

Abstract
Background & Aims:
The mechanisms by which macrophages regulate intestinal epithelial cell (IEC) barrier properties are poorly understood. Protein tyrosine phosphatase non-receptor type 2 (PTPN2) protects the IEC barrier from inflammation-induced disruption and regulates macrophage functions. We investigated whether PTPN2 controls interactions between IECs and macrophages to maintain intestinal barrier function.
Methods:
Human IEC (Caco-2BBe/HT-29.cl19a cells) and mouse enteroid monolayers were co-cultured with human macrophages (THP-1, U937, primary monocyte-derived macrophages from patients with inflammatory bowel disease [IBD]) or mouse macrophages, respectively. We assessed barrier function (transepithelial electrical resistance [TEER] and permeability to 4 kDa fluorescently labeled dextran [FD4] or rhodamine B-dextran 70 kDa) and macrophage polarization. We analyzed intestinal tissues from mice with myeloid cell-specific deletion of PTPN2 (Ptpn2-LysMCre mice) and mice without disruption of Ptpn2 (controls); some mice were given injections of a neutralizing antibody against interleukin 6 (IL6). Proteins were knocked down in macrophages and/or IECs with small hairpin RNAs.
Results:
Knockdown of PTPN2 in either macrophages and/or IECs increased the permeability of IEC monolayers, had a synergistic effect when knocked down from both cell types, and increased development of inflammatory macrophages in macrophage–IEC co-cultures. Colon lamina propria from Ptpn2-LysMCre mice had significant increases in inflammatory macrophages; these mice had increased in vivo and ex vivo colon permeability to FD4 and reduced ex vivo colon TEER. Nanostring analysis revealed significant increases in expression of IL6 in colon macrophages from Ptpn2-LysMCre mice. An IL6 blocking antibody reversed the effects of PTPN2-deficient macrophages, reducing permeability of IEC monolayers in culture and in Ptpn2-LysMCre mice. Macrophages from patients with IBD carrying a single-nucleotide polymorphism associated with the disease (PTPN2 rs1893217) had the same features of PTPN2-deficient macrophages from mice, including reduced TEER and increased permeability in co-cultures with human IEC or mouse enteroid monolayers, which were restored by anti-IL6.
Conclusions:
PTPN2 is required for interactions between macrophages and IECs; loss of PTPN2 from either cell type results in intestinal barrier defects, and loss from both cell types has a synergistic effect. We provide a mechanism by which PTPN2 gene variants compromise intestinal epithelial barrier function and increase risk of inflammatory disorders such as IBD.
Keywords: TCPTP, Innate Immune Cells, Tight Junction, Claudin-2
Lay Summary:
The authors identified a mechanism by which alterations in PTPN2 that have been found in many patients with IBD affects interactions between intestinal epithelial cells and immune cells. Disruption of this interaction can affect intestinal barrier properties and the immune response.
Introduction
The intestinal epithelium is lined by a single layer of intestinal epithelial cells (IECs), which form a tight, but selective barrier that allows for electrolyte and nutrient uptake but prevents luminal bacteria and undigested food particles from unrestricted access to the body1, 2. To form and maintain this barrier, IECs are interconnected via tight junctions (TJs), that restrict paracellular passage of molecules across the epithelium1, 2. TJs constitute several transmembrane molecules, including claudins, junctional adhesion molecules (e.g. JAM-A), occludin, and tricellulin. These membrane-spanning molecules are anchored to the cytoskeleton via zonula occludens (ZO)-1, which is important in maintaining TJ functionality2. While most claudins function as barrier-sealing molecules, some members of the claudin family, such as claudin-2 and claudin-15 form ion selective pores3, 4 and their upregulation in response to pro-inflammatory cytokines mediates increased cation and water flux across the epithelium resulting in diarrhea5. In addition to this pore-dependent, fast, and charge/size-selective ion exchange across the epithelial border, displacement of barrier sealing proteins away from the TJ, described as the leak pathway6, enables slow paracellular passage of larger, non-charged molecules, including bacterial products1, 2. Adequate regulation of the pore and leak pathways enables normal intestinal function, and disturbed TJ protein expression/localization and epithelial barrier function has been linked to a number of intestinal and systemic disorders, including inflammatory bowel disease (IBD), celiac disease, gastrointestinal infections7, chronic liver disorders8, autoimmune arthritis9, multiple sclerosis10, type 1 diabetes11, and Parkinson’s disease12, thus clearly linking barrier defects with aberrant systemic immune responses.
Directly beneath the epithelial cell layer resides the body’s largest pool of immune cells, which maintain intestinal homeostasis via removal of invading bacteria and dying cells, secretion of anti-inflammatory cytokines, and induction/maintenance of tolerance towards commensal bacteria and food particles13. Macrophages constitute a considerable fraction of intestinal immune cells that initiate and orchestrate effective immune responses towards bacteria that breach the epithelial barrier14. Most resident intestinal macrophages are constantly replenished from blood monocytes15, 16, although long-lasting, self-renewing intestinal macrophages have been described14. When recruited to the intestine during physiological conditions, blood-derived monocytes gradually acquire a tissue-resident phenotype with a high proportion of anti-inflammatory macrophages15, often referred to as alternatively activated or M2 macrophages which secrete high levels of tolerance-inducing cytokines, including interleukin (IL)-10 and tumor growth factor (TGF)β17, 18. If recruited during inflammation, blood-derived monocytes/macrophages acquire a more pro-inflammatory phenotype and produce inflammatory cytokines, including IL-6, IL-1β, IL-12, and tumor necrosis factor (TNF)-α, a phenotype often described as classically activated, or M1 macrophages15, 17. M1 and M2 macrophages represent two extremes of macrophage polarization and in both inflamed and non-inflamed settings, macrophages can modify their phenotype along the M1-M2 continuum and several M2 macrophage sub-types have been described15, 19.
Loss of function variants in the protein tyrosine phosphatase non-receptor type 2 (PTPN2) gene, which encodes for T cell tyrosine phosphatase (TCPTP) have been associated with an increased risk of developing chronic inflammatory disorders, including rheumatoid arthritis20, type 1 diabetes21, celiac disease21 and the two sub-forms of IBD, ulcerative colitis (UC) and Crohn’s disease (CD)21, 22. TCPTP dephosphorylates several cellular substrates, including JAK/STAT molecules23, 24, the EGF receptor25, T cell receptor kinases26 and thereby restricts inflammatory responses27. In IECs, loss of TCPTP potentiates interferon (IFN)-γ-induced leak and pore pathways5, 28, promotes EGF-induced suppression of chloride secretion25, and promotes secretion of pro-inflammatory cytokines29, 30. Defective TCPTP function promotes inflammatory T cell subsets31, enhances secretion of MCP-1, IL-6 and IL-1β from monocytes/macrophages24 and deletion of TCPTP in T cells or macrophages promotes susceptibility to experimental colitis31, 32. However, it is currently not known how TCPTP affects the polarization/development of pro-inflammatory (M1-like) and anti-inflammatory (M2-like) macrophages.
Macrophages play an essential role in promoting and maintaining an anti-inflammatory state in the intestine33, and barrier defects upon loss of JAM-A compromise macrophage-mediated neutrophil recruitment34. However, the impact of macrophages on IEC barrier function remains poorly understood and factors that regulate macrophage – IEC crosstalk are poorly defined. The aim of this study was to investigate how TCPTP contributes to normal macrophage - IEC cross regulation and how this impacts intestinal permeability.
Methods
Cell lines and cell culture.
Caco-2BBe, HT-29.cl19a, THP-1 and U937 cells were maintained as previously described28, 31. Efficient PTPN2 knockdown (Supplementary Figure S1) was introduced using shRNA constructs as detailed in the supplementary methods. Caco-2BBe and HT-29.cl19a cells were seeded on 12mm permeable transwell inserts (Corning Inc., Corning NY) and differentiated for 9 days before start of basolateral co-culture with 500,000 monocytes/macrophages. For non-polarized (M0) macrophage differentiation, THP-1 and U937 cells were pulsed with 50 ng/ml phorbol myristate acetate (PMA; Sigma-Aldrich, St. Louis, MO) for 3 h, washed and seeded into 12 well plates. After 48 h, medium was changed to serum-free DMEM (Corning Inc.) and transwells containing polarized IECs were transferred into the same wells. For polarization into “M1” or “M2” macrophages, non-polarized macrophages were incubated with 400 IU/ml IFN-γ (Roche, Basel, Switzerland) + 100 ng/ml lipopolysaccharide (LPS; Invivogen, San Diego, CA), or 50 ng/ml IL-4 (Peptrotech, Rocky Hill, NJ, respectively, for an additional 24h. For studies with IL-6 inhibition or IL-6 supplementation, a blocking rat IgG1κ anti-human IL-6 antibody (1 μg/ml; Clone MQ2–13A5; Thermo Fisher Scientific, Waltham, MA) or recombinant IL-6 (20 ng/ml, previously established dose-response, own unpublished data; Peprotech Inc, Rocky Hill, NJ) were added basolaterally at the start of the co-cultures. For production of THP-1 or U937 conditioned medium, PMA-pulsed THP-1 or U937 were differentiated for 48 h, medium was replaced and macrophages incubated for 24 h prior to supernatant collection.
