

Abstract
Delayed-release dosage forms are mainly manufactured as batch processes and include coated tablets, pellets, or particles with gastric resistant polymers. Authors propose a novel approach using the hot-melt extrusion technique to prepare delayed release dosage forms via a continuous manufacturing process, a new trend in the pharmaceutical industry. A full factorial design was employed to correlate input variables, including stearic acid (SA) content, drug content, and pellet size with drug release properties of the pellets. PLS fit method suitably elaborated the relationship between input and output variables with reasonably good fit and goodness of prediction. All three input factors influenced drug release in enzyme-free simulated gastric fluid (SGF) after 120 min; however, SA content did not significantly affect drug dissolution in the enzyme-free simulated intestinal fluid (SIF). An optimized formulation and design space were determined by overlaying multiple contours established from regression equations. The continuous manufacturing process was successfully monitored using inline near-infrared (NIR) and inline particle size analysis, with drug load and pellet size being well-controlled within the design space. The obtained pellets released less than 5% after 120 min in SGF and more than 85% and 95% after 30 min and 45 min, respectively, after switching to SIF.
Keywords: Continuous manufacturing, Delayed-release, FT-NIR, Inline particle size analysis, Hot melt extrusion
1. Introduction
Encouraged by both regulatory bodies and pioneer manufacturers, continuous manufacturing (CM) is emerging as a new trend in the pharmaceutical industry, owing to its advantages.1, 2, 3, 4, 5 By the end of 2018, five drug products manufactured using CM from four pharmaceutical companies had been approved by the US FDA.6 In addition, 20 other companies are working with the FDA to develop and implement CM processes.7 Adopting CM for pharmaceutical production is expected to reduce drug product quality issues, lower manufacturing costs, ease scale-up, and improve availability of quality medicines to patients.8 CM requires in-depth understanding of the process, and the critical quality attributes are continuously tested in real time; therefore, a large amount of information is acquired and available for quality evaluation, allowing better control of drug quality.9 There is no interruption, and human involvement in activities is limited; hence, human-associated mistakes are minimized. Another advantage of CM is the ease of scaling up, which is currently a bottleneck in process development for batch type production.10 The scaled up production performs on the same instrumentation by increasing throughput, running for a longer duration, or adding up operation units instead of enlarging the volume of the machines.11 More importantly, it allows faster response to changes in demand, and it can bring down the cost of products and capital investment. CM uses relatively small operation units, has fewer steps, and does not need volume holders for semi-finished products; thus, it requires a smaller footprint facility that can reduce the required investment.12
In CM, critical quality attributes and processing parameters need to be monitored and controlled inline, online, or at-line throughout the manufacturing process. Two important components of CM are design space and process analytical technology (PAT) tools.13, 14 Unlike batch-wise manufacturing, CM processes are dynamic systems where, during normal operation, critical process parameters and quality attributes are controlled within predefined operation ranges. PAT tools collect information and provide information related to the process in real time, which is utilized to judge product quality. The information can also be used to control the process through a feedback mechanism where returning information is used to calculate adjustments needed to bring the process back to center levels. Subsequently, the corresponding operation machine performs the calculated adjustments that ultimately minimize fluctuation and make the process more stable.15 Many PAT tools have demonstrated the capability for real-time release testing such as near-infrared (NIR), Raman spectroscopy, spatial filter velocimetry, focused beam reflectance measurement (FBRM), high-speed imaging, photometric imaging, microwave resonance sensing, acoustic emission, and laser diffraction.16
Hot-melt extrusion (HME) is widely appreciated as a robust technique to formulate highly viscous formulations without any solvents.17 HME is an inherently continuous process where multiple functions can be performed on a single operation unit that allows reducing operation steps and shortening manufacturing time.18 The application of design space and incorporation of PAT tools for process monitoring enable the development of HME as a CM platform.19, 20 HME is established as a versatile technique for various drug delivery strategies, by which many products have reached the market.21 Production of pellets is a predominant example showing the advantage of HME among successfully investigated dosage forms. HME enables the production of uniformly high drug-load pellets in fewer steps. Moreover, pellets can be manufactured continuously by coupling HME with die-surface cutting pelletization17, 22, 23 or extruded strands cutting palletization.24, 25
Nonsteroidal anti-inflammatory drugs are a widely used category of drugs worldwide, and are reported to have high risk of gastric issues such as mucosal irritation, gastrointestinal (GI) tract ulceration, and gastrointestinal hemorrhage.25, 26, 27, 28 Formulation of nonsteroidal anti-inflammatory drugs as delayed release dosage forms is the most popular approach to mitigate gastro duodenal ulcers and minimize other adverse effects of the active pharmaceutical ingredients (APIs). Usually, APIs are formulated in the form of pellets or tablets, followed by coating with a gastric resistant polymer such as shellac, Eudragit® L100, hydroxypropyl methylcellulose phthalate (HPMCP), or hydroxypropyl methylcellulose acetate succinate (HPMCAS). Such a process is time consuming, uses solvents, and requires intensive control to ensure the formation of uniform thin films on every unit dose. Multi-unit dosage forms such as pellets, particles, and microspheres are safer and more effective than monolithic dosage forms, as they can avoid “all or nothing” effects, and drugs can be quickly released and spread in the intestinal tract.29
HME has been utilized to formulate various gastric resistant dosage forms such as HME matrices loaded with pre-prepared delayed release granules,30 HME gastric resistance capsules,31 gastric resistance sustained release pellets,32, 33 and gastric resistance sustained release HME tablets.34 However, some of these processes require multiple steps including coating granules or pellets that prevent them from being integrated into a continuous production line. Others show a slow drug release in intestinal dissolution media making these dosage forms resemble sustained release forms rather than real delayed release dosage forms. This study proposed a novel approach to formulate delayed release matrix pellets via one step using HME platform. A new paradigm of continuous manufacturing process was developed systematically combining design space, inline monitoring and real time control. An experimental design was used to elaborate the impact of critical attributes on gastric resistance potential and dissolution in the intestinal medium. HME palletization was developed as a CM process where an inline FT-NIR and a Parsum™ particle size analyzer were used as PAT tools to monitor the process. This study also focused on the application of quality by design (QbD) design space and real-time release testing strategies to monitor the manufacturing process.
2. Materials and Methods
2.1. Materials:
Ketoprofen USP standard (KTP), a nonsteroidal anti-inflammatory drug, was procured from ScienceLab Inc. (Houston, TX, USA). Stearic acid (SA) (USP/NF standard) was purchased from Alfa Aesar Chemicals (Tewksbury, MA, USA). Eudragit® L100 and Eudragit® L100-55, polyacrylic acids (USP/NF standard), were generously gifted by Evonik Industries AG (Essen, Germany). Other high-performance liquid chromatography (HPLC) grade solvents and analytical-grade chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA)
2.2. Thermal analysis
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to confirm the compatibility of the API and the excipients as well as their thermal stability during extrusion. The pure components and physical mixtures were subjected to TGA using a Pyris 1 system (Perkin Elmer, Waltham, MA, USA). A sample amount of 5 – 10 mg was loaded into a platinum pan. Samples were then heated from 25 °C to 200 °C at a ramp rate of 10 °C/min under an inert atmosphere created by purging ultrahigh-purity nitrogen at a flow rate of 20 mL/min. The sample weight was monitored and plotted against the chamber temperature.
DSC experiments were performed using a Discovery DSC 2500 system (TA Instrument, New Castle, DE, USA) to confirm the TGA results. Samples weighing 5–10 mg were loaded and sealed in hermetic aluminum pans. Samples were equilibrated at 0 °C for 2 min, followed by heating from 0 °C to 200 °C at a ramp rate of 10 °C/min in an ultrahigh purity nitrogen environment at a cell purge flow of 50 mL/min and a base purge flow of 300 mL/min. The heat flow difference between the sample pan and empty pan was monitored and plotted against the temperature change using Trios software (TA Instrument, DE, USA). The thermograms were analyzed for unanticipated thermal events.
