

Abstract
Background & Aims:
Pancreatic ductal adenocarcinomas (PDACs) are hypovascular, resulting in the upregulation of hypoxia inducible factor 1 alpha (HIF1A), which promotes survival of cells under low-oxygen conditions. We studied the roles of HIF1A in development of pancreatic tumors in mice.
Methods:
We performed studies with KrasLSL-G12D/+;Trp53LSL-R172H/+; Pdx1-Cre (KPC) mice, KPC mice with labeled pancreatic epithelial cells (EKPC), and EKPC mice with pancreas-specific depletion of HIF1A. Pancreatic and other tissues were collected and analyzed by histology, and immunohistochemistry. Cancer cells were cultured from PDACs from mice and analyzed in cell migration and invasion assays and by immunoblots, real-time PCR, and liquid chromatography– mass spectrometry. We performed studies with the human pancreatic cancer cell lines PATU-8988T, BxPC-3, PANC-1 and MiaPACA-2, which have no or low metastatic activity, and PATU-8988S, AsPC-1, SUIT-2 and Capan-1, which have high-metastatic activity. Expression of genes was knocked down in primary cancer cells and pancreatic cancer cell lines using small hairpin RNAs; cells were injected intravenously into immune-competent and NOD/SCID mice and lung metastases were quantified. We compared levels of mRNAs in pancreatic tumors and normal pancreas in the Cancer Genome Atlas.
Results:
EKPC mice with pancreas-specific deletion of HIF1A developed more advanced pancreatic neoplasias and PDACs with more invasion and metastasis, and had significantly shorter survival times, than EKPC mice. Pancreatic cancer cells from these tumors had higher invasive and metastatic activity in culture than cells from tumors of EKPC mice. HIF1A-knockout pancreatic cancer cells had increased expression of protein phosphatase 1 regulatory inhibitor subunit 1B (PPP1R1B). There was an inverse correlation between levels of HIF1A and PPP1R1B in human PDAC tumors; higher expression of PPP1R1B correlated with shorter survival times of patients. Metastatic human pancreatic cancer cell lines had increased levels of PPP1R1B and lower levels of HIF1A compared with non-metastatic cancer cell lines; knockdown of PPP1R1B significantly reduced the ability of pancreatic cancer cells to form lung metastases in mice. PPP1R1B promoted degradation of p53 by stabilizing phosphorylation of MDM2 at Ser166.
Conclusions:
HIF1A can act a tumor suppressor by preventing expression of PPP1R1B and subsequent degradation of the p53 protein in pancreatic cancer cells. Loss of HIF1A from pancreatic cancer cells increases their invasive and metastatic activity.
Keywords: Tumor Progression, Cell Motility, Signal Transduction, Carcinogenesis
Graphical Abstract
Introduction:
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal human cancers becoming the fourth-leading cause of cancer-related death in the US with a 5-year survival of only 8%.1 PDAC often develops without early symptoms, and therefore most patients are diagnosed with metastatic disease, a point where chemotherapeutic or surgical intervention have minimal benefit. Unlike the comprehensive understanding of genetic mutations that initiate PDAC, the molecular mechanisms that promote the metastatic spread of pancreatic cancer are less clear. In addition to the high prevalence of KRAS mutations2, PDACs are characterized by extensive desmoplastic stroma and severe hypovascularity3,4 resulting in extreme intra-tumoral hypoxic microenvironment.5 In response to hypoxic stress, cancer cells set off many adaptive responses through the stabilization and activation of the hypoxia inducible factor (HIF) family of transcription factors.6 In PDAC, hypoxia and consequent HIF1A stabilization have been associated with the transcriptional activation of multiple pathways, involved in the regulation of metabolism, angiogenesis, cell survival, and inflammation.7,8,9 Elevated HIF1A expression is usually linked with higher patient mortality in many cancer types, including breast and colorectal cancers.10,11 However, emerging evidences propose that HIF1A may not be universally tumor promoting. In certain malignancies, HIF1A appears to have a tumor suppressive role and correlates with lower cancer or diminished patient mortality, implicating contrasting, context-dependent functions for HIF1A.12,13,14
The role of HIF1A in pancreatic cancer has been evolving in the recent years. Previously published reports showed inconsistent results on associations between expression of HIF1A and clinical outcomes in PDAC.15,16,17 Therefore, the precise role of HIF1A in PDAC pathogenesis is not completely comprehended. In part, a lack of detailed understanding of HIF1A’s role in PDAC is due to use of RNA interference of HIF1A in in vitro assays or xenograft tumor models,16,18 which cannot truly recapitulate the complex relationship of neoplastic epithelial cells with their microenvironment. A previous study, using p48-Cre; LSL-KrasG12D mouse model of PDAC showed that HIF1A deficiency promotes PDAC initiation through enhanced B cell infiltration.14 In the present study, we sought to identify the functional role of HIF1A in pancreatic cancer using a well characterized autochthonous model of PDAC that harbors the lox-stop-lox (LSL)-KrasG12D; LSL-p53R172H and Pdx-Cre (KPC) alleles that faithfully recapitulates all of the extant features of the human PDAC.19 We demonstrate that ablation of HIF1A in these mice resulted in drastically increased invasion, metastasis and substantially reduced survival. We further uncovered that loss of HIF1A leads to increased expression of PPP1R1B and subsequent degradation of the p53 protein that promotes invasion and metastasis in both mouse and human PDACs.
Material and Methods
Mouse Strains
KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre (KPC) mice have been previously described in detail19. ROSA26LSL-EYFP mice 20 (obtained from Jackson Laboratory) were crossed with KPC mice to label pancreatic epithelial cells expressing Cre (EKPC). Pancreas specific depletion of HIF1A was attained by crossing EKPC with HIF1A flox/flox conditional strain 21 referred as HIF1A−/−EKPC.
Cell Lines
Mouse pancreatic cancer cells (EKPC and HIF1A−/−EKPC) were grown in DMEM with supplements as previously described.22 Human pancreatic cancer cell lines were grown in their required growth medium as per the ATCC description (SI Materials and Methods).
In Vivo lung colonization assay
For lung colonization assays, mouse and human pancreatic cancer cells were stably transduced with luciferase transgene injected intravenously into the tail vein of C57BL/6 mice and bioluminescence imaging was initiated on day seven post transplantation.
LC-MS/MS and Data Analysis
Proteomics analysis was performed by the Proteomics Core, Sanford Burnham Prebys Medical Discovery Institute and data were analyzed as described in SI Materials and Methods.
Human Data Sets
The gene expression data and survival analyses of PDAC patients with annotated clinical outcomes were downloaded from The Cancer Genome Atlas. We used cBioPortal23, 24 and GEPIA25 for data visualization, analysis and download of large-scale cancer genomics data sets.
Quantification and Statistical Analysis
Statistical analyses for figures were performed using GraphPad Prism software. Data are presented as the mean ± s.e.m unless otherwise specified. All experiments were performed at least three independent times, unless otherwise noted. A log-rank (Mantel-Cox) test was used to evaluate statistical significance for the Kaplan-Meier survival plots.