Organoid – macrophage co-cultures.
Bone marrow macrophages were generated from WT or Ptpn2-LysMCre mice (see below) as described in supplementary methods. Crypts were isolated from WT mice and organoids generated as detailed in supplementary methods. 600 organoids were seeded per cell culture insert and maintained in mouse minimal medium. After 3 days, the organoid-derived monolayer containing inserts were transferred onto wells containing 500,000 bone marrow-derived macrophages and cultured for 24 h.
Protein and RNA extraction, Western blotting, qPCR.
Proteins and RNA were isolated using standard procedures described in the supplementary methods and subjected to Western blot and qPCR analyses, respectively, as described in detail in the supplementary methods
H&E, Immunofluorescence, and TUNEL staining.
For immunofluorescent imaging, IECs were grown on glass cover slides until confluency prior to incubation with THP-1 or U937-conditioned medium for 24h. Cells were washed 3 times in ice cold PBS and staining for ZO-1/claudin-2 performed as described in the supplementary methods. Paraffin-embedded or OCT-embedded frozen colonic sections were stained as described in the supplementary methods.
Mice.
Generation of mice lacking Ptpn2 in myeloid cells is described in supplementary methods. Ptpn2fl/fl-LysMCre− (“WT”) and Ptpn2-LysMCre+/− (“KO” or “Ptpn2-LysMCre”) 8–12 week old, sex and age-matched littermates were used for all experiments. For studies with IL-6 inhibition, a rat IgG1κ neutralizing anti-IL-6 antibody or isotype control (0.25mg/day; Clone MP5–20F3 and BE0088, respectively; BioXcell; West Lebanon, NH) was injected intraperitoneally daily for 7 days. All mouse experiments were performed according to local animal welfare legislation and approved UCR IACUC protocols.
Ussing chamber.
Ussing chamber experiments were performed as previously described35 and as detailed in the supplementary methods.
Flow cytometry.
For flow cytometry, lamina propria immune cells were isolated as described32, and staining/acquisition/analysis performed according to standard procedures as detailed in the supplementary methods.
Multiplex gene expression profiling (Nanostring®).
Colon lamina propria immune cells were isolated as described30 and macrophages sorted directly into RLT buffer (Qiagen) supplemented with 40mM DTT (Sigma-Aldrich) on a MoFlo cell sorter (Beckman Coulter, Brea, CA) and total RNA extracted using the RNeasy mini kit from Qiagen. Using 50 ng total RNA from each sample as input, the digital multiplexed NanoString nCounter Analysis System (NanoString Technologies, Seattle, WA, USA) was used for gene expression profiling according to the manufacturer’s instructions. RNA samples were analyzed using the nCounter Autoimmune Profiling Panel consisting of 770 human immune-related genes supplemented with a set of 30 custom genes (Nanostring Technologies, Seattle, WA). Sample preparation and measurements were performed according to the manufacturer’s instructions on a Digital Analyzer (Nanostring Technologies) and results analyzed using the nSolver Analysis Software (NanoString Technologies).
ELISA and LDH assay.
IL-6 and IL-1β DuoSet ELISAs were obtained from R&D Systems (Minneapolis, MN) and performed according to the manufacturer’s guidelines. Lactate dehydrogenase (LDH) was measured using the CyQUANT™ LDH Cytotoxicity Assay from Thermo Fisher Scientific (Waltham, MA).
Patient-derived macrophages.
Whole blood samples were obtained from previously genotyped, sex and age-matched patients from the Swiss IBD cohort and healthy volunteers. All patients presented with quiescent disease at time of sample collection. Supplementary table S1 summarizes patient characteristics and medication at the time of blood collection. All patients and healthy controls signed informed consent before study inclusion and approved by the local ethics commission (Cantonal Ethics Commision Zurich, Switzerland; Approval number EK-1755). Peripheral blood monocytes were isolated as described in supplementary methods and differentiated to macrophages for 7 days using macrophage colony stimulating factor (20ng/ml, Peprotech), washed and incubated in serum free medium for 8 h prior to co-culture with Caco-2BBe IECs.
Statistical Analysis.
Statistical analysis was performed using GraphPad Prism. Statistical tests and number of repetitions (n) are given in the figure legends. P-values below 0.05 were considered significant.
Results
Alternatively activated M2-like macrophages selectively promote intestinal epithelial cell barrier function.
To understand how macrophages affect IEC barrier function, HT-29.cl19a and Caco-2BBe cells were grown for 9 days on cell culture inserts to allow the formation of TJs. IECs were co-cultured up to 48 h with non-differentiated THP-1 monocytes, PMA-differentiated non-polarized (M0), IFN-γ and LPS polarized (classically activated, M1), or IL-4 polarized (alternatively activated, M2) THP-1 macrophages. When HT-29.cl19a or Caco-2BBe cells were co-cultured with non-differentiated THP-1 monocytes or with M1 macrophages, we observed a significant drop of transepithelial electrical resistance (TEER; Figure 1A+B). Co-culture with M0 or M2 macrophages, however, significantly enhanced TEER (Figure 1A+B). Monocytes and M1 macrophages promoted 4kDa FITC-dextran (FD4) permeability, while co-culture with M0 or M2 macrophages reduced FD4 permeability (Figure 1C+D). Consistent with functional changes, monocytes and M1 macrophages promoted expression of the pore-forming protein claudin-2, and phosphorylation of myosin light chain (MLC), which is associated with actin ring contraction and increased macromolecule permeability2, while markers of cell death were unaffected (Supplementary Figure S2). In contrast, M0 and M2 macrophages suppressed claudin-2 but enhanced barrier sealing claudin-4 expression (Figure 1E). M2 macrophages additionally promoted expression of the barrier sealing proteins JAM-A and tricellulin as well as E-cadherin, that facilitates TJ assembly, while occludin levels were not affected (Figure 1E; quantitative analysis in Supplementary File 2_Densitometry). Collectively, this indicates that non-polarized macrophages and alternatively activated/M2 macrophages promote tightness of the intestinal epithelial barrier, while monocytes and pro-inflammatory/M1 macrophages induce barrier permeability.
Figure 1. Non-polarized and M2-like macrophages promote barrier function.

HT-29.cl19a and Caco-2BBe cells were cultured for 9 days on transwells to allow the formation of an epithelial barrier. THP-1 cells were left untreated (monocytes, Mono), pulsed with PMA (50ng/ml, 3h) to induce macrophage differentiation, and after 48 h left untreated (M0), treated with IFN-γ + LPS (M1), or IL-4 (M2) for 24 h prior to culture on the basolateral side of IEC monolayers. A+B: Transepithelial electrical resistance (TEER), C+D) FD4 flux into the basolateral medium, and E) representative Western blot images for the indicated proteins after 24 h. *P < .05, **P < 0.01, 1-way ANOVA. Representative results from one out of three independent experiments with n = 3–4.
Co-culture with macrophages promotes epithelial expression of TCPTP.
We previously demonstrated that TCPTP protects IEC barrier function and restricts claudin-2 expression in vitro5, 28. Hence, we assessed whether epithelial permeability changes arising from co-culture with monocytes or macrophages could be attributed to altered epithelial TCPTP levels. Co-culture with monocytes and macrophages promoted PTPN2 mRNA and TCPTP protein expression in IECs with no significant difference between M0/M2 macrophages vs. monocytes/M1 macrophages (Figure 2A+B). In turn, co-culture with IECs induced PTPN2 mRNA expression in non-polarized (M0) but not in M2/M1 macrophages, or in monocytes (Figure 2C). Of note, M0 and M2 macrophages expressed higher baseline levels of TCPTP than monocytes or M1 macrophages (Figure 2C, P < .05, each).
Figure 2. Co-culture with macrophages promotes TCPTP expression in IECs.
