

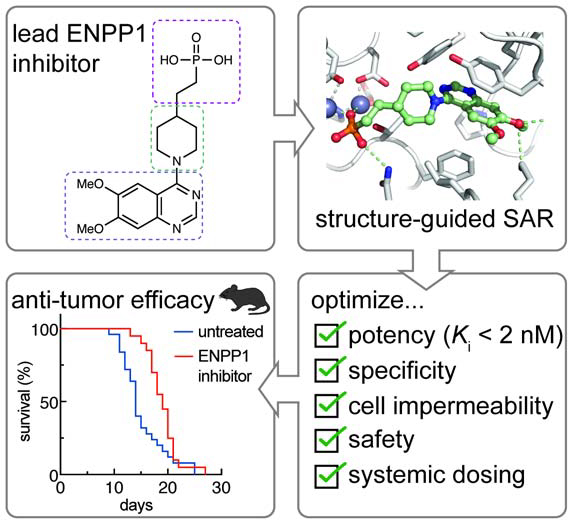
Summary
Cancer cells initiate an innate immune response by synthesizing and exporting the small molecule immunotransmitter cGAMP, which activates the anti-cancer Stimulator of Interferon Genes (STING) pathway in the host. An extracellular enzyme, ectonucleotide pyrophosphatase phosphodiesterase 1 (ENPP1), hydrolyzes cGAMP and negatively regulates this anti-cancer immune response. Small molecule ENPP1 inhibitors are much needed as tools to study basic biology of extracellular cGAMP and as investigational cancer immunotherapy drugs. Here, we surveyed structure-activity relationships around a series of cell-impermeable and thus extracellular-targeting phosphonate inhibitors of ENPP1. Additionally, we solved the crystal structure of an exemplary phosphonate inhibitor to elucidate the interactions that drive potency. This study yielded several best-in-class compounds with Ki < 2 nM and excellent physicochemical and pharmacokinetic properties. Finally, we demonstrate that an ENPP1 inhibitor delays tumor growth in a breast cancer mouse model. Together, we have developed ENPP1 inhibitors that are excellent tool compounds and potential therapeutics.
eTOC
As the dominant hydrolase of the immunotransmitter cGAMP, the extracellular enzyme ENPP1 dampens the anti-cancer immune response. Carozza et al. solved the crystal structure of ENPP1 in complex with a lead inhibitor, followed by structure-activity relationship studies. The resulting potent and specific extracellular ENPP1 inhibitor delays tumor growth in mice.
Graphical Abstract
Introduction
Adaptive immune checkpoint inhibitors such as anti-PD-1, anti-PD-L1, and anti-CTLA-4 are now curing cancer patients who were previously considered terminally ill (Topalian et al., 2012). These inhibitors work by removing the immunological brakes that cancer cells place on tumor-infiltrating lymphocytes (TILs), therefore increasing the cancer-killing efficacy of TILs. However, only “hot” tumors – those that already have high numbers of TILs – respond to checkpoint inhibitor therapy. Most tumors do not exhibit this TIL inflammation and thus are immunologically “cold” (Corrales et al., 2017; Fuertes et al., 2011; Woo et al., 2015). Turning “cold” tumors “hot” by activating the innate immune detection of cancer, which is upstream of the recruitment of adaptive immune TILs, could revolutionize cancer immunotherapy.
The cytosolic double-stranded DNA (dsDNA) sensing-Stimulator of Interferon Genes (STING) pathway is the key innate immune pathway that responds to cancerous cells. Chromosomal instability and extrachromosomal DNA are hallmarks of cancer that can lead to leakage of dsDNA into the cytosol (Bakhoum and Cantley, 2018; Bakhoum et al., 2018; Harding, 2017; Mackenzie et al., 2017; Turner et al., 2017). The cytosolic dsDNA is detected by the enzyme cyclic-GMP-AMP synthase (cGAS) (Sun et al., 2013), which synthesizes the cyclic dinucleotide 2’,3’-cyclic GMP-AMP (cGAMP) (Ablasser et al., 2013; Wu et al., 2013). cGAMP then binds and activates STING, which leads to production of Type I interferons (IFNs) and downstream TIL infiltration. We recently discovered that cancer cell lines basally synthesize cGAMP and export it to the extracellular space (Carozza et al., 2020). Extracellular cGAMP is then internalized by host cells to trigger anti-cancer innate immune responses (Carozza et al., 2020; Lahey et al., 2020; Marcus et al., 2018; Ritchie et al., 2019; Schadt et al., 2019; Zhou et al., 2020b). However, we also discovered that the ubiquitously expressed extracellular enzyme ENPP1, which was previously known only as an ATP hydrolase, is the dominant hydrolase for extracellular cGAMP and dampens innate responses to cancer (Carozza et al., 2020; Li et al., 2014). We previously developed phosphorothioate cGAMP analogs that are resistant to ENPP1 hydrolysis (Li et al., 2014). Since then, other stable cGAMP analogs have entered clinical trials in combination with anti-PD-1 checkpoint blockers (Trial IDs NCT03172936 and NCT03010176, respectively). However, these cGAMP analogs need to be injected directly into the tumors to achieve efficacy and to avoid systemic interferon response. Alternatively, we propose to inhibit ENPP1 to boost the efficacy of the endogenous extracellular cGAMP that is only locally exported by cancer cells. Indeed, genetic knockout and pharmacological inhibition of ENPP1 both increased tumor-infiltrating dendritic cells and controlled tumor growth, without any safety concerns (Carozza et al., 2020). This work suggests that ENPP1 inhibitors could turn “cold” tumors “hot” and render them more sensitive to adaptive immune checkpoint inhibitors.
Outside of its role in cancer immunotherapy, cGAMP has shown excellent adjuvant activities in vaccination against influenza (Wang et al., 2020) and may aid in the current urgent demand to develop vaccines against SARS-CoV-2. We, therefore, propose that ENPP1 inhibitors may also stabilize cGAMP when administered as an adjuvant.
ENPP1 is a single-pass transmembrane protein that is anchored with the catalytic domain outside of the cell. It can also be exported extracellularly in a soluble form that is abundant in the circulation (Belli et al., 1993; Jansen et al., 2012; Meyre et al., 2005). ENPP1 was known to convert nucleotide triphosphates (preferentially ATP) to nucleotide monophosphates (cite something). We reported that ENPP1 also converts cGAMP to AMP and GMP by hydrolyzing first the 2’−5’ phosphodiester bond, and then the 3’−5’ (Li et al., 2014). Similar to the other NPP family members, its catalytic site coordinates two zinc ions that hold the substrate phosphate in place for threonine-mediated nucleophilic attack. The adenosine base of ENPP1 substrates stacks with aromatic residues that form a tight pocket in the active site (Kato et al., 2012a). Later co-crystal structures of ENPP1 with pApG, the degradation intermediate of 2’3’-cGAMP, and with 3’3’-cGAMP provided mechanistic insight into why ENPP1 degrades 2’3’-cGAMP, but not 3’3’-cGAMP (Kato et al., 2018). However, all natural ENPP1 substrates have Km values higher than 20 μM (Kato et al., 2012a; Li et al., 2014).
Despite ENPP1 being a highly sought-after target (Chang et al., 2014; Forcellini et al., 2018; Lee et al., 2017, 2016; Lefebvre et al., 2017; Patel et al., 2009; Shayhidin et al., 2015; Supe et al., 2018; Zelikman et al., 2018), developing drug-like ENPP1 inhibitors has proved difficult. Here, we report the development of the most potent ENPP1 inhibitors to date and the co-crystal structure of an exemplary phosphonate inhibitor with ENPP1. Inspired by the molecular scaffold of a previous inhibitor, QS1,(Patel et al., 2009; Shayhidin et al., 2015) which lacks potency at physiological conditions, we build structure-activity relationships (SAR) around the three sections of the molecule – the zinc-binding head, the core, and the tail – and develop several inhibitors with nanomolar Ki values. Our crystal structure revealed extensive interactions between the inhibitor and ENPP1 and explains its 1000–10,000-fold improvement in affinity over the natural substrates. Finally, these inhibitors have desirable physicochemical and pharmacokinetic properties, which enables their systemic use in mouse studies. Indeed, we demonstrate that treatment with one of our top ENPP1 inhibitors delays tumor growth in a breast cancer mouse model.
Results
cGAMP is rapidly degraded in human and mouse plasma
Given that a soluble form of ENPP1 is present in the circulation, we first asked how much ENPP1 activity is present in freshly drawn mouse and human plasma by measuring the half-life of radiolabeled cGAMP. cGAMP was degraded rapidly (t1/2 = 16 minutes for mouse; t1/2 = 30–60 minutes for human, based on 5 healthy donors) (Fig 1a–c). Detectable hydrolysis of cGAMP ex vivo occurs only in WT, and not Enpp1−/− mouse plasma (Li et al., 2014), and inhibiting ENPP1 activity using EDTA to chelate the catalytic zinc ions abrogated cGAMP degradation in both mouse and human plasma (Fig 1a–c). Rapid degradation of extracellular cGAMP by circulating ENPP1 underscores the need to develop potent and systemic ENPP1 inhibitors.
