

Abstract
Dysregulated Wnt signaling is linked to major neurodegenerative diseases, including Alzheimer disease (AD). In mouse models of AD, activation of the canonical Wnt signaling pathway improves learning/memory, but the mechanism for this remains unclear. The decline in brain function in AD patients correlates with reduced glucose utilization by neurons. Here, we test whether improvements in glucose metabolism mediate the neuroprotective effects of Wnt in AD mouse model. APPswe/PS1dE9 transgenic mice were used to model AD, Andrographolide or Lithium was used to activate Wnt signaling, and cytochalasin B was used to block glucose uptake. Cognitive function was assessed by novel object recognition and memory flexibility tests. Glucose uptake and the glycolytic rate were determined using radiotracer glucose. The activities of key enzymes of glycolysis such as hexokinase (HK) and phosphofructokinase (PFK), Adenosine triphosphate (ATP)/ Adenosine diphosphate (ADP) levels and the pentose phosphate pathway (PPP) and activity of glucose-6 phosphate dehydrogenase were measured. Wnt activators significantly improved brain glucose utilization and cognitive performance in transgenic mice. Wnt signaling enhanced glucose metabolism by increasing the expression and/or activity of HK, PFK and AMP-activated protein kinase (AMPK). Inhibiting glucose uptake partially abolished the beneficial effects of Wnt signaling on learning/memory. Wnt activation also enhanced glucose metabolism in cortical and hippocampal neurons, as well as brain slices derived from APPswe/PS1E9 transgenic mice. Combined, these data provide evidence that the neuroprotective effects of Wnt signaling in AD mouse models result, at least in part, from Wnt-mediated improvements in neuronal glucose metabolism.
Keywords: Glucose metabolism, Wnt signaling, Alzheimer disease, Neuroprotection
Text for Schematic Abstract
Dysregulated Wnt signaling is linked to major neurodegenerative diseases, including Alzheimer disease (AD). The decline in brain function in AD patients correlates with reduced glucose utilization by neurons. Here, we test whether improvements in glucose metabolism mediate the neuroprotective effects of Wnt in AD mouse model. APPswe/PS1dE9 transgenic mice were used to model AD, andrographolide or lithium was used to activate Wnt signaling. Wnt activators significantly improved brain glucose utilization and cognitive performance in transgenic mice. The neuroprotective effects of Wnt signaling in AD mouse models result, at least in part, from Wnt-mediated improvements in neuronal glucose metabolism.
Introduction
Described over 100 years ago, AD has become the most relevant age-related neurodegenerative disorder. Mainly characterized by a progressive cognitive impairment, current therapeutical approaches are limited by the uncertainties about its precise etiology (Querfurth and LaFerla 2010; Ballard et al. 2011). Histopatholgically, AD considers two pathognomonic alterations, the extracellular formation of amyloid plaques, constituted by aggregated forms of amyloid-β (Aβ) peptide; and the intraneuronal accumulation of neurofibrillary tangles, composed by hyperphosphorylated tau protein. Relevantly, additional molecular events are also related with the progression of the disease and include the increase in the reactive oxygen species (ROS) production, mitochondrial dysfunction, inflammation, and a decrease in cerebral glucose uptake/utilization (Chen and Zhong 2013; Kapogiannis and Mattson 2011; Cisternas and Inestrosa 2017; Serrano-Pozo et al. 2011). Different research groups have evidenced that impaired glucose metabolism verifies in the brains of AD patients, being of particular interest the involvement of cortex and hippocampus, brain areas related with learning and memory and among the critical ones affected during the progression of AD (Chen and Zhong 2013; Doraiswamy et al. 2012). Relevantly, even when the impaired glucose utilization is close related with several physiological and molecular alterations, including the abnormal expression profile of the glucose transporters (GLUTs) 1, 3 and 4, brain insulin resistance and altered mitochondrial respiration (Bubber et al. 2005; Harr et al. 1995; Schapira 2012; Simpson et al. 1994; Cisternas and Inestrosa 2017). It has been demonstrated that the exogenous stimulation of the glucose metabolism can rescue the cognitive performance in AD patients (Craft et al. 2012; Baker et al. 2011; Parthsarathy and Holscher 2013). Likewise, the ability of increased glucose metabolism to improve cognitive performance or rescue neuronal cells against the toxicity of Aβ has been described in several model organisms, including mice, rats and Drosophila (Liu et al. 2017; Niccoli et al. 2016). This further demonstrates the close relationship between the deregulation of cerebral glucose metabolism and the cognitive failures described in AD. However, the molecular mechanisms and crosstalk between the different signaling pathways involved in the regulation of glucose metabolism and cognitive improvements, or the mechanisms by which these pathways are deregulated in AD, have not been identified.
Wnt signaling has recently been implicated in regulating glucose metabolism in the brain, and this pathway is down-regulated in AD (Bayod et al. 2015; Cisternas et al. 2016a; Cisternas et al. 2016b). Wnt signaling plays diverse roles in many organ systems; its loss- or gain-of-function is linked to cancer, type 2 diabetes and neurodegenerative diseases such as Parkinson’s disease (Oliva et al. 2018; Rios et al. 2014). In the CNS, Wnt stimulates adult hippocampal neurogenesis, promotes the establishment of synapses, increases neuronal firing activity, enhances neuronal plasticity and nerve transmission, and stimulates mitochondrial dynamics (Oliva and Inestrosa 2015; Farias et al. 2010; Godoy et al. 2014a). The relationship between Wnt signaling and glucose metabolism in the brain, however, has not been clearly established (Cisternas and Inestrosa 2017). We previously demonstrated that canonical and non-canonical Wnt signaling stimulate glucose metabolism in cultured hippocampal neurons by promoting glucose uptake and utilization (Cisternas et al. 2016a; Cisternas et al. 2016b), but whether this mediates the neuroprotective effect of Wnt in AD mouse models remains unknown. Here, we provide evidence that the canonical Wnt signaling activators Andrographolide (ANDRO) (Tapia-rojas et al. 2015; Serrano et al. 2014) and Lithium (Li) (Toledo and Inestrosa 2010; Forlenza et al. 2014), both inhibiting the activity of glycogen synthase kinase 3 beta (GSK-3β) and promoting the accumulation of β-catenin, improve glucose metabolism and cognitive performance in the APPswe/PS1E9 double transgenic mouse model of AD by increasing the activity of AMPK, a recently described target of Wnt3a signaling (Juvenal A. Ríos et al. 2018), as well as the activity of key metabolic enzymes, including HK and PFK. The beneficial effects of Wnt activators in cultured primary neurons, brain slices, and in vivo were blocked by the co-administration of Cyt B, a glucose uptake inhibitor, at IC50 dose. Our results suggest that enhancing glucose metabolism in the brain mediates, at least in part, the Wnt-induced cognitive improvements seen in an AD mouse model.
Methods
Animals and ethical standards.
The primary neuronal cultures were obtained from pregnant rats Sprague Dawley (n: 9, RRID: RGD_734476). Male APPswe/PS1dE9 (RRID: MMRRC_34829-JAX) (4-month-old, APP/PS1, as asymptomatic model before start the treatment) and control C57BL/6 wild-type (4-month-old, Wt, RRID: IMSR_JAX:000664) mice were used in this study. APP/PS1 animals co-express the Swedish (K594M/N595L) mutation of a chimeric mouse/human APP (Mo/ HuAPP695swe) together with the human exon-9-deleted variant of PS1 (PS1-dE9); these mice secrete elevated levels of human Aβ peptide (Jaworski et al. 2010; Jankowsky et al. 2004). This strain was obtained from The Jackson Laboratory (USA). Animals were maintained at the Animal Facility of the Pontificia Universidad Católica de Chile under sanitary barrier in ventilated racks and in closed colonies, no sample calculation was performed. Experimental procedures were approved by the Bioethical and Biosafety Committee of the Faculty of Biological Sciences of the P. Universidad Católica de Chile with the ethical approval CBB-158/2014. A total of 9 rats and 63 mice were used and handled according to the National Institutes of Health guidelines (NIH, Baltimore, MD). We used simple randomization to allocate mice to different cages. Intraperitoneal (i.p.) injections of Andrographolide (CAS Number 5508-58-7, cat: 365645, ANDRO) (2 groups, 2 mg/kg), Li2CO3 (cat: 62470, Li, 2 groups, 4 mg/kg) or saline solution as vehicle (2 groups) were carried out three times per week for 16 weeks. After 14 weeks of treatments with ANDRO, Li, or saline control, we administered Cytochalasin B (cat: C6762, Cyt B) at the IC50 dose (2 μg/Kg) for two weeks (2 injections per week) by intraventricular injection, just in one group of ANDRO and one group of Li (Figure 4 A) (Yamazaki et al. 2014; Ledo et al. 2012). All the reagents were purchased from Sigma-Aldrich, USA. Also, this study was not pre-registered. The inclusion/exclusion criteria for this study were the health of the animals after treatment, in this study no animals were excluded. All the animals that finish correctly the treatment and remain healthy were used to continue the analysis. The animals that showed sign of illness were excluded. For this inclusion/exclusion criterion we evaluated the weight of the animals, the lipid and hepatic parameters and the visual inspection. To avoid animal suffering the animals were reviewed by technical personal every day to look evidence of suffering (NIH tables of supervision), also, we used isoflurane (4% for induction) before the utilization of animals for removing the brain. All the cognitive test (training and experiment) and the slices experiments were performed in a double-blind manner, between the operator (MM, V.I.T and J.M.Z) and the designer of the experiment (P.C).
Figure 4. The chronic administration both ANDRO and Li improved cognitive performance in a transgenic model for AD.

(A) Schematic of the protocol for assaying the correlation between Wnt3a signaling and glucose metabolism in the brain. After the indicated treatments, cognitive tests related to hippocampal function were performed: Large open field (B), NOR (C), NOL (D) and memory flexibility (E). Data obtained from APP/PS1 mice (under blue line). Data represent the mean ± SEM of n = 9 (number of animals), *p < 0.05; **p < 0.01, Bonferroni test.
