

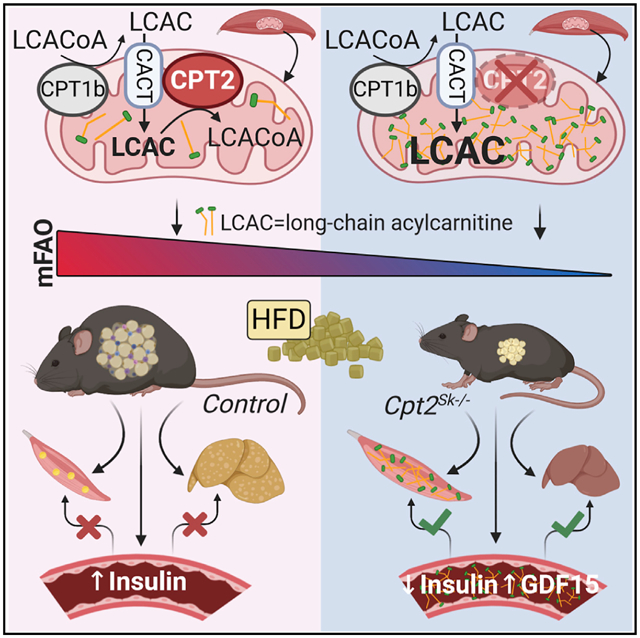
SUMMARY
To assess the effects of acylcarnitine accumulation on muscle insulin sensitivity, a model of muscle acylcarnitine accumulation was generated by deleting carnitine palmitoyltransferase 2 (CPT2) specifically from skeletal muscle (Cpt2Sk−/− mice). CPT2 is an irreplaceable enzyme for mitochondrial long-chain fatty acid oxidation, converting matrix acylcarnitines to acyl-CoAs. Compared with controls, Cpt2Sk−/− muscles do not accumulate anabolic lipids but do accumulate ~22-fold more long-chain acylcarnitines. High-fat-fed Cpt2Sk−/− mice resist weight gain, adiposity, glucose intolerance, insulin resistance, and impairments in insulin-induced Akt phosphorylation. Obesity resistance of Cpt2Sk−/− mice could be attributed to increases in lipid excretion via feces, GFD15 production, and energy expenditure. L-carnitine supplement intervention lowers acylcarnitines and improves insulin sensitivity independent of muscle mitochondrial fatty acid oxidative capacity. The loss of muscle CPT2 results in a high degree of long-chain acylcarnitine accumulation, simultaneously protecting against diet-induced obesity and insulin resistance.
Graphical Abstract
In Brief
Pereyra et al. show that loss of muscle mitochondrial long-chain fatty acid oxidation results in a large accumulation of long-chain acylcarnitines in muscle and plasma that do not cause insulin resistance. Rather, the inability to flux fatty acids through β-oxidation prevents diet-induced obesity and insulin resistance.
INTRODUCTION
Obesity and type 2 diabetes are caused by a combination of poor diet quality, high caloric intake, and physical inactivity. The skeletal muscle accounts for 50% of total energy expenditure (EE), and because it is highly bioenergetically demanding and insulin responsive, it greatly affects systemic metabolism in physiological and pathological scenarios. Disrupted skeletal muscle mitochondrial oxidative capacity is a consequence of overnutrition and obesity and is a potential cause of insulin resistance. However, obesity-induced mitochondrial dysfunction occurs at many levels, and dissecting the differential junctures of mitochondrial metabolism to determine the key disruptions leading to metabolic diseases remains ongoing. Among potential metabolic disruptors are medium-chain acylcarnitines (MCACs) and long-chain acylcarnitines (LCACs), fatty acid oxidative intermediates that when accumulate are implicated as causative in insulin resistance (Adams et al., 2009; Aguer et al., 2015; Bouchouirab et al., 2018; Keung et al., 2013; Kim et al., 2014; Koves et al., 2008; Li et al., 2015; Liepinsh et al., 2016, 2017; Mai et al., 2013; McCoin et al., 2015b; Power et al., 2007; Ringseis et al., 2012; Samimi et al., 2016; Vavrova et al., 2016; Warfel et al., 2017). LCACs accumulate during numerous physiological and non-physiological stresses, including fasting, ketogenic diet, exercise, overnutrition, heart disease, and insulin resistance (Adams et al., 2009; Bouchouirab et al., 2018; Mai et al., 2013; McCoin et al., 2015a; Sampey et al., 2012; Schooneman et al., 2013, 2014; Soeters et al., 2009; Su et al., 2005; Xu et al., 2016; Zhang et al., 2017). We previously demonstrated a high degree of LCAC accumulation in a mouse model of heart and muscle carnitine palmitoyltransferase 2 (CPT2) deficiency (Cpt2M−/−) (Pereyra et al., 2017). CPT2 is a critical, autonomous enzyme required for carnitine-mediated oxidation of long-chain fatty acids in the mitochondria by converting CPT1-generated ACs into acyl-coenzyme A (CoA) coenzymes within the mitochondrial matrix. The Cpt2M−/− mice develop severe cardiac hypertrophy and ultimately heart failure (Pereyra et al., 2017) and do not respond well to high-fat-diet feeding. Therefore, we generated a skeletal muscle-specific CPT2-deficient mouse model (Cpt2Sk−/−) to investigate the role of muscle fatty acid oxidation and LCAC accumulation in obesity and insulin sensitivity. Cpt2Sk−/− mice accumulate substantial levels of LCAC in plasma and muscle, ~20-fold greater than control mice, thus positioning the Cpt2Sk−/− mice as an excellent model to study the effects of LCAC accumulation. Loss of muscle CPT2 had minimal effects on systemic metabolic parameters of mice fed a low-fat diet; however, upon high-fat-diet feeding, Cpt2Sk−/− mice resisted diet-induced weight gain and adiposity. Resistance to obesity coincided with improved glucose homeostasis and Akt phosphorylation in response to insulin in Cpt2Sk−/− mice compared with control mice. Thus, despite a high degree of LCAC accumulation in skeletal muscle and plasma, the loss of fatty acid oxidation capacity in the muscle resulted in retained or heightened insulin sensitivity even under a high-fat dietary challenge. Moreover, the improvements in glucose tolerance and insulin sensitivity in Cpt2Sk−/− mice after long-term L-carnitine (L-carn) supplementation in the drinking water suggests that the carnitine-driven rescue mechanisms occur independent of muscle mitochondrial fatty acid oxidative flux.
RESULTS
Skeletal-Muscle-Specific CPT2-Deficient Mice Accumulate LCACs
Skeletal muscle-specific CPT2-deficient mice were generated by crossing Cpt2lox/lox mice (Lee et al., 2015) with Cre-recombinase-expressing mice driven by the human α-actin 1-Cre (ACTA1/HSA) transgene. Specific deletion was confirmed by Cpt2 mRNA and protein that was absent from Cpt2Sk−/− muscle (Figure 1A) while being preserved in liver (Figure S1K). As expected, the loss of CPT2 reduced the rates of long-chain fatty acid oxidation into carbon dioxide in muscle homogenates (by 65%) (Figure 1B). However, no significant difference was observed for palmitate oxidation in acid-soluble metabolites, in agreement with the formation of acid-soluble ACs by CPT1 that are not further oxidized in the absence of CPT2 (Figure 1C). Cpt2Sk−/− muscle homogenates maintained the capacity to oxidize the medium-chain fatty acid free octanoate, which can bypass the CPT-carnitine shuttle for mitochondrial fatty acid oxidation (Figure 1D), suggesting that Cpt2Sk−/− mitochondria are intact and capable of oxidizing fatty acids in a carnitine-independent manner. Pyruvate oxidation was increased 54% in Cpt2Sk−/− muscle compared with control, suggesting compensatory mechanisms to increase the use of non-fatty acid substrates for energy production (Figure 1E). Next, the specific AC species and the extent of its accumulation was evaluated. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) quantitative assessment of both skeletal muscle and plasma revealed that the tibialis anterior (TA) muscle of Cpt2Sk−/− mice had increased LCACs, such as AC18 and AC18:1 by ~5- and ~7.6-fold, respectively (Figure 1F). Conversely, short-chain ACs like butyryl-carnitine (AC4) were decreased in Cpt2Sk−/− mice by 86% (Figure 1F). Plasma of CPT2-deficient mice did not have reduced AC4 but showed a similar profile of LCAC accumulation, with increased AC18 and AC18:1 by 1.5- and 5.2-fold, respectively (Figure 1F). To obtain a broad and comprehensive assessment of the many ACs affected in Cpt2Sk−/− mice, we performed direct injection mass spectrometry (DIMS). DIMS showed that although nearly all LCACs 14 carbons and greater in length were increased in the Cpt2Sk−/− TA muscle relative to controls; the most abundant species were AC18:1 (56-fold), AC18:2 (36-fold), AC18:0 (31-fold), and AC16:0 (14-fold) (Figure 1G). In contrast, short- and medium-chain ACs were significantly reduced in Cpt2Sk−/− muscle by ~61% (Figure 1H). Thus, the loss of CPT2 reduced medium-and short-chain ACs and caused accumulation of LCACs. In plasma, the same LCACs were elevated in Cpt2Sk−/− mice compared to controls, albeit to different degrees: AC18:1 (6.5-fold), AC18:2 (6.2-fold), AC18:0 (5.5-fold), and AC16:0 (4.2-fold) (Figure 1I). Unlike the reductions observed in muscle for short-and medium-chain ACs, no genotype effect was observed in the plasma for these metabolites (Figure 1J). These data confirm that the lack of CPT2 in the skeletal muscle results in a high degree of LCAC accumulation, especially 16- and 18-carbon-long ACs, but not medium- or short-chain ACs, in plasma and muscle.
Figure 1. Skeletal-Muscle-Specific CPT2-Deficient Mice Accumulate LCACs.
