cholesterol through the late endosomal/lysosomal (E/L) compartment for subsequent delivery to the endoplasmic reticulum [1,2]. Mutations in either NPC1 or NPC2 result in continual E/L entrapment of unesterified cholesterol (UC) and glycosphingolipids [3]. This sequestration of lipids, particularly of UC, can lead to varying degrees of neurodegeneration, pulmonary dysfunction, splenomegaly and liver damage [4]. The number of mutations in NPC1 far exceeds that in NPC2 [5]. Globally, the incidence of NPC disease (NPCD) is estimated to be at least 1:120,000 live births [4].
As recently discussed in a comprehensive review [6], a diverse array of treatment strategies for NPC1 deficiency has been evaluated in cell and animal models as well as patients. One particular compound, 2-hydroxypropyl-β-cyclodextrin (2HPβCD), has shown the best prospects for the effective management of NPC1 disease in multiple organs including the central nervous system. Historically, the laboratory of Dr. Robert Erickson was the first to investigate the effects of intraperitoneal 2HPβCD administration on liver cholesterol storage and neurological symptoms in Npc1−/− mice [7,8]. However, it was the outcome of later research directed by Dr. John Dietschy showing a single subcutaneous (sc) injection of 2HPβCD in 7-day old Npc1−/− mice significantly slowed neurodegeneration and increased lifespan [9] that is now acknowledged as the first description of the use of cyclodextrin as a potential therapeutic agent for NPC1 disease [10]. Other studies at that time by Davidson et al. [11] demonstrated additional benefit from chronic cyclodextrin administration in Npc1−/− mice, thus further propelling research into the potential of this agent for treating NPCD in humans. Over the ensuing years, research in numerous laboratories worldwide defined how 2HPβCD impacted cholesterol metabolism at a cellular, biochemical and molecular level in multiple organ systems of animal models of NPC1 and NPC2 disease [12,13]. Of particular significance was the demonstration that intracerebroventricular administration of 2HPβCD to very young Npc1−/− mice [14], and intracisternal delivery of this agent in a feline model of NPC1 disease [15], prevented Purkinje cell death and delayed neurodegeneration. A subsequent non-randomised, open-label, phase 1–2 trial in NPC1 patients demonstrated that intrathecal delivery of 2HPβCD decreased neurological disease progression [16].
The mechanism(s) by which 2HPβCD facilitates release of entrapped UC is not fully understood [13] but in one proposed model it is delivered via pinocytosis to the late E/L compartment where it performs the functions of NPC2 and NPC1 in facilitating the exit of UC [17]. Irrespective of how the release is orchestrated, cells must respond expeditiously to the acute rise in their UC content because of its potential toxicity [18]. While suppression of local cholesterol synthesis provides a rapid and efficient defense mechanism, other adaptations can also come into play. These include esterification [19] and various cellular efflux mechanisms [20]. In tissues there are two cholesterol esterifying enzymes, one of which is sterol O-acyltransferase (SOAT1, previously called ACAT1) present in splenic macrophages, steroidogenic organs, kidneys, and sebaceous glands [21–23]. The other enzyme is SOAT2 (previously called ACAT2) which is expressed in enterocytes and hepatocytes [22–25]. The tissue distribution and functions of SOAT1 and 2 are discussed in a 2012 review [26]. Ordinarily in most tissues, with the exception of organs like the adrenal gland, only a small proportion of the cholesterol present is esterified [27,28]. However, tissue EC levels can rise appreciably under various conditions as seen for example in the liver during periods of high dietary cholesterol intake [29]. Esterified cholesterol can also become greatly elevated in rare diseases such as Tangier disease [30], Wolman disease (WD) and Cholesteryl Ester Storage Disease (CESD) [31]. In a mouse model of CESD the elimination of SOAT2 resulted in a marked reduction in EC sequestration in the liver and small intestine [32]. Other studies demonstrated that in Npc1−/− mice lacking SOAT2 there was less hepatomegaly and UC sequestration, and lower liver transaminase levels [33]. In these models the elimination of SOAT2-driven EC formation likely diminished the EC content of nascent very low density lipoproteins (VLDL) and chylomicrons to the extent that the remnants of these particles ultimately cleared from the circulation carried less cholesterol for potential sequestration in the E/L compartment because of mutations in LIPA, NPC1 or NPC2.
In the formative studies that explored in vivo the impact of acute 2HPβCD treatment on cholesterol metabolism in Npc1−/− mice, the most striking changes seen 24 h later occurred in the liver and spleen where a dramatic drop in the UC level was accompanied by a decisive rise in tissue EC concentration, together with a profound fall in the rate of cholesterol synthesis [34–37]. However, it has not been determined whether in the liver and spleen, or indeed in most organs, these two adaptive mechanisms together are largely responsible for offsetting the release of entrapped UC over the first 24 h after treatment starts. To address this question two sets of young adult mice of four genotypes (Npc1+/+ : Soat2+/+, Npc1+/+ : Soat2−/−, Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/−) that had 24 h earlier received a bolus subcutaneous injection of 2HPβCD in saline, or saline only, were used for the measurement of either UC and EC concentrations in liver and spleen, or the absolute rate of sterol synthesis in these organs. The data show that in the livers of the treated Npc1−/− : Soat2+/+ mice the defensive changes in EC formation and cholesterol synthesis together amounted to 64% of the UC mass that was mobilized during the 24 h after 2HPβCD administration. In the treated Npc1−/− : Soat2−/− mice, where the pre-treatment hepatic levels of UC were appreciably less than in their Npc1−/− : Soat2+/+ counterparts, almost no new EC was generated but the adaptive fall in cholesterol synthesis equated to 85% of the mass of UC that was liberated. A very different result was seen in the spleen where the corresponding figures for the Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice were only 13% and 23%, respectively. These findings, especially for the liver may have particular relevance to the actions of systemically administered 2HPβCD in NPC1 patients given there is evidence that the level of hepatic SOAT2 activity in humans generally may be far lower than it is in mouse models.
2. Materials and methods
2.1. Animals
Npc1+/− mice (pure BALB/c) originally obtained from a colony at the National Institute of Neurological Diseases and Stroke [38] were source of the NPC1 mutation (Npc1−/− nih) used in this project. Soat2+/− mice (mixed 129Sv : C57BL/6 background) were purchased from Jackson Laboratories. Thus in the current studies all mice were littermates of a mixed strain background (BALB/c : 129Sv : C57BL/6) derived from Npc1+/− : Soat2+/− breeding stock as described previously [33]. This resulted in progeny of multiple genotypes, four of which were selected for study. These were Npc1+/+ : Soat2+/+, Npc1+/+ : Soat2−/−, Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/−. The pups were weaned at 21 days on to a low-cholesterol, cereal-based, chow diet with a fat content of ~4% (No. 7001, Envigo :Teklad, Madison, WI). The mice were group housed in plastic colony cages containing wood shavings, and had continual access to drinking water. They were kept in a light-cycled room with 12 h of light (1200–2400 h) and darkness (0–1200 h). All mice were studied in the fed state at about 1100h. The studies were carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory Animals, and the protocols used were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center.