Results
Loss of HIF1A leads to adverse outcome in PDAC
To study the role of HIF1A in PDAC in vivo, we used the KPC mouse, a well characterized autochthonous model of PDAC that harbors the lox-stop-lox (LSL)-KrasG12D; LSL-p53R172H and Pdx-Cre (KPC) alleles.19 Additionally, we introduced an LSL-Rosa-EYFP allele20 into this model to facilitate identification of the carcinoma cells (EKPC). Finally, we added a conditional allele of HIF1A21 to generate murine PDACs lacking HIF1A (HIF1A−/−EKPC, Figure 1A, Supplementary Figure 1A-E). Loss of HIF1A expression was confirmed by immunohistochemistry of PDACs obtained from HIF1A−/−EKPC mice. (Supplementary Figure 1F). Since HIF1A is most commonly thought to contribute to poor outcome in cancer patients,10,11, 26, 27 we expected HIF1A−/− EKPC mice to have diminished disease burden and better survival rate than the EKPC cohort. Surprisingly, HIF1A−/−EKPC mice exhibited significantly worse outcome. While the median survival of the EKPC mice (n=25 ) was 159 days, similar to that reported previously,19, 28 the HIF1A−/−EKPC mice (n=24) had a significantly abbreviated median survival of 113 days (p<0.005) (Figure1B). Comprehensive necropsy of HIF1A−/−EKPCs and EKPCs revealed more aggressive disease in HIF1A−/−EKPC mice (Table 1). Careful analysis of pancreatic intraepithelial neoplasias (PanINs) in the EKPC and HIF1A−/−EKPC mice revealed that most of the evaluable PanINs in the pancreata of HIF1A−/−EKPC mice had more diffuse boundaries that suggest more advanced PanIN lesions that have progressed closer to, but have not yet achieved, focal invasion (i.e. progression to frank PDAC), compared with more defined boundaries in the PanINs found in EKPC mice (Figure1C). Additionally, 75% of the PDACs found in HIF1A−/−EKPC mice showed evidence of direct local invasion into the adjacent organs (i.e. locally invasive PDAC), compared with 24% of those in the EKPC cohort (Figure 1D,E, Table 1). While 60% of EKPC mice display metastatic dissemination, 91.6% of HIF1A−/−EKPCs displayed incidences of metastasis (Figure 1D,F, Table 1). Additionally, 75% of HIF1A−/−EKPC mice displayed macro metastatic lesions compared with only 36% found in the EKPC cohort (Figure 1G,H, Table 1). HIF1A−/−EKPC mice also exhibited higher percentage of lymph node metastasis and increased dissemination of metastasis in multiple organs (Figure 1I,J, Supplementary Figure 2, Table 1). We next examined the effects of HIF1A depletion on tumor differentiation. Although there was no significant difference in overall histological grades between the EKPC and HIF1A−/−EKPC PDACs, we observed higher percentage of PanINs in HIF1A−/−EKPC tumors compared to EKPC tumors, indicating that the loss of HIF1Afacilitates the initiation and progression of PDAC precursors (Supplementary Figure 3). To gain insight into how HIF1A modulates intra-tumoral hypoxia and stroma during the PDAC development, we performed immunohistochemical staining for carbonic anhydrases-9 (a surrogate marker of tumor hypoxia) and stromal contents including Collagen type I, type III and α-smooth muscle actin (α-SMA). IHC staining and histologic analysis showed no difference in these markers between EKPC and HIF1A−/−EKPC PDAC sections indicating that HIF1A is largely dispensable for the intra-tumoral stromal content in mouse PDAC. (Supplementary Figure 4). We further analyzed the composition of immune cell infiltration in EKPC and HIF1A-/EKPC tumors by immunohistochemistry (Supplementary Figure 5). We observed significant increase in CD19+ B cell population in HIF1A−/−EKPC tumors, similar to an earlier report that HIF1Adeficiency enhances B-cell infiltration in PDACs.14 We further analyzed the effect of HIF1A inhibition after the PDACs have been established in KPC mice by using pharmacological inhibitor of HIF1A (PX-478). Based on Ki67 immunostaining, we observed pronounced increase in cell proliferation in tumor sections from KPC mice treated with HIF1A inhibitor (P = 0.008) (Figure 1K,L), suggesting that inhibition of HIF1A either genetically or pharmacologically can promote tumorigenesis in these mice. Taken together, these data clearly demonstrate that loss of HIF1A in mouse PDACs resulted in markedly increased invasion and metastasis, with reduced survival rate.
Figure 1: Loss of HIF1A leads to adverse outcome in PDAC.

(A) Schematics of EKPC (Blue) mouse model of pancreatic cancer, which employs LSLKrasG12D/+ (K), LSLp53R172H/+ (P), Pdx1-Cre (C), and LSLRosa EYFP (E) alleles. Pancreas specific depletion of HIF1A was attained by crossing EKPC with HIF1Aflox/flox mouse strain and referred as HIF1A−/−EKPC (Orange). (B) Kaplan-Meier analysis comparing survival of EKPC (n = 25) and HIF1A−/−EKPC (n = 24) mice; **P<0.005 Mantel-Cox test. (C) Hematoxylin and eosin (H&E) staining of pancreata from EKPC and HIF1A−/−EKPC mice. Loss of HIF1A promotes more advanced PanINs and locally invasive and metastatic PDAC. Representative PanIN lesions in EKPC and HIF1A−/−EKPC mice (i). The duodenum-PDAC interface in EKPC and HIF1A−/−EKPC mice showing a distinct example of an intact boundary without local invasion in EKPC mice, whereas PDACs from HIF1A−/−EKPC mice frequently show local invasion into adjacent organs (ii & iii) (D) Table showing comparative median survival (P<0.005), and incidences of local invasion (P<0.0005) and metastasis (P<0.005) in EKPC and HIF1A−/−EKPC mice; by Fisher’s exact test. (E) Comparative Invasion score (total number of organs showing local invasion by primary tumor divided by total number of animals) in EKPC (n=25) and HIF1A−/−EKPC mice (n=24), ****P<0.0001 by unpaired t test (F) Metastatic score (total number of organs showing Mets divided by total number of animals) in EKPC (n=25) and HIF1A−/−EKPC mice (n=24), ** P<0.005 by unpaired t test (G) Representative H&E stained images of the liver and lung mets (marked by arrows) in EKPC and HIF1A−/−EKPC cohorts. (Scale bar:10μm C,F) (H) Percentage of Macro-mets in EKPC and HIF1A−/−EKPC, ****P<0.0001 by Fisher’s exact test (I) Percentage of EKPC and HIF1A−/−EKPC mice with lymph node involvement; ****P<0.0001 by Fisher’s exact test (J) Percentage of EKPC and HIF1A−/−EKPC mice with metastasis in more than one organ; ***P < 0.001 by Fisher’s exact test. (K) Representative Ki-67 staining for proliferating cells in PDAC from KPC mice treated with HIF1Ainhibitor (PX-478) and vehicle control (L) Quantification of Ki-67 staining as total number of cells per field of vision in tumor sections from control and treated KPC mice. 10 FOV from each treatment group were randomly chosen and Ki67 positive cells were calculated, **P<0.005 by unpaired t test.