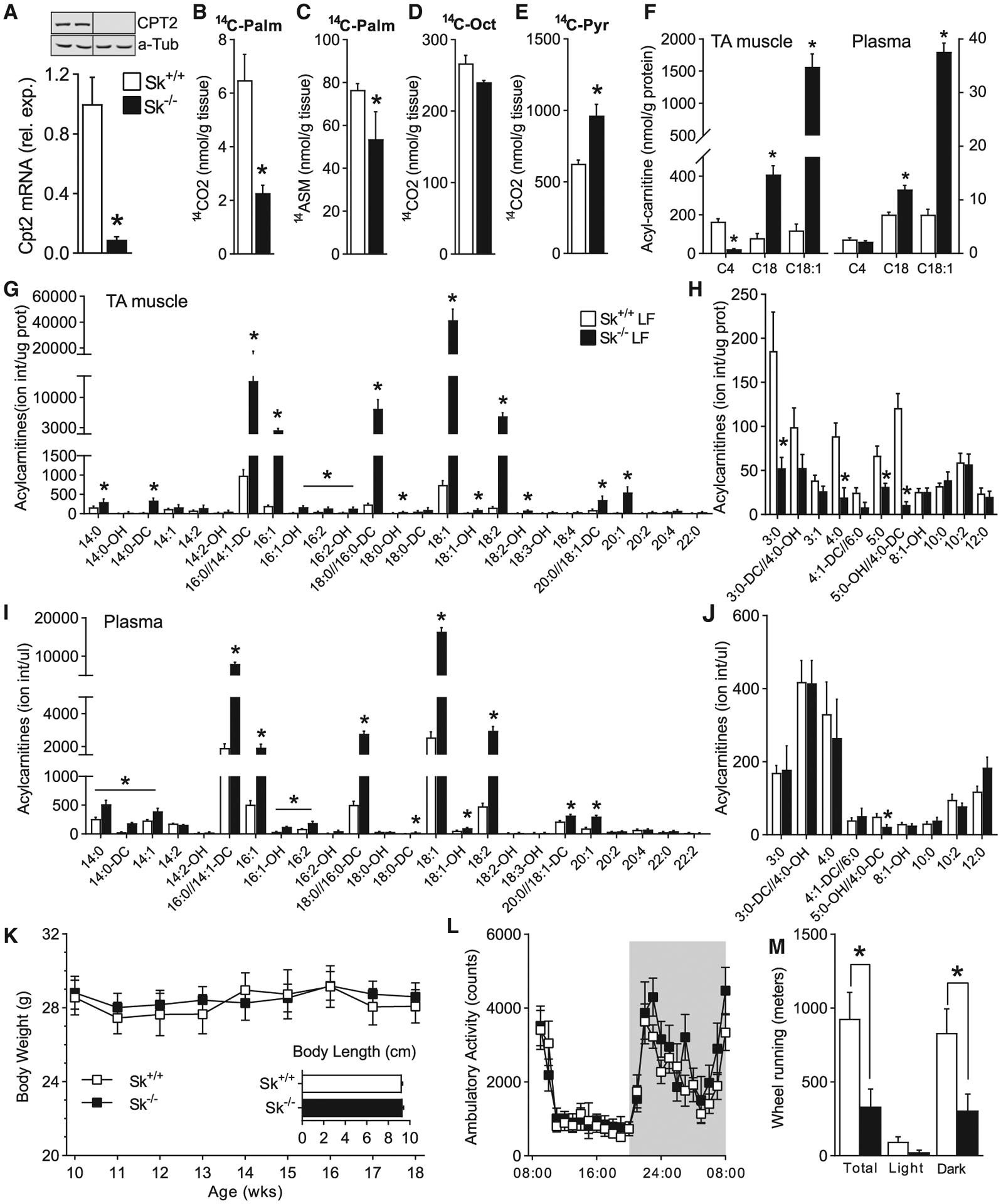
(A) mRNA and protein expression of Cpt2 in skeletal muscle of Cpt2Sk+/+ and Cpt2Sk−/− mice (n = 6).
(B–E) Complete and incomplete oxidation of 14C-palmitate, 14C-octanoate, and 14C-pyruvate in skeletal muscle homogenates of Cpt2Sk+/+ and Cpt2Sk−/− mice (n = 3).
(F) Acylcarnitine quantification in TA muscle and plasma of Cpt2Sk+/+ and Cpt2Sk−/− female mice (n = 6).
(G and H) Ion intensity per microgram of protein of LCACs and of medium- and short-chain ACs in skeletal muscle of Cpt2Sk+/+ and Cpt2Sk−/− female mice, 24 weeks of age (n = 6).
(I and J) Ion intensity per microliter of plasma of LCACs and of medium- and short-chain ACs in plasma of Cpt2Sk+/+ and Cpt2Sk−/− female mice, 24 weeks of age (n = 6).
(K) Body weight and length of Cpt2Sk+/+ and Cpt2Sk−/− male mice, 12 weeks of age (n = 7–12).
(L) Spontaneous, home-cage locomotor activity during light and dark cycles of male mice, 12 weeks of age (average of 2 recording days, n = 8).
(M) Voluntary wheel activity during light and dark cycles of male mice, 12 weeks of age (average of 2 recording days, n = 8).
Data are presented as mean ± SEM. *p < 0.05 determined by Student’s t test. See also Figure S1.
Cpt2Sk−/− Mice Are Protected from Obesity Induced by a High-Fat Diet
When Cpt2Sk−/− mice were fed a low-fat, standard diet, the loss of mitochondrial long-chain fatty acid oxidation in skeletal muscle did not affect body weight, body length, body fat, or lean mass (Figures 1K and S1A). Specifically, inguinal and gonadal fat depots and gastrocnemius (GA) and TA muscles had similar mass between genotypes; however, the soleus (SOL) muscle was 32% heavier than in controls (Figure S1B). Cpt2Sk−/− mice exhibited similar whole-body EE, food intake, and ambulatory activity compared with control mice (Figures 1L, S1C–S1E, and S1I). However, when given access to a running wheel, voluntary running in Cpt2Sk−/− mice was ~75% less than in controls (Figure 1M). These data suggest that the loss of muscle mitochondrial long-chain fatty acid oxidation does not overtly affect whole-body energetic homeostasis but that Cpt2Sk−/− mice avoid voluntary wheel exercise.
Next, we exposed Cpt2Sk−/− mice to a chronic high-fat dietary challenge that resulted in complete resistance to weight gain in Cpt2Sk−/− mice (Figure 2A). The lack of weight gain in Cpt2Sk−/− mice was attributed to adiposity resistance, because total body fat was ~90% below that of high-fat-fed controls (Figure 2B). In agreement, Cpt2Sk−/− inguinal and gonadal white adipose tissue (iWAT and gWAT, respectively) fat depot mass was ~70%–90% less than that of controls (Figure 2C). Histological assessment of gWAT revealed that Cpt2Sk−/− mice were refractory to adipocyte expansion and adipose inflammation induced by the high-fat diet, as depicted by the lack of macrophage infiltrates in classic crown-like structures (Figure 2D). Total lean mass was not significantly different between genotypes that spent up to 5 weeks on a high-fat diet but became ~20% reduced in the Cpt2Sk−/− mice by 14 weeks of feeding, likely the consequence of resistance to hepato- and cardio-megaly induced by the high-fat diet (Figures 2E and 2F). Despite the lack of weight gain in high-fat-fed Cpt2Sk−/− mice, they consumed similar food and water compared with control mice (Figure 2G). Thus, obesity resistance in Cpt2Sk−/− mice results from lack of increased adiposity.
Figure 2. Cpt2Sk−/− Protection from Obesity Induced by a High-Fat Diet.
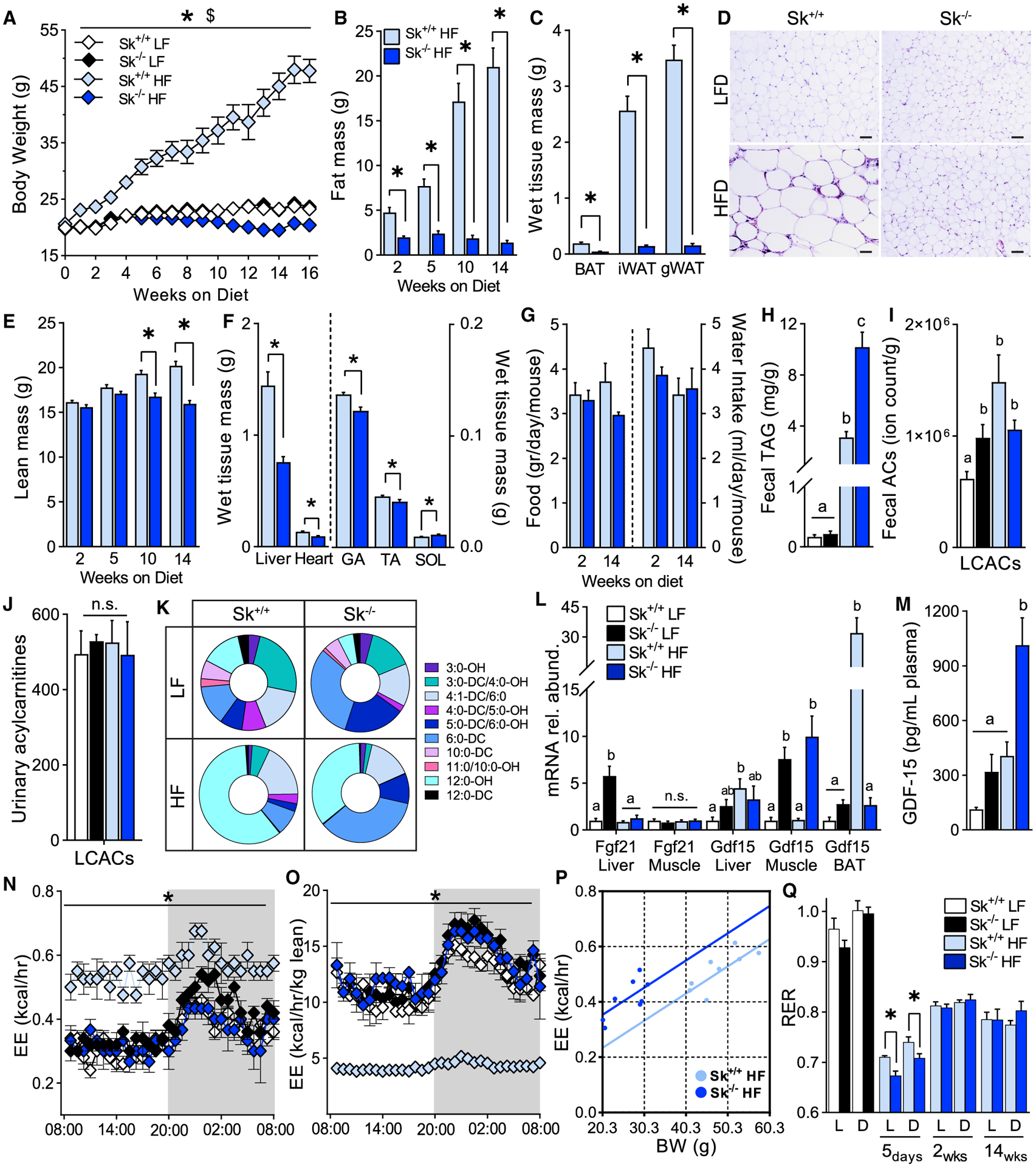
(A) Body weight of Cpt2Sk+/+ and Cpt2Sk−/− female mice fed a low-fat (LF) or a high-fat (HF) diet for 16 weeks (n = 7–12).
(B) Whole-body fat mass after 2, 5, 10, and 14 weeks of HF diet fed to female mice (n = 7–12).
(C) Wet tissue mass of BAT, iWAT, and gWAT individual depots after 16 weeks of HF diet fed to female mice (n = 7–12).
(D) Representative histological image of gWAT after 8 weeks of HF diet, H&E stain. Scale bar, 100 μm.
(E) Whole-body lean mass after 2, 5, 10, and 14 weeks of HF diet fed to female mice (n = 7–12).
(F) Wet tissue mass of liver, heart, GA, TA, and SOL muscle after 16 weeks of HF diet fed to female mice (n = 7–12).
(G) Food and water intake at 2 and 14 weeks of HF diet fed to female mice (n = 7–12).
(H) Fecal TAGs of male mice after 8 weeks of LF- and HF-diet feeding (n = 5).
(I) Fecal LCACs of male mice after 8 weeks of LF- or HF-diet feeding (n = 5).
(J) Urinary levels of LCACs before and after a 10-day-long HF-diet feeding of male mice (n = 6).
(K) Distribution of the most abundant hydroxylated and dicarboxylated short- and medium-chain species in male mice before and after a 10-day-long HF-diet feeding (n = 6).
(L) Liver, muscle, and BAT mRNA abundance of genes involved in regulation of systemic metabolism of female mice fed a LF or HF diet for 16 weeks (n = 6).
(M) Plasmatic GDF15 of male mice fed a LF or HF diet for 8 weeks (n = 6).
(N and O) Energy expenditure (EE) at 14 weeks of LF- or HF-diet feeding presented as both non-normalized and normalized to body lean mass of female mice (n = 3–5).
(P) Regression plot of EE versus body weight (BW) at 14 weeks of HF-diet feeding. Each regression line represents the ANCOVA predicted mean EE per value of BW (n = 8).