2.3. Extrusion process
Prior to weighing, raw materials were sieved separately through a USP #45 mesh screen to remove aggregates and clumps. For each formulation, the extrusion process was performed with 80 g of the physical mixture. SA in the formulations was first ground and mixed with Eudragit® L100-55 following the equal mass mixing principle using a mortar and pestle. The mixture was then mixed with KTP using a V-shell blender (Maxiblend, GlobePharma, NJ, USA) at 20 rpm for 10 min before loading into a volumetric feeder. Pellets manufacturing was performed using a twin-screw co-rotating extruder (Process 11™, Thermo Fisher Scientific, TX, USA) equipped with a Thermo Scientific standard screw configuration, a volumetric feeder, a conveyor belt, and a pelletizer (Varicut Pelletizer, Thermo Fisher Scientific GmbH, Germany), as shown in Figure S1. Except the feeding zone, which was cooled with water recirculation from a chiller set at 15 °C, all other zones on the barrel and die were set at 110 °C. The extrusion was performed at a screw speed of 100 rpm and feed rate of 100 g/h. Prior to processing, the extruder barrel and die were heated to 110 °C and allowed to reach thermal equilibrium for 15 min. For each experiment, the first 30 g of extrudate was discarded, and pellets were collected during a steady state of extrusion. About 30 g pellets of each formulation was collected for further characterization. The pellet size was controlled by the diameter of the die insert, conveyor speed, and pellet cut length. Once leaving the die, the hot-softened extrudate was stretched by adjusting the conveyor belt and the pelletizing speed to synchronize with the extrudate formation rate. When passing through the conveyor belt, the extrudate strand was cooled and hardened prior to entering the pelletizer. The extruded pellets were stored in tight plastic bags at 20–25 °C until further characterization.
2.4. Drug load monitoring
The drug content in the extrudate was monitored in real time using an FT-NIR spectroscopy method previously developed in our lab.35 Briefly, NIR spectra of the melt extrudate were acquired inline using an FT-NIR spectrometer (Antaris II MDS, Thermo Fisher Scientific, USA) equipped with a diffuse reflectance probe mounted on the die of the extruder to collect near-infrared light reflected/scattered back from the in-process formulations. Because the melted extrudates were relatively transparent, the return signals were weak and did not stand out from the noise; hence, a stainless steel reflector was customized and positioned opposite to and distanced 2 mm from the NIR probe to enhance intensity of the collected signal. A background reference spectrum was acquired using the transmission sampling module at the beginning of each experiment set. NIR spectra were collected every 20 s at a spectral range of 4000–10 000 cm−1, 8 cm−1 spectral resolution, and 16 scans co-added (8 s scan time). The raw spectra were first transformed using the Norris second derivative to remove the effects of baseline drift and then subjected to a standard normal variant to minimize the effects of spectral path length variation. A partial least squares (PLS) model was derived to estimate the ketoprofen concentration using TQ Analyst™ software. All spectral data were mean-centered in the PLS regression, and the correlation was established using a spectral range from 5500 to 6650 cm−1. Significant model factors of the derived PLS were cross-validated using the leave-one-out approach.
2.5. Pellet size monitoring
The pellet size distribution was determined using the spatial filtering velocimetry technique, which measures the chord length of particles passing through an optical analysis window. Briefly, after pelletization, the obtained pellets are led to a particle size analysis system via a perforated incline functioning as a dedusting station. The pellet size was determined using an inline particle size analysis probe (Parsum™ IPP 80-P, Parsum GmbH, Germany) equipped with a pneumatic dilution unit activated by an internal air flow. The pellets were sucked into the dispersion unit and accelerated by the internal air flow. The air supply for the inline particle size analysis system was set at a flow rate of 10 L/min. The bursting air interval was set to 1 s every 2 min to unblock any obstruction in the dispersion unit. The coincidence and buffer size parameters were set at 5% and 1500 counts, respectively. Sample loading was equal to the pellet production throughput (i.e., 1.7 g/min) as all the pellets were introduced into the analysis window. The adjustment factor converting chord length to pellet size was evaluated and set at 1.2. The signals were acquired using ParsumRuntime V8.01 and processed using the LogAnalyzer macro (Parsum GmbH, Germany) to calculate volume-based particle size distribution expressed by the percentile of pellet population (e.g., D10, D50, D90).
2.6. Experimental design
A full factorial design was applied to investigate the effects of drug load, SA content, and pellet size on the drug release profile of the extruded pellets (drug release after 2 h in pH 1.2 medium and dissolution after 30 min and 45 min in pH 6.8 buffer medium). The variation ranges of drug load (40–60%), SA content (3.0–6.0%) and pellet size (0.5–1.5 mm) were determined through a series of preliminary experiments (more information can be found at Supplement Table S1). The correlation between the independent variables and output variables was determined by fitting experimental data to a mathematical model, and the goodness of fit was evaluated by statistical analysis. The regression equation is expressed as follows:
where Yi is the dependent variable number i, Xj is the encoded independent variable number j, Ai is the intercept, and Bij and Cijk are coefficients of variables Xj and Xj.Xk, respectively, corresponding to Yi. One-way analysis of variance (ANOVA) was used to evaluate the goodness of fit, goodness of model prediction, and the significance of each independent variable. Design space was determined by overlaying multiple contours established under the constraints of the output variables to find a common range of independent variables satisfying all performance requirements of the dosage form. The experiment plan and data analysis were performed using Modde 8.0 (Umetrics Inc., Sweden).
2.7. Assay
Drug content in the formulations was determined following the Ketoprofen capsule monograph of USP 35. Briefly, 2 g of the prepared pellets was ground into a fine powder using a mortar and pestle. A sample amount equivalent to approximately 100 mg of KTP was accurately weighed and transferred into a 100-mL volumetric flask. Mobile phase (70 mL) was added into the flask and sonicated for 5 min (Branson 2510, Branson Ultrasonic Corp., Danbury, CT, USA). The mobile phase was added up to the flask volume and agitated. Samples were then centrifuged at 12 000 rpm (equal to centrifugal force of 15 000 g) for 10 min at 25 °C (Centrifuge 5415R, Eppendorf AG, Germany), and diluted 50 times with the mobile phase before performing HPLC analysis.
HPLC analysis was performed at ambient temperature (20–25 °C) using a Waters 600 HPLC system (Waters Corp., Milford, MA, USA) equipped with an autosampler, UV/VIS detector, and a Phenomenex® Luna C18 column (5 mm, 250 mm × 4.6 mm). The HPLC mobile phase was a mixture of acetonitrile, water, and glacial acetic acid at a 90:110:1 ratio (v/v). HPLC parameters were set as follows: elution rate 1.0 mL/min, injection volume 20 mL, and detection wavelength 254 nm. Data were acquired and processed using the Power software suite.
2.8. Dissolution
The delayed release pellets (unit dose equal to 50 mg KTP) were subjected to a bi-phase dissolution study as required by USP for delayed release dosage forms (USP/<711> Dissolution/Delayed-Release Dosage Forms). The first step was conducted with 750 mL of fasted-state simulated gastric fluid (SGF; solution HCl 0.1 N, pH 1.2) for the first 120 min and then switched to 1000 mL simulated intestinal fluid (SIF; phosphate buffer pH 6.8) for another 60 min. The test was performed using USP dissolution apparatus type II (Hanson SR8; Hanson Research, Chatsworth, CA, USA) at a temperature of 37 ± 0.5 °C and paddle rotation speed of 50 rpm. Dissolution samples (2.0 mL) were withdrawn at each predetermined time point, and 2.0 mL fresh dissolution medium was added for compensation. The dissolution samples were subsequently filtered through 0.2 mm, 13 mm PTFE membrane filters (Whatman Inc., MA, USA). After discarding the first 0.5 mL, filtered samples were diluted with an equal volume of mobile phase, and then, 20 μL diluted sample was injected into the HPLC system for determination of KTP concentration following the HPLC method as described above.
3. Results and Discussion
3.1. Preliminary study
Eudragit® L100 and Eudragit® L100-55 gastric resistant polymers, are widely used to formulate delayed release dosage forms by coating on tablets, pellets, or granules. These polymers are thermally stable and suitable for hot-melt extrusion up to 200 °C.36 Different from its sibling, Eudragit® L100-55 has a lower glass transition temperature (Tg) (111 °C compared with 195 °C), which allows the process of Eudragit® L100-55-based HME formulations at lower temperatures. In addition, Eudragit® L100-55 is soluble at lower environmental pH, which could facilitate drug dissolution more quickly after being dislodged from the stomach. SA is a low-melting-point fatty acid (about 70 °C) that serves as a lubricant/processing aid agent in hot-melt extrusion.37 It can also be utilized to modify the hydrophobicity of matrixes, and thus, to tailor the drug release profile.29
The extrusion process was performed at the lowest processable temperature (110 °C) aiming to minimize thermal degradation of the formulation. This temperature was still higher than the melting temperature of KTP (92 °C), hence the extrusion process follows a miscible regime17. When the operation temperature ≤ 100 °C the extruded strand became less stretchable causing the pellet size was difficult to control. The feeding rate was set at 100 g/h because a higher feed rate caused the physical mixture to slowly accumulate at the extruder feeding hopper, which at a certain level prevented the formulation from flowing properly into the barrel. Screw speed was set at 100 rpm, and a feed rate of 100 g/h allowed sufficient time to manually synchronize extrusion with pelletization. The pellet size was controlled by the extrudate conveying speed, die opening diameter, and stretching force. The diameter of the extruded strands is usually larger than that of the opening because of extrudate expansion at the die surface. The pellet size was controlled by the cut length by varying extrudate conveying speed, which governed the stretch applied on extruded strands, and a 1 mm die was able to produce 0.5–1.5 mm pellets.