2.2. Administration of 2HPβCD
A 20% (w/v) solution of 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich product H107) was prepared by dissolution in sterile saline at room temperature. During the late dark phase the mice were administered a single subcutaneous injection of 2HPβCD at the scruff of the neck. The dose was 4000 mg/kg body weight. Matching mice injected with saline only served as controls. After injection all mice were returned to their cages until used for metabolic measurements 24 h later. Two sets of mice were established, one for the measurement of tissue UC and EC concentrations and the other for determining absolute rates of sterol synthesis in the liver and spleen. The small intestines were also taken from these mice for such measurements as described in an earlier publication [39]. All groups contained a varying proportion of male and female mice as noted in Table 1.
Table 1.
Body and organ weights of mice deficient in either NCP1 or SOAT2, or both genes, that were given a single treatment with 2-hydroxypropyl-β-cyclodextrin in saline, or saline alone 24h before study
| Genotype | Treatment | No. of mice | Body weight | Liver weight | Spleen weight |
|---|---|---|---|---|---|
| ♂ / ♀ | (g) | (g) | (g) | ||
| Npc1+/+ : Soat2+/+ | Saline | 7 / 5 | 23.1 ± 1.0 | 1.29 ± 0.08a | 0.067 ± 0.003a |
| Npc1+/+ : Soat2+/+ | 2HPβCD | 7 / 5 | 23.6 ± 1.2 | 1.32 ± 0.09a | 0.077 ± 0.003a |
| Npc1+/+ : Soat2−/− | Saline | 5 / 5 | 21.7 ± 1.2 | 1.18 ± 0.09a | 0.068 ± 0.003a |
| Npc1+/+ : Soat2−/− | 2HPβCD | 5 / 5 | 22.1 ± 1.4 | 1.14 ± 0.07a | 0.064 ± 0.004a |
| Npc1−/− : Soat2+/+ | Saline | 7 / 5 | 20.7 ± 1.0 | 1.79 ± 0.07b | 0.107 ± 0.007b |
| Npc1−/− : Soat2+/+ | 2HPβCD | 8 / 4 | 20.8 ± 1.0 | 1.76 ± 0.10b | 0.114 ± 0.009b |
| Npc1−/− : Soat2−/− | Saline | 7 / 5 | 19.7 ± 0.8 | 1.43 ± 0.10a | 0.113 ± 0.010b |
| Npc1−/− : Soat2−/− | 2HPβCD | 8 / 4 | 20.8 ± 1.0 | 1.56 ± 0.11a,b | 0.137 ± 0.017b |
All mice were 48–51 days old at the time of study. Approximately one half of the mice in each group were used for tissue cholesterol concentration measurements. The remaining mice were used specifically for determining rates of cholesterol synthesis in vivo. Values are mean ± SEM for the total number of mice indicated. Different letters denote statistically significant differences as determined by one-way ANOVA using Tukey’s multiple comparison test.
2.3. Esterified and unesterified cholesterol concentrations
Approximately 24 h after they had received their respective treatments the mice were anesthetized and exsanguinated from the inferior vena cava, with 0.8 to 1.0 ml of blood being obtained. The liver and spleen were excised, rinsed in cold saline, blotted and weighed. Aliquots of liver totaling 300 to 350 mg and the whole spleen were added to ~40 ml of chloroform:methanol (2:1 v/v). The absolute concentrations of esterified and unesterified cholesterol were measured by established methods using a combination of column and gas chromatography [33]. The concentrations were expressed as mg/g wet tissue weight. No corrections were made for the presence of EC and UC contained in residual blood in the tissue aliquots. In addition, as all mice were in the fed state at exsanguination, plasma total cholesterol concentrations and lipoprotein compositions were not determined.
2.4. Cholesterol synthesis rates
The cholesterol synthesis measurements were made towards the end of the dark phase over a 1-h period starting 23 h after the 2HPβCD, or saline only, had been administered. Custom-generated tritiated water, with an activity of 5 Ci/ml (PerkinElmer Life Sciences, Waltham, MA) was diluted in sterile saline to an activity of approximately 200 mCi/ml. The mice were weighed, given an intraperitoneal injection of [3H]water (~2 mCi/g body weight), and then kept in a warm environment under a well ventilated fume hood for the next 60 min. They were then anesthetized and exsanguinated into a heparinized syringe. The liver and spleen were excised, rinsed in cold saline, blotted and weighed. The liver was cut into multiple pieces and an aliquot (~300–350 mg) was placed in 5 ml of ethanolic KOH as was the entire spleen. From each mouse, a 100 μl aliquot of plasma was taken through a series of dilutions for the measurement of the plasma water specific activity. The labeled sterols in the digested tissues were extracted and quantitated [40].The rate of cholesterol synthesis was expressed in two ways. The first was the nmol of water incorporated into sterol per hour per gram wet weight of tissue (nmol/h/g).These data, which did not include a correction for the presence of labeled sterol in the residual blood present in the tissues were subsequently used to calculate the absolute quantity of sterol produced over 24 h (mg of sterol synthesized/day/g). The terms used in the equation to make this calculation are defined elsewhere [41,42]. It should be emphasized that digitonin precipitates not only cholesterol but also several of the intermediates in the biosynthetic pathway including desmosterol, lathosterol and lanosterol [43]. Their respective positions in the synthetic pathway are well documented [44].
2.5. Analysis of data
All values are presented as the mean ± SEM of data from the specified number of animals. GraphPad Prism 6.02 software (GraphPad, San Diego, CA) was used to perform all statistical analyses. Differences between means were tested for significance (p < 0.05) using a one-way analysis of variance (ANOVA) and applying Tukey’s multiple comparison test.
3. Results
3.1. In the livers of the Npc1−/− : Soat2+/+ mice the reduction in UC levels induced by 2HPβCD resulted in both a decisive rise in tissue EC levels and a marked suppression of sterol synthesis but in their Npc1−/− : Soat2−/− counterparts only the latter response occurred
In reviewing the organ weight data in Table 1 it should be noted that the reduction in hepatomegaly but not splenomegaly in Npc1−/− : Soat2−/− mice has been previously documented [31]. Those earlier studies also showed that the absence of SOAT2 alone caused a marked reduction in hepatic EC levels in mice with or without NPC1. The data from the new studies demonstrate how much SOAT2 function influences the impact of acute 2HPβCD treatment on hepatic cholesterol levels and rates of synthesis in mice lacking NPC1. The concentration of UC in the Npc1−/− : Soat2+/+ fell on average by 5.8 mg/g over 24 h (Fig. 1A). In the matching Npc1−/− : Soat2−/− mice where there was less UC entrapment at baseline the reduction was 2.6 mg/g (p > 0.05). Even more striking are the data for hepatic EC levels, particularly in the treated Npc1−/− : Soat2−/− vs Npc1−/− : Soat2+/+ mice (Fig. 1B). Consistent with the results of past studies the mice deficient in NPC1 alone exhibited the prototypical marked increase in hepatic EC levels. In the matching treated Npc1−/− : Soat2−/− mice EC levels did not change. The rates of hepatic sterol synthesis, measured in vivo in a different set of mice, are presented in Fig. 1C. Here the two most notable findings pertain to the mice lacking NPC1, with and without SOAT2. One is that the rate of hepatic sterol synthesis at baseline in the Npc1−/− : Soat2−/− mice was significantly higher than in their in Npc1−/− : Soat2+/+ counterparts. The other is that despite this difference 2HPβCD treatment resulted in a profound reduction in the rate of synthesis in both types of mice.