Primary neuronal cell culture.
Hippocampal neurons isolated from the forebrains of 17-day-old rat embryos, as described in (Arrazola et al. 2009), were seeded in poly-D-lysine-coated culture dishes at a density of 5×106 cells/cm2 and cultured in Dulbecco’s modified Eagle’s medium (cat: D5030, DMEM, Invitrogen, USA) containing 10 % (v/v) fetal bovine serum (cat: 16000044, FBS, Thermo Fisher Scientific Inc., USA). After 30 min, the culture medium was changed to Neurobasal medium (cat: 21103049, NeuB, Thermo Fisher Scientific Inc., USA) supplemented with B27 serum-free supplement for neural cell culture (cat: 15504044, Thermo Fisher Scientific Inc., USA), 2 mM L- glutamine (cat: 21051024, Thermo Fisher Scientific Inc., USA), 100 U/mL penicillin plus 100 mg/mL streptomycin (cat: 15070063, Thermo Fisher Scientific Inc., USA), and 2.5 mg/mL fungizone (cat: 15240112, Thermo Fisher Scientific Inc., USA),. Cells were incubated in 5 % CO2 in a humidified environment at 37 °C. Neuronal cells were used after 14 days of in vitro culture (DIV 14) in all experiments. Cytosine arabinoside (cat: C1768, Ara-C, Sigma-Aldrich, USA) was used to prevent non-neuronal cell contamination.
Slice preparation.
Hippocampal slices were prepared as previously described (Cerpa et al. 2010). Briefly, transverse slices (350 μm) from the dorsal hippocampus were sectioned in cold artificial cerebrospinal fluid (ACSF: 119 mM NaCl, 26.2 mM NaHCO3, 2.5 mM KCl, 1 mM NaH2PO4, 1.3 mM MgCl2, 10 mM glucose, 2.5 mM CaCl2) and incubated in ACSF for 1 h at room temperature. After incubation, slices were treated with activator/inhibitor for different length of time (0–60 min). The slices were then used in different glucose metabolism assays (glucose uptake, glycolytic rate, ATP/ADP levels, enzyme activity (AMPK, HK and PFK).
D-[1-14C] glucose biodistribution.
Upon the completing the cognitive tests, six mice from each group were injected with D-[1-14C] glucose (cat: NEC043X001MC, Perkin Elmer Perkinelmer, USA) via tail vein. Briefly, mice were anesthetized with isoflurane and injected intravenously via the tail with 50 μCi of tracer diluted to a final volume of 20 μL in isotonic saline. Following a 15 min uptake, animals were killed and tissues were collected. Tissue radioactivity was quantified by liquid scintillation. D-[1-14C] glucose levels were normalized to the weight of resected tissue and expressed as the percent injected dose (Tsytsarev et al. 2012; Cox et al. 2015).
Formation of amyloid-β oligomers.
Synthetic Aβ1–42 peptide corresponding to wild-type human Aβ were obtained from Genemed Synthesis, Inc. (custom made, LOT#105053, USA). An Aβ peptide stock solution was prepared by dissolving freeze-dried aliquots of Aβ in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, cat: H-8508, Sigma-Aldrich), incubating at room temperature for 1 h and lyophilized. For oligomer preparation, the peptide film was dissolved in dimethyl sulfoxide (DMSO, cat: W387520, Sigma-Aldrich USA) and diluted in distilled water to a final concentration of 100 μM. The preparation was incubated overnight for Aβos formation. Aβos were visualized by electron microscopy and analyzed by Tris-Tricine SDS gel electrophoresis as described previously (Silva-Alvarez et al. 2013).
Analysis of apoptosis by nuclear staining.
After treatments, cells were washed with PBS and fixed with 4 % PFA (cat: 1040051000, Merck, USA). Cells were then incubated with the nuclear stain Hoechst (1 mg/mL, cat: H3570, Thermo Fisher Scientific Inc., USA) for 30 min at room temperature. Cells were washed with PBS and analyzed by fluorescence microscopy. For each condition, a total of 200 nuclei were counted in three different cultures, and the results were expressed in proportion to normal nuclei and nuclei that showed chromatin condensation (Tapia-rojas et al. 2015).
Drug treatment.
The neurons and slices were treated with recombinant Wnt3a (rWnt3a, 300 ng/mL, 0–24 h, cat: 1324-WN/CF, R&D Systems, USA), Aβ (5 μM), dickkopf-1 (Dkk1, 300 ng/mL, 0–24 h, antagonist of Wnt3a, as internal control, cat: 5897-DK/CF R&D System, USA), cytochalasin B (Cyt B, 1 μM, 3 h, inhibitor of GLUT transporters, this concentration is the IC50 value, in the uptake we used 20 μM, Sigma-Aldrich USA), AZD5356 (20 nM, 12 h, an inhibitor of all Akt isoforms, cat: 5773, TOCRIS, UK), andrographolide (ANDRO, 50 μM, 12 h, agonist of Wnt3a signaling, Sigma-Aldrich USA), carbonate of lithium (Li, 10 mM, 12 h, agonist of Wnt3a signaling, Sigma-Aldrich USA), oligomycin (2 mM, inhibitor of ATP synthase, cat: 861944, Sigma-Aldrich USA) and 2-deoxy-D-glucose (2-DG, 7 mM, as a competitive inhibitor of hexokinase, Sigma-Aldrich USA). Neurons were carefully examined under the microscope to ensure that only plates showing uniform neuronal growth were used. After different treatments, cells were used in different metabolic assays.
Glucose uptake analysis.
After activator/inhibitor treatments, cells were washed with incubation buffer (15 mM HEPES (cat: H3375), 135 mM NaCl (cat: s3014), 5 mM KCl (cat: P5405), 1.8 mM CaCl2 (cat: C1016), and 0.8 mM MgCl2 (cat: 208337), all the reagents were purchased from Sigma-Aldrich, USA) supplemented with 0.5 mM glucose (Cisternas et al. 2014a). Cells were then incubated for 15 s with 1–1.2 μCi 2-deoxy-D-[1,2-(N)3H] (cat: NET328250UC, PerkinElmer, USA) or glucose ([2-3H]-DG (cat: NET328A250UC, PerkinElmer, USA) at a final specific activity of 1–3 disintegrations/min/pmol (~1 mCi/mmol). Glucose uptake was arrested by washing the cells with ice-cold PBS supplemented with 1 mM HgCl2 (cat: 203777, Sigma-Aldrich, USA). The incorporated radioactivity was quantified by liquid scintillation counting. In separate experiments, brain slices were incubated with different drugs for 1 h, then treated with [2-3H]-DG for 15 min and processed as described above. The kinetic parameters were determined using a single rectangular hyperbola of the form 𝑉max ∗ [glc]/(𝐾𝑚 + [glc]), adjusted to the data by nonlinear regression using SigmaPlot 12 (Barros et al. 2009).
Determination of the glycolytic rate.
Glycolytic rates were determined as previously described (Cisternas et al. 2014a; Herrero-Mendez et al. 2009). Briefly, after activator/inhibitor treatments, neurons were placed in tubes containing 5 mM glucose and then washed twice in Krebs Henseleit solution (11 mM Na2HPO4, 122 mM NaCl, 3.1 mM KCl, 0.4 mM KH2PO4, 1.2 mM MgSO4, and 1.3 mM CaCl2, pH 7.4) containing the appropriate concentration of glucose. After equilibration in 0.5 mL of Hank’s balanced salt solution/glucose (cat: 14025076, Thermo Fisher Scientific Inc., USA). at 37 °C for 10 min, 0.5 mL of Hank’s balanced salt solution (containing various concentrations of [3-3H] glucose (cat: NET331C250UC, PerkinElmer, USA) was added, with a final specific activity of 1–3 disintegrations/min/pmol (~1 mCi/mmol). Aliquots of 100 μL were then transferred to another tube, placed inside a capped scintillation vial containing 0.5 mL of water, and incubated at 45 °C for 48 h. After this vapor-phase equilibration step, the tube was removed from the vial, a scintillation mixture was added, and the 3H2O content was determined by counting over a 5-min period. For brain slice experiments, slices were treated with different activators/inhibitors, and the glycolytic rate was determined as described above.
Quantification of hexokinase (HK) activity.
After treatments with activators/inhibitors, cultured neurons or brain slices were washed with PBS, treated with trypsin/EDTA, and centrifuged at 500 g for 5 min at 4 °C. Then, the cells or tissue was resuspended in isolation medium (250 mM sucrose (cat: S9378), 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1mM EDTA (cat: E6758), 1mM DTT (cat: D0632), 2 mg/mL aprotinin (cat: A1153), 1 mg/mL pepstatin A (cat: 77170), and 2 mg/mL leupeptin (cat: L8511) at a 1:3 dilution, sonicated at 4 °C, and then centrifuged at 1,500 g for 5 min at 4 °C. HK activity of the supernatant was quantified. For the assay, purified fraction was mixed with the reaction medium (25 mM Tris-HCl (cat: T5941), 1 mM DTT, 0.5 mM NADP/Na+ (cat: N8035), 2 mM MgCl2, 1 mM ATP (cat: A1852), 2 U/mL G6PDH (cat: G6378), and 10 mM glucose (cat: G8270), and the mixture was incubated at 37 °C for 30 min. The reaction was stopped by the addition of 10 % trichloroacetic acid (cat: T6399, TCA), and the generation of NADPH was measured at 340 nm (Cisternas et al. 2016a). All the reagents were purchased from Sigma-Aldrich, USA.
Measurement of glucose oxidation through the pentose phosphate pathway (PPP).
