

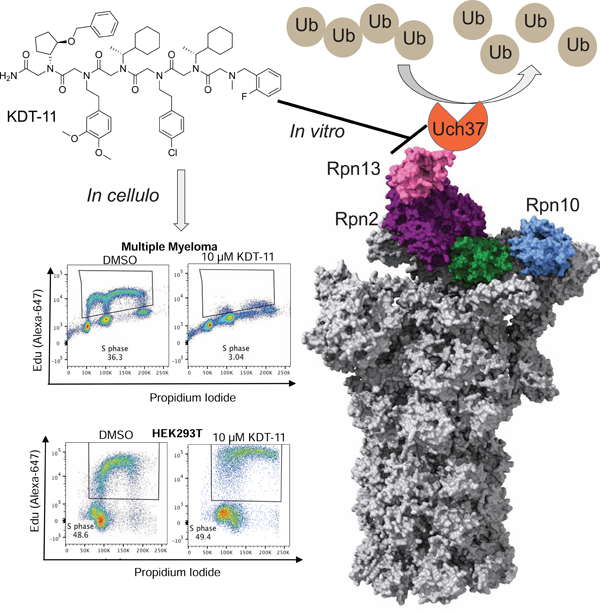
Summary
We previously reported a peptoid ligand for the proteasomal ubiquitin receptor Rpn13 called KDT-11 and demonstrated that this compound is toxic to multiple myeloma (MM) cells, but not non-malignant cells. We show here that KDT-11 decreases the viability of a variety of cancer cell lines, especially melanomas and various blood cancers. The peptoid induces selective G1 cell cycle arrest, resulting in eventual apoptosis. While KDT-11 does not antagonize any of the known protein-protein interactions involving Rpn13, the peptoid inhibits the ability of Rpn13 to stimulate the activity of an associated deubiquitylase Uch37/UCHL5 in vitro, suggesting a high level of Uch37 activity might be important for cancer cell proliferation. However, a variety of experiments in SK-MEL-5 melanoma cells suggest that KDT-11’s cytotoxic effects are mediated by interactions with proteins other than Rpn13.
Graphical Abstract
eTOC:
Dickson et al. characterize the mechanism of action of peptidomimetic KDT-11, previously reported to bind Rpn13 and reduce viability of cancer cells. Through biochemical and cellular approaches, the group demonstrates that KDT-11 does not disrupt Rpn13’s known protein interactions, nor is cellular sensitivity to KDT-11 correlated with Rpn13 levels.
Introduction
Most non-lysosomal protein degradation is executed by the ubiquitin proteasome system (UPS) (DeMartino and Slaughter, 1999). The majority of proteasome substrates are post-translationally modified by poly-ubiquitin (Ub) chains (Hershko and Ciechanover, 1998), which are recognized by several ubiquitin receptor subunits on the 19S regulatory particle of the 26S proteasome (Yu and Matouschek, 2017). Substrate proteins are then unfolded by a hexameric AAA ATPase ring, which allows the unstructured polypeptide to access the interior of the barrel-like 20S core particle where it is degraded (Groll et al., 2005). Typical of most post-translational modifications (PTMs), ubiquitylation Is a dynamic event that is opposed by deubiquitylase enzymes. There are over 100 annotated deubiquitylases in the human proteome, three of which (Uch37, Usp14, and Rpn11) are associated with the proteasome (Reyes-Turcu et al., 2009).
Proteasome active site inhibitors such as bortezomib and carfilzomib are used clinically to treat multiple myeloma (MM), which is a cancer of plasma cells (Rajkumar and Kumar, 2016). These cells are particularly sensitive to proteasome inhibition. This is thought to be due, at least in part, to the high-level production of immunoglobulins that places considerable stress on the folding quality control apparatus, including the proteasome . Unfortunately, bortezomib has been unsuccessful as a single agent to treat solid tumors, partly because of its dose-limiting toxicity (Huang et al., 2014). All cells require some level of proteasome function, so it is unsurprising that inhibiting the catalytic activity of the complex impairs the viability of non-malignant cells. The limitations of currently available proteasome inhibitors led our group to explore an alternative subunit of the proteasome as a potential anti-cancer target. Rpn13 is a 19S regulatory particle subunit that recruits polyubiquitinated substrates to the complex (Husnjak et al., 2008). It also recruits the deubiquitylase Uch37 (also known as UCHL5) to the 26S proteasome and strongly stimulates its enzymatic activity (Yao et al., 2006). Rpn13 is unique among proteasome subunits in that it is encoded by a non-essential gene (Al-Shami et al., 2010; Hamazaki et al., 2015). On the other hand, there are multiple reports linking Rpn13 expression to cancer cell proliferation, suggesting that some malignant cells depend on Rpn13 activity for growth and survival (Chen et al., 2009; Fejzo et al., 2013; Fejzo et al., 2008; Fejzo et al., 2011; Jang et al., 2014; Jiang et al., 2017; Shenoy, 2015). This apparent difference in the requirement for Rpn13 in some malignant and non-malignant cells suggests that it might be an interesting chemotherapeutic target.
To explore this idea, our laboratory previously screened a peptoid library for ligands to Rpn13 in vitro. One of the compounds that resulted from this effort, KDT-11 (Fig. 1A), was shown to engage Rpn13 selectively with a modest KD of about 3 μM in vitro (Trader et al., 2015). When MM cells were exposed to KDT-11, a dose-dependent inhibition of their proliferation was observed with an IC50 that matched the KD of the peptoid-Rpn13 complex in vitro. Moreover, Western blotting showed that KDT-11 treatment resulted in a build-up of poly-ubiquitylated proteins, consistent with the compound having some inhibitory effect on the ubiquitin-proteasome pathway. In line with the fact that Rpn13 is non-essential in healthy cells, no effect on proliferation of the non-malignant HEK293T cell line was observed, even at the solubility limit of KDT-11. These preliminary data suggested to us that a deeper exploration of the mechanism of action of the mechanism of KDT-11 is justified, which is reported here.
Figure 1. KDT-11 broadly inhibits cancer cell growth.
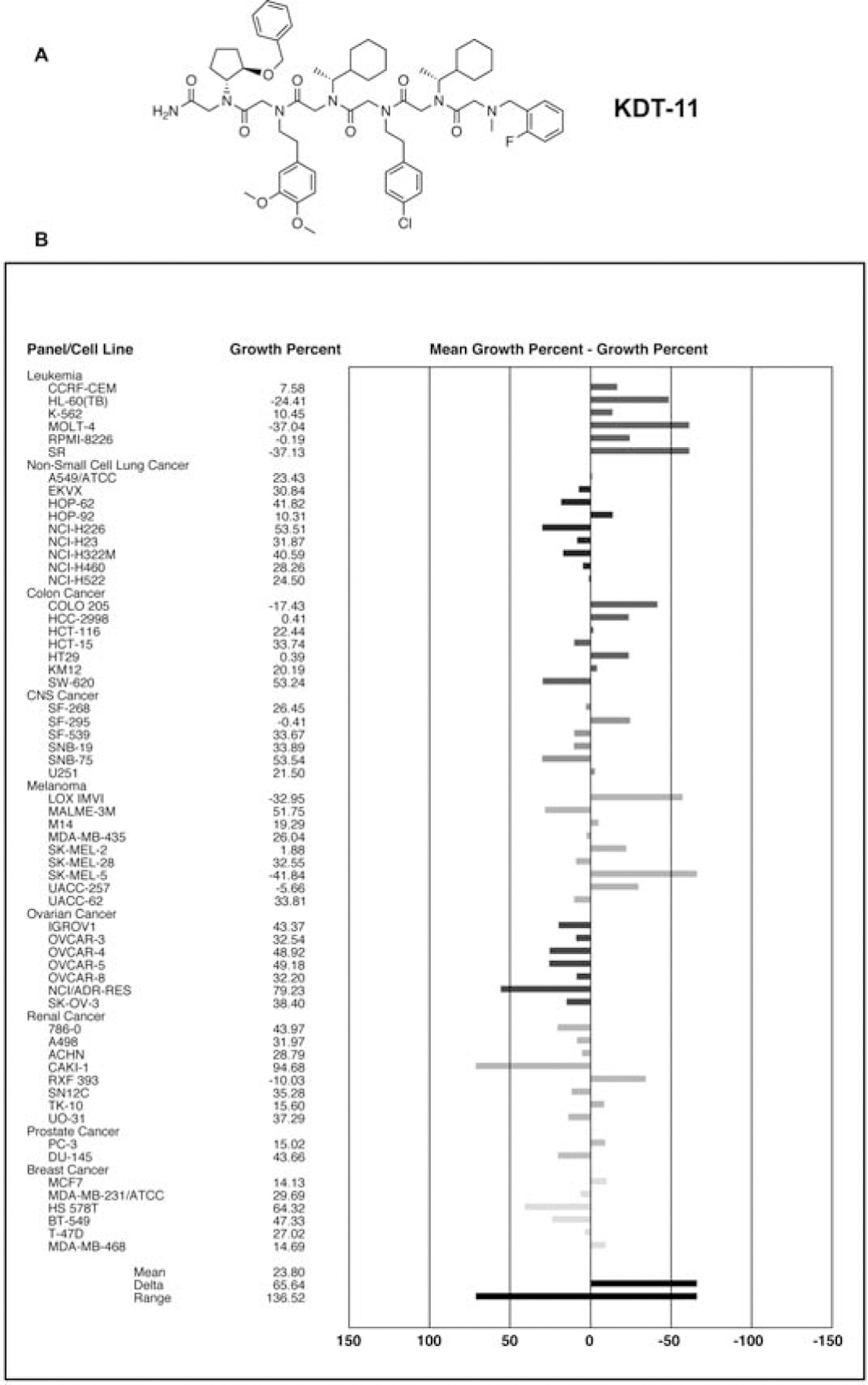
(A) Structure of KDT-11. (B) The NCI60 panel of cell lines was exposed to KDT-11 (10 μM). Growth was assessed 48 hours after compound treatment. See also Supplemental Figure 1.
Results
KDT-11 has activity against a broad range of cancer cell lines
Our previous work showed that KDT-11 was toxic to multiple myeloma (MM) cells, but not a non-malignant HEK293T cell line (Trader et al., 2015). To determine the generality of this result we exposed the NCI60 lines to 10 μM KDT-11 and monitored their growth after 48 hours. As shown in Figure 1B, almost all of these lines were growth inhibited by KDT-11, though to significantly different extents. Many displayed a high level of dead cells, particularly leukemia, colorectal and melanoma cell lines. Further analysis using five different does of KDT-11 against some of the NCI60 cell lines confirmed the result shown in Figure 1B, with the IC50 being approximately 5 μM in the most sensitive lines (see Supplemental Figure 1), which is close to the KD of the KDT-11-Rpn13 complex in vitro. The relatively broad, albeit modestly potent, activity of KDT-11 justified a further analysis of its mechanism of action.
