

SUMMARY
Rpn13 is one of several ubiquitin receptors in the 26S proteasome. Cys88 of Rpn13 has been proposed to be the principal target of RA190, an electrophilic small molecule with interesting anti-cancer activities. Here, we examine the claim that RA190 mediates its cytotoxic effects through engagement with Rpn13. We find no evidence that this is the case. In vitro, RA190 is has no measurable effect on any of the known interactions of Rpn13. In cellulo, we see no physical engagement of Rpn13 by RA190, either on C88 or any other residue. However, chemical proteomics experiments in two different cell lines reveal that dozens of other proteins are heavily engaged by RA190. Finally, increasing or reducing the level of Rpn13 in HeLa and melanoma cells had no effect on the sensitivity of HeLa or melanoma cells to RA190. We conclude that Rpn13 is not the physiologically relevant target of RA190.
In Brief
Dickson et al. used proteome-wide and targeted analysis of a cytotoxic molecule, RA190. Despite previous reports of RA190’s selectivity for proteasomal ubiquitin receptor Rpn13, the group finds no evidence for cellular engagement. Instead, a number of proteins were found to be liganded by RA190, suggesting that polypharmacology drives its anti-cancer activity.
Graphical Abstract
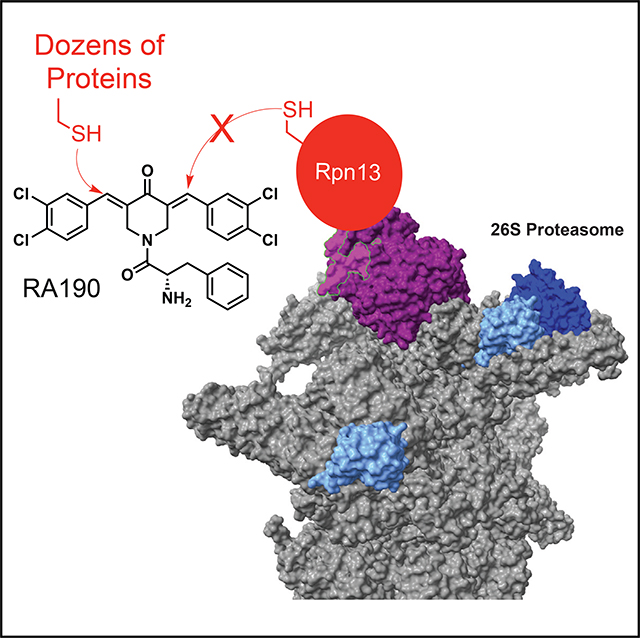
INTRODUCTION
The 26S proteasome is an approximately 2.5-MDa complex responsible for the degradation of many proteins within the cell, regulating numerous cellular pathways and playing a critical role in protein folding quality control (Voges et al., 1999). It is comprised of two sub-complexes: the 19S regulatory particle, which recognizes and unfolds a polyubiquitinated substrate protein, and the barrel-like 20S catalytic particle (CP) (Coux et al., 1996), which degrades the unfolded polypeptide using three distinct active sites. The proteasome is a validated target for the treatment of certain cancers that display an unusually high dependence on protein turnover (Goldberg, 2012; Kisselev and Goldberg, 2001). In 2003, the first-in-class proteasome inhibitor bortezomib (Paramore and Frantz, 2003), a peptide-boronic acid hybrid that targets the active sites of the 20S CP, was approved for treatment of multiple myeloma (MM), a cancer of plasma B cells under extreme proteotoxic stress caused by uncontrolled immunoglobulin production (Rajkumar and Kumar, 2016). However, resistance to bortezomib usually appears within 1–2 years and the utility of this drug for most cancers is compromised by dose-limiting toxicity (de Wilt et al., 2012; Franke et al., 2012; Huang et al., 2014). These limitations have spurred interest in targeting other units of the proteasome in hopes of achieving a greater therapeutic window and delaying the emergence of resistance (Li et al., 2017; Perez et al., 2017).
Of particular interest in this regard is Rpn13, one of several proteasomal ubiquitin receptors responsible for recruiting a polyubiquitinated substrate to the 26S proteasome (Husnjak et al., 2008). Rpn13 also recruits the deubiquitinase Uch37 to the proteasome and strongly stimulates its enzymatic activity (Yao et al., 2006). Several published reports show that some cancer cells overexpress ADRM1/Rpn13 and indicate that its depletion using genetic methods leads to reduced cancer cell viability (Chen et al., 2009; Fejzo et al., 2008, 2011, 2013, 2015). On the other hand, deletion of Rpn13 in yeast does not result in a severe phenotype (Seong et al., 2007), suggesting that Rpn13 not an essential protein. There are conflicting reports of Rpn13’s essentiality in mice, with one group reporting viable, but infertile, Rpn13 null pups (Al-Shami et al., 2010) and another group reporting postnatal lethality of Rpn13 knockout mice (Hamazaki et al., 2015). This stands in contrast to other sub-units of the proteasome, which are encoded by absolutely essential genes. Taken together, these data suggest the intriguing possibility that Rpn13-targeted anti-cancer drugs might have a large therapeutic window with few side effects.
The first small molecule reported to target Rpn13 was the bisbenzylidinepiperidinone RA190 (Figure 1A) (Anchoori et al., 2013). It is a more potent and soluble derivative of a compound called RA1 that was identified in a high-throughput phenotypic screen for molecules that were substantially more toxic to cervical cancer cells than non-malignant cells (Anchoori et al., 2011; Bazzaro et al., 2011). Treatment of cultured cells with RA190 results in an accumulation of polyubiquitinated proteins, suggesting inhibition of some facet of the ubiquitin-proteasome pathway. The molecule also has activity in mouse models of cancer (Anchoori et al., 2013; Song et al., 2016; Soong et al., 2020). The highly electrophilic nature of the α,β-unsaturated ketone in RA190 suggested that it might engage cysteine thiols irreversibly, and indeed pre-incubation of the compound with thiols or modification of the ketone moiety reduced its activity greatly (Anchoori et al., 2013). When biotin-RA190 (Figure 1A) was incubated with a HeLa cell extract, the major protein engaged by the compound, as determined by denaturing SDS-PAGE and blotting with streptavidin, was Rpn13 (Anchoori et al., 2013). This prompted a structural investigation of the RA190-Rpn13 complex by NMR. The largest spectral differences between free and RA190-adducted Rpn13 were clustered around cysteine 88 (C88) in the ubiquitin- and Rpn2-binding Pru domain of Rpn13, suggesting that this residue is modified covalently by the compound (Anchoori et al., 2013). Therefore, the model proposed is that alkylation of this residue in Rpn13 inhibits some function of Rpn13 that is essential to some cancer cells.
Figure 1. Physical and Functional Interactions of RA190 and Rpn13 In Vitro.

(A) Structures of RA190 and the functionalized derivatives used in this study.
(B) Analysis of the alkylation of purified Rpn13 by biotin-RA190. Wild type and C88A His6-Rpn13 were incubated with 1 μM of biotin-RA190 for 1 h at 4°C then analyzed by immunoblotting. In the reactions containing both the biotinylated RA190 and the parent compound, 10 μM RA-190 was pre-incubated for 1 h at 4°C.
(C) SK-MEL-5 cell lysate was treated with RA190-alkyne, followed by click chemistry with TAMRA azide, resolved by SDS-PAGE, and visualized for TAMRA signal (right), and then stained with Coomassie dye (left).
(D) His6-Rpn13 was incubated with MBP-Uch37 in the presence of RA190 or vehicle for 1 h at 4°C. MBP-Uch37 was pulled down with amylose resin and the amount of protein precipitated was evaluated by SDS-PAGE and Coomassie staining.
(E) RA190 (100 μM) or vehicle (1% DMSO) was incubated with 6His-Rpn13 (100 μM) in PBS buffer for 1 h at 4°C before addition of an equal volume of a solution containing ubiquitin 48-fluorescein (50 nM). Aliquots of 2-fold serial dilutions of this solution were prepared and the level of fluorescence polarization was determined. Error bars represent the standard deviation of technical triplicate measures.
Subsequent studies, however, seem to be incompatible with this model. Song et al. (2016) reported that HCT-116 cells, which are moderately sensitive to RA190, are still viable when ADRM1 is knocked out. This argues against the idea that some activity of Rpn13 is essential in this cell line and that inhibition of Rpn13 is the cause of RA190-inudced cytotoxicity. However, these authors also reported that the ADRM1−/− HCT-116 cells are much less sensitive to RA190 than the wild-type cells. RA190 sensitivity was restored when Rpn13, but not Rpn13C88A, was expressed in this genetic background. These data, taken at face value, certainly argue that the presence of Rpn13, and specifically C88, are somehow important for RA190-mediated cytotoxicity. But combined with the fact that Rpn13 is not essential for cell growth, the only model consistent with these data is that RA190 interaction with Rpn13 Cys88 (either direct or indirect) somehow induces the protein to have a dominant negative effect on cell growth, although the authors did not seem to acknowledge this.
Recent structural and biophysical studies also seem incompatible with the original model of RA190 activity. Two crystal structures of Rpn13 bound to the C-terminal domain of Rpn2 that tethers Rpn13 to the proteasome (Lu et al., 2015) showed that C88 is not solvent exposed and thus not accessible to RA190 (Lu et al., 2017; VanderLinden et al., 2017). Competition binding experiments confirmed the prediction from these structures that RA190 and Rpn2 compete for binding to Rpn13 (Lu et al., 2017). Thus, RA190 cannot alkylate Rpn13 at this residue when it is incorporated into the proteasome.
Our group has also been interested in testing the hypothesis that Rpn13 is a good target for chemotherapy. Taking a more focused approach, we conducted a screen for Rpn13 ligands in vitro and identified a peptoid (KDT11) that engages the protein selectively with a modest KD of 3 μM (Trader et al., 2015). Treatment of cells with KDT11 also causes a buildup of polyubiquitinated proteins. Furthermore, the peptoid is selectively toxic to MM, melanoma and several other cancer cell lines with a half maximal inhibitory concentration (IC50) that corresponds closely to the KD of the KDT11-Rpn13 complex (Trader et al., 2015). Surprisingly, however, our recent studies of the cellular mechanism of action of KDT11 have cast doubt upon the idea that it drives cancer cell-selective cytotoxicity through interaction with Rpn13 (Dickson et al., 2020 [this issue of Cell Chemical Biology]). These data, combined with the rather confusing picture of RA190’s mechanism of action, call into question whether there are really any cancer cell-specific activities of Rpn13 that can be inhibited by a small molecule. In this study, we use a variety of chemical biology approaches to ask if RA190 engages Rpn13 in several cancer cell types either physically or functionally. As reported below, we find no evidence for engagement of Rpn13 by RA190 in cells, nor do we find a correlation between Rpn13 levels and the degree of RA190 sensitivity. Biochemically, RA190 has no effect on any of the known activities of Rpn13. However, we show that RA190 acts as a promiscuous alkylator in cellulo, efficiently coupling to dozens of different proteins, none of which are Rpn13. Based on these data, we argue that Rpn13 is not the physiologically relevant target of RA190 and speculate that the compound probably evinces its cytotoxicity through a “polypharmacology” mechanism that involves many different targets.