Glucose oxidation via the PPP was measured as previously described based on the difference in 14CO2 production from [1-14C] glucose (cat: NEC043X001MC, PerkinElmer, USA) decarboxylated in the 6-phosphogluconate dehydrogenase-catalyzed reaction and in the Krebs cycle) and [6-14C] glucose (cat: NEC045X050UC, PerkinElmer, USA) only decarboxylated in the Krebs cycle) (Cisternas et al. 2014b). After treatments with activators/inhibitors, the medium was removed, and cells or brain slices were washed with ice-cold PBS and collected by trypsinization. Cell pellets were resuspended in O2-saturated Krebs Henseleit buffer and 500 μL of this suspension (~ 106 cells) was placed in Erlenmeyer flasks with another 0.5 mL of the Krebs Henseleit solution containing 0.5 μCi D-[1-14C] glucose or 2 μCi D-[6-14C] glucose and 5.5 mM D-glucose (final concentration). The Erlenmeyer flasks were equipped with a central well containing an Eppendorf tube with 500 μL of benzethonium hydroxide (cat: B2156, Sigma-Aldrich, USA). The flasks were flushed with O2 for 20 s, sealed with rubber caps, and incubated for 60 min in a 37 °C water bath with shaking. The incubations were stopped by the injection of 0.2 mL of 1.75 M HClO4 (cat: 311421, Sigma-Aldrich, USA) into the main well, and shaking was continued for another 20 min to facilitate the trapping of 14CO2 by benzethonium hydroxide. Radioactivity was quantified by liquid scintillation spectrometry (Bolaños et al. 2008; Herrero-Mendez et al. 2009).
ATP Content.
After cultured neurons or brain slices were treated with activators/inhibitors, ATP levels were measured using an ATP determination kit (cat: A22066, Invitrogen/Molecular Probes) (Calkins et al. 2011).
Activity of PFK and AMPK.
We used hippocampal slices obtained from treated animals and determined the activity of PFK and AMPK one hour after slice preparation. As a proxy, AMPK activity was measured using an antibody specific to the phosphorylated T-172 (active) form of AMPK-α. Detection was obtained using ELISA following the manufacturer’s protocol. Experiments were conducted in triplicate and repeated at least three times (cat: KHO0651, Thermo Fisher Scientific Inc., USA) (Moreno-Navarrete et al. 2011; Stow et al. 2016). PFK activity was measured using the PFK Colorimetric Assay Kit from (cat: K776, BioVision, USA ) according to the manufacturer’s instructions (Thurley et al. 2017).
Large open-field (LOF) test.
A 120 × 120 cm transparent Plexiglas platform with 35-cm-high transparent walls was used to study locomotor and stress behavior in our mouse model. The open field, which measured 40 × 40 cm, was defined as the center area of the field. Data were collected using an automatic tracking system (HVS Imagen, UK). Each mouse was placed alone in the center of the open field, and its behavior was tracked for 20 min. At the end of the session, the mouse was returned to its home cage. The parameters measured included total time moving and number of times the mouse crossed the center area of the platform (Cummins and Walsh 1976; Cisternas et al. 2015a).
Novel object recognition (NOR) and novel object localization (NOL).
The NOR and NOL tasks were performed as previously described (Bevins and Besheer 2006; Vargas et al. 2014). Mice were habituated to the experimental room in the experimental cages for 3 consecutive days for 30 min per day (3 consecutive days) and for 1 h on the testing day. The task occurred in a 120×120 cm transparent Plexiglas platform with 35-cm-high transparent walls containing two identical objects places at specific locations. For object familiarization, mice were allowed to explore the platform for 10 min. The animals were subsequently returned to their home cages for 1 h, followed by a 5-min exposure to a novel localization of one of the familiar objects (NOL). The mice were again returned to their home cages for 1 h and were subsequently exposed to a novel object (NOR) for 5 min. The mice had no observed baseline preference for the different objects. An object preference index was determined by calculating the time spent near the relocated/novel object divided by the cumulative time spent with both the familiar and relocated/novel objects. The cages were routinely cleaned with ethanol following mouse testing/habituation of the mice.
Memory flexibility test.
This test was performed as previously described (Chen et al. 2000; Salazar et al. 2017), and the pool conditions of the pool were the same as those of a Morris Water Maze (Cisternas et al. 2015b). Each animal was trained for one pseudo-random location of the platform per day, for 5 days, with a new platform location each day. Training was conducted for up to 10 trials per day, until the criterion of 3 successive trials with an escape latency of 20 s was achieved. When testing was completed, the mouse was removed from the maze, dried and returned to its cage. Animals were tested for the next location on the following day. Data were collected using a video tracking system (HVS Imagen).
Quantitative real-time PCR (qRT-PCR).
After drug treatment, mRNA was obtained from the neuronal cultures or hippocampal tissue and used to generate cDNA. Quantitative real time RT–PCR (qRT–PCR) was conducted using SYBR master mix (cat: 4368577, Thermo Fisher Scientific Inc., USA) and 18S mRNA as a control, with the program recommended by the manufacture (Cisternas et al. 2016a). As a housekeeping gene, we used cyclophilin, and the values were calculated using the delta Ct, in comparison with the control gene. Duplicated control reactions for every sample without reverse transcription were included to ensure that PCR products were not due to amplification of contaminant genomic DNA. We used the following sets of primers: 18S gene forward 5’-TCAACGAGGAATGCCTAGTAAGC-3’ and reverse 5’-ACAAAGGGCAGGGACGTAGTC-3’. As a housekeeping gene, we used cyclophilin: forward 5’-TGGAGATGAATCTGTAGGAGGAG-3’ and reverse 5’- TACCACATCCATGCCC TCTAGAA-3; Camk4: forward 5’- TTATGCAACTCCAGCCCCTG-3’ and reverse 5’-AGCCT CGGAGAATCTCAGGT-3’; Glut1: forward 5′-ATGGATCCCAGCAGCAAGAAG-3’ and reverse 5′-AGAGACCAAAGCGTGGTGAG-3’; Glut3: forward 5′-GGATCCCTTGTCCTTCT GCTT-3’ and reverse 5′-ACCAGTTCCCAATGCACACA-3’; Glut4: forward 5′-CGGCTCTGA CGATGGGGAA-3’ and reverse 5′-TTGTGGGATGGAATCCGGTC-3’; Cyclin D1: forward 5′-AAAATGCCAGAGGCGGATGA-3’ and reverse 5′-GCAGTCCGGGTCACACTTG-3’; c-Myc: forward 5′-GGAGTGGTTCAGGATTGGGG-3’ and reverse 5′-GGGTAGCTTACCAGAGTCG C-3’; Hexokinase-1: forward 5′-GGATGGGAACTCTCCCCTG-3’ and reverse 5′-GCATACGT GCTGGACCGATA; Phosphofructokinase-1: forward 5′-AGGGCCTTGTCATCATTG GG-3’ and reverse 5′-ACTGCTTCCTGCCTTCCATC-3’; Akt-1: forward 5′-TCACGTGAGCCC TTCTCCTA-3’ and reverse 5′-CTCCCACCCACTAACAAGGC-3’; Ampk (alpha subunit): forward 5′-GTGAAGATCGGCCACTACATCC-3’ and reverse 5′-GGCTTTCCTTTTCGTCCA ACC-3’; Pyruvate kinase-1 (Pk1): forward 5′-CAGCATCATTGCCACCATCG-3’ and reverse 5′-GACTCCAGTGCGTATCTCGG-3’. Al the primers were purchased from IDT Integrated DNA Technologies, USA.
ADP content.
ADP levels in whole-cell lysates of primary neurons and slices were measured using an ADP Assay Kit (cat: ab83359, Abcam, UK), according to the manufacturer’s instructions (Chen et al. 2000).
Determination of Glucose-6-phosphate dehydrogenase (G6PDH) activity.
After activators/inhibitors treatments, cells were washed with PBS, collected by trypsinization (0.25% trypsin-0.2% EDTA (w/v), cat: T4049, Sigma-Aldrich USA), and pelleted. Cells were then resuspended in isolation medium (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 2 mg/mL aprotinin, 1 mg/mL pepstatin A, and 2 mg/mL leupeptin) at a 1:3 dilution, sonicated at 4 °C, and then centrifuged for 5 min at 1500 g at 4 °C. Subsequently, the pellet was discarded, and the supernatant was further separated by centrifugation at 13000 g for 30 min at 4° C. Finally, the supernatant was used to quantify the G6PDH activity in a reaction buffer containing 1 mM ATP and 10 mM glucose- 6-phosphate (G6P) for 30 min at 37 °C. The reaction was stopped by the addition of 10 % TCA. The generation of NADPH was measured at 340 nm (Cs 2018).
Statistical analysis.
All experiments were performed at least 3 times, with triplicates for each condition in each experimental run. The results are expressed as means ± standard errors. Data were analyzed by one-way or two-way analysis of variance (ANOVA), followed by Bonferroni’s post hoc test; *p ≤ 0.05 and **p ≤ 0.01 were considered significant differences. Statistical analyses were performed using Prism software (GraphPad, USA). For testing normality, we used the SPSS Statistics software (IBM, USA) using the numerical method. In our study we not detected outliers data, for this we used the Prism software (GraphPad, USA), using boxplot graphical way.
Results
The neuroprotective effect of Wnt signaling depends on the activation of glucose metabolism.
Cultured hippocampal neurons were treated with 5 μM Aβ1–42 oligomers (0–24 h) and cell viability was measured by nuclear staining. The quality of the Aβ oligomer was confirmed by electron microscopy (Figure 1A, i). Aβ oligomer-treatment reduces neuronal viability over time (56 ±6 % after 12 h and 42 ± 4 % after 24 h, p=0.01) (Figure 1A, ii–iv). Recombinant rWnt3a significantly attenuated cell death induced by Aβ. Ιn the presence of Aβ and rWnt3a, viability of neurons was 72 ± 7 % (at 12 h) and 69 ± 6 % (at 24 h) (Figure 1A, v–vi). The neuroprotective effect of rWnt3a at 12 h was abrogated by co-incubation with 1 μM Cyt B, an inhibitor that blocks glucose uptake by glucose transporters (GLUTs) (Figure 1B). At the IC50 dose used (1 μM), Cyt B did not affect cell viability (Figure 1B) (Augustin 2010; Kapoor et al. 2016). The protective effects of rWnt3a were also abolished by co-incubating cells with AZD5356, an inhibitor of AKT (Figure 1B).