Fig. 1.

In the livers of Npc1−/− : Soat2+/+ mice the 2HPβCD-induced release of entrapped UC resulted in both increased EC formation along with marked inhibition of cholesterol synthesis whereas in their Npc1−/− : Soat2−/− counterparts only the latter response was seen. For the study in which the data for tissue UC and EC concentrations (panels A and B, respectively) were obtained there were 6 mice per group. The sterol synthesis measurements (panel C) were made in separate mice with 6 per group in all cases except for the Npc1+/+ : Soat2−/− mice where there were 4 given either saline alone or 2HPβCD in saline. Values are the mean ± SEM. Different letters denote statistically significant (p < 0.05) differences as determined by one-way ANOVA.
3.2. In the spleens of both the Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice treatment with 2HPβCD led to increased cholesterol esterification as well as suppression of sterol synthesis
The splenomegaly seen in the Npc1−/− : Soat2+/+ mice was equally evident in their Npc1−/− :Soat2−/− littermates and was not impacted by 2HPβCD treatment in either case (Table 1). This treatment also did not change the concentrations of UC or EC in the spleens of either the Npc1+/+ : Soat2+/+ or Npc1+/+ : Soat2−/− mice (Figs 2A and 2B, respectively). However, there was a decisive reduction in the UC concentration after 2HPβCD was given in both the Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice (Fig. 2A). The splenic EC concentrations in these same two groups of mice increased markedly and to about the same degree after 2HPβCD administration (Fig 2B). The pattern of genotypic and treatment-related changes in cholesterol synthesis rates in the spleen (Fig. 2C) paralleled those found in the liver (Fig. 1C), with the main finding being a profound reduction in cholesterol synthesis rates in both the Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice in response to 2HPβCD.
Fig. 2.

In the spleens of both the Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice the 2HPβCD-induced reduction in UC levels resulted in comparable increments in rates of cholesterol esterification as well as similar degrees of cholesterol synthesis inhibition. The data in panels A-C were obtained from the same respective sets of mice used for the corresponding measurements in the liver. Values are the mean ± SEM. Different letters denote statistically significant (p < 0.05) differences as determined by one-way ANOVA.
3.3. The quantitative importance of cholesterol synthesis suppression either alone or with a concomitant rise in cholesterol esterification in countering the 2HPβCD-driven release of sequestered UC was much greater in the liver than in the spleen
The objective of Fig. 3 is to present a side-by-side comparison of how much an increase in SOAT2-driven cholesterol esterification on the one hand and a suppression of local cholesterol synthesis on the other compare quantitatively in accommodating the 2HPβCD-mediated release of UC in the liver and spleen over the 24 h following its acute administration. These data are presented for just the Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice because 2HPβCD treatment did not impact UC levels in the corresponding groups that were Npc1+/+. The magnitude of the drop in UC levels in both organs after 2HPβCD was less pronounced in the Npc1−/− : Soat2−/− mice than in their Npc1−/− : Soat2+/+ counterparts, especially in the liver (Fig. 3A). This was not unexpected given that Npc1−/− mice without SOAT2 have lower baseline levels of UC especially in the liver. The data in Fig. 3B are particularly striking. For the liver there was a substantial rise in hepatic EC levels in the Npc1−/− : Soat2+/+ mice after treatment. This accounted for about 38% of the fall in UC after 2HPβCD was given. In the matching Npc1−/− : Soat2−/− mice essentially none of the released UC underwent esterification. The data for the spleen sharply contrast with those for the liver. Thus in both the Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice there were comparable but small increments in EC levels in spleen after 2HPβCD. In the case of the Npc1−/− : Soat2+/+ mice the rise in EC levels accounted for only about 7% of the amount of UC lost from the spleen over the 24-h period. For their treated Npc1−/− : Soat2−/− littermates this figure was about 10%.
Fig. 3.

Comparison of the net change in UC concentration with that in the EC concentration and the absolute rate of sterol synthesis in the liver and spleen of Npc1−/− : Soat2+/+ versus Npc1−/− : Soat2−/− mice in the 24-h period following acute 2HPβCD treatment. Corresponding data for the matching Npc1+/+ : Soat2+/+ and Npc1+/+ : Soat2−/− mice are not shown because 2HPβCD treatment did not result in statistically significant changes in any of the three parameters measured in either the liver (Fig. 1 A–C) or spleen (Fig. 2 A–C). The rates of sterol synthesis shown in panel C of both Fig 1 and Fig 2 represent the nmol of water incorporated into sterol per g of wet weight tissue per h. As described in Materials and Methods, these rates were converted to an approximate absolute amount of sterol generated, with the units of mg synthesized per day per g wet weight of tissue. Then, as was the case for estimating the net change in UC and EC levels following 2HPβCD treatment (panels A and B, respectively), the magnitude of the net change in sterol synthesis (panel C) was determined as the difference in the mean values between the 2HPβCD- and saline only-treated groups.
The data in Fig. 3C similarly show a decisive difference between the two organs in the extent to which adaptive reductions in the absolute amount of cholesterol synthesized offset the release of UC after 2HPβCD administration. In the livers of the Npc1−/− : Soat2+/+ mice the reduction of 1.41 mg/g in newly synthesized sterol over 24 h equated to 24% of the 5.78 mg/g of UC released. When combined with the amount of released UC that was esterified in these particular mice over this time (2.19 mg/g, which was equal to 38% of the drop in the UC level) it is clear that together these two mechanisms represented the primary defense against the surge in unsequestered UC after the 2HPβCD was given. For their Npc1−/− : Soat2−/− counterparts the reduction of 2.16 mg/g in the amount of sterol synthesized by the liver over 24 h equated to 83% of the 2.60 mg/g fall in the UC level. There was a negligible rise in hepatic esterification of UC in these mice thus making sterol synthesis inhibition the overwhelming defensive mechanism in the absence of SOAT2-mediated esterification.