(Q) Resting energy requirements (RERs) at several time points during LF- or HF-diet feeding of female mice (n = 5–10).
Data are presented as mean ± SEM. *p < 0.05 determined by Student’s t test for Cpt2Sk+/+ HF diet versus Cpt2Sk−/− HF diet; $p < 0.05 for Cpt2Sk+/+ LF diet versus Cpt2Sk+/+ HF diet. Statistical analysis by 2-way ANOVA for 4 groups and by ANCOVA for EE. Means depicting a different letter indicate significant differences between groups (p < 0.05) (K–M). See also Figures S1 and S2.
In a search for possible contributors to obesity resistance, we found that high-fat-fed Cpt2Sk−/− mice had fecal triacylglycerol (TAG) content elevated by ~3-fold compared with control mice on the high-fat diet (Figures 2H and S2E). Fecal free fatty acid, cholesterol ester, or acylcarnitine content was not elevated by the high-fat diet between genotypes (Figures 2I and S2F–S2H). We next considered another form of carbon wasting that is specific to the CPT2 deficiency model: urinary secretion of carbons in the form of ACs. Plasma and urinary ACs were determined before and after 10 days of high-fat-diet feeding, and despite the high levels in the plasma, LCACs in the urine of Cpt2Sk−/− mice were not changed relative to controls before or after the high-fat diet (Figure 2J). The total abundance of short-, medium-, and acetyl-ACs was not changed in urine of Cpt2Sk−/− mice before or after the high-fat diet (Figures S2A and S2B), yet the distribution of these metabolites was significantly regulated by both diet and genotype. Specifically, high-fat-diet feeding increased the percentage of dicarboxylic and hydroxylated ACs in both control and Cpt2Sk−/− mice (Figure S2C). Furthermore, loss of muscle CPT2 increased the percentage of medium-chain dicarboxylated ACs (Figure 2K), biomarkers of increased fatty acid oxidation at the endoplasmic reticulum (ER) and peroxisome and often elevated in the urine of individuals with metabolic myopathies (Vissing et al., 2019; Wanders et al., 2011). Altogether, these data suggest that muscle CPT2 deficiency alone is sufficient to drive increased urinary excretion of omega-oxidation products. These data also suggest that high levels of plasma ACs in Cpt2Sk−/− mice are not reflected in urine or feces, acute high-fat-diet feeding does not increase urinary ACs, and increased lipid excretion via feces might contribute to obesity resistance in Cpt2Sk−/− mice.
The inability of Cpt2Sk−/− mice to gain weight on the high-fat diet is consistent with models of impaired muscle mitochondrial metabolism, an effect that has been linked to release of myomitokines such as FGF21 and GFD15 that contribute to obesity resistance (Bowman et al., 2016; Chung et al., 2017; Vandanmagsar et al., 2016). Surprisingly, the mitokine FGF21, which is commonly upregulated in mitochondrial metabolic disruption models (Kliewer and Mangelsdorf, 2019), was not increased in muscle or liver of Cpt2Sk−/− high-fat-fed mice (Figure 2L). In contrast, Gdf15 gene expression was upregulated in muscle of Cpt2Sk−/− mice and corresponded with a 2.5-fold increase in GDF15 plasma levels compared with high-fat-fed controls (Figures 2L and 2M). GDF15 expression can be induced in muscle by the integrated stress response; however, the lack of elevation in Cpt2Sk−/− muscle of eIF2A phosphorylation and of upregulation of its downstream effectors, such as Chop and Atf4, suggests an alternate mechanism for GDF15 induction (Figures S2I and S2J).
To assess the early metabolic adaptations to high-fat feeding, indirect calorimetry was performed during the low- to high-fat-diet transition. Inability to use fatty acids as fuel was evident immediately after the dietary switch in Cpt2Sk−/− mice, with respiratory exchange ratio (RER) values dropping below 0.7 by day 5 of high-fat-diet feeding, reflecting an acute switch to gluconeogenesis and ketogenesis (Schutz and Ravussin, 1980) (Figures 2Q and S1F–S1H). Next, whole-body metabolism was assessed across the time course of chronic low- and high-fat-diet feeding. As expected (Kaiyala and Schwartz, 2011; Tschöp et al., 2011), larger mice had significantly higher EE, oxygen consumption (VO2), and carbon dioxide release (VCO2) relative to smaller, thinner mice that became evident toward the end of the 16-week-long intervention (Figures 2N, 2O, and S1I). As such, the leanness of Cpt2Sk−/− mice on the high-fat diet was reflected in lower EE relative to the heavier high-fat-fed controls. To remove the cofounding effect of body weight between high-fatfed control and Cpt2Sk−/− mice, we performed analysis of covariance (ANCOVA) to estimate the group difference in EE controlling for body mass. This analysis revealed that high-fat-fed Cpt2Sk−/− mice have higher EE (0.53 ± 0.032 kcal/h) than controls (0.4 ± 0.032 kcal/h; p = 0.05) (Figure 2P). After two weeks of high-fat-diet feeding, RER was changed only in a diet-dependent manner (Figure 2Q). Altogether, these data suggest that a 32% increase in EE, increased fecal lipid excretion, and increased myomitokines contributed to resistance to diet-induced adiposity in Cpt2Sk−/− mice.
Loss of Muscle CPT2 Regulates Systemic Metabolism
Next, systemic metabolic homeostasis was assessed to determine how the lack of fatty acid mitochondrial oxidative energy production in muscle affected peripheral metabolism. Cpt2Sk−/− mice had reduced plasma glucose and increased free fatty acids and ketones, an effect exacerbated by the high-fat diet (Figures 3A–3C). Plasma TAG and cholesterol were not significantly affected by genotype on the low-fat diet; moreover, Cpt2Sk−/− mice resisted increases in circulating TAG and cholesterol induced by the high-fat diet (Figures 3D and 3E). Adrenergic-driven phosphorylation of hormone-sensitive lipase (HSL) at protein kinase A (PKA) target sites in adipose tissue increased ~2-fold in high-fat-fed Cpt2Sk−/− mice when compared with controls (Figure 3F). Models of obesity resistance and persistent adrenergic stimuli often undergo browning of white adipose tissue; however, Ucp1 mRNA was not changed between genotypes in inguinal white adipose tissue or in interscapular brown adipose tissue (BAT), and no histological evidence of browning was observed in gonadal adipose (Figures 2D, 3G, S3C, and S3D). Lack of inflammation induced by the high-fat diet in Cpt2Sk−/− white adipose tissue, as initially observed in tissue sections, was confirmed by gene expression of Tnfα, F4/80, and Mcp1 (Figures 2D and 3H). Liver gene expression profiles related to ketogenesis, β-oxidation, carnitine synthesis, gluconeogenesis, and lactate catabolism were increased, whereas glycolytic genes were decreased in Cpt2Sk−/− mice compared with control mice, particularly on the high-fat diet, in agreement with elevated circulating fatty acids and ketones and reduced RER below 0.7 (Schutz and Ravussin, 1980) (Figures 3I–3N). Increase in Cpt2Sk−/− mice of key ketogenesis enzyme 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2) was confirmed at the protein level (Figure S3E). These data suggest that loss of muscle fatty acid oxidation increases adrenergic tone, ketogenesis, and gluconeogenesis in the absence of white adipose browning.
Figure 3. Loss of Muscle CPT2 Regulates Systemic Metabolism.
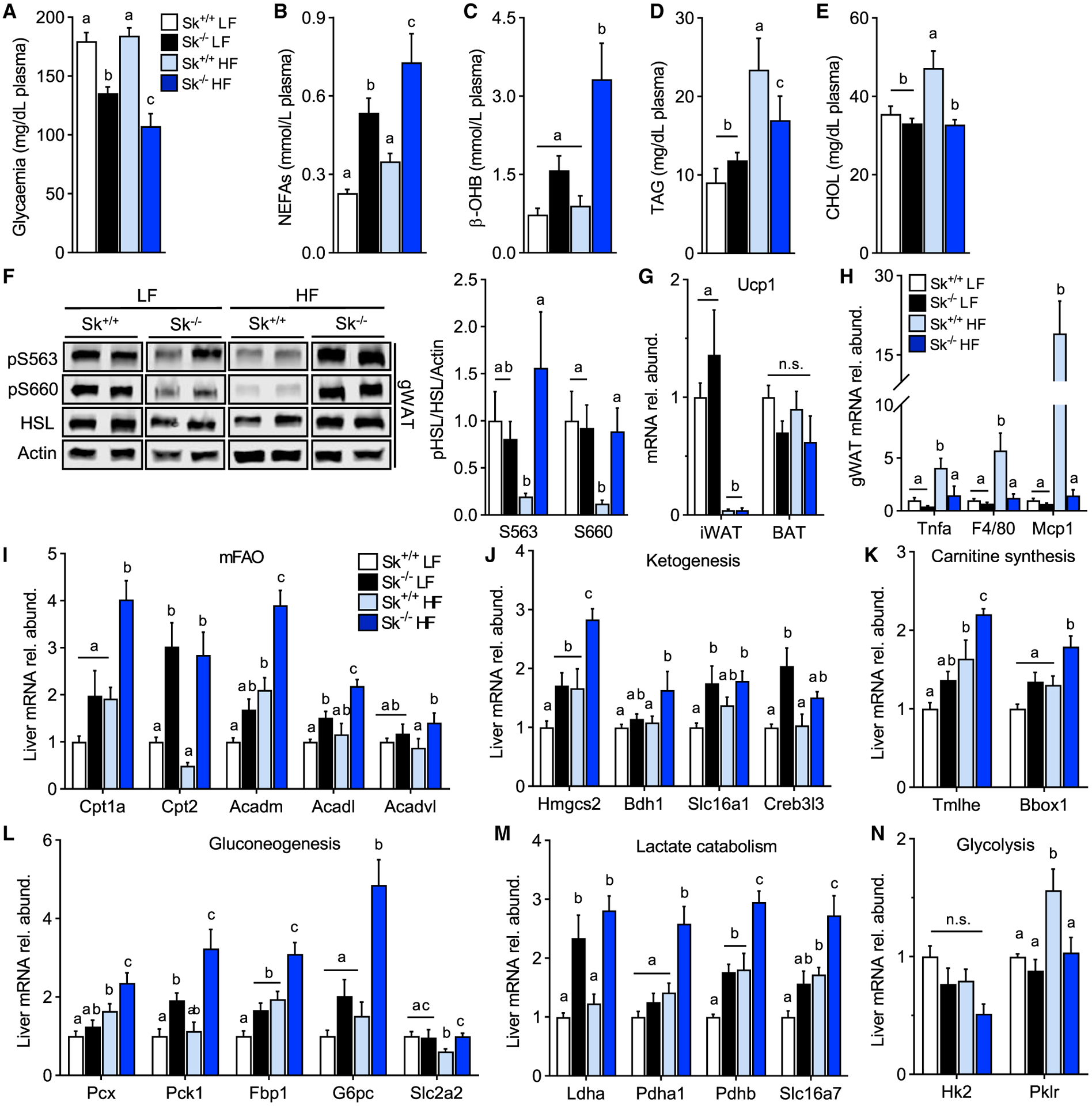
(A) Plasma glucose levels of Cpt2Sk+/+ and Cpt2Sk−/− female mice fed a LF or HF diet for 16 weeks (n = 6–14).
(B) Non-esterified fatty acids (NEFAs) of female mice fed a LF or HF diet for 16 weeks (n = 6–14).
(C) β-hydroxybutyrate (β-OH) of female mice fed a LF or HF diet for 16 weeks (n = 6–14).
(D) Triacylglycerol (TAG) of female mice fed a LF or HF diet for 16 weeks (n = 6–14).