Figure 1. Glucose uptake facilitated the neuroprotective effect of Wnt3a on Aβ toxicity.

(A) Aβ oligomer structure determined by electron microscopy, treatment with Aβ reduced cell viability in a time-dependent manner, and the decrease in cell viability induced by Aβ was partially blocked by co-incubation with Wnt3a (arrow indicates apoptotic nucleus), n:3. (B) In contrast, co-incubation with Wnt3a+Aβ and Cyt B diminished the neuroprotective effect of the Wnt ligand. (C) Incubation with Aβ induced a dramatic decrease in 2-DG uptake. Wnt3a blocked this decrease, and Dkk partially inhibited the protective effect of Wnt3a. (D) The treatment of hippocampal neurons with ANDRO and Li stimulated the uptake of 2DG and this is inhibited by Cyt B. Data represent the mean ± SEM of n = 3 (independent experiments), each performed in triplicate. *p < 0.05; **p < 0.01, Bonferroni test.
Next, we determined the effects of Aβ and rWnt3a on glucose uptake in cultured hippocampal neurons. In untreated controls, we observed 24.4 ± 2.7 nmol 2-DG glucose uptake per million cells. Treatment with 5 μM Aβ for 12 h significantly decreased 2-DG uptake (11.4 ± 2.1 nmol/106 cells, p=0.05) (Figure 1C). rWnt3a alone stimulated a robust increase in 2-DG uptake and also prevented the reduction in glucose uptake induced by Aβ (Figure 1C). The positive effect of rWnt3a on glucose uptake was abolished by Dkk1, AZD5363, and Cyt B (Figure 1C).
ANDRO and Li are known activators of Wnt signaling in vivo and in vitro (Toledo and Inestrosa 2010; Serrano et al. 2014). Therefore, we also determined the effects of both molecules on glucose uptake. Treatments of cultured hippocampal neurons with ANDRO and Li alone enhanced 2-DG uptake (Figure 1D), and these Wnt signaling activators also prevented the reduction in glucose uptake induced by Aβ; these effects were abrogated by Cyt B (Figure 1D).
To further examine the effects of Aβ on glucose metabolism and on the neuroprotection induced by rWnt3a, we measured additional metabolic parameters in cultured hippocampal neurons. Exposure to Aβ for 12 h markedly reduced (~2.7 fold) the glycolytic rate of cultured neurons (Figure 2A). Treatment with rWnt3a blocked the Aβ-induced decrease in the glycolytic rate, and this neuroprotective effect was abolished by Dkk1 (Figure 2A). The activity of HK that catalyzes the first reaction in glycolysis, however, was not affected by Aβ treatment (Figure 2B). Glucose entering neurons can be channeled into glycolysis or PPP. Thus, we measured the effect of Aβ on PPP pathway activity and observed no difference in the presence of Aβ or rWnt3a (Figure 2C). However, Aβ exposure significantly reduced cellular ATP levels (Figure 2D). The presence of rWnt3a prevented this decrease, and the neuroprotective effect of rWnt3a was abolished by Dkk1 and 2-DG (Figure 2D). Cellular ADP levels and ATP/ADP ratios were also determined. In the presence of Aβ, we observed a marked decrease in the ATP/ADP ratio (45 %), and this reduction was blocked by rWnt3a (Figure 2E). This protective effect of Wnt3a was abrogated by Dkk1 and 2-DG (Figure 2E).
Figure 2. Wnt3a treatment protected against the Aβ-induced decrease in glucose metabolism.

(A) A time course of treatments showing that Aβ decreases the glycolytic rate in hippocampal neurons, in vitro. This effect was abolished by co-incubation with Wnt3a and this restoration was blocked by Dkk. (B) The activity of HK after 12 h of treatment with Aβ and/or the indicated compounds. (C) The activity of the PPP pathway after treatment with Aβ and the indicated compounds. This pathway remained unaltered. (D) ATP levels after Aβ treatment were dramatically reduced but rescued by co-treatment with Wnt3a. (E) The decrease in the levels of ATP correlated with the ATP/ADP ratio. Both ATP (E) and the ATP/ADP ratio increased in the presence of Wnt3a. Data represent the mean ± SEM of n = 3 (independent experiments), each performed in triplicate. *p < 0.05; **p < 0.01, Bonferroni test.
To validate our results from cultured neurons, we used hippocampal slices obtained from wild type (Wt) and APPswe/PS1E9 (APP/PS1) transgenic mice. Consistent with our neuronal culture data, glucose uptake was reduced ~50 % in brain slices from APP/PS1 mice (Figure 3A). Glucose uptake in APP/PS1 brain slices was restored Wt levels when Wnt signaling was activated by rWnt3a, Li, or ANDRO (Figure 3A). The effect of the Wnt agonists on brain slices was abolished by oligomycin, an inhibitor of ATP synthase (Figure 3A). The glycolytic rate of APP/PS1 brain slices was also reduced ~50 % relative to that of Wt brain slices; this reduced rate was restored to Wt levels by rWnt3a, Li, or ANDRO. Again, this effect of the Wnt agonists was abolished by oligomycin (Figure 3B). Given the reduction in glycolytic rate, we measured the activity of HK and PFK, two key enzymes of glycolysis. HK and PFK activities in APP/PS1 brain slices were ~50 % of the Wt control activities; and this reduction could not be restored by Li or ANDRO treatments (Figure 3C–D).
Figure 3. Wnt signaling promoted utilization of glucose to enhance production of ATP in hippocampal slices.

(A) The uptake of radioactive glucose in slices obtained from Wt and APP/PS1 mice (under blue line). Slices were treated with the indicated drugs for 1 h and then glucose uptake was measured. Treatment with Wnt3a and Wnt agonists increased the uptake of glucose in APP/PS1 slices and this was blocked by oligomycin. (B) The glycolytic rate after treatment with the indicated drugs. The decreased glycolytic rate of APP/PS1 slices was rescued by Wnt signaling. The activity of two key regulatory glycolytic enzymes, HK and PFK (C and D, respectively). The treatments, with the exception of rWnt3a affecting PFK, did not rescue the APP/PS1 mediated decreases in HK or PFK activity. Both ATP and the ATP/ADP ratio were decreased in slices from APP/PS1 mice, and both were increased in the presence of the agonists of Wnt3a signaling (E and F, respectively). Data represent the mean ± SEM of n = 3 (independent experiments), each performed in triplicate. *p < 0.05; **p < 0.01, Bonferroni test.
Consistent with the reduced glucose uptake and glycolytic rate, cellular ATP levels and the ATP/ADP ratio in APP/PS1 brain slices were markedly reduced (Figure 3E–F). rWnt3a and ANDRO, but not Li, treatments were able to partially restore cellular ATP and the ATP/ADP ratio in APP/PS1 brain slices. The positive effect of rWnt3a and ANDRO was blocked by the AKT inhibitor, AZD5363 (Figure 3E–F).
In vivo stimulation of Wnt signaling activates glucose metabolism and improves cognitive performance in an AD mouse model
Next, we asked if in vivo activation of Wnt signaling can improve glucose metabolism in the APP/PS1 model of AD. We administrated ANDRO and Li to APP/PS1 mice, with or without Cyt B (IC50 level), to determine the link between Wnt signaling activation and glucose metabolism (Figure 4A). The IC50 dose of Cyt B was used to ensure at least 50 % reduction in glucose transport without generalized toxicity. Open field tests were performed to determine the general behavioral status of the animals. We observed no differences between control and treated groups (Figure 4B i–ii) in several parameters, including the percentage of moving time and the number of lines that the animals cross at the center of the box. To examine the functionality of short-term memory, we performed NOR and NOL tests. We observed a decrease in the preference index in APP/PS1 mice relative to Wt controls (Figure 4C–D). Injection of ANDRO and Li significantly improved performance in both the NOR and NOL tests, with the treated animals performing as well as the Wt mice, suggesting a rescue of short-term memory. The effects of both ANDRO and Li were abolished in animals co-treated with Cyt B (Figure 4C–D). These results suggest that the effect of Wnt agonists depends on the activation of glucose metabolism. Next, we performed a memory flexibility test to measure learning and memory function. Control animals took an average of 5 trials to fulfill the completion criteria after 5 days of testing. In contrast, the APP/PS1 mice took an average of 12 trials to meet the same criteria, indicating impaired learning and memory. The APP/PS1 mice treated with ANDRO and Li took an average of 6 and 7 trials, respectively, to fulfill the completion criteria, indicating improved learning and memory. The beneficial effects of ANDRO on learning and memory were abrogated by when animals were co-treated with Cyt B (Figure 4E i and ii).
Wnt agonists increase glucose uptake in the brain
Wnt agonists significantly improved cognitive performance in APP/PS1 mice, so we next tested whether this is due to increased glucose uptake in the brain. We injected radioactive glucose (D-[1-14C] glucose) via the tail vein and 15 min post-injection measured the amount of radioactivity in the whole brain, as well as in the hippocampus and cortex, two brain regions most affected in AD. Glucose uptake in the whole brain of APP/PS1 mice was 60 % of that observed in the Wt mice (Figure 5A). Treatments with ANDRO and Li improved glucose uptake, and these effects were abolished in animals co-injected with Cyt B (Figure 5A). In the hippocampus, glucose uptake was reduced ~45 % in APP/PS1 mice relative to Wt controls (Figure 5B). Treatment with ANDRO restored glucose uptake in the hippocampus to that of Wt controls, and this effect was abolished in animals co-injected with Cyt B (Figure 5B). In contrast to ANDRO, treatment with Li did not increases glucose uptake in the hippocampus of APP/PS1 mice. (Figure 5B).
Figure 5. Treatment with ANDRO or Li increased the in vivo uptake of glucose.