In marked contrast to what occurred in the livers, in the spleens of both the Npc1−/− : Soat2+/+ and Npc1−/− : Soat2−/− mice the pronounced reduction in sterol synthesis over the 24-h post treatment period represented, in absolute terms, only a minor adaptive response to the substantial amounts of UC released after 2HPβCD administration. Here it must be noted however that the rate of sterol synthesis in the spleen across all genotypes and treatments was much lower than seen for the livers in these same animals. Thus in the spleens of the Npc1−/− : Soat2+/+ mice the 2HPβCD-facilitated release of 5.09 mg of UC /g resulted in a fall in cholesterol synthesis of just 0.31 mg/day/g, along with a rise in EC formation amounting to only 0.38 mg/g. Together, these adaptive changes were equivalent to only 13% of the amount of UC released after 2HPβCD was given. In the spleens of their treated Npc1−/− : Soat2−/− counterparts, the liberation of 3.98 mg of UC/g was offset by a reduction in sterol synthesis of only 0.42 mg/day/g and an increment in the EC level of just 0.50 mg/g. The sum of these defensive changes combined represented only 23% of the amount of UC level liberated by 2HPβCD. Taken together these various sets of data show that at least in this Npc1−/− mouse model the 2HPβCD-facilitated release of UC in the spleen is offset primarily through a mechanism(s) of greater quantitative importance than that derived from a combination of cholesterol synthesis inhibition and increased cholesterol esterification.
4. Discussion
The impetus for the development of 2HPβCD as a therapy for NPC1 disease derives from a plethora of data generated in studies using mouse and cat models with varying mutations in NPC1 [12,13,45]. The first clinical trial investigated the impact of intrathecal delivery of 2HPβCD on neurological disease progression [16]. Several other trials, either at or near completion (ClinicalTrials.gov Identifier NCT02939547 and NCT02912793) or currently enrolling patients (ClinicalTrials.gov Identifier NCT03887533), have or will evaluate repeated systemic administration of 2HPβCD on multiple parameters including specific indices of hepatic cholesterol metabolism in NPC1 patients over a broad age range. Questions will nevertheless remain concerning the relative quantitative importance of cholesterol synthesis suppression and increased esterification as first-line defense mechanisms in protecting the liver and indeed other organs like the spleen against the toxic effects of a surge in cellular UC content caused by high doses of systemically administered 2HPβCD. It is important to note here that while the Npc1−/− nih model has been widely used, newer models have been developed including the Npc1I1061T mouse which expresses the most common human NPC1 mutation [46], and more recently the Npc1em1Pav mouse [47]. A comprehensive review of animal models for NPC disease research appeared in 2019 [10]. The current and earlier studies on the potential benefit of eliminating SOAT2 function in NPC1 disease used the Npc1−/− nih model mainly because its cholesterol metabolism has been characterized at a biochemical and molecular level far more extensively than is the case for other murine models with disrupted NPC1 function.
In evaluating the findings presented here several points should be made regarding potential sources of error in the measurements and the conclusions drawn from them. The first of these pertains to two aspects of the sterol synthesis data. These were measured as the rates of incorporation of water into newly generated sterol over a 1-h interval corresponding to the 24th hour after the acute administration of 2HPβCD or saline alone. These rates were subsequently converted to an absolute amount of sterol synthesized over 24 h using established equations (Fig. 3C). Thus the question this calculation raises is whether the rate measured in the 24th hour faithfully reflected the amount of sterol generated during the course of the preceding 23 h given that hepatic sterol synthesis rates usually show a marked diurnal variation [48,49]. In our early studies with Npc1−/− mice we found that, unlike their Npc1+/+ littermates, they did not exhibit diurnal changes in their rate of hepatic sterol synthesis [50]. The second point to consider in evaluating the sterol synthesis data is that past studies showed the suppressive impact of the liberated UC on the biosynthetic pathway in this model does not become maximal in either the liver or spleen until 3 to 6 h after the 2HPβCD is administered [37]. Thus the conversion of rates of incorporation of labeled water into new sterol measured in the 24th hour following treatment (Fig. 1C and 2C) into absolute amounts of sterol synthesized over 24 h (Fig. 3C) results in an overestimation of the true amount of new sterol generated per day. The magnitude of this error, which is likely to be small, would be the same for both the liver and spleen.
With regard to the tissue UC and EC data one potential source of error warranting comment relates to the presence of UC and EC in residual blood within the tissue aliquots taken for analysis. Earlier studies using 59Fe to assess residual tissue blood content (% of wet weight) in multiple organs of adult C57BL6 mice after 400ul of blood was removed found values for the spleen and liver of 22.1 and 5.3%, respectively [51]. Whole blood contains UC in cell membranes as well as varying levels of UC and EC in lipoproteins in the plasma compartment, with this EC having been generated mainly through the actions of SOAT2 and also of lecithin-cholesterol acyltransferase (LCAT) in plasma [52]. Given that in the current studies the amount of blood removed at exsanguination was at least 0.8 ml across all experimental groups, the degree of error stemming from residual blood contamination is believed to not significantly impact the calculations for the treatment-driven changes in UC or EC levels for either organ in the Npc1−/− : Soat2+/+ mice and their Npc1−/− : Soat2−/− counterparts (Fig. 3A and 3B). Perhaps the most striking point about the UC concentration data is that even though the baseline levels in the spleen of the Npc1−/− : Soat2+/+ mice (Fig. 2A) were lower than those for the liver (Fig. 1A), the magnitude by which they fell in the 24 h after 2HPβCD administration was about the same in both organs (Fig. 3A). In the case of the matching Npc1−/− : Soat2−/− mice the extent of the fall in the liver UC levels after 2HPβCD treatment was not as much as that for the spleens (Fig. 3A). This reflected the fact that the absence of SOAT2 in itself drove down baseline UC levels in the livers but not the spleens of Npc1−/− mice. In regards to the EC data for the liver (Fig. 1B) it should be noted that past studies demonstrated high levels of SOAT2 activity in the livers of chow-fed mice of a mixed background (C57BL/6 : 129Sv) mice [53]. Thus the marked rise in EC levels in the 24 h after 2HPβCD administration seen in the Npc1−/− : Soat2+/+ mice was anticipated. However, the finding that hepatic EC levels in the matching treated Npc1−/− : Soat2−/− mice did not change might be considered surprising given that macrophage presence increases in several organs including the liver when NPC1 function is disrupted [54]. Although our past studies in Npc1−/− : Soat2−/− mice showed less of an increase in hepatic mRNA levels for SOAT1 than seen in their Npc1−/− : Soat2+/+ counterparts, they were still appreciably elevated compared to those found in matching Npc1+/+ : Soat2+/+ and Npc1+/+ : Soat2−/− mice [33]. One might speculate therefore that in the Npc1−/−nih mouse model the proportion of total hepatic cholesterol esterifying activity attributable to SOAT1 is just a fraction of that occurring via SOAT2 as has been reported for the African Green monkey [25]. Irrespective of how much of the liberated UC is esterified by each of these enzymes, ultimately the hepatic pool of EC is hydrolyzed with some, if not most, of the resulting UC appearing in the bile and later the fecal neutral sterol fraction [35,55].