Table 1:
Clinical spectrum of disease in HIF1A−/−EKPC mice
Clinical spectrum of disease in EKPC mice
| No. | ID | Genotype | Gender | DOB | DOD | Age (days) | PDA | Liver | Lung | Dia | Lymph | Others | Organs involved in invasion | Total Organ met burden |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ES33 | EKPC Hif1α fl/fl | F | 11/16/13 | 04/03/14 | 138 | Y (+++) | Ym | N | N | Y | Dd; Ld, Adherent to K | 2 | 1 |
| 2 | ES110 | EKPC Hif1α fl/fl | F | 01/08/14 | 05/29/14 | 141 | Y(+) | N | N | N | N | 0 | 0 | |
| 3 | ES388 | EKPCHif1α fl/fl | M | 07/14/14 | 10/24/14 | 102 | Y(++) | N | N | YM | Y | Ld; Adherent to Sp | 1 | 1 |
| 4 | ES469 | EKPC Hif1α fl/fl | M | 09/13/14 | 11/18/14 | 66 | Y(+++) | Ym | N | N | Y | Lpd | 1 | 1 |
| 5 | ES593 | EKPC Hif1α fl/fl | M | 12/14/14 | 03/15/15 | 91 | Y(+++) | Ym | N | YM | Y | Dd; Ld | 3 | 2 |
| 6 | ES596 | EKPC Hif1α fl/fl | M | 12/14/14 | 04/28/15 | 135 | Y(+) | N | Ym | N | Y | Id | 1 | 1 |
| 7 | ABA614 | EKPCHif1α fl/fl | F | 01/16/13 | 08/05/13 | 199 | Y(+++) | YM | YM | YM | Y | Dd; Std; Id | 3 | 3 |
| 8 | ES643 | EKPC Hif1α fl/fl | M | 01/06/15 | 05/15/15 | 129 | Y(+++) | Ym | Ym | Ym | N | 0 | 3 | |
| 9 | ABA618 | EKPC Hif1α fl/fl | M | 01/16/13 | 05/19/13 | 123 | Y(+++) | Ym | N | YM | Y | Dd; Kd; GBd | 3 | 2 |
| 10 | ES658 | EKPCHif1α fl/fl | F | 01/24/15 | 05/22/15 | 118 | Y(+) | N | Ym | N | Y | Spm; GBM | 1 | 3 |
| 11 | ABA664 | EKPC Hif1α fl/fl | M | 02/16/13 | 07/25/13 | 159 | Y (+++) | YM | N | N | Y | 0 | 1 | |
| 12 | ES696 | EKPC Hif1α fl/fl | F | 02/14/15 | 07/24/15 | 160 | Y(++) | Ym | YM | N | N | Stm | 0 | 3 |
| 13 | ES717 | EKPC Hif1α fl/fl | M | 03/03/15 | 05/22/15 | 80 | Y(++) | Ym | YM | Ym | N | Dd; Id | 2 | 3 |
| 14 | ABA742 | EKPCHif1α fl/fl | F | 03/17/13 | 07/09/13 | 114 | Y(+++) | Ym | N | N | Y | Dd; Std; Adherent to I | 2 | 1 |
| 15 | ABA763 | EKPC Hif1α fl/fl | F | 03/06/13 | 07/25/13 | 141 | Y(+++) | Ym | YM | Ym | Y | Dd; Ld; LpM | 2 | 4 |
| 16 | ABA784 | EKPC Hif1α fl/fl | M | 03/25/13 | 07/04/13 | 99 | Y(++) | Ym | Ym | N | Y | Dd; Ld | 2 | 1 |
| 17 | ABA 869 | EKPCHif1α fl/fl | M | 04/24/13 | 08/15/13 | 113 | Y (+++) | YM | N | N | Y | Id; Ld | 2 | 1 |
| 18 | ABA909 | EKPC Hif1α fl/fl | M | 05/06/13 | 09/10/13 | 127 | Y(+++) | YM | N | N | Y | Dd; Ld | 2 | 1 |
| 19 | ABA933 | EKPC Hif1α fl/fl | M | 05/18/13 | 08/26/13 | 100 | Y(+++) | Ym | N | YM | Y | Dd; Ld | 2 | 2 |
| 20 | ABA950 | EKPC Hif1α fl/fl | M | 05/24/13 | 08/15/13 | 83 | Y(+++) | YM | YM | YM | Y | Spm, Km, Hm, Diad, Ld, Dd,LgD | 4 | 5 |
| 21 | ABA954 | EKPCHif1α fl/fl | M | 05/30/13 | 09/13/13 | 106 | Y(+++) | N | N | N | Y | Dd | 1 | 0 |
| 22 | ABA973 | EKPC Hif1α fl/fl | M | 07/01/13 | 10/17/13 | 108 | Y(+) | Ym | Ym | N | Y | 0 | 3 | |
| 23 | ABA1029 | EKPC Hif1α fl/fl | F | 08/28/13 | 12/02/13 | 94 | Y(+) | N | Ym | N | Y | Ld | 1 | 1 |
| 24 | ABA1031 | EKPCHif1α fl/fl | M | 08/28/13 | 10/08/13 | 41 | Y(+++) | N | YM | N | Y | Std | 1 | 1 |
| No. | ID | Genotype | Gender | DOB | DOD | Age (days) | PDA | Liver | Lung | Dia | Lymph | Others | Organs involved in invasion | Total Organ met burden |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ES149 | EKPC | F | 02/05/14 | 07/30/14 | 175 | Y(+++) | YM | YM | YM | Y | Dd; Ld; Spd | 3 | 3 |
| 2 | ES154 | EKPC | M | 02/05/14 | 10/29/14 | 177 | Y(++) | N | N | N | Y | 0 | 0 | |
| 3 | ES236 | EKPC | M | 03/24/14 | 05/29/14 | 66 | Y(+) | Ym | N | N | N | 0 | 1 | |
| 4 | ES273 | EKPC | M | 04/19/14 | 10/14/14 | 175 | Y(++) | N | N | N | N | 0 | 0 | |
| 5 | ES368 | EKPC | M | 07/05/14 | 02/08/15 | 213 | Y(++) | N | N | N | Y | Dd | 1 | 0 |
| 6 | ABA336 | EKPC | M | 08/16/12 | 12/12/12 | 116 | Y(+++) | N | N | N | N | 0 | 0 | |
| 7 | ABA405 | EKPC | F | 10/15/12 | 05/13/13 | 210 | Y(+) | Ym | N | YM | Y | 0 | 2 | |
| 8 | ABA407 | EKPC | F | 10/15/12 | 04/08/13 | 175 | Y(+) | YM | N | N | N | 0 | 1 | |
| 9 | ABA541 | EKPC | F | 12/10/12 | 03/17/13 | 97 | Y(+) | N | N | N | N | 0 | 0 | |
| 10 | ABA567 | EKPC | F | 12/27/12 | 09/06/13 | 249 | Y(++) | YM | N | N | Y | 0 | 1 | |
| 11 | ABA571 | EKPC | M | 12/27/12 | 07/25/13 | 208 | Y(++) | N | YM | N | N | 0 | 1 | |
| 12 | ABA642 | EKPC | F | 02/11/13 | 05/01/13 | 79 | Y(+++) | Ym | N | N | N | Id | 1 | 1 |
| 13 | ABA775 | EKPC | M | 03/24/13 | 08/06/13 | 135 | Y(+) | YM | N | N | Y | 0 | 1 | |
| 14 | ABA806 | EKPC | F | 04/06/13 | 07/12/13 | 97 | Y(++) | Ym | N | N | Y | 1 | 1 | |
| 15 | PP451 | KPC | M | 01/10/14 | 08/20/14 | 222 | Y(+++) | N | Ym | N | N | 0 | 1 | |
| 16 | PP509 | KPC | M | 02/01/14 | 09/30/14 | 241 | Y(+++) | N | N | N | Y | 0 | 0 | |
| 17 | PP793 | KPC | M | 04/19/14 | 08/28/14 | 131 | Y(+++) | N | Ym | N | N | Ld | 1 | 1 |
| 18 | PP866 | KPC | M | 05/16/14 | 10/22/14 | 159 | Y(++) | Ym | Ym | N | N | Ld | 1 | 2 |
| 19 | PP901 | KPC | F | 05/24/14 | 09/18/14 | 117 | Y (+++) | N | N | N | Y | 0 | 0 | |
| 20 | PP1011 | KPC | F | 06/19/14 | 12/19/14 | 183 | Y (+++) | N | Ym | N | Y | 0 | 2 | |
| 21 | PP1012 | KPC | F | 06/19/14 | 10/03/14 | 106 | Y(+) | N | N | N | N | 0 | 0 | |
| 22 | PP1034 | KPC | M | 06/21/14 | 12/21/14 | 183 | Y(+++) | YM | Ym | Ym | Y | 0 | 3 | |
| 23 | PP1041 | KPC | M | 06/27/14 | 09/10/14 | 75 | Y(+) | N | N | N | N | 0 | 0 | |
| 24 | PP1051 | KPC | F | 06/30/14 | 10/03/14 | 95 | Y(++) | N | N | N | N | 0 | 0 | |
| 25 | PP1091 | KPC | F | 08/13/14 | 11/13/14 | 90 | Y(+++) | N | N | Ym | N | 0 | 1 |
Y, disease detected [ >50% normal acini (+); 20–50% normal acini (++) and <20 % normal acini (+++)]; N, no disease detected; M, macrometastasis; m, micrometastasis; d, direct invasion; Ab, abdominal wall; L, liver; Lp, lymph node, I, intestine; Sp, spleen; St, stomach; GB, gall bladder; K, kidney; D, Duodenum
Depletion of HIF1A enhances invasion and migration of Pancreatic Cancer cells