A series of preliminary studies were performed to determine the variation in ranges of the independent variables. The drug-polymer ratio affected the processing temperature and integrity of the extruded strands. Increasing the drug load could lower the processing temperature, but it also compromised the integrity of the extruded strands. When the drug load exceeded 60%, extruded strands were poorly stretchable, impeding the pelletization process. The second variable, SA, was used to aid the extrusion process as well as moderate drug release from the matrix. It demonstrated a noticeable lubricant effect starting from 3% but exhibited a significant effect on drug dissolution in the SIF when its ratio was higher than 6%. Therefore, a range from 3% to 6% was chosen to facilitate the extrusion process, whereas its effect on drug dissolution in the SIF was minimized. The third variable, pellet size, was targeted at 1 mm for ease of processing and reasonable drug release in both SGF and SIF dissolution media. Formulating large pellets reduced drug release in the SGF, but it also prolonged release in the SIF. In contrast, small pellets compromised the gastric resistance of the pellets.
The thermal behavior of the materials and formulation was evaluated using DSC and TGA. The results confirmed that individual ingredients and their physical mixture were thermally stable up to 170 °C, which was 60 °C higher than the extrusion temperature (110 °C). The thermal analysis results concurred with those of a previous study on HME of KTP. In addition, the drug load of the extrudates was comparable with the theoretical values and there was no new or abnormal peak in the HPLC chromatograms of the extrudate compared with that of the physical mixture, which further confirmed the stability of the formulations during the extrusion process. The inferred materials and formulations were stable during the extrusion process. Melted API (melting temperature 92 °C, Figure S2) also helped decrease the extrusion temperature; otherwise, an operation temperature higher than 165 °C was required.
Within the experimental ranges, the extrusion process ran smoothly for all 11 design of experiment (DOE) formulations (Table 1) with relatively low torque (14–18%) and die pressure (19–24 bar). The DSC results suggested that crystalline KTP was transformed to an amorphous form during the melt extrusion process. Pellets were collected at a steady state, indicated by the stable die pressure, torque, and strand stretch. The pellet length was automatically controlled by synchronization between the extruded strand uptake and rotation speed of the bed-knife rotor. The diameter of the cylindrical pellets was targeted comparable to the pellet length. It was controlled by the die opening diameter and stretch applied on extruded strands, which were adjusted via conveyor speed. Such low aspect ratio pellets could be filled into capsules more consistently, and low bulk density depended on the pellet orientation, which ultimately facilitated the manufacturing process.
Table 1.