In addition to esterification another potential mechanism for rapid handling of the liberated UC, particularly in the absence of SOAT2, might be its ABCA1-mediated efflux onto ApoA1 to form nascent HDL. Studies by Pedrelli et al. using mice injected with an antisense oligonucleotide targeted to hepatic SOAT2 detected increased hepatic ABCA1 protein levels [56]. However, a study by Taylor et al. using Npc1−/−nih mice given 2HPβCD found hepatic ABCA1 mRNA levels to be mostly unchanged except for a transient rise 6 h after treatment [37]. This was attributed to ABCA1 not being an LXR target gene in hepatocytes where a specific ABCA1 promoter is required.
Irrespective of the comparative levels of SOAT1 and 2 activity in the liver of the Npc1−/− nih mouse and other murine models of NPCD, the EC data for the spleen (Fig.2B) clearly show that 2HPβCD administration stimulated esterification of UC in this organ in the absence of SOAT2 thus confirming a significant presence of SOAT1.This finding is consistent with data showing a comparatively high mRNA expression level for SOAT1 in human spleen [23].The more striking finding for the spleen in the current studies was that the 2HPβCD-driven release of UC was offset to only a modest degree through the combined defensive actions of increased esterification and suppression of synthesis of cholesterol (Fig. 3B and 3C). In the studies of Taylor et al. a marked and sustained increase in mRNA levels for ABCAI in the spleen was found as early as 2 h after 2HPβCD administration [37]. Thus it is attractive to speculate from those data that ABCA1-mediated efflux of UC represents a quantitatively important pathway for removing liberated UC from the spleen in concert with suppression of synthesis and raised esterification rates of cholesterol.
A major question arising from the data presented here is their relevance to hepatic cholesterol metabolism in NPC1 patients before and following systemic 2HPβCD treatment, with a focus on SOAT2-driven cholesterol esterification. It is important to first point out what is known about hepatic cholesterol levels in patients with either NPC1 or NPC2 disease. There appear to be no published data specifically for hepatic UC and EC levels in either NPC1 or NPC2 patients. However, more than a decade before the discovery of mutations in NPC2 Vanier reported total cholesterol levels in the livers and spleens of patients with NPC disease [57]. For a total 15 NPC patients and 6 control subjects, hepatic total cholesterol concentrations (mean ± SEM) of 9.7 ± 0.6 and 4.6 ± 0.5 mg/g wet weight, respectively, were found. The latter value is consistent with data from two other studies that reported the total cholesterol concentration in normal human liver to be 3.9 ± 0.2 mg/g (n=12) [58] and 5.4 ± 1.1 mg/g (n=11) [59], respectively. In one of those studies the fraction of cholesterol present in the esterified form averaged 25% [58]. While further data for hepatic cholesterol levels in NPC1 patients might arise from future studies, it is interesting that the values found in the Vanier study are appreciably less than those seen in animal models of NPC1 disease that were maintained on low-cholesterol rodent chow diets. This is best exemplified by data for untreated adult Npc1−/−nih mice on a pure BALB/c background where total cholesterol levels in liver were generally in the range of ~20–30 mg/g which represented at least a 7-fold increase over those found in matching wild-type controls [60–62]. This remained the case even in the mixed strain Npc1−/− : Soat2+/+ mice in the present studies (Fig. 1A). Comparable findings have also been reported for the Npc1nmf164 model on a pure C57BL/6 background [63]. It is also noteworthy that early research using a feline model of NPC1 disease found hepatic total cholesterol concentrations of ~18 and ~2.3 mg/g, respectively in the mutants and normal controls [64].
Such comparative data raise many more questions than can be addressed here. One has to do with the link between SOAT2-driven esterification of cholesterol in enterocytes and hepatocytes and the levels of hepatic UC entrapment resulting from NPC1 dysfunction. The amount of lipoprotein cholesterol cleared daily from the circulation by the liver far exceeds that of any other organ. Thus not only is it the site of clearance of chylomicron remnants containing cholesterol absorbed from the small intestine [65] but it ordinarily is also the principal organ for removing LDL and related apoB-containing lipoproteins from the circulation [66]. Hypothetically, when NPC1 function is impaired or lost, the concurrent elimination of SOAT2 activity by either genetic or pharmacological manipulation should lower the level of hepatic UC sequestration because less EC would be generated in hepatocytes for incorporation into nascent VLDL and ultimately mature LDL particles. This should in turn result in lower levels of UC entrapment when these particles are cleared from the circulation. Such a scenario is borne out by the lower hepatic UC levels and reduced plasma activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in Npc1−/− : Soat2−/− vs Npc1−/− : Soat2+/+ mice [33]. It must be emphasized however that although it is widely agreed there is a high level of expression of SOAT2 in the small intestine of humans [23,24], a nonhuman primate model [21], and mice of a mixed genetic background (C57BL/6 : 129 Sv) [53], available data suggest that SOAT2 activity in human hepatocytes is very low [25] compared to that detected in the livers of a primate model [25] and of C57BL/6 : 129Sv mice [53]. Even if this were not the case the selective and potent inhibitors of SOAT2 that have a decisive therapeutic benefit in mouse models of CESD [67] and atherosclerosis [68] have not yet been approved for use in humans. This raises the question of what other type of agent that targets cholesterol handling by the small intestine and subsequently the liver could serve as an adjunct to systemic 2HPβCD administration aimed primarily at ameliorating liver disease in specific NPC1 patients. Interest in this question is driven partly by the results of studies in humans [69,70] and a feline NPC1 model [15] indicating adverse effects of high doses of systemic 2HPβCD administration on the lungs. One possible candidate for combination therapy is ezetimibe, a potent inhibitor of cholesterol absorption [71] which is FDA-approved for use in pediatric patients with heterozygous familial hypercholesterolemia who are 10 years and older [72]. Ezetimibe also has been shown to reduce the severity of liver disease not only in the Npc1−/− nih mouse [73] but also in a mouse model for CESD [74] as well as in patients with CESD [75].
5. Conclusion
The sequestration of unesterified cholesterol (UC) in tissues resulting from mutations in the NPC1 gene can be overcome by acute systemic treatment with 2-hydroxypropyl-β-cyclodextrin (2HPβCD). However, compensatory mechanisms are needed to offset the resulting surge in cellular UC content which can be cytotoxic. In the current studies using NPC1-deficient mice, with and without the cholesterol esterifying enzyme SOAT2, it was found that in the liver but not the spleen the suppression of new cholesterol synthesis with or without increased esterification provided the major immediate defenses against the 2HPβCD-facilitated release of UC. This was not the case in the spleen where we conclude cholesterol transporters may have facilitated the cellular exit of much of this excess UC.
Highlights.