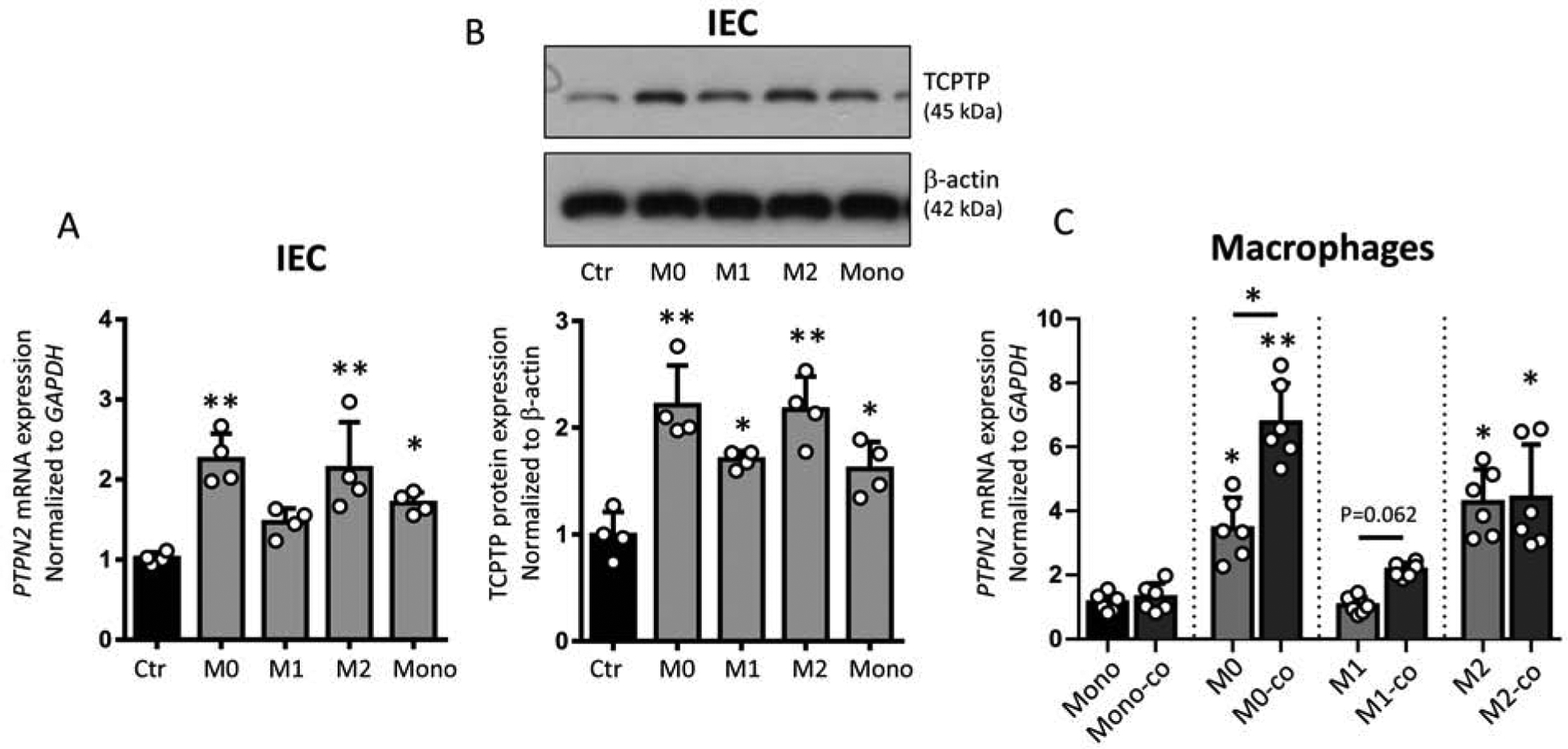
HT-29.cl19a cells were cultured on inserts as in Figure 1 and THP-1 monocytes left untreated (monocytes, Mono) or differentiated into M0, M1 or M2 macrophages as in Figure 1. A) PTPN2 mRNA levels and B) representative Western blot images and densitometry of TCPTP in HT-29.cl19a IECs, n = 4, each; and C) PTPN2 mRNA expression in THP-1 cells with or without co-culture with IECs, n = 6. In A + B, asterisks denote statistical significances compared to nonco-cultured controls (Ctr), and in C compared to monocytes or as indicated by the lines above the bars (*P < .05, **P < .01, 1-way ANOVA).
Knockdown of PTPN2 compromises macrophage-IEC crosstalk resulting in increased IEC barrier permeability.
Given the high PTPN2 expression in IECs co-cultured with macrophages, we next investigated whether knockdown of PTPN2 affects IEC-macrophage interactions. Consistent with our previous reports28, PTPN2 knockdown in Caco-2BBe and HT-29.cl19a cells resulted in a marked decrease of steady-state TEER (Figure 3A + Supplementary Figure S3). Macrophages transfected with non-targeting control shRNA increased TEER in Ctr-transfected IEC, but not in PTPN2-KD IECs (Figure 3A + Supplementary Figure S3A), indicating that the presence of TCPTP is required to maintain a normal IEC barrier and to mediate the barrier sealing effect of macrophages. Further, knockdown of PTPN2 in IECs as well as in macrophages exacerbated this IEC barrier defect in a synergistic manner (Figure 3A + Supplementary Figure S3). Similar effects were observed in IECs cultured in THP-1 conditioned medium (Supplementary Figure S4) indicating that these barrier-altering effects do not require direct macrophage-IEC cell-cell contact. Consistent with reduced TEER and increased FD4 permeability in PTPN2-KD IECs and IECs co-cultured with PTPN2-KD macrophages, claudin-2 expression was enhanced, claudin-4, E-cadherin, JAM-A, and tricellulin levels were reduced, while occludin approached a significant decrease (P=0.073) (Figure 3A+ Supplementary Figure S3C, supplementary File 2). In line with increased FD4 permeability, ZO-1 localization to the cell border was compromised in IECs cultured in PTPN2-KD THP-1-macrophage-conditioned medium (Supplementary Figure S5A). PTPN2-KD in Caco-2BBe cells per se affected ZO-1 localization at cell borders, resulting in a ruffled appearance of the cell-cell border, low-level ZO-1 internalization, small gaps between neighboring cells, and increased claudin-2 expression/membrane localization, effects that were further increased upon culture in PTPN2-KD THP-1-conditioned medium (Supplementary Figure S5). These barrier-disrupting effects were not due to reduced viability, since levels of the cell death markers cleaved caspase-3 and LDH were unaffected (Supplementary Figure S6). The barrier-compromising effect of TCPTP-deficient macrophages was confirmed in vivo in Ptpn2-LysMCre mice, which lack PTPN2 in myeloid cells32. FD4 flux from the intestinal lumen to blood was significantly increased in Ptpn2-LysMCre mice, while creatinine flux, used to functionally measure the ‘pore’ pathway36, remained unaffected (Figure 3B). In contrast, Ussing chamber studies on ex vivo proximal and distal colonic mucosae indicated a significant decrease in TEER (Figure 3C), identifying the presence of a ‘pore’ barrier defect in Ptpn2-LysMCre mice. In line with our in vitro data, permeability to FD4, but not to 70 kDa Rhodamine-dextran was enhanced (Figure 3E) indicating that increased FD4 translocation was not associated with tissue damage. Ex vivo tissue viability was confirmed by functional ion transport responses to Ca2+ or cAMP-dependent electrogenic changes in short-circuit current (Supplementary Figure S7). In line with our in vitro data, colonic IECs from Ptpn2-LysMCre mice expressed higher levels of claudin-2, but reduced levels of claudin-4, and JAM-A, while occludin verged on a significant decrease (P=0.0585) (Figure 3D, Supplementary File 2). We detected increased levels of claudin-2 but reduced/more diffuse E-cadherin localization in proximal colon from Ptpn2-LysMCre mice (Figure 3E), and more diffuse ZO-1 localization (Supplementary Figure S8A). In contrast, cell viability and epithelial morphology were not affected in Ptpn2-LysMcre mice (Supplementary Figure S8B+C). Consistent with previous reports32, unchallenged Ptpn2-LysMCre did not show signs of diarrhea, but they were more susceptible to chemical-induced colitis (Supplementary Figure S8D). Finally, the effects of PTPN2-deficient macrophages on IEC permeability were recapitulated in mouse small intestinal organoids co-cultured with macrophages from WT or Ptpn2-LysMCre mice (Figure 3F). These data confirm that loss of TCPTP in myeloid cells compromises intestinal epithelial barrier function in vivo and validate our in vitro data demonstrating that TCPTP plays an essential role in regulating macrophage – IEC interactions to support intestinal barrier function.