(E) Cholesterol esters (CHOLs) of female mice fed a LF or HF diet for 16 weeks (n = 6–14).
(F) Phosphorylation status of hormone-sensitive lipase (HSL) at serine residues S563 and S660 in gWAT of female mice fed a LF or HF diet for 16 weeks. Data are normalized to control mice on a LF diet (n = 6).
(G) iWAT and BAT mRNA abundance thermogenesis marker of female mice fed a LF or HF diet for 16 weeks. Data are normalized to control mice on a LF diet (n = 6).
(H) gWAT mRNA abundance inflammatory markers of female mice fed a LF or HF diet for 16 weeks. Data are normalized to control mice on a LF diet (n = 6).
(I–N) Liver mRNA abundance of female mice fed a LF or HF diet for 16 weeks. Data are normalized to control mice on a LF diet (n = 6).
Data are presented as mean ± SEM. Statistical analysis by 2-way ANOVA. Means depicting a different letter indicate significant differences between groups (p < 0.05).
Improved Glucose Tolerance in Cpt2Sk−/− Mice
Because LCACs have been implicated in interfering with insulin-induced Akt signaling (Aguer et al., 2015; Blackburn et al., 2020; Liepinsh et al., 2016, 2017; McCoin et al., 2015b), the effect of LCAC accumulation on insulin responsiveness was assessed. In the low-fat diet paradigm, Akt phosphorylation in muscle was not affected by CPT2 deficiency, with or without insulin stimulation (Figure 4A). Although high-fat-diet feeding trended toward reduced insulin-induced Akt phosphorylation in muscle of control mice, Cpt2Sk−/− mice retained a high degree of insulin-dependent Akt phosphorylation (Figure 4A). In Cpt2Sk−/− liver and gonadal white adipose, Akt phosphorylation was suppressed at basal conditions in both low- and high-fat-diet feeding (Figures 4B and 4C); however, in response to insulin stimulation, Akt phosphorylation increased to similar levels across the groups. Thus, the net effect of insulin-induced Akt phosphorylation was increased in Cpt2Sk−/− liver and adipose compared with control mice independent of diet (Figures 4B and 4C).
Figure 4. Systemic and Tissue-Specific Insulin Sensitivity in Cpt2Sk−/− Mice.
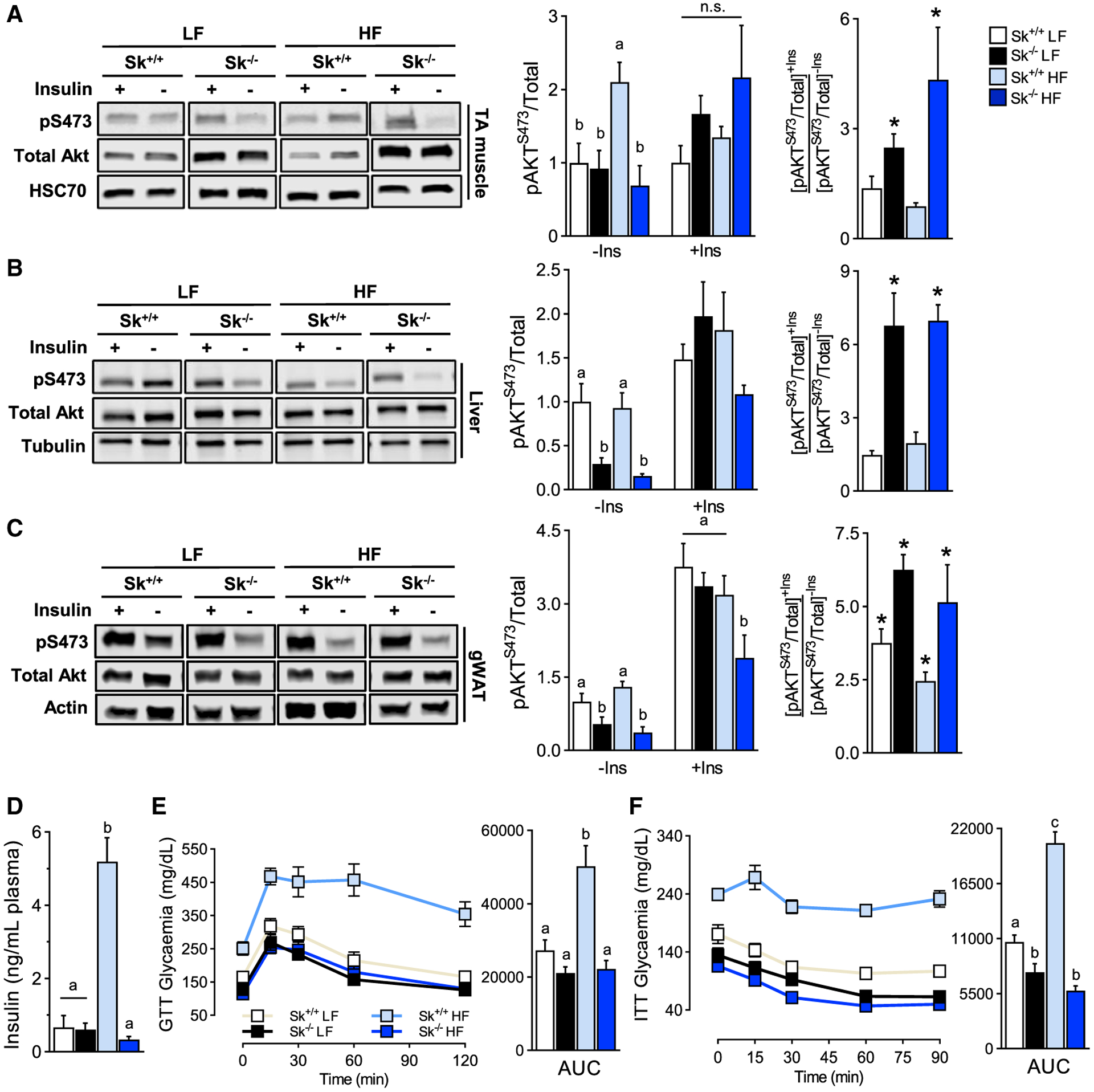
(A) Phosphorylation status of Akt protein in TA muscle of Cpt2Sk+/+ and Cpt2Sk−/− female mice fed a LF or HF diet for 16 weeks with (+Ins) or without (−Ins) insulin stimuli. Data are normalized to control mice on a LF diet without insulin (n = 6).
(B) Phosphorylation status of Akt protein in liver of female mice fed a LF or HF diet. Data are normalized to control mice on a LF diet without insulin (n = 6).
(C) Phosphorylation status of Akt protein in gWAT of female mice fed a LF or HF diet. Data are normalized to control mice on a LF diet without insulin (n = 6).
(D) Fasting insulinemia of male mice fed a LF or HF diet for 8 weeks (n = 6).
(E) Glucose tolerance test of male mice fed a LF or HF diet for 8 weeks. Glycaemia over time (left) and the corresponding area under the curve (right) (n = 8–10).
(F) Insulin tolerance test of male mice fed a LF or HF diet for 8 weeks. Glycaemia over time (left) and the corresponding area under the curve (right) (n = 8–10).
Data are presented as mean ± SEM. Statistical analysis by 2-way ANOVA. Means depicting a different letter indicate significant differences between groups (p < 0.05). *p < 0.05 determined by Student’s t test between the −Ins versus the +Ins group for each genotype and diet.
Despite reduced fasting glycaemia (Figure 3A), Cpt2Sk−/− mice on the high-fat diet have normal fasting insulin plasma and resist hyperinsulinemia induced by the high-fat diet (Figure 4D). These features are highly indicative of retained insulin sensitivity and supported by Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) index values (Figure S3F). In agreement, during a glucose tolerance test, Cpt2Sk−/− male mice responded similarly to controls when fed a low-fat diet while resisting impairments in glucose tolerance induced by the high-fat diet (Figure 4E). In response to a systemic insulin challenge, low-fat-fed Cpt2Sk−/− mice, compared with control mice, had greater glycemic reductions and resisted high-fat diet-induced insulin resistance (Figure 4F). Altogether, these data suggest that in Cpt2Sk−/− mice, glucose tolerance is preserved because of retained insulin sensitivity despite LCAC accumulation.
Loss of Muscle CPT2 Does Not Increase Muscle Complex Lipids
A metabolic consequence of limiting fatty acid catabolic flux is the accumulation of anabolic lipid metabolites, such as phospholipids, TAG, and ceramides (Kim et al., 2014; Vandanmagsar et al., 2016). Muscle lipidomic profiling revealed that despite the inability to oxidize long-chain fatty acids, Cpt2Sk−/− mice do no accumulate excess of TAG or ceramides and resist increases in muscle TAG and ceramides induced by the high-fat diet relative to controls (Figures 5A and 5B). To determine whether lack of TAG accumulation was a result of increased lipolytic activity, HSL phosphorylation at two PKA target sites (S563 and S660) was assessed in muscle but was not changed by genotype on either the low- or the high-fat diet (Figure 5C). We next questioned whether the AC species that most significantly accumulate (AC16:0, AC16:1, AC18:0, AC18:1, and AC18:2) (Figure 5D) could flux back to acyl-CoAs for repurposing into complex lipid synthesis. Therefore, the acyl-chain composition of complex lipids was profiled, and within the TAG pool, no change in the composition of acyl chains was observed between genotypes; rather, increase in all species was evident in high-fat-fed control mice (Figure 5E). Total phospholipid content was relatively higher in Cpt2Sk−/− mice compared with controls on the low-fat diet; however, after high-fat diet feeding, total phospholipids decreased by ~60% (Figure 5F), as reflected by significant changes in all classes of glycerophospholipids and sphingomyelin (Figure 5G). Membrane phospholipid acyl-chainacyl-chain composition affects membrane dynamics (Harayama and Riezman, 2018). Analysis of the phospholipid acyl-chain composition revealed several genotype-driven alterations. Specifically, although most phosphatidylcholines (PCs) containing 1 unsaturated acyl chain were not changed in Cpt2Sk−/− muscle relative to controls, PCs containing 2 and 3 unsaturated bonds were ~4-fold more abundant (Figures 5H and S4A). Conversely, in Cpt2Sk−/− muscle compared with controls, PCs containing 0, 4, 5, 6, and >7 unsaturated bonds were between 15% and 70% less abundant (Figures 5H and S4A). This shift from highly unsaturated acyl chains in membrane lipids was observed across all phospholipids, including PC, phosphatidylethanolamine, phosphatidylglycerol, phosphatidylinositol, and phosphatidylserine, as well as sphingomyelin (Figures S4B–S4F). The reported changes in PC acyl-chain composition were augmented by high-fat diet, especially the reductions in highly unsaturated phospholipids of 5, 6, and >7 unsaturated bonds that decreased in Cpt2Sk−/− muscle by ~90% (Figures 5H and S4A). Altogether, these data demonstrate that the loss of CPT2 in muscle reduces highly unsaturated fatty acids in membrane phospholipids, potentially affecting membrane dynamics. In addition, these data suggest that fatty acid oxidative metabolic flux proceeds to acylcarnitine formation by CPT1b but does not revert back to acyl-CoA for subsequent complex lipid synthesis.
Figure 5. Complex Lipid Profile in Cpt2Sk−/− TA Muscle.