NPC1 is a cholesterol transporter in the lysosomal/endosomal compartment of cells
NPC1 deficiency causes entrapment of unesterified cholesterol (UC) and other lipids
2-hydroxypropyl-β-cyclodextrin releases the sequestered UC which can be cytotoxic
Adaptive responses include less synthesis and more esterification of UC
In Npc1−/− mice these are the major adaptations in the liver but not the spleen
Acknowledgements
This research was supported by National Institutes of Health Grants R01 HL 009610 (SDT) and DK 078592 (JJR) and the Ara Parseghian Medical Research Foundation (JJR). Dr.Turley also received partial salary support from the Department of Internal Medicine at UT Southwestern. We thank the late Dr. Lawrence Rudel of Wake Forest University School of Medicine for informative discussions about possible species differences in the activities of cholesterol esterifying enzymes in various organs. The assistance of Monti Schneiderman with the maintenance of the mouse colony is gratefully acknowledged.
Abbreviations
- ABCA1
ATP-binding cassette transporter A1
- bw
body weight
- EC
esterified cholesterol
- E/L
endosomal / lysosomal
- 2HPβCD
2-hydroxypropyl-β-cyclodextrin
- LAL
lysosomal acid lipase
- NPC1
Niemann-Pick type C1
- NPC2
Niemann-Pick type C2
- SOAT1
sterol O-acyltransferase 1
- SOAT2
sterol O-acyltransferase 2
- UC
unesterified cholesterol
Footnotes
Conflict of Interest
None of the authors have any conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS, Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease, J. Biol. Chem. 250 (1975) 8487–8495. [PubMed] [Google Scholar]
- [2].Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE, Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol, Cell 137 (2009) 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Peake KB, Vance JE, Defective cholesterol trafficking in Niemann-Pick C-deficient cells, FEBS Lett.584 (2010) 2731–2739. [DOI] [PubMed] [Google Scholar]
- [4].Vanier MT, Niemann-Pick disease type C, Orphanet J. Rare Dis. 5 (2010) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Patterson MC, Clayton P, Gissen P, Anheim M, Bauer P, Bonnot O, Dardis A, Dionisi-Vici C, Klϋnemann H-H, Latour P, Lourenço CM, Ory DS, Parker A, Pocoví M, Strupp M, Vanier MT, Walterfang M, Marquardt T, Recommendations for the detection and diagnosis of Niemann-Pick disease type C. An update, Neurol.Clin.Pract. 7 (2017) 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hammond N, Munkacsi AB, Sturley SL. The complexity of a monogenic neurodegenerative disease: More than two decades of therapeutic driven research into Niemann-Pick type C disease, Biochim.Biophy.Acta Mol. Cell. Biol. Lipids 1864 (2019) 1109–1123. [DOI] [PubMed] [Google Scholar]
- [7].Camargo F, P Erickson R, Garver WS, Hossain GS, Carbone PN, Heidenreich RA, Blanchard J, Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 70 (2001) 131–142. [DOI] [PubMed] [Google Scholar]
- [8].Coisne C, Tilloy S, Monlier E, Wils D, Fenart L, Gosselet F, Cyclodextrins as emerging therapeutic tools in the treatment of cholesterol-associated vascular and neurodegenerative diseases, Molecules 21 (2016) 1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu B, Li H, Repa JJ, Turley SD, Dietschy JM, Genetic variations and treatments that affect the lifespan of the NPC1 mouse, J.Lipid Res. 49 (2008) 663–669. [DOI] [PubMed] [Google Scholar]
- [10].Fog CK, Kirkegaard T, Animal models for Niemann-Pick type C: implications for drug discovery and development, Expert Opin.Drug Discov. 14 (2019) 499–509. [DOI] [PubMed] [Google Scholar]
- [11].Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU, Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression, PLoS One 4 (2009) e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu B, Therapeutic potential of cyclodextrins in the treatment of Niemann-Pick type C disease, Clin.Lipidol. 7 (2012) 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vance JE, Karten B, Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin, J.Lipid Res. 55 (2014) 1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aqul A, Liu B, Ramirez CM, Pieper AA, Estill SJ, Burns DK, Liu B, Repa JJ, Turley SD, Dietschy JM, Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment, J.Neurosci. 31 (2011) 9404–9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vite CH, Bagel JH, Swain GP, Prociuk M, Sikora TU, Stein VM, O’Donnell P, Ruane T, Ward S, Crooks A, Li S, Mauldin E, Stellar S, De Meulder M, Kao ML, Ory DS, Davidson C, Vanier MT, Walkley SU, Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease, Sci.Transl.Med. 7 (2015) 276ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ory DS, Ottinger EA, Farhat NY, King KA, Jiang X, Weissfeld L, Berry-Kravis E, Davidson CD, Bianconi S, Keener LA, Rao R, Soldatas A, Sidhu R, Walters KA, Xu X, Thurm A, Solomon B, Pavan WJ, Machielse BN, Kao M, Silber SA, McKew JC, Brewer CC, Vite CH, Walkley SU, Austin CP, Porter FD, Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial, Lancet 390 (2017) 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rosenbaum AI, Maxfield FR, Niemann-Pick type C disease; molecular mechanisms and potential therapeutic approaches, J.Neurochem. 116 (2011) 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tabas I, Consequences of cellular cholesterol accumulation: basic concepts and physiological implications, J.Clin.Invest. 110 (2002) 905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Warner GJ, Stoudt G, Bamberger M, Johnson WJ, Rothblat GH, Cell toxicity induced by inhibition of acyl coenzyme A:cholesterol acyltransferase and accumulation of unesterified cholesterol, J.Biol.Chem. 270 (1995) 5772–5778. [DOI] [PubMed] [Google Scholar]
- [20].Phillips MC, Molecular mechanisms of cellular cholesterol efflux, J.Biol.Chem. 289 (2014) 24020–24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee RG, Willingham MC, Davis MA, Skinner KA, Rudel LL, Differential expression of ACAT1 and ACAT2 among cells within liver, intestine, kidney and adrenal of nonhuman primates, J.Lipid Res. 41 (2000) 1991–2001. [PubMed] [Google Scholar]
- [22].Lee J-Y, Carr TP, Dietary fatty acids regulate acyl-CoA:cholesterol acyltransferase and cytosolic cholesteryl ester hydrolase in hamsters, J.Nutr. 134 (2004) 3239–3244. [DOI] [PubMed] [Google Scholar]