Dissolution of the DOE formulations in SGF and SIF (n = 3)
| Form | Input variables |
Output variables (% Drug release) |
||||
|---|---|---|---|---|---|---|
| Drug load (%) | SA content (%) | Pellet Size (mm) | At 120 min in pH 1.2 | At 30 min in pH 6.8 | At 45 min in pH 6.8 | |
| 1 | 40 | 3.0 | 0.5 | 11.56 ± 0.71 | 92.10 ± 5.3 | 95.40 ± 3.59 |
| 2 | 60 | 3.0 | 0.5 | 12.94 ± 0.82 | 97.25 ± 4.46 | 100.78 ± 5.41 |
| 3 | 40 | 6.0 | 0.5 | 5.76 ± 0.58 | 83.87 ± 6.17 | 97.06 ± 2.77 |
| 4 | 60 | 6.0 | 0.5 | 9.17 ± 1.93 | 90.26 ± 4.77 | 99.42 ± 5.14 |
| 5 | 40 | 3.0 | 1.5 | 6.18 ± 0.09 | 68.49 ± 3.02 | 84.16 ± 4.43 |
| 6 | 60 | 3.0 | 1.5 | 7.39 ± 0.14 | 83.84 ± 4.95 | 91.98 ± 3.72 |
| 7 | 40 | 6.0 | 1.5 | 0.85 ± 0.52 | 72.18 ± 2.77 | 87.01 ± 4.79 |
| 8 | 60 | 6.0 | 1.5 | 1.27 ± 0.81 | 76.60 ± 4.27 | 86.90 ± 2.94 |
| 9 | 50 | 4.5 | 1.0 | 6.61 ± 0.98 | 82.59 ± 5.4 | 94.33 ± 5.22 |
| 10 | 50 | 4.5 | 1.0 | 6.20 ± 0.67 | 85.16 ± 3.66 | 93.39 ± 2.48 |
| 11 | 50 | 4.5 | 1.0 | 6.36 ± 0.4 | 81.05 ± 4.86 | 93.12 ± 4.04 |
3.2. Effect of independent variables on performance of dosage form
All 11 DOE formulations exhibited a delayed-release dissolution pattern where drug release was controlled below 15% after 2 hours in SGF and more than 60% drug release after 30 min switching to SIF (Figure 1). Drug load, SA, and pellet size significantly influenced the drug release in SGF dissolution medium (p < 0.05). However, SA lost significance, whereas drug content gained more significance for drug dissolution in the SIF (Figure S3). In addition to the role in aiding processing, SA also improved the performance of the dosage form by minimizing drug release in the gastric medium but did not significantly interfere with drug dissolution in the SIF. The effect of input variables on drug dissolution at various time points were visualized by responses surfaces shown in Figure 2 and contours as shown in Figure 3.
Figure 1:
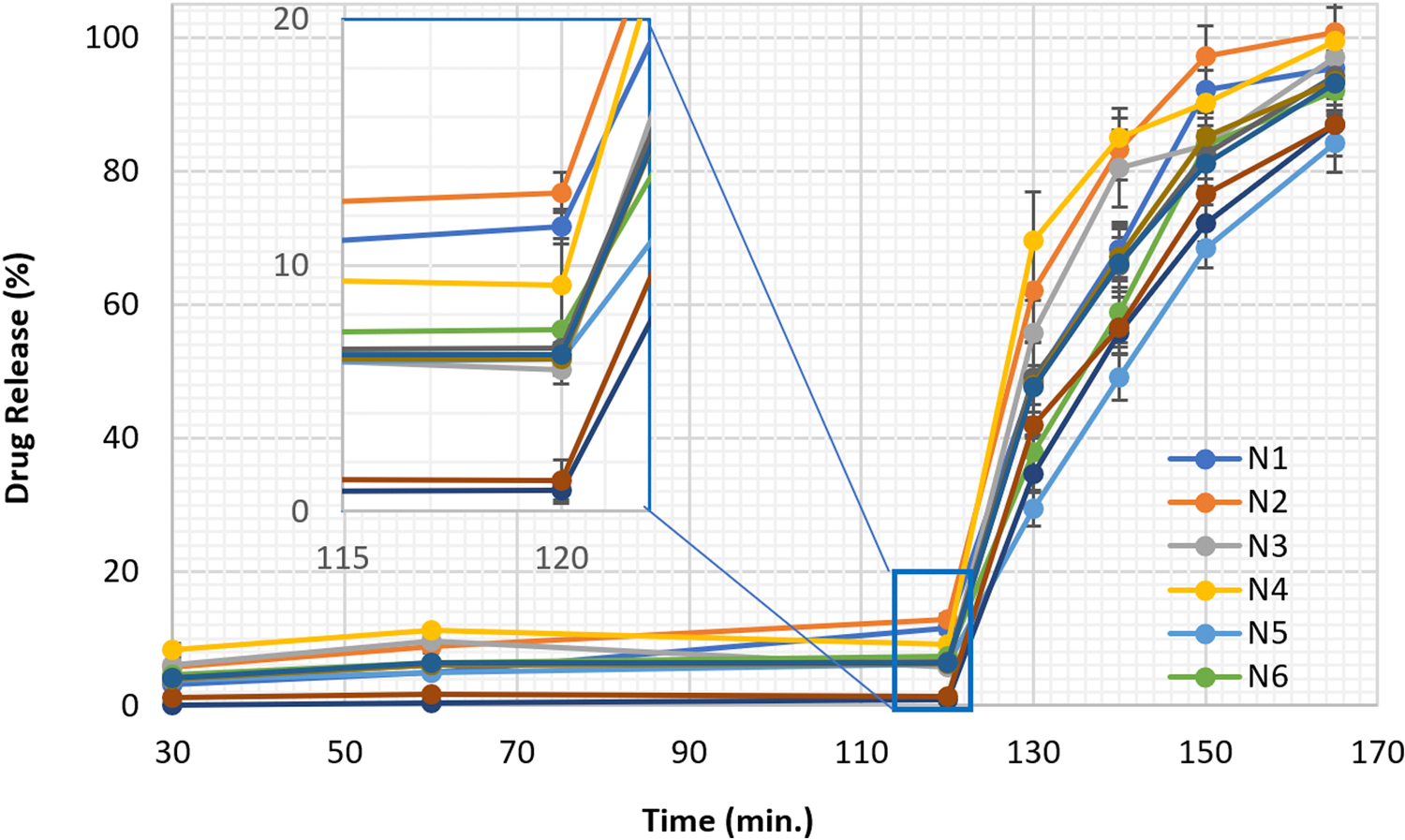
Two-step dissolution profiles of the DOE formulations
Figure 2:
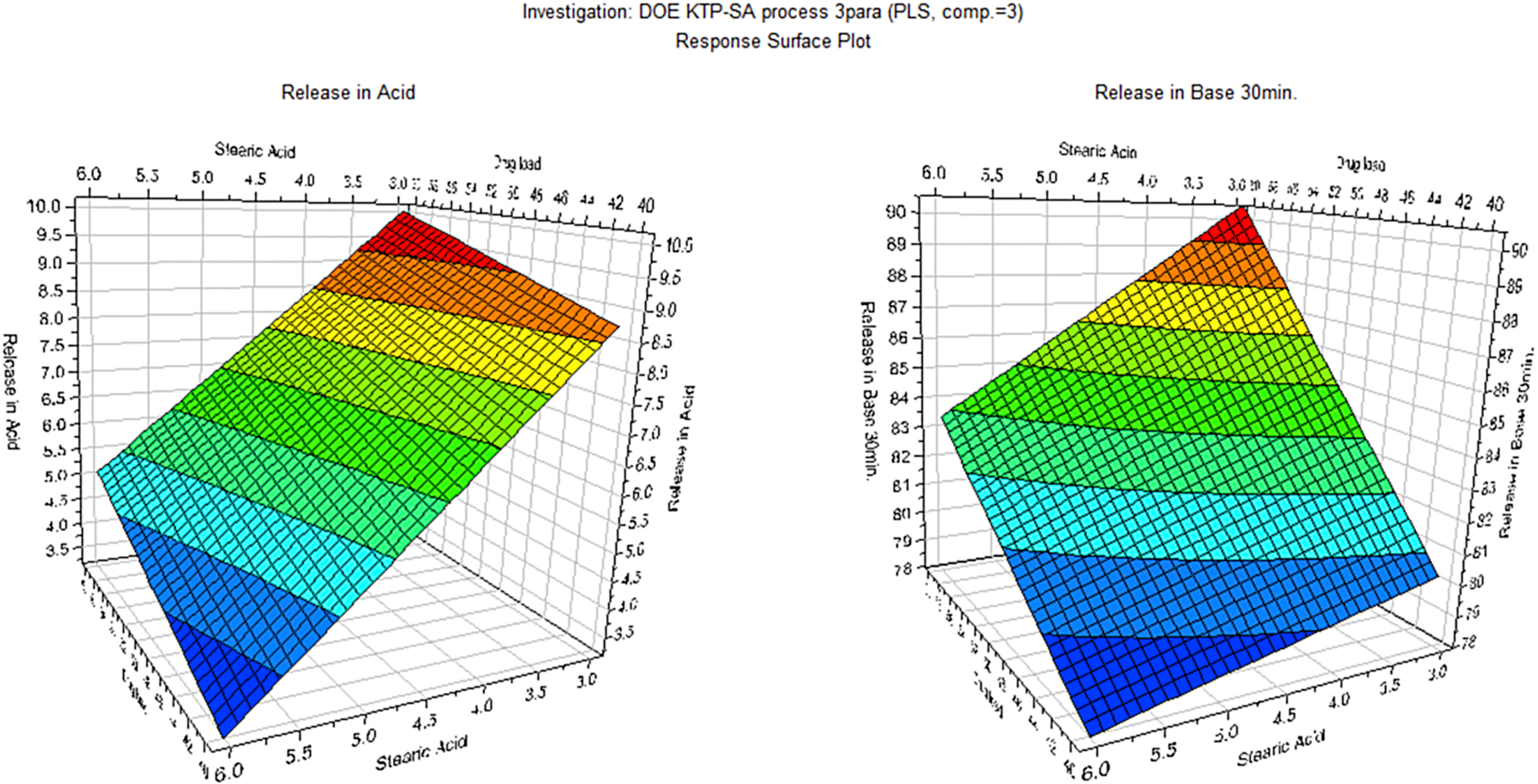
Response surfaces demonstrating the effect of SA content and drug load on dissolution of the pellets in the SGF (after 2 h) and SIF (at 30 min)
Figure 3:
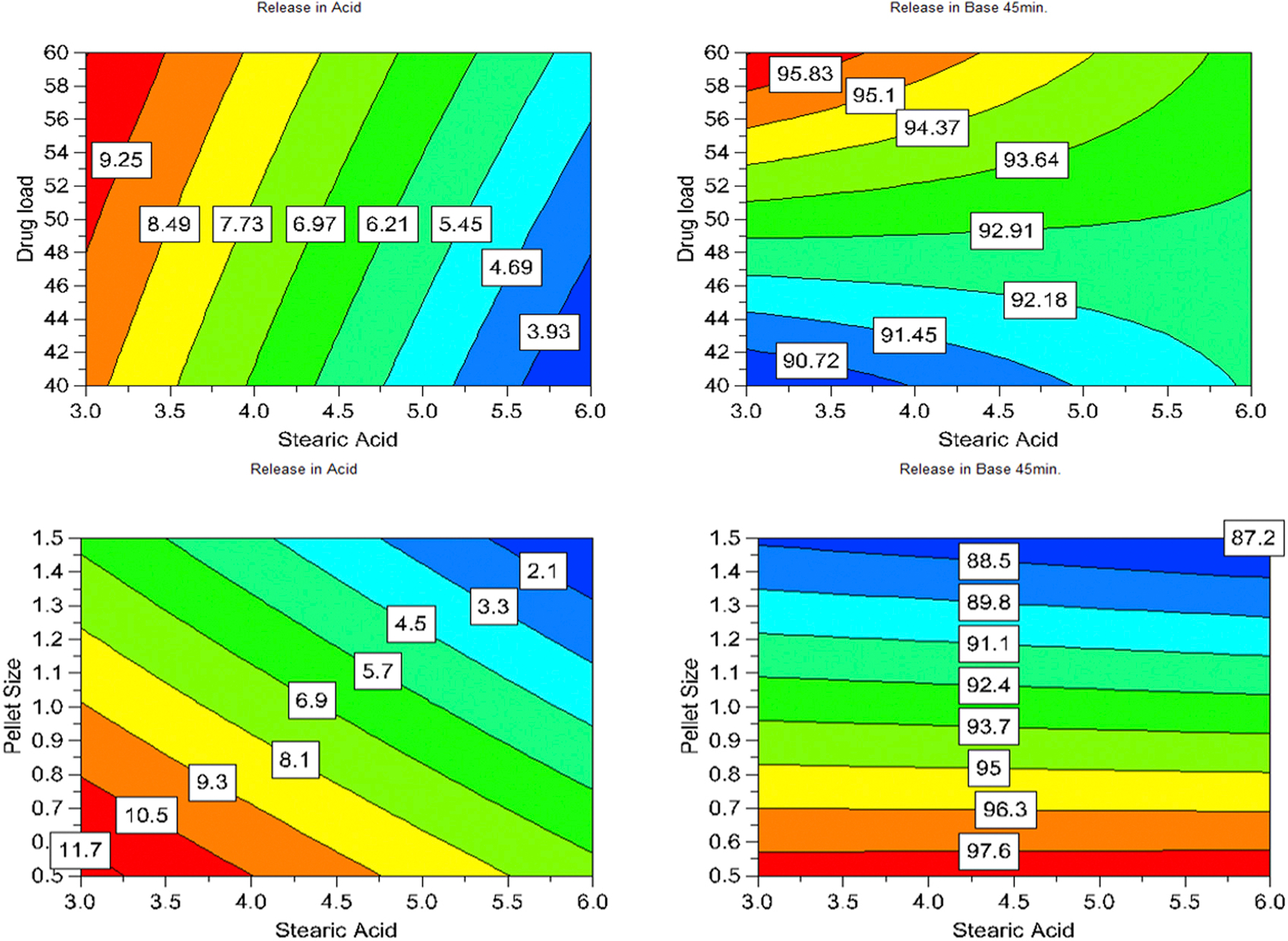
Contours demonstrated the effect of input variables on drug release of the pellets in the SGF (after 2 h) and drug dissolution in the SIF (at 45 min)
3.3. Effect of drug load
Regression results showed that drug load significantly influenced drug release in both SGF and SIF media (p < 0.05). As shown in Table 2, this factor had a positive effect on drug release in both gastric and intestinal media, meaning higher drug load would facilitate faster drug release. This phenomenon stemmed from the fact that drug load proportionally influenced the amount of API exposed to the dissolution media. Drug load affected drug dissolution in the SIF more than that in the SGF. In comparison to SA content and pellet size, the effect of drug load on dissolution in SGF was relatively low, but it increased in the SIF.