After the indicated treatments, radioactive glucose was tail-vein injected, and 15 min later, glucose uptake in the whole brain (A), hippocampus (B), and cortex (C) was measured. The APP/PS1 mice exhibited decreased uptake of glucose. Wnt agonists improved glucose uptake in the APP/PS1 animals. Data from APP/PS1 mice (under blue line). The effects of the various treatments on the mRNA levels of the GLUT 1 (D), 3 (E) and 4 (F) glucose transporter proteins, which are known to be affected in AD. Data represent the mean ± SEM of n = 6 (number of animals for treatment), *p < 0.05; **p < 0.01, Bonferroni test.
Similar to the hippocampus, glucose uptake in the cortex of APP/PS1 mice was ~57 % less than that of Wt controls (Figure 5C). Administration of ANDRO and Li significantly improved glucose uptake in the cortex of APP/PS1 mice, and these effects were abolished when animals were co-injected with Cyt B (Figure 5C).
Impaired cognitive performance has been correlated with decreased expression of glucose transporters in the hippocampus (Shah et al. 2012; Emmanuel et al. 2013). We therefore examined the mRNA expression level of Glut1, 3 and 4 in the hippocampus. Expression of Glut1 did not differ between APP/PS1 mice and Wt controls, nor was its expression altered by Wnt agonists (Figure 5D). Expression of Glut3, the main glucose transporter in neurons, was reduced 68 % in the hippocampus of APP/PS1 mice relative to Wt control levels (Figure 5E). Expression of Glut3 in the hippocampus of APP/PS1 mice was restored to that of Wt controls by ANDRO and Li treatments, and the positive effects of Wnt agonists were abolished by Cyt B (Figure 5E). Interestingly, while the expression of Glut4 was reduced 80 % in the hippocampus of APP/PS1 mice relative to Wt control levels, neither ANDRO nor Li treatment were able to restore its expression (Figure 5F).
Wnt agonists stimulates glucose utilization in the hippocampus
After ANDRO or Li treatment, we obtained hippocampal slices from the treated and control groups to determine parameters of glucose utilization. In non-treated animals, the glycolytic rate of hippocampal slices from APP/PS1 mice was reduced ~42 % relative to Wt control levels (Figure 6A). Treatment of APP/PS1 mice with ANDRO and Li restored the glycolytic rate of APP/PS1 animals to that of Wt controls, and this effect was abolished in animals co-treated with Cyt B (Figure 6A). HK and PFK activities in the hippocampal slices from APP/PS1 mice were reduced 49 % and 57 %, respectively, relative to Wt control levels. ANDRO and Li treatments restored HK and PFK activities to Wt levels (Figure 6B–C). Co-treatment of Cyt B abolished the effect of ANDRO and Li on HK, but not PFK, activity. We observed an 87 % reduction in PPP activity in hippocampal slices of APP/PS1 mice. Treatments of APP/PS1 mice with ANDRO and Li led to a significant increase of PPP activity, and this effect was abolished in animals treated with Cyt B (Figure 6D). Since PPP pathway activity was reduced in APP/PS1 mice, we determined the activity of G6PDH, the first and rate-limiting enzyme of the PPP. In hippocampal slices from APP/PS1 mice, the activity of G6PDH was reduced 73 % relative to Wt control levels (Figure 6E). Treatments of APP/PS1 mice with ANDRO and Li restored G6PDH activity to that of Wt levels. Again, these effects were abolished in animals co-injected with Cyt B (Figure 6E). AMPK is one of the key serine/threonine kinases that regulate cellular ATP levels by modulating catabolic processes that generate ATP (Ruderman et al. 2013). For this reason, we also measured the activation state of AMPK. In hippocampal slices from APP/PS1 mice, AMPK activity (as a metric of phospho-AMPKα) was reduced 48 % relative to Wt control levels, and ANDRO or Li treatment restored AMPK activity to Wt levels (Fig. 6F). Consistent with the glycolytic rate and enzyme activity, cellular ATP levels and the ATP/ADP ratio were significantly reduced in hippocampal slices from APP/PS1 mice and restored nearly to Wt levels by ANDRO and Li treatments. Again, these effects were abolished by Cyt B (Figure 6G and H).
Figure 6. Treatment with ANDRO and Li induced the utilization of glucose promoting the generation of ATP and NAPDH.

Parameters related to glucose metabolism, including the glycolytic rate, were measured in the brains of control or treated mice, Data from APP/PS1 mice (under blue line). (A) Treatment with ANDRO or Li stimulated the glycolytic rate. Treatment with ANDRO or Li stimulated the enzymatic activity of two check point proteins of glycolysis: HK (B) and PFK (C). ANDRO and Li stimulated PPP (D) and the activity of the first control point of the PPP pathway: G6PD (E). Effects of treatments on the levels of AMPK, an important metabolic sensor of glucose metabolism (F). ATP (G) and the ATP/ADP ratio (H) were measured to correlate the previous parameters with the energy status of cells. ANDRO and Li restored the levels of both parameters in APP/PS1 mice. Data represent the mean ± SEM of n = 3 (independent experiments), each performed in triplicate. *p < 0.05; **p < 0.01, Bonferroni test.
Finally, we examined whether Wnt-mediated enhancement in glucose metabolism also involved transcriptional upregulation of key metabolic genes. The known Wnt3a-target genes (Camk4, Cyclin D1, and c-Myc) were down-regulated in the hippocampus of APP/PS1 mice relative to Wt animals, and ANDRO and Li treatments restored expression of these genes to that of Wt levels (Figure 7A–C). The expression of Hk, Pk-1 and Pfk-1 was also down-regulated in the APP/PS1 mice relative to Wt control levels, and ANDRO treatment significantly up-regulated expression to that of Wt levels (Figure 7D–F). We found that the expression of Akt was reduced 60 % in the APP/PS1 mice relative to Wt control levels and ANDRO treatment restored its expression to that of Wt controls (Figure 7G). In contrast to Akt, the mRNA expression of AMPK was not affected in the APP/PS1 mice (Figure 7H).
Figure 7. ANDRO and Li changed the expression pattern of several genes involved in the control of glucose metabolism.

The effects of the indicated treatments on the brain mRNA levels of genes encoding several proteins involved in glucose metabolism, including Campkiv (A), Cyclin D1 (B), c-Myc (C), which are known target genes of Wnt3a signaling. These target genes were increased by ANDRO and Li. The effect of treatments on the mRNA levels encoding additional metabolic proteins: Hk (D), Pk1 (E), Pfk1 (F), Akt (G) and Ampk (H). The mRNA levels of Hk, Pk1, Pfk1 and Akt increased after treatment with ANDRO. While the treatment with Li just recover the mRNA levels of Hk, Pkf1. Data from APP/PS1 mice (under blue line). Data represent the mean ± SEM of n = 3 (samples obtained from 3 different animals, by treatment), each performed in triplicate, *p < 0.05; **p < 0.01, Bonferroni test.
Discussion
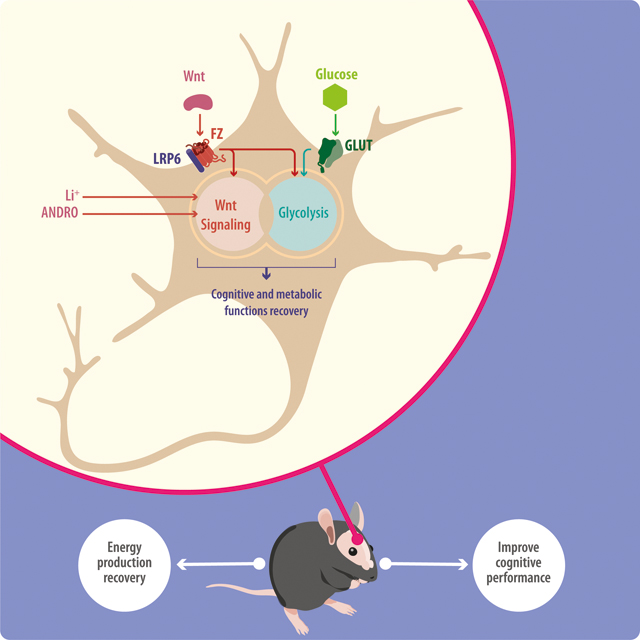
We have previously shown that Wnt signaling can stimulate glucose metabolism in cultured neurons and hippocampal slices (Cisternas et al. 2016c; Cisternas et al. 2016a). The current study extends these findings to more complex biological systems, such as ex vivo brain slices and an in vivo AD mouse model. Our most relevant and novel finding is that Wnt signaling improves cognitive performance in a mouse model of AD by enhancing glucose metabolism. This has important implications for the treatment of AD and possibly other brain disorders (Figure 8).
Figure 8. General model of Wnt-Glucose interactions in the brain.

Treatment with ANDRO/Li triggers activation of canonical Wnt signaling promoting the activations of several Wnt target genes, many related to cellular metabolism. Activation of Wnt signaling also enhances several glucose-related parameters, including glucose uptake, the glycolytic rate, and the production of ATP. Wnt signaling stimulates the enzymatic activities of several key metabolic enzymes and sensors such as hexokinase (HK), phosphofructokinase (PFK), and AMP-activated protein kinase (AMPK). The interaction of Wnt signaling and glucose metabolism promotes the recovery of cognitive performance in a mouse model of Alzheimer’s disease.
Both Wnt3a, ANDRO and Li are able to turn on the Wnt canonical pathway, constituting a potentially relevant therapeutic approach to upregulate Wnt canonical signaling in vivo. Indeed, both molecules promote the accumulation of β-catenin by preventing the degradation of this protein through the inhibition of GSK-3β (Toledo and Inestrosa 2010; Tapia-rojas et al. 2015; Rivera et al. 2018; Liang et al. 2017; Xia et al. 2017). Relevantly, the canonical Wnt signaling has emerged as one of the more relevant pathways to be addressed when approaching to neurodegenerative disorders, overall because it has been related to several brain pathologies, such as AD, autism, schizophrenia, and Parkinson disease. In fact, it has been recently described that a loss in the activity of Wnt canonical signaling accelerates the appearance of pathological hallmarks of AD in a transgenic mice model and in the Wt mice (Tapia-Rojas and Inestrosa 2018). On the other hand, it is well-known that the activation of the Wnt/β-catenin pathway has huge effects in cell transcriptome being a possibility that can also have regulatory effects on glucose metabolism; however, until now, there has been no report of the effects of chronic Wnt agonist administration on glucose metabolism and on the enzymes involved in this process. Whether key metabolic enzymes are also target genes of canonical Wnt signaling has also not been explored. Here, we hypothesized that activation of canonical Wnt signaling is vital for regulating neuronal metabolism and this effect is important for proper functioning of the neuronal network. The present study confirms and extends our previous findings, further supporting the notion that Wnt-mediated improvements in glucose metabolism are linked to its neuroprotective effects in vivo, in a chronic manner.