- [23].Lee JW, Huang J-D, Rodriguez IR, Extra-hepatic metabolism of 7-ketocholesterol occurs by esterification to fatty acids via cPLA2α and SOAT1 followed by selective efflux to HDL, Biochim.Biophys.Acta 1851 (2015) 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chang CCY, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, Lin S, Lee C-YG, Strom SC, Kashyap R, Fung JJ, Farese RV Jr., Patoiseau J-F, Delhon A, Chang TY, Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine, J.Biol.Chem. 275 (2000) 28083–28092. [DOI] [PubMed] [Google Scholar]
- [25].Parini P, Davis M, Lada AT, Erickson SK, Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin B, Tomoda H, Omura S, Willingham MC, Rudel LL, ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver, Circulation 110 (2004) 2017–2023. [DOI] [PubMed] [Google Scholar]
- [26].Pramfalk C, Eriksson M, Parini P, Cholesteryl esters and ACAT, Eur.J.Lipid Sci.Technol. 114 (2012) 624–633. [Google Scholar]
- [27].Goodman DS, Cholesterol ester metabolism, Physiol.Rev. 45 (1965) 747–839. [DOI] [PubMed] [Google Scholar]
- [28].d’Hollander F, Chevallier F, Qualitative and quantitative estimation of free and esterified sterols in whole rat and in 23 of its tissues and organs, Biochim.Biophys. Acta 176 (1969) 146–162. [PubMed] [Google Scholar]
- [29].Turley SD, Spady DK, Dietschy JM, Identification of a metabolic difference accounting for the hyper- and hypo-responder phenotypes of cynomolgus monkey, J.Lipid Res. 38 (1997) 1598–1611. [PubMed] [Google Scholar]
- [30].Oram JF, Lawn RM, ABCA1: the gatekeeper for eliminating excess tissue cholesterol, J.Lipid Res. 42 (2001) 1173–1179. [PubMed] [Google Scholar]
- [31].Ghishan FK, Inborn errors of metabolism that lead to permanent liver injury in: Boyer TD, Manns MP, Sanyal AJ(Eds.), Zakim and Boyer’s Hepatology : A Textbook of Liver Disease, Saunders/Elsevier, Philadelphia, PA: 2012, pp. 1155–1201. [Google Scholar]
- [32].Lopez AM, Chuang J-C, Turley SD, Impact of loss of SOAT2 function on disease progression in the lysosomal acid lipase-deficient mouse, Steroids 130 (2018) 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lopez AM, Jones RD, Repa JJ, Turley SD, Niemann-Pick C1-deficient mice lacking sterol O-acyltransferase 2 have less hepatic cholesterol entrapment and improved liver function, Am.J.Physiol.Gastroent.Liver Physiol. 315 (2018) G454–G463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, Dietschy JM, Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse, Proc.Natl.Acad.Sci.USA 106 (2009) 2377–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu B, Ramirez CM, Miller AM, Repa JJ, Turley SD, Dietschy JM, Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid, J.Lipid Res. 51 (2010) 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ramirez CM, Liu B, Aqul A, Taylor AM, Repa JJ, Turley SD, Dietschy JM, Quantitative role of LAL, NPC2, and NPC1 in lysosomal cholesterol processing defined by genetic and pharmacological manipulations. J.Lipid Res. 52 (2011) 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Taylor AM, Liu B, Mari Y, Liu B, Repa JJ, Cyclodextrin mediates rapid changes in lipid balance in Npc1−/− mice without carrying cholesterol through the bloodstream, J.Lipid Res. 53 (2012) 2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pentchev PG, Niemann-Pick C research from mouse to gene, Biochim.Biophys.Acta 1685 (2004) 3–7. [DOI] [PubMed] [Google Scholar]
- [39].Lopez AM, Ramirez CM, Taylor AM, Jones RD, Repa JJ, Turley SD, Ontogenesis and modulation of intestinal unesterified cholesterol sequestration in a mouse model of Niemann-Pick C1 disease, Dig.Dis.Sci. 65 (2020) 158–167. [DOI] [PubMed] [Google Scholar]
- [40].Lopez AM, Chuang J-C, Posey KS, Turley SD, Suppression of brain cholesterol synthesis in male Mecp2-deficient mice is age dependent and not accompanied by a concurrent change in the rate of fatty acid synthesis, Brain Res. 1654 (2017) 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dietschy JM, Spady DK, Measurement of rates of cholesterol synthesis using tritiated water, J.Lipid Res. 25 (1984) 1469–1476. [PubMed] [Google Scholar]
- [42].Lopez AM, Chuang J-C, Turley SD, Measurement of rates of cholesterol and fatty acid synthesis in vivo using tritiated water, Methods Mol.Biol. 1583 (2017) 241–256. [DOI] [PubMed] [Google Scholar]
- [43].Dietschy JM, Siperstein MD, Effect of cholesterol feeding and fasting on sterol synthesis in seventeen tissues of the rat, J.Lipid Res. 8 (1967) 97–104. [PubMed] [Google Scholar]
- [44].Sharpe LJ, Brown AJ, Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), J.Biol.Chem. 288 (2013) 18707–18715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ottinger EA, Kao ML, Carillo-Carrasco N, Yanjanin N, Shankar RK, Janssen M, Brewster M, Scott I, Xu X, Cradock J, Terse P, Dehdashti S, Marugan J, Zheng W, Portilla L, Hubbs A, Pavan WJ, Heiss J, Vite CH, Walkley SU, Ory DS, Silber SA, Porter FD, Austin CP, McKew JC, Collaborative development of 2-hydroxypropyl-β-cyclodextrin for the treatment of Niemann-Pick Type C1 disease, Curr.Top.Med.Chem. 14 (2014) 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Praggastis M, Tortelli B, Zhang J, Fujiwara H, Sidhu R, Chacko A, Chen Z, Chung C, Lieberman AP, Sikora J, Davidson C, Walkley SU, Pipalia NH, Maxfield FR, Schaffer JE, Ory DS, A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele, J.Neurosci.35 (2015) 8091–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rodriguez-Gil JL, Watkins-Chow DE, Baxter LL, Elliot G, Harper UL, Wincovitch SM, Wedel JC, Incao AA, Huebecker M, Boehm FJ, Garver WS, Porter FD, Broman KW, Platt FM, Pavan WJ, Genetic background modifies phenotypic severity and longevity in a mouse model of Niemann-Pick disease type C1, Dis.Model Mech. 13 (2020) dmm042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Edwards PA, Muroya H, Gould RG, In vivo demonstration of the circadian rhythm of cholesterol biosynthesis in the liver and intestine of the rat, J.Lipid Res.13 (1972) 396–401. [PubMed] [Google Scholar]
- [49].Jurevics H, Hostettler J, Barrett C, Morell P, Toews AD, Diurnal and dietary-induced changes in cholesterol synthesis correlate with levels of mRNA for HMG-CoA reductase, J.Lipid Res. 41 (2000) 1048–1053. [PubMed] [Google Scholar]