KDT-11 does not inhibit any of the known protein-protein interactions involving Rpn13
Since KDT-11 was identified simply for its ability to bind selectively to Rpn13 in vitro, rather than in a functional screen, it is of interest to ask if the peptoid modulates one or more of the known activities of Rpn13, at least in vitro. As mentioned above, Rpn13 is a ubiquitin receptor and is tethered to the proteasome via an interaction with the extreme C-terminus of the Rpn2 protein (Lu et al., 2017; VanderLinden et al., 2017). Disruption of either of these interactions would presumably prevent Rpn13 from contributing to the capture of poly-ubiquitylated substrates by the proteasome. In addition, Rpn13 recruits the Uch37 deubiquitylase to the proteasome and strongly stimulates its activity (Yao et al., 2006).
To assess the effect of the peptoid on these binding events, His6-Rpn13 (1 μM) was pre-incubated with KDT-11 (50 μM) or vehicle (0.5% DMSO) for one hour followed by addition of either ` or MBP-Uch37. After a two-hour incubation the complexes were pulled down and analyzed by denaturing SDS-PAGE and staining with Coomassie Brilliant Blue. As shown in Figure 2 (A and B) the peptoid had no effect on either of these interactions.
Figure 2. KDT-11 does not perturb Rpn13’s known protein-protein interactions, but blunts the ability of Rpn13 to activate the deubiquitylase activity of Uch37 in vitro.
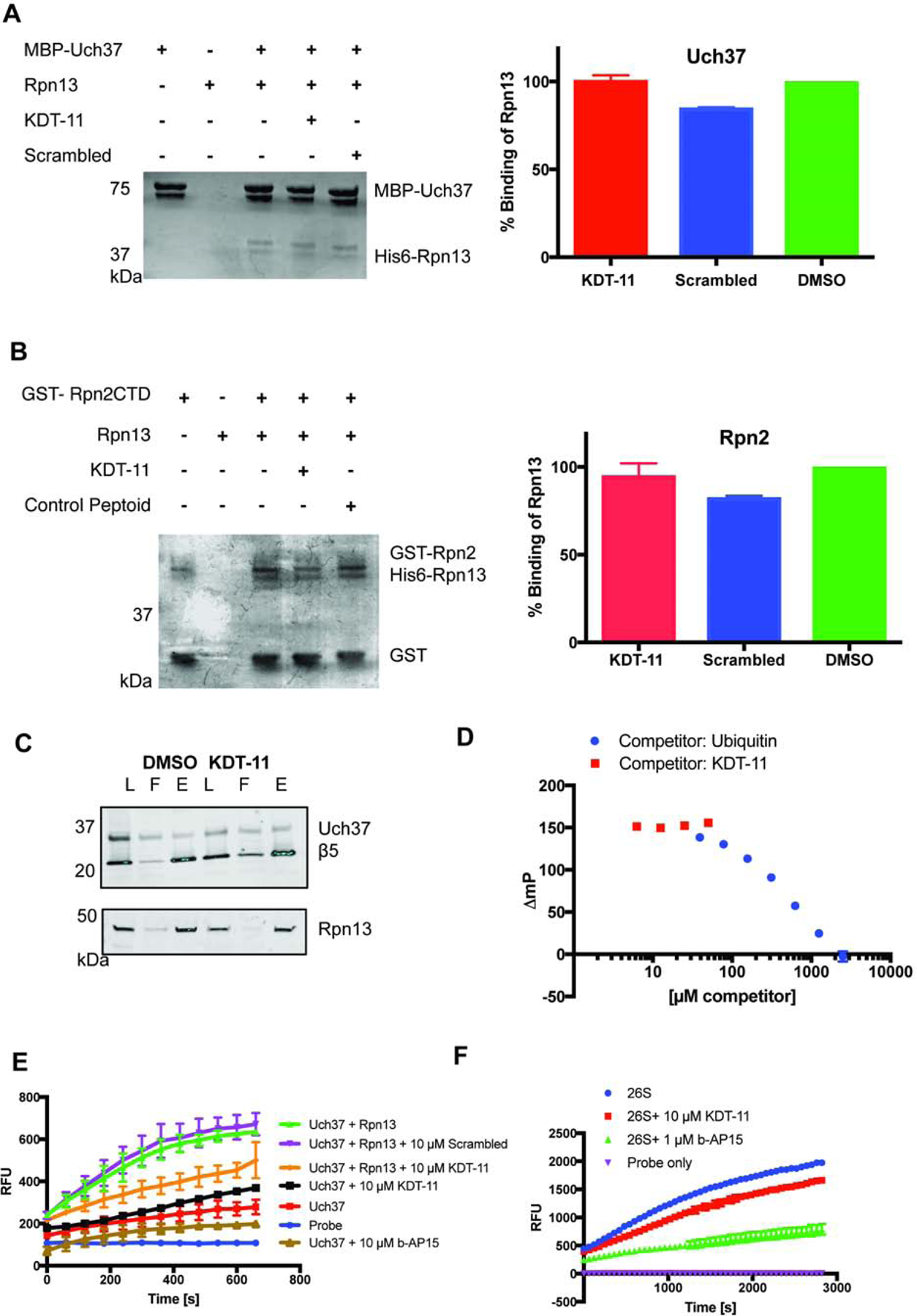
(A) Effect of KDT-11 on the interaction of recombinant Rpn13 and Uch37. His6-Rpn13 and MBP-Uch37 (1 μM each) were incubated with KDT-11 (50 μM), a scrambled peptoid (50 μM) or vehicle (0.5% DMSO). MBP-Uch37 was pulled down using amylose resin. The proteins present in the precipitate were analyzed by SDS-PAGE and Coomassie Brilliant Blue staining. The bar graphs to the right of the gels represent the quantitative values obtained from biological triplicate experiments and the error bars represent the standard deviation of these readings. The structure of the scrambled control peptoid is shown in Supplementary Figure 2. (B) Effect of KDT-11 on the interaction of recombinant Rpn13 and Rpn2. This experiment was conducted exactly as described above for Rpn13-Uch37, except that a GST-Rpn2 fusion protein containing the Rpn13-interacting domain of Rpn2 was substituted for MBP-Uch37 and the proteins were pulled down using glutathione-agarose beads. (C) Effect of KDT-11 on the Rpn13-Uch37 interaction in the context of the proteasome. SK-MEL-5 cells were treated with KDT-11 (10 μM) or 0.1% DMSO (vehicle) for 16 hours, lysed and the proteasome was immunoprecipitated with anti-β5 antibody. The precipitated proteins were analyzed by SDS-PAGE and Western blotting using antibodies against β5, Rpn13, and Uch37. The western blot for DMSO and KDT-11 treated SK-MEL-5 cells included cell lysate (L=load), unbound material (F=flow through), and the resin-bound eluate (E = elution). (D) 1 μM Rpn13 was mixed with an equal volume of 50 nM ubiquitin K48C-fluorescein. This complex was then challenged with increasing amounts of KDT-11 or unlabeled Ubiquitin and the change in the FP signal was measured. The addition of KDT-11 had no effect on the complex, whereas increasing unlabeled ubiquitin (self-challenge) resulted in a decrease in the FP signal. The polarization values are presented as ∆mp, which indicates that the mP value of unliganded Ubiquitin K48C-Fluorescin has been subtracted from all of the values.
(E) The effect of KDT-11 on the deubiquitylase activity of recombinant Uch37 was monitored in the presence and absence recombinant Rpn13. The assay followed the Uch37-mediated hydrolysis of the fluorogenic substrate Ubiquitin-AMC fluorogenic probe. His6-Rpn13 (50 nM), if present, was incubated with KDT-11 (10 μM) or vehicle in assay buffer for 30 minutes at room temperature, after which recombinant Uch37 was added to a final concentration of 10 nM. After a further 30 minute incubation, AMC probe (0.5 μM) was added. Fluorophore release was measured every 60 seconds for 30 minutes. Points and error bars represent the mean and standard deviation, respectively, of biological triplicate readings. (F) Effect of KDT-11 and b-AP15 on the deubiquitylase activity of purified proteasome, as monitored by hydrolysis of Ub-AMC. Purified 26S proteasome was pre-incubated with vehicle, KDT-11 (10 μM), or b-AP15 (1 μM) followed by addition of Ub-AMC. Points and error bars represent the mean and standard deviation, respectively, of biological triplicate readings.
To assess this point in a more physiological setting, 26S proteasome was immunoprecipitated (using anti-β5 (a 20S component) antibody) from lysates of SK-MEL-5 melanoma cells that had or had not been treated with KDT-11 overnight. Rpn13 and Uch37 levels in each immunoprecipitate were determined by immunoblotting (Figure 2C). The data show that KDT-11 has no effect on the amount of Rpn13 or Uch37 associated with the proteasome. From these data, we conclude that KDT-11 does not antagonize Rpn13-Rpn2 nor Rpn13-Uch37 binding.
To assess the effect of KDT-11 on Ubiquitin binding to Rpn13, we employed a previously established fluorescence polarization assay (Du and Strieter, 2018). Briefly, KDT-11, unlabeled ubiquitin or vehicle were mixed with 1μM Rpn13 and a constant amount of a Ubiquitin-fluorescein conjugate (50 nM). Ubiquitin-Rpn13 association was followed by the change in fluorescence polarization. Ubiquitin was able to compete with its fluorescein-labeled counterpart in a concentration-dependent manner, whereas KDT-11 did not have any observable effect on Ubiquitin-Fluorescein binding up to saturating concentrations of the ligand (Figure 2D). Rpn13’s affinity for Ubiquitin is ~40 μM according to Hill et al 2017, while its affinity for KDT-11 is ~3 μM (supplemental figure 3), which means the concentration of these two competitors required for loss of fluorescent conjugate binding will be different based on their respective affinities. Therefore, we would expect KDT-11 to compete with ubiquitin-fluorescein at a lower concentration than what is required for self-competition using unlabeled ubiquitin. Although we could not saturate our self-competition with ubiquitin, we can be fairly confident that the concentrations of KDT-11 used in this experiment would be sufficient to displace Ubiquitin binding if its binding site was competitive.