RESULTS
Physical and Functional Interactions of RA190 and Rpn13 In Vitro
As discussed above, RA190 can alkylate C88 of purified Rpn13 in vitro, but published data suggests that this is unlikely to reflect a selective alkylation of this site. Using intact protein mass spectrometry (MS), Walters and co-workers showed that other cysteines on Rpn13 can be alkylated as well (Lu et al., 2017). In our hands using an alternative approach, this is also the case. Purified 6His-Rpn13 or 6His-Rpn13C88A (1 μM) was incubated in PBS buffer for 1 h at 4°C with equimolar biotin-RA190 (Figure 1A) or vehicle control (0.1% DMSO). Covalent adduct formation was assessed by SDS-PAGE and blotting with labeled streptavidin. As shown in Figure 1B, both wild-type Rpn13 and the C88A mutant were labeled by biotin-RA190 with approximately equal efficiency. In both cases, pre-incubation of the protein with RA190 reduced the yield of the biotinylated product greatly, indicating that the biotinylated compound recapitulates the binding behavior of the unmodified drug faithfully. Lu et al. (2017) also reported that RA190 can alkylate purified Uch37 as well, suggesting significant promiscuity, at least in vitro. To test this further, we incubated alkyne-RA190 (Figure 1A) with a crude cell lysate for 1 h, labeled the alkylated proteins with rhodamine azide using click chemistry, then assessed the scope of alkylation using SDS-PAGE and fluorescence imaging. As shown in Figure 1C, a large number of proteins were dose-dependently alkylated by alkyne-RA190. There did not appear to be any proteins that were preferentially engaged with high efficiency at lower alkyne-RA190 concentrations. These data, along with those published previously (Lu et al., 2017), demonstrate that, while RA190 can certainly alkylate Rpn13 in vitro, this does not appear to a selective process. Instead, RA190 behaves in as a promiscuous alkylator in vitro.
We proceeded to ask if RA190 alkylation affects any of the known protein-protein interactions of Rpn13. Equimolar amounts of a Maltose Binding Protein (MBP)-Uch37 fusion protein and 6His-Rpn13 (1 μM) were incubated in PBS supplemented with 50 μM RA190 or vehicle control (0.5% DMSO) for 1h at 4°C. Amylose resin was added to the solution to capture MBP-Uch37. The amount of Rpn13 also retained on the resin was assessed by SDS-PAGE and Coomassie staining (Figure 1D). There was no observable change in the amount of Rpn13 associated with Uch37 in the presence or absence of RA190. This result is in agreement with the report of Lu et al. (2017) who also found that RA190 had no effect on Uch37-Rpn13 interactions using a different assay.
To determine if RA190 affects interaction of Rpn13 with ubiquitin, we used a previously described fluorescence polarization-based assay (Du and Strieter, 2018). 6His-Rpn13 (100 μM) was pre-incubated with RA190 (100 μM) or vehicle (1% DMSO) for 1h at 4°C, before the addition of a ubiquitin-fluorescein conjugate. Complex formation between Rpn13 and the labeled ubiquitin results in an increase in fluorescence polarization relative to that of the free ubiquitin. A series of 2-fold diluted solutions of this master mix were prepared, and the level of fluorescence polarization was measured. As shown in Figure 1E, the presence of RA190 had no effect on the interaction of Rpn13 and the labeled ubiquitin. Since it has already been shown that RA190 cannot compete with Rpn2 for binding to Rpn13 (Lu et al., 2017), we conclude that RA190 has no effect on any of the known protein-protein interactions of Rpn13 in vitro.
Rpn13 Levels Do Not Correlate with RA190 Sensitivity in Cells
One expects that the IC50 of a high-affinity drug will vary depending on the concentration of its cellular target protein. Thus, evaluating the effect of increasing or decreasing the level of Rpn13 on the sensitivity of cells to RA190 constitutes a simple test to determine if Rpn13 is indeed the physiologically relevant target of RA190. Since much of the work with RA190 has been done in MM cells, we initially tried to carry out this study in that cell line, but in our hands these cells are difficult to transfect and we were unsuccessful in achieving efficient small interfering RNA (siRNA)-mediated knockdown of Rpn13. Therefore, we evaluated the toxicity of RA190 in a variety of other lines. As shown in Figure S2A, RA190 is dose-dependently toxic to a number of cancer cells, including breast, blood, lung, cervical, and melanoma lines, all displaying an IC50 in the 1–10 μM range. Interestingly, in our hands non-malignant cells were similarly sensitive to RA190 (Figure S2A). We chose HeLa cervical cancer cells and SK-MEL-5 melanoma cells for further experiments since these are readily transfectable. We also showed that treatment of SK-MEL5 cells inhibits cell-cycle progression in the melanoma line at the G2/M transition (Figure S2B), as is already known for HeLa cells (Randles et al., 2016).
The level of Rpn13 protein was knocked down to about 14% of normal using siRNA in each cell line (Figures 2A and 2C). This large reduction in the amount of Rpn13 did not substantially affect cell viability in either case. Twenty-four hours after transfection, the cells were treated with the indicated concentration of RA190 for 24 h, then cell viability was assessed using the Cell-Titer-Glo 2.0 Assay, which quantifies total ATP levels within a cell population. A vehicle control sample was used as a baseline. As shown in Figures 2B and 2D, the IC50 of RA190 in both cell lines was unaffected by the reduction in the level of the Rpn13 protein.
Figure 2. Analysis of the Dependence of Rpn13 Protein Level on RA190 Toxicity.

(A) SK-MEL-5 cells were transfected with siRNA targeting Rpn13, or a control siRNA for 24 h. The levels of Rpn13 were measured by western blotting with anti-Rpn13 and anti-GAPDH antibodies.
(B) SK-MEL-5 cells shown above were then exposed to the indicated concentration of RA9190 for 24 h, after which time the viability of the cells was assessed using the CellTiter-Glo 2.0 Assay. Error bars represent standard deviation of technical triplicate. This experiment was conducted with three biological replicates.
(C) HeLa cells were transfected with siRNA targeting Rpn13, or a control siRNA for 24 h. The levels of Rpn13 were measured by western blotting with anti-Rpn13 and anti-GAPDH antibodies.
(D) Viability of HeLa cells was performed as described for SK-MEL-5 cells using the CellTiter-Glo 2.0 Assay.
To probe this issue in a different way, we examined the effect of increasing Rpn13 levels in SK-MEL-5 cells. If Rpn13 is indeed the target of RA190, then overexpression of the protein should make the cells less sensitive to the compound. We carried out this experiment by overexpressing FLAG-tagged Rpn13 either on top of native Rpn13 or in cells where the native protein had been knocked down using an siRNA targeted to an untranslated region (UTR) of the message. As shown in Figure 3A, the level of FLAG-Rpn13 in these cells was in all cases 8- to 10-fold higher than the level of native Rpn13. However, this large increase in the level of Rpn13 had no effect on the sensitivity of the cells to RA190 (Figure 3B).
Figure 3. Determination RA190’s Dependence on Rpn13 Cys88 for Cellular Toxicity.

(A) Effect of Rpn13 overexpression on the sensitivity of SK-MEL-5 cells to RA190. SK-MEL-5 cells were transfected with non-targeting siRNA or siRNA targeting the Rpn13’s 5′ UTR for 24 h. The cells were then transfected with pcDNA5_FLAG-Rpn13. The levels of FLAG-Rpn13 were determined by western blotting with Rpn13 and FLAG antibody, with GAPDH as the loading control.
(B) The sensitivity of these cells to RA190 was determined 24 h after addition of the indicated concentration of RA190. The error bars in all of the graphs represent the standard deviation of the mean of triplicate measures.
(C) Effect of Rpn13C88A overexpression on the sensitivity of SK-MEL-5 cells to RA190. SK-MEL-5 cells were transfected with non-targeting siRNA or Rpn13 UTR siRNA for 24 h. The cells were then transfected with pcDNA5_FLAG-Rpn13C88A. Protein level were determined by western blotting with Rpn13 and FLAG antibody, with GAPDH as the loading control.
(D) The viability of these cells was determined 24 h after addition of the indicated concentration of RA190. The error bars in all of the graphs represent the standard deviation of the mean of triplicate measures.
The same experiment was repeated, but with overexpressed Rpn13C88A instead of the wild-type protein Figure 3C. Again, if Rpn13 is the target of RA190, overexpression of the target should render the cells less sensitive to RA190. But, in this case, if C88 is indeed the site alkylated by RA190, then cells overexpressing Rpn13C88A should be impervious to the compound. However, the same result was obtained as with the wild-type protein. The cells expressing Rpn13C88A were just as sensitive to RA190 as SK-MEL-5 cells expressing native Rpn13 (Figure 3D).
We conclude from these experiments that the level of Rpn13 in SK-MEL-5 or HeLa cells has no effect on the sensitivity of the cells to RA190. This is not the result one would expect if Rpn13 is the physiological target of RA190.
Analysis of Biotin-RA190-Rpn13 Engagement In Cellulo
Whereas RA190 was reported to alkylate Rpn13 in a cell lysate (Anchoori et al., 2013), physical engagement of RA190 with Rpn13 has not, to our knowledge, been demonstrated unequivocally in cellulo. To examine this issue, we first asked if treatment of cells with biotin-RA190 results in alkylation of Rpn13. To ensure that the biotinylated compound was active and penetrated cells, the sensitivity of SK-MEL-5 cells to this compound was compared with their sensitivity to unmodified RA190 after 24 h of treatment (Figure 4A). Biotin-RA190 was about 5-fold less potent than the parent compound, likely due to reduced cell permeability, but was still quite toxic to the cells at 10 μM. SK-MEL-5 melanoma cells were treated with biotin-RA190 (10 or 30 μM for 2 h) and covalent adduct formation was assessed by analyzing cell lysates via SDS-PAGE and blotting with labeled streptavidin. Several major biotin-containing bands appeared upon biotin-RA190 treatment, yet none of them co-migrated with Rpn13 (visualized by blotting with anti-Rpn13 antibody) (Figure 4B).