- [50].Xie C, Turley SD, Dietschy JM, Centripetal cholesterol flow from the extrahepatic organs through the liver is normal in mice with mutated Niemann-Pick type C protein (NPC1), J.Lipid Res. 41 (2000) 1278–1289. [PubMed] [Google Scholar]
- [51].Schϋmann K, Szegner B, Kohler B, Pfaffl MW, Ettle T, A method to assess 59Fe in residual tissue blood content in mice and its use to correct 59Fe-distribution kinetics accordingly, Toxicology 241 (2007) 19–32. [DOI] [PubMed] [Google Scholar]
- [52].Kunnen S, Van Eck M, Lecithin:cholesterol acyltransferase: old friend or foe in atherosclerosis? J.Lipid Res. 53 (2012) 1783–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Buhman KK, Accad M, Novak S, Choi RS, Wong JS, Hamilton RL, Turley S, Farese RV Jr., Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice, Nat. Med. 6 (2000) 1341–1347. [DOI] [PubMed] [Google Scholar]
- [54].Liu B, Xie C, Richardson JA, Turley SD, Dietschy JM, Receptor-mediated and bulk- phase endocytosis cause macrophage and cholesterol accumulation in Niemann-Pick C disease, J.Lipid Res. 48 (2007) 1710–1723. [DOI] [PubMed] [Google Scholar]
- [55].Lopez AM, Terpack SJ, Posey KS, Liu B, Ramirez CM, Turley SD, Systemic administration of 2-hydroxypropyl-β-cyclodextrin to symptomatic Npc1-deficient mice slows cholesterol sequestration in the major organs and improves liver function, Clin. Exp. Pharmacol. Physiol. 41 (2014) 780–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Pedrelli M, Davoodpour P, Degirolamo C, Gomaraschi M, Graham M, Ossoli A, Larsson L, Calabresi L, Gustafsson J-A, Steffensen KR, Eriksson M, Parini P, Hepatic ACAT2 knock down increases ABCA1 and modifies HDL metabolism in mice, PloS One 9 (2014) e93552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Vanier MT, Biochemical studies in Niemann-Pick disease. Major sphingolipids of liver and spleen. Biochim.Biophys.Acta 750 (1983) 178–184. [DOI] [PubMed] [Google Scholar]
- [58].Kwiterovich PO Jr, Sloan HR, Fredrickson DS, Glycolipids and other lipid constituents of normal human liver, J.Lipid Res. 11 (1970) 322–330. [PubMed] [Google Scholar]
- [59].Frantz ID Jr, Carey JB Jr, Cholesterol content of human liver after feeding of corn oil and hydrogenated coconut oil, Proc.Soc.Exp.Biol.Med. 106 (1961) 800–801. [DOI] [PubMed] [Google Scholar]
- [60].Morris MD, Bhuvaneswaran C, Shio H, Fowler S, Lysosome lipid storage disorder in NCTR-BALB/c mice. 1. Description of the disease and genetics, Am.J.Pathol. 108 (1982) 140–149. [PMC free article] [PubMed] [Google Scholar]
- [61].Xie C, Turley SD, Pentchev PG, Dietschy JM, Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein, Am.J.Physiol.Endocrinol.Metab. 276 (1999), E336–E344. [DOI] [PubMed] [Google Scholar]
- [62].Amigo L, Mendoza H, Castro J, Quiñones V, Miquel JF, Zanlungo S, Relevance of Niemann-Pick Type C1 protein expression in controlling plasma cholesterol and biliary lipid secretion in mice, Hepatology 36 (2002) 819–828. [DOI] [PubMed] [Google Scholar]
- [63].Maue RA, Burgess RW, Wang B, Wooley CM, Seburn KL, Vanier MT, Rogers MA, Chang CC, Chang T-Y Harris BT, Graber DJ, Penatti CAA, Porter DM, Szwergold BS, Henderson LP, Totenhagen JW, Trouard TP, Borbon IA, Erickson RP, A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations, Hum. Mol Genet. 21 (2012) 730–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Somers KL, Brown DE, Fulton R, Schultheiss PC, Hamar D, Smith MO, Allison R, Connally HE, Just C, Mitchell TW, Wenger DA, Thrall MA, Effects of dietary cholesterol restriction in a feline model of Niemann-Pick type C disease, J.Inherit.Metab.Dis. 24 (2001) 427–436. [DOI] [PubMed] [Google Scholar]
- [65].Cooper AD, Hepatic uptake of chylomicron remnants, J. Lipid Res. 38 (1997) 2173–2192. [PubMed] [Google Scholar]
- [66].Dietschy JM, Turley SD, Spady DK, Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans, J.Lipid Res.34 (1993) 1637–1659. [PubMed] [Google Scholar]
- [67].Lopez AM, Chuang J-C, Posey KS, Ohshiro T, Tomoda H, Rudel LL, Turley SD, PRD125, a potent and selective inhibitor of sterol O- acyltransferase 2 markedly reduces hepatic cholesteryl ester accumulation and improves liver function in lysosomal acid lipase-deficient mice, J. Pharmacol. Exp. Ther. 355 (2015) 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ohshiro T, Ohtawa M, Nagamitsu T, Matsuda D, Yagyu H, Davis MA, Rudel LL, Ishibashi S, Tomoda H, New pyripyropene A derivatives, highly SOAT2-selective inhibitors, improve hypercholesterolemia and atherosclerosis in atherogenic mouse models, J. Pharmacol. Exp. Ther 355 (2015) 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chien Y-H, Shieh Y-D, Yang C-Y, Lee N-C, Hwu W-L, Lung toxicity of hydroxypropyl-β-cyclodextrin infusion, Mol.Genet.Metab. 109 (2013) 231–232. [DOI] [PubMed] [Google Scholar]
- [70].Matsuo M, Togawa M, Hirabaru K, Mochinaga S, Narita A, Adachi M, Egashira M, Irie T, Ohno K, Effects of cyclodextrin in two patients with Niemann-Pick type C disease, Mol.Genet.Metab.108 (2013) 76–81. [DOI] [PubMed] [Google Scholar]
- [71].Davis HR Jr., Altmann SW, Niemann-Pick C1 Like1 (NPC1L1) an intestinal transporter, Biochim.Biophys.Acta 1791 (2009) 679–683. [DOI] [PubMed] [Google Scholar]
- [72].Kusters DM, Caceres M, Coll M, Cuffie C, Gagnè C, Jacobson MS, Kwiterovich PO, Lee R, Lowe RS, Massaad R, McCrindle BW, Musliner TA, Triscari J, Kastelein JP, Efficacy and safety of ezetimibe monotherapy in children with heterozygous familial or nonfamilial hypercholesterolemia, J.Pediatr. 166 (2015) 1377–1384. [DOI] [PubMed] [Google Scholar]
- [73].Beltroy EP, Liu B, Dietschy JM, Turley SD, Lysosomal unesterified cholesterol content correlates with liver cell death in murine Niemann-Pick type C disease, J.Lipid Res. 48 (2007) 869–881. [DOI] [PubMed] [Google Scholar]
- [74].Chuang J-C, Lopez AM, Posey KS, Turley SD, Ezetimibe markedly attenuates hepatic cholesterol accumulation and improves liver function in the lysosomal acid lipase-deficient mouse, a model for cholesteryl ester storage disease, Biochem.Biophys.Res.Commun. 443 (2014) 1073–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Di Rocco M, Pisciotta L, Madeo A, Bertamino M, Bertolini S, Long term substrate reduction therapy with ezetimibe alone or associated with statins in three adult patients with lysosomal acid lipase deficiency, Orphanet J. Rare Dis. 13 (2018) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]