Other than protein-protein interactions, the only other known activity of Rpn13 is to stimulate the deubiquitylase activity of Uch37 (Yao et al., 2006). To evaluate the effect of the peptoid on this activity, KDT-11 or vehicle was added to a solution containing Uch37 (10 nM) and the fluorogenic substrate Ubiquitin-aminomethylcoumarin (Ub-AMC) in the presence or absence of saturating Rpn13 (50 nM). Uch37-catalyzed hydrolysis of the Ub-AMC bond was followed by monitoring the increase in fluorescence. As shown in Figure 2E, Uch37 alone has weak deubiquitinase activity, which is not inhibited by the addition of excess KDT-11 (10 μM). This is consistent with the fact that KDT-11 shows no detectable affinity for Uch37 (Supplemental Figure 3). As expected, addition of Rpn13 to the Uch37-catalyzed reaction resulted in a stimulation of the rate of hydrolysis. When this experiment was repeated in the presence of KDT-11 (10 μM), significant inhibition of activity was observed, whereas addition of a peptoid with the same residues as KDT-11, but with their order scrambled (Supplementary Figure 2), had no effect. Using a two-tailed t-test, we have determined the p-value for 10 μM KDT-11 (compared to DMSO control) to be p<0.0001, for 1 μM p=0.014, and for 10 μM scrambled p=0.638.
The rate of Uch37-mediated hydrolysis observed in the presence of Rpn13 and KDT-11 was intermediate between the unstimulated and Rpn13-stimulated rate. In the buffer employed for this experiment, 10 μM was the highest concentration of KDT-11 that could be achieved. Given the KD of the complex (≈ 3 μM (Trader et al., 2015)), we would expect Rpn13 to be slightly more than 50% occupied with KDT-11 under these conditions. This suggests that if full occupancy of Rpn13 by the peptoid could have been achieved, the rate of hydrolysis would be reduced back to that of the unstimulated reaction, though validation of this hypothesis will have to await the development of more potent and/or soluble analogues of KDT-11. Unfortunately, as described below, our efforts to do so have so far met with little success.
We also evaluated the effect of KDT-11 on the deubiquitylase activity of the proteasome. Purified 26S human proteasome was treated with KDT-11 (10 μM) or vehicle, and the rate of Ub-AMC hydrolysis was measured. As shown in Fig. 2F, KDT-11 inhibited the rate of hydrolysis modestly, as expected. We did not compare unscrambled KDT-11 given the demonstrated lack of activity in the recombinant protein assay shown in Fig. 2E, however the calculated p-value for KDT-11 inhibition of Uch37 is p=0.0003.
The proteasome has other deubiquitylases besides Uch37, including USP14 and Rpn11, that would not be expected to be inhibited by KDT-11, so the remaining activity probably reflects the activity of these other enzymes. This is supported by the fact that treatment of the proteasome with the dual Uch37/USP14 inhibitor b-AP15 (D’Arcy et al., 2011; D’Arcy et al., 2015) resulted in greater inhibition (Figure 2F). Taken together, these data are consistent with the idea that KDT-11 can inhibit the Rpn13-stimulated activity of Uch37 in the context of the full proteasome, at least in vitro.
KDT-11 blocks cell cycle progression at the G0/G1 to S boundary and drives subsequent apoptosis
We reported previously that KDT-11 dose-dependently inhibits the proliferation of MM cells (Trader et al., 2015). To better understand the nature of this effect, we asked whether this reduction in proliferation was mediated through a cytostatic effect, a cytotoxic effect, or both. MM1.R cells exposed to KDT-11 or vehicle were treated with Cell ToxGreen, a cell impermeable DNA-intercalating dye that serves as a marker of cell death since it can only access the genome upon disruption of the membrane. As shown in Figure 3A, MM cell staining with this reagent increased steadily over the course of 48 hours, indicating slow induction of cell death. As anticipated from our previous study, no such effect was observed when HEK293T cells were treated with the peptoid. Induction of cell death displayed the expected dose-dependence (Figure 3B)
Figure 3. KDT-11 slowly induces apoptosis in MM1.R cells but not HEK293T cells.

(A) Time course for cell killing by KDT-11 (50 μM), as measured by binding of the cell impermeable dye CellTox Green to DNA. Bars represent the mean of technical triplicate readings, with error bars corresponding to the standard deviation of these readings. (B) Dose dependence of cell killing by KDT-11 as measured using the CellTox Green (Promega) assay. Points represent the mean of triplicate readings, and error bars represent the standard deviation of these measures (n=3). (C) Dose dependence of the induction of apoptosis by KDT-11. Apoptosis induction was measured by caspase 3/7 activity via the Caspase-Glo substrate (Promega). Background signal from vehicle control treated cells was subtracted as a baseline. The mean and standard deviation of experimental triplicates were plotted as a function of compound concentration.
To determine if the pathway for cell death involves apoptosis, Caspase-3/7 activity was quantified (Figure 3C). Indeed, treatment of MM1.R cells with KDT-11 for 24 hours resulted in a dose-dependent increase in caspase activity, whereas no caspase induction was seen in HEK293T cells. We observed large variability in the apoptotic induction of MM1.R cells at 10 μM KDT-11 (see this point in Figure 3C), which may be due to a slow and uneven induction of apoptosis, which can be corroborated by the single-cell level flow cytometry data in Figure 4 (vide infra).
Figure 4. Time course of apoptosis induction in MM1.R cells by KDT-11.

MM1.R cells were treated with 10 μM KDT-11 (10 μM) or vehicle (0.1% DMSO). At the times indicated, cell death was assessed by staining the cells with PI and Annexin-V Brilliant Violet 510 followed by flow cytometry. Apoptotic cells are Annexin-V positive and PI negative. Plots are representative of triplicate experiments. The data show a slow induction of apoptosis in the KDT-11-treated cells. For bortezomib control data, see supplemental figure 4.
To further examine the induction of apoptosis upon compound treatment, Annexin V and propidium iodide (PI) staining was performed on MM.1R and HEK293T cells at 8, 16, and 24 hours following compound treatment. Cells undergoing apoptosis will be detected by extracellular Annexin V staining yet remain unstained by PI until cell death occurs. The prototypical proteasome inhibitor Bortezomib induces apoptosis and cell death rapidly. (Supplementary Figure 4). Almost all of the Bortezomib-treated cells are heavily stained with both dyes within 16 hours. In contrast, whereas KDT-11 also increases the number of cells that stain positive for Annexin V and PI, the effect is far less dramatic and much slower (Figure 4). Only ≈ 6% of the cells were both Annexin V- and PI-positive for at 16 hours, compared to ≈ 85% in the Bortezomib-treated population (Supplementary Figure 4). Even at 24 hours, most of the cells stain poorly with PI, indicating that their membranes remain intact (Figure 4). Obviously, KDT-11 and Bortezomib have very different effects in MM cells, arguing that the peptoid does not broadly antagonize proteasome activity.
Taken together, these data suggest that KDT-11 has a rapid cytostatic effect on cell proliferation in cancer cells, followed by a slower apoptotic response. The proteasome is well known to be intimately involved in cell cycle regulation (Glotzer et al., 1991). While the role of Rpn13 in proteasome-mediated degradation of cell cycle proteins is unclear, a recent study reported at least a modest effect on cell cycle progression when Rpn13 was knocked down using siRNA in HeLa cells (Randles et al., 2016). A similar result was observed when Uch37 was knocked down as well. Therefore, we assessed the effect of KDT-11 on cell cycle progression in HEK293T and MM.1R cells using flow cytometry (Figure 5). Total DNA content was measured by propidium iodide (PI) staining after cell fixation and newly synthesized DNA was measured by monitoring incorporation of the thymidine analog 5-ethynyl-2’-deoxyuridine (Edu). Vehicle treated MM.1R cells show a robustly cycling population, including cells with an 8n chromosome content as determined by PI staining. This is characteristic of some cancers, and is enhanced in those who have developed resistance to treatment (Thompson and Compton, 2011). MM.1R is a patient-derived, dexamethasone-resistant multiple myeloma cell line (Goldman-Leikin et al., 1989) that contains a distinct binucleated population. When MM.1R was treated with KDT-11 (10μM) for 24 hours, the S phase population was almost completely absent (Figure 5A). In stark contrast, there was no significant difference in the percentage of HEK293T cells in S phase whether they were treated with vehicle or KDT-11 (Figure 5B).
Figure 5. Effect of KDT-11 on cell cycle progression.

The cells indicated were treated with vehicle (0.1% DMSO) or KDT-11 (10 μM) for 24 hours. They were then incubated with 10 μM EdU for 90 minutes to label S-phase cells, followed by labeling the newly incorporated DNA with Alexa647-azide and staining with propidium iodide for total DNA content. The cells were then analyzed by flow cytometry. Bar graphs represent the mean of triplicate readings, error bars represent standard deviation of these readings. (A) MM1.R cell cycle profile (B) HEK293T cell cycle profile (C) SK-MEL-5 cell cycle profile. The data show that KDT-11 blocks entry into S phase rapidly and almost completely in the two cancer cell lines but has almost no effect on the fraction of HEK293T cells in S phase. See Supplemental Figure 5 for additional SK-MEL-5 data. (D) MM.1R cells were treated with KDT-11 (10 μM) or vehicle (DMSO) for 24 hours. P21 and p27 levels (relative to GAPDH) were then assessed by SDS-PAGE and Western blotting. Quantitation of the Western blots using ImageJ. Error bars represent the standard deviation of triplicate readings. Protein was quantified using ImageJ analysis and Excel. A two-tailed Student’s t-test was performed between DMSO and KDT-11. p - value for p27 levels = 0.0001. p-value for p21 = 0.0249.
For reasons elaborated below, we also wished to assess the effect of KDT-11 on the cell cycle in SK-MEL-5 cells, a melanoma-derived cell line that is also highly sensitive to KDT-11 (Figure 1 and Supplementary Figure 5). As shown in Figure 5C, treatment of these cells with KDT-11 again virtually eliminated the S phase population (two-tailed t-test, p=0.0005). In this case, it was clear that there was additionally a decrease in the G2/M population (two-tailed t-test, p=0.01) as well and an increase in the G0/G1 population (two-tailed t-test, p=0.037). These data argue that KDT-11 introduces a strong block to cell cycle progression at the G0/G1 to S phase transition.
One possible explanation for this observation is that KDT-11 impairs the proteolysis of certain proteins that must be turned over in order for cells to proceed into S phase. For example, p27kip1 and p21waf1 are proteins that function to inhibit cyclin-dependent kinases that mediate the G1/S transition (Shirane et al., 1999) and are consumed by the proteasome at the appropriate time. Therefore, lysates prepared from MM.1R cells incubated with vehicle or KDT-11 (10 μM) for 24 hours were analyzed by SDS-PAGE and Western blotting using anti-p27 or anti-p21 antibodies. As shown in Figure 5D, KDT-11 indeed drove a 2- to 2.5-fold increase in p27kip1 and p21waf1 levels compared to cells treated with vehicle.