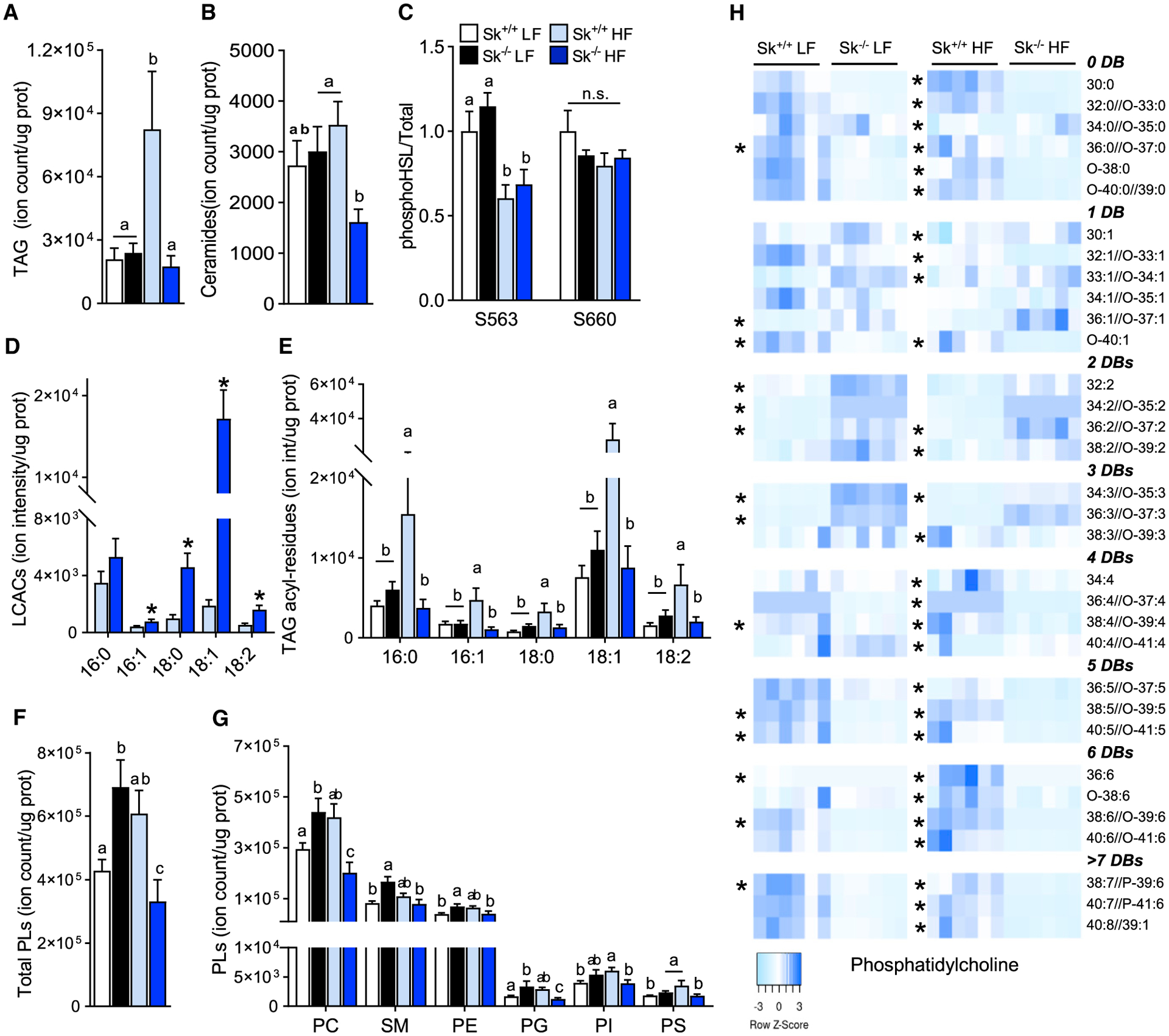
(A) Ion intensity per microgram of protein for TAGs as measured by direct injection mass spectrometry (DIMS) on Cpt2Sk+/+ and Cpt2Sk−/− female mice fed a LF or HF diet for 16 weeks (n = 6).
(B) Ion intensity per microgram of protein for ceramides of female mice fed a LF or HF diet (n = 6).
(C) Phosphorylation status of HSL at serine residues S563 and S660 in TA muscle of female mice fed a LF or HF diet (n = 6).
(D) Ion intensity per microgram of protein for LCACs of female mice on a HF diet (n = 6).
(E) Ion intensity per microgram of protein for fatty acyl residues used as product ions for the DIMS scans used for TAGs screening of female mice fed a LF or HF diet (n = 6).
(F) Ion intensity per microgram of protein for total phospholipids of female mice fed a LF or HF diet (n = 6).
(G) Ion intensity per microgram of protein for phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylserine (PS), and sphingomyelin (SM) of female mice fed a LF or HF diet (n = 6).
(H) Ion intensity per microgram of protein for PC species arranged by unsaturation degree as number of double bonds (DBs) of female mice fed a LF or HF diet (n = 6).
Data are presented as mean ± SEM. *p < 0.05 determined by Student’s t test. Statistical analysis by 2-way ANOVA. Means depicting a different letter indicate significant differences between groups (p < 0.05). See also Figures S2 and S3.
Carnitine Supplementation Lowers LCACs Independent of Fatty Acid Oxidative Metabolism
Increasing free carnitine via exogenous supplementation is well known to improve glucose homeostasis in animal models. It is predicted that the mechanism through which this protection occurs is mediated partly by lowering ACs (Morand et al., 2014; Noland et al., 2009; Power et al., 2007; Ringseis et al., 2012). Here, we questioned the requirement of muscle fatty acid oxidative flux in the ability of free carnitine to improve glucose homeostasis. Therefore, control and Cpt2Sk−/− mice on the low- and high-fat diets received L-carn supplementation in the drinking water for 16 weeks. L-carn did not affect weight gain or adiposity relative to the non-carnitine-treated mice on the low- or high-fat diet; however, L-carn did prevent liver TAG accumulation induced by the high-fat diet (Figures 6A–6C). In Cpt2Sk−/− muscle, L-carn modestly rescued free carnitine and acetyl-carnitine levels in the high-fat-diet condition (Figure 6D). Short- and medium-chain ACs were increased by L-carn in control low-fat-fed mice and Cpt2Sk−/− high-fat-fed mice (Figures 6E and 6F). Despite lower acetylcarnitine in Cpt2Sk−/− muscle, acetyl sequestration on lysine residues (Davies et al., 2016) was not different from that of control mice (Figure S2D). High-fat diet feeding, a known physiological inducer of LCAC accumulation, increased control mice LCACs by ~3-fold in muscle; surprisingly, however, the high-fat diet did not increase but rather significantly decreased Cpt2Sk−/− muscle LCACs by 40% relative to low-fat diet conditions (Figure 6G). Importantly, L-carn was highly effective at lowering LCACs in both control and Cpt2Sk−/− muscle by 50%–86% (Figure 6G). L-carn normalized total phospholipids across all diets and genotypes, driven mainly by significant reductions in total PC, sphingomyelin, and phosphatidylethanolamine in low-fat-fed Cpt2Sk−/− mice and high-fat-fed control mice (Figures 5F, 6G, S4G, and S4H). Similar to skeletal muscle without L-carn (Figure 5H), Cpt2Sk−/− mice receiving L-carn for 16 weeks retained elevations in 2 and 3 unsaturated phospholipids and reductions in highly unsaturated phospholipids, regardless of diet (Figure 6I). Thus, L-carn supplementation affected muscle acyl-carnitine levels, but not membrane acyl-chain composition, in a CPT2-independent manner.
Figure 6. Carnitine Supplementation Lowers LCACs Independent of Fatty Acid Oxidative Metabolism.
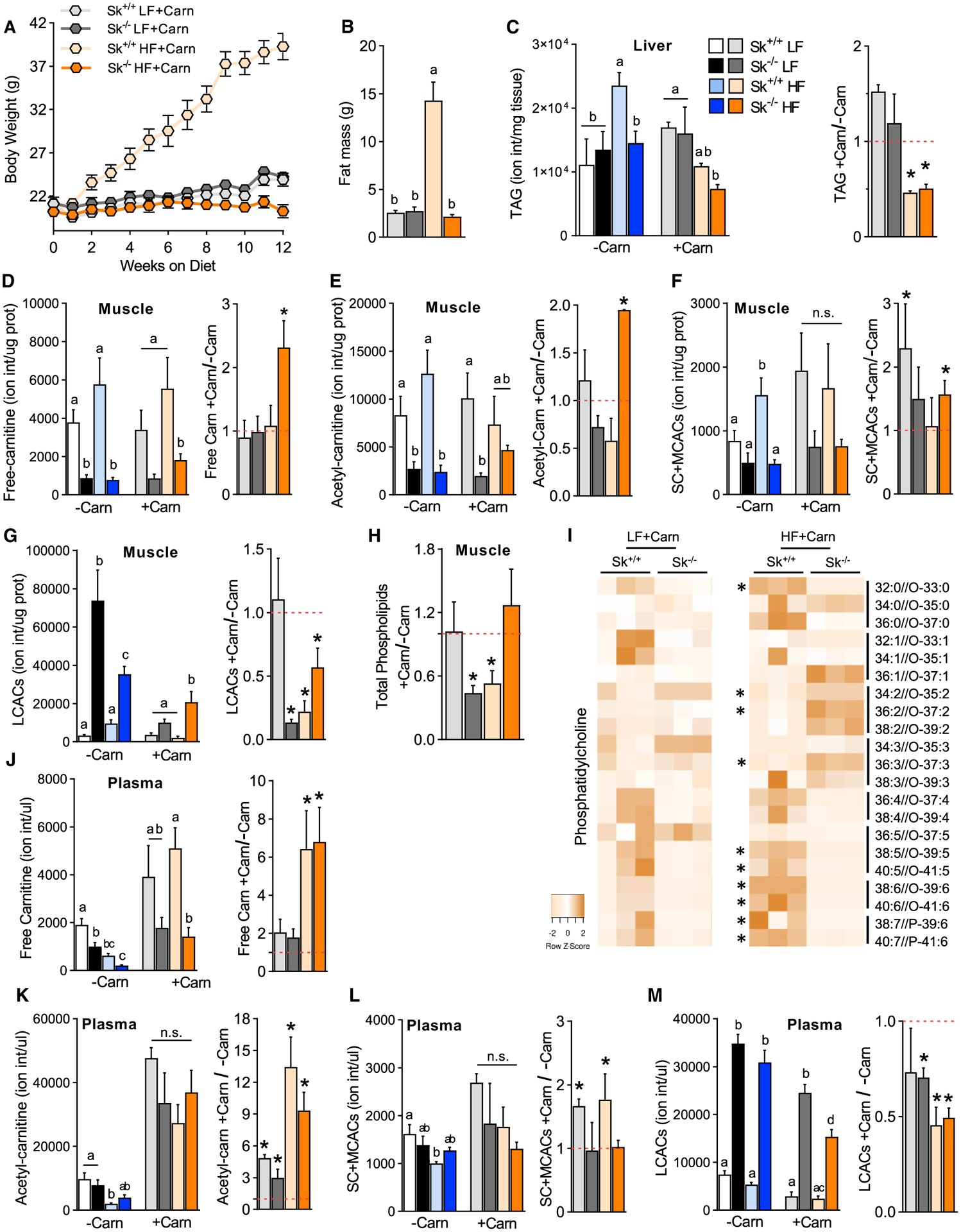
(A) Body weight of Cpt2Sk+/+ and Cpt2Sk−/− female mice fed a LF or HF diet supplemented with (+Carn) or without (−Carn) L-carnitine for 16 weeks (n = 6–9).
(B) Whole-body fat mass of female mice fed a LF or HF diet with L-carnitine supplementation (n = 6–9).
(C) Ion intensity per milligram of liver for TAGs of female mice fed a LF or HF diet with and without L-carnitine (n = 6 for −Carn and n = 3–4 for +Carn).