In the slices of APP/PS1 mouse brain, we observed a dramatic decrease in glucose utilization and ATP production. To study if the administration of ANDRO or Li restore glucose utilization in the APP/PS1 mice to wild-type levels in vivo we used radioactive D-[1–14C] glucose to measure the global capacity of brain to uptake glucose. Similar to AD patients, APP/PS1 mice have decreased glucose uptake in the cortex and hippocampus (near to 50 %), two major brain regions involved in learning/memory and commonly affected in AD patients (Vlassenko et al. 2012). We showed that administration of ANDRO and Li increase glucose uptake in the brain. Interestingly, Li only increased glucose uptake in the cortex, whereas ANDRO promoted glucose uptake in both the cortex and hippocampus. The direct correlation between glucose metabolism and AD has been recently studied in genetic AD patients. In these patients, downregulation of glucose metabolism occurs mainly in the grey matter after the aggregation and accumulation of Aβ, and this is considered the second hallmark of AD progression in humans (Gordon et al. 2018; Jeong et al. 2017).
Treatment with ANDRO or Li increases both cellular ATP and ADP in the brain and neurons, suggesting an overall increase in mitochondrial function and metabolic activity. In this regard, mitochondrial metabolism not only drives ATP production, but also plays a remarkable role in additional cellular homeostatic mechanisms, including ROS balance and Ca2+ buffering. Moreover, mitochondrial are able to determine the cellular fate because of its involvement in the apoptotic cascade (Detmer and Chan 2007). Accordingly, it is not surprising that several chronic-degenerative disorders, including AD, be related with the mitochondrial dysfunction (Martin 2012). Although the relevance of the mitochondria is out of question, it is quite astonishing the limited advances about our understanding of the mechanisms able to modulate the mitochondrial activity, especially under pathological conditions affecting the central nervous system (Lin and Beal 2006; Reddy 2009). Regarding our work, it is necessary to mention that the relationship between the Wnt signaling and the mitochondria it has been studied previously in cancer. In some of these studies, it has been demonstrated that the stimulation of MCF-7 cells with the Wnt3a or Wnt1 ligands blocks mitochondrial respiration through cytochrome C inhibition by a mechanism involving the activation of the Wnt/Snail axis. Relevantly, the Wnt/Snail signaling can modulate several aspects of the cell metabolism, including glucose usage by the cells (Lee et al. 2012). Indeed, Pate and cols. (Pate et al. 2014), evidenced that the blockade of the Wnt signaling induces the modulation of several enzymes related with the glucose metabolism, including the pyruvate dehydrogenase kinase (PDK1), which ultimately will cause the inhibition of the pyruvate dehydrogenase (PDH) complex within the mitochondria, limiting the availability of substrates to be used in the tricarboxylic acid (TCA) cycle. At the end, the Wnt blockade will cause the downregulation of the oxidative phosphorylation leading to an altered cellular metabolism. In the context of AD, we showed that Wnt3a treatment prevents mitochondrial damage induced by Aβ, possibly by inhibiting opening of the mitochondrial permeability transition pore (mPTP) (Arrázola et al. 2017). Wnt signaling also stimulates glucose metabolism in neurons and this could promote the generation of ROS. We observed that chronic administration of ANDRO and Li increases the activity of PPP, elevating NADPH levels critical for regenerating ascorbate and glutathione needed for the anti-oxidative defenses of cells (Reiter 1995; Rice 1999). In the current study, we found that ANDRO and Li treatments increase the activity of G6PDH, the key regulated enzyme of the PPP, in part accounting for the enhanced PPP activity, leading to increased cellular levels of NADPH. The mechanism that modulates G6PDH activity is unclear. Several studies have suggested that inhibiting the activity of cAMP-dependent protein kinase A (PKA) could enhance G6PDH activity (Gallegos et al. 2012; Dehydrogenase et al. 2013; Zhang et al. 2012). However, this mechanism has not been described in brain cells. Nevertheless, Wnt-mediated enhancement in G6PDH activity could be through inactivating PKA activity.
In brain slices and whole brains of APP/PS1 AD mice, we observed a decrease in the mRNA levels of Glut3 and Glut4 but not the ubiquitously expressed Glut1. Administration in vivo of Wnt3a agonist ANDRO and Li restored Glut3 expression to that of wild-type levels, leading to improved glucose uptake. Interestingly, Wnt agonist treatments did not restore Glut4 expression. Despite the importance of this transporter in the CNS, the localization, expression and function of GLUT4 in hippocampal neurons are not well defined, nor are the expression of other GLUT isoforms in different neuronal populations (Ashrafi et al. 2018; Reno et al. 2017). We did not observed changes in the expression of mRNA of GLUTs transporters. Consequently, we hypothesize that ANDRO and Li most likely modulate the activity of downstream metabolism related proteins to increase glucose uptake.
We observed that the APP/PS1 mice have reduced AMPK activity, and this correlated with a decrease in cellular ATP levels and in the ATP/ADP ratio. Treatments with ANDRO or Li were able to restore AMPK activity and cellular ATP levels, consistent with recent data that suggest that AMPK is a downstream target of Wnt3a signaling (Juvenal A. Ríos et al. 2018). Future studies are needed to identify additional cellular proteins modulated by Wnt signaling that can have an impact on glucose metabolism. The reactions catalyzed by HK, PFK and PK1 regulate the flux of glucose through the glycolytic pathway. A decrease in the activity of these key regulated enzymes will reduce the overall substrates available for complete glucose oxidation via the TCA cycle in the mitochondria. We observed that APP/PS1 mouse brains have reduced HK and PFK activities, and this could account for their decreased cellular ATP levels and reduced glucose utilization. Both Li and ANDRO enhanced or completely restored the activity of HK and PFK in APP/PS1 mouse brains to that of wild-type levels.
Whether and how HK is being regulated in brain cells is largely unknown. In other tissues, mostly in the context of cancer models, activation of tyrosine kinases (e.g., c-Src) positively modulate the activity of HK. Interestingly, c-Src kinase activity also enhances the expression of Lef/TCF, effectors of canonical Wnt signaling (Ramiere et al. 2014; Zhang et al. 2017; Karni et al. 2005; Yokoyama and Malbon 2009). It remains to be determined whether Wnt signaling increases HK activity through c-Src tyrosine kinase. In the case of PFK, Wnt signaling may enhance its activity through the activation of the PI3K/Akt/mTOR pathway, as this pathway has been shown to modulate PFK activity (Brown et al. 2017; Zhang et al. 2016; Godoy et al. 2014b). Given the complexity of metabolic regulation, further studies are needed to better understand how Wnt signaling regulates the activity of metabolic enzymes (Aleshin et al. 1998; Mor et al. 2011). In the present study, we noted that the recovery in enzyme activity was correlated with an increase in mRNA expression key metabolic regulators (e.g., Hk, Pk-1, Pfk-1 and Akt) in the hippocampus. These results suggest that ANDRO and Li administration could also promote glucose metabolism via transcriptional mechanism as well.
Consistent with these protein expression/activity data, we observed a significant decrease in all metabolic measures related to the utilization of glucose in our in vitro and in vivo models, with the production of ATP being a primary outcome of glucose utilization. In hippocampal neurons, we observed that Aβ treatment strongly decreased the glycolytic rate, the activity of HK, and the production of ATP. This is a critical finding since the high energy demand of neurons supports normal brain function, and a modest decrease in ATP levels can induce neuronal death. The ability of Aβ to reduce cellular ATP production in vitro was blocked by Wnt3a treatment and this protective effect was abolished by an ATP synthase inhibitor. This suggests that the major deleterious effects of Aβ are due, at least in part, to its effect on mitochondrial energy metabolism and that Wnt signaling exerts its neuroprotective role by restoring mitochondrial function in neurons (Arrázola et al. 2017).
In the APP/PS1 AD mouse model, treatment with either ANDRO or Li improved cognitive performance and short-term memory, functions of the hippocampus affected in AD patients. This cognitive improvement was abolished by co-administration, directly into brain, of the glucose transport inhibitor, Cyt B. The positive effect of Wnt signaling on cognitive performance has been described previously. However, we show here for first time that this effect depends, in part, by Wnt-mediated improvements in glucose metabolism in neurons. These data suggest that activating Wnt signaling promotes generation of both ATP and NADPH, thus increasing both energy availability and the defense against oxidative stress, both of which are dysregulated in various brain disorders. Our results help explain, in part, why Wnt agonists can improve cognitive performance in the context of neurological diseases (Figure 8) (Reger et al. 2008; Reger et al. 2006; Freiherr et al. 2013).
In summary, we have provided evidence that activation of Wnt signaling stimulates glucose metabolism in both cultured neurons and in the brain. Wnt-mediated improvements in glucose metabolism correlate with improved cognitive performance in a mouse model of AD. Currently, ANDRO and Li are used in patients as anti-inflammatory drugs and mood stabilizer, respectively, described as general mechanism to their action the effect as agonist of canonical Wnt signaling (Tobergte and Curtis 2013; Chiu et al. 2013). Our results suggest that the utilization of ANDRO or Li could delay the progression of neuropathological hallmarks associated with AD through a partial recovery in neuronal glucose metabolism. Additional future works are clearly needed to further illuminate the mechanisms by which these drugs exert their beneficial effects in the context of neurodegenerative diseases (Figure 8).