Genetic manipulation of Rpn13 does not phenocopy the cellular effects of KDT-11
The data presented above show that KDT-11 inhibits Rpn13’s ability to stimulate the deubiquitylase activity of Uch37 in vitro (without disrupting the physical interaction of the two proteins) and that it selectively arrests cell cycle progression in myeloma and melanoma cell lines, (MM1.R and SK-MEL-5) but not in a non-malignant line. This suggests that Rpn13 stimulation of Uch37 is important for cell cycle progression in these cancer cells. To test this idea, we asked if knockdown of Rpn13 protein levels using genetic means recapitulates the phenotype driven by KDT-11.
Unfortunately, we were unable to achieve efficient knockdown of Rpn13 in MM cells, which, in our hands, were difficult to transfect efficiently. Therefore, we turned to the SK-MEL-5 melanoma cell line, which is sensitive to KDT-11 (Supplementary Figure 5) and exhibits the same KDT-11-driven blockade to entry of the cells into S phase as MM.1R cells (Figure 5C). Pooled sets of 4 targeting siRNAs per gene (Dharmacon) were transiently transfected into SK-MEL-5 cells to knock down Rpn13 Uch37, or two control proteins, GAPDH and USP14. After 48 hours, protein levels were quantified by SDS-PAGE and Western blotting (Figure 6A). Rpn13 and Uch37 levels were reduced to 14% and 11%, respectively, of those found in cells treated with a control siRNA. Knockdown of GAPDH and USP14 was also efficient. We also observed a reduction in Uch37 protein level upon Rpn13 siRNA treatment, which was also observed previously in HeLa cells (Randles et al., 2016). We then monitored cell viability at 24, 48 and 72 hours post-transfection. As shown in Figure 6B, all of the cells exhibited approximately equal viability regardless of the siRNA with which they were treated. Clearly, neither knockdown of Rpn13 nor Uch37, at least to the levels achieved in these experiments, recapitulates the phenotype exhibited by KDT-11 treatment in melanoma cells. A possible explanation for this surprising result is that the Rpn13 or Uch37 remaining after knockdown was sufficient to support cancer cell proliferation. We attempted to use CRISPR/Cas9-based methods to generate Rpn13 and Uch37 knockout derivatives of SK-MEL-5, but unfortunately, these efforts did not result in viable deletion clones.
Figure 6. Analysis of the effect of si-RNA-mediated knockdown of Rpn13 on melanoma cell viability and KDT-11 sensitivity.
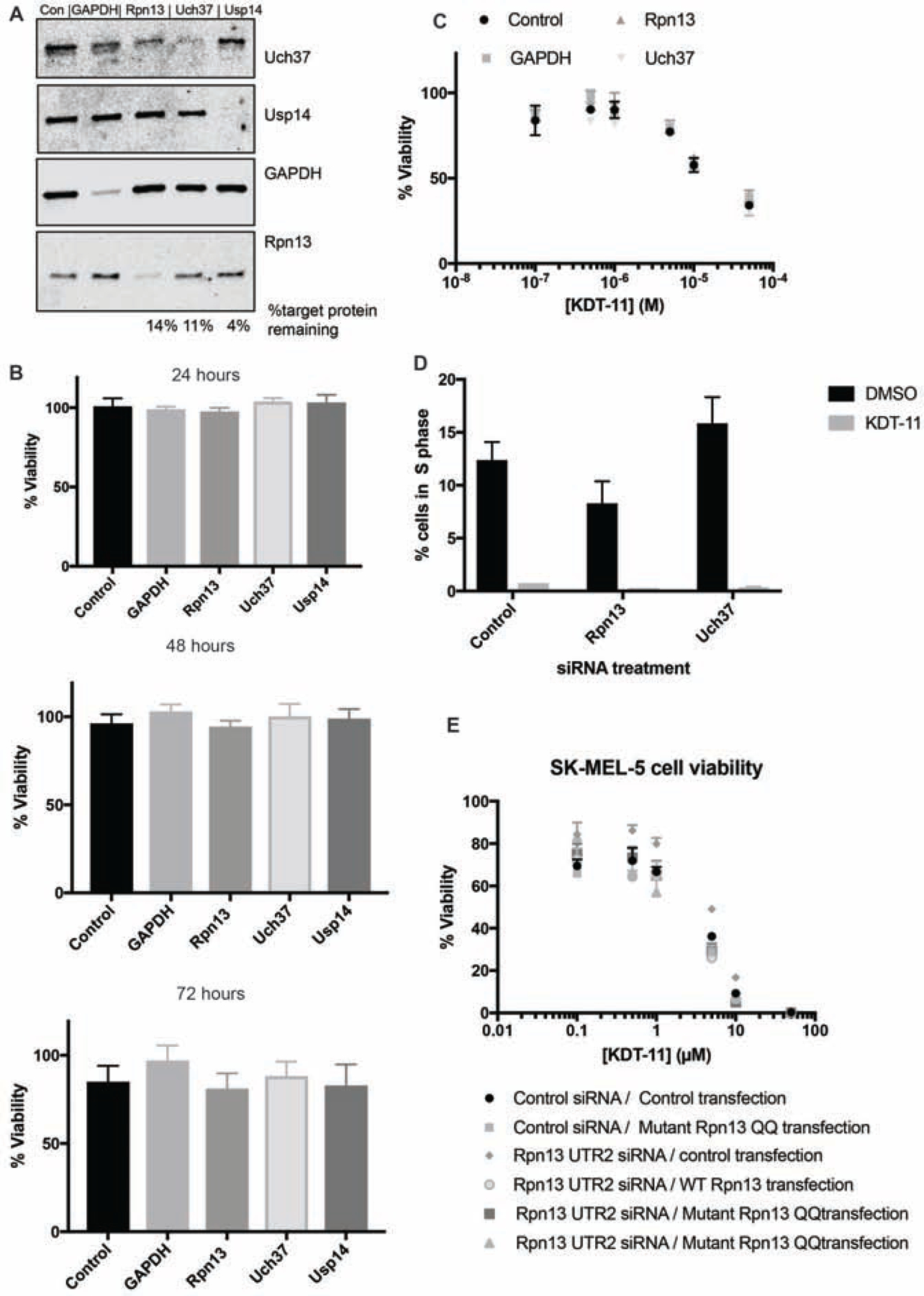
(A) SDS-PAGE/Western blot analysis of protein levels in cells treated for with the indicated siRNAs (top of gel). Lysates were prepared 48 hours after transfection with the siRNAs. Band intensities were quantified using ImageJ. (B) Viability of cells of cells treated with the siRNAs indicated was determined using the Cell Titer Glo 2.0 at the time indicated. Bars and error bars represent the mean and standard deviation of experimental triplicate readings. The data show that cell viability was unaffected by knockdown of Rpn13 or Uch37.
(C) Cells were treated with 50 nM of pooled siRNA targeting Rpn13 ,Uch37 or GAPDH for 48 hours. They were then exposed to varying concentrations of KDT-11, or vehicle. Cell viability was measured using the Cell Titer Glo 2.0 viability assay 24 hours later. The assays were conducted in technical triplicate, with error bars representing the standard deviation of the mean of the measurements. (D) Analysis of the cell cycle in cells with the indicated protein knocked down. Cells were treated with 10 μM KDT-11 for 24 hours, followed up cell cycle analysis. Flow cytometry-based cell cycle analysis was carried out as described in Figure 5 and the percentage of cells in S phase was quantified. The assays were conducted in technical triplicate, with error bars representing the standard deviation of the mean of the measurements. (E) Endogenous Rpn13 was depleted in SK-MEL-5 cells by UTR-targeting siRNA for 24 hours, followed by transfection with plasmids that drive the expression of wild-type Rpn13 or Rpn13Q337A/Q338A (Rpn13QQ), which is defective in Uch37 activation. Cells were then treated with indicated concentrations of KDT-11 for 24 hours. Viability was assessed by Cell Titer Glo 2.0 in technical triplicate and normalized to vehicle control. The experiment was done in biological duplicate. The error bars represent the standard deviation of the mean of these measurements. The data show that expression of Rpn13Q337A/Q338A does not affect the viability or KDT-11 sensitivity of SK-MEL-5 cells, see supplemental figure 6 for blotting data.
Another classical chemical biology test for target validation is to determine if the sensitivity of cells to the bioactive compound is sensitive to the level of the presumed target. We therefore proceeded to ask if manipulation of the level of the Rpn13 protein alters the sensitivity of SK-MEL-5 cells to KDT-11. As shown in Figure 6C, knockdown of Rpn13 had no effect on the sensitivity of the cells to KDT-11. The same result was observed in the cells where Uch37 had been knocked down. Half-maximal inhibition of viability was still observed at about 10 μM KDT-11 in each case. We also examined cell cycle progression in SK-MEL-5 cells transfected with control siRNA, or siRNAs targeted to Rpn13 or Uch37. As shown in Figure 6D, these manipulations had little effect on the fraction of the population in S phase. However, when the cells were treated with KDT-11 (10 μM), the S phase population was lost almost completely. Thus, contrary to our initial expectation, cells do not become more sensitive to KDT-11 when Rpn13 levels are decreased by almost 10-fold, whether assessed by gross viability or entry into the S phase of the cell cycle.
Given that KDT-11 blunts the Rpn13-mediated activation of Uch37 in vitro, we also asked if mutations in Rpn13 known to dampen its ability to activate Uch37 might phenocopy the effect of KDT-11. The double mutant Rpn13Q337A/Q338A only stimulates Uch37 about 30% as well as wild-type Rpn13, but the mutations do not affect the physical interaction of the two proteins (VanderLinden et al., 2015). We first knocked down native Rpn13 to low levels using siRNAs targeted to the UTR of Rpn13 (Supplementary Figure 6A). These cells were also transfected with a plasmid directing the expression of FLAG-tagged Rpn13Q337A/Q338A or, as a control, FLAG-tagged wild-type protein. Both expressed well (Supplementary Figure 6B). The cells were then tested for viability. As shown in Figure 6E, expression of the Rpn13 double mutant that only poorly activates Uch37 had no significant effect on viability relative to cells in which wild-type Rpn13 was expressed or to cells in which Rpn13 had not been knocked down at all. Clearly, overexpression of Rpn13Q337A/Q338A does not phenocopy the effect of treating cells with KDT-11. Moreover, when these cells were treated with KDT-11 all exhibited approximately equal sensitivity to the peptoid (Fig. 6E).