Figure 4. Assessment of RA190 Binding to Rpn13 in Cells.

(A) RA190 and its alkyne and biotin derivatives, as depicted in Figure 1A, were incubated with SK-MEL-5 cells for 24 h followed by CellTiter-Glo 2.0 viability analysis. Cell viability was normalized to vehicle control. Error bars represent the standard deviation of the technical triplicate measures. The curve was fit using the four-parameter (variable slope) non-linear regression of Prism 7.
(B) SK-MEL-5 cells were treated with the indicated concentration of biotin-RA190 for 2 h. Lysates were prepared and analyzed by SDS-PAGE and blotting with anti-Rpn13 antibody (green) or streptavidin (red). Specific labeling of Rpn13 by RA190 would result in a single band that stained both red and green. This was not observed. The RA190-dependent bands did not co-migrate with Rpn13.
(C) Cellular biotin-RA190 binding was assessed by proteasome co-immunoprecipitation (coIP). Proteasome coIP was performed on biotin-RA190-treated MM1.R cell lysate. This experiment was conducted in biological triplicate. One replicate, including load, flowthrough (FT) and elution (EL) are shown.
(D) Effect on Rpn13-proteasome (Rpn2) interaction in cellulo. HEK293T cells treated with the indicated compounds (10 μM) were lysed and immunoprecipitated using anti-β5 antibody. CoIP of Rpn13 with the proteasome was assessed by SDS-PAGE and immunoblotting. KDT11 is an Rpn13-binding peptoid that we have shown previously to have no effect on the Rpn13-Rpn2 interaction and thus serves as a negative control. In the lane labeled “Rpn2,” the cells were transfected with an expression vector for a Rpn2-derived peptide known to compete binding of Rpn13 and Rpn2. Bort, bortezomib.
To look for Rpn13 alkylation with higher sensitivity, we decided to repeat this type of experiment but enrich the Rpn13 protein by immunoprecipitation of the proteasome before analysis. Moreover, we conducted the assay in MM1.R, which are somewhat more sensitive to RA190 than SK-MEL-5 cells. MM1.R cells were incubated in the absence (0.1% DMSO) or presence of biotin-RA190 (5 μM) for 2 h. A lysate was prepared and incubated with resin-bound anti-β5 antibody to immunoprecipitate the proteasome. After washing, bound proteins were eluted with SDS-PAGE loading buffer. The immunoprecipitated proteins were analyzed by SDS-PAGE and blotting with labeled streptavidin, anti-Rpn13 antibody, or anti-β5 antibody. The results are shown in Figure 4C. MM1.R cells have a single major natively biotinylated protein (lane 1). Treatment with biotin-RA190 results in the biotinylation of many other proteins (lane 4); however, all of them fail to be retained by the immobilized anti-b5 antibody (compare lanes 4 and 5). Blotting for β5 and Rpn13 shows that both proteins were highly enriched by antibody pull-down, but that the immunoprecipitated Rpn13 was not detectably biotinylated (lane 6).
We conclude that biotin-RA190 does not alkylate Rpn13 to a detectable degree in melanoma or MM cells. Moreover, the fact that biotin-RA190 has no effect on the amount of Rpn13 that co-immunoprecipitates with the proteasome argues that the compound does not inhibit Rpn13 association with Rpn2 in cellulo. To confirm this conclusion using unmodified RA190, we treated HEK293T cells with RA190 itself, or various control compounds, immunoprecipitated the proteasome using immobilized anti-β5 antibody, and assessed the amount of Rpn13 in the precipitate. As shown in Figure 4D, RA190 had no effect on the amount of Rpn13 associated with the proteasome (compare lanes 1 and 3). However, in cells where the domain of Rpn2 that engages Rpn13 was overexpressed, no detectable Rpn13 co-immunoprecipitated with the proteasome (compare lanes 1 and 6). Furthermore, treatment of the cells with bortezomib, which is known to destabilize the association of the 20S core complex and the 19S regulatory particle (Welk et al., 2016), reduced the level of Rpn13 in the immunoprecipitate relative to the negative controls (compare lanes 1 and 5). These positive controls validate that, if RA190 had inhibited association of Rpn13 with the proteasome, it would have been detected using this assay. These data are in accord with the results of a similar experiment reported by Lu et al. (2017) using a different anti-proteasome antibody.
Global Analysis of RA190 Engagement with the Proteome
To probe interaction of RA190 with the proteome more deeply, we turned to global chemoproteomic methods. First, we used a label-free quantitation (LFQ) workflow (Figure 5A) to assess the proteome-wide binding of RA190 in MM cells. MM1.R cells were treated with RA190 (5 and 10 μM) or vehicle (DMSO) for 2 h, then lysed. The cell lysates were incubated with 30 μM alkyne-RA190 for 1 h, resulting in the alkylation of hundreds of proteins (Figures 1C and S3). A biotin azide label was then clicked (Kolb and Sharpless, 2003) onto the alkynylated proteins. These were enriched using streptavidin-coated beads, digested, and analyzed by liquid chromatography-tandem MS (LC-MS/MS)-based proteomics using label-free quantification (Figure 5A) (Cox and Mann, 2008). With this protocol, labeling of proteins by alkyne-RA190 in vitro will be blocked if they are already engaged by RA190 in cellulo. Figure 5B shows the ratio of recovery from cells treated with DMSO or RA190 for the MM1.R cysteine-containing proteome.
Figure 5. MS-Based Analysis of RA190-Protein Interactions in MM1.R Cells.

(A) Schematic of the label-free quantitative proteomic workflow. See text for details.
(B) Label-free quantitation (LFQ) ratios of proteins from cells treated with 5 μM RA190 or DMSO.
(C) Table of proteins found to be most highly engaged by RA190 and the LFQ ratios of the signals in the RA190- and DMSO-treated samples (a low ratio represents a high level of interaction with RA190 in cellulo). Biological triplicates were analyzed in experimental duplicate, as indicated by standard deviations (represented as ±). Stringency filters were applied to remove potential false positive results. Proteins in green indicate the targets are within the 0.5 cutoff for positive hit identification.
RA190 was found to dose-dependently engage a plethora of proteins in cellulo with high confidence. A total of 158 of the 1,246 proteins detected in this experiment displayed average ratios of ≤0.5 (Figure 5B; Table S1). Thirty-six3 proteins were highly engaged by RA190, as defined by a ratio of ≤0.2 (Figure 5B; Table S1). Rpn13, however, was not engaged detectably by RA190, nor was its associated deubiquitinase Uch37 (Figures 5B and 5C).
The same experiment was conducted with SK-MEL-5 cells. Again, a large number of proteins were found to be engaged by RA190 in cellulo (see Figure S3; Table S2). Again, neither Rpn13 nor Uch37 were among this group, with average ratios around 1, as was the case in MM1.R cells.
There was considerable overlap between the two datasets. Four of the 12 (33.3%) proteins found to be most highly engaged by RA190 in SK-MEL-5 cells were also heavily engaged in the myeloma line, specifically CYB5B, which was tested in downstream assays (vide infra), and MPV17L2, NUP210, and MUL1. This overlap shows there are consensus interactors of RA190 in MM1.R and SK-MEL-5 cells, although perhaps not surprisingly there are appreciable numbers of distinct target proteins in each cell type. What is consistent across both cell types, however, is the absence of RA190 engagement with Rpn13 or Uch37.
We sought to corroborate the label-free proteomics data with an isotopic tandem orthogonal proteolysis-activity-based protein profiling (isoTOP-ABPP) experiment (Backus et al., 2016). In brief, compound- and vehicle-treated cells are lysed separately and treated with highly reactive iodoacetamide to label cysteines globally. In the RA190-treated sample, all sites occupied by the ligand will not be labeled by iodoacetamide, allowing for quantitation when compared with a vehicle-treated control. The iodoacetamide label contains an alkyne handle suitable for subsequent click chemistry. This alkyne handle is then coupled to a biotin azide that contains two functional groups to enable analysis—an isotope-based barcode and a TEV protease site for release after enrichment. After click chemistry to install the biotin tag, the labeled proteomes are pooled, proteins are enriched using streptavidin beads, and protease digested for LC/LC-MS/MS analysis (Figure 6A). The ratio of tryptic peptides from the treated and untreated samples is assessed to reveal ligand-binding hotspots. A ratio of 1 means a peptide is equally observed in both samples, indicating that a ligand does not engage the cysteine in that peptide. A ratio of 2 corresponds to 50% ligand occupancy, and a ratio of 20 indicates essentially 100% ligand binding.
Figure 6. Chemical Proteomic Analysis of RA190-Cysteine Interactions in MM Cells.

(A) Schematic of cysteine reactivity analysis using the isoTOP-ABPP method. isoTOP-ABPP ratios (R) (DMSO/RA190; blue/red) reflect RA190-dependent reductions in reactivity of cysteines with the broad-spectrum IA-alkyne probe.
(B) isoTOP-ABPP R values for quantified cysteines in MM1.R cells treated with DMSO (0.1%) or RA190 (5 μM, 2 h). Sites with R ≥ 4 were considered efficiently engaged by RA190 and are highlighted in green (see inset). Averaged R values from biological replicates (n = 2–3) are shown.
(C) Representative MS1 chromatograms for cysteine-containing peptides of Rpn13 (ADRM1) in DMSO-treated (blue) and RA190-treated (red) MM1.R cells.
MM1.R cells were treated with 5 μM RA190 or vehicle (0.1% DMSO) in biological triplicate for 2 h, followed by isotope-ABPP processing. From stringency filtered data, 2,746 cysteines were quantified in at least 2 of 3 replicates, and 55 of these cysteines (2.0%) exhibited ratios showing they were strongly engaged by RA190 (ratio of 4.0 or higher for DMSO/RA190; Figure 6B). The large number of enriched peptides shows that RA190 is a rather promiscuous alkylator in MM1.R cells. The tryptic peptide of Rpn13 containing C88 was only observed to be weakly liganded (R ≈ 1.8, z40% labeled) in one of the three experiments (Figure 6C). The peptide containing C80 of Rpn13 was detected with higher confidence and was clearly unliganded by RA190 with an average ratio of 1.05 ± 0.11.