Figure 1 |. ENPP1 rapidly degrades cGAMP but current inhibitors are not potent enough as tools to investigate ENPP1 function.

(A)–(C) cGAMP (5 μM, with trace radiolabeled [32P]cGAMP) was incubated with C57B6/J mouse plasma (A, B) or human plasma (C) at 37 °C. 50 mM EDTA was included in the indicated reactions. At indicated times, degradation was assessed by separation of cGAMP from degradation products by thin layer chromatography (TLC) and autoradiography. (A) shows a representative experiment for mouse plasma. For (B), data are from 4 independent experiments, mean + SD shown. For (C), data are from 3 donors, each with 2 independent experiments; replicates are plotted individually. (D) Assay for measuring ENPP1 inhibitor Ki values. cGAMP (5μM) was incubated with increasing concentrations of ENPP1 inhibitor (maximum = 10 μM) and ENPP1 (3 nM) in a buffer containing 50 mM Tris, 150 mM NaCl, 1 μM ZnCl2, and 500 μM CaCl2 at the indicated pH value. The minimum measurable Ki is 2 nM (determined by enzyme concentration); the maximum is 10 μM. (E) Chemical structures and Ki values of compounds 1 and 2 (mean of at least 2 independent replicates; evaluated in assay described in D).
Previous ENPP1 inhibitors are not potent enough as tools to investigate ENPP1 function
Assays used to assess the potency of previously attempted ENPP1 inhibitors are inconsistent (Chang et al., 2014; Forcellini et al., 2018; Lee et al., 2017, 2016; Lefebvre et al., 2017; Patel et al., 2009; Shayhidin et al., 2015; Supe et al., 2018; Zelikman et al., 2018); however, developing an appropriate assay is key to determining the utility of the molecules in inhibiting cGAMP degradation under physiological conditions. The most common model substrate is p-nitrophenyl-5’-TMP (p-NPTMP), but IC50 values measured using this substrate have been shown to deviate significantly from IC50 values measured using a natural substrate such as ATP (Lee et al., 2017). In addition, perhaps to increase the speed of the assay, most reported assays are conducted at pH 9, where ENPP1 is most active. However, since ENPP1 is active in serum at physiological pH (Fig. 1a–c), an effective ENPP1 inhibitor needs to be potent at pH 7.4 or even lower, as can occur in the tumor microenvironment (Boedtkjer and Pedersen, 2020; Vaupel et al., 1989). Therefore, we evaluated inhibitors with an assay using cGAMP as a substrate at pH 7.4 (Fig. 1d) (Mardjuki et al., 2020).
There were only two non-nucleotide inhibitor hits from the literature which had the potential to be developed into a lead compound for inhibiting ENPP1. First, a thioacetamide inhibitor (compound 1, Fig. 1e) was reported to have a Ki of 5 nM against human ENPP1 using p-nitrophenyl-5’-TMP (p-NPTMP) as the substrate, but the Ki increased to 5 μM when using ATP as the substrate, both assays conducted at pH 9 (Chang et al., 2014). When we tested the potency of compound 1 using mouse ENPP1 and cGAMP as a substrate at both pH 7.4 and pH 9, we detected no activity at concentrations below 10 μM (Fig. 1e), suggesting that it cannot block cGAMP degradation activity specifically compared to other substrates, or that it may be specific to human ENPP1. Regardless, lack of efficacy against cGAMP and/or mouse ENPP1 disqualifies this molecule as a scaffold to use for further development.
Second, Patel et al reported a quinazoline-piperidine-sulfamide inhibitor (QS1, compound 2) with an IC50 of 36 nM against ENPP1 using ATP as a substrate (Patel et al., 2009). However, we found that the potency of QS1 dropped 100-fold when adjusted the pH down to 7.4 instead of pH 9 (Ki = 1.6 μM using cGAMP as a substrate, Fig. 1e), making it unsuitable for experiments at physiological pH and in vivo. In addition, we have previously shown that QS1 non-specifically blocks cGAMP export, limiting its use as a tool for studying extracellular cGAMP biology (Carozza et al., 2020). We, therefore, sought to develop more potent and specific ENPP1 inhibitors.
ENPP1 inhibitors with a zinc-binding phosphonate head group exhibit high potency at physiological pH
We hypothesized that QS1 lacked potency at pH 7.4 because deprotonation of the sulfamide group (pKa around 7–8) is important for efficient binding to the zinc atoms at the catalytic site of ENPP1. Therefore, we tested several other classes of zinc-binding head groups on the QS1 scaffold (Fig. 2a–b). We also tried methylene, ethyl, and/or propyl linkage between the head groups and the piperidine core. Ureas (compounds 4 and 5) and carboxylic acid (compound 6) were less potent compared to sulfamides (compounds 2 and 3), but we observed improvement in potency with boronic acid (compound 7), hydroxamic acids (compounds 8 and 9), and especially phosphates (compounds 10 and 11), the natural zinc binding group of the ENPP1 substrates cGAMP and ATP. To increase compound stability while preserving the core properties of phosphates, we tested thiophosphates (compounds 12 and 13), phosphonates (compounds 14–16), and a thiophosphonate (compound 17), all of which are known to be more stable against enzymatic degradation compared to phosphates. The phosphonates showed Ki values less than 50 nM independent of the pH (Fig. 2a–b).
Figure 2 |. ENPP1 inhibitors with a zinc-binding phosphonate head group exhibit high potency at physiological pH.

(A) Chemical structure of the R group (core = piperidine, tail = 6,7-dimethoxy quinazoline). (B) Chemical structures of zinc-binding heads with corresponding Ki values (mean of at least 2 independent replicates) at pH 7.4 and pH 9.0. Ki values were determined using 3 nM ENPP1 and 5 μM cGAMP.
We chose to proceed with phosphonates because they are potent, stable, and synthetically tractable. In addition, there are several phosphonate drugs on the market including Tenofovir and Pradefovir as antivirals, Fosfomycin as an antibiotic, and bisphosphonates (Pamidronic aicd, Zoledronic acid, Alendronic acid) for osteoporosis and bone disease. Finally, they are negatively charged at all physiological pH values, which is crucial for zinc binding and keeping them cell impermeable to act on the extracellular target ENPP1. We also chose the ethylene linker (compound 15, Ki = 33 nM, previously reported as STF-1084 (Carozza et al., 2020)) because it has been shown previously that for the sulfamides, analogs with shorter linker lengths have less affinity for the cardiac potassium channel hERG, a detrimental off target (Forcellini et al., 2018; Shayhidin et al., 2015).
Co-crystal structure of ENPP1 and compound 15 reveals molecular determinants of potency
ENPP1 is a >100 kDa multidomain glycoprotein with three glycosylation chains. It has a catalytic domain, a nuclease-like domain which provides structural support important for catalysis, and two disordered SMB domains. We used a mouse ENPP1 construct where the transmembrane anchor was truncated and replaced by a signal peptide, and expressed the construct in HEK293S GnT1− cells to generate secreted soluble ENPP1 with simplified glycosylation chains as previously described (Kato et al., 2012a, 2012b, 2018). We then determined the structure of ENPP1 in complex with compound 15 to 3.2 Å resolution using X-ray crystallography (Fig. 3, Fig. S1, Tables S2–4). Compound 15 occupies the substrate-binding pocket (Fig. 3a–d) and forms extensive interactions with zinc ions and residues in the catalytic site, demonstrating that compound 15 is a competitive inhibitor. The phosphonate oxygens bind both zinc ions, and the third phosphonate oxygen forms a hydrogen bond with N259, a residue previously determined to be important for catalytic activity. The piperidine group is engaged in hydrophobic interactions with L272, and the quinazoline group sits between Y322 and F239 to form π-π interactions with both (Fig. 3b–d). Although compound 15 adopts a similar binding conformation as the reaction product AMP (Kato et al., 2012a) (PDB 4GTW, Fig. 3e–f), there are two significant differences between the ligands that suggest why compound 15 has a much higher affinity than the substrates ATP or cGAMP. First, the quinazoline ring of compound 15 sits ~2.5 A further back in the catalytic pocket compared to the purine ring of AMP, perhaps facilitating hydrophobic interactions with residues in the back of the pocket, and possibly a polar interaction between N3 of the quinazoline and Y353. Second, the 7-methoxy group of compound 15 makes a direct hydrogen bond with D308, whereas AMP makes a water-mediated hydrogen bond (Fig. 3e). In addition, residue K277 forms a hydrogen bond with the 7-methoxy oxygen, further strengthening this direct hydrogen bond network. Together, the potency of compound 15 is driven by its zinc ion binding, hydrophobic interactions with the back of the binding pocket, and direct hydrogen bonds with active side resides of ENPP1. In addition, this structure guided our SAR for the core and tail parts of the inhibitor.