Analysis of KDT-11 analogs
Efforts were undertaken to identify analogs of KDT-11 with increased activity and perhaps more favorable physical properties such as a lower clogP value (predicted to be 14.6 for KDT-11). These efforts will be reported in detail elsewhere. Briefly, dozens of analogs of KDT-11 were created by parallel solid-phase synthesis. The molecules were tagged with fluorescein on-resin, then released into solution where their affinity for Rpn13 was measured by a change in fluorescence polarization after exposure to the protein. No compounds with improved affinity for Rpn13 were identified. We used Rpn13 binding as our optimization assay because Uch37 inhibition by KDT-11 is not extremely strong. Given these molecules lack of improved binding to Rpn13, we did not test these analogs in the Ub-AMC assay, as we do not expect any change in their ability to inhibit Uch37 activity. We did find a few somewhat less hydrophobic molecules, such as KPD-11 and KPD-12 (Figure 7A) that retained the ability to bind Rpn13, albeit with modestly reduced affinities: KDT-11 and KPD-12 have similar binding affinities of 3.5 μM and 3.3 μM, respectively, while KPD-11 is crippled to 40.6 μM (Fig. 7B). We compared the toxicity of these compounds with that of KDT-11 in the MM.1R and SK-MEL-5 cell lines (Figure 7 C). In the myeloma cell line, KPD-12 displayed weak activity corresponding to a 13.2 μM IC50 , while KPD-11 was slightly more cytotoxic than KDT-11(2.4 μM as compared to 6 μM). In the melanoma line, KPD-11 (IC50 = 6.8 μM). was considerably more active than KDT-11 while KPD-12 (IC50 =34.2 μM) displayed activity almost identical to that of KDT-11(IC50 = 14.7 μM). These data show that the toxicity of KDT-11 derivatives does not correlate well with their affinity for Rpn13 in vitro.
Figure 7. Determination of Rpn13 affinity and cellular cytotoxicity of two KDT-11 analogs.

A. Structures of KPD-11 and KPD-12, with the structural differences from KDT-11 indicated in the lighter shade. B. Titration of the indicated fluorescein-compound conjugates with Rpn13. The increase in fluorescence polarization was monitored. C. Measurement of the effect of the three compounds on the viability of MM.1R and SL-MEL-5 cells after 24 hours. See supplemental figure 7 for characterization data.
Discussion
We reported previously that KDT-11 (Figure 1A), a peptoid that binds Rpn13 with modest affinity (KD = 3 μM) but good selectivity in vitro, is selectively toxic to multiple myeloma (MM) cells (Trader et al., 2015). Furthermore, treatment of multiple myeloma cells with this molecule results in a buildup of polyubiquitylated proteins, suggesting that the molecule inhibits their proteasome-mediated turnover and/or deubiquitylation (Trader et al., 2015). This study was focused on better characterizing the mechanistic basis of the cancer cell-selective cytotoxicity of the molecule and testing if this interesting phenotype is indeed the result of modulation of Rpn13 function in cellulo.
We find that KDT-11 inhibits the growth of a wide variety of cancer cell lines (Figure 1B), not simply MM cells, and is particularly toxic to leukemia, melanoma and colon cancer lines. In both MM.1R and SK-MEL-5 melanoma cell lines, growth inhibition is due to rapid establishment of a cytostatic effect, followed by a slower induction of apoptosis (see Figures 4 and 5 and Supplemental Figure 5). Inhibition of cell growth is due to the inability of KDT-11-treated MM or SK-MEL-5 cells to enter into S phase of the cell cycle (Figure 5). In stark contrast to the almost complete G0/G1 to S phase block induced in the cancer cell lines by KDT-11, the peptoid had no effect on cell cycling in HEK293T cells (Figure 5).
In vitro, we found that KDT-11 has no measurable effect on any of the protein-protein interactions in which Rpn13 engages, including interactions with Uch37, Ub and Rpn2 (Figure 2). However, the peptoid does interfere with the ability of Rpn13 to activate the deubiquitylase activity of Uch37 (Figure 2E). Unfortunately, in the buffer employed for this assay, KDT-11 was only soluble up to a concentration of 10 μM, meaning that Rpn13 was not fully engaged with the peptoid. Thus, it was not possible to determine if the peptoid, when saturating Rpn13, would have completely squelched the activity of Uch37 or simply returned it to the rather slow, non-Rpn13-stimulated rate. In any case, this experiment suggests a model in which KDT-11 evinces its interesting cellular effects through an allosteric inhibition of Uch37-mediated deubiquitylation. This would be consistent with the aforementioned observation that KDT-11 treatment of cells results in a buildup of poly-ubiquitylated proteins, though to a more modest level than is observed when proteasome activity is blocked completely with Bortezomib (Trader et al., 2015). Taking this line of thought a step further, these data suggest that a high level of Uch37 activity is essential for entry of at least some cancer cells into S phase, whereas non-malignant cells such as the HEK293T line do not display such a dependence. Interestingly, Jackson and colleagues have recently demonstrated that Uch37’s deubiquitylase activity plays an important role in DNA double-stranded break repair through homologous recombination (Nishi et al., 2014) and it is known that self-inflicted double-stranded breaks are important for sustaining tumorigenicity and a high level of “stemness” in some cancer cells (Liu et al., 2017). This could conceivably provide a mechanism for the striking cancer cell-selective toxicity of KDT-11.
However, this model is predicated on the idea that KDT-11 blocks cell cycle progression in myeloma and melanoma cells by targeting Rpn13 and that this results in lower Uch37 deubiquitylase activity in cellulo, as is observed in vitro. One approach to test this assumption is to ask if knockdown of the suspected target protein at the genetic level recapitulates the effect of small molecule treatment. This was not the case here. In the SK-MEL-5 line, which is highly sensitive to KDT-11, knockdown of Rpn13 to a level approximately 9- to 10-fold lower than that that observed in untreated cells did not result in detectable growth inhibition or an effect on cell cycle progression (Figure 6). The same result was obtained when Uch37 was knocked down. Another common test is to determine if altering the level of the presumed target protein alters the sensitivity of the cells to the probe molecule. This was also not the case for KDT-11 and Rpn13. Cells in which Rpn13 and Uch37 were knocked down displayed the same sensitivity to KDT-11 as the untreated cells or those in which control proteins had been knocked down (Figure 6A–C). Additionally, the expression of a mutant Rpn13 protein that only weakly activates Uch37 on top of the knockdown of wild-type Rpn13 also did not recapitulate the effects of KDT-11 (Figure 6E). Finally, the cytotoxicity of two KDT-11 analogs did not correlate with their affinity for Rpn13 (Figure 7).
These surprising data call into question the idea that KDT-11 mediates its cellular effects through interaction with Rpn13. Indeed, they would suggest that the MM- and melanoma-specific cell cycle blockade is due to interaction with one or more off targets. However, there are some caveats. First, it is possible that the levels of Rpn13 and Uch37 remaining after siRNA-mediated knockdown are sufficient to support melanoma cell growth, though this seems unlikely, and is certainly at odds with the idea that a very high level of Rpn13-stimulated Uch37 deubiquitylase activity is essential for cell cycle progression in cancer cells. To address this issue decisively, a complete knockout of Rpn13 or Uch37 would be necessary. As mentioned above, we were unsuccessful in attempts to do so in SK-MEL-5 cells using CRISPR technology, though whether this reflects a requirement for some level of Rpn13 or was simply due to technical issues is unclear. Second, the fact that knockdown of Rpn13 does not sensitize cells to KDT-11 (see Figure 6) should be interpreted with caution. While it is routine in chemical biology to anticipate that manipulation of target protein levels should alter sensitivity to a drug, this is only perfectly clear for highly potent compounds in which essentially every molecule that enters the cell engages its target. In other words, binding is stoichiometric. For a modest affinity ligand such as KDT-11 (KD = 3 μM), binding would only be stoichiometric if the intracellular concentration of Rpn13 is far above this concentration. This number is not known. If the native Rpn13 concentration is well below the KD then it will be half-saturated at about 3 μM KDT-11 irrespective of whether the protein is knocked down or not. Having said this, such arguments do not rationalize why the cytotoxicity of KDT-11 analogues KPD-11 and KPD-12 in MM and SK-MEL-5 cells do not track with their affinity for Rpn13 if that protein is truly the physiologically relevant target.
While the details are not reported here, we attempted to identify mutants that were resistant to KDT-11, but the mismatch repair deficient HCT116 cell line used for this approach was not sufficiently sensitive to KDT-11 to allow for selection. We also probed for KDT-11-Rpn13 engagement in the cell by asking if the protein was more thermally stable when cells were treated with KDT-11. No effect was observed, but this result is difficult to interpret since Rpn13 ligands had no effect on the thermal stability of purified protein (data not shown). The upshot of the data reported in this study and these additional efforts mentioned above, is that we have no evidence to indicate that KDT-11 is indeed engaging Rpn13 in cellulo.
As discussed in the introduction we and others have been attracted to Rpn13 as a possible target for cancer chemotherapy because it has been reported to be strongly up-regulated in certain tumors (Chen et al., 2009; Fejzo et al., 2013; Fejzo et al., 2008; Fejzo et al., 2011; Jang et al., 2014; Jiang et al., 2017; Shenoy, 2015) yet is less essential for non-malignant cell survival (Al-Shami et al., 2010; Hamazaki et al., 2015). This idea was apparently supported by the work of Anchoori, et al., who reported that the electrophilic compound RA190 engages Rpn13 selectively in vitro and displays interesting anti-cancer activity in cellulo and in vivo (Anchoori et al., 2013), as well as our own work that showed KDT-11 binds Rpn13 in vitro and is selectively toxic to MM cells (Trader et al., 2015). However, the data presented here strongly call into question the idea that KDT-11 drives this interesting phenotype through modulation of Rpn13 activity, at least in the MM.1R and SK-MEL-5 cell lines. In addition, we have recently conducted a comprehensive analysis of RA190-proteome interactions in two different cancer cell lines and find that Rpn13 is not engaged by this compound in cellulo (Dickson, et al., submitted). Thus, the question of whether Rpn13 is indeed an attractive cancer target has yet to be addressed decisively using pharmacological methods.
RESOURCE AVAILABILITY
LEAD CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for materials should be directed to the Lead Contact Thomas Kodadek (Kodadek@scripps.edu).
MATERIALS AVAILABILITY
Mutant plasmid pcDNA5_FLAG-ADRM1Q337AQ338A created in this study will be available for distribution through the Lead Contact.