We also scrutinized the dataset for evidence of RA190 binding to the Rpn13-associated deubiquitylase Uch37 as well. Similar to Rpn13, the overall enrichment of these peptides was quite low, suggesting inherently low probe accessibility/reactivity or protein abundance. In a single replicate, Uch37’s active site (Cys88) was observed with a ratio of 1.2, while Cys191 was observed in three replicates with an average ratio of 1.3 ± 0.15. Thus, there are no compelling data from the isoTOP-ABPP experiment to indicate RA190 binding to Uch37 in cellulo.
From the 36 proteins previously identified to be highly engaged by RA190 in MM1.R cells in the LFQ experiment, 18 appear in the isoTOP-ABPP experiment, validating the ability of these approaches to identify true binding interactions with RA190.
Genetic Knockdown of Individual Proteins Engaged by RA190 In Cellulo Does Not Recapitulate the Cytotoxic Effects of RA190
As described above, while we find no evidence for physical engagement of Rpn13 by RA190, this highly electrophilic compound does label a large number of other proteins. We proceeded to ask whether one or more of these interactions could be responsible for the cytotoxicity of RA190. In particular, several proteins identified in the MM1.R proteomics experiment were also enriched in the SK-MEL-5 dataset, and some have known roles in proliferation (Wang et al., 2010). We selected OCIAD1, c18orf32, CYB5B, and REEP5 for testing. Since RA190 induces a block to the cell cycle at the G2/M transition (Figure S2) (Randles et al., 2016), we tested the effect of siRNA-mediated knockdown of these proteins on cell-cycle progression. While all of the targeted proteins were knocked down substantially (Figure 7A), in no case was there a substantial effect on cell-cycle progression (Figure 7B). Furthermore, the cells in which the target proteins had been knocked down exhibited identical sensitivity to RA190 as cells treated with a control siRNA (Figure 7C), arguing that RA190 engagement with these proteins does not, by itself, drive cytotoxicity.
Figure 7. Analysis of the Effect of Genetic Knockdown of RA190-Engaged Proteins on SK-MEL-5 Cells.

SK-MEL-5 cells were transfected with 50 nM siRNA for 48 h.
(A) SDS-PAGE and western blotting was used to assess the level of knockdown of the various proteins.
(B) Cell-cycle progression was determined by the Click-it EdU Flow Cytometry Assay (see Figure S4 for the raw data). Biological duplicate experiments were performed for each siRNA treatment, and error bars represent the standard deviation of these two readings.
(C) Analysis of the effect on various gene knockdowns on the sensitivity of SK-MEL-5 cells to RA190. The experiment was performed in triplicate. The error bars represent the standard deviation of the three replicates.
DISCUSSION
RA190 is an electrophilic compound reported to have interesting anti-cancer properties in a variety of mouse models. As mentioned in the Introduction, it has been claimed that the target of RA190 is the ubiquitin receptor Rpn13, and specifically the C88 residue on this protein (Anchoori et al., 2013). This was claimed to result in inhibition of some activity of Rpn13 important for proteasome function in some cancer cells, thus explaining the observed buildup of polyubiquitylated proteins in RA190-treated cells. There are three major pieces of evidence in the literature to support this idea. First, Anchoori et al. (2013) found that biotin-RA190 alkylates Rpn13 preferentially in a crude cell extract. Second, when RA190 and purified Rpn13 are mixed together, the greatest perturbation of NMR chemical shifts occurs in the region of C88 (Anchoori et al., 2013). Third, Song et al. (2016) reported that HCT-116 cells lose their sensitivity to RA190 when the gene encoding Rpn13 (ADRM1) is knocked out. They also reported that expression of wild-type Rpn13, but not Rpn13C88A, in this background re-sensitized the cells to RA190.
Recent studies from other laboratories have shed doubt on this model, particularly with respect to the idea that RA190 alkylates Rpn13 in the context of the proteasome. Two structural studies of the Rpn13-Rpn2 complex demonstrated that Cys88 of Rpn13 is not solvent exposed in the complex and that RA190 and Rpn2 cannot co-occupy Rpn13 (Lu et al., 2017; VanderLinden et al., 2017). Moreover, biochemical analysis showed conclusively that the small molecule cannot compete with the high-affinity Rpn2 domain that interacts with Rpn13. Therefore, RA190 cannot access this residue while Rpn13 is associated with the proteasome. Of course, it remains possible that RA190 engages Rpn13 on a different cysteine, contrary to the expectation from the NMR data, and indeed Lu et al. (2017) presented evidence that at least one other cysteine in Rpn13 is alkylated by RA190 in vitro, a result that is in agreement with our experiment showing that purified wild-type Rpn13 and Rpn13C88A are alkylated with approximately equal efficiency (Figure 1B). However, they also found that RA190 alkylates Uch37 as well, raising the possibility that RA190 is a more promiscuous alkylator than the previous literature suggested.
There are apparent disconnects between the model and cellular data as well. As mentioned above, while deletion of ADRM1 has been claimed to eliminate the sensitivity of HCT116 cells to RA190, these cells remain viable, which would seem incompatible with the idea that RA190 evinces its cytotoxicity by inhibiting some function of Rpn13. Another issue is that knockdown of Rpn13 or Uch37 with siRNA in HeLa cells affects the cell cycle by inhibiting the G0/G1 to S transition. However, RA190 treatment causes a far more pronounced cell-cycle defect is due to a block at the G2/M transition (Randles et al., 2016). Indeed, based on the data currently available in the literature, we would argue that, if RA190 does mediate its cytotoxic effects through Rpn13, then this must be due to the protein-small molecule complex acquiring some type of dominant negative function, which would explain why deletion or knockdown of Rpn13 protein level does not phenocopy the effect of treating cells with the compound. Moreover, even the dominant negative model would have to postulate that RA190 alkylates Rpn13 on a cysteine other than C88, or, if that is indeed the target residue, that the interaction takes place outside the context of the proteasome.
Given this clouded picture, and because we are also interested in evaluating Rpn13 as a chemotherapeutic target (Trader et al., 2015), we applied a series of classical chemical biology methods to ask if Rpn13 is indeed the physiologically relevant target of RA190. In short, we find no evidence whatsoever for physical or functional engagement of Rpn13 by RA190 in MM, melanoma, and cervical cancer cell lines. Instead, the data clearly show RA190 to be a promiscuous alkylator of many different proteins in the cellular proteome, none of which are Rpn13.
On a functional level, one expects that the sensitivity of cells to a potent bioactive molecule will be affected by the level of that molecule’s target protein in cells. Reduced levels should increase sensitivity and increased target protein levels should reduce sensitivity. This is clearly not the case in SK-MEL-5 or HeLa cells, as knockdown of Rpn13 had no effect on the sensitivity of the cells to RA190 (Figure 2), nor did the cells containing this lower level of Rpn13 display any reduced viability. When Rpn13 was overexpressed, again no change in sensitivity to RA190 was observed (Figure 3). This was also the case when we overexpressed Rpn13 C88A (Figure 3). This result seems completely irreconcilable with the idea that RA190’s cytotoxicity inhibits some critical function of Rpn13 through alkylation of C88.
We used a number of approaches to determine if RA190 engages Rpn13 physically in cells. As shown in Figure 4, treatment of SK-MEL-5 cells with biotin-RA190 resulted in the alkylation of many proteins, as visualized by SDS-PAGE analysis of a lysate made from these cells followed by blotting with labeled streptavidin. However, none of these bands co-migrated with Rpn13 (Figure 4B). Even when the proteasome was immunoprecipitated from biotin-RA190 cells to concentrate Rpn13, no detectable alkylation was observed (Figure 4C).
To gain a more global view of RA190-proteome interactions, three MS-based analyses were carried out. An LFQ experiment was used to identify proteins that were alkylated by RA190 in MM (Figure 5) and melanoma (Figure S3) cells, as defined by a reduced level of subsequent alkylation with excess alkyne-RA190 in a lysate prepared from cells that had or had not been treated with RA190. These data showed clearly that Rpn13 was not occupied by RA190 in cellulo, nor was its associated deubiquitinase Uch37. In striking contrast, however, we found dozens of proteins that were heavily engaged by RA190 in a dose-dependent fashion (Figures 5 and S3; Table S1 and S2).
To supplement the LFQ analysis, we also used a different technique, isoTOP-ABPP, to assess RA190 interaction with the MM proteome. The results were similar, in that there was no evidence for significant engagement of Rpn13, but several dozen other proteins were heavily alkylated by RA190 (Figure 6; Table S3). Taken together, the LFQ and isoTOP-ABPP proteomic data do not support the idea that this compound engages Rpn13 in cellulo to any significant extent, and instead reveal RA190 to be a highly promiscuous alkylator. It is interesting in this vein, that a recent chemical proteomics study of the deubiquitylase inhibitors bAP15 and VLX1570, which share the same core pharmacophore with RA190, were also revealed to be promiscuous alkylators (Ward et al., 2020).
Finally, we asked if some of the proteins most heavily alkylated by RA190 in both cell types might be the functionally relevant targets of RA190. To do so we knocked down these proteins in SK-MEL-5 cells and assessed the effect on cell viability and cell-cycle progression. In no case was any significant effect observed (Figure 7). While it is understandable to wish to assign the bioactivity of a small molecule to a single target, we believe that the data presented here strongly suggest that the cytotoxicity of RA190 is probably due to complex polypharmacology involving many different targets and perhaps even broad effects on the redox balance in the cell.
In summary, we believe that the data reported here and elsewhere (Lu et al., 2017; VanderLinden et al., 2017) argue strongly against the model that RA190 evinces its cytotoxic effects through Rpn13, and certainly rule out the idea that the small molecule engages C88 of Rpn13 in the context of the proteasome. We find no evidence for physical or functional engagement with Rpn13 in at least three different cancer cell lines using a variety of different techniques. Indeed, not only are our data incompatible with the idea that RA190 inhibits some function of Rpn13, they also argue strongly against even the more esoteric model mentioned above that RA190-Rpn13 engagement endows the protein with some sort of dominant negative activity. This would almost certainly require physical interaction of RA190 and Rpn13, for which we see no evidence, and is also incompatible with the fact that knockdown or overexpression of Rpn13 has no effect on the sensitivity of cells to RA190. We do not understand the apparent requirement for wild-type Rpn13 in order for HCT116 cells to exhibit sensitivity to RA190 (Song et al., 2016). It is possible, although in our view unlikely, that the situation is fundamentally different in this cell line, or that this effect is the result of some complex, indirect mechanism.
STAR⋆METHOD
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for materials should be directed to the Lead Contact Thomas Kodadek (kodadek@scripps.edu).