Table 2.
Summary of PLS regression results for encoded independent variables
| Variable | Drug release (%) |
|||||
|---|---|---|---|---|---|---|
| 120 min in pH 1.2 |
30 min in pH 6.8 |
45 min in pH 6.8 |
||||
| Coeff. | P | Coeff. | P | Coeff. | P | |
| Constant | 6.75 | 0.000 | 83.04 | 0.000 | 93.05 | 0.000 |
| Drug load | 0.72 | 0.024 | 3.50 | 0.013 | 1.73 | 0.009 |
| SA content | −2.35 | 0.000 | −2.10 | 0.064 | −0.22 | 0.579 |
| Pellet size | −2.65 | 0.000 | −6.97 | 0.001 | −4.77 | 0.000 |
| (Drug load)*(SA content) | 0.12 | 0.529 | −0.97 | 0.259 | −1.10 | 0.027 |
| (Drug load)*(Pellet size) | −0.32 | 0.154 | 0.82 | 0.327 | 0.00 | 0.995 |
| (SA content)* (Pellet size) | −0.19 | 0.356 | 1.17 | 0.189 | −0.25 | 0.473 |
| Goodness of prediction (Q2) | 0.672 | 0.511 | 0.737 | |||
| Goodness of fit (R2) | 0.988 | 0.962 | 0.982 | |||
| P-Model Correctness | 0.001 | 0.008 | 0.002 | |||
3.4. Effect of SA
In contrast to drug loading, SA exhibited negative effects on the dissolution of the dosage form, meaning that an increase in SA content would result in retarding drug release in both dissolution environments. SA content had a great effect on dissolution in the SGF (p = 0.00), however, it lost its significance gradually in the SIF (p > 0.05). In the SGF, it elevated the hydrophobicity of the insoluble matrix, hence, reducing drug release. The encoded regression results (Table 2) exhibited that effect of SA content on KTP dissolution in the SGF was comparable with that of pellet size and 3 time greater than that of drug load. However, in the alkaline environment, acid functional groups of the polymer and KTP were ionized and became more hydrophilic. Such effects also occurred with the SA molecule, making it less hydrophobic. In addition, the polymer became soluble in SIF ensuring drug release along with dissolution of the polymer, unlike in the SGF, where the API release relied on the diffusion process. The SA ratio was also relatively smaller than that of the polymer; thus, the erosion rate of matrices in the SIF was dominated by polymer dissolution. Therefore, the effect of SA content on dissolution of the dosage form in the SIF was minimal. Unlike in the SGF, effect of SA on dissolution in the SIF was much smaller compared with the other two factors. Its effect was 22fold smaller and 8fold smaller than that of pellet size and drug load, respectively. Therefore, effect of SA content on dissolution in the SIF can be ignored. There was a potential interaction between the variables drug load and SA content regarding the effect on dissolution at 45 min (p < 0.05), indicating that the effect of drug load depended on the SA content, even though SA alone did not significantly affect drug dissolution at 45 min (p > 0.5).
3.5. Effect of pellet size
Pellet size had the greatest absolute effect on drug dissolution in both dissolution media, exhibiting the greatest regression coefficients. This factor governed the specific surface area of the dosage form, and thus controlled the amount of drug exposed to dissolution media as well as drug diffusion from the matrix. Similar to the SA content, the pellet size had a negative impact on drug release. As shown in Table 2, in SGF, the pellet size effect was comparable to that of the SA content, but approximately 4 times greater than that of the drug load. However, its effect on dissolution at 30 min and 45 min in the SIF was more than 3 times and 20 times greater, respectively, than that of SA content.
3.6. Determination of the design space
The analysis of regression results revealed that dosage form dissolution was governed by multiple factors; hence, the dissolution profile could be customized by carefully choosing the input factors. One prominent advantage of the DOE application is that correlations between input factors and dependent variables can be expressed by regression equations, which allows the prediction of experimental outcomes from the input variables. In the QbD context, DOE is a viable approach to perform risk assessment and determination of design space. These also provide a tool to determine the disparity range of input variables within which product quality is maintained as its design. In addition, adjustments regarding formulation and processing parameters within the approved design space are not considered as change, and thus, are not mandatory to be filed with regulatory bodies.38
In CM, critical properties and parameters are monitored and controlled in real time for the entire production process. These variables tend to fluctuate to a certain extent (within the state of control) during the manufacturing process rather than being ideally constant.10 Therefore, it is necessary to define variation ranges for variables (design space) within which fluctuations of variables do not lead to a change in final product quality. If a critical attribute falls out of the design space, the system will provide a warning or halt the process, which then prompts operators to perform adjustments or start rejecting products. At advanced levels, the process can be automatically controlled based on the feedback mechanism where information collected by PAT tools is used to calculate and perform necessary adjustments to bring critical attributes back to their target values. This enables the control of product quality for the entire manufacturing process rather than performing endpoint quality control as in batch production.
This study focused on the assurance of drug release attributes, a critical quality attribute of the delayed dosage form, which was governed by drug content, SA content, and pellet size. The design space was first determined to define the variation ranges of the critical attributes within which the dissolution of the dosage form meets predetermined requirements. Dissolution requirements of the dosage form were set at stricter levels than those of the references, including USP requirements for delayed release dosage forms (<711> Dissolution, USP) and various delayed release drug monographs. These constraints were determined as follows: 1) drug release after 120 min in SGF shall not more than 8% and 2) drug dissolution at 30 min and 45 min in SIF shall not less than 80% and 90%, respectively.
The obtained DOE regression results allowed us to plot contours where each point was marked with a color based on the number of dissolution constraints satisfied. The collection of points representing the same number of constraints satisfied formed a distinct region on the plots. The contour plots were divided into regions showing where one, two, three (sweet spot), or no constraints, were satisfied (Figure 4A, B). Each contour plot describes the experimental results for the variation of two variables, whereas the third variable was fixed at a certain value. By overlaying multiple contour plots, a common sweet spot satisfying all three constraints was identified (Figure 4C). Subsequently, the design space was determined within the common sweet spot in consideration of other characteristics of the manufacturing process. The variation range of SA content, pellet size, and drug content (i.e., design space) were chosen as listed in Table 3, where drug load and SA content could vary by ± 5% and pellet size could vary by ± 20% compared with their targeted values.