Figure 3. Loss of PTPN2 results in reduced IEC barrier function in vitro and in vivo.

A) Caco-2BBe cells expressing either non-targeting control shRNA (shCtr Caco-2BBe) or PTPN2-specific shRNA (shPTPN2 Caco-2BBe) were seeded on transwells and left to differentiate for 9 days prior to co-culture with PMA-differentiated THP-1 macrophages expressing either non-specific control (Ctr) or PTPN2-specific (KD) shRNA, for up to 24 h. The graphs show TEER over time, FD4 flux and representative Western blot pictures of the indicated proteins after 24h. B) Ptpn2-LysMCre mice (KO) and WT littermates (WT) were gavaged with 80 mg/ml FD4 and 100mg/ml creatinine, and serum levels assessed 5 h later. C) Mucosa from proximal (PC) and distal (DC) colon was mounted in Ussing chambers, TEER measured, FD4/Rhodamine B-dextran 70 kDa (RD70) flux from the mucosal to the serosal media measured 2 h later. D) IECs from Ptpn2-LysMCre mice were analyzed by Western blot for the indicated proteins, each lane represents one individual mouse. E) Representative images from proximal colon sections of WT and Ptpn2-LysMCre littermates stained for Claudin-2 and E-cadherin. Scale bar: 50mm (overview) and 25mm (magnification). F) Mouse small intestinal organoid monolayers were co-cultured with bone-marrow-derived macrophages from WT or Ptpn2-LysMCre (KO) mice for 24 h and TEER/FD4 flux measured as in A).*P < .05, **P < .001, 1-way ANOVA. Representative results from one out of three independent experiments with n = 3–4, each (A-D), or as indicated within the Figure (E+F).
PTPN2-deficient macrophages preferentially differentiate into pro-inflammatory M1-like macrophages and express increased IL-6.
Given the permeability-inducing effect of classically activated/M1 macrophages as opposed to the barrier-sealing effect of alternatively activated/M2 macrophages, combined with the barrier-disrupting effects of loss of TCPTP, we next assessed how KD/loss of PTPN2 affected macrophage phenotypes. While control (Ctr) THP-1 macrophages only expressed high levels of CD86 upon IFN-γ/LPS treatment (M1 induction), and down-regulated CD86 expression upon culture with IL-4 (M2 induction), PTPN2-KD THP-1 macrophages expressed high levels of CD86 regardless of their polarization. Conversely, Ctr THP-1 macrophages readily up-regulated the M2 marker CD206 upon culture with IL-4, which was suppressed in PTPN2-KD THP-1 macrophages (Figure 4A). Consistently, the frequency of CD86+ pro-inflammatory (M1-like) macrophages was elevated in tissue-resident macrophages from Ptpn2-LysMCre mice, while CD206+ anti-inflammatory (M2-like) macrophages were reduced (Figure 4B). Analysis of the monocyte-macrophage continuum revealed higher proportions of Ly6Cint, MHCIIhigh recently infiltrating, but reduced proportions of Ly6C−, MHCIIhigh tissue resident macrophages (Supplementary Figure S9). To further investigate the differences between WT and PTPN2-deficient macrophages, a customized NanoString® nCounter multiplex RNA analysis was performed on colonic lamina propria macrophages from WT and Ptpn2-LysMCre mice (Figure 4C). Ptpn2-deficient macrophages expressed increased mRNA levels of the M1 marker Cd80, while M2 markers Cd206, Cd163 and Arg1 were decreased. In addition, Il6 was highly upregulated in PTPN2-deficient macrophages, while mRNA levels of the IL-6 and IL-4 receptor subunits Il6ra and Il4ra were reduced. In line with increased Il6 mRNA expression in PTPN2-deficient macrophages, mRNA and protein expression of IL-6 was higher in colon tissue from Ptpn2-LysMCre mice when compared to their WT littermates (Figure 4D+E). Moreover, in our co-culture system, PTPN2-KD THP-1 cells expressed highly increased levels of IL6 mRNA and secreted more IL-6 protein (Figure 4F+G). They also expressed higher levels of TNFA and CD86, but decreased levels of CD206 and IL10 mRNA (Supplementary Figure S10), further indicating that PTPN2-deficient macrophages shift towards a pro-inflammatory/M1-like phenotype.
Figure 4. PTPN2-deficient macrophages show a pro-inflammatory M1-like phenotype.
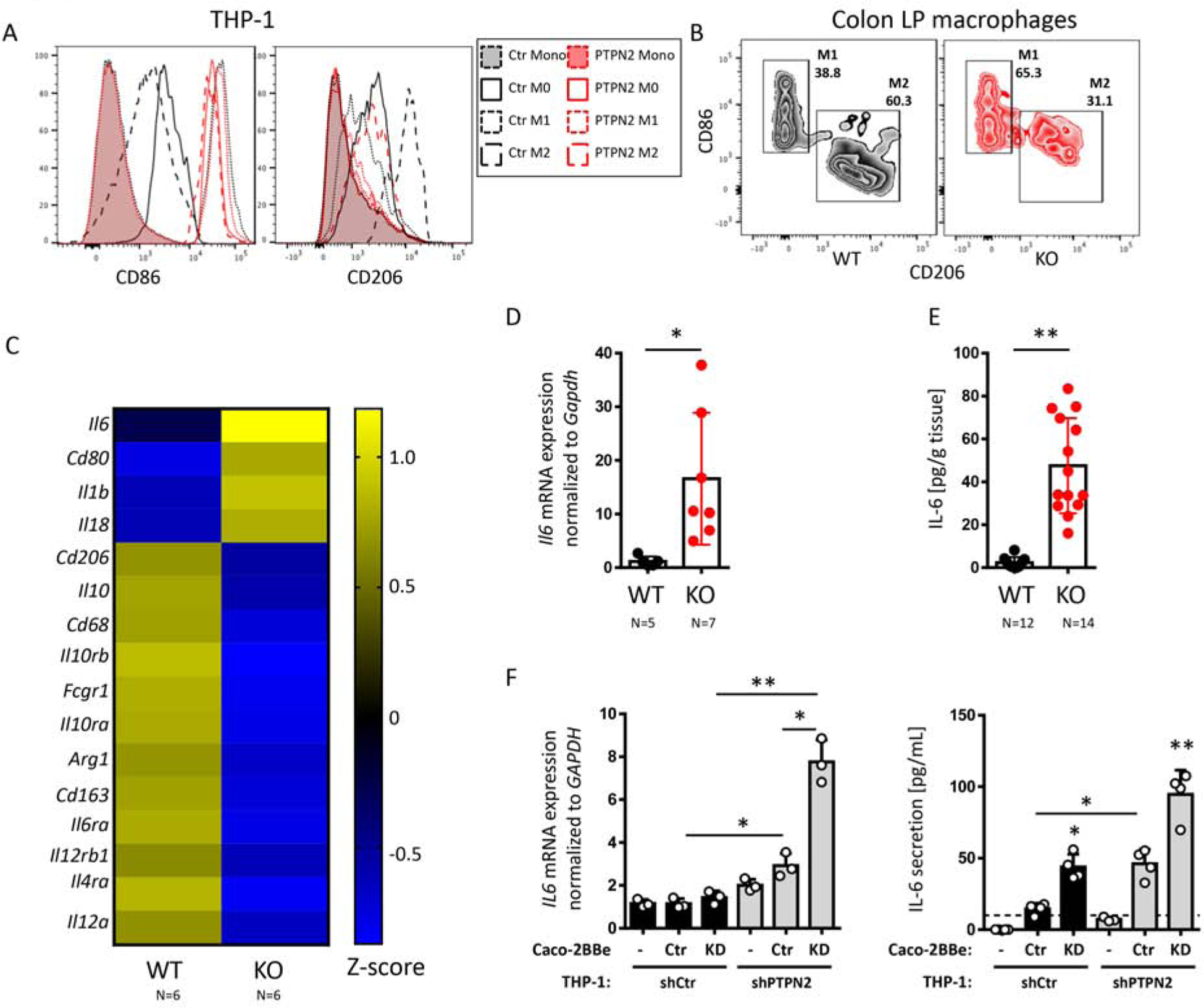
A) THP-1 cells expressing either non-targeting control (Ctr) or PTPN2-specific (PTPN2) shRNA were left untreated (monocytes, Mono), differentiated into non-polarized macrophages (M0), or treated with IFN-γ + LPS (M1) or with IL-4 (M2) for 24 h and analyzed for surface expression of CD86 and CD206. B) Colonic lamina propria immune cells were isolated from WT and Ptpn2-LysMCre mice and CD11b+/CD64+, Ly6C-MHC-II+ macrophages analyzed for CD86+ (M1-like) and CD206+ (M2-like) macrophages. Gating as shown in Supplementary Figure S9A. C) Nanostring analysis on colonic lamina propria macrophages from WT and Ptpn2-LysMCre mice. Shown are differentially expressed macrophage-related genes (color-coded average z-score values in each group). D) Il6 mRNA levels and E) IL-6 protein levels in distal colon segments from Ptpn2-LysMCre (KO) and WT littermates. F: THP-1 macrophages expressing control (shCtr) or PTPN2-specific (shPTPN2) shRNA were co-cultured with Caco-2BBe IECs expressing non-targeting control (Ctr) or PTPN2-specific (KD) shRNA. Depicted are F) IL6 mRNA expression and IL-6 protein levels in the basolateral medium. *P < .05, **P < .001, 1-way ANOVA. Representative results from one out of three independent experiments with n = 3–4 (A, B, F), each, or as indicated within the Figure (C, D, E).