To further investigate whether HIF1A impacts the invasive and the metastatic behavior of the PDACs in a cell autonomous manner, we derived multiple cell lines from primary PDACs in EKPC and HIF1A−/−EKPC mice (Supplementary Figure 6). Interestingly, while cell lines derived from EKPC primary tumors showed considerable phenotypic heterogeneity ranging from epithelial to mixed mesenchymal, all the tumor lines derived from HIF1A−/−EKPC PDACs showed consistent and noticeable mesenchymal state with elongated morphology, suggesting an invasive phenotype (Figure 2A). To better understand the role of other HIFs in the context of loss of HIF1A in PDACs, we analyzed expression of other HIFs at transcriptional and protein level in EKPC and HIF1A−/−EKPC cells, in hypoxic and normoxic conditions. As expected we observed increased mRNA and protein expression of HIF1A, HIF1B and HIF2A in the EKPC and HIF1B, and HIF2A expression in the HIF1A−/−EKPC cells under hypoxic conditions (Figure 2B,C). We did not detect any expression of HIF3A transcripts in normoxic or hypoxic conditions in any of these cells lines. However, we did not observe any compensatory increase in HIF1B and HIF2A in HIF1A−/−EKPC cells (Figure 2B). Furthermore, we analyzed expression of HIF1A and HIF2A in both EKPC and HIF1A−/−EKPC PDACs by immunohistochemistry. Consistent with our cell line data, we did not observed any significant increase in HIF2A expression in PDACs derived from HIF1A−/−EKPC mice (Figure 2D,E). Next, we examined the invasive and migrartory capacity of EKPC and HIF1A−/−EKPC cells by in vitro transwell matrigel asaays. We observed that HIF1A−/−EKPC cells manifested significantly higher invasion and migration compared with the EKPC cells (Figure 2F,G). Epithelial–mesenchymal transition (EMT) has been shown to play a critical role in promoting metastasis in carcinomas. Indeed, HIF1A−/−EKPC cells exhibited elevated EMT, including reduced expression of the epithelial marker E-Cadherin and increased expression of the mesenchymal markers Snail and Slug at the transcriptional as well as the protein levels (Figure 2H, Supplementary Figure 7A). We further investigated the effect of HIF2A on EMT regulators in HIF1A−/−EKPC cells by knocking down its expression using siRNA. We did not observe any reduction in E-Cadherin, SNAIL and SLUG expression in HIF2A depleted HIF1A−/−EKPC cells (Supplementary Figure 7B). To investigate how EMT markers like Snail and Slug is upregulated in absence HIF1A, we found that all the three HIF1A−/−EKPC biological replicates exhibited higher expression of c-MYC protein compared to the EKPC counterparts (Figure 2G). We further investigated the role of c-MYC on Snail and Slug expression by depleting c-MYC in HIF1A−/−EKPC cells. Knockdown of c-Myc in HIF1A−/−EKPC cell lines impaired both Snail and Slug expression (Supplementary Figure 7C), indicating that the regulation of EMT markers in HIF1A−/−EKPC may be controlled by pathways independent of HIF1A, c-MYC being one of them. To further confirm that the loss of HIF1A resulted into increased EMT features in HIF1A−/−EKPC, we performed the rescue experiment by ectopically expressing a mutant version of HIF1AP402A/P577A/N813A (m-HIF1A) which is stable and active in normoxia,29 which resulted in more epithelial morphology and increased E-Cadherin and decreased N-Cadherin expression in HIF1A−/−EKPC cells (Supplementary Figure 7D,E).
Figure 2: Depletion of HIF1A enhances invasion and migration of Pancreatic Cancer cells.

(A) Bright field image of primary cell lines derived from EKPC (ES149, PP142 & ES507) and HIF1A−/− EKPC (ES595, ES658 & ES469) tumors. (B) Expression of HIF1A, HIF1B, and HIF2A mRNA under hypoxic/normoxic conditions in EKPC and HIF1A−/−EKPC cell lines. Relative gene expression level for HIF1A, HIF1B, and HIF2A in EKPC (blue) and HIF1A−/− EKPC (orange) cells were normalized with GAPDH expression; n=3 biologically independent experiments; *P<0.05 and **P<0.005 by unpaired t test. (C) Western blot analysis of HIF1A, HIF2A and HIF1B under hypoxic/normoxic conditions in EKPC and HIF1A−/−EKPC cell lines. Beta-actin was used as loading control. (D) Representative images of immunohistochemistry staining for HIF1A and HIF2A in EKPC and HIF1A−/−EKPC tumor sections. Scale bar, 50 μm (E) Quantification of HIF1A and HIF2A positive cells per field of vision in EKPC and HIF1A−/−EKPC tumor sections. The data are shown as the mean ± s.e.m. P values by unpaired t test. ns, not significant, ****P < 0.0001. (F&G) Representative images and quantitative analysis of transwell migration and matrigel invasion assay of EKPC and HIF1A−/−EKPC cells. Total number of cells per field were counted. n=3 biologically independent experiments ***P < 0.001, ****P < 0.0001 by t test. (H) Immunoblot analysis of EMT markers (E-cad, Snail and Slug) and c-Myc protein in EKPC and HIF1A−/−EKPC cells. (I) Schematics of in vivo lung colonization assay for metastatic burden. EKPC and HIF1A-/EKPC cells expressing Luciferase were injected into the lateral tail vein of immunocompetent C57BL/6 mice (n=5 for each group), and (J&K) Representative bioluminescence imaging and quantitative analysis of lung metastasis by BLI plot **p<0.001 by Mann Whitney test. (L) KaplanMeier analysis of mice injected with EKPC and HIF1A−/−EKPC cells. **p< 0.005 by Log-rank (Mantel–Cox) test.
To investigate if HIF1A impedes metastatic potential of the PDAC in vivo, equal number of luciferase expressing HIF1A−/−EKPC and EKPC cells were injected via tail vein into immunocompetent C57BL/6 mice (Figure 2I) and their dissemination was evaluated at various time points by bioluminescence imaging (BLI). Mice injected with HIF1A−/−EKPC cells had significantly more luciferase signal compared to the ones injected with EKPC cells (Figure 2J,K, Supplementary Figure 8). Moreover, mice inoculated with HIF1A−/−EKPC cells had reduced survival rate compared to those injected with EKPC cells (Figure 2L). Taken together, the results suggest more efficient lung colonization by HIF1A−/−EKPC cells compared to EKPC cells. Indeed, enumeration of lung nodules in the two cohorts show HIF1A−/−EKPC cells formed significantly more tumor nodules than the EKPC cells (Supplementary Figure 8B,C), thus phenocopying the metastatic effect observed in HIF1A−/−EKPC mice. These data provide evidence that genetic inhibition of HIF1A in the pancreatic cancer cells induces EMT and significantly increases their invasive and metastatic potential in a cell autonomous manner.