Figure 4:
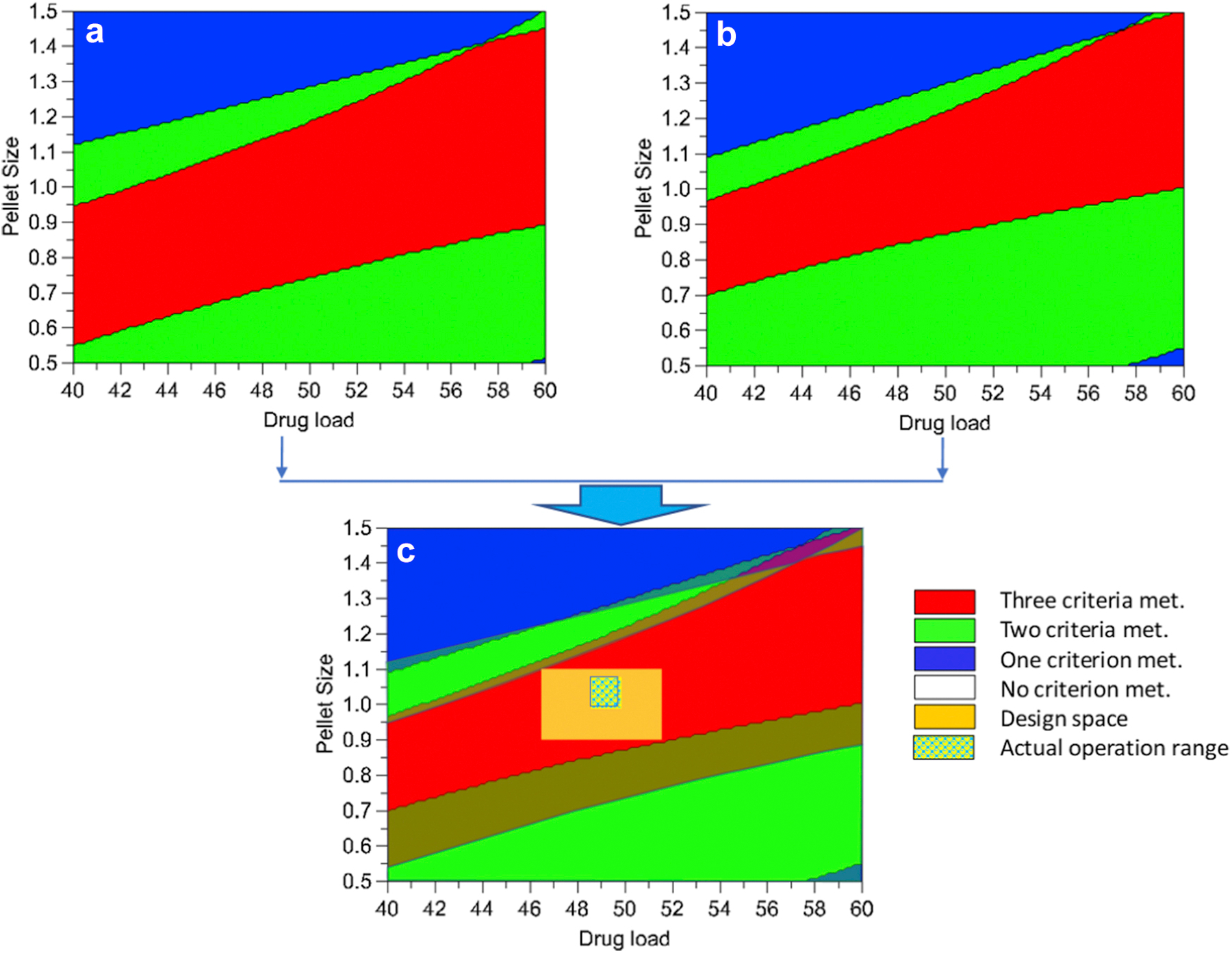
Schematic of development of design space from sweet spot contours and actual variation in the monitored attributes during a continuous process of the optimized formulation
Table 3.
Design space and actual variation in the manufacturing process for the optimized formulation
| Variable | Unit | Design space |
Actual variation |
|||
|---|---|---|---|---|---|---|
| Targeted value | Lower limit | Upper limit | Lowest value | Highest value | ||
| SA content | % | 4.5 | 4.28 | 4.73 | N/A | N/A |
| Drug load | % | 49 | 46.5 | 51.5 | 48.9 | 50.4 |
| Pellet size | mm | 1.0 | 0.8 | 1.2 | 0.94 | 1.15 |
3.7. Monitoring the continuous manufacturing process
To demonstrate the capacity to develop a continuous process, pellets were manufactured with the optimized formulation. Critical attributes were continuously monitored and controlled using two evaluated PAT tools, namely inline FT-NIR spectrometry and inline particle size analysis. The inline FT-NIR analysis and particle size analysis responded sensitively to drug load and pellet size changes, respectively. It took approximately 20 min to fill the extruder barrel, to stabilize the extrusion process, and to synchronize pelletization with extrusion throughput. The process ran relatively smoothly and steadily after the initial fluctuation. During the manufacturing process, real-time pellet size monitoring results were used to manually adjust the speed of extrudate conveying. Along with monitoring drug load, the inline FT-NIR spectrometry also reflected the uniformity of the dosage form. Analytical results showed that the actual values were close to the predictions. As shown in Figure 5, drug content variation was well below 5% and D50 particle distribution varied less than 15% throughout the process. The actual variation in monitored parameters was well within the design space (Figure 4).
Figure 5:
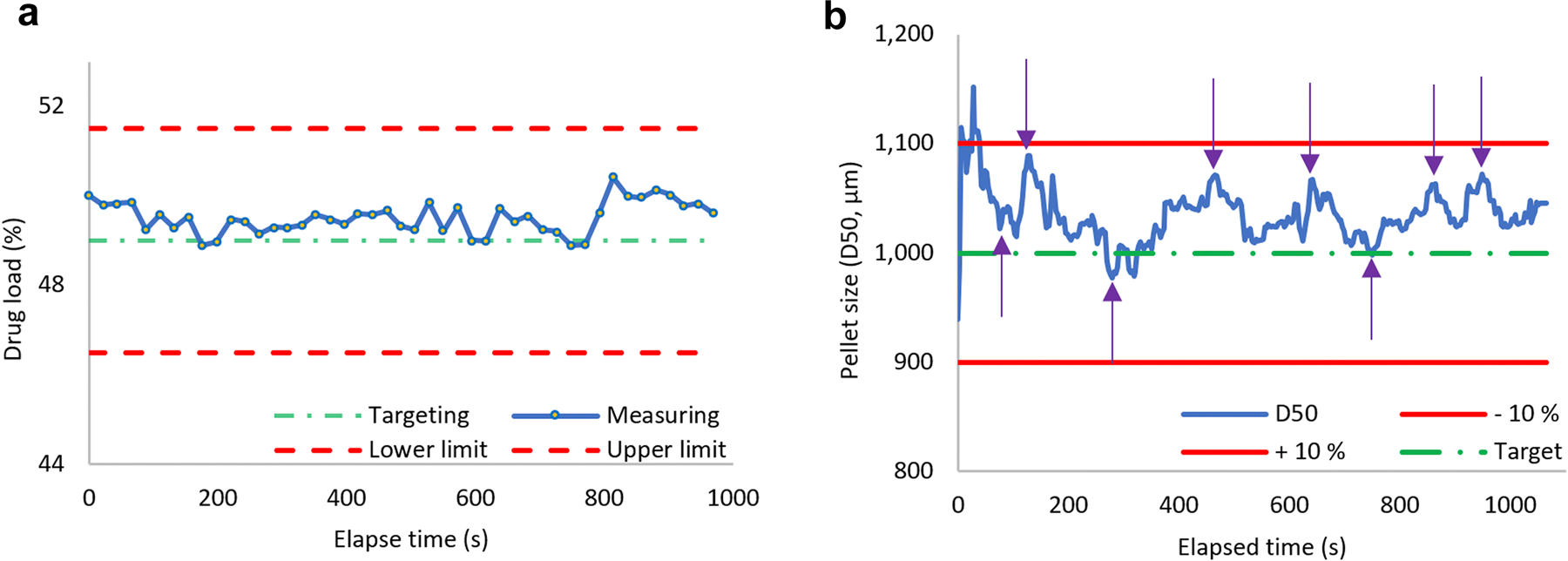
Inline monitoring of the continuous process of the optimized formulation A) drug load monitoring and B) pellet size monitoring. The purple arrows in B) mark the time points pelletizing and conveying speed were adjusted.
In this study, a pre-mixed physical mixture was fed into the hopper via a volumetric feeder. Inside the extruder barrel, the mixture was further blended in a molten state to further improve the formulation homogeneity. This implied that the drug content would be consistent throughout the manufacturing process. The inline FT-NIR monitoring showed that the drug content deviated from the average value by less than 1% throughout the run time. However, feeding a premixed mixture, i.e., with a fixed ratio of composition, leaves no room for in process adjustment if the drug content is wrong. For industrial application, materials or a group of materials can be fed separately using multiple feeders, such that the feeding ratio can be adjusted by increasing or decreasing the feed rate of the API when the drug load is lower or higher, respectively, than the target values.39, 40 The response time of the inline FT-NIR was less than 30 s, and the residence time of the formulation in the extruder barrel was approximately 10 min.35 Compared with drug content, particle size was found to fluctuate more, and the pelletizing speed was required to be adjusted frequently in order to keep the particle size within the design space. This issue stems from the volumetric feeder, the feed rate fluctuation of which is widely known. The density of materials in the feeder was influenced by gravity compression (e.g., feeder is full or low filled) or feeder refill, which ultimately affected extrusion throughput, and thus, pellet size. Therefore, in general, volumetric feeders are inappropriate for pharmaceutical CM.41 Using gravimetric feeders is a practical approach to solve this issue42, 43. As shown in Figure S4, the inline particle size distribution (PSD) measurement response to the changes is relatively quick (less than 1 min). This allows the adjustment of pellet size almost immediately. This response time is governed by the loading buffer setting and the throughput of the pelletization. Setting a low buffer would result in a quick response; however, it would also be too sensitive to noises that may trigger unnecessary alarms.