Recombinant IL-6 recapitulates increased STAT3 activation and barrier defects.
Consistent with STAT3 being a major downstream signaling molecule of the IL-6 receptor, and elevated IL-6 release from PTPN2-deficient macrophages, co-culture with PTPN2-deficient macrophages promoted STAT3 activation in IECs, while PTPN2-KD IEC showed increased basal STAT3 phosphorylation levels that was further increased by co-culture with PTPN2-deficient macrophages (Figure 5A). Similarly, STAT3 phosphorylation levels were also elevated in colonic IECs isolated from Ptpn2-LysMCre mice (Figure 5B), and in Caco-2BBe co- cultured with THP-1 monocytes and M1 macrophages (Figure 5C). Adding IL-6 to the cell culture media (alone or even more pronounced with Ctr macrophage-conditioned medium) promoted STAT3 phosphorylation (Figure 5D) and induced a similar phenotype to culture with PTPN2-deficient macrophages, i.e. increased FD4 permeability, reduced TEER (Figure 5E), and internalization of ZO-1 (Figure 5F), indicating that exogenous IL-6 recapitulates the IEC barrier-disrupting phenotype of PTPN2-deficient macrophages. This further suggests that enhanced IL-6 secretion might mediate the leaky barrier phenotype observed upon loss of PTPN2 in macrophages and/or IECs.
Figure 5. Loss of PTPN2 in IECs or macrophages promotes STAT3 phosphorylation in IECs.

A) Caco-2BBe cells were co-cultured with polarized THP-1 macrophages for 24 h and lysates analyzed for STAT3 phosphorylation. B) IECs from Ptpn2-LysMCre and WT littermates were analyzed by Western blot for phosphorylated and total STAT3. C) Caco-2BBe expressing control (shCtr) or PTPN2-specific (shPTPN2) shRNA were co-cultured for 24 h with THP-1 macrophages expressing control (Ctr) or PTPN2-specific (KD) shRNA and Caco-2BBe lysates were analyzed for STAT3 phosphorylation. D+E: Caco-2BBe cells were co-cultured with Ctr or PTPN2-knockdown (KD) THP-1 macrophages, with recombinant IL-6, or with Ctr THP1 macrophages + recombinant IL-6 for 24 h and D) IEC cell lysates analyzed for STAT3 phosphorylation; E) analysis of FD4 flux after 24 h and TEER over time. F) Caco-2BBe cells were grown on coverslips and incubated with supernatant from Ctr or KD THP-1 macrophages, recombinant IL-6, or supernatant from Ctr THP-1 macrophages + rIL-6 and stained for ZO-1 and claudin-2. White arrowheads: internalized ZO-1, white asterisks: gaps between neighboring IECs, yellow arrowheads: claudin-2 membrane localization, dashed line: z-stack cut. Scale bars: 10 mm. *P < .05, 1-way ANOVA. Representative results from one out of three independent experiments with n = 2–3 (A-D) or n = 6 (E+F).
Despite high IL-6 in the co-culture environment, we only detected minimal STAT3 phosphorylation in PTPN2-KD macrophages, while phosphorylation of STAT1 and NF-κB p65 was clearly increased (Supplementary Figure S11), suggesting a reduced/altered response to IL-6 in PTPN2-KD macrophages.
Inhibition of IL-6 rescues the leaky barrier phenotype in vitro.
Consistent with IL-6 being responsible for the observed effects, inhibition of IL-6 completely restored the ability of PTPN2-KD macrophages to promote barrier function, normalized TEER and FD4 flux in PTPN2-KD IECs (Figure 6A), reversed the upregulation of claudin-2, restored claudin-4 and tricellulin expression, and reduced STAT3 phosphorylation in IECs (Figure 6B). Similar effects were observed in co-cultures of organoid monolayers with PTPN2-deficient macrophages (Supplementary Figure 12). Furthermore, ZO-1 internalization and intercellular gap formation upon loss of epithelial TCPTP was mitigated by IL-6 inhibition (Figure 6C). This strongly indicates that elevated IL-6 secretion drives the increased permeability phenotype observed upon loss of TCPTP in IEC and/or macrophages. IL-6 inhibition also prevented monocyte and M1 macrophage-induced barrier disruption, while inhibition of IL-4 had no effect on the barrier-inducing capacity of M2 macrophages (Supplementary Figure S13A+B), and IL-1β, a pro-inflammatory cytokine elevated in Ptpn2-LysMCre mice32, was very low in our co-culture system (Supplementary Figure S13C). Furthermore, STAT3 siRNA transfection confirmed that elevated claudin-2 expression upon IL-6 addition or co-culture with PTPN2-deficient macrophages was STAT3 dependent (Supplementary Figure S14).
Figure 6. IL-6 mediates TCPTP-dependent barrier defects in vitro and in vivo.