Figure 3 |. Co-crystal structure of ENPP1 and compound 15 reveals molecular determinants of potency.

(A) The 3.2 Å crystal structure of compound 15 (green spheres) bound to mouse ENPP1 (gray surface; catalytic and nuclease-like domains). (B) Expanded view of compound 15 (green sticks/spheres) bound in the active site of ENPP1 (gray sticks). Zincs are shown as dark gray spheres. Hydrogen bonds and metal coordination are shown as dashed lines. (C) Chemical structure of compound 15. (D) Schematic drawing of interactions formed between compound 15 (green) and the ENPP1 active site (black). Residues within 5 Å are shown. Metal coordination shown as gray dashed lines, hydrogen bonds shown as black dashed lines, aromatic interactions shown as black wedged lines, and hydrophobic or polar interactions shown as spokes. (E) Overlay of compound 15 (green) with the AMP (orange, PDB 4GTW), both bound to ENPP1. Ligands are shown as sticks/spheres, protein residue D308 is shown as sticks, and water is shown as sticks/spheres. Density for water molecule is present only in AMP-ENPP1 crystal structure. (F) Chemical structure of AMP. See also Figure S1 and Tables S2–S4.
Piperidine and benzyl amine cores are optimal molecular linkers
Since our crystal structure suggests that the zinc-binding phosphonate head and quinazoline tail form the most important interactions with ENPP1, we next sought to explore the core region to achieve optimal geometry between these two functional groups (Fig. 4a–b). Inverting the nitrogen in the piperidine from aryl to alkyl (compound 18) resulted in more than a 200-fold loss in potency, as did installing a piperazine (compound 19). The benzyl group in compound 20 resulted in approximately a 3-fold loss in potency compared to the piperidine in compound 15. When we installed a nitrogen to form aminobenzyls (compounds 21 and 22), we again observed small losses in potency. Extending the length to a benzyl amine (compounds 23 and 24) improved Ki values to 12 nM and 26 nM, respectively. We anticipate that the core region itself does not make significant contacts with ENPP1, but rather dictates the angle between head and tail. The piperidine (compound 15) and benzyl amine (compounds 23 and 24) cores have unique geometry but both result in low nanomolar Ki values, suggesting that these linkers yield favorable binding poses.
Figure 4 |. Piperidine and benzyl amine cores are optimal molecular linkers.

(A) Chemical structures of the R1 group (phosphonate head) and the R2 group (6,7-dimethoxy quinazoline tail). (B) Chemical structures of cores with corresponding Ki values (mean of at least 2 independent replicates) at pH 7.4. Ki values were determined using 3 nM ENPP1 and 5 μM cGAMP.
8-methoxy quinazoline tails achieve high potency compared to other quinazoline substitutions
Using the phosphonate zinc-binding head and the piperidine core as a scaffold, we then sought to determine optimal substitution of the quinazoline tail (Fig. 5a–b). Based on our co-crystal structure of ENPP1 with compound 15, we predicted that the 7-methoxy would be critical for binding to ENPP1, while the 6-methoxy would be dispensable since it is solvent-exposed. Indeed, when we deleted the methoxy groups individually, we found that the 6-methoxy alone (compound 25) was 100-fold less potent, while the 7-methoxy alone (compound 27) displayed similar Ki to the 6,7-methoxy (compound 15). Substituting a hydrogen bond donor/electron-donating group in the 7 position (compounds 29 and 30) led to a loss of potency compared to the 7-methoxy (compound 27), suggesting that the methyl group makes favorable interactions with the protein. Extending to a 7-ethoxy (compound 28) or 7-isopropyloxy (compound 31) was not tolerated, presumably due to steric constraints.
Figure 5 |. 8-methoxy quinazoline and 3-nitrile quinoline tails achieve high potency.

(A) Chemical structures of the R group (head = phosphonate, core = piperidine). (B)–(C) Chemical structures of quinazoline (B) and quinoline (C) tails with corresponding Ki values (mean of at least 2 independent replicates) at pH 7.4. Ki values were determined using 3 nM ENPP1 and 5 μM cGAMP.
We then moved to substituents at the 8-position of the quinazoline ring. The 8-methoxy (compound 32) and 8-ethoxy (compound 33) quinazolines displayed impressive potency with a Ki values less than 2 nM, which is the limit of quantification of the assay when using a 3 nM concentration of ENPP1. We reasoned that combining other methoxy groups with the 8-methoxy could make additional interactions with the binding pocket, since the 7-methoxy (compound 27) alone was also potent. However, combinations of 8-methoxy with substituents in either the 5, 6, or 7 positions (compounds 36–38 and 40–43) all decreased potency. From these data and the crystal structure, we hypothesize that the 8-methoxy (compound 32) and 8-ethoxy (compound 33) quinazolines make hydrogen bonds with both D208 and/or K277 similar to the 7-methoxy in compound 15. This would suggest that compounds 32 and 33 shift in the pocket to accommodate these interactions. Adding more substituents to the quinazoline does not necessarily lower potency further, as the inhibitor-interacting residues may already be occupied.
In addition, similar to previous SAR (Forcellini et al., 2018; Patel et al., 2009), the 2-substituted vinyl-3-pyridyl combined with the 6,7-dimethoxy (compound 44) showed identical potency to its parent 6,7-dimethoxy (compound 15). This is surprising, since the crystal structure shows limited space in this area of the pocket, suggesting that the pyridine may make a specific interaction with a protein residue. However, this trend did not hold for the 8-methoxy (compound 45), where the addition of the 2-substituted vinyl-3-pyridyl decreased potency by more than 100-fold. This further supports the hypothesis that different substitutions cause shifts in the inhibitor binding to the pocket, possibly excluding space that was previously available.
3-nitrile quinolines substitute for quinazolines in the tail
From the crystal structure of compound 15 bound to ENPP1, we noted that the quinazoline tail of compound 15 is close to residues near the back of the binding pocket. We perturbed the tail aromatic ring structure itself to investigate these possible interactions (Fig. 5a,c). Working from the quinazoline compound 15, we deleted the nitrogens on the ring on by one. Deleting N3 (quinoline, compound 46) led to merely a 3-fold decrease in potency, but deleting N1 (isoquinoline, compound 51) led to a more than 100-fold decrease in potency. We tested whether we could replace the inhibitor-protein interaction that involved the N3 or N1 with a nitrile, a more electronegative group and possible hydrogen bond acceptor, as has been previously suggested (Tsou et al., 2005). Indeed, the 6,7-dimethoxy, 3-nitrile quinoline (compound 47) was more potent than the quinazoline itself (compound 46), and this pattern repeated with all of the other methoxy substituents (compounds 48–50). Compound 50 was the most potent molecule with a Ki less than our limit of quantification of 2 nM. When we attempted to substitute back the missing nitrogen with a nitrile group on the isoquinoline scaffold (compound 52), we lost potency, possibly due to molecular clashes between inhibitor and protein. In summary, the N1 nitrogen is critical for potency, possibly due to polar interactions with the protein backbone ~4 Å away. It cannot be replaced by a nitrile since space is limited. The N3 nitrogen can be replaced by a more electronegative nitrile, leading to much more potent compounds.
Hybrid ENPP1 inhibitors combine the most potent heads, cores, and tails
Taking into account of all of our SAR data, we then synthesized hybrid molecules composed of the most potent heads, cores, and tails (8-methoxy quinazoline, e.g., compound 32, and 8-methoxy quinoline 3-nitrile, e.g., compound 50) (Fig. 6). For all of these molecules, Ki values fell below 100 nM. Combining the benzyl amine core with the 8-methoxy quinoline 3-nitrile tail (compound 57) yielded another inhibitor with a Ki less than our limit of quantification of 2 nM. Attaching other zinc binding head groups, including thiophosphate (compound 53), boronic acid (compounds 60 and 61), and hydroxamic acid (compound 62), to the most potent core/tail combinations also yielded potent inhibitors.
Figure 6 |. Hybrid ENPP1 inhibitors combine the most potent heads, cores, and tails.

Chemical structures of hybrid ENPP1 inhibitors connecting the most potent heads, cores, and tails with corresponding Ki values (mean of at least 2 independent replicates) at pH 7.4. Ki values were determined using 3 nM ENPP1 and 5 μM cGAMP.
Compound 32 leads to tumor growth delay in mouse breast cancer model after optimizing in vitro ADME and in vivo pharmacokinetics.