3.8. Evaluation of pellets obtained from the CM process
The optimized pellets were subjected to evaluation in terms of sieve passing, drug content, and the dissolution test. The majority of the pellets passed through the USP #16 mesh screen (> 85%) but were retained on the USP #25 mesh screen (> 95%), which was comparable with the results of the inline particle size analysis. The drug load of the pellets determined by HPLC was 49.8% ± 1.3% (n = 3). This was slightly higher than the theoretical value, which might be owing to water/solvent evaporation during the extrusion process. The dissolution profile of the optimized pellets exhibited two phases: less than 5% drug released in SGF after 120 min and more than 85% drug dissolved in the SIF after 30 min. As shown in the Figure 6 the experimental results were comparable to the prediction values and well within the constraints. This confirmed that continuous control of drug load and pellet size variation within the design space could assure product quality.
Figure 6:
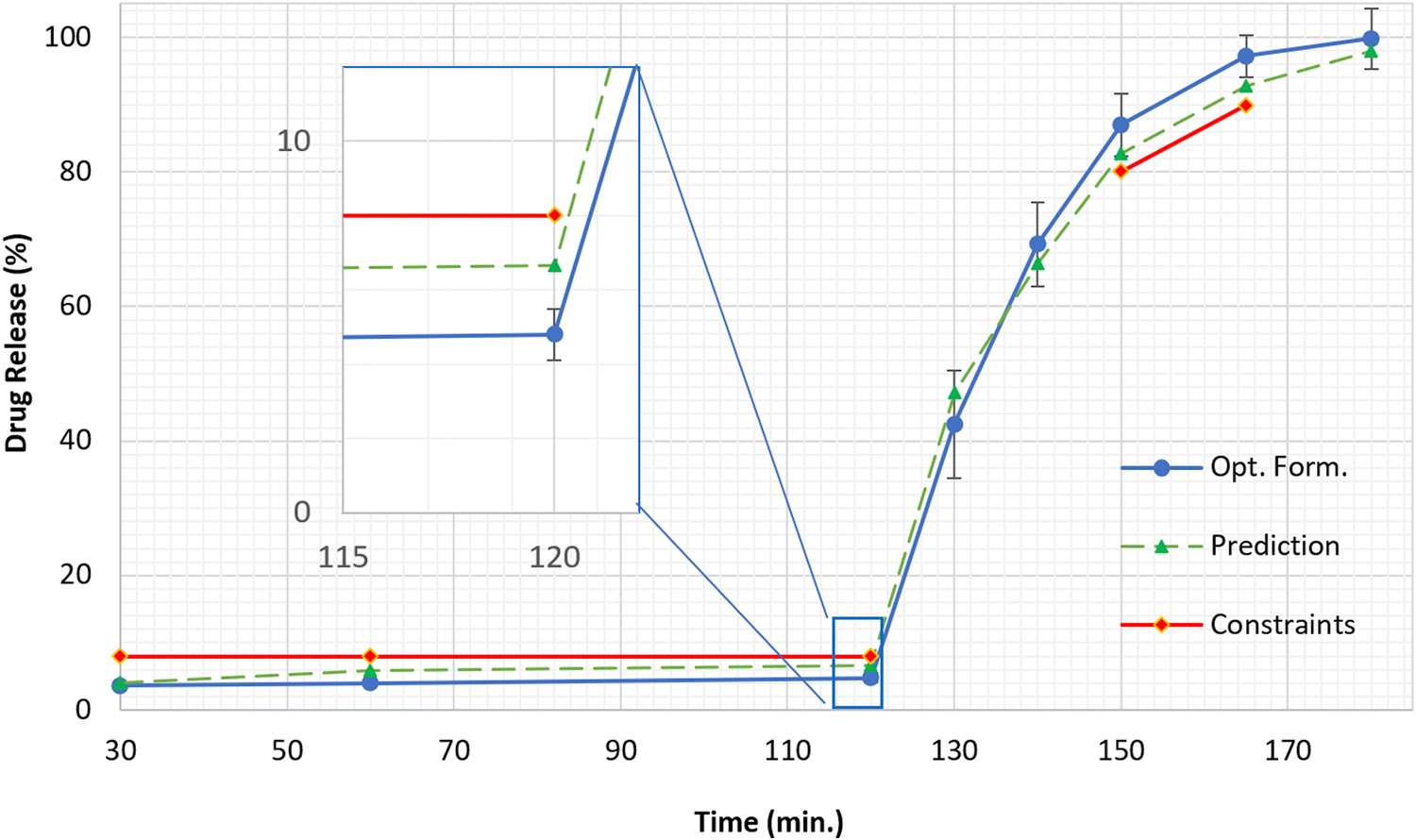
Dissolution profile of the optimized formulation running within design space in comparison with the prediction based on the regression model and dissolution constraints
4. Conclusion
Delayed release pellets of ketoprofen were successfully prepared by a continuous melt extrusion process. Drug release of the dosage form in SGF was controlled by SA content, pellet size, and drug load, whereas the dissolution in SIF was governed by pellet size and drug load. The optimal formulation and design space were systematically developed using the DOE approach with constraints on drug release in the SGF and dissolution in the SIF satisfying the requirements of the USP. The inline FT-NIR and particle size analysis were demonstrated to be sensitive tools for real-time monitoring of critical attributes of the dosage form, namely drug content and pellet size. The obtained pellets released less than 5% after 120 min in SGF and more than 85% and 95% after switching to SIF dissolution medium for 30 min and 45 min, respectively. The process is a viable prototype to develop a CM process where critical attributes are monitored in real time and processing parameters are controlled automatically within the design space in response to the feedback mechanism.
Supplementary Material
Acknowledgements
The authors would like to thank Malvern Instruments and Thermo Fisher Scientific for their generous support for this project. This work was also partially supported by Grant Number P30GM122733 from the National Institute of General Medical Sciences (NIGMS), a component of the National Institutes of Health as one of its Centers of Biomedical Research Excellence (COBRE)
Glossary
- CM
continuous manufacturing
- DSC
differential scanning calorimetry
- HME
hot-melt extrusion
- SA
stearic acid
- SGF
simulated gastric fluid
- SIF
simulated intestinal fluid
- TGA
thermogravimetric analysis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: None.
References
- 1.Badman C, Trout BL. Achieving Continuous Manufacturing. J Pharm Sci 2015;104(3):779–780. [DOI] [PubMed] [Google Scholar]
- 2.Poechlauer P, Manley J, Broxterman R, Gregertsen B, Ridemark M. Continuous processing in the manufacture of active pharmaceutical ingredients and finished dosage forms: An industry perspective. Org Process Res Dev 2012;16(10):1586–1590. [Google Scholar]
- 3.Plumb K Continuous processing in the pharmaceutical industry: Changing the mind set. Chem Eng Res Des 2005;83(6):730–738. [Google Scholar]
- 4.U.S. Food and Drug Administration (FDA). Pharmaceutical cGMPs for the 21s Century - A risk-based approach. Available at: https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/pharmaceutical-quality-21st-century-risk-based-approach-progress-report. September 2004.