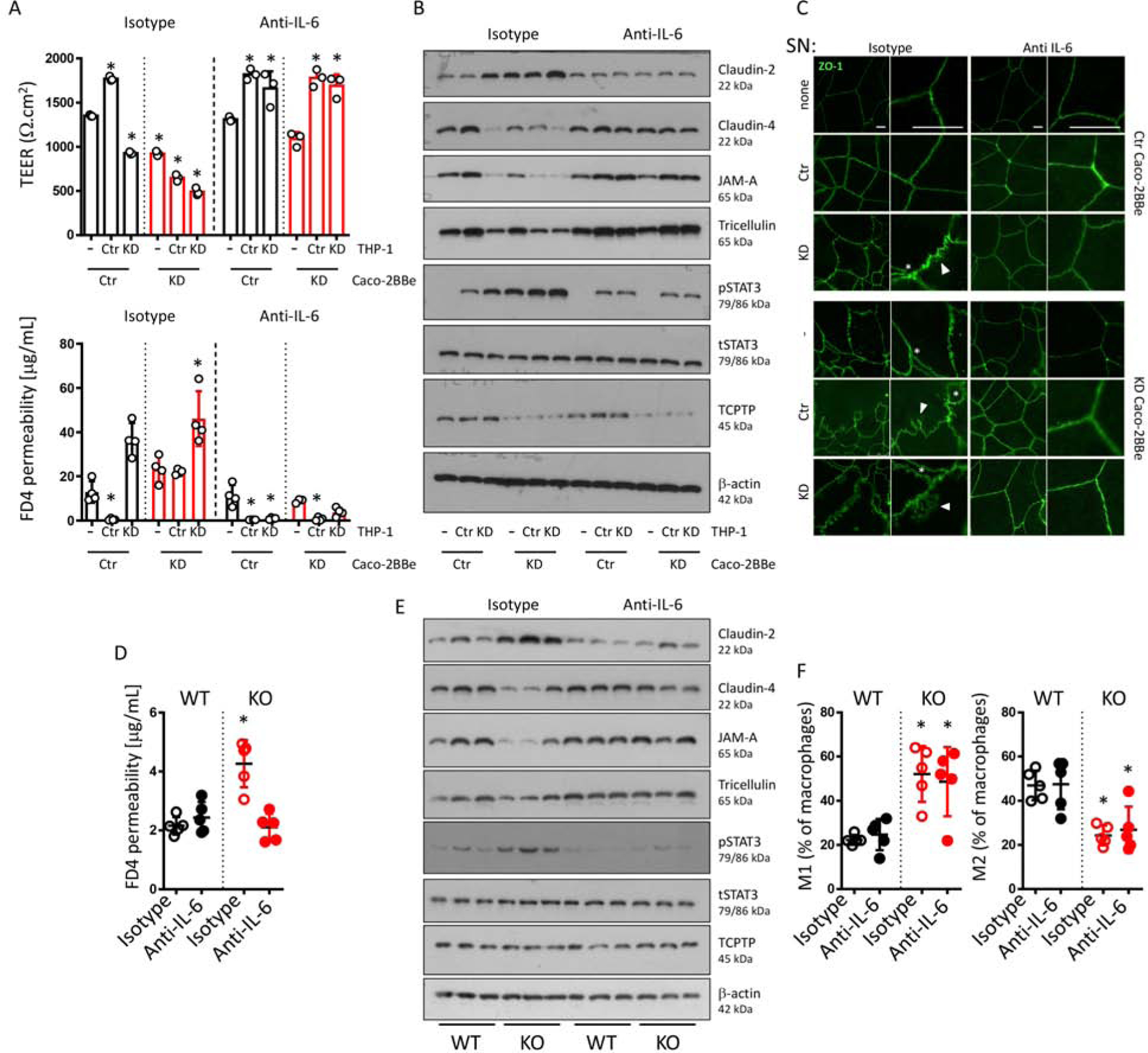
A+B) Caco-2BBe cells expressing either non-targeting control shRNA (Ctr) or PTPN2-specific shRNA (KD) were co-cultured with THP-1 macrophages expressing either non-specific control (Ctr) or PTPN2-specific (KD) shRNA in the presence of an isotype control, or an inhibitory anti-IL-6 antibody for 24 h. A) TEER (top), FD4 flux (bottom), and B) representative Western blot pictures for the indicated proteins. C) Caco-2BBe cells were grown on cover slides and incubated with normal medium (−), supernatant from THP-1 macrophages expressing control shRNA (Ctr) or PTPN2-specific shRNA (KD). White arrowheads point to internalized ZO-1, white asterisks indicate gaps between neighboring IECs. Scale bars: 10mm. D-F: Ptpn2-LysMCre mice (KO) and WT littermates (WT) were treated with anti-IL-6 antibody for 7 days. D) Mice were gavaged with 80 mg/ml FD4 and permeability into the serum assessed after 5 h. E) Colonic IEC were analyzed for the indicated proteins by Western blot, each lane represents one individual mouse. F) Colonic lamina propria immune cells were analyzed by flow cytometry for the proportion of M1 (CD86high, CD206low) and M2 (CD86int, CD206high) macrophages (gated on CD11b+, Ly6G−, CD64+ cells). *P < .05, **P < .001, 1-way ANOVA. Representative results from one out of three independent experiments with n = 3–4, each (A-C), or one out of 2 independent experiments with n = 5 per group (D-F).
The underlying barrier defect in Ptpn2-LysMCre mice is rescued by IL-6 inhibition.
In line with our in vitro co-culture data, IL-6 inhibition by injection of anti-IL-6 antibody for 7 days, reduced FD4 permeability to normal levels in Ptpn2-LysMCre mice with no effect in WT littermates (Figure 6D). Anti-IL-6 treatment also normalized the increased STAT3 phosphorylation and claudin-2 protein expression in IEC from Ptpn2-LysMCre mice (Figure 6E), while promoting expression of the barrier-sealing molecules claudin-4 and tricellulin (Figure 6F). This indicates that in vivo, the barrier defects resulting from loss of TCPTP in macrophages are dependent on elevated IL-6 levels. Of interest, IL-6 inhibition did not reduce the frequency of inflammatory macrophages in Ptpn2-LysMCre mice, nor did it affect M2 macrophage proportions (Figure 6F), indicating that a normalization of macrophage function was not necessary for restoration of epithelial barrier function.
IBD-patient derived macrophages carrying the disease-associated PTPN2-variant promote barrier dysfunction
To assess the relevance of our findings for human IBD, we used monocyte-derived macrophages from patients of the Swiss IBD cohort, which have previously been genotyped for various IBD-associated genes, including the loss-of-function PTPN2 SNP rs189321737. Co-culture of Caco-2BBe cells with macrophages from healthy control subjects increased TEER and reduced FD4 permeability (Figure 7A+B). These effects were significantly mitigated in Caco-2BBe cells co-cultured with macrophages derived from PTPN2 WT IBD patients. Co-culture with macrophages from patients carrying the disease-associated variant in SNP rs1893217, however, resulted in a marked decrease in TEER and enhanced FD4 permeability, indicating that presence of this variant severely compromises macrophage-induced barrier sealing (Figure 7A + B). Furthermore, PTPN2-variant macrophages promoted epithelial STAT3 phosphorylation and claudin-2 expression, while levels of claudin-4 were decreased (Figure 7C). No changes in markers of IEC viability (caspase-3 cleavage; LDH) were observed (Figure 7C).
Figure 7. Monocyte-derived macrophages from IBD patients carrying the disease-associated PTPN2 SNP compromise barrier function.
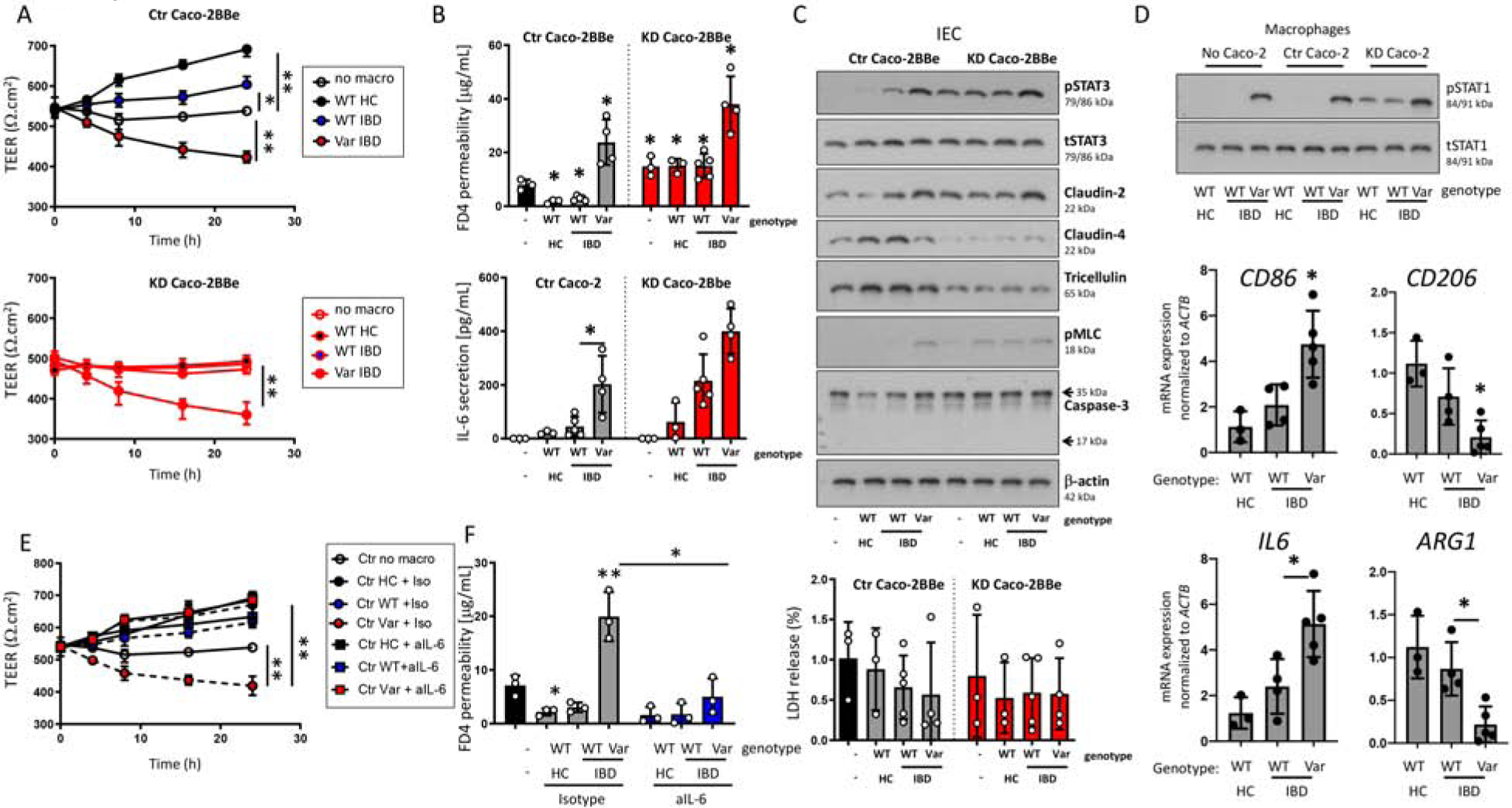
A-D: Monocyte-derived macrophages from healthy controls (WT HC, n = 3; WT for SNP rs1893217), IBD patients not-carrying the disease-associated variant (WT IBD; n = 5) or IBD patients heterozygous for SNP rs1893217 (Var IBD, n = 4) were co-cultured with Caco-2BBe cells expressing non-targeting control (Ctr) or PTPN2-specific (KD) shRNA. The graphs show A) TEER over time, B) FD4 flux and IL-6 secretion after 24 h, C) representative Western blot pictures for the indicated proteins in Caco-2BBe lysates, and LDH levels in the basolateral supernatant, D) representative Western blot pictures for the indicated proteins and mRNA expression of indicated genes normalized to ACTB and the median of WT HC in macrophages. E+F: Caco-2BBe were co-cultured with patient-derived macrophages as in A-D in the presence of an isotype control or a neutralizing anti-IL-6 antibody. The graphs show E) TEER over time and F) FD4 flux after 24 h co-culture. *P < .05, **P < .01; 1-way ANOVA
Consistent with our results in cell lines, PTPN2-variant macrophages showed elevated STAT1 phosphorylation and enhanced markers of classical macrophage activation/M1 polarization (i.e. increased mRNA expression of CD86, but reduced Arg1 and CD206 mRNA expression; Figure 7D). Patient-derived macrophages carrying the PTPN2-variant secreted higher levels of IL-6, an effect further potentiated in the presence of PTPN2-KD Caco-2BBe cells (Figure 7B). Consistent with our other in vitro and in vivo data, the decrease in TEER and increase in FD4 permeability arising from SNP rs1893217 was reversed by inhibition of IL-6 (Figure 7E+F). The clinical relevance of our findings is emphasized by the PTPN2 SNP rs1893217 severely compromising macrophage-IEC regulation, ultimately promoting epithelial barrier defects.