We further evaluated the potency and in vitro ADME properties of the seven most potent inhibitors. First, we evaluated the protein-shifted IC50, which can help predict what the efficacy will be in vivo, as this value is generally higher than that observed in vitro due to protein binding. We observed that all of the inhibitors displayed a shift in potency when we did the assay in the presence of human serum albumin, although several still stayed below 50 nM (Fig. 7a, Table S1, Fig. S2). We then tested their potency in both mouse and human plasma, and obtained similar values, confirming that mid-nanomolar concentrations are sufficient in biological fluids to prevent degradation of cGAMP (Fig. 7b, Table S1). Although ENPP1 protein is present in our circulation, these data demonstrate that we can completely block serum ENPP1 activity with as little as 100 nM of our most potent inhibitors, showing that protein binding does not negate the inhibition, and suggesting that serum ENPP1 will not sequester all the systemically administered ENPP1 inhibitor. Finally, all of our inhibitors are stable and cell impermeable. Although we expect these phosphonate inhibitors to have few intracellular off-targets due to their impermeability, we also confirmed that the inhibitors are non-toxic to primary human peripheral blood mononuclear cells (PBMCs) (Table S1, Fig. S3). Since QS1 has an off-target hERG liability (Patel et al., 2009), we tested two phosphonate inhibitors (compounds 15 and 32) and observed no inhibition at 25 μM (Table S1).
Figure 7 |. Optimized phosphonate ENPP1 inhibitors prevent cGAMP degradation in mouse and human plasma at nanomolar concentrations.

(A) In vitro dose-inhibition curves for selected top ENPP1 inhibitors. Ki values were determined using 3 nM ENPP1 and 5 μM cGAMP and (where indicated) in the presence of 40 μM human serum albumin at pH 7.4. Dotted line represents the minimum Ki value (2 nM) measurable with the given enzyme concentration. (B) In mouse and human plasma, IC50 values were determined using 5 μM cGAMP with trace radiolabeled [32P]cGAMP. In vitro dose-response curves replotted from (A). Chemical structures of inhibitors are displayed below. Dots represent the mean of two independent replicates, and shaded areas around the fitted curves represent the 95% confidence interval of the fit. See also Figure S2. (C) Concentrations of compound 32 in mouse serum, kidney, and liver after one 10 mg/kg dose via intravenous (IV) or subcutaneous (SC) route. IC95 = 40 nM; LOQ = 10 nM. Mean ± SEM is plotted, n = 2 mice for each point except the following, where n = 1: liver SC 4 h (point removed as an outlier using the ROUT method with Q = 1%) and serum 8 h (concentration below LOQ). Data were fit with one-phase decay with 1/Y2 weighting to obtain half-life. (D) Concentrations of compound 32 in mouse serum after consecutive once daily dosing at 300 mg/kg via SC route for 6 days. Serum was collected for analysis 24 hours after last dose, immediately before the next dose. (E) Tumor volumes (left; each line represents one mouse) and survival (right) of mice bearing E0771 breast cancer tumors. Mice were treated with vehicle (no treatment, n = 25 mice) or treated with compound 32 (n = 20 mice) dosed once daily at 300 mg/kg via SC route for seven consecutive days after tumors reached 80–120 mm3. For survival, P = 0.0006 (Gehan-Breslow-Wilcoxon test).
We then assessed the pharmacokinetic (PK) profile of one of the top inhibitors, compound 32, in mice. Since compound 32 has an IC50 value of 4 nM in mouse plasma, we aim to achieve serum concentrations of 40 nM (IC95 value). First, we administered intravenous (IV) and subcutaneous (SC) doses of compound 32 at 10 mg/kg to mice and analyzed the concentration of compound 32 in the serum, kidney, and liver for the next 8 hours. We found that serum concentrations declined to 10 nM or less within 8 hours for both IV and SC administration, and the half-life was only 10–15 minutes. Concurrent with the rapid decline in serum concentrations, we saw concentration plateaus in the kidney and liver in the micromolar range, suggesting that compound 32 is rapidly excreted through these organs. This is in agreement with our previously reported PK study of compound 32 performed at 300 mg/kg via SC administration where we observed that the serum concentration drops to ~100 nM at the end of 24 hours (Carozza et al., 2020). Therefore, we were only able to test the efficacy of compound 32 in that study dosing via an osmotic pump to maintain serum concentration of compound 32 above its IC95. To achieve sustained serum concentrations without surgically implanting osmotic pumps, we repeated the 300 mg/kg SC dosing daily for 6 days. We collected serum 24 hours after the previous dose, which corresponds to the expected lowest point in the trough. We observed on average ~100 nM of compound 32 across several days, and the lowest concentration measured was 40 nM. We also observed no adverse effects and the mice maintained healthy weights, suggesting that this dosage amount and timing is tolerated well. Together, we succeeded in achieving convenient, systemic, once daily dosing of compound 32 that results in sustained serum concentrations above the IC95.
After optimization of compound 32 PK, we then asked if compound 32 has anti-tumor efficacy in the orthotopic and syngeneic E0771 triple negative breast cancer model. We chose this model because E0771 cells basally export cGAMP in cell culture. In addition, we previously saw that E0771 cells implanted into mice grow more slowly in Enpp1−/− mice than in wild type mice (Carozza et al., 2020). We treated mice with established E0771 tumors (~100 mm3) with compound 32 using the optimized dosing schedule for seven consecutive days. Remarkably, we observed a delay in tumor growth in mice treated with compound 32 relative to the controls, which also led to prolonged survival. The tumor growth delay observed with pharmacological inhibition of ENPP1 mirrors that from genetic ablation of ENPP1 we previously reported (Carozza et al., 2020), demonstrating that our ENPP1 inhibitors are therapeutically beneficial.
Discussion
Here we report the development of highly potent phosphonate ENPP1 inhibitors. We demonstrate that they are active against the physiological substrate cGAMP under physiological conditions, including an in vitro assay at pH 7.4 and an ex vivo assay against ENPP1 in mouse and human plasma. Our compounds are by far the most potent inhibitors reported to date. Although some of the boronic acids (compounds 60 and 61) and hydroxamic acids (compound 62) are also potent, further investigation of their cell permeability and possible off-target effects would be needed. The lead phosphonate compounds, however, are cell impermeable through passive diffusion, avoiding all potential intracellular off-targets. We, therefore, nominate them as specific tool compounds to study ENPP1 and extracellular cGAMP biology.
To understand the potency of our compounds, we solved the crystal structure of ENPP1 with compound 15. We found that compound 15 adopts a similar binding pose as AMP, the product of cGAMP and ATP hydrolysis, but there are extra interactions that explain its enhanced potency including a direct hydrogen bond between the 7-methoxy and D308 (instead of water-mediated), extra hydrogen bond interactions with K277, and other hydrophobic and polar interactions with L272 and Y353. Our SAR of other phosphonate inhibitors allowed us to build a model of the drivers of potency. The π-π stacking interactions are key, as only a couple of the cores we tried resulted in potent inhibitors and the core region could be important for positioning of the zinc-binding phosphonate with respect to the π-π stacking tail. We hypothesize that moving the methoxy groups to different positions on the ring would also engage D308 and/or K277 in hydrogen bonding. Even though some of the positions on the ring are solvent-exposed in the compound 15 structure in complex with ENPP1, combinations of more substituents are detrimental to potency, suggesting that these compounds could be shifted in the binding pocket leading to steric hinderance when extra substituents are added. It is interesting to note that ENPP1 makes better interactions with the methoxy group, in contrast to the hydroxy or amine (e.g., compare compound 27 to compounds 29 and 30; compare compound 32 to compound 34), suggesting that aliphatic carbons are important in addition to hydrogen bonding. In addition, we can speculate about the importance of the 3-nitrile group off the quinoline, e.g. compound 47 and related analogs. Compared to the N3 on the quinazoline, it is possible that the nitrile can make stronger polar interactions with the protein backbone or surrounding residues (e.g., D200, Y353, or E355), act as a hydrogen bond acceptor, or polarize the quinoline ring for better π-π stacking.(Fleming et al., 2010) Previous modeling showed that nitriles can replace water-mediated azomethine-protein interactions (Tsou et al., 2005). This could extend to our scenario in replacing a weak polar interaction with a stronger one. In addition, it is surprising that the 2-vinyl-3-pyridyl substituent (compound 44) is potent, given that the 2 position faces the protein backbone. It is possible that this large substituent can slide into the narrow pocket between H242 and Y353 or could make a specific interaction with one of the residues. It also suggests that this space is not as available with the 8-methoxy substituent, as compound 45 (with 2-vinyl-3-pyridyl) was more than one hundred-fold less potent than compound 32 (without 2-vinyl-3-pyridyl).