DATA AND CODE AVAILABILITY
The published article includes all datasets generated or analyzed during this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human embryonic kidney (female, HEK293T Cat#CRL-3216; RRID: CBCL_0063) cells were cultured at 37°C with 5% CO2 in DMEM (10566016, Gibco) supplemented with 10% FBS (16140–071, ThermoFisher). MM.1R (female, Cat#CRL-2975; RRID: CVCL_8794)and MM.1S (female, Cat#CRL-2974; RRID: CVCL_8792) cells were cultured at 37°C with 5% CO2 in in RPMI1640 (A1049101, ThermoFisher) supplemented with 10% FBS (16140–071,ThermoFisher). SK-MEL-5 (female, Cat#HTB-70; RRID: CVCL_0527) melanoma cells were cultured at 37°C with 5% CO2 in EMEM (30–2003, ATCC) supplemented with 10% FBS (16140–071, ThermoFisher). Adherent cells (HEK293T, SK-MEL-5) were passaged using TrypLE (12605–010, ThermoFisher) every 3 days. Suspension cells (MM.1R, MM.1S) were passaged every 4 days without the use of trypsinizing agent. Cells were immediately lysed for experiments or stored at −80°C until use. Cells were routinely tested for mycoplasma (
Materials and Methods
METHOD DETAILS
Compounds
Peptoids were synthesized as described previously, purified using reverse-phase HPLC, and characterized by MALDI-TOF MS (Trader et al., 2015). Briefly, KDT-11, scrambled KDT-11, KPD-11, and KPD-12 were prepared by solid phase synthesis using Rink amide MHBA resin (160 μm, 0.7mmol/g, 50 mg). Resin was swollen in 1:1 dimethylformamide (DMF), Fmoc protecting group was removed with 20% piperidine in dimethylformamide (DMF) two times for 10 minutes, resin was washed with DMF five times, and molecules synthesis proceeded using standard peptoid synthesis methods. Resin-displayed NH2 is activated with a solution of 1M bromoacetic acid in DMF and 1M diisopropylcarbodiimide (DIC) for 5 minutes at 37°C. Resin was washed five times and displacement proceeded with 1M primary amine in DMF for 1 hour at 37°C. This cycle continued until molecule synthesis was completed, at which point resin was washed with DCM and material was cleaved using 95% TFA/2.5% TIPS/2.5%DCM for 2 hours at room temperature. KPD-12 was prepared in the same way, until the final reaction step, where acid coupling was performed to complete the synthesis using 5 equivalents of 4-(3-Methyl-5-oxo-2-pyrazolin-1-yl)benzoic acid (AK Scientific, CAS # 60875–16-3), 7.5 equivalents of DIC, 5 equivalents of collidine, and 5 equivalents of oxyma. Preparative HPLC was performed on a Waters 1525 pump system with Vydac C18 column with acetonitrile and water mobile phases. Collected fractions were assessed by Shimadzu Axima MALDI-TOF mass spectrometry in positive reflectron mode. Analytical data for KPD-11 and KPD-12 can be found in supplemental figure 7. All other compounds were purchased from listed sources. Chemical structures were drawn using ChemDraw (RRID: SCR_016768).
Recombinant protein production
Plasmid for bacterial expression of 6xHis-Rpn13 was obtained from Addgene as a kind gift from the Conaway lab (Addgene #19423; RRID: Addgene_19423). Plasmid for bacterial expression of MBP-His8-Uch37 fusion protein was obtained from DNASU (#HsCD00084019). The GST-Rpn2 expression plasmid was a kind gift from Kylie Walters’ laboratory. These plasmids were transformed into BL21 DE3 pLysS cells (Cat#L1195), which were induced at 1.0 OD600 with 1mM IPTG. Cells were collected after 4 hours of induction at 16°C followed at 4,000g. MBP-His8-Uch37 and His6-Rpn13 pellets were resuspended in lysis buffer (300 mM sodium chloride, 50 mM sodium phosphate, 3% glycerol, 1% triton X-100, 1 mM PMSF) followed by probe sonication at 50% power 3 times for 30 seconds each. Following clarification at 8,000g for 30 minutes, soluble supernatant was incubated with Ni-NTA resin (Qiagen) for one hour at 4°C. GST-Rpn2 was prepared for purification using GST lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.1% triton X-100, 5 mM DTT, and 1 mM PMSF) and incubated with glutathione resin (ThermoFisher). Ni-NTA purification proceeded by washing with sodium phosphate buffer and eluting with increasing concentrations of buffered imidazole. Gluthatione purification proceeded with a 100 mM Tris pH 8 resin wash followed by elution with reduced glutathione in buffered Tris pH 8. Pure protein fractions were verified by SDS-PAGE and collected for dialysis. Following overnight dialysis in PBS, protein was quantified by Nanodrop, snap frozen and stored at −80°C until use.
Immunoblotting
10 cm dishes of confluent cells (5×106 cells) were collected and washed 2x with DPBS (Cat#14190250, Thermo Fisher). Cells were then lysed with 1 mL Mammalian Permeabilization Reagent (MPER) (78501, Thermo Fisher) and incubated at RT for 10 minutes. Soluble protein supernatant was clarified by centrifugation at 15,000g for 15 minutes at 4°C in a benchtop microcentrifuge (Eppendorf). Protein concentration was determined by UV/Vis (Nanodrop) and normalized with additional MPER. Lysates were diluted with 5x SDS Page loading dye (Bio-Rad) and 50 μg were loaded onto 12–20% gradient gels (BioRad). SDS-Page gels were run at 180V for 40 minutes, and transfer to nitrocellulose membrane was executed according to manufacturer’s instructions (1704270, Bio-Rad). Membranes were blocked in 5% Blot-Quickblocker modified milk protein (786–011, G-Biosciences) in PBST. Antibody was used at the recommended dilution and incubated with membrane for 16 hours at 4°C to reduce non-specific binding. Proteins were visualized using Licor Odyssey instrument after 1 hour RT incubation with IR dye conjugated antibodies: Donkey anti-mouse 680RD (Licor Cat#925–32212; RRID: AB_27116622) and Goat anti-rabbit 800CW (Licor Cat#925–32211; RRID: AB_2651127) diluted 1:10,000 in Odyssey blocking buffer (Licor). The following antibodies were used for western blotting at the dilutions indicated: Rpn13 (ADRM1, Novus Cat#NBP1–30447; RRID: AB_2225663), p21 Waf1/Cip1 (CST Cat#CST #2947; RRID: AB_823586, 1:500), p27/Kip1 (Novus Cat#NBP1–32213; RRID: AB_10003918, 1:1000), GAPDH (Cat#Sc-32233; RRID: AB_627679, 1:1000), β-tubulin (Cat#SC-5274; RRID: AB_2288090, 1:1000), Rpn2 (Cat#NBP1–58221; RRID: AB_11041020, 1:500), β5 (PSMB5 Cat#BML-PW8895–0025; RRID: AB_2052392, 1:1000), Uch37 (Abcam Cat#Ab133508 ; RRID: AB_2814821, 1:1000), Usp14 (Bethel Laboratories Cat#A300–920A; RRID: AB_2304343, 1:1000), FLAG (Cat#F1804; RRID: AB_262044, 1:1000). Biological triplicate was performed for all experiments, unless otherwise noted. Blots were quantified using ImageJ (RRID: SCR_003070) after imaging with LI-COR Oddysey system and LI-COR image studio software (RRID: SCR_015795). For cell cycle substrate immunoblotting, p-values were generated from a two-tailed t-test between treated and untreated samples.
Dose response viability assay
Cellular viability analyzed by Cell Titer Glo 2.0 (Promega Cat#G9241). Briefly, 4,000 cells/well were seeded into 96-well white plates (Costar) and incubated with compound in 1% DMSO as indicated. At the time of readout, 100 μL of Cell Titer Glo 2.0 reagent was added and plates were shaken at 800 rpm for 5 minutes. Luminescence was quantified by Tecan plate reader at 250ms. Data were plotted using Graphad Prism 8 (RRID:SCR_000306) and non-linear regression using a inhibitor dose response was performed.
Cell toxicity assay
Cell death was assessed by Cell ToxGreen reagent (Promega Cat#G8741). 8,000 cells/well were seeded into 96-well black plates (Grenier) and incubated with compound on 1% DMSO as indicated. Toxicity reagent was diluted 1:1000 as indicated by the manufacturer. Probe signal was quantified by fluorescence at 380 nm excitation and 460 nm emission (Tecan plate reader).
Caspase3/7 apoptosis assay
Induction of pro-apoptotic enzymes due to compound treatment was assessed by Caspase-Glo 2.0 (Promega Cat#G8090), in which 4,000 cells/well were seeded into 96-well white plates (Costar) and incubated with compound in 1% DMSO as indicated. At the time of readout, 100 μL of Caspase-Glo 2.0 reagent was added and plates were shaken at 800 rpm for 5 minutes. Luminescence was quantified by Tecan plate reader at 250ms.
Apoptosis flow cytometry assay
MM.1R cells were treated as indicated and apoptosis was measured by staining with Annexin V-510 (Biolegend) and Propidium Iodide (Sigma). Briefly, cells were pelleted and washed 2 times with cold DPBS. Cells were then resuspended in cold Annexin V staining buffer (HEPES, EDTA, CaCl2) and detection reagents were added (5 μg/mL Annexin V-510 and 50 μg/mL Propidium Iodide). After 15 minutes of room temperature incubation in the dark, 106 cells were passed through a cell strainer (BD Falcon) and analyzed by flow cytometry on a BD FACSCanto II flow cytometer (BDBiosciences) using the violet and blue laser. Experiments were performed with three biological replicates, and data was processed using FlowJo (RRID: SCR_008520).
Cell Cycle flow cytometry
HEK293T, MM.1R, and SK-MEL-5 cells were treated as indicated and then incubated with 10 μM 5-ethynyl-2’-deoxyuridine (EdU, Invitrogen ) for 90 minutes. Cells were subsequently harvested, fixed, and stained with Alexa647-azide according to the manufacturer’s instructions (Click-iT EdU Flow Cytometry assay kit Cat#C10424). Cells were resuspended in DPBS containing propidium iodide (PI, Cat#P4864) and 106 cells were passed through a cell strainer (BD Falcon) and analyzed by flow cytometry on a BD FACSCanto II flow cytometer (BDBiosciences) using the red and blue laser. A two-tailed t-test was performed on biological triplicate average of the respect percentage of cells in S phase. Samples were gated using FlowJo (RRID: SCR_008520). Error bars on the graph represent the standard deviation.
siRNA knockdown
Validated, pooled siRNA towards the nontargeting control(Cat#D-001810–10), GAPDH (Cat#D-001830–10), Rpn13(Cat#L-012340–01), Uch37(Cat#L-006060–00-0020), and Usp14(Cat#L-006065–00-0005) were purchased from Dharmacon. Custom UTR targeting siRNA towards Rpn13 were designed using Dharmacon’s siDesign Center(Cat#CTM-485557, Cat#CTM-485558). siRNA was transfected (50 nM) used according to the manufacturer’s protocol for a 6-well dish. Cells were incubated with knockdown reagent for 48 hours prior to analysis.