Discussion
We demonstrate that TCPTP is a crucial regulator of macrophage-IEC crosstalk and defective TCPTP expression compromises normal barrier function resulting in sub-clinical barrier defects that might promote the development of intestinal inflammation. These effects were recapitulated using macrophages from IBD-patients that carry the disease-associated SNP in the PTPN2 gene, indicating a clear clinical relevance of our findings.
Alternatively activated/M2 macrophages promoted barrier tightness via induction of barrier-sealing tight junction molecules in epithelial cells, while classically activated/M1 macrophages and monocytes, which typically infiltrate the intestine in large numbers upon infections/inflammation15, promoted the expression of the pore-forming tight junction molecule, claudin-2, and triggered internalization and degradation of barrier-sealing molecules, such as claudin-4 and E-cadherin, ultimately causing a leaky barrier. Notably, PTPN2-deficient macrophages were intrinsically prone to express higher levels of pro-inflammatory/M1 macrophage markers, which might explain their failure to promote barrier properties in IECs.
The increase in barrier permeability arising from loss of TCPTP expression and function in macrophages was likely mediated by increased expression of IL-6, since inhibition of IL-6 rescued the increased paracellular passage of electrolytes and macromolecules. However, inhibition of IL-6 did not revert the M1 macrophage phenotype, indicating that the macrophage-induced barrier leak effects resulted from an IEC-specific effect of IL-6. In IECs, IL-6 promotes expression of the pore-forming claudin-2 and MLC-kinase38, which might explain the decreased TEER in PTPN2-deficient IECs and enhanced MLC phosphorylation enabling gap formation and increased FD4 permeability. The clear effect of anti-IL-6 on IECs without affecting macrophage polarization indicates that the correction of a barrier defect induced by pro-inflammatory macrophages does not require macrophage normalization, which is of great interest when considering potential treatment strategies to restore epithelial barrier function.
In addition to the intrinsic preference of PTPN2-deficient macrophages to polarize into inflammatory subsets, loss of PTPN2 in IECs also promoted inflammatory macrophage polarization. In contrast to the clear role of IL-6 in macrophage-mediated effects on IECs, PTPN2-deficient IECs promoted a pro-inflammatory phenotype in macrophages in an IL-6-independent manner. In macrophages, IL-6 and subsequent STAT3 activation enhances alternative activation/M2 polarization, although it does not induce it on its own39, 40. While IL-6 levels were highly elevated upon deletion of PTPN2 in either IEC or macrophages, the macrophage phenotype was shifted towards a pro-inflammatory/M1 phenotype, an effect possibly due to reduced IL-4 receptor expression in colonic macrophages. Despite high levels of IL-6, STAT3 activation was moderate in PTPN2-deficient macrophages, which might result from the marked decrease in IL-6 receptor expression in those cells. In contrast to moderate STAT3 activation, we observed high levels of STAT1 phosphorylation, and increased NF-κB activation. STAT1 as well as NF-κB have been shown to promote classical macrophage activation/M1 polarization41. Therefore, the overall balance between STAT3 and STAT1/NF-κB activation might decide whether a macrophage develops towards a classically activated/M1 or towards an alternative/M2 phenotype. TCPTP dephosphorylates both STAT1 and STAT342, but has a particularly high affinity for STAT143 and thus may fine-tune the cellular response to molecules that activate both pathways. Besides elevated IL-6 levels upon loss of TCPTP in IEC and/or macrophages, and in line with previous reports29, we observed an increase in TNFα production. TNFα is a potent activator of NFkB, which in turn promotes classical macrophage activation/M1 polarization44. Thus, elevated TNFα levels together with decreased responsiveness to IL-4 and IL-6, and increased STAT1 phosphorylation, might contribute to the enhanced M1 polarization we observed upon loss of TCPTP. While IL-6 also drives T-cell activation/differentiation32, T-cells were not significantly altered in Ptpn2-LysMCre mice. However, we cannot exclude a potential intermediary role for other immune cells in the FD4 flux response to macrophage IL-6 in vivo.
We demonstrate that IEC and macrophages critically influence each other to maintain a functional intestinal epithelial barrier and altered function of one cell type fundamentally affects barrier promoting/disrupting capabilities of the other. Normal IECs educate macrophages to develop a more tolerogenic phenotype, an effect completely lost upon reduction of TCPTP activity in IEC. The barrier-promoting effects of M0/M2 macrophages are intriguing, and while not the focus of our study, worthy of follow-up investigations. Notably, PTPN2-deficient macrophages were intrinsically prone to develop into an inflammatory phenotype and produced high levels of inflammatory molecules IL-6 and TNF with reduced levels of the anti-inflammatory cytokine IL-10. This inflammatory environment created by PTPN2-deficient macrophages promoted the expression of pore-forming tight junction proteins and internalization of barrier sealing molecules, resulting in a more permeable barrier. Cumulatively, the loss of functional TCPTP in both macrophages and IECs potentiated the barrier-compromising effect of a loss of TCPTP in either cell type. Interestingly, despite the effect of PTPN2-deficient macrophages on the intestinal barrier, Ptpn2-LysMCre mice do not develop spontaneous intestinal inflammation, but they are more susceptible to colitis induction32. While previously reported colitis-exacerbating effects in Ptpn2-LysMCre mice were driven by excessive IL-1β32 production, the underlying barrier defect in unchallenged Ptpn2-LysMCre mice was mediated by IL-6, and IL-1β did not play a significant role.
Epithelial barrier regulation is a complex and highly integrated process involving both tight and adherens junctions. While E-cadherin deficiency is embryonically lethal45, and its loss in adult IECs promotes barrier disruption46, loss of JAM-A enhances permeability and mediates aberrant macrophage function in the intestine34, which is consistent with our findings that PTPN2-deficiency decreased JAM-A expression and compromises macrophage-IEC interactions. Claudin-2 enhances ion/water permeability, is a marker of disrupted barrier function in IBD, and is negatively regulated by TCPTP5. Here, we demonstrated the essential role of macrophages in regulating the fine-tuned expression of these molecules, and identified PTPN2/TCPTP as a critical factor synchronizing macrophage-mediated barrier promotion.