Materials Availability
Mutant plasmids (pET19b_Rpn13Cys88Ala and pcDNA5_FLAG-ADRM1Cys88Ala) created in this study will be available for distribution through the Lead Contact.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Cell Lines
HEK293T (Female, RRID: CVCL_0063) and HeLa (Female, RRID: CVCL_0030) cells were cultured at 37°C with 5% CO2 in DMEM (#10566016, Gibco) supplemented with 10% FBS (Cat#16140–071, Life Technologies). MM.1R(Female, RRID: CVCL_8794), MM.1S(Female, RRID: CVCL_8792), and MDA-MB-231(Female, RRID: CVCL_0062) cells were cultured at 37°C with 5% CO2 in in RPMI1640 (A1049101, ThermoFisher) supplemented with 10% FB. SK-MEL-5 (Female, RRID: CVCL_0527) melanoma cells were cultured at 37°C with 5% CO2 in EMEM (30–2003, ATCC) supplemented with 10% FBS. HEK293T, SK-MEL-5, A549, and HeLa cells were passaged using TrypLE (12605–010, ThermoFisher) every 3 days. Suspension cells (MM.1R, MM.1S) were passaged every 4 days without the use of trypsinizing agent. A549 (Male, RRID: CVCL_0023) cells were grown in F-12K media supplemented with 10% FBS. PBMCs (Cat#70025.1) and CLL2007 (Chiorazzi lab, Feinstein Medical Institute) were thawed from a cryostock by incubating in a 37°C bead bath until thawed, followed by dilution into 10 mL of warm RPMI + 10% FBS to remove residual cryopreservant. Cells were then resuspended in fresh, warm media and prepared for analysis. We do not know the gender of primary cell sources. Cells were routinely tested for mycoplasma using a PCR-based method according to manufacturer’s protocol (ABM Biosciences Cat#G238).
METHOD DETAILS
Protein Expression and Purification
Rpn13 was expressed and purified using the previously reported protocol (Trader et al., 2015) with minor modifications. Briefly, DE3-PLysS cells (Cat#L1195) carrying plasmid pET19b-His6-Rpn13 (RRID: Addgene_19423) were grown in Luria-Broth with 100 μg/mL carbenicillin at 37°C. When OD600 reached 1.0, the culture was induced with 1 mM isopropyl β-D-1thiogalactopyranoside (IPTG) for 4 hours at 37°C. Cells were pelleted at 4000g for 10 minutes and resuspended in lysis buffer (50 mM sodium phosphate, 300mM sodium chloride, 3% glycerol, 1% Triton X-100, pH 8, 1 mM PMSF), followed by three probe sonication cycles at 50% power for 1 minute. Soluble supernatant was separated by centrifugation at 10,000g for 30 minutes at 4°C and applied to pre-equilibrated nickel-NTA resin (Qiagen Cat#30210, 5 mL of 50% slurry) and incubated for 1 hour at 4°C. The resin was washed three times with equilibration buffer (50 mM sodium phosphate, 300mM sodium chloride, pH 8), and eluted from the resin using a stepwise gradient of imidazole. The equilibration buffer was supplemented with increasing concentrations of imidazole from 50 mM to 500 mM. Fractions were run on an SDS-PAGE gel and pure fractions were collected for dialysis and concentration. Protein was dialyzed in phosphate buffered saline (PBS, 4L) overnight and concentrated using an Amicon conical concentrator (10kDa cutoff) to micromolar concentration, at which point it was snap frozen in liquid nitrogen and stored at −80○C. Mutant C88A Rpn13 and MBP-Uch37(DNASU ID#84019) were purified using the same procedure.
In Vitro Binding Assay
Purified His6-Rpn13 (1 μM) was incubated with 1 μM RA190 hour at 4°C. Samples were quenched with SDS PAGE loading dye (250 mM Tris-HCl pH 6.8, 10% SDS, 30% glycerol, 10 mM DTT, 0.05% (w/v) Bromophenol blue) and boiled for 5 minutes. Ligand-protein complex was resolved by SDS-PAGE and western blotting was performed as described.
Generation of Cys88Ala Mutant
pET19b-His6-Rpn13(RRID: Addgene_19423) and pcDNA5 FLAG-ADRM1(RRID: Addgene_19417) plasmid was PCR-amplified using mutagenic primers designed using the PIPE site-directed mutagenesis method using KOD HotStart polymerase master mix (EMD Millipore Cat#71842) (Klock and Lesley, 2009). Plasmid (1 ng) was mixed with 2x PCR master mix (25 μL), forward and reverse primer (IDT, STAR Methods, 0.3 μL, 10 μM stock), and diluted to 50 μL with nuclease-free water. Thermal cycling was conducted using the following method: 95°C for 2 minutes, 40 cycles of 95°C for 30 seconds, 72°C for 20 seconds, 68°C for 3 minutes, followed by a 5 minute 72°C final extension. PCR products (5 μL) were mixed with 6x DNA loading dye (1 μL) were verified on a 1% agarose gel run in 1x Tris-Acetate EDTA (TAE) buffer for 40 minutes at 100V. DNA was purified using the QiaQuick PCR Purification clean up kit, followed by transformation into chemically competent XL10 Gold bacterial cells (Cat#200314). DNA (2 μL) was incubated with cells (100 μL) and beta-mercaptoethanol(1 μL) for 30 minutes on ice. Cells were heat-shocked at 42°C for 45 seconds, followed by recovery on ice for two minutes. Warm SOC media (350 μL ) was added and cells were allowed to recover at 37°C for 1 hour with shaking. Cells were plated on LB+Agar supplemented with 100 μg/mL carbenicillin for plasmid selection. Mutation was confirmed by Sanger sequencing (Genewiz) and analysis of sequences using Geneious (RRID: SCR_010519)
Knockdown by siRNA
Validated, pooled siRNA towards the GAPDH (Cat#D-001830–10), CYB5B(Cat#L-014633–02), C18orf32 Cat# (L-034916–02), Rpn13(Cat#L-012340–01), REEP5 (Cat#L-019467–01), and OCIAD1(Cat#L-020825–02) were purchased from Dharmacon. Custom UTR targeting siRNA towards Rpn13 (Cat#CTM-485557) were designed using Dharmacon’s siDesign Center. Non-targeting siRNA (Cat#D-001810–10) was used as a transfection control. siRNA was transfected (50 nM) used according to the manufacturer’s protocol for a 6-well dish. Cells were incubated for 48 hours prior to analysis. Protein quantitation was performed using ImageJ (RRID: SCR_003070) with biological triplicate experiments.
Plasmid Transfection
Mammalian expression plasmid pcDNA5-FLAG-Adrm1 (RRID: Addgene_19417) and mutant C88A construct were transiently transfected into SK-MEL-5 using Omnifect transfection reagent (Cat#OTR1003) according to the manufacturer’s protocol. For a well dish transfection, Opti-MEM reduced serum media (200 μL) was incubated with Omnifect (4 μL) and plasmid (2 μg) for 10 minutes followed by addition to SK-MEL-5 cells at 70–80% confluency. Cells were collected and prepared for respective analyses after 24 hours of transfection.
Western Blotting
Cell lysates were diluted with 4x Laemmli Sample buffer (Biorad) and run on 4–20% SDS-PAGE denaturing gel (Biorad Cat#4561096) under reducing conditions (180V for 40 minutes). Protein was transferred to 0.2 μm nitrocellulose membrane (Bio-Rad) using the Trans-Blot Turbo semi-dry transfer apparatus. Blots were blocked in 5% milk in PBST and primary antibodies were diluted into this solution for staining. Primary polyclonal antibodies were purchased from the following suppliers and used at the indicated dilutions: CYB5B (RRID AB_2230349, 1:1000), C18orf32 (RRID AB_2638853, 1:500), Rpn13/ADRM1 (RRID: AB_2225663, 1:1000), REEP5 (RRID AB_2178440, 1:1000), OCIAD1 (RRID AB_11035850, 1:1000), GAPDH (RRID: AB_627679, 1:1000), FLAG (RRID: AB_262044, 1:1000),β5 proteasome subunit (RRID: AB_2052392, 1:1000), Uch37 (RRID: AB_2814821, 1:1000). Primary antibodies were incubated with membrane overnight at 4°C. Secondary antibodies IRDye 680RD donkey anti-mouse IgG (RRID: AB_27116622) and IRDye 800CW goat anti-rabbit IgG (RRID: AB_2651127) were used at 1:10,000 dilution with a 1 hour incubation at room temperature. Streptavidin IR680 RD (Licor #926–68079) was used at a 1:5000 dilution to detect biotin-RA190 protein adducts. Secondary antibody was diluted into Licor Oddysey blocking buffer. Blots were resolved using Licor Oddysey and Licor Image Studio Software (RRID: SCR_015795)
Cell Viability
Cell Titer Glo 2.0 (Cat#G9241) reagent was used to analyze cellular viability in technical triplicate. Briefly, 4,000 cells/well were seeded into 96-well white plates (Costar) and incubated with compound in 1% DMSO at 100 μL volume. At the time of readout, 100 μL of Cell Titer Glo 2.0 reagent was added and plates were shaken at 800 rpm for 5 minutes. Luminescence was quantified by Tecan plate reader at 250ms in experimental triplicate for two independent experiments.
In Vitro Uch37 Association Assay
Recombinant proteins were incubated at 1 μM each in PBS, with 0.5% DMSO or 50 μM RA190. Rpn13/Uch37 complex was pulled down using amylose resin (NEB Cat#E8021S) to bind the MBP tag on Uch37. Complexes were washed three times with PBS, followed by elution from the resin with 1x SDS Reducing Dye at 95°C for 10 minutes, separation on a 12–20% gradient gel (Biorad) and visualization with Coomasie staining (Gel Code Blue, Thermo Fisher).
Proteasome Co-immunoprecipitation
HEK293T cells were plated at 70% confluence in 10 cm dishes and treated with KDT-11, RA190, or bortezomib (Cat#B140825MG) as indicated overnight. Cell lysates were prepared through 10 minutes of passive lysis with M-PER reagent (Thermo Fisher) and clarified at 17,000 rpm at 4°C for 15 minutes. 1 mg of protein lysate was applied to 50 μL of proteasome affinity resin (Cat#BML-PW1075A-0001) and immunoprecipitated according to the manufacturer’s protocol, followed by western blot analysis.