Acknowledgements
This work was supported by grants from the Basal Center of Excellence in Aging and Regeneration (CONICYT-AFB 170005) to N.C.I., FONDECYT (no. 1160724) to N.C.I., FONDECYT (no. 11160651) to P.C, and the National Institute of Health (DK084171) to G.W.W. We also thank the Sociedad Química y Minera de Chile (SQM) for the special grants “The role of K+ on Hypertension and Cognition” and “The role of Lithium in Human Health and Disease”.
Abbreviations
- AD
Alzheimer disease
- AMPK
AMP-activated protein kinase
- Aβ
Amyloid-β (Aβ)
- ROS
Reactive oxygen species
- GLUTs
Glucose transporters
- GLP-1
Glucagon like peptide 1
- CNS
Central nervous system
- ANDRO
Andrographolide
- Li
Lithium
- CytB
Cytochalasin B
- HK
Hexokinase
- PPP
Pentose phosphate pathway
- PFK
Phosphofructokinase
- LOF
Large open-field
- NOR
Novel object recognition
- NOL
Novel object localization
- qRT-PCR
Quantitative real-time PCR
- G6PDH
Glucose-6-phosphate dehydrogenase
- RRID
Resource Identification Initiative
Footnotes
Competing interests
The authors declare that they have no Competing interests the contents of this article.
References
- Aleshin AE, Zeng C, Bourenkov GP, Bartunik HD, Fromm HJ, Honzatko RB (1998) The mechanism of regulation of hexokinase: New insights from the crystal structure of recombinant human brain hexokinase complexed with glucose and glucose-6-phosphate. Structure 6, 39–50. [DOI] [PubMed] [Google Scholar]
- Arrázola MS, Ramos-Fernández E, Cisternas P, Ordenes D, Inestrosa NC (2017) Wnt signaling prevents the aβ oligomer-induced mitochondrial permeability transition pore opening preserving mitochondrial structure in hippocampal neurons. PLoS One 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrazola MS, Varela-Nallar L, Colombres M, Toledo EM, Cruzat F, Pavez L, Assar R, et al. (2009) Calcium/calmodulin-dependent protein kinase type IV is a target gene of the Wnt/beta-catenin signaling pathway. J Cell Physiol 221, 658–667. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Wu Z, Farrell RJ, Ryan TA, Medicine WC (2018) synapses. 93, 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin R. (2010) The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life 62, 315–333. [DOI] [PubMed] [Google Scholar]
- Baker LD, Cross DJ, Minoshima S, Belongia D, Stennis Watson G, Craft S. (2011) Insulin resistance and alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol 68, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. (2011) Alzheimer’s disease. Lancet 377, 1019–1031. [DOI] [PubMed] [Google Scholar]
- Barros LF, Bittner CX, Loaiza A, Ruminot I, Larenas V, Moldenhauer H, Oyarzun C, Alvarez M. (2009) Kinetic validation of 6-NBDG as a probe for the glucose transporter GLUT1 in astrocytes. J Neurochem 109 Suppl, 94–100. [DOI] [PubMed] [Google Scholar]
- Bayod S, Felice P, Andrés P, Rosa P, Camins A, Pallàs M, Canudas AM (2015) Downregulation of canonical Wnt signaling in hippocampus of SAMP8 mice. Neurobiol. Aging 36, 720–729. [DOI] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. (2006) Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study “recognition memory.” Nat Protoc 1, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Bolaños JP, Delgado-Esteban M, Herrero-Mendez A, Fernandez-Fernandez S, Almeida A. (2008) Regulation of glycolysis and pentose-phosphate pathway by nitric oxide: Impact on neuronal survival. Biochim. Biophys. Acta - Bioenerg 1777, 789–793. [DOI] [PubMed] [Google Scholar]
- Brown K, Yang P, Salvador D, Kulikauskas R, Ruohola-Baker H, Robitaille AM, Chien AJ, Moon RT, Sherwood V. (2017) WNT/β-catenin signaling regulates mitochondrial activity to alter the oncogenic potential of melanoma in a PTEN-dependent manner. Oncogene 36, 3119–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE (2005) Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol 57, 695–703. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH (2011) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet 20, 4515–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa W, Farias GG, Godoy JA, Fuenzalida M, Bonansco C, Inestrosa NC (2010) Wnt-5a occludes Abeta oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol Neurodegener 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, et al. (2000) A learning de cit related to age and b -amyloid plaques in a mouse model of Alzheimer ‘ s disease. 2940, 975–979. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhong C. (2013) Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol 108, 21–43. [DOI] [PubMed] [Google Scholar]
- Chiu C-T, Wang Z, Hunsberger JG, Chuang D-M (2013) Therapeutic Potential of Mood Stabilizers Lithium and Valproic Acid: Beyond Bipolar Disorder. Pharmacol. Rev 65, 105–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisternas P, Inestrosa NC (2017) Brain glucose metabolism: Role of Wnt signaling in the metabolic impairment in Alzheimer’s disease. Neurosci. Biobehav. Rev 80. [DOI] [PubMed] [Google Scholar]
- Cisternas P, Lindsay CB, Salazar P, Silva-Alvarez C, Retamales RM, Serrano FG, Vio CP, Inestrosa NC (2015a) The increased potassium intake improves cognitive performance and attenuates histopathological markers in a model of Alzheimer’s disease. Biochim. Biophys. Acta - Mol. Basis Dis 1852, 2630–2644. [DOI] [PubMed] [Google Scholar]
- Cisternas P, Lindsay CB, Salazar P, Silva-Alvarez C, Retamales RM, Serrano FG, Vio CP, Inestrosa NC (2015b) The increased potassium intake improves cognitive performance and attenuates histopathological markers in a model of Alzheimer’s disease. Biochim. Biophys. Acta - Mol. Basis Dis 1852. [DOI] [PubMed] [Google Scholar]
- Cisternas P, Salazar P, Silva-Álvarez C, Barros LF, Inestrosa NC (2016a) Activation of Wnt signaling in cortical neurons enhances glucose utilization through glycolysis. J. Biol. Chem 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisternas P, Salazar P, Silva-Álvarez C, Barros LF, Inestrosa NC (2016b) Wnt5a Increases the Glycolytic Rate and the Activity of the Pentose Phosphate Pathway in Cortical Neurons. Neural Plast. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisternas P, Silva-Alvarez C, Martínez F, Fernandez E, Ferrada L, Oyarce K, Salazar K, Bolaños JP, Nualart F. (2014b) The oxidized form of vitamin C, dehydroascorbic acid, regulates neuronal energy metabolism. J. Neurochem 129, 663–671. [DOI] [PubMed] [Google Scholar]
- Cox BL, Mackie TR, Eliceiri KW (2015) The sweet spot : FDG and other 2-carbon glucose analogs for multi-modal metabolic imaging of tumor metabolism. 5, 1–13. [PMC free article] [PubMed] [Google Scholar]
- Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, et al. (2012) Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol 69, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cs T. (2018) Purification and kinetic characterization of hexokinase and glucose 6 phosphate dehydrogenase from Schizosaccharomyces pombe. [DOI] [PubMed] [Google Scholar]
- Cummins RA, Walsh RN (1976) The Open-Field. 482–504. [PubMed] [Google Scholar]
- Dehydrogenase G phosphate, Survival C, Stanton RC (2013) NIH Public Access. NIH Public Access 64, 362–369. [Google Scholar]
- Detmer SA, Chan DC (2007) Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8, 870–879. [DOI] [PubMed] [Google Scholar]
- Doraiswamy PM, Sperling RA, Coleman RE, Johnson KA, Reiman EM, Davis MD, Grundman M, et al. (2012) Amyloid-beta assessed by florbetapir F 18 PET and 18-month cognitive decline: a multicenter study. Neurology 79, 1636–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanuel Y, Cochlin LE, Tyler DJ, Jager C. A. de, David Smith A, Clarke K. (2013) Human hippocampal energy metabolism is impaired during cognitive activity in a lipid infusion model of insulin resistance. Brain Behav. 3, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias GG, Godoy JA, Cerpa W, Varela-Nallar L, Inestrosa NC (2010) Wnt signaling modulates pre- and postsynaptic maturation: therapeutic considerations. Dev Dyn 239, 94–101. [DOI] [PubMed] [Google Scholar]
- Forlenza OV, Coutinho AMN, Aprahamian I, Prando S, Mendes LL, Diniz BS, Gattaz WF, Buchpiguel CA (2014) Long-term lithium treatment reduces glucose metabolism in the cerebellum and hippocampus of nondemented older adults: An [18F]FDG-PET study. ACS Chem. Neurosci 5, 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiherr J, Hallschmid M, Frey WH 2nd, Brunner YF, Chapman CD, Holscher C, Craft S, Felice FG De Benedict C. (2013) Intranasal insulin as a treatment for Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs 27, 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos TF, Kouznetsova V, Kudlicka K, Sweeney DE, Bush KT, Willert K, Farquhar MG, Nigam SK (2012) A protein kinase A and Wnt-dependent network regulating an intermediate stage in epithelial tubulogenesis during kidney development. Dev. Biol 364, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy JA, Arrazola MS, Ordenes D, Silva-Alvarez C, Braidy N, Inestrosa NC (2014a) Wnt-5a ligand modulates mitochondrial fission-fusion in rat hippocampal neurons. J. Biol. Chem 289, 36179–36193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy JA, Rios JA, Zolezzi JM, Braidy N, Inestrosa NC (2014b) Signaling pathway cross talk in Alzheimer’s disease. Cell Commun. Signal 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, Flores S, Christensen J, et al. (2018) Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol. 4422, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr SD, Simonian NA, Hyman BT (1995) Functional alterations in Alzheimer’s disease: decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J Neuropathol Exp Neurol 54, 38–41. [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11, 747–752. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, et al. (2004) Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet 13, 159–170. [DOI] [PubMed] [Google Scholar]
- Jaworski T, Dewachter I, Seymour CM, Borghgraef P, Devijver H, Kügler S, Leuven F Van (2010) Alzheimer’s disease: Old problem, new views from transgenic and viral models. Biochim. Biophys. Acta - Mol. Basis Dis 1802, 808–818. [DOI] [PubMed] [Google Scholar]