In addition to unmatched potency, our ENPP1 inhibitors also have desirable ADME profiles in vitro, and we have measured the pharmacokinetics of one of the top inhibitors to demonstrate efficacious concentrations in mice with once daily systemic dosing. Compared to jump starting the anti-cancer innate immune response by treating with direct STING agonists, our ENPP1 inhibitors provide two advantages. First, although endogenous extracellular cGAMP enhances the anti-tumor immune response (Carozza et al., 2020), the same may not be true for cGAMP analogs. Because cGAMP is specifically transported into different cell types through different transporters (Cordova et al., 2020; Lahey et al., 2020; Luteijn et al., 2019; Ritchie et al., 2019; Zhou et al., 2020a), it is difficult to design cGAMP analogs that match the cell-targeting profile of cGAMP itself. It is important to target specific cells because STING activation in cancer cells promotes metastasis,(Bakhoum et al., 2018) and STING activation in T cells leads to T cell death (Cerboni et al., 2017; Gulen et al., 2017; Larkin et al., 2017), both of which would be detrimental to cancer patients. In contrast, ENPP1 inhibitors should increase the half-life of endogenous cGAMP and thus enhance the natural anti-tumor response. Indeed, treatment of mice with compound 32 delayed tumor growth in the E0771 breast cancer mouse model, which is a promising result for ENPP1 inhibitors as cancer therapeutics. Second, STING agonists can only be introduced intratumorally to achieve efficacy and to avoid systemic inflammation, which limits treatment to palpable and injectable tumors. Since our ENPP1 inhibitors can be administered systemically, we hypothesize that ENPP1 inhibitors could treat a wide variety of cancers and may even be effective against undetectable micrometastasis. They could also avoid causing widespread toxic interferon production, since they only enhance endogenous cGAMP. This hypothesis is supported by the fact that ENPP1-inactivating mutations in both humans and mice do not cause interferonopathy (Okawa et al., 1998; Rutsch et al., 2001). Our ENPP1 inhibitors are biological tools as well as candidate investigational drugs that have shown efficacy in mouse tumor models, and they hold the promise to potentiate the efficacy of radiation and other immune checkpoint inhibitors. Finally, since cGAMP has shown stunning results as an adjuvant for influenza vaccination (Wang et al., 2020), our lead ENPP1 inhibitor that prevents its extracellular degradation may have the potential to maximize cGAMP’s adjuvancy effect in developing vaccines for pandemic threats.
STAR Methods Text
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Dr. Lingyin Li (lingyinl@stanford.edu).
Materials Availability
This study did not generate any unique biological reagents. Synthesis of all chemicals is detailed within the manuscript.
Data and Code Availability
Coordinates and structure factors have been deposited and are available at the PDB website (https://www.rcsb.org/) under the accession PDB code 6XKD.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
Five- to nine-week-old female C57BL6/J mice (Jackson Laboratories) were maintained at Stanford University in compliance with Stanford University Institutional Animal Care and Use Committee regulations and procedures were approved by the Stanford University administrate panel on laboratory animal care. Mice were co-housed at 2–5 mice per cage and were maintained under a 12 hour light cycle (light from 7 am to 7 pm) and given unrestricted food and water supply.
Mammalian cell culture
All cells were maintained in a humidified incubator at 37 °C and 5% CO2. Primary human peripheral blood mononuclear cells (PBMCs) were isolated by subjecting enriched buffy coat from whole blood (obtained de-identified from the Stanford Blood Center) to a Percoll density gradient and cultured in RPMI (Cellgro) supplemented with 10% heat-inactivated FBS (Atlanta Biologicals), 100 U/mL penicillin-streptomycin (ThermoFisher), 50 μM 2-mercaptoethanol (Sigma) and 10 ng/mL M-CSF (PeproTech). HEK 293S GnT1− cells stably expressing secreted mouse ENPP1 (a gift from Dr. Osamu Nureki, University of Tokyo, Japan) were adapted to suspension and maintained in 2 L spinner flasks in media consisting of 50% [Freestyle (ThermoFisher) media] and 50% [DMEM with 10% FBS and 100 U/mL penicillin-streptomycin]. E0771 cells were cultured in RPMI (Cellgro) supplemented with 10% FBS, 100 U/mL penicillinstreptomycin, and 10 mM HEPES (Gibco). Caco-2 cells were cultured in DMEM (Cellgro) supplemented with 10% FBS, 100 U/mL penicillin-streptomycin, and MEM non-essential amino acids (Gibco).
METHOD DETAILS
Plasma kinetics and IC50 value determination
Blood from C57B6/J mice was obtained by cardiac puncture and deposited in heparin-coated tubes (BD Microtainer with PST additive). All procedures to obtain plasma after blood draw were performed at 4 °C. The blood was centrifuged at 2000 rcf for 20 minutes and plasma was isolated from the top layer. Human plasma was obtained de-identified from Innovative Research (sodium heparin anticoagulant). Plasma reactions included the following: 80% plasma, PBS (or PBS + 50 mM EDTA), and 5 μM cGAMP with trace [32P]cGAMP. [32P]cGAMP was synthesized enzymatically by incubating the following overnight at room temperature: 1 mM GTP, 1.67 μM (250 μCi) [32P]ATP, 50 μM recombinant sscGAS protein, and 100 μg/mL dsDNA (from herring testes; Sigma Aldrich) in a buffer containing 50 mM Tris pH 7.4 and 20 mM MgCl2. Following synthesis, the [32P] cGAMP product was separated from the reactants by prep TLC using a silica TLC plate (EMD Millipore) and 85% ethanol + 5 mM NH4HCO3 mobile phase. The eluate was subsequently run through a 3 kDa MWCO filter. For IC50 value assays, 5-fold dilutions of inhibitor in PBS were also included. At indicated times, reaction was quenched by spotting on a silica gel TLC plate (EMD Millipore). Plates were run in 85% ethanol + 5 mM NH4HCO3 mobile phase, dried, exposed on a phosphor screen (GE BAS IP MS), imaged on a Typhoon 9400, and quantified with ImageJ (Schneider et al., 2012). Half-life values were fit with Graphpad Prism 7.03.
Expression and purification of mouse ENPP1
The extracellular region (residues 92–905) of mouse ENPP1 was expressed and purified according to previous publication (Kato et al., 2012a). ENPP1 was expressed in suspension 293S GnT1− cells, removed from the media using affinity chromatography with the P20.1 antibody, and eluted with 0.2 mg/mL TARGET peptide (YPGQx5 + V) in 20 mM Tris pH 7.4, 150 mM NaCl. The protein was dialyzed into 50 mM Tris pH 8, 0.5 mM EDTA, and 1 mM DTT and cleaved with TEV protease before concentration to 500 μL and further purification using a Superose 12 10/300 GL column with buffer including 5 mM Tris pH 7.4, 150 mM NaCl. The protein was concentrated to 5 mg/mL, flash frozen, and stored at −80 °C.
Synthesis and purification of cGAMP
cGAMP was synthesized enzymatically (Ritchie et al., 2019) by incubating the following overnight at room temperature: 2 mM GTP, 2 mM ATP, 1 μM recombinant sscGAS protein, and 100 μg/mL dsDNA (from herring testes; Sigma Aldrich) in a buffer containing 50 mM Tris pH 7.4 and 20 mM MgCl2. The reaction was heated at 95C for 3 min and filtered through a 3-kDa filter. cGAMP was purified from the reaction mixture using a PLRP-S polymeric reversed phase preparatory column (100 Å, 8 mm, 300 3 25 mm; Agilent Technologies) on a preparatory HPLC (1260 Infinity LC system; Agilent Technologies) connected to UV-vis detector (ProStar; Agilent Technologies) and fraction collector (440-LC; Agilent Technologies). The flow rate was set to 25 mL/min. The mobile phase consisted of 10 mM triethylammonium acetate in water and acetonitrile. The mobile phase started as 2% acetonitrile for first 5 min. Acetonitrile was then ramped up to 30% from 5–20 min, then to 90% from 20–22 min, maintained at 90% from 22– 25 min, and then ramped down to 2% from 25–28 min. Fractions containing cGAMP were lyophilized and resuspended in water. The concentration was determined by measuring absorbance at 280 nm.
In vitro ENPP1 activity assays
Mouse ENPP1 (3 nM) was incubated with cGAMP (5 μM) and 5-fold serial dilutions of compounds in buffer containing 50 mM Tris pH 7.4, 250 nM NaCl, 500 uM CaCl2, and 1 uM ZnCl2 (total reaction volume = 10 μL). To determine the protein-shifted potency, 40 μM human serum albumin (Sigma) was included in the reaction. Reactions were incubated at room temperature for 3 hours and then heat inactivated at 95°C for 10 minutes. The AMP degradation product was converted to ATP using an enzyme mixture of polyphosphate:AMP phosphotransferase (PAP) and myokinase (MilliporeSigma), which was detected using luciferase (CellTiterGlo, Promega) according to previous publication (Mardjuki et al., 2020). PAP was diluted to 2 mg/mL in buffer containing 50 mM Tris pH 7.5, 0.1% NP-40. Myokinase was diluted to 2 KU/mL in buffer containing 3.2 mM ammonium sulfate pH 6.0, 1 mM EDTA, and 4 mM polyphosphate.