Rpn13 Immunoprecipitation
T75 flasks of MM1.R cells were treated with vehicle (0.1% DMSO) or 5 μM biotin-RA190 for 2 hours, followed by lysis with M-PER, dilution to 0.5mg/mL with PBS, and incubation overnight with 10 μg/mg lysate anti-Rpn13 antibody at 4°C. Protein G plus agarose (Thermo Cat#22851) was washed once with PBS and 100 μL of slurry was applied to the lysate-antibody mixture. After 30 minutes of incubation at 4°C, agarose was washed three times with PBS and eluted with 1x SDS PAGE sample loading buffer. Protein was resolved by western blot analysis as described below.
Compound Synthesis
RA190 and biotin-RA190 were synthesized as previously described (Anchoori et al., 2013). Briefly, a suspension of 4-piperidone hydrochloride monohydrate in glacial acetic acid was mixed with 2 equivalents of 3,4-dicholorobenzyaldehyde were mixed. Dry hydrochloric acid gas was passed through the mixture and allowed to sit for 1 day. Precipitate was then coupled with phenylananine amino acid via standard amide bond forming conditions and purified by chromatography. Biotin RA190 was prepared through subsequent acylation reaction with free biotin. Alkyne RA190 was prepared through an acylation of the free amine of RA190’s phenylalanine appendage. 0.11 mmol 5-hexynoic acid (12.1 μL) was dissolved in 1 mL of DMF and pre-activated with Oxyma (1 equiv, 16.5 mg) and diisopropylcarbodiimide (1 equiv, 16.5 mg). 0.11 mmol RA190 (65 mg) was dissolved in 1mL DMF and added to pre-activated 5-hexynoic acid. The reaction was stirred overnight. The product was worked up once with ethyl acetate, five times with saturated sodium bicarbonate, and once with brine. The product was then separated by flash column chromatography (Biotage) with hexanes and ethyl acetate and confirmed the product by NMR and LC/MS. Product yield was 38.1 mg (0.058 mmol, 53% yield). For analytical data see Figure S1 (1H NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.60 – 7.50 (m, 4H), 7.35 (d, J = 2.1 Hz, 1H), 7.29 (dd, J = 8.3, 2.1 Hz, 1H), 7.25 – 7.20 (m, 3H), 7.07 (dd, J = 8.3, 2.1 Hz, 1H), 7.00 – 6.89 (m, 2H), 6.30 (d, J = 8.0 Hz, 1H), 4.97 (td, J = 8.3, 6.3 Hz, 1H), 4.80 – 4.69 (m, 2H), 4.43 (d, J = 16.8Hz, 1H), 4.20 (d, J = 16.8Hz, 1H), 2.87 (dd, J = 13.2, 8.4 Hz, 1H), 2.79 (dd, J = 13.2, 8.4 Hz, 1H), 2.26 (t, J = 7.4 Hz, 2H), 2.22 – 2.06 (m, 2H), 1.95 (t, J = 2.6 Hz, 1H), 1.80 – 1.72 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 185.11, 171.67, 170.42, 136.08, 135.94, 135.80, 134.46, 134.36, 134.12, 133.91, 133.55, 133.36, 132.17, 132.08, 132.05, 131.72, 131.19, 131.02, 129.40, 129.28, 129.15 (two carbons), 128.89 (two carbons), 127.46, 83.45, 69.41, 49.99, 46.26, 43.21, 39.90, 34.83, 24.01, 17.87.) 13C NMR (101 MHz, CDCl3) δ 185.11, 171.67, 170.42, 136.08, 135.94, 135.80, 134.46, 134.36, 134.12, 133.91, 133.55, 133.36, 132.17, 132.08, 132.05, 131.72, 131.19, 131.02, 129.40, 129.28, 129.15 (two carbons), 128.89 (two carbons), 127.46, 83.45, 69.41, 49.99, 46.26, 43.21, 39.90, 34.83, 24.01, 17.87.) Structures and scheme were drawn in Chemdraw (RRID: SCR_016768).
Rpn13-Ub Fluorescence Polarization Assay
100 μM RA190 or vehicle (1% DMSO) was incubated with 100 μM Rpn13 in PBS for 1 hour at 4°C prior to addition of an equal volume of 50 nM Ubiquitin48-fluorescein in buffer plus 100 μM RA190. Ubiquitin-fluorescein was prepared as described by Du et al, using a bacterially purified single cysteine variant of ubiquitin (K48C) that has been labeled by fluorescein-maleimide. Serial two-fold dilutions of Rpn13 were prepared to observe potential shifts in ubiquitin binding to Rpn13 using a fluorescent plate reader in polarization format in technical triplicate. Graphs were prepared in Graphad Prism 8 (RRID:SCR_000306) and curves were fit to a one-binding site Hill slope equation.
Flow Cytometry Cell Cycle Analysis
SK-MEL-5 cells were treated as indicated and incubated with 10 μM 5-ethynyl-2’-deoxyuridine (EdU, Invitrogen) for 90 minutes. Cells were subsequently harvested, fixed, and stained with Alexa647-azide according to the manufacturer’s instructions (Click-iT EdU Flow Cytometry assay kit, Cat#C10424). Cells were resuspended in DPBS and 106 cells were passed through a cell strainer (BD Falcon) and analyzed by flow cytometry on a BD FACSCanto II flow cytometer (BDBiosciences) using the red and blue laser. Data was exported and cell cycle phases were gated using FlowJo (RRID: SCR_008520). Population percentages were plotted in Prism 8 and a t-test with unequal variance assumption was performed. See Figure S4 for representative plots.
Isotope-Labeling Quantitative Proteomics
MM1.R cells were treated with 5 μM RA190 or 0.1% DMSO for 2 hours with incubation at 37°C at 5% CO2. Cells were collected and washed with PBS one time before snap freezing. Cells were subsequently lysed, treated with iodoacetamide alkyne and installation of a trifunctional tag (isotope labeled biotin-TEV-azide) through copper catalyzed click reaction. Vehicle (heavy tag) and RA190 treated (light tag) samples were combined, enriched using streptavidin beads, and digested prior to LC/LC-MS/MS analysis. Biological triplicates were performed in order to ensure robust target identification, and filters were applied. Representation in two of three replicates was required, replicates were averaged if standard deviation was <60% of the mean. If SD >60% of mean, the lowest value was taken if any value was R<4, and if all R values were above four, the average was taken. Manual verification of cysteines with high standard deviation was performed.
SDS-PAGE Based In Vitro Labeling with RA190-Alkyne
SK-MEL-5 lysate (2 mg/mL, 25 μL) was obtained via sonication in PBS and treated with indicated concentrations of RA190-alkyne (1 μL of 25x stock in DMSO) or DMSO for one hour at r.t. Click chemistry was initiated by the addition of TAMRA azide (Cat#AZ109, 50 μM, 25x stock in DMSO), tris(2-carboxyethyl hydrochloride (TCEP, Cat#C4706, 1 mM, fresh 50x stock in water), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, Cat#1061, 100 μM, 16x stock in DMSO:tButanol 1:4), and copper(II) sulfate (Cat#C1297, 1 mM, 50x stock in water) to the lysate and incubated in the dark for one hour at r.t. SDS-PAGE reducing loading buffer (4x) was added and proteins were separated using a 11% SDS-PAGE gel. Gel was visualized using a Sapphire Biomolecular Imager (Azure Biosystems), then stained using Coomassie.
In-situ Labeling Experiment with RA190
SK-MEL-5 cells were seeded (1e6 cells/mL) in a 6-well plate and left overnight to attach and grow. Cells were treated with indicated concentrations of RA190 for two hours at 37°C in culture media. Cells were washed two times with PBS, harvested by scraping, centrifuged at 1,500 x g for five minutes at 4°C and resuspended in PBS. Cells were lysed by sonication to form cell lysates and protein concentration was determined using the Bradford assay. Lysate (2 mg/mL, 25 μL) were treated with 30 μM of RA190-alkyne (1 μL of 25x stock in DMSO) or DMSO for one hour at r.t. Click chemistry, reducing SDS-PAGE and visualization were performed as described above.
In Situ Competitive Experiment for Mass SpectrometrySK-MEL-5 Cells
SK-MEL-5 cells were seeded in 10 cm petri dishes, grown to 90 % confluence and were subsequently treated with DMSO, 10 μM RA190 or 30 μM RA190 (1000x stock in DMSO) for two hours at 37°C in cell culture media. Cells were washed twice with PBS, harvested by scraping, centrifuged at 1,500 x g for five min. at 4°C and resuspended in PBS. Cells were lysed by sonication to form cell lysates. Protein concentration was determined using the Bradford assay (Bio-Rad). 1.5 mg (2 mg/mL) of proteins was treated with 30 μM RA190-alkyne (100x stock in DMSO) for one hour at room temperature and then protein was subjected to click chemistry. Biotin azide (60 μM, 50x stock in DMSO), tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (1 mM, 50x fresh stock in water), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (100 μM, 16x stock in DMSO:tBuOH 1:4), and copper(II) sulfate (1 mM, 50x stock in water) were added to the proteome and left to react for one hour at r.t. Protein was precipitated by adding MeOH (4 vol.), CHCl3 (1 vol.) and water (3 vol.) to the reaction mixture and the turbid mixture was centrifuged for five min. at 14,000 x g at 4°C yielding a protein layer between the aqueous and organic layers. The protein layer was isolated, dried and solubilized in 2 % SDS in PBS via sonication. Tubes were centrifuged at 4,700 x g for five min. and soluble fraction was transferred to a new tube. PBS was added to give a final SDS concentration of 0.2 %. 160 μL of streptavidin agarose beads (ProteoChem) were added and the mixture was rotated overnight at r.t. Beads were washed with 1 % SDS in PBS (1× 10 mL), PBS (3× 10 mL), and water (3× 10 mL). Beads were resuspended in 6 M urea in PBS (500 μL), reduced with 10 mM neutralized TCEP (20x fresh stock in water) for 30 min. at r.t., and alkylated with 25 mM iodoacetamide (400 mM fresh stock in water) for 30 min. at r.t. in the dark. Beads were pelleted by centrifugation (1’400 x g, two minutes) and resuspended in 150 μL of 2 M urea in 50 mM NH4HCO3, 1 mM CaCl2 (100x stock in water) and trypsin (Thermo Scientific, 1 μL of 0.5 μg/μL). The digestion was performed for six hours at 37°C. Samples were acidified to a final concentration of 5 % acetic acid, desalted over a self-packed C18 spin column and dried. Samples were analyzed by LC-MS/MS (see below) and the MS data were processed with MaxQuant(RRID:SCR_014485) (see below).