- Jeong YJ, Yoon HJ, Kang D-Y (2017) Assessment of change in glucose metabolism in white matter of amyloid-positive patients with Alzheimer disease using F-18 FDG PET. Medicine (Baltimore). 96, e9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos J, Godoy J, Inestrosa NC (2018) Wnt3a ligand facilitates autophagy in hippocampal neurons by modulating a novel GSK-3β-AMPK axis. Cell Commun Signal, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, Mattson MP (2011) Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol 10, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor K, Finer-Moore JS, Pedersen BP, Caboni L, Waight A, Hillig RC, Bringmann P, et al. (2016) Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci 113, 4711–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni R, Gus Y, Dor Y, Meyuhas O, Levitzki A. (2005) Active Src Elevates the Expression of β-Catenin by Enhancement of Cap-Dependent Translation Active Src Elevates the Expression of β-Catenin by Enhancement of Cap-Dependent Translation. Mol. Cell. Biol 25, 5031–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, Felice FG De, Ferreira ST (2012) Amyloid- b oligomers link depressive-like behavior and cognitive deficits in mice Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol. Psychiatry 18, 1053–1054. [DOI] [PubMed] [Google Scholar]
- Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G, Han SI, Park HG, Kang HS (2012) Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res 72, 3607–3617. [DOI] [PubMed] [Google Scholar]
- Liang Y, Li M, Lu T, Peng W, Wu J. (2017) Andrographolide Promotes Neural Differentiation of Rat Adipose Tissue-Derived Stromal Cells through Wnt / β -Catenin Signaling Pathway. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- Liu W, Zhuo P, Li L, Jin H, Lin B, Zhang Y, Liang S, et al. (2017) Activation of brain glucose metabolism ameliorating cognitive impairment in APP/PS1 transgenic mice by electroacupuncture. Free Radic. Biol. Med 112, 174–190. [DOI] [PubMed] [Google Scholar]
- Martin LJ (2012) Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci 107, 355–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor I, Cheung EC, Vousden KH (2011) Control of glycolysis through regulation of PFK1: Old friends and recent additions. Cold Spring Harb. Symp. Quant. Biol 76, 211–216. [DOI] [PubMed] [Google Scholar]
- Moreno-Navarrete JM, Ortega FJ, Rodriguez-Hermosa JI, Sabater M, Pardo G, Ricart W, Fernandez-Real JM (2011) OCT1 expression in adipocytes could contribute to increased metformin action in obese subjects. Diabetes 60, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccoli T, Cabecinha M, Tillmann A, Kerr F, Wong CT, Cardenes D, Vincent AJ, et al. (2016) Increased Glucose Transport into Neurons Rescues Ab Toxicity in Drosophila. Curr. Biol 26, 2291–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva CA, Inestrosa NC (2015) A novel function for Wnt signaling modulating neuronal firing activity and the temporal structure of spontaneous oscillation in the entorhinal-hippocampal circuit. Exp Neurol 269, 43–55. [DOI] [PubMed] [Google Scholar]
- Oliva CA, Montecinos-Oliva C, Inestrosa NC (2018) Wnt Signaling in the Central Nervous System: New Insights in Health and Disease. 81–130. [DOI] [PubMed] [Google Scholar]
- Parthsarathy V, Holscher C. (2013) Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PLoS One 8, e58784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, McQuade MM, et al. (2014) Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J 33, 1454–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N. Engl. J. Med 362, 329–344. [DOI] [PubMed] [Google Scholar]
- Ramiere C, Rodriguez J, Enache LS, Lotteau V, Andre P, Diaz O. (2014) Activity of Hexokinase Is Increased by Its Interaction with Hepatitis C Virus Protein NS5A. J. Virol 88, 3246–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH (2009) The role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectr 14, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Frey WH 2nd, Baker LD, Cholerton B, Keeling ML, Belongia DA, et al. (2006) Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging 27, 451–458. [DOI] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, Fishel MA, et al. (2008) Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 70, 440–448. [DOI] [PubMed] [Google Scholar]
- Reiter RJ (1995) Oxidative processes and antioxidative defense mechanisms in the aging brain. FASEB J 9, 526–533. [PubMed] [Google Scholar]
- Reno CM, Puente EC, Sheng Z, Daphna-iken D, Bree AJ, Routh VH, Kahn BB, Fisher SJ (2017) Brain GLUT4 Knockout Mice Have Impaired Glucose Tolerance, Decreased Insulin Sensitivity, and Impaired Hypoglycemic Counterregulation. 66, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME (1999) Ascorbate compartmentalization in the CNS. Neurotox Res 1, 81–90. [DOI] [PubMed] [Google Scholar]
- Rios JA, Cisternas P, Arrese M, Barja S, Inestrosa NC, Ríos J, Cisternas P, Arrese M, Barja S, Inestrosa NC (2014) Is Alzheimer’s disease related to metabolic syndrome? A Wnt signaling conundrum. Prog Neurobiol 121, 125–146. [DOI] [PubMed] [Google Scholar]
- Rivera DS, Lindsay C, Codocedo JF, Morel I, Pinto C, Bozinovic F, Inestrosa NC (2018) A ll C o n t en t S ea r ch Andrographolide recovers cognitive impairment in a natural model of Alzheimer ‘ s disease ( Octodon degus ) Related Articles. 4580, 1–2. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Carling D, Prentki M, Cacicedo JM (2013) AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Invest 123, 2764–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar P, Cisternas P, Codocedo JF, Inestrosa NC (2017) Induction of hypothyroidism during early postnatal stages triggers a decrease in cognitive performance by decreasing hippocampal synaptic plasticity. Biochim. Biophys. Acta - Mol. Basis Dis 1863, 870–883. [DOI] [PubMed] [Google Scholar]
- Schapira AH (2012) Targeting mitochondria for neuroprotection in Parkinson’s disease. Antioxid Redox Signal 16, 965–973. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano FG, Tapia-rojas C, Carvajal FJ, Hancke J, Cerpa W. (2014) Andrographolide reduces cognitive impairment in young and mature A β PPswe / PS-1 mice. 9, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Desilva S, Abbruscato T. (2012) The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimeŕs Disease. Int J Mol Sci 13, 12629–12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Alvarez C, Arrazola MS, Godoy JA, Ordenes D, Inestrosa NC (2013) Canonical Wnt signaling protects hippocampal neurons from Abeta oligomers: role of non-canonical Wnt-5a/Ca(2+) in mitochondrial dynamics. Front Cell Neurosci 7, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P. (1994) Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol 35, 546–551. [DOI] [PubMed] [Google Scholar]
- Stow LR, Jacobs ME, Wingo CS, Cain BD (2016) Endothelin 1 gene regulation. 25, 16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Rojas C, Inestrosa NC (2018) Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer’s disease in J20-APP transgenic and wild-type mice. J. Neurochem 144, 443–465. [DOI] [PubMed] [Google Scholar]
- Tapia-rojas C, Sch A, Lindsay CB, Ureta RC, Hancke J, Melo F, Inestrosa NC (2015) Andrographolide activates the canonical Wnt signalling pathway by a mechanism that implicates the non-ATP competitive inhibition of GSK-3 β : autoregulation of GSK-3 β in vivo. 430, 415–430. [DOI] [PubMed] [Google Scholar]
- Thurley K, Herbst C, Wesener F, Koller B, Wallach T, Maier B, Kramer A, Westermark PO (2017) Principles for circadian orchestration of metabolic pathways. Proc. Natl. Acad. Sci 114, 1572–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobergte DR, Curtis S. (2013) Andrographolide: A Review of its Anti-inflammatory Activity via Inhibition of NF-kappaB Activation from Computational Chemistry Aspects. J. Chem. Inf. Model 53, 1689–1699.23800267 [Google Scholar]
- Toledo EM, Inestrosa NC (2010) Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1ΔE9 mouse model of Alzheimer’s disease. Mol. Psychiatry 15, 272–285. [DOI] [PubMed] [Google Scholar]
- Tsytsarev V, Maslov KI, Yao J, Parameswar AR, Demchenko AV, Wang LV (2012) In vivo imaging of epileptic activity using 2-NBDG, a fluorescent deoxyglucose analog. J. Neurosci. Methods 203, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JY, Fuenzalida M, Inestrosa NC (2014) In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model. J Neurosci 34, 2191–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, Benzinger TL, Morris JC (2012) PET amyloid-beta imaging in preclinical Alzheimer’s disease. Biochim Biophys Acta 1822, 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Zhao X, Huang Q, Sun H, Sun C, Yuan J, He C. (2017) Activation of Wnt / β -catenin signaling by lithium chloride attenuates D -galactose-induced neurodegeneration in the auditory cortex of a rat model of aging. 7, 759–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Hirai Y, Miyake K, Shimada T. (2014) Targeted gene transfer into ependymal cells through intraventricular injection of AAV1 vector and long-term enzyme replacement via the CSF. 1–7. Sci Rep. 4:5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama N, Malbon CC (2009) Dishevelled-2 docks and activates Src in a Wnt-dependent manner. J. Cell Sci 122, 4439–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Qian X, Qin C, Lin Y, Wu H, Chang L, Luo C, Zhu D. (2016) Phosphofructokinase-1 Negatively Regulates Neurogenesis from Neural Stem Cells. Neurosci. Bull 32, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang S, Jiang B, Huang L, Ji Z, Li X, Zhou H, et al. (2017) C-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat. Commun 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yang Z, Zhu B, Hu J, Liew CW, Zhang Y, Leopold JA, Handy DE, Loscalzo J, Stanton RC (2012) Increasing Glucose 6-Phosphate Dehydrogenase Activity Restores Redox Balance in Vascular Endothelial Cells Exposed to High Glucose. PLoS One 7(11): e49128. 10.1371/journal.pone.0049128 [DOI] [PMC free article] [PubMed] [Google Scholar]