Mutant Rpn13 Q337A Q338A generation
Plasmid pcDNA5-FLAG-Adrm1 was obtained from Addgene (#19417) and site directed mutagenesis was performed through the PIPE cloning method. PCR was used to incorporate the Q337A and Q338A mutations with the following cycling conditions: 1 ng plasmid was mixed with 2x KOD HotStart PCR master mix (25 μL), primers (STAR Methods, 0.3 μL, 10 μM stock), and water to 50 μL. PCR conditions were as follows: 95°C for 2 minutes, 35 cycles of 95°C for 30 seconds, 75°C for 20 seconds, 68°C for 3.5 minutes, followed by a 5 minute 72°C final extension. PCR products (5 μL) were mixed with 6x DNA loading dye (1 μL) were verified on a 1% agarose gel run in 1x Tris-Acetate EDTA (TAE) buffer for 40 minutes at 100V. DNA was purified and transformed into chemically competent XL10 Gold bacterial cells (Agilent Cat#200314). DNA (2 μL) was incubated with cells (100 μL) and beta-mercaptoethanol(1 μL) for 30 minutes on ice. Cells were heat-shocked at 42°C for 45 seconds, followed by cooling on ice for two minutes. Warm SOC media was added and recovery proceeded at 37°C for 1 hour with shaking. Cells were plated on LB+Agar with 100 μg/mL carbenicillin. Single colonies were picked and grown in 5mL cultures in liquid LB+carbenicillin for subsequent DNA extraction using the Qiagen Miniprep kit. Mutation was confirmed by Sanger sequencing (Genewiz) and trace visualization using Geneious (RRID: SCR_010519.
Plasmid transfection
Mammalian expression plasmid pcDNA5-FLAG-Adrm1 (Addgene#19417; RRID: Addgene_19417) and mutant Q337AQ338A construct were was transiently transfected into SK-MEL-5 using Omnifect transfection reagent (Transomic Cat#OTR1003) according to the manufacturer’s protocol. For a 6-well dish transfection, Opti-MEM reduced serum media (200 μL) was incubated with Omnifect (4 μL) and plasmid (2 μg) for 10 minutes followed by addition to SK-MEL-5 cells at 70–80% confluency. Cells were collected and prepared for respective analyses after 24 hours of transfection.
In vitro pulldown assays
Recombinant proteins were incubated at 1 μM each in PBS, with 0.5% DMSO or 50 μM KDT11. Rpn13/Uch37 complex was pulled down using amylose resin (NEB) to bind the MBP tag on Uch37. Complexes were washed three times with PBS mimicking incubation conditions with or without compound. Protein was eluted from the resin with SDS Reducing Dye and PBS at 95C for 10 minutes, ran on a 12–20% gradient gel (Biorad) and visualized with Coomassie staining (Gel Code Blue, Thermo Fisher). This experiment was conducted in biological triplicate with graph error bars representing standard deviation. A two-tailed t-test was performed to generate the p-values of treated samples relative to vehicle treatment.
In vitro deubiquitinase activity assay
Recombinant Uch37 was assayed for activity using a ubiquitin-AMC fluorogenic probe (Boston Biochem Cat#U-550). 10 nM Uch37 was incubated with KDT-11 or bAP-15(Cat#: 11324) in 0.5% DMSO in Ub-AMC assay buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 0.1% BSA) for 30 minutes at room temperature. 50 nM Rpn13 was added during incubation where indicated. Reactions were carried out in 384-well low volume black assay plates (Grenier). Probe was added (0.5 μM) and plate was read out immediately in a Tecan plate reader at 385 nm excitation and 360 emission. Functional enzyme was characterized by comparing activity to reported kinetic values. To assess 26S and 19S proteasome deubiquitinase activity, 1 nM proteasome was incubated in modified Ub-AMC assay buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 5 mM ATP, 0.1% BSA) and experiment was performed. The experiment was performed in biological triplicate with statistical analysis including a two-tailed t-test performed between compound a vehicle treated samples.
Ubiquitin fluorescence polarization
100 μM KDT-11 or vehicle (1% DMSO) was incubated with 100 μM Rpn13 in PBS for 1 hour at 4°C prior to addition of an equal volume of 50 nM UbiquitinK48C-fluorescein. Ubiquitin-fluorescein was prepared through methods reported in Du, et al 2018: by mutating lysine 48 to cysteine, reacting protein with fluorescein-maleimide, and separating unbound material. Rpn13 was serially diluted two-fold into buffer containing Ubiquitin-fluorescein and ligand (or vehicle, in the case of control). This ensured that the final concentration of both probe and ligand remained consistent, while varying only the Rpn13 concentration. Data was collected on a Tecan fluorescent plate reader in fluorescence polarization format with excitation of 470 and emission of 518 nm. Data were collected in technical triplicate with two experimental replicates. Competition fluorescence polarization was performed with unlabeled bovine ubiquitin (Sigma Cat#: U6253)
KDT-11 fluorescence polarization
50 nM fluorescein-ligand in PBST was mixed with an equal volume of 200 μM Rpn13, followed by serial dilution into 25 nM FL-KDT11 solution to generate a dilution series of protein. Incubation was allowed to proceed for 15 minutes at room temperature to allow equilibration. Fluorescence polaraization was measured on a Tecan InfinitePro plate reader with 470 nm excitation and 518 nm emission. Data were collected in technical triplicate with two experimental replicates.
Proteasome co-immunoprecipitation
HEK293T cells were plated at 70% confluence in 10 cm dishes and treated with compound as indicated overnight. Cell lysates were prepared through 10 minutes of passive lysis with M-PER reagent supplemented with MgCl2, ATP, and glycerol (Thermo Fisher) and clarified at 17,000 rpm at 4°C for 15 minutes. Proteasome was immunoprecipitated according to manufacturer’s instructions using 20S-antibody crosslinked resin (Enzo, Cat#BML-PW1075A-0001).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data quantitation and statistical analysis is described in the methods where applicable. All experiments were performed in biological triplicate unless otherwise noted. Error bars on graphs represent standard deviation of readings.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ADRM1 (hRpn13) | Novus Biologics | Cat#NBP1–30447; RRID: AB_2225663 |
| Rabbit monoclonal anti-p21 waf1/cip1 (12D1) | Cell Signaling Technology | Cat#CST #2947; RRID: AB_823586 |
| Rabbit polyclonal anti-p27/kip1 | Novus Biologics | Cat#NBP1–32213; RRID: AB_10003918 |
| Mouse monoclonal anti-GAPDH 6C5 | Santa Cruz Biotechnology | Cat#Sc-32233; RRID: AB_627679 |
| Rabbit polyclonal anti-20S proteasome beta 5 subunit | Enzo Life Sciences | Cat#BML-PW8895–0025; RRID: AB_2052392 |
| Rabbit polyclonal anti-USP14 | Bethyl Laboratories | Cat#A300–920A; RRID: AB_2304343 |
| Rabbit monoclonal anti-UCH37 [EP4897] | Abcam | Cat#Ab133508 ; RRID: AB_2814821 |
| Monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| Goat anti-rabbit 800CW | Licor | Cat#925–32211; RRID: AB_2651127 |
| Donkey anti-mouse 680RD | Licor | Cat#925–32212; RRID: AB_27116622 |
| Mouse monoclonal Anti-β-tubulin | Santa Cruz Biotechnology | Cat#SC-5274; RRID: AB_2288090 |
| Rabbit polyclonal anti-Rpn2 | Novus Biologicals | Cat#NBP1–58221; RRID: AB_11041020 |
| Bacterial and Virus Strains | ||
| BL21(DE3)pLysS competent cells | Promega | Cat#L1195 |
| XL-10 Gold Ultracompetent cells | Agilent | Cat#200314 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| KDT-11 | Synthesized as reported | Trader, et al. 2015 |
| KPD-11 | This paper | N/A |
| KPD-12 | This paper | N/A |
| Ubiquitin-AMC | Boston Biochem | Cat#U-550 |
| Ubiquitin-fluorescein | Prepared as reported | Du, et al. 2017 |
| Omnifect transfection reagent | Transomic | Cat#OTR1003 |
| Ubiquitin (from bovine erythrocytes) | Sigma | CAS: 79586–22-4 Cat#: U6253 |
| bAP-15 | Cayman Chemical | Cat#: 11324 |
| Propidium iodide | Sigma Aldrich | Cat#P4864 |
| Critical Commercial Assays | ||
| CellTiter-Glo 2.0 Cell Viability Assay | Promega | Cat#G9241 |
| Caspase-Glo 3/7 Assay | Promega | Cat#G8090 |
| CellTox Green Cytotoxicity Assay | Promega | Cat#G8741 |
| Click-iT EdU Alexa Fluor 647 Flow cytometry assay | Life Technologies | Cat#C10424 |
| Proteasome purification kit | Enzo Life Sciences | Cat#BML-PW1075A-0001 |
| Experimental Models: Cell Lines | ||
| MM1.R myeloma cell line, Homo sapiens | ATCC | Cat#CRL-2975; RRID: CVCL_8794 |
| SK-MEL-5 melanoma cell line, Homo sapiens | ATCC | Cat#HTB-70; RRID: CVCL_0527 |
| HEK293T kidney cell line, Homo sapiens | ATCC | Cat#CRL-3216; RRID: CBCL_0063 |
| MM1.S myeloma cell line, Homo sapiens | ATCC | Cat#CRL-2974; RRID: CVCL_8792 |
| Oligonucleotides | ||
| ON-TARGETplus Rpn13 (11047) siRNA SMARTpool | Dharmacon | Cat#L-012340–01 |
| ON-TARGETplus GAPDH siRNA SMARTpool | Dharmacon | Cat#D-001830–10 |
| ON-TARGETplus nontargeting siRNA pool | Dharmacon | Cat#D-001810–10 |
| ON-TARGETplus UCHL5/Uch37 (51377) siRNA pool | Dharmacon | Cat#L-006060–00-0020 |
| ON-TARGETplus Usp14 (9097) siRNA pool | Dharmacon | Cat#L-006065–00-0005 |
| Custom ADRM1 (Rpn13) duplex ON-TARGET siRNA - UTR1 Rpn13 Sense:5’AGGAAGAGCGAGCCCGGACUU3’ Antisense:5’GUCCGGGCUCGCUCUUCCUUU3’ |
Dharmacon | Cat#CTM-485557 |
| CustomADRM1(Rpn13)duplexON-TARGETsiRNA- UTR2 Rpn13 Sense: 5’ UCCGAGGAACUGGGCGCUUUU 3’ Antisense: 5’ AAGCGCCCAGUUCCUCGGAUU 3’ |
Dharmacon | Cat#CTM-485558 |
| Site directed mutagenesis primers: For: 5’ CCTCGCCCCAGTTCGCGGCGGCCCTGGGCATGTTCAGCGCAG 3’ | IDT | N/A |
| Site directed mutagenesis primers: Rev: 5’ CATGCCCAGGGCCGCCGCGAACTGGGGCGAGGTCAGGGTATTC 3’ | IDT | N/A |
| Recombinant DNA | ||
| pcDNA5 FLAG-ADRM1 | Yao, et al 2006 | Addgene#19417; RRID: Addgene_19417 |
| Pet19b His6 ADRM1 | Yao, et al 2006 | Addgene #19423; RRID: Addgene_19423 |
| Mammalian expression FLAG-Rpn2(938–952) | Kylie Walters lab National Cancer Institute |
Lu, et al. 2015 |
| Bacterial expression GST-Rpn2(938–952) | Kylie Walters lab National Cancer Institute |
Lu, et al. 2015 |
| pVP16 MBP-Uch37 | DNASU | ID#84019 |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | RRID: SCR_003070 |
| Licor Image Studio Software | LI-COR Image Studio Software | RRID: SCR_015795 |
| Prism 8 | Graphpad | RRID:SCR_000306 |
| Geneious | Geneious | RRID: SCR_010519 |
| ChemDraw | PerkinElmer | RRID: SCR_016768 |
| FlowJo | FlowJo | RRID: SCR_008520 |
| Other | ||
| Amylose resin | NEB | Cat#E8021S |
| Ni-NTA resin | Qiagen | Cat#30210 |
| Pierce Glutathione agarose | Thermo Fisher | Cat#16100 |
| 4–20% Mini-PROTEAN Gels | Biorad | Cat#4561096 |
| Fetal Bovine Serum, Heat inactivated | Life technologies | Cat#10082147 |
| KOD HotStart PCR Master Mix | EMD Millipore | Cat#71842 |
| Mycoplasma PCR Test Kit | ABM Good | Cat#G238 |
| Annexin V BV510 Reagent | BioLegend | Cat#640937 |
Highlights.