In Situ Competitive Experiment for Mass Spectrometry- MM1.R Cells
MM1.R cells were seeded to confluency in T150 flasks and were subsequently treated with 0.1% DMSO, 5 μM RA190, or 10 μM RA190 for two hours. Cells were washed twice with PBS, centrifuged at 500 x g for 5 minutes at 4°C and snap frozen in liquid nitrogen until further processing. Cells were lysed by sonication to form cell lysates. Protein concentration was determined using the Bradford assay (Biorad). 0.8 mg (2mg/mL) of protein was treated with 30 μM RA190-alkyne (100x stock in DMSO) for one hour at room temperature and then subjected to click chemistry as described above. Protein was processed for LC-MS/MS analysis as described with SK-MEL-5 cells.
LC-MS/MS Analysis
Peptides were resuspended in water with 0.1 % formic acid (FA) and analyzed using EASY-nLC 1200 nano-UHPLC coupled to Q Exactive HF-X Quadrupole-Orbitrap mass spectrometer (Thermo Scientific). The chromatography column consisted of a 40 cm long, 75 μm i.d. microcapillary capped by a 5 μm tip and packed with ReproSil-Pur 120 C18-AQ 2.4 μm beads (Dr. Maisch GmbH). LC solvents were 0.1 % FA in H2O (Buffer A) and 0.1 % FA in 90 % MeCN: 10 % H2O (Buffer B). Peptides were eluted into the mass spectrometer at a flow rate of 300 nL/min. over a 240 min. linear gradient (5–35 % Buffer B) at 65°C. Data were acquired in data-dependent mode (top-20, NCE 28, R = 7’500) after full MS scan (R = 60’000, m/z 400–1’300). Dynamic exclusion was set to 10 s, peptide match to prefer and isotope exclusion was enabled.
MaxQuant Analysis
The MS data were analyzed with MaxQuant (V1.6.1.0) ( RRID:SCR_014485) and searched against the human proteome (Uniprot) and a common list of contaminants (included in MaxQuant). The first peptide search tolerance was set at 20 ppm, 10 ppm was used for the main peptide search and fragment mass tolerance was set to 0.02 Da. The false discovery rate for peptides, proteins and sites identification was set to 1%. The minimum peptide length was set to 6 amino acids and peptide re-quantification and label-free quantification (MaxLFQ) were enabled. The minimal number of peptides per protein was set to two. Methionine oxidation was searched as a variable modification and carbamidomethylation of cysteines was searched as a fixed modification.
QUANTIFICATION AND STATISTICAL ANALYSIS
Each experiment was performed in technical triplicate and with three biological replicates, unless otherwise noted throughout the figure captions and methods. All graphing and statistical analyses were performed in Prism 8 (Graphpad, RRID:SCR_000306), with error bars representing the standard deviation of each experiment. Dose-repsonse curves were generated using Prism 8 non-linear inhibitor dose-response regression Significance between treated and untreated samples was performed using a two-tailed Student’s t-test with the Welch’s correction, which does not require the assumption of equal variances. Western blots were quantified by ImageJ according to NIH guidelines. Proteomics data statistical analysis was described in the Maxquant Analysis methods section.
Data and Software Availability
The published article includes all of the mass spectrometry data generated during this study (Tables S1, S2, and S3).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ADRM1 (hRpn13) | Novus Biologics | Cat#NBP1–30447; RRID: AB_2225663 |
| Mouse monoclonal anti-GAPDH 6C5 | Santa Cruz Biotechnology | Cat#Sc-32233; RRID: AB_627679 |
| Rabbit polyclonal anti-20S proteasome beta 5 subunit | Enzo Life Sciences | Cat#BML-PW8895–0025; RRID: AB_2052392 |
| Rabbit monoclonal anti-UCH37 [EP4897] | Abcam | Cat#Ab133508; RRID: AB_2814821 |
| Monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| REEP5 antibody | Proteintech Cat#14643–1-AP | Cat#14643–1-AP; RRID AB_2178440 |
| OCIAD1 antibody | Novus NBP1–76242 | Cat#NBP1–76242; RRID AB_11035850 |
| CYB5B antibody | Proteintech Cat# 15469–1-AP | Cat#15469–1-AP; RRID AB_2230349 |
| C18orf32 antibody | Thermo Fisher PA5–59354 | Cat#PA5–59354; RRID AB_2638853 |
| Goat anti-rabbit 800CW | Licor | Cat#925–32211; RRID: AB_2651127 |
| Donkey anti-mouse 680RD | Licor | Cat#925–32212; RRID: AB_27116622 |
| Bacterial and Virus Strains | ||
| BL21(DE3)pLysS competent cells | Promega | Cat#L1195 |
| XL-10 Gold Ultracompetent cells | Stratagene | Cat#200314 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Streptavidin 680RD | Licor | Cat#926–68079 |
| RA190 | Synthesized as reported | Anchoori, et al. 2013 |
| Biotin-RA190 | Synthesized as reported | Anchoori, et al. 2013 |
| Alkyne-RA190 | This paper | N/A |
| KDT-11 | Synthesized as reported | Trader, et al. 2015 |
| Bortezomib | LC Laboratories | Cat#B140825MG |
| Ubiquitin-fluorescein | Prepared as reported | Du, et al. 2017 |
| Omnifect transfection reagent | Transomic | Cat#OTR1003 |
| TAMRA azide | Click Chemistry Tools | Cat#AZ109 |
| Tris(2-carboyxlethyl)phosphine- HCl (TCEP) | Sigma | Cat#C4706 |
| Tris(benzyltriazolylmethyl)amine (TBTA) | Click Chemistry Tools | Cat#1061 |
| Copper (II) sulfate | Sigma | Cat#C1297 |
| Critical Commercial Assays | ||
| CellTiter-Glo 2 0 Cell Viability Assay | Promega | Cat#G9241 |
| Click-iT EdU Alexa Fluor 647 Flow cytometry assay | Life Technologies | Cat#C10424 |
| Proteasome purification kit | Enzo Life Sciences | Cat#BML-PW1075A-0001 |
| Experimental Models: Cell Lines | ||
| MM1.R myeloma cell line, Homo sapiens | ATCC | Cat#CRL-2975; RRID: CVCL_8794 |
| MM1.S myeloma cell line, Homo sapiens | ATCC | Cat#CRL-2974; RRID: CVCL_8792 |
| SK-MEL-5 melanoma cell line, Homo sapiens | ATCC | Cat#HTB-70; RRID: CVCL_0527 |
| HEK293T kidney cell line, Homo sapiens | ATCC | Cat#CRL-3216; RRID: CVCL_0063 |
| MDA-MB-231 breast cancer cell line, Homo sapiens | ATCC | Cat#HTB-26; RRID: CVCL_0062 |
| HeLa cervical cancer cell line, Homo sapiens | ATCC | Cat#CCL-2; RRID: CVCL_0030 |
| A549 lung cancer cell line, Homo sapiens | ATCC | Cat#CCL-185; RRID: CVCL_0023 |
| Peripheral blood mononuclear cells (PBMCs) | Stem Cell | Cat#70025.1 |
| Chronic Lymphocytic Leukemia cells (CLL3007) | Nicholas Chiorazzi lab, Feinstein Institute for Medical Research | Sarkar, et al. 2016 |
| Oligonucleotides | ||
| ON-TARGETplus Rpn13 (11047) siRNA SMARTpool | Dharmacon | Cat#L-012340–01 |
| ON-TARGETplus GAPDH siRNA SMARTpool | Dharmacon | Cat#D-001830–10 |
| Custom ADRM1 (Rpn13) duplex ON-TARGET siRNA - UTR1 Rpn13 Sense:5'AGGAAGAGCGAGCCCGGACUU3' Antisense:5'GUCCGGGCUCGCUCUUCCUUU3' |
Dharmacon | Cat#CTM-485557 |
| ON-TARGETplus nontargeting siRNA pool | Dharmacon | Cat#D-001810–10 |
| ON-TARGETplus CYB5B (80777) siRNA SMARTpool | Dharmacon | Cat#L-014633–02 |
| ON-TARGETplus C18orf32 (497661) siRNA SMARTpool | Dharmacon | Cat#L-034916–02 |
| ON-TARGETplus OCIAD1 (54940) siRNA SMARTpool | Dharmacon | Cat#L-020825–02 |
| ON-TARGETplus REEP5 (7905) siRNA SMARTpool | Dharmacon | Cat#L-019467–01 |
| Site directed mutagenesis forward primer 5’ GCTGGGGGCCTGCGGCACCCGCTTGAACTCACAGTCGTC 3’ |
IDT | N/A |
| Site directed mutagenesis reverse primer 5’ CCGCAGGCCCCCAGCGGGAGGGTCTACGTGCTGAAGTTC 3’ |
IDT | N/A |
| Recombinant DNA | ||
| pcDNA5 FLAG-ADRM1 | Yao, et al., 2006 | Addgene #19417; RRID: Addgene_19417 |
| Pet19b His6 ADRM1 | Yao, et al., 2006 | Addgene #19423; RRID: Addgene_19423 |
| pcDNA5 FLAG-Rpn13_C88A | This paper | N/A |
| Pet19b His6 Rpn13_C88A | This paper | N/A |
| Rpn2 plasmid FLAG-Rpn2 | Kylie Walters lab, National Cancer Institute | Lu, et al. 2015 |
| pVP16 MBP-Uch37 | DNASU | ID#84019 |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | RRID: SCR_003070 |
| Licor Image Studio Software | LI-COR Image Studio Software | RRID: SCR_015795 |
| Prism 8 | Graphpad | RRID:SCR_000306 |
| FlowJo | FlowJo LLC | RRID: SCR_008520 |
| MaxQuant V1.6.1.0 | Cox and Mann, 2008 | RRID:SCR_014485 |
| Geneious | Geneious | RRID: SCR_010519 |
| ChemDraw | PerkinElmer | RRID: SCR_016768 |
| Other | ||
| Amylose resin | NEB | Cat#E8021S |
| Ni-NTA resin | Qiagen | Cat#30210 |
| Protein G Plus agarose | Thermo Fisher | Cat#22851 |
| 4–20% Mini-PROTEAN Gels | Biorad | Cat#4561096 |
| Fetal Bovine Serum, Heat inactivated | Life technologies | Cat#16140–071 |
| KOD HotStart PCR Master Mix | EMD Millipore | Cat#71842 |
| Mycoplasma PCR Test Kit | ABM Good | Cat#G238 |
SIGNIFICANCE.