Taken together, our findings demonstrate the importance of bidirectional macrophage-IEC signaling for IEC barrier maintenance and reveal that the IBD risk gene, PTPN2, crucially contributes to normal macrophage-IEC interactions governing macrophage differentiation and epithelial barrier function that represent targets for therapeutic intervention in IBD. Specifically, our results indicate that IBD patients with aberrant macrophage-IEC interactions, and especially those harboring PTPN2 loss-of-function variants, might benefit from therapeutics that target IL-6, or the JAK/STAT pathway to correct compromised intestinal barrier function.
Supplementary Material
What You Need to Know.
Background and Context:
Interactions between epithelial cells and macrophages are important for intestinal barrier homeostasis. The effects of variants in genes associated with increased risk of inflammatory bowel disease (IBD) on intestinal permeability is poorly understood.
New Findings:
In co-cultures of mouse or human intestinal epithelial cells and macrophages from mice or patients with IBD respectively, the product of a gene associated with increased risk of IBD, PTPN2, regulated interaction between these cell types to control epithelial barrier properties and macrophage polarization.
Limitations:
This study was performed with mice, and human cells.
Impact:
Loss of function variants in PTPN2 can compromise pathways regulating epithelial barrier and innate immune functions. Strategies to alter these pathways might be developed for personalized treatment of IBD.
Funding:
This study was supported by the NIH (2R01DK091281, 1R01AI153314; to DFM) and a research stipend from the Swiss National Science Foundation (to MRS). The funding institutions had no role in the study design in the collection, analysis, and interpretation of data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no conflicts of interest.
References
- 1.McCole DF, Barrett KE. Varied role of the gut epithelium in mucosal homeostasis. Curr Opin Gastroenterol 2007;23:647–54. [DOI] [PubMed] [Google Scholar]
- 2.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 2009;9:799–809. [DOI] [PubMed] [Google Scholar]
- 3.Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev 2013;93:525–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Itallie CM, Holmes J, Bridges A, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 2008;121:298–305. [DOI] [PubMed] [Google Scholar]
- 5.Krishnan M, McCole DF. T cell protein tyrosine phosphatase prevents STAT1 induction of claudin-2 expression in intestinal epithelial cells. Ann N Y Acad Sci 2017;1405:116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen L, Weber CR, Raleigh DR, et al. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol 2011;73:283–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol 2010;28:573–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mao JW, Tang HY, Zhao T, et al. Intestinal mucosal barrier dysfunction participates in the progress of nonalcoholic fatty liver disease. Int J Clin Exp Pathol 2015;8:3648–58. [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards CJ. Commensal gut bacteria and the etiopathogenesis of rheumatoid arthritis. J Rheumatol 2008;35:1477–14797. [PubMed] [Google Scholar]
- 10.Westall FC. Abnormal hormonal control of gut hydrolytic enzymes causes autoimmune attack on the CNS by production of immune-mimic and adjuvant molecules: A comprehensive explanation for the induction of multiple sclerosis. Med Hypotheses 2007;68:364–9. [DOI] [PubMed] [Google Scholar]
- 11.Makela M, Vaarala O, Hermann R, et al. Enteral virus infections in early childhood and an enhanced type 1 diabetes-associated antibody response to dietary insulin. J Autoimmun 2006;27:54–61. [DOI] [PubMed] [Google Scholar]
- 12.Clairembault T, Leclair-Visonneau L, Coron E, et al. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol Commun 2015;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014;14:667–85. [DOI] [PubMed] [Google Scholar]
- 14.De Schepper S, Verheijden S, Aguilera-Lizarraga J, et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018;175:400–415 e13. [DOI] [PubMed] [Google Scholar]
- 15.Bain CC, Scott CL, Uronen-Hansson H, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol 2013;6:498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bain CC, Bravo-Blas A, Scott CL, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol 2014;15:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantovani A, Sica A, Sozzani S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004;25:677–86. [DOI] [PubMed] [Google Scholar]
- 18.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010;32:593–604. [DOI] [PubMed] [Google Scholar]
- 19.Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cobb JE, Plant D, Flynn E, et al. Identification of the tyrosine-protein phosphatase non-receptor type 2 as a rheumatoid arthritis susceptibility locus in europeans. PLoS One 2013;8:e66456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glas J, Wagner J, Seiderer J, et al. PTPN2 gene variants are associated with susceptibility to both Crohn’s disease and ulcerative colitis supporting a common genetic disease background. PLoS One 2012;7:e33682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simoncic PD, Lee-Loy A, Barber DL, et al. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr Biol 2002;12:446–53. [DOI] [PubMed] [Google Scholar]
- 24.Scharl M, Hruz P, McCole DF. Protein tyrosine phosphatase non-receptor Type 2 regulates IFN-gamma-induced cytokine signaling in THP-1 monocytes. Inflamm Bowel Dis 2010;16:2055–64. [DOI] [PubMed] [Google Scholar]
- 25.Scharl M, Rudenko I, McCole DF. Loss of protein tyrosine phosphatase N2 potentiates epidermal growth factor suppression of intestinal epithelial chloride secretion. Am J Physiol Gastrointest Liver Physiol 2010;299:G935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiede F, Shields BJ, Chew SH, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest 2011;121:4758–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiede F, Chew SH, van Vliet C, et al. Strain-dependent differences in bone development, myeloid hyperplasia, morbidity and mortality in ptpn2-deficient mice. PLoS One 2012;7:e36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scharl M, Paul G, Weber A, et al. Protection of epithelial barrier function by the Crohn’s disease associated gene protein tyrosine phosphatase n2. Gastroenterology 2009;137:2030–2040 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scharl M, McCole DF, Weber A, et al. Protein tyrosine phosphatase N2 regulates TNF{alpha}-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut 2011;60:189–97. [DOI] [PubMed] [Google Scholar]
- 30.Scharl M, Mwinyi J, Fischbeck A, et al. Crohn’s disease-associated polymorphism within the PTPN2 gene affects muramyl-dipeptide-induced cytokine secretion and autophagy. Inflamm Bowel Dis 2011. [DOI] [PubMed] [Google Scholar]
- 31.Spalinger MR, Kasper S, Chassard C, et al. PTPN2 controls differentiation of CD4(+) T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol 2015;8:918–29. [DOI] [PubMed] [Google Scholar]
- 32.Spalinger MR, Manzini R, Hering L, et al. PTPN2 Regulates Inflammasome Activation and Controls Onset of Intestinal Inflammation and Colon Cancer. Cell Rep 2018;22:1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol 2003;3:331–41. [DOI] [PubMed] [Google Scholar]
- 34.Luissint AC, Williams HC, Kim W, et al. Macrophage-dependent neutrophil recruitment is impaired under conditions of increased intestinal permeability in JAM-A-deficient mice. Mucosal Immunol 2019;12:668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCole DF, Rogler G, Varki N, et al. Epidermal growth factor partially restores colonic ion transport responses in mouse models of chronic colitis. Gastroenterology 2005;129:591–608. [DOI] [PubMed] [Google Scholar]
- 36.Tsai PY, Zhang B, He WQ, et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe 2017;21:671–681 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yilmaz B, Spalinger MR, Biedermann L, et al. The presence of genetic risk variants within PTPN2 and PTPN22 is associated with intestinal microbiota alterations in Swiss IBD cohort patients. PLoS One 2018;13:e0199664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem 2011;286:31263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin Z, Ma T, Lin Y, et al. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J Cell Biochem 2018;119:9419–9432. [DOI] [PubMed] [Google Scholar]
- 40.Choi JW, Kwon MJ, Kim IH, et al. Pyropia yezoensis glycoprotein promotes the M1 to M2 macrophage phenotypic switch via the STAT3 and STAT6 transcription factors. Int J Mol Med 2016;38:666–74. [DOI] [PubMed] [Google Scholar]
- 41.Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol 2013;33:1135–44. [DOI] [PubMed] [Google Scholar]
- 42.Grohmann M, Wiede F, Dodd GT, et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018;175:1289–1306 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol 2002;22:5662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu CP, Zhang X, Tan QL, et al. NF-kappaB pathways are involved in M1 polarization of RAW 264.7 macrophage by polyporus polysaccharide in the tumor microenvironment. PLoS One 2017;12:e0188317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larue L, Ohsugi M, Hirchenhain J, et al. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A 1994;91:8263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grill JI, Neumann J, Hiltwein F, et al. Intestinal E-cadherin Deficiency Aggravates Dextran Sodium Sulfate-Induced Colitis. Dig Dis Sci 2015;60:895–902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.