The heat-inactivated ENPP1 reaction was incubated with PAP (0.01 μg/μL) and myokinase (0.0075 U/μL) in buffer containing 40 mM Tris pH 7.5, 0.05 mg/mL Prionex, 5 mM MgCl2, 20 μM polyphosphate, and 0.15 g/L phenol red (for ease of pipetting) for 3 hours (total reaction volume = 20 μL). CellTiterGlo (20 μL) was added to the reaction according to manufacturer’s protocol at 1:1 ratio and luminescence was measured. Data were normalized to 100% enzyme activity (no compound) and 0% enzyme activity (no enzyme) before being fit to the function 100 / (1 + ([compound] / IC50) ). Ki values were calculated according to the Cheng-Prusoff equation Ki = IC50 / (1 + [substrate] / Km). Km values were determined by incubating increasing concentrations of cGAMP with trace [32P]cGAMP and ENPP1 and measuring initial rates of reaction by TLC and autoradiography (described above in “Plasma kinetics”). Data were plotted as substrate concentration vs. initial velocity (Michaelis-Menten plot) and fit for Km (Km = 240 μM for mouse ENPP1, substrate cGAMP, at pH 7.4) with Graphpad Prism 7.03.
Cell viability
Primary human PBMCs were plated in 384 well plates at 2000 cells/well incubated with 5-fold dilutions of compounds for 16 hours. Viability was measured with CellTiterGlo (Promega) and normalized to 100% viability (no compound) in Graphpad Prism 7.03.
Protein production and crystallography
Crystals of ENPP1 in complex with compound 15 were grown at 20 °C by the hanging-drop vapor-diffusion method by mixing 1 μL of the protein solution [5 mg/mL in 5 mM Tris·HCl (pH 8.0), 150 mM NaCl, and 0.2 mM ZnSO4] and 1 μL of the reservoir solution [17–19% (v/v) PEG600, 50 mM sodium acetate (pH 4.5), 50 mM magnesium acetate, 0.5% (w/v) polyvinylpyrrolidone and 1 mM compound 15]. Crystals were harvested and cryo-cooled in liquid nitrogen.
Multiple needle-like crystals were tested for X-ray diffraction which, when X-ray exposed, showed weak diffracting power. One needle crystal was isolated and used to collect a data set to a minimum d-spacing of around 3.2 Å. Data was collected at cryogenic temperature (100 K) at beamline 5.0.1 of the Advanced Light Source (ALS) Synchrotron (Berkeley, CA, USA) at a single 0.97741 Å wavelength. Data was reduced with Mosflm (Leslie and Powell, 2007) and scaled with SCALA (Evans, 2011) within the CCP4 suite (Winn et al., 2011). The crystal belonged to the trigonal space group P 31 and contained two polypeptide chains per asymmetry unit. Data collection and refinement statistics are listed in Table S3. The structure was solved by the molecular replacement method with Phaser (McCoy et al., 2007) using mouse ENPP1 (PDB code: 4GTW) polypeptide chain stripped from ligands, ions, and glycan chains, as the search model. Structural refinement was done using REFMAC (Murshudov et al., 1997) iteratively with visual inspection of electron density maps and manual adjustment of atomic coordinates in COOT(Emsley et al., 2010) until progression to convergence. The final refined structure shows an excellent agreement with reference protein data as shown by Ramachandran statistics (Table S4). Data collection statistics are derived from SCALA.(Evans, 2011) To calculate Rfree, 5% of the reflections were excluded from the refinement. Rsym is defined as Rsym = ΣhklΣi|Ii(hkl) - <I(hkl)>| / ΣhkIΣiIi(hkl). Data refinement statistics are derived from REFMAC (Murshudov et al., 1997). The final quality check was done with Procheck (Vaguine et al., 1999). Graphic renderings were prepared with Pymol.(DeLano, 2002)
As previously observed (Kato et al., 2012a), the structure could be traced unambiguously from residue 170 to 902. No electron density is observed for the two SMB-like domains (residues 92–169). The two independent polypeptide chains in the asymmetric unit consist of a catalytic domain (residues 190–578), a nuclease-like domain (residues 629–902), and two long linker regions, L1 and L2 (residues 170–189 and 579–628, respectively). Portions of the structure not seen in the electron density map and not modeled belong predominantly to the nuclease-like domain and linker L2 region: chain A, residues 506–512; 609–628; 681–689; 708–715; 725–735; 799–803; 848–855, and chain B, residues 507–511; 607–629; 681–692; 709–715; 726–734; and 848–855. The structure has N-glycosylated sites at Asn267, Asn323, and Asn567. The two zinc ions are bound within the active site of ENPP1 and coordinated by Asp358, His362, and His517 at one site, and by Asp200, Thr238, Asp405, and His406 at the other. In the nuclease domain, a calcium ion is coordinated by the side chains of Asp780, Asp782, Asp784, and Asp788 and the main chain carbonyl group of Arg786, forming an EF hand-like motif. Extra electron density unaccounted for by the model allowed placement of one copy of compound 15 in the substrate-binding pocket with the dimethoxyquinazoline ring sandwiched between the side chains of Phe239 and Tyr322 and the phosphonic acid group between the zinc atoms. In the final rounds of refinement, the ligand was modeled at 0.5 occupancy in the binding pocket of chain A. The very weak electron density of the cycloalkyl carbon atoms prevented to accurately model its pucker and therefore the piperidine ring was tentatively assigned a chair shape with the nitrogen and the 4-carbon lying out-of-plane. This seemed to be a reasonable choice as a survey of the Cambridge Structural Database(Groom et al., 2016) revealed that in three available quinazolinylpiperidinyl heterocycle crystal structures (CCDC codes: LUMVIA, PASNOP, ZAYQIC) the piperidine ring is in the chair conformation.
ADME assays
Cell permeability: Caco-2 cells were seeded at 50,000 cells/well into a 12-well plate with polycarbonate filter inserts (Costar) in DMEM supplemented with 20% FBS and allowed to grow and differentiate for 25 ± 4 days to form monolayers. Apparent permeability coefficients were determined for apical to basolateral and basolateral to apical directions. Test articles and reference compounds were dissolved in Hank’s balanced salt solution (HBSS) containing 25 mM HEPES to yield a final concentration of 10 μM. The assays were performed in HBSS at pH 7.4 for the basolateral side and pH 6.5 for apical side at 37 °C. Prior to the study, the monolayers were washed in prewarmed HBSS. At the start of the experiments, prewarmed HBSS containing the test articles was added to the donor side of the monolayer, and HBSS without test articles was added to the receiver side. Aliquots of the receiver side were taken over the 2-hour incubation period; aliquots of the donor side were taken at 0-hour and 2-hour time points. Aliquots were diluted with an equal volume of methanol/water with 0.1% formic acid containing the internal standard. The mixture was analyzed by LC−MS. The apparent permeability coefficients (Papp) were calculated using the formula: Papp = (dCrec/dt)/(A × C0,donor)] × 106 with dCrec/dt being the change in concentration in the receiver compartment with time; C0, donor the concentration in the donor compartment at time 0; and A the area of the cells monolayer.
Microsome stability: Incubation mixtures consisted of liver microsomes (BD, 0.5 mg microsomal protein/ml), substrates (1.0 mM), MgCl2 (10 mM), and NADPH (1 mM) in a total volume of 0.5 ml in potassium phosphate buffer (100 mM, pH 7.5). Reactions were started with the addition of NADPH and shaken in a water bath open to the air at 37°C. Aliquots (50 μL) were removed and quenched with acetonitrile containing internal standard at time points between 0 and 40 minutes. The samples were mixed and centrifuged; the resulting supernatant was diluted with mobile phase and analyzed by LC-MS. Plasma protein binding: Frozen plasma was thawed, mixed, and centrifuged. Plasma supernatant pH was adjusted to 7.5 with phosphoric acid. Compound was spiked into human or mouse plasma at 1 mM, mixed, and loaded into the sample chamber of the rapid equilibrium device (Thermo Fisher); PBS was loaded into the corresponding receiver chamber. Each compound was analyzed in triplicate. The plate was sealed and incubated on an orbital shaker at 37 °C for five hours. After the incubation period, the seal was removed, and chambers were visually checked to confirm no change in volumes. Aliquots from the chambers were removed and mixed with the corresponding matrix, plasma with PBS and PBS with blank plasma, to create analytically matched samples. To each sample, acetonitrile containing internal standard was added. Samples were mixed and centrifuged; the resulting supernatant was diluted with mobile phase and analyzed by LC-MS.
hERG channel inhibition assay: This assay was performed by Cyprotex. https://www.cyprotex.com/admepk/cardiotoxicity/hergsafety).
Mouse pharmacokinetics
Mice were injected via SC or retro-orbital IV route with 10 mg/kg of compound 32 (200 μL of 2 mg/mL dissolved in PBS). At indicated time points, mice were sacrificed and blood, kidneys, and livers were collected. Alternatively, mice were injected via SC with 300 mg/kg of compound 32 (200 μL of 60 mg/mL dissolved in PBS). At indicated time points, 50–100 μL of blood was collected from live mice via cheek puncture. Blood was allowed to clot at room temperature for 30 minutes before centrifuging at 1000×g to isolate serum. Organs were homogenized with 2.0 mm disruption beads (RPI) in Sarstedt tubes (Fisher, 50–809-242) using a tissue homogenizer (Bertin Precellys 24) in PBS. Organ homogenates and serum were precipitated with acetonitrile, centrifuged at 16,000×g, and resuspended in a matrix of 2:1 0.1% formic acid:acetonitrile with clemizole as the internal standard for analysis by LC-MS.
LC-MS Analysis
The compounds were separated with a gradient of water containing 0.1% formic acid (mobile phase A) and acetonitrile containing 0.1% formic acid (mobile phase B); starting at 0% B for 1 minute, then ramping to 65% B by 2.5 minute, holding at 65% B until 3.5 minute, and returning to 0% B by 3.6 minute. Total run time was 5 minutes, and the flow rate was at 0.5 mL/minute. The reverse phase Synergi Hydro-RP 30 × 2 mm column (Phenomenex, Torrance, CA) was at ambient temperature, connected to a binary pump and autosampler (Shimadzu Corp, Pleasanton, CA) and mass spectrometer (AB Sciex QTRAP 4000, Redwood City, CA). Compounds were detected in electrospray positive mode. Compounds were analyzed using Analyst 1.6.3 (AB Sciex, Redwood City, CA) by dividing compound peak area by the internal standard peak area.
Mouse tumor models
Five- to nine-week-old female C57BL6/J mice (Jackson Laboratories) were inoculated with 5 × 104 E0771 cells suspended in 50 μL of PBS in the fifth mammary fat pad. When tumor volume (determined by length2 × width/2) reached 100 ± 20 mm3, mice were randomized to achieve groups with equal average tumor volumes and were injected via SC route with 300 mg/kg of compound 32 as described above, once daily for seven consecutive days. Tumor volumes were recorded up to the humane endpoint (tumor volume exceeding 1000 mm3), when mice were sacrificed. Animal death was plotted with GraphPad Prism 7.03, and statistical significance was assessed by the Gehan-Breslow-Wilcoxon test.
QUANTIFICATION AND STATISTICAL ANALYSIS
In ENPP1 inhibition assays, the half maximal inhibitory concentrations (IC50) were obtained by sigmoidal dose-response fitting with Prism software. In cGAMP degradation assays, half-life was obtained by one phase exponential decay fitting with Prism software. Graphs show means and standard deviation (±SD) or standard error of the mean (±SEM), or individual data points as indicated in the figure legends. Statistical significance, group size, and experimental details are described in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Human plasma, single donor | Innovative Research | Cat#IPLASNAH2ML |
| Human buffy coat | Stanford Blood Center | N/A |
| UltraPool human liver microsomes | BD | Cat#452117 |
| Male mouse microsomes (CD-1) | BD | Cat#452701 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FreeStyle 293 Expression Medium | ThermoFisher | Cat#12338018 |
| TARGET peptide (YPGQx5 + V) | custom synthesis by Stanford PAN facility | N/A |
| DMEM | Corning | Cat#MT10013CV |
| RPMI 1640 | Corning | Cat#10-040-CV |
| FBS | Atlanta Biologics | Cat#S11150 |
| Penicillin/streptomycin | ThermoFisher | Cat#15140163 |
| HEPES (1 M) | Gibco | Cat#15-630-080 |
| 2’3’-cGAMP | This paper | N/A |
| Recombinant sscGAS | (Ritchie et al., 2019) | N/A |
| [α-32P]ATP, 250 μCi | PerkinElmer | Cat#BLU003H250UC |
| GTP, disodium salt hydrate | Sigma Millipore | Cat#G8877-1G |
| Herring testes DNA | Sigma Millipore | Cat#D6898-1G |
| HP-TLC silica gel aluminum | EMD Millipore | Cat#1.05548.0001 |
| Human serum albumin | Sigma Millipore | Cat#A3782-500MG |
| polyphosphate:AMP phosphotransferase (PAP) | (Mardjuki et al., 2020) | N/A |
| Myokinase | Sigma Millipore | Cat#M3003-2.5KU |
| Mouse ENPP1 | This paper | N/A |
| 2-mercaptoethanol | Fisher Scientific | Cat#AC125472500 |
| Human GM-CSF | PeproTech | Cat#300-03-5ug |
| Compounds 1–62, detailed synthesis described in Supplemental Information, Data S1 | This paper | N/A |
| Critical Commercial Assays | ||
| CellTiterGlo | Promega | Cat#G8461 |
| Deposited Data | ||
| ENPP1-compound 15 structure | This paper | PDB: 6XKD |
| ENPP1-AMP structure | (Kato et al., 2012) | PDB: 4GTW |
| Experimental Models: Cell Lines | ||
| HEK293S GnT1− stably expressing mouse ENPP1 | (Kato et al., 2012) | N/A |
| E0771 | CH3 BioSystems | Cat#940001 |
| Caco-2 | ATCC | Cat#HTB-37 |
| Software and Algorithms | ||
| Prism 7.03 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Mnova 14.1 | Mestrelab | https://mestrelab.com/software/mnova/ |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Mosflm | (Leslie and Powell, 2007) | https://www.mrc-lmb.cam.ac.uk/harry/imosflm/ver721/imosflm_link/ver730/introduction.html |
| CCP4 | (Winn et al., 2011) | http://www.ccp4.ac.uk/ |
| SCALA | (Evans, 2011) | http://www.ccp4.ac.uk/html/scala.html |
| REFMAC | (Murshudov et al., 1997) | http://www.ccp4.ac.uk/html/refmac5.html |
| COOT | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Procheck | (Vaguine et al., 1999) | https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ |
| Pymol | (DeLano, 2002) | https://www.pymol.org/2/ |
| Other | ||
| Superose 12 10/300 GL | GE Healthcare | GE17-5173-01 |
| Rapid Equilibrium Device | Thermo Fisher | Cat#99006 |
Significance.
The extracellular enzyme ENPP1 is a promising immunotherapy target for treating cancers. ENPP1 degrades the extracellular immunotransmitter cGAMP, thus dampening the anti-cancer innate immune response. We developed the first cell-impermeable phosphonate ENPP1 inhibitors to target this extracellular enzyme. Their impermeability avoids intracellular off-targets while retaining the inhibitor in the extracellular space to stay on-target. Our compounds are the most potent reported and have high specificity. We also solved the co-crystal structure of ENPP1 with compound 15, one of our phosphonate inhibitors, demonstrating that the inhibitor makes contact with active site residues not involved in binding natural substrates. Finally, our inhibitor compound 32 can be dosed systemically and has efficacy in murine tumor models. These inhibitors will be invaluable as tools to study regulation of extracellular cGAMP by ENPP1 and as potential therapeutics.
Highlights.
ENPP1 inhibitors are designed as extracellular cell-impermeable inhibitors.
SAR studies lead to highly potent ENPP1 inhibitors at physiological conditions.
Crystal structure of ENPP1 : inhibitor complex reveals key interactions
Systemic dosing of ENPP1 inhibitor delays tumor growth in mice
Acknowledgments
This work was performed in collaboration with the ChEM-H Medicinal Chemistry Knowledge Center. We thank Dr. Sam Banister, Stephen R. Stabler, Dr. Phil Thomson, and members of the ACME Biosciences synthetic chemistry team for compound synthesis; Khanh Nguyen and Grace Lam for in vitro ADME assays and LC-MS/MS analysis; beamline staff at the Advanced Light Source for technical support during diffraction data collection; and Dr. Dan Herschlag and all members of the Li lab and Medicinal Chemistry Knowledge Center for helpful discussions. This research was supported by the Stanford ChEM-H Medicinal Chemistry Knowledge Center and Macromolecular Structure Knowledge Center and the National Institutes of Health award DP2CA228044 (L.L.), DOD grant W81XWH-18–1-0041 (L.L.), Ono Pharma Foundation Grant (L.L.), NIH fellowship F31CA239510 (J.A.C.), Xu Family Foundation Stanford Interdisciplinary Graduate Fellowship affiliated with ChEM-H (J.A.C.), and National Science Foundation graduate fellowship DGE-1656518 (J.A.B. and R.E.M.). This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02–05CH11231.
Footnotes
Declaration of Interests
M.S. and L.L. are scientific cofounders of Angarus Therapeutics, which has exclusive licensing rights to patents PCT/US2018/50018 and PCT/US2020/015968. J.A.C., V.B., M.S., and L.L. are inventors on patents PCT/US2018/50018 and PCT/US2020/015968.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner K-P, Ludwig J, and Hornung V (2013). cGAS produces a 2′−5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, and Cantley LC (2018). The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 174, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belli S, van Driel I, and Goding J (1993). Identification and characterization of a soluble form of the plasma cell membrane glycoprotein PC-1. Eur. J. Biochem 217, 421–428. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, and Pedersen SF (2020). The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 82, annurev-physiol-021119–034627. [DOI] [PubMed] [Google Scholar]
- Carozza JA, Böhnert V, Nguyen KC, Skariah G, Shaw KE, Brown JA, Rafat M, von Eyben R, Graves EE, Glenn JS, et al. (2020). Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nat. Cancer 1, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg M-C, Picard C, Neven B, Fischer A, Amigorena S, et al. (2017). Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med. 214, 1769–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Lee SY, Leonczak P, Rozenski J, De Jonghe S, Hanck T, Müller CE, and Herdewijn P (2014). Imidazopyridine- and purine-thioacetamide derivatives: Potent inhibitors of nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1). J. Med. Chem. 57, 10080–10100. [DOI] [PubMed] [Google Scholar]
- Cordova AF, Ritchie C, Böhnert V, Mardjuki RE, and Li L (2020). Murine M1 macrophages are among the direct responders to tumor-derived extracellular cGAMP and their human counterparts use SLC46A2 to import cGAMP. BioRxiv https://www.biorxiv.org/content/10.1101/2020.04.15. [Google Scholar]
- Corrales L, Matson V, Flood B, Spranger S, and Gajewski TF (2017). Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 27, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002). Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 40, 82–92. [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR (2011). An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming FF, Yao L, Ravikumar PC, Funk L, and Shook BC (2010). Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcellini E, Boutin S, Lefebvre CA, Shayhidin EE, Boulanger MC, Rhéaume G, Barbeau X, Lagüe P, Mathieu P, and Paquin JF (2018). Synthesis and biological evaluation of novel quinazoline-4-piperidinesulfamide derivatives as inhibitors of NPP1. Eur. J. Med. Chem. 147, 130–149. [DOI] [PubMed] [Google Scholar]
- Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, and Gajewski TF (2011). Host type I IFN signals are required for antitumor CD8 + T cell responses through CD8α + dendritic cells. J. Exp. Med. 208, 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom CR, Bruno IJ, Lightfoot MP, and Ward SC (2016). The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, and Ablasser A (2017). Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen S, Perrakis A, Ulens C, Winkler C, Andries M, Joosten RP, Van Acker M, Luyten FP, Moolenaar WH, and Bollen M (2012). Structure of NPP1, an ectonucleotide pyrophosphatase/phosphodiesterase involved in tissue calcification. Structure 20, 1948–1959. [DOI] [PubMed] [Google Scholar]
- Kato K, Nishimasu H, Okudaira S, Mihara E, Ishitani R, Takagi J, Aoki J, and Nureki O (2012a). Crystal structure of Enpp1, an extracellular glycoprotein involved in bone mineralization and insulin signaling. Proc. Natl. Acad. Sci. U. S. A. 109, 16876–16881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Nishimasu H, Mihara E, Ishitani R, Takagi J, Aoki J, and Nureki O (2012b). Expression, purification, crystallization and preliminary X-ray crystallographic analysis of Enpp1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Nishimasu H, Oikawa D, Hirano S, Hirano H, Kasuya G, Ishitani R, Tokunaga F, and Nureki O (2018). Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat. Commun. 9, 4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahey LJ, Xi Wen., Mardjuki RE, Böhnert V, Ritchie C, Carozza JA, Hess GT, Maduke M, Bassik MC, and Li L. (2020). The LRRC8A:C heteromeric channel is a cGAMP transporter and the dominant cGAMP importer in human vasculature cells. BioRxiv https://www.biorxiv.org/content/10.1101/2020.02.13. [Google Scholar]
- Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, and Poltorak A (2017). Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J. Immunol. 199, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-Y, Sarkar S, Bhattarai S, Namasivayam V, De Jonghe S, Stephan H, Herdewijn P, El-Tayeb A, and Müller CE (2017). Substrate-dependence of competitive nucleotide pyrophosphatase / phosphodiesterase1 (NPP1) inhibitors. Front. Pharmacol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Perotti A, De Jonghe S, Herdewijn P, Hanck T, and M??ller CE. (2016). Thiazolo[3,2-a]benzimidazol-3(2H)-one derivatives: Structure-activity relationships of selective nucleotide pyrophosphatase/phosphodiesterase1 (NPP1) inhibitors. Bioorganic Med. Chem. 24, 3157–3165. [DOI] [PubMed] [Google Scholar]
- Lefebvre C-A, Forcellini E, Boutin S, Côté M-F, Gaudreault CR, Mathieu P, Lagüe P, and Paquin J-F. (2017). Synthesis of novel substituted pyrimidine derivatives bearing a sulfamide group and their in vitro cancer growth inhibition activity. Bioorg. Med. Chem. Lett. 27, 299–302. [DOI] [PubMed] [Google Scholar]
- Leslie AGW, and Powell HR (2007). Evolving Methods for Macromolecular Crystallography: Processing Diffraction Data with Mosflm. [Google Scholar]
- Li L, Yin Q, Kuss P, Maliga Z, Millán JL, Wu H, and Mitchison TJ (2014). Hydrolysis of 2’3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 10, 1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, Mcwhirter SM, Katibah GE, Corn JE, Woodward JJ, et al. (2019). SLC19A1 transports immunoreactive cyclic dinucleotides. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, and Raulet DH (2018). Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardjuki RE, Carozza JA, and Li L (2020). Development of cGAMP-Luc, a sensitive and precise coupled enzyme assay to measure cGAMP in complex biological samples. J. Biol. Chem. jbc.RA119.012170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyre D, Bouatia-Naji N, Tounian A, Samson C, Lecoeur C, Vatin V, Ghoussaini M, Wachter C, Hercberg S, Charpentier G, et al. (2005). Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat. Genet. 37, 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, and Ikegawa S (1998). Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Genet. 19, 271–273. [DOI] [PubMed] [Google Scholar]
- Patel SD, Habeski WM, Cheng AC, de la Cruz E, Loh C, and Kablaoui NM (2009). Quinazolin-4-piperidin-4-methyl sulfamide PC-1 inhibitors: Alleviating hERG interactions through structure based design. Bioorganic Med. Chem. Lett. 19, 3339–3343. [DOI] [PubMed] [Google Scholar]
- Ritchie C, Cordova AF, Hess GT, Bassik MC, and Li L (2019). SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol. Cell 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B, Schauerte P, Kalhoff H, Sano K, Boisvert WA, Superti-Furga A, et al. (2001). PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am. J. Pathol. 158, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, Yagita H, Guggisberg E, Saba S, Nascakova Z, et al. (2019). Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 29, 1236–1248.e7. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayhidin EE, Forcellini E, Boulanger M-C, Mahmut A, Dautrey S, Barbeau X, Lagüe P, Sévigny J, Paquin J-F, and Mathieu P (2015). Quinazoline-4-piperidine sulfamides are specific inhibitors of human NPP1 and prevent pathological mineralization of valve interstitial cells. Br. J. Pharmacol. 172, 4189–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science (80-. ). 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supe L, Afzal S, Mahmood A, Ejaz SA, Hein M, Iaroshenko VO, Villinger A, Lecka J, Sévigny J, Iqbal J, et al. (2018). Deazapurine Analogues Bearing a 1H-Pyrazolo[3,4-b]pyridin-3(2H)-one Core: Synthesis and Biological Activity. European J. Org. Chem. 2018, 2629–2644. [Google Scholar]
- Topalian SL, Hodi SM, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. NEJM 366, 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou HR, Overbeek-Klumpers EG, Hallett WA, Reich MF, Floyd MB, Johnson BD, Michalak RS, Nilakantan R, Discafani C, Golas J, et al. (2005). Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 48, 1107–1131. [DOI] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaguine AA, Richelle J, and Wodak SJ (1999). SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. Sect. D Biol. Crystallogr. 55, 191–205. [DOI] [PubMed] [Google Scholar]
- Vaupel P, Kallinowski F, and Okunieff P (1989). Blood Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human Tumors: A Review. Cancer Res. 49, 6449–6465. [PubMed] [Google Scholar]
- Wang J, Li P, Yu Y, Fu Y, Jiang H, Lu M, Sun Z, Jiang S, Lu L, and Wu MX (2020). Pulmonary surfactant–biomimetic nanoparticles potentiate heterosubtypic influenza immunity. Science (80-. ). 367, eaau0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo S-R, Corrales L, and Gajewski TF (2015). Innate Immune Recognition of Cancer. Annu. Rev. Immunol. 33, 445–474. [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (80-. ). 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelikman V, Pelletier J, Simhaev L, Sela A, Gendron F, Arguin G, Senderowitz H, and Se J (2018). Highly Selective and Potent Ectonucleotide Pyrophosphatase-1 (NPP1) Inhibitors Based on Uridine 5’-Pa,a-Dithiophosphate Analogues. J. Med. Chem. 1. [DOI] [PubMed] [Google Scholar]
- Zhou C, Chen X, Planells-Cases R, Chu J, Wang L, Cao L, Li Z, López-Cayuqueo KI, Xie Y, Ye S, et al. (2020. a). Transfer of cGAMP into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-viral Immunity. Immunity 52, 1–15. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Fei M, Zhang G, Liang WC, Lin WY, Wu Y, Piskol R, Ridgway J, McNamara E, Huang H, et al. (2020. b). Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 52, 357–373.e9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited and are available at the PDB website (https://www.rcsb.org/) under the accession PDB code 6XKD.