HIF1A regulates PPP1R1B in pancreatic cancer
To delineate a pathway whereby HIF1A inhibits invasion and metastasis in PDAC, we performed a comparative proteomic analysis between EKPC and HIF1A−/−EKPC cells (Figure 3A). Ingenuity Pathway Analysis (IPA) of the proteomics data revealed that the most significantly activated cellular functions in HIF1A−/−EKPC cells included cell movement (P=3.84E11 −09 −06), invasion (P=2.15E ) and migration (P=1.73E ), whereas the most significantly suppressed cellular functions were cell-cell adhesion (P=2.67E−04), binding (P=3.03E−04) and cell-cell contact (P=1.01E−03) (Figure 3B, Supplementary Figure 9, Supplementary Table 1). These results further validate our conclusion that inhibition of HIF1A promotes invasion and metastasis in PDAC. Intriguingly, one of the top differentially expressed proteins was a dual kinase/phosphatase inhibitor named protein phosphatase 1 regulatory inhibitor subunit 1B (PPP1R1B) (Figure 3C, Supplementary Table 2). PPP1R1B was first shown to be expressed in the adult brain in regions that are responsive to dopamine, consistent with its alternate name Dopamine And cAMP Regulated Phosphoprotein 32 (DARPP-32).30 At the mechanistic level, PPP1R1B/DARPP-32 regulates downstream signaling of a variety of kinases through its ability to bind and inhibit the activity of Protein Phosphatase 1. This, in turn, controls the activity of hundreds of phosphorylated proteins.31 Increased levels of PPP1R1B transcripts have been reported in several cancers including breast, colon, esophagus, lung, and prostate32–36 and elevated expression of PPP1R1B has been associated with advanced disease and poor prognosis in several cancers33, 37 (Supplementary Figure 10). However, its role in PDAC and in metastasis remains poorly understood.
Figure 3: HIF1A regulates PPP1R1B in pancreatic cancer.

(A) Schematic workflow of proteomics analysis of EKPC and HIF1A−/−EKPC cells (B) The Disease and Function heatmap generated by IPA of the proteomics data reflecting the significantly enhanced (orange) or decreased (blue) cellular functions in HIF1A−/−EKPC compared with the EKPC cells. The positive and negative z scores are indicated by orange and blue color respectively. The size of the boxes reflects the predicted direction of change for the function. p values are calculated by Fisher’s Exact Test (C) Scatter plot depicting the differentially expressed proteins (DEP) in HIF1A−/−EKPC cells compared with the EKPC cells. Proteins were organized by log2 fold change (x-axis) and p-value (y-axis). The cutoff lines (pvalue of 0.05 and log2 fold change ±1.5) are shown in black. PPP1R1B protein is marked exhibiting 8.426 log2fold change; p=0.016. (D) qPCR analysis showing Ppp1r1b mRNA levels in EKPC (blue) and HIF1A−/−EKPC (orange) cells. Gapdh was used as control. ***p < 0.001 by t test analysis. (E) Immunoblot analysis of HIF1A, Ppp1r1b in EKPC and HIF1A−/−EKPC cells. (F) Western blot analysis of HIF1A and PPP1R1B expression in untreated and CoCl2 treated (100 μM for 12 hr) EKPC and HIF1A−/−EKPC cells. (G,H) IHC staining and quantification for PPP1R1B+ cells in EKPC and HIF1A−/−EKPC tumors, Scale:200 μm, (n = 5 for each group, n = 3 field of vision). ***p < 0.001 by t test analysis. (I-J) q-PCR analysis of HIF1A and PPPP1R1B transcripts in non/low-metastatic (BxPC-3, PANC-1, Mia PACA-2 and PATU8988T) and high-metastatic (AsPC-1, PATU8988S, SUIT-2 and Capan-1) human pancreatic cancer cell lines. ****p < 0.0001 by ordinary one-way ANOVA (K) Immunoblot analysis of HIF1A and PPPP1R1B in non/low-metastatic and high-metastatic human pancreatic cancer cell lines (L) Comparative expression analysis of PPP1R1B, HIF1A, HIF1B, HIF2A and HIF3A between non/low-metastatic (BxPC-3, PANC-1, Mia PACA-2 and PATU8988T) and high-metastatic (AsPC-1, PATU8988S, SUIT-2 and Capan-1) human pancreatic cancer cell lines. ns, not significant **p < 0.005, ****p < 0.0001 by unpaired t test analysis. (M) Western blot analysis of PPP1R1B and HIF1A of MIA-PaCa-2 and PATU8988T cells transfected with sh.Ctrl and sh.HIF1A (N) Heat map of HIF1A and PPP1R1B expression in PDAC patients from TCGA. (O) Paired analysis of PPP1R1B and HIF1A expression in PDAC patients (n=36) ranked based on Z scores (≥0.5). ****p<0.0001 by paired t test. All experiments were carried out in triplicate. All bar graphs represent mean and error bars are s.e.m.
We observed significant upregulation of Ppp1r1b transcript and protein in HIF1A−/−EKPC cells compared to EKPC cells (Figure 3D,E) suggesting Ppp1r1b is directly regulated by HIF1A. EKPC cells treated with the hypoxia-mimetic agent CoCl2 resulted into decreased expression of Ppp1r1b, but no such reduction of Ppp1r1b expression was observed in the HIF1A−/−EKPC cells (Figure 3F). Conversely, overexpression of m-HIF1A in HIF1A−/−EKPC cells resulted into reduced expression of Ppp1r1b (Supplementary Figure 11A). Immunohistochemistry using antibody against PPP1R1B revealed intense signal in the frank PDACs as well as in metastatic lesions in the HIF1A−/−EKPC mice as compared with the EKPC counterparts (Figure 3 G,H, Supplementary Figure 11B-D). To determine whether the observed inverse relationship between HIF1α and PPP1R1B is true in human pancreatic cancers, we carried out similar analysis in human metastatic and non-metastatic pancreatic cancer cell lines. For our analysis, we chose the well-studied human pancreatic cancer cell lines, BxPC-3, PANC-1, MIAPaCa-2, PATU8988T (Non/low-metastatic) and AsPC-1, PATU8988S, SUIT-2 and Capan-1 (High-metastatic). Notably, PATU8988T and PATU8988S are derived from the same patient.38 We performed comparative expression analysis of HIF1A and PPP1R1B in these non/low-metastatic and high-metastatic human pancreatic cancer cell lines (Figure 3I-K). Consistent with our observation, we found significant inverse correlation between HIF1A and PPP1R1B, with increased PPP1R1B and decreased HIF1A being a prominent signature in all the human pancreatic cancer cells known for high metastatic dissemination (AsPC-1, PATU8988S, SUIT-2 and Capan-1). We did not find any significant change in HIF1B, HIF2A and HIF3A expression between metastatic and nonmetastatic pancreatic cancer cells (Figure 3L). To further validate that HIF1A can directly regulate PPP1R1B expression in human pancreatic cancer cells, we carried out shRNA mediated knockdown of HIF1A in two human pancreatic cancer cell lines, PATU8988T and MIAPaCa-2. Ablation of HIF1A in these cells led to increased PPP1R1B protein levels, clearly demonstrating that HIF1A negatively regulates PPP1R1B expression in human pancreatic cancer cells (Figure 3M). We further analyzed expression of HIF1A and PPP1R1B transcripts in a dataset of human PDAC patients39 obtained from the TCGA and human pancreatic cancer cell lines from CCLE database.40 We found that the expression of HIF1A and PPP1R1B are inversely correlated (Pearson coefficient −0.184; P=0.016) (Figure 3N, Supplementary Figure 12). Further, a paired analysis among PDAC patients (n=38) ranked based on Z scores (≥0.5) showed significant inverse relationship between PPP1R1B and HIF1A expression (P=0.0001) (Figure 3O, Supplementary Table 3). These results suggest that PPP1R1B is regulated by HIF1A and is more abundant in the metastatic pancreatic cancer cell lines, suggesting their potential role in pancreatic cancer metastasis.