(D–G) Ion intensity per microgram of protein for free carnitine, acetyl-carnitine, short- and medium-chain ACs, and LCACs in skeletal muscle of female mice fed a LF or HF diet with and without L-carnitine (n = 6 for −Carn and n = 3–4 for +Carn).
(H) Fold change (+Carn/−Carn) for the sum of total phospholipids in TA muscle after L-carnitine supplementation of female mice (n = 6 for −Carn and n = 3–4 for +Carn).
(I) Heatmap for normalized abundance of PC species arranged by unsaturation degree of female mice fed a LF or HF diet with L-carnitine (n = 3).
(J–M) Ion intensity per microliter of plasma for free carnitine, acetyl-carnitine, short- and medium-chain ACs, and LCACs of female mice fed a LF or HF diet with and without L-carnitine (n = 6 for −Carn and n = 3–4 for +Carn).
Data are presented as mean ± SEM. *p < 0.05 determined by Student’s t test. Statistical analysis by 2-way ANOVA. Means depicting a different letter indicate significant differences between groups (p < 0.05). See also Figures S4 and S5.
Next, we assessed how L-carn affected circulating ACs in the context of CPT2 muscle deficiency. In the plasma of Cpt2Sk−/− mice, endogenous free carnitine was reduced ~48% with the low-fat diet and ~67% with the high-fat diet, suggesting a further systemic free carnitine deficiency in Cpt2Sk−/− by high-fat feeding (Figure 6J). L-carn rescued the plasma free carnitine deficit in Cpt2Sk−/− mice on both diets but had greater effects in high-fat-fed mice, increasing it by ~8-fold (Figure 6J). Cpt2Sk−/− mice resisted the reductions in plasma acetyl-, short-, and medium-chain ACs induced by the high-fat diet that were observed in control mice (Figures 6K and 6L). Upon supplementation with L-carn, plasma acetylcarnitine increased in all groups, whereas short- and medium-chain ACs increased in control mice, but not in Cpt2Sk−/− mice (Figures 6K and 6L). Plasma LCACs were 5- to 6-fold greater in Cpt2Sk−/− mice, independent of diet, and L-carn significantly reduced plasma LCACs by ~25%–50%, regardless of genotype and diet (Figure 6M). Thus, plasma acetyl-, short-, and medium-chain ACs did not mirror but LCACs did mirror the genotype-driven effects observed in muscle. These data show that L-carn supplementation rescued plasma free carnitine deficiency and effectively lowered LCACs with a greater effect in high-fat fed-mice and that these outcomes occurred independent of muscle fatty acid oxidation.
Although excess circulating ACs in Cpt2Sk−/− mice are not excreted in urine (Figure 2J) or in feces (Figure 2I), the metabolic fate of circulating ACs remains unclear. The hepatic lipid profile was assessed to determine whether shifting concentrations of plasma ACs and free carnitine affected the liver. Surprisingly, neither high-fat-diet feeding nor loss of CPT2 in skeletal muscle affected hepatic levels of free-carnitine, short- or medium- ACs, or LCACs (Figures S5A–S5D). L-carn supplementation increased hepatic free carnitine by ~2- to 3-fold in control and Cpt2Sk−/− mice fed a low-fat diet, with no effect on high-fat-fed mice (Figures S5A–S5D). In control mice, hepatic phospholipids and ceramides were not altered between low- and high-fat diet conditions; however, in Cpt2Sk−/− mice, high-fat diet feeding increased total hepatic phospholipids, each phospholipid class, and ceramides by ~40% relative to low-fat-fed mice, independent of L-carn supplementation (Figures S5F and S5G). These data suggest that the liver lipid homeostasis is refractory to systemic effects caused by loss of muscle fatty acid oxidation, dysregulated plasma ACs, high-fat diet feeding, and carnitine supplementation. Altogether, these data demonstrate that carnitine supplementation was highly effective at lowering LCACs in plasma and muscle in a manner that does not depend on shuttling fatty acids into muscle phospholipids or toward oxidative flux, suggesting that carnitine supplementation may lower ACs in muscle by facilitating their flux out of cells, potentially for catabolic metabolism in peripheral tissues.
Insulin Sensitivity Is Improved by L-Carn Supplementation Independent of Fatty Acid Oxidative Flux
L-carn supplementation is well established to improve fatty acid oxidative flux in carnitine-deficient patients and in healthy individuals and to improve insulin sensitivity (Müller et al., 2002). The question remains as to whether the beneficial effects of carnitine supplementation on glucose homeostasis depend on fatty acid oxidative flux in muscle. To address this question, we first assessed circulating metabolites important to metabolic disease outcomes. L-carn slightly reduced plasma glucose ~10%–15% across genotypes and diets and prevented hypertriglyceridemia induced by the high-fat diet in control mice without affecting adiposity (Figures 3D, 7A, and 7B). Although carnitine supplementation did not affect circulating free fatty acids, the major regulatory mechanism driving ketogenesis, it did increase circulating ketones in control mice on the low-fat diet by ~3.5-fold, an effect attributable to L-carn-induced increases in genes regulating hepatic fatty acid oxidation, ketogenesis, lactate catabolism, and gluconeogenesis (Figures 7C–7E). In control mice on the low-fat diet, L-carn supplementation did not affect response to a glucose tolerance test but did improve response to insulin tolerance test and insulin-induced Akt phosphorylation under high-fat diet (Figures 7F and 7G). Remarkably, in Cpt2Sk−/− mice fed either the low- or the high-fat diet, L-carn had a greater impact compared with control mice, improving both glucose tolerance and insulin sensitivity even further (Figures 7F and 7G). Carnitine supplementation elicited greater effects during the insulin tolerance test, rather than the glucose tolerance test, suggesting that most affects insulin responsiveness. In muscle, L-carn significantly increased insulin-stimulated Akt phosphorylation compared with non-carnitine in both high-fat-fed groups, by ~1.8-fold in controls and ~3.2-fold in Cpt2Sk−/− mice (Figure 7H). Thus, L-carn supplementation prevented glucose intolerance and insulin resistance induced by the high-fat diet independent of effects on adiposity and enhanced insulin-dependent Akt phosphorylation in muscle. L-carn also enhanced ketogenic and gluconeogenic gene expression profiles in the liver in both control and Cpt2Sk−/− mice, an effect elicited concomitant to reductions in muscle and plasma LCACs but independent of muscle mitochondrial long-chain fatty acid oxidative metabolism.
Figure 7. Carnitine Supplementation Improves Systemic Metabolism and Insulin Sensitivity in Cpt2Sk−/− Mice.
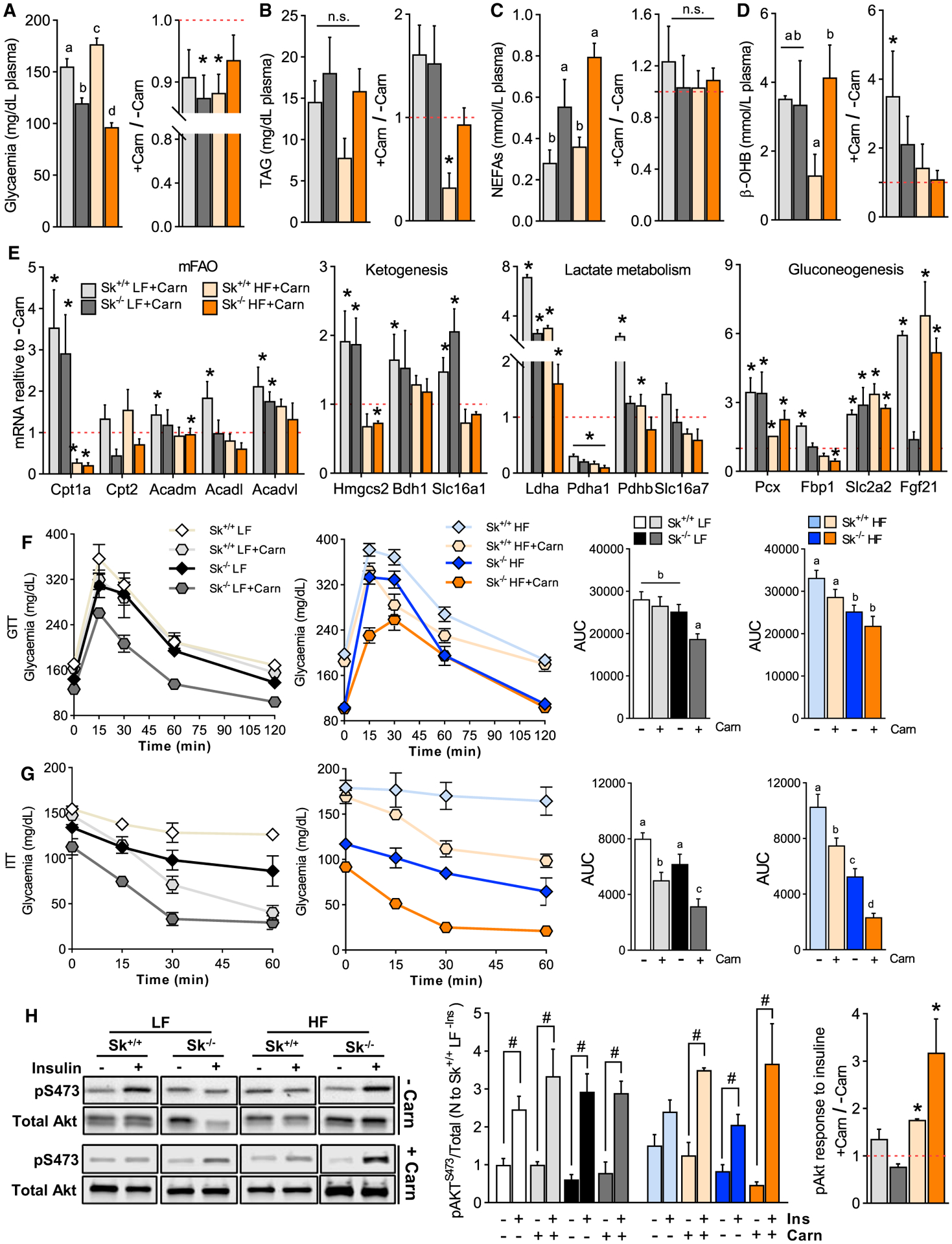
(A) Plasma glucose of Cpt2Sk+/+ and Cpt2Sk−/− female mice fed a LF or HF diet supplemented with L-carnitine for 16 weeks (n = 12).
(B) Plasma triacylglycerol of female mice fed a LF or HF diet with L-carnitine (n = 12).
(C) Plasma free fatty acids of female mice fed a LF or HF diet with L-carnitine (n = 12).
(D) Plasma β-OH of female mice fed a LF or HF diet with L-carnitine (n = 12).
(E) Liver mRNA relative abundance of female mice fed a LF or HF diet with L-carnitine. Data are normalized to the −Carn respective group (n = 3–4).
(F) Glucose tolerance test of female mice fed a LF or HF diet with and without L-carnitine (n = 6 for −Carn and n = 3–4 for +Carn).
(G) Insulin tolerance test of female mice fed a LF or HF diet with and without L-carnitine (n = 6 for −Carn and n = 3–4 for +Carn).
(H) Phosphorylation status of Akt protein in TA muscle of female mice fed a LF or HF diet with and without L-carnitine (n = 6 for −Carn and n = 3–4 for +Carn).
Data are presented as mean ± SEM. #p < 0.05 between the −Ins versus the +Ins group for each genotype and diet determined by Student’s t test; *p < 0.05 between +Carn versus −Carn for each genotype and diet determined by Student’s t test. Statistical analysis by 2-way ANOVA. Means depicting a different letter indicate significant differences between groups (p < 0.05).
DISCUSSION
Although the consumption of high-fat diets and the obesity epidemic have revealed that disruptions in fat metabolism are mechanistic drivers of metabolic disease, it is unclear which specific metabolites and metabolic processes are the major etiological contributors. Among the proposed mechanistic culprits are LCACs, which accumulate as intermediates of incomplete mitochondrial fatty acid oxidation during metabolic disease. Interest in studying non-canonical, lipotoxic roles for ACs has largely arisen from an increasing number of reports that observe differences in LCACs during numerous stresses, including fasting, ketogenic diet, exercise, overnutrition, heart disease, and insulin resistance (Adams et al., 2009; Bouchouirab et al., 2018; Mai et al., 2013; McCoin et al., 2015a; Sampey et al., 2012; Schooneman et al., 2013, 2014; Soeters et al., 2009; Su et al., 2005; Xu et al., 2016; Zhang et al., 2017). LCACs are accused of interfering with insulin signaling (Adams et al., 2009; Aguer et al., 2015; Bouchouirab et al., 2018; Keung et al., 2013; Kim et al., 2014; Koves et al., 2008; Li et al., 2015; Liepinsh et al., 2016, 2017; Mai et al., 2013; McCoin et al., 2015b; Power et al., 2007; Ringseis et al., 2012; Samimi et al., 2016; Vavrova et al., 2016; Warfel et al., 2017), and they accumulate at particularly high levels in the skeletal muscle, compared with other tissues (Pradas et al., 2018). However, the lack of mouse models with consistent and specific increases in endogenously generated muscle LCACs has hampered the assessment of their biological effects. Here, we developed a model of LCAC accumulation in skeletal muscle. The Cpt2Sk−/− muscle and plasma have the capacity to accumulate up to 22-fold more total LCACs, with species such as AC18:1 up to 56 times higher, than in control mice. The high degree of LCAC accumulation in the muscle and heart (Pereyra et al., 2017), but less so in the adipose or liver of CPT2-deficient mouse models (Lee et al., 2015), suggests that striated muscle provides a stable environment for LCAC accumulation, the rate of LCAC production is greater in these tissues, LCACs are metabolically stable in muscle, or the proteins responsible for export of cellular LCACs regulate the degree of accumulation. LCACs cannot freely pass through membrane bilayers, unlike free fatty acids, and thus require protein-mediated transport. Although, the accumulation of LCACs in the circulation reflects protein-mediated export from muscle, the regulators of their export out of muscle and subsequent import into peripheral tissues and the downstream metabolic handling remains unclear.
We have generated a muscle-specific model of CPT2 deficiency to block mitochondrial long-chain fatty acid oxidation; however, CPT1, not CPT2, is the frequent target in fatty acid oxidation research, because it is the regulated step of the pathway. Genetically deficient Cpt1b mice, manipulations of Cpt1’s allosteric inhibitor malonyl-CoA, and chemical inhibitors such as etomoxir have been used to study the effects of CPT1 inhibition of mitochondrial fatty acid oxidation in muscle (Timmers et al., 2012; Vandanmagsar et al., 2016; Warfel et al., 2016, 2017). Although targeting CPT1b and CPT2 blocks the same pathway, the biochemical distinction between inhibiting fatty acid oxidation either before (at CPT1) or after (at CPT2) the formation of ACs results in numerous distinct metabolic and molecular signatures. Biochemically, the inhibition of CPT1 results in accumulation of its substrate acyl-CoAs, which are toxic and readily ciphered into complex lipids, resulting in accumulation of TAG in the CPT1b-deficient mice (Warfel et al., 2017; Wicks et al., 2015). Conversely, blocking CPT2 results in accumulation of its substrate LCACs, which unlike acyl-CoAs, accumulate extensively in muscle and do not trigger increases in complex lipids (Figure 5). Another biochemical distinction is the lack of CPT2 isoenzymes, whereas in CPT1b-deficient muscle, CPT1a is shown to be upregulated to partially compensate for the loss of CPT1b (Wicks et al., 2015). Thus, the loss of CPT2, compared with CPT1b, stands to more severely block mitochondrial long-chain fatty acid oxidation and result in the accumulation of LCACs, but not complex lipids. Phenotypically, both models demonstrate resistance to weight gain on high-fat diets and improved glucose tolerance. However, there are several phenotypical distinctions between the models. For example, CPT1b deficiency, but not CPT2 deficiency, induces an increase in FGF21 in muscle and serum and browning of white adipose. In the CPT2 muscle-deficient mice, but not in CPT1b-deficient mice, insulin tolerance is improved and LCACs accumulate. It is unknown whether muscle CPT1b deficiency similarly affects fecal lipid loss, modulates changes in membrane lipids, or is responsive to L-carn supplementation. In summary, targeting different steps in the muscle mitochondrial long-chain fatty acid oxidation pathway (Cpt1b versus Cpt2) results in overlapping (weight resistance) and distinct (lipid accumulation and insulin sensitivity) outcomes.
Resistance of Cpt2Sk−/− mice to obesity under the high-fat diet agrees with several other muscle mitochondrial metabolically dysfunctional mouse models (Ghosh et al., 2019; Kim et al., 2014; Morrow et al., 2017; Vandanmagsar et al., 2016; Zhang et al., 2010; Zhao et al., 2019). The mechanistic explanation for obesity resistance in these models has been attributed to FGF21-mediated modifications of systemic metabolism and the induction of browning of white adipose tissue. However, the Cpt2Sk−/− mice do not exhibit browning or the induction of Fgf21 while on the high-fat diet. The lack of Fgf21 induction is particularly surprising, because it is often elevated in response to a myriad of metabolic stresses (Kliewer and Mangelsdorf, 2019), including CPT2 deficiency in the liver and adipose tissue (Lee et al., 2015, 2016), but is in agreement with reports of normal FGF21-serum levels in patients with CPT2 deficiency (Motlagh Scholle et al., 2019). Moreover, the increase in hepatic Fgf21 gene expression in response to L-carn supplementation yielded no change in adiposity resistance but stands to contribute to improvements in glucose tolerance. The higher EE, higher fecal lipid loss, and mitohormetic induction of GDF15 are likely the major contributors to Cpt2Sk−/− obesity resistance.
Contrary to reports that AC accumulation and/or exogenous AC exposure to cells in vitro or muscle in vivo results in impaired insulin-induced signaling (Aguer et al., 2015; Blackburn et al., 2020; Liepinsh et al., 2016, 2017), we find that high LCAC accumulation in plasma and muscle does not impair basal or insulin-mediated Akt signaling. In agreement, whole-body glucose homeostasis was improved in Cpt2Sk−/− mice. Although glucose homeostasis in a CPT2-deficient mouse is inherently intertwined with the lack of fatty acid oxidative metabolism as a major source of energy for the muscle, we used carnitine supplementation to target AC accumulation in our model and assess insulin-responsive outcomes. Surprisingly, L-carn supplementation in the drinking water for 16 weeks not only managed to lower ACs in the absence of fatty acid oxidative flux but also improved insulin responsiveness in Cpt2Sk−/− mice. Thus, we have discovered that L-carn supplementation lowers ACs in a manner that is independent of muscle mitochondrial fatty acid oxidation. The question arises as to how carnitine lowers ACs in the absence of fatty acid oxidation. It is possible that carnitine supplementation inhibits fatty acid oxidation, thereby preventing the formation of new ACs. However, this concept is in contradiction to reports that carnitine supplementation increases fatty acid oxidative flux (Center et al., 2012; Müller et al., 2002). Conversely, the carnitine-acylcarnitine translocase (CACT) is a bidirectional transporter moving ACs into and free carnitine out of the mitochondria, and free carnitine supplementation could drive backward flux because of carnitine accumulation in the cytosolic compartment or gradient-based limitations could prevent flux of AC transport into the mitochondria. Ultimately, this type of compartmentalization could allow cytosolic accumulation of ACs available for shuttling out of the cell. After exit from the cell or tissue, our data suggest that some LCACs are likely taken up by the liver to fuel ketogenesis in Cpt2Sk−/− mice. In addition, the same acyl chains of the accumulated ACs were increased in phospholipids of the liver, particularly on the high-fat diet, suggesting some of these acyl chains are fluxing into liver phospholipids. Although it has been speculated that circulating ACs are partially secreted in the urine, a factor that could contribute to obesity resistance in Cpt2Sk−/− mice, we did not observe significant increases in urinary ACs with or without high-fat-diet exposure. Another potential destination for the ACs is that they are fluxing into muscle phospholipids; however, muscle phospholipids were not increased in Cpt2Sk−/− mice, yet the acyl-chain composition was highly irregular and deficient in highly unsaturated fatty acids. It is possible that the observed reduction in muscle phospholipid unsaturation reflects greater flux of acyl chains from LCACs into phospholipids. Saturated fatty acids are the predominant accumulating LCACs, yet membrane saturated acyl chains are not increased in the Cpt2Sk−/− mice, casting doubt on this theory. An additional consideration is the unknown spatial and biological format of LCAC accumulation in muscle. The amphipathic nature of ACs could allow them to reside within membranes, and if so, they may interfere with membrane properties. The remarkable reduction in highly unsaturated fatty acids in the muscle membranes of Cpt2Sk−/− mice could reflect cell biology-mediated modifications that restore critical properties of acylcarnitine-enriched membranes. Lastly, the reduction in highly unsaturated acyl-residues in membrane lipids could reflect increased flux of these fatty acids through alternative oxidation pathways, such as peroxisomal oxidation.
Altogether, our work solves a fundamental and long-standing mystery in metabolic control of insulin sensitivity and the role of ACs therein. The importance of muscle fatty acid metabolism has remained a central, but contested, player in metabolic disease and insulin resistance. These data put forth the concept that ACs do not result in insulin resistance; rather, it seems that mice unable to flux fatty acids through mitochondrial β-oxidation are refractory to diet-induced insulin resistance, suggesting a role for downstream fatty acid catabolism, beyond AC formation, in the etiology of insulin resistance. The disconnect between AC accumulation and anabolic lipid accumulation suggests that LCAC accumulates are non-toxic in the context of insulin sensitivity. In addition, L-carn supplementation elicited beneficial effects on insulin sensitivity independent of muscle fatty acid oxidation; thus, although carnitine supplementation remains a promising therapy for insulin-resistant individuals, its use in individuals with fatty acid oxidation disorders should be managed with care given to glycemic monitoring.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and fulfilled by the Lead Contact, Jessica M. Ellis (ellisje18@ecu.edu).
Materials Availability
This study did not generate unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
CPT2 conditionally, Cre-dependent, deficient mice were generated as described (Lee et al., 2015). CPT2-floxed mice were then crossed with the skeletal muscle-specific Cre Recombinase expressing mice driven by the human alpha-skeletal actin promoter (The Jackson Laboratory; Stock No: 006149). Littermates lacking the Cre gene (Cpt2Sk+/+) were used as controls. All mice were of C57BL/6J genetic background. Both, female and male mice were used ranging from 8 weeks of age to 26 weeks of age. Details for each experiment are provided in corresponding figure legend. All procedures were performed in accordance with the Animal Care and Use Committee, Purdue Assurance (Assurance A3231–01) and East Carolina University (Assurance A3469–01).
METHOD DETAILS
Dietary intervention and indirect calorimetry
Mice were given free access to water and standard chow (PicoLab 5053, Lab Diets, Richmond, IN), in pathogen-free housing under 12-hour light-dark cycles. Indirect calorimetry was determined in either Oxymax chambers (Columbus Instruments) in Figure 3 (14 week) or by TSE Metabolic Cages in Figures 1 and 3 (2 weeks) with or without free access to a running wheel attached to the indirect calorimetry recording system. Fat and lean mass was determined by EchoMRI-700™ (EchoMRI LLC, Houston, TX). For diet studies, mice were given ad libitum access to either high-fat diet (Bio-Serv 3282, 60% fat calories) or the matched low-fat diet (Bio-Serv 4031). For carnitine studies, powdered L-carnitine (L(−)-Carnitine, 99+%, ACROS Organics) was directly dissolved into the drinking water at 300 mg/kg/day.
Blood/plasma assays
Glucose and insulin tolerance tests were performed after 4-hour food deprivation and initiated with intraperitoneal injections of glucose (Sigma G7021; 2 g/kg) or insulin (Sigma I0516; 0.7 U/kg). Blood glucose was determined by glucometer (NovaMax, Billerica, MA). Blood was collected in 5%–10% 0.5M EDTA and plasma NEFAs (Wako, Richmond, VA), triacylglycerol (Sigma Aldrich, St. Louis, MO), and beta-hydroxybutyrate (Cayman Chemical, Ann Arbor, MI) were determined by colorimetric assay per manufacturer’s instruction. GDF15 levels in plasma were determined using the Mouse/Rat GDF-15 Quantikineâ ELISA kit (R&D Systems, Inc., Minneapolis, MN) per manufacturer’s instruction. Basal insulinemia was determined in plasma collected after 3–4 hours of fasting using the Mouse Ultrasensitive Insulin ELISA assay (ALPCO, Salem, NH) per manufacturer’s instruction.
Lipid analysis
AC quantitation in muscle and plasma was performed by LC-MS/MS analysis. Briefly, each samples were was spiked with a 25μL of a mix of deuterated acylcarnitine internal standards (Cat#NSK-B-1, Cambridge Isotope Laboratories, Inc, Andover, MA) as well as 100ng of Stearoyl-L-carnitine-d3 (Cat#26580, Cayman Chemicals, Ann Arbor, MI) and 100ng of Oleoyl-L-carnitine-d3 (Cat#26578, Cayman Chemicals, Ann Arbor, MI) before lipids acylcarnitines were extracted with 1.5mL acetonitrile (ACN). Supernatant was recovered, dried in a speedvac concentrator, reconstituted in 75% ACN and analyzed using an Agilent 6460 6470 QQQ coupled to a 1290 Infinity II UPLC system (Agilent Technologies, Palo Alto, CA)1200 Rapid Resolution LC. A Waters BEH HILIC column was used for separation and data was collected in multiple reaction monitoring mode. Mobile phase A was water with 0.1% formic acid and mobile phase B was acetonitrile with 0.1% formic acid. Initial conditions were 100:0 A:B, held for 3 minutes, followed by a linear gradient to 5:95 at 13.5 min. Column re-equilibration was performed by returning to 100:0 A:B at 14 minutes and held until 15 minutes. Column flow rate was 0.3 mL/min. Concentration of endogenous acylcarnitines was determined based on ratio to corresponding internal standard and further normalized to sample protein concentration measured in advance.
High-throughput lipid profiling of tissue, feces, plasma, and urine was performed by direct injection mass spectrometry (DIMS) as previously described (Pereyra et al., 2017). Briefly, lipids were extracted using Bligh and Dyer Method (Bligh and Dyer, 1959) and both, the lipid and the polar phases were dried separately, resuspended in ACN:MeOH:NH4Ac and directly injected via a micro-autosampler (G1377A) into a triple quadrupole mass spectrometer (QQQ6410 from Agilent Technologies, San Jose, CA) operated in the positive ion mode and equipped with ESI ion source Data was collected in multiple reaction monitoring mode (de Lima et al., 2018; Franco et al., 2018; Pereyra et al., 2017). Ion intensities acquired by an in-house script were further normalized to sample protein concentration for tissues or extraction volume for biological fluids.
Total TAGs and cholesterol in plasma and feces were measured by colorimetric enzymatic assays per manufacturer’s instructions (Stanbio, Boerne, TX) and NEFAs (Wako, Richmond, VA). For this, fecal lipids were extracted using a modified Folch method (Kraus et al., 2015; Mataki et al., 2007). Briefly, feces were collected from individually house mice over 24 hours and homogenized in normal saline (ratio 1:12, gr/mL) using a Potter-Elvehjem grinder and drill. Then, same volume of chloroform:methanol (2:1) was added and vigorously vortexed. Suspension was centrifuged at 1000 g for 10 min and the lower, organic phase was recovered and dried. Lipid extracts were resuspended in 1% Triton-X in water.
Adipose histology
Immediately after euthanasia the gonadal white adipose tissue was collected, fixed in 4% PFA in PBS overnight and kept in 70% ethanol in water until paraffin embedded. 10–12μM-thick sections were collected and stained with Hematoxylin-Eosin mixture. Images were acquired at 20X using a ZEISS Axio Observer microscope.
Gene expression
RNA was isolated using Trizol (Invitrogen, Waltham, MA) and RNA was converted to cDNA (Applied Biosystems High Capacity cDNA RT Kit) and used for SYBR Green (Bio-Rad, Hercules, CA) based real time PCR. Results were normalized to housekeeping gene and expressed as arbitrary units of 2−ΔCT. Primers were purchased from Integrated DNA Technologies (https://www.idtdna.com/pages). For sequence details see Key Resources Table and Table S1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HSP70 | Cell Signaling | Cat#4872S; RRID: AB_2279841 |
| Anti-Beta Actin | Cell Signaling | Cat#3700S; RRID: AB_2242334 |
| Anti-Alpha Tubulin | Sigma | Cat#T0198; RRID: AB_477556 |
| Anti-CPT2 | Millipore | Cat#ABS85; RRID: AB_11204869 |
| Mouse anti-pan Akt | Cell Signaling | Cat#2920S; RRID: AB_1147620 |
| Anti-phospho Akt Ser 473 | Cell Signaling | Cat#4060S; RRID: AB_2315049 |
| Anti-total HSL | Cell Signaling | Cat#4107S; RRID: AB_2296900 |
| Anti-phospho HSL Ser 563 | Cell Signaling | Cat#4139S; RRID: AB_2135495 |
| Anti-phospho HSL Ser 660 | Cell Signaling | Cat#4126S; RRID: AB_490997 |
| Anti-eIF2A | Cell Signaling | Cat#9722S; RRID: AB_2230924 |
| Anti-phospho eIF2A Ser 51 | Cell Signaling | Cat#3398S; RRID: AB_2096481 |
| Rabbit anti-acetylated lysine | Cell Signaling | Cat#9441S; RRID: AB_331805 |
| Anti-HMGCS2 | Cell Signaling | Cat#40364S; RRID: AB_2799175 |
| Goat anti-rabbit IgG (H+L) IRDye 800CW | LI-COR | Cat#926–32211; RRID: AB_621843 |
| Goat anti-rabbit IgG (H+L) IRDye 680RD | LI-COR | Cat#926–68071; RRID: AB_10956166 |
| Goat anti-mouse IgG (H+L) IRDye 800CW | LI-COR | Cat#926–32212; RRID: AB_621847 |
| Goat anti-mouse IgG (H+L) IRDye 680RD | LI-COR | Cat#926–68070; RRID: AB_10956588 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Insulin solution from bovine pancreas | Sigma | Cat#I0516 |
| Glucose | Sigma | Cat#G7021 |
| Labeled Carnitine Standards Set B | Cambridge Isotope Laboratories | Cat#NSK-B-1 |
| Stearoyl-L-carnitine-d3 | Cayman | Cat#26580 |
| Oleoyl-L-carnitine-d3 | Cayman | Cat#26578 |
| Palmitic Acid, [1-14C] | American Radiolabeled Chemicals | Cat#ARC0172A |
| Octanoic Acid, Sodium Salt, [1-14C] | American Radiolabeled Chemicals | Cat#ARC0149–50 |
| Pyruvic Acid, Sodium Salt, [2-14C] | Perkin Elmer | Cat# NEC256050UC |
| Critical Commercial Assays | ||
| HR Series NEFA-HR (2) | FUJIFILM Wako | Cat#999–35691, 999–34791, 999–34891, 999–35191, 276–76491 |
| Triglyceride and Free Glycerol Kits and Reagents | Sigma-Aldrich | Cat#F6428 and T2449 |
| β-Hydroxybutyrate (Ketone Body) Colorimetric Assay Kit | Cayman Chemical | Cat# 700190 |
| Mouse/Rat GDF-15 Quantikine® ELISA kit | R&D Systems | Cat#MGD150 |
| Mouse Ultrasensitive Insulin ELISA assay | ALPCO | Cat#80-INSMSU-E01 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Cpt2tm1Mwol/Cpt2tm1Mwol | PMID: 25578732 | MGI: 6283566; RRID:MGI:6283566 |
| Mouse: B6.Cg-Tg(ACTA1-cre)79Jme/J | The Jackson Laboratory | JAX: 006149; RRID:IMSR_JAX:006149 |
| Oligonucleotides | ||
| See also Table S1 for more primers | ||
| Cpt2 F: CAACTCGTATACCCAAACCCAGTC | Integrated DNA Technologies | https://www.idtdna.com/pages |
| Cpt2 R: GTTCCCATCTTGATCGAGGACATC | Integrated DNA Technologies | https://www.idtdna.com/pages |
| Fgf21 F: CTGCTGGGGGTCTACCAAG | Integrated DNA Technologies | https://www.idtdna.com/pages |
| Fgf21 R: CTGCGCCTACCACTGTTCC | Integrated DNA Technologies | https://www.idtdna.com/pages |
| Gdf15 F: GCTATGCTGCCCTCTGGCGG | Integrated DNA Technologies | https://www.idtdna.com/pages |
| Gdf15 R: TCGCCCACGCACATGCTCAG | Integrated DNA Technologies | https://www.idtdna.com/pages |
| Tnfa F: GACCCTCACACTCAGATCAT | Integrated DNA Technologies | https://www.idtdna.com/pages |
| Tnfa R: TTGAAGAGAACCTGGGAGTA | Integrated DNA Technologies | https://www.idtdna.com/pages |
| F4/80 F: TGACTCACCTTGTGGTCCTAA | Integrated DNA Technologies | https://www.idtdna.com/pages |
| F4/80 R: CTTCCCAGAATCCAGTCTTT CC | Integrated DNA Technologies | https://www.idtdna.com/pages |
| Software and Algorithms | ||
| Heatmapper | Wishart Research Group, University of Alberta, Edmonton, Canada | http://www.heatmapper.ca/ |
| Energy Expenditure Analysis | NIDDK Mouse Metabolic Phenotyping Centers | https://www.mmpc.org/shared/regression.aspx |
| Prism 8.0 | GraphPad Software | https://www.graphpad.com/ |
| Other | ||
| High Fat Diet (60% calories from fat; 5.49 kcal/gm) | Bio-Serv | Cat# F3282 |
| Low Fat Diet (3.93 kcal/gm) | Bio-Serv | Cat# F4031 |
Immunoblots
Tissues were harvested after 3–4 hours of food deprivation with or without insulin injected intraperitoneal (1 U/kg) 10 min prior to tissue harvest. All data obtained from female mice were fed either a low-fat (LF) or a high-fat (HF) diet for 16 weeks, unless otherwise specified. Total homogenates were collected in sucrose media (10mM Tris, 1mM EDTA, 250mM sucrose) with protease and phosphatase inhibitors. Lysates were collected in lysis buffer (50mM Tris-HCl, 150mM NaCl, 1mM