Peptidomimetic KDT-11 induces cancer-selective G1/S cell cycle arrest
KDT-11 does not disrupt its reported target Rpn13’s known protein interactions
Cellular sensitivity to KDT-11 does not relate to Rpn13 protein level
Significance.
The peptoid KDT-11, which was identified through a high-throughput in vitro screening campaign a Rpn13 ligand, enforces a nearly complete block to cell cycle progression in certain cancer cells, but not in a non-malignant line. In vitro, KDT-11 inhibits the Rpn13-stimulated deubiquitylase activity of Uch37, suggesting that this activity is critical for progression into S phase for some cancer cells. However, genetic manipulation of Rpn13 or Uch37 levels do not phenocopy the effects of KDT-11 in a melanoma cell line and the cytoxicity of two KDT-11 analogues do not correlate with their affinity for Rpn13, suggesting that the cell cycle arrest driven by these compounds is due to interaction with one ro more off-targets rather than engagement with Rpn13 in cellulo. These data, along with recent findings that another putative Rpn13 inhibitor, RA-190, does not engage Rpn13 in cells (Dickson, et al., submitted), argue that there are currently no compelling pharmacological data to support the idea that Rpn13 is a good target for cancer chemotherapy.
Acknowledgements
This research was supported by a grant from the NIH (GM133041).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors have no interests to declare.
References
- Al-Shami A, Jhaver KG, Vogel P, Wilkins C, Humphries J, Davis JJ, Xu N, Potter DG, Gerhardt B, Mullinax R, et al. (2010). Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS One 5, e13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anchoori RK, Karanam B, Peng S, Wang JW, Jiang R, Tanno T, Orlowski RZ, Matsui W, Zhao M, Rudek MA, et al. (2013). A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell 24, 791–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Hu XT, Shi Q-L, Zhang F-B, and He C (2009). Knockdown of the novel proteasome subunit Adrm1 located on the 20q13 amplicon inhibits colorectal cancer migration, survivability and tumorigenicity. Oncol Rep 21, 531–537. [PubMed] [Google Scholar]
- D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, et al. (2011). Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 17, 1636–1640. [DOI] [PubMed] [Google Scholar]
- D’Arcy P, Wang X, and Linder S (2015). Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther 147, 32–54. [DOI] [PubMed] [Google Scholar]
- DeMartino G, and Slaughter CA (1999). The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem 274, 22123–22126. [DOI] [PubMed] [Google Scholar]
- Du J, and Strieter ER (2018). A fluorescence polarization-based competition assay for measuring interactions between unlabeled ubiquitin chains and UCH37*RPN13. Anal Biochem 550, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejzo MS, Anderson L, von Euw EM, Kalous O, Avliyakulov NK, Haykinson MJ, Konecny GE, Finn RS, and Slamon DJ (2013). Amplification Target ADRM1: Role as an Oncogene and Therapeutic Target for Ovarian Cancer. Int J Mol Sci 14, 3094–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejzo MS, Dering J, Ginther C, Anderson L, Ramos L, Walsh C, Karlan B, and Slamon DJ (2008). Comprehensive analysis of 20q13 genes in ovarian cancer identifies ADRM1 as amplification target. Genes Chromosomes Cancer 47, 873–883. [DOI] [PubMed] [Google Scholar]
- Fejzo MS, Ginther C, Dering J, Anderson L, Venkatesan N, Konecny G, Karlan B, and Slamon DJ (2011). Knockdown of ovarian cancer amplification target ADRM1 leads to downregulation of GIPC1 and upregulation of RECK. Genes Chromosomes Cancer 50, 434–441. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, and Kirschner MW (1991). Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138. [DOI] [PubMed] [Google Scholar]
- Goldman-Leikin RE, Salwen HR, Herst CV, Variakojis D, Bian ML, Le Beau MM, Selvanayagan P, Marder R, Anderson R, Weitzman S, et al. (1989). Characterization of a novel myeloma cell line, MM.1. J Lab Clin Med 113, 335–345. [PubMed] [Google Scholar]
- Groll M, Bochtler M, Brandstetter H, Calusen T, and Huber R (2005). Molecular machines for protein degradation. ChemBioChem 6, 222–256. [DOI] [PubMed] [Google Scholar]
- Hamazaki J, Hirayama S, and Murata S (2015). Redundant Roles of Rpn10 and Rpn13 in Recognition of Ubiquitinated Proteins and Cellular Homeostasis. PLoS Genet 11, e1005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, and Ciechanover A (1998). The ubiquitin system. Ann Rev Biochem 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Huang Z, Wu Y, Zhou X, Xu J, Zhu W, Shu Y, and Liu P (2014). Efficacy of therapy with bortezomib in solid tumors: a review based on 32 clinical trials. Future Oncol 10, 1795–1807. [DOI] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, and Dikic I (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SH, Park JW, Kim HR, Seong JK, and Kim HK (2014). ADRM1 gene amplification is a candidate driver for metastatic gastric cancers. Clin Exp Metastasis 31, 727–733. [DOI] [PubMed] [Google Scholar]
- Jiang RT, Yemelyanova A, Xing D, Anchoori RK, Hamazaki J, Murata S, Seidman JD, Wang TL, and Roden RBS (2017). Early and consistent overexpression of ADRM1 in ovarian high-grade serous carcinoma. J Ovarian Res 10, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li F, Huang Q, Zhang Z, Zhou L, Deng Y, Zhou M, Fleenor DE, Wang H, Kastan MB, et al. (2017). Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res 27, 764–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Nowicka U, Sridharan V, Liu F, Randles L, Hymel D, Dyba M, Tarasov SG, Tarasova NI, Zhao XZ, et al. (2017). Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat Commun 8, 15540–15553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi R, Wijnhoven P, le Sage C, Tjeertes J, Galanty Y, Forment JV, Clague MJ, Urbe S, and Jackson SP (2014). Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat Cell Biol 16, 1016–1026, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar SV, and Kumar S (2016). Multiple Myeloma: Diagnosis and Treatment. Mayo Clin Proc 91, 101–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randles L, Anchoori RK, Roden RB, and Walters KJ (2016). The Proteasome Ubiquitin Receptor hRpn13 and Its Interacting Deubiquitinating Enzyme Uch37 Are Required for Proper Cell Cycle Progression. J Biol Chem 291, 8773–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, and Wilkinson KD (2009). Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78, 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy N (2015). Is ADRM1 a Good Target for Cancer Therapy? Acta Haematol 134, 86–87. [DOI] [PubMed] [Google Scholar]
- Shirane M, Harumiya Y, Ishida N, Hirai A, Miyamoto C, Hatakeyama S, Nakayama K, and Kitagawa M (1999). Down-regulation of p27(Kip1) by two mechanisms, ubiquitin-mediated degradation and proteolytic processing. J Biol Chem 274, 13886–13893. [DOI] [PubMed] [Google Scholar]
- Thompson SL, and Compton DA (2011). Chromosomes and cancer cells. Chromosome Res 19, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trader DJ, Simanski S, and Kodadek T (2015). A reversible and highly selective inhibitor of the proteasomal ubiquitin receptor rpn13 is toxic to multiple myeloma cells. J Am Chem Soc 137, 6312–6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLinden RT, Hemmis CW, Schmitt B, Ndoja A, Whitby FG, Robinson H, Cohen RE, Yao T, and Hill CP (2015). Structural basis for the activation and inhibition of the UCH37 deubiquitylase. Mol Cell 57, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLinden RT, Hemmis CW, Yao T, Robinson H, and Hill CP (2017). Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J Biol Chem 292, 9493–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, Washburn MP, Conaway RC, Conaway JW, and Cohen RE (2006). Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol 8, 994–1002. [DOI] [PubMed] [Google Scholar]
- Yu H, and Matouschek A (2017). Recognition of Client Proteins by the Proteasome. Annu Rev Biophys 46, 149–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.