Rpn13, one of the ubiquitin receptors in the proteasome, has been suggested to be a good target for the development of chemotherapeutics. Over the last few years two compounds, RA190 and the peptoid KDT11, which bind Rpn13 in vitro, have been shown to have interesting cancer cell toxicity and RA190 has shown efficacy in several mouse models of cancer. However, the lack of unequivocal evidence for Rpn13 engagement by RA190 or KDT11 in cells and the absence of a defined mechanism of action have left open the possibility that the phenotypes driven by these compounds are instead the result of off-target interactions. In this study we probe this issue for RA190. A variety of physical and functional assays show conclusively that RA190 does not engage Rpn13 in at least three cancer cell types and, in fact, is a highly promiscuous alkylator of dozens of proteins in the proteome. In a separate study we show that it is highly unlikely that the cytotoxicity of KDT11 is the result of interaction with Rpn13. Thus, the idea that this ubiquitin receptor may be an interesting target for the development of new chemotherapeutics remains to be tested pharmacologically.
Highlights.
Small-molecule RA190 is a highly promiscuous alkylating agent
Chemical proteomics reveal a wide interaction network with RA190
Previously reported target Rpn13 is not identified in cellular experiments
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (GM 131420). The authors acknowledge financial support from The Scripps Research Institute.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chembiol.2020.08.007.
REFERENCES
- Al-Shami A, Jhaver KG, Vogel P, Wilkins C, Humphries J, Davis JJ, Xu N, Potter DG, Gerhardt B, Mullinax R, Shirley CR, Anderson SJ, and Oravecz T (2010). Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS One 5, e13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anchoori RK, Khan SR, Sueblinvong T, Felthauser A, Iizuka Y, Gavioli R, Destro F, Isaksson Vogel R, Peng S, Roden RB, et al. (2011). Stressing the ubiquitin-proteasome system without 20S proteolytic inhibition selectively kills cervical cancer cells. PLoS One 6, e23888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anchoori RK, Karanam B, Peng S, Wang JW, Jiang R, Tanno T, Orlowski RZ, Matsui W, Zhao M, Rudek MA, et al. (2013). A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell 24, 791–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzaro M, Anchoori RK, Mudiam MK, Issaenko O, Kumar S, Karanam B, Lin Z, Isaksson Vogel R, Gavioli R, Destro F, et al. (2011). alpha,beta-Unsaturated carbonyl system of chalcone-based derivatives is responsible for broad inhibition of proteasomal activity and preferential killing of human papilloma virus (HPV) positive cervical cancer cells. J. Med. Chem 54, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Hu XT, Shi Q-L, Zhang F-B, and He C (2009). Knockdown of the novel proteasome subunit Adrm1 located on the 20q13 amplicon inhibits colorectal cancer migration, survivability and tumorigenicity. Oncol. Rep. 21, 531–537. [PubMed] [Google Scholar]
- Coux O, Tanaka K, and Goldberg AL (1996). Structure and function of the 20S and 26S proteasomes. Ann. Rev. Biochem 65, 801–847. [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dickson P, Simanski S, Ngundu JM, and Kodadek T (2020). Mechanistic studies of the multiple myeloma and melanoma cell-selective toxicity of the Rpn13-binding peptoid KDT-11. Cell Chem. Biol 10.1016/j.chembiol.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, and Strieter ER (2018). A fluorescence polarization-based competition assay for measuring interactions between unlabeled ubiquitin chains and UCH37-RPN13. Anal Biochem. 550, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejzo MS, Dering J, Ginther C, Anderson L, Ramos L, Walsh C, Karlan B, and Slamon DJ (2008). Comprehensive analysis of 20q13 genes in ovarian cancer identifies ADRM1 as amplification target. Genes Chromosomes Cancer 47, 873–883. [DOI] [PubMed] [Google Scholar]
- Fejzo MS, Ginther C, Dering J, Anderson L, Venkatesan N, Konecny G, Karlan B, and Slamon DJ (2011). Knockdown of ovarian cancer amplification target ADRM1 leads to downregulation of GIPC1 and upregulation of RECK. Genes Chromosomes Cancer 50, 434–441. [DOI] [PubMed] [Google Scholar]
- Fejzo MS, Anderson L, von Euw EM, Kalous O, Avliyakulov NK, Haykinson MJ, Konecny GE, Finn RS, and Slamon DJ (2013). Amplification target ADRM1: role as an oncogene and therapeutic target for ovarian cancer. Int. J. Mol. Sci 14, 3094–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejzo MS, Anderson L, Chen HW, Anghel A, Zhuo J, Anchoori R, Roden R, and Slamon DJ (2015). ADRM1-amplified metastasis gene in gastric cancer. Genes Chromosomes Cancer 54, 506–515. [DOI] [PubMed] [Google Scholar]
- Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, Zweegman S, Chan ET, Kirk CJ, Geerke DP, et al. (2012). Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 26, 757–768. [DOI] [PubMed] [Google Scholar]
- Goldberg AL (2012). Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol 199, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki J, Hirayama S, and Murata S (2015). Redundant roles of Rpn10 and Rpn13 in recognition of ubiquitinated proteins and cellular homeostasis. PLoS Genet. 11, e1005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wu Y, Zhou X, Xu J, Zhu W, Shu Y, and Liu P (2014). Efficacy of therapy with bortezomib in solid tumors: a review based on 32 clinical trials. Future Oncol. 10, 1795–1807. [DOI] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, and Dikic I (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev AF, and Goldberg AL (2001). Proteasome inhibitors: from research tools to drug candidates. Chem. Biol 8, 739–758. [DOI] [PubMed] [Google Scholar]
- Klock HE, and Lesley SA (2009). The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol 498, 91–103. [DOI] [PubMed] [Google Scholar]
- Kolb HC, and Sharpless KB (2003). The growing impact of click chemistry on drug discovery. Drug Discov. Today 8, 1128–1137. [DOI] [PubMed] [Google Scholar]
- Li J, Yakushi T, Parlati F, Mackinnon AL, Perez C, Ma Y, Carter KP, Colayco S, Magnuson G, Brown B, et al. (2017). Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat. Chem. Biol 13, 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Liu F, Durham SE, Tarasov SG, and Walters KJ (2015). A high affinity hRpn2-derived peptide that displaces human Rpn13 from proteasome in 293T cells. PLoS One 10, e0140518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Nowicka U, Sridharan V, Liu F, Randles L, Hymel D, Dyba M, Tarasov SG, Tarasova NI, Zhao XZ, et al. (2017). Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat. Commun 8, 15540–15553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramore A, and Frantz S (2003). Bortezomib. Nat. Rev. Drug Discov 2, 611–612. [DOI] [PubMed] [Google Scholar]
- Perez C, Li J, Parlati F, Rouffet M, Ma Y, Mackinnon AL, Chou TF, Deshaies RJ, and Cohen SM (2017). Discovery of an inhibitor of the proteasome subunit Rpn11. J. Med. Chem 60, 1343–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar SV, and Kumar S (2016). Multiple myeloma: diagnosis and treatment. Mayo Clin. Proc 91, 101–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randles L, Anchoori RK, Roden RB, and Walters KJ (2016). The proteasome ubiquitin receptor hRpn13 and its interacting deubiquitinating enzyme Uch37 are required for proper cell cycle progression. J. Biol. Chem 291, 8773–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar M, Liu Y, Qi J, Peng H, Morimoto J, Rader C, Chiorazzi N, and Kodadek T (2016). Targeting stereotyped B cell receptors from chronic lymphocytic leukemia patients with synthetic antigen surrogates. J. Biol. Chem 291, 7558–7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong KM, Baek J, Yu M, and Kim J (2007). Rpn13p and Rpn14p are involved in the recognition of ubiquitinated Gcn4p by the 26S proteasome. FEBS Lett. 581, 2567–2573. [DOI] [PubMed] [Google Scholar]
- Song Y, Ray A, Li S, Das DS, Carrasco RD, Chauhan D, and Anderson KC (2016). Targeting proteasome ubiquitin receptor Rpn13 in multiple myeloma. Leukemia 9, 1877–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong R-S, Anchoori RK, Roden RBS, Cho R-L, Chen Y-C, Tseng S-C, Huang Y-L, Liao P-C, and Shyu Y-C (2020). Bis-benzylidine piperidone RA190 treatment of hepatocellular carcinoma via binding RPN13 and inhibiting NF-kB signaling. BMC Cancer 20, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trader DJ, Simanski S, and Kodadek T (2015). A reversible and highly selective inhibitor of the proteasomal ubiquitin receptor rpn13 is toxic to multiple myeloma cells. J. Am. Chem. Soc 137, 6312–6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLinden RT, Hemmis CW, Yao T, Robinson H, and Hill CP (2017). Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J. Biol. Chem 292, 9493–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voges D, Zwicki P, and Baumeister W (1999). The 26S proteasome: a molecular machine designed for controlled proteolysis. Ann. Rev. Biochem 68, 1015–1068. [DOI] [PubMed] [Google Scholar]
- Wang C, Michener CM, Belinson JL, Vaziri S, Ganapathi R, and Sengupta S (2010). Role of the 18:1 lysophosphatidic acid-ovarian cancer immunoreactive antigen domain containing 1 (OCIAD1)-integrin axis in generating late-stage ovarian cancer. Mol. Cancer Ther 9, 1709–1718. [DOI] [PubMed] [Google Scholar]
- Ward JA, Pinto-Fernandez A, Cornelissen L, Bonham S, Díaz-Sáezz L, Riant O, Huber KVM, Kessler BM, Feron O, and Tate EW (2020). Re-evaluating the mechanism of action of α,β-unsaturated carbonyl DUB inhibitors b-AP15 and VLX1570: a paradigmatic example of unspecific protein cross-linking with Michael acceptor motif-containing drugs. J. Med. Chem 63, 3756–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welk V, Coux O, Kleene V, Abeza C, Trumbach D, Eickelberg O, and Meiners S (2016). Inhibition of proteasome activity induces formation of alternative proteasome complexes. J. Biol. Chem 291, 13147–13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilt LH, Jansen G, Assaraf YG, van Meerloo J, Cloos J, Schimmer AD, Chan ET, Kirk CJ, Peters GJ, and Kruyt FA (2012). Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem. Pharmacol 83, 207–217. [DOI] [PubMed] [Google Scholar]
- Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, Washburn MP, Conaway RC, Conaway JW, and Cohen RE (2006). Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat. Cell Biol 8, 994–1002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.
The published article includes all of the mass spectrometry data generated during this study (Tables S1, S2, and S3).