Increased expression of PPP1R1B correlates with poor survival and its knockdown inhibits malignant properties of PDAC cells
We then investigated the clinical significance of PPP1R1B expression in patients with PDAC using the TCGA database. We found elevated expression of PPP1R1B in PDAC samples compared to normal pancreas (Figure 4A). Kaplan-Meier analysis suggested significantly better overall survival of patients with low PPP1R1B expression, P=0.0355 (Figure 4B). Immunohistochemistry for PPP1R1B using human tumor microarray confirmed elevated expression of PPP1R1B in PDAC and distal metastasis compared to benign pancreas and other normal tissues (Figure 4C). We next validated the metastatic ability of PATU8988S cell line showing HIF1Alo/ PPP1R1Bhi ratio as compared to its syngeneic counterpart, PATU8988T cell line exhibiting HIF1Ahi/ PPP1R1Blo ratio in in vivo lung colonization assay (Supplementary Figure 13A-C). Following the observation that high expression of PPP1R1B promotes lung colonization of human PDAC cell lines, we investigated whether PPP1R1B is critical for accelerated metastasis in human pancreatic cancer. We knocked down PPP1R1B in PATU8988S cells using shRNA (Figure 4D). PATU8988S cells transfected with shPPP1R1B or sh.Control (sh.Ctrl) were injected intravenously into NOD/SCID mice and were subsequently assessed for lung metastasis. Whereas PATU8988S +sh.Ctrl cells formed tumor nodules in the lungs at expected frequency, PPP1R1B depletion led to significantly reduced lung metastasis (Figure 4E,F, Supplementary Figure 13D,E). This data clearly demonstrated that increased expression of PPP1R1B results into higher metastatic dissemination of pancreatic cancer cells.
Figure 4: Increased expression of PPP1R1B correlates with poor survival and its knockdown inhibits malignant properties of PDAC cells.

(A). Comparative expression analysis of PPP1R1B in 179 PDAC tissue samples with that of 171 normal pancreas using the TCGA database (GEPIA). *P<0.05 (B) Kaplan–Meier survival analysis of pancreatic cancer patients with high (n=55) and low (n=63) PPP1R1B expression (TCGA), *P<0.05 by Mantel-Cox test (C) IHC staining of PPP1R1B in human tissue array which includes normal kidney, placenta, benign pancreatic duct, PDACs and nodal metastasis. (D) Western blot analysis of PPP1R1B in PATU8988S cells transfected with sh.Ctrl or sh.PPP1R1B. (E&F) Lung colonization assay of PATU8988S+sh.PPP1R1B and PATU8988S+sh.Ctrl cells in NOD/SCID mice by bioluminescence, n=4 for each group. *p<0.05 by t test.
PPP1R1B promotes metastasis in pancreatic cancer by reducing p53
We next investigated the pathway through which PPP1R1B promotes metastasis in PDAC. In our proteomics analysis, we observed significantly reduced levels of p53 protein (−8.80 log2fold change) in HIF1A−/−EKPC cells compared to EKPC cells (Figure 5A, Supplementary Table 2), indicating that p53 protein levels are HIF1A dependent in EKPC tumors. P53 is known to be stabilized by HIF1A.41 Western blot analysis revealed reduced p53 protein in the HIF1A−/−EKPC cells as well as in HIF1Alo/ PPP1R1Bhi cancer cell lines, AsPC-1 and PATU8988S (compared to HIF1Ahi/ PPP1R1Blo cells) (Figure 5B,C). Since the reduction or loss of p53 protein was evident in the cells with high levels of PPP1R1B, we investigated whether elevating PPP1R1B expression reduces p53 levels. Ectopic expression of PPP1B1B in HIF1Ahi/ PPP1R1Blo cell lines (BxPC-3 and PATU8988T), resulted into reduced p53 protein compared to the control (Figure 5D). Next, we wanted to investigate how expression of PPP1R1B may reduce p53 protein. Intracellular p53 levels are regulated by the E3 ubiquitin ligase, MDM2.42 Phosphorylation of MDM2 at Ser166 maintains its stability by preventing self-ubiquitination.43 De-phosphorylation of MDM2Ser166 is critical for its degradation and concomitant stability of p53 (Figure 5E). We hypothesized that PPP1R1B, a protein phosphatase 1 inhibitor, can stabilize MDM2 by inhibiting dephosphorylation of MDM2Ser166 and thereby effecting p53 degradation. Ectopic expression of PPP1R1B should then increase the phosphorylation of MDM2Ser166. Indeed, expression of PPP1R1B increased the phosphorylation of MDM2Ser166 in PATU8988T and BxPC-3 (Figure 5F), suggesting that PPP1R1B potentiates MDM2 via increased MDM2Ser166 phosphorylation, which in turn leads to less stable p53 protein. The effect of PPP1R1B on p53 stability was measured in both PATU8988T and BxPC-3 cell lines by overexpressing PPP1R1B and performing a cycloheximide chase assay. The stability of p53 protein was significantly less (P < 0.05) in PPP1R1B overexpressing PATU8988T and BxPC-3 cells (Figure 5G,H and Supplementary Figure 14A,B), thus suggesting that increased PPP1R1B expression in PDAC cells results in p53 degradation.
Figure 5: PPP1R1B promotes metastasis in pancreatic cancer by reducing p53.

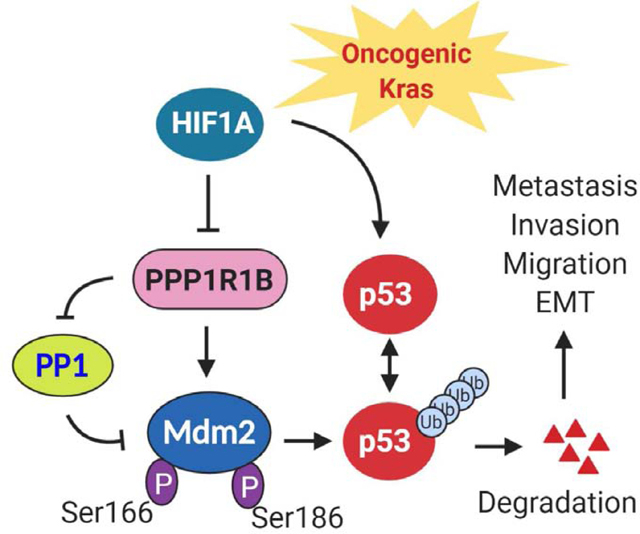
(A) Scatter plot depicting the most significantly up or down regulated proteins (> ± log28) in HIF1A−/−EKPC cells compared with the EKPC cells. Proteins were organized by log2 fold change (Y-axis). PPP1R1B and p53 are indicated by red and blue respectively. (B,C) Immunoblot analysis of PPP1R1B and p53 levels in mouse and human pancreatic cancer cell lines. β ACTIN was used as control. (D) Western blot analysis of ectopic expression of PPP1R1B and its effect on p53 level in BxPC-3 and PATU8988T cells. GFP was used as control. (E) Schematic model of MDM2 and p53 feedback regulatory loop (F) Western blot analysis of phospho-MDM2Ser186, total MDM2 and β ACTIN in BxPC-3 and PATU-8988T cells overexpressing PPP1R1B. GFP was used as control. (G & H) Cycloheximide chase assay and quantification for p53 in PATU8988T cells overexpressing PPP1R1B or GFP. β-ACTIN was used as a loading control. (I) Western blot analysis of p53 and β ACTIN levels in EKPCLuc cells stably transfected with sh.Ctrl and two independent sh.p53. (J & K) Lung colonization of EKPCLuc + sh.p53 or EKPCLuc + sh.Ctrl cells bioluminescence imaging. N=5 for each group. ***p<0.0005 by t test (L) Schematic of the mechanistic link between loss of HIF1A, increased expression of PPP1R1B and degradation of the p53 protein that promotes invasion and metastasis in PDAC.
Based on these observations, we hypothesized that PPP1R1B mediated loss of residual p53 in the PDACs leads to increased metastatic potential of these cells. To test this, we sought to determine whether loss of p53 in EKPC cells enhanced their metastatic ability, as observed in HIF1A−/−EKPC cells (Figure 2I,J). We knocked down p53 in EKPC cell lines by stably expressing shRNA against p53 (Figure 5I). Equal number of EKPC+shp53 and EKPC+sh.Ctrl cells were injected into the lateral tail vein of immunocompetent C57/BL6 mice, and their lung colonization was evaluated at various times by bioluminescence. Loss of residual p53 in the EKPC cells resulted in a significant increase in metastatic potential compared to the control cells (Figure 5J, K). Taken together, these results support a model in which depletion of HIF1A promotes metastasis in pancreatic cancer cells, in part, via de-regulation of PPP1R1B, leading to increased phosphorylation at the MDM2Ser166 and reduction of p53 in pancreatic cancer (Figure 5L).
Discussion
In spite of the expression of pro-angiogenic factors, pancreatic cancer remains persistently hypo vascular. This results in acute hypoxia and upregulation of HIF1A, a key transcription factor responsible for inducing changes in the tumor metabolism that allows it to adapt to insufficient oxygen.44 Hypoxia has been shown to induce the Hedgehog signaling,16 stimulate cancer-associated fibroblasts45–47 and has been suggested to contribute to poor outcome in several cancers, including PDAC.48–50 Thus, there have been considerable efforts to understand and target HIF1A for clinical intervention, especially in hypoxic cancers like PDAC.7
Our results, however, support the emerging evidence that the role of HIF1A is not universally oncogenic. The ability of HIF1A to function as a tumor suppressor was first shown in VHL-deficient renal carcinoma, where patients harboring a focal deletion of HIF1A at chromosome 14q presented a more aggressive form of the cancer.12 Subsequently, tumor suppressive role of HIF1A has also been shown in acute myeloid leukemia and IDH-mutant gliomas.13,51 Genetic deletion of HIF1A has also been shown to promote pro-tumorigenic role for B cells in a pancreatic cancer model.14 Additionally, Leppänen et al. demonstrated that weak HIF1A expression correlated with poor prognosis in resectable PDAC.17 These studies, along with the results reported here, suggest that the role of HIF1A in cancer is context dependent. Although in some cases it appears to promote tumorigenesis, loss of HIF1A seems to drive a more malignant phenotypes in other cases. Here, we explored the functional consequences of the loss of HIF1A in pancreatic cancer and provide multiple lines of evidence that the deficiency of HIF1A promotes invasive and metastatic phenotype of PDAC cells. These results have great significance in our understanding of HIF1A as well as the potential treatment of pancreatic cancer.
Enhanced expression of other HIFs due to loss of HIF1A expression, mostly HIF2A, has been reported in some malignancies, particularly in renal cancer.52 However, we did not observe any compensatory increase in HIF2A in HIF1A−/−EKPC cells. A number of other studies also stated no such compensatory link between HIF1A and HIF2A.53 Loss of HIF1A have also been reported to result in enhanced cell proliferation during KrasG12D-driven PanIN progression, but no apparent compensatory role of HIF2A was observed.14 Taken together, these data suggest that differential activation of HIF2A and/or other HIFs in the context of tumor progression due to loss of HIF1A may be dependent upon the mutational landscape of the tumor cells, cell types and the stability of other cofactors.54 However, in order to address a role of HIF2A in the context of HIF1A deficient PDAC in an unambiguous manner, an autochthonous mouse model of PDAC needs to be studied in which both HIF1A and HIF2A can be genetically manipulated.
This study also identified PPP1R1B, a gene negatively regulated by HIF1A, as a key promoter of metastasis in pancreatic cancer. Elevated expression of PPP1R1B have been associated with advanced disease and poor prognosis in several cancers including breast and lung cancer.33–35 Also in our analyses, elevated levels of PPP1R1B correlated with poorer outcome in pancreatic cancer patients. We also found inverse correlation between HIF1Aand PPP1R1B, (high PPP1R1B and low HIF1A levels) being a prominent signature in several human pancreatic cancer cells known for high metastatic dissemination. We show that genetic inhibition of PPP1R1B in human pancreatic cancer cells significantly reduces their invasive and metastatic spread in vivo.
We also noticed a significant reduction in p53 protein stability in HIF1A compromised human and mouse PDACs. Earlier studies have suggested that HIF1A can stabilize p53.41, 55 We propose that stabilization of p53 by HIF1A can be mediated by HIF1A’s ability to down-regulate PPP1R1B. Loss of HIF1A and concommitant de-regulation of PPP1R1B results in loss of p53 in pancreatic cancer cells. Our data clearly suggest that loss of residual p53 in PDAC, which may include a mutant p53 allele, can lead to a significant increase in the metastatic potential of pancreatic cancer cells. This observation is in line with recent studies which implicate loss of p53 as a major driver of metastasis in KRas driven cancers.56–58 Finally, inactivation of protein phosphatases, namely PP1 and PP2A, has been shown to be prevalent in several cancers, including PDACs.59, 60 Activation of these protein phosphatases by exploiting their regulators are emerging as possible therapeutic intervention for these cancers.61, 62 Our study identifies PPP1R1B as a potential target to mitigate pancreatic cancer metastasis and provides a detailed molecular understanding of how inhibiting HIF1A may influence the outcome of pancreatic cancer unfavorably in clinical setting.
Supplementary Material
What you need to know:
Background and Context:
Pancreatic ductal adenocarcinomas (PDACs) are hypovascular, resulting in the upregulation of hypoxia inducible factor 1 alpha (HIF1A), which promotes survival of cells under low-oxygen conditions. We studied the roles of HIF1A in development of pancreatic tumors in mice.
New Findings
HIF1A can act a tumor suppressor by preventing expression of PPP1R1B and subsequent degradation of the p53 protein in pancreatic cancer cells. Loss of HIF1A from pancreatic cancer cells increases their invasive and metastatic activity.
Limitations
This study was performed in mice and human cell lines and tissues. Further studies are needed of this pathway in human pancreatic tumor progression.
Impact
HIF1A is a tumor suppressor in pancreatic cancer cells; strategies to restore its activity might be developed for treatment of PDAC.
Lay Summary
The researchers identified a protein that is lost from pancreatic cancer cells that allows them to become more invasive and promote tumor metastasis.
Acknowledgements
We thank Robert Wechsler-Reya for generously sharing lab reagents including the sh.p53 construct and providing critical feedback on the manuscript. We thank animal core facility staff for help with mouse genotyping, proteomics and immunohistochemistry core services at the Sanford Burnham Prebys Medical Discovery Institute. This work was funded in part by grants from the National Cancer Institute (1R01CA200643–01A1 to A.B., R01CA181385 to P.P.P.), the Department of Defense (PC160884 to A.B.), the American Cancer Society Research Scholar Awards (125627-RSG-14–074-01-TBG to A.B., RSG-14–171-01-CSM to P.P.P.), National Institute of Health (U54CA210190 University of Minnesota Physical Sciences in Oncology Center Project 2 to P.P.P. and Core 2: Cell and Whole Animal Genetic Engineering to A.B.), the Randy Shaver Research and Community Fund (P.P.P.) and the Masonic Cancer Center Brainstorm award (to A.B. and P.P.P.)
Footnotes
Conflicts of interest: The authors disclose no conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 3.Cannon A, Thompson C, Hall BR, et al. Desmoplasia in pancreatic ductal adenocarcinoma: insight into pathological function and therapeutic potential. Genes Cancer 2018;9:78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craven KE, Gore J, Korc M. Overview of pre-clinical and clinical studies targeting angiogenesis in pancreatic ductal adenocarcinoma. Cancer Lett 2016;381:201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koong AC, Mehta VK, Le QT, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys 2000;48:919–22. [DOI] [PubMed] [Google Scholar]
- 6.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 2010;40:294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017;32:71–87 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008;30:393–402. [DOI] [PubMed] [Google Scholar]
- 9.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ 2008;15:678–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schindl M, Schoppmann SF, Samonigg H, et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin Cancer Res 2002;8:1831–7. [PubMed] [Google Scholar]
- 11.Baba Y, Nosho K, Shima K, et al. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am J Pathol 2010;176:2292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen C, Beroukhim R, Schumacher SE, et al. Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov 2011;1:222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, et al. HIF-1alpha can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood 2014;124:3597–607. [DOI] [PubMed] [Google Scholar]
- 14.Lee KE, Spata M, Bayne LJ, et al. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Discov 2016;6:256–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun HC, Qiu ZJ, Liu J, et al. Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. Int J Oncol 2007;30:1359–67. [PubMed] [Google Scholar]
- 16.Spivak-Kroizman TR, Hostetter G, Posner R, et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res 2013;73:3235–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leppänen J, Helminen O, Huhta H, et al. Weak HIF-1alpha expression indicates poor prognosis in resectable pancreatic ductal adenocarcinoma. World Journal of Surgical Oncology 2018;16:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaika NV, Gebregiworgis T, Lewallen ME, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A 2012;109:13787–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50. [DOI] [PubMed] [Google Scholar]
- 20.Srinivas S, Watanabe T, Lin C-S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Developmental Biology 2001;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan HE, Poloni M, McNulty W, et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res 2000;60:4010–5. [PubMed] [Google Scholar]
- 22.Schreiber FS, Deramaudt TB, Brunner TB, et al. Successful growth and characterization of mouse pancreatic ductal cells: functional properties of the Ki-RAS(G12V) oncogene. Gastroenterology 2004;127:250–60. [DOI] [PubMed] [Google Scholar]
- 23.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Research 2017;45:W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuo Y, Ding Q, Desaki R, et al. Hypoxia inducible factor-1 alpha plays a pivotal role in hepatic metastasis of pancreatic cancer: an immunohistochemical study. J Hepatobiliary Pancreat Sci 2014;21:105–12. [DOI] [PubMed] [Google Scholar]
- 27.Yang M-H, Wu M-Z, Chiou S-H, et al. Direct regulation of TWIST by HIF-1α promotes metastasis. Nature Cell Biology 2008;10:295. [DOI] [PubMed] [Google Scholar]
- 28.Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu CJ, Sataur A, Wang L, et al. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell 2007;18:4528–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berger B, Febvret A, Greengard P, et al. DARPP-32, a phosphoprotein enriched in dopaminoceptive neurons bearing dopamine D1 receptors: distribution in the cerebral cortex of the newborn and adult rhesus monkey. J Comp Neurol 1990;299:327–48. [DOI] [PubMed] [Google Scholar]
- 31.Avanes A, Lenz G, Momand J. Darpp-32 and t-Darpp protein products of PPP1R1B: Old dogs with new tricks. Biochem Pharmacol 2019;160:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebihara Y, Miyamoto M, Fukunaga A, et al. DARPP-32 expression arises after a phase of dysplasia in oesophageal squamous cell carcinoma. British Journal of Cancer 2004;91:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alam SK, Astone M, Liu P, et al. DARPP-32 and t-DARPP promote non-small cell lung cancer growth through regulation of IKKα-dependent cell migration. Communications Biology 2018;1:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beckler A, Moskaluk CA, Zaika A, et al. Overexpression of the 32-kilodalton dopamine and cyclic adenosine 3’,5’-monophosphate-regulated phosphoprotein in common adenocarcinomas. Cancer 2003;98:1547–51. [DOI] [PubMed] [Google Scholar]
- 35.Hamel S, Bouchard A, Ferrario C, et al. Both t-Darpp and DARPP-32 can cause resistance to trastuzumab in breast cancer cells and are frequently expressed in primary breast cancers. Breast Cancer Res Treat 2010;120:47–57. [DOI] [PubMed] [Google Scholar]
- 36.Vangamudi B, Peng D-F, Cai Q, et al. t-DARPP regulates phosphatidylinositol-3-kinase-dependent cell growth in breast cancer. Molecular Cancer 2010;9:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lenz G, Hamilton A, Geng S, et al. t-Darpp Activates IGF-1R Signaling to Regulate Glucose Metabolism in Trastuzumab-Resistant Breast Cancer Cells. Clin Cancer Res 2018;24:1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elsasser HP, Lehr U, Agricola B, et al. Establishment and characterisation of two cell lines with different grade of differentiation derived from one primary human pancreatic adenocarcinoma. Virchows Arch B Cell Pathol Incl Mol Pathol 1992;61:295–306. [DOI] [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32:185–203 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghandi M, Huang FW, Jané-Valbuena J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An WG, Kanekal M, Simon MC, et al. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature 1998;392:405–408. [DOI] [PubMed] [Google Scholar]
- 42.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res 2003;1:1001–8. [PubMed] [Google Scholar]
- 43.Feng J, Tamaskovic R, Yang Z, et al. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem 2004;279:35510–7. [DOI] [PubMed] [Google Scholar]
- 44.Eales KL, Hollinshead KER, Tennant DA. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016;5:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Ahrens D, Bhagat TD, Nagrath D, et al. The role of stromal cancer-associated fibroblasts in pancreatic cancer. Journal of Hematology & Oncology 2017;10:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalluri R The biology and function of fibroblasts in cancer. Nature Reviews Cancer 2016;16:582. [DOI] [PubMed] [Google Scholar]
- 47.Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Rev 2012;31:195–208. [DOI] [PubMed] [Google Scholar]
- 48.Moeller BJ, Dreher MR, Rabbani ZN, et al. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell 2005;8:99–110. [DOI] [PubMed] [Google Scholar]
- 49.Semenza GL. Targeting HIF-1 for cancer therapy. Nature Reviews Cancer 2003;3:721–732. [DOI] [PubMed] [Google Scholar]
- 50.Zhao X, Gao S, Ren H, et al. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res 2014;74:2455–64. [DOI] [PubMed] [Google Scholar]
- 51.Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012;483:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 2005;25:5675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu CJ, Wang LY, Chodosh LA, et al. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 2003;23:9361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol 2014;49:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen D, Li M, Luo J, et al. Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function. J Biol Chem 2003;278:13595–8. [DOI] [PubMed] [Google Scholar]
- 56.Powell E, Shao J, Yuan Y, et al. p53 deficiency linked to B cell translocation gene 2 (BTG2) loss enhances metastatic potential by promoting tumor growth in primary and metastatic sites in patient-derived xenograft (PDX) models of triple-negative breast cancer. Breast Cancer Research 2016;18:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Powell E, Piwnica-Worms D, Piwnica-Worms H. Contribution of p53 to metastasis. Cancer Discov 2014;4:405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ignatius MS, Hayes MN, Moore FE, et al. tp53 deficiency causes a wide tumor spectrum and increases embryonal rhabdomyosarcoma metastasis in zebrafish. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tohmé R, Izadmehr S, Gandhe S, et al. Direct activation of PP2A for the treatment of tyrosine kinase inhibitor–resistant lung adenocarcinoma. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allen-Petersen BL, Risom T, Feng Z, et al. Activation of PP2A and Inhibition of mTOR Synergistically Reduce MYC Signaling and Decrease Tumor Growth in Pancreatic Ductal Adenocarcinoma. Cancer Res 2019;79:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sangodkar J, Perl A, Tohme R, et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. The Journal of Clinical Investigation 2017;127:2081–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tomiyama A, Kobayashi T, Mori K, et al. Protein Phosphatases-A Touchy Enemy in the Battle Against Glioblastomas: A Review. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.