- 5.Rantanen J, Khinast J. The Future of Pharmaceutical Manufacturing Sciences. J Pharm Sci 2015;104(11):3612–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burcham CL, Florence AJ, Johnson MD. Continuous Manufacturing in Pharmaceutical Process Development and Manufacturing. Annu Rev Chem Biomol Eng 2018;9:253–281. [DOI] [PubMed] [Google Scholar]
- 7.U.S. Food and Drug Administration (FDA). FDA statement on FDA’s modern approach to advanced pharmaceutical manufacturing. FDA Center for Drug Evaluation and Research (CDER), Silver Spring, MD: Published 2019. Available at: https://www.fda.gov/news-events/press-announcements/fda-statement-fdas-modern-approach-advanced-pharmaceutical-manufacturing. Accessed September 19, 2019. [Google Scholar]
- 8.U.S. Food and Drug Administration (FDA). Quality Considerations for Continuous Manufacturing Guidance for Industry. FDA Center for Drug Evaluation and Research (CDER), Silver Spring, MD: Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/quality-considerations-continuous-manufacturing. February 2019. [Google Scholar]
- 9.Silva AF, Vercruysse J, Vervaet C, et al. In-Depth Evaluation of Data Collected During a Continuous Pharmaceutical Manufacturing Process: A Multivariate Statistical Process Monitoring Approach. J Pharm Sci 2019;108(1):439–450. [DOI] [PubMed] [Google Scholar]
- 10.Lee SL, O’Connor TF, Yang X, et al. Modernizing Pharmaceutical Manufacturing: from Batch to Continuous Production. J Pharm Innov 2015;10:191–199. [Google Scholar]
- 11.Allison G, Cain YT, Cooney C, et al. Regulatory and quality considerations for continuous manufacturing May 20–21, 2014 continuous manufacturing symposium. J Pharm Sci 2015;104(3):803–812. [DOI] [PubMed] [Google Scholar]
- 12.Schaber SD, Gerogiorgis DI, Ramachandran R, Evans JMB, Barton PI, Trout BL. Economic analysis of integrated continuous and batch pharmaceutical manufacturing: A case study. Ind Eng Chem Res 2011;50(17):10083–10092. [Google Scholar]
- 13.Su Q, Ganesh S, Moreno M, et al. A perspective on Quality-by-Control (QbC) in pharmaceutical continuous manufacturing. Comput Chem Eng 2019;125:216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vargas JM, Nielsen S, Cárdenas V, et al. Process analytical technology in continuous manufacturing of a commercial pharmaceutical product. Int J Pharm 2018;538(1–2):167–178. [DOI] [PubMed] [Google Scholar]
- 15.Mascia S, Heider PL, Zhang H, et al. End-to-end continuous manufacturing of pharmaceuticals: Integrated synthesis, purification, and final dosage formation. Angew Chemie - Int Ed 2013;52(47):12359–12363. [DOI] [PubMed] [Google Scholar]
- 16.Fonteyne M, Vercruysse J, De Leersnyder F, et al. Process Analytical Technology for continuous manufacturing of solid-dosage forms. TrAC - Trends Anal Chem 2015;67:159–166. [Google Scholar]
- 17.Repka MA, Bandari S, Kallakunta VR, et al. Melt extrusion with poorly soluble drugs – An integrated review. Int J Pharm 2018;535(1–2):68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markl D, Wahl PR, Menezes JC, et al. Supervisory control system for monitoring a pharmaceutical hot melt extrusion process. AAPS PharmSciTech 2013;14:1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maniruzzaman M, Nokhodchi A. Continuous manufacturing via hot-melt extrusion and scale up: regulatory matters. Drug Discov Today. 2017;22(2):340–351. [DOI] [PubMed] [Google Scholar]
- 20.Treffer D, Wahl P, Markl D, Koscher G, Roblegg E, Khinast JG. Hot Melt Extrusion as a Continuous Pharmaceutical Manufacturing Process. In: Repka M, Langley NDJ, ed. Melt Extrusion. AAPS Advances in the Pharmaceutical Sciences Series.; 2013:363–396. [Google Scholar]
- 21.Simões MF, Pinto RMA, Simões S. Hot-melt extrusion in the pharmaceutical industry: toward filing a new drug application. Drug Discov Today. 2019;24(9):1749–1768. [DOI] [PubMed] [Google Scholar]
- 22.Treffer D, Wahl PR, Hörmann TR, et al. In-line implementation of an image-based particle size measurement tool to monitor hot-melt extruded pellets. Int J Pharm 2014;466(1–2):181–189. [DOI] [PubMed] [Google Scholar]
- 23.Bialleck S, Rein H. Preparation of starch-based pellets by hot-melt extrusion. Eur J Pharm Biopharm 2011;79(2):440–448. [DOI] [PubMed] [Google Scholar]
- 24.Hörmann TR, Rehrl J, Scheibelhofer O, et al. Sensitivity of a continuous hot-melt extrusion and strand pelletization line to control actions and composition variation. Int J Pharm 2019;566:239–253. [DOI] [PubMed] [Google Scholar]
- 25.Follonier N, Doelker E, Cole ET. Various ways of modulating the release of diltiazem hydrochloride from hot-melt extruded sustained release pellets prepared using polymeric materials. J Control Release. 1995;36(3):243–250. [Google Scholar]
- 26.Fosslien E Adverse effects of nonsteroidal anti-inflammatory drugs on the gastrointestinal system. Ann Clin Lab Sci 1998;28(2):67–81. [PubMed] [Google Scholar]
- 27.Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol 2010;24(2):121–132. [DOI] [PubMed] [Google Scholar]
- 28.Bjarnason I, Scarpignato C, Holmgren E, Olszewski M, Rainsford KD, Lanas A. Mechanisms of Damage to the Gastrointestinal Tract From Nonsteroidal Anti-Inflammatory Drugs. Gastroenterology. 2018;154(3):500–514. [DOI] [PubMed] [Google Scholar]
- 29.Vo AQ, Feng X, Morott JT, et al. A novel floating controlled release drug delivery system prepared by hot-melt extrusion. Eur J Pharm Biopharm 2016;98:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schilling SU, McGinity JW. Novel application of hot-melt extrusion for the preparation of monolithic matrices containing enteric-coated particles. Int J Pharm 2010;400(1–2):24–31. [DOI] [PubMed] [Google Scholar]
- 31.Mehuys E, Remon JP, Vervaet C. Production of enteric capsules by means of hot-melt extrusion. Eur J Pharm Sci 2005;24(2–3):207–212. [DOI] [PubMed] [Google Scholar]
- 32.Schilling SU, Lirola HL, Shah NH, Waseem Malick A, McGinity JW. Influence of plasticizer type and level on the properties of Eudragit® S100 matrix pellets prepared by hot-melt extrusion. J Microencapsul 2010;27(6):521–532. [DOI] [PubMed] [Google Scholar]
- 33.Schilling SU, Shah NH, Waseem Malick A, McGinity JW. Properties of melt extruded enteric matrix pellets. Eur J Pharm Biopharm 2010;74(2):352–361. [DOI] [PubMed] [Google Scholar]
- 34.Yang R, Wang Y, Zheng X, Meng J, Tang X, Zhang X. Preparation and evaluation of ketoprofen hot-melt extruded enteric and sustained-release tablets. Drug Dev Ind Pharm 2008;34(1):83–89. [DOI] [PubMed] [Google Scholar]
- 35.Vo AQ, He H, Zhang J, Martin S, Chen R, Repka MA. Application of FT-NIR Analysis for In-line and Real-Time Monitoring of Pharmaceutical Hot Melt Extrusion: a Technical Note. AAPS PharmSciTech 2018;19:3425–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parikh T, Gupta SS, Meena A, Serajuddin ATM. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion - III: Polymethacrylates and polymethacrylic acid based polymers. J Excipients Food Chem 2014;5(1):56–64. [Google Scholar]
- 37.Desai D, Sandhu H, Shah N, et al. Selection of Solid-State Plasticizers as Processing Aids for Hot-Melt Extrusion. J Pharm Sci 2018;107(1):372–379. [DOI] [PubMed] [Google Scholar]
- 38.Yu LX. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm Res 2008;25:781–791. [DOI] [PubMed] [Google Scholar]
- 39.Van Snick B, Holman J, Cunningham C, et al. Continuous direct compression as manufacturing platform for sustained release tablets. Int J Pharm 2017;519(1–2):390–407. [DOI] [PubMed] [Google Scholar]
- 40.Taipale-Kovalainen K, Karttunen AP, Niinikoski H, Ketolainen J, Korhonen O. The effects of unintentional and intentional process disturbances on tablet quality during long continuous manufacturing runs. Eur J Pharm Sci 2019;129:10–20. [DOI] [PubMed] [Google Scholar]
- 41.Blackshields CA, Crean AM. Continuous powder feeding for pharmaceutical solid dosage form manufacture: a short review. Pharm Dev Technol 2018;23(6):554–560. [DOI] [PubMed] [Google Scholar]
- 42.Engisch WE, Muzzio FJ. Method for characterization of loss-in-weight feeder equipment. Powder Technol 2012;228:395–403. [Google Scholar]
- 43.Hanson J Control of a system of loss-in-weight feeders for drug product continuous manufacturing. Powder Technol 2018;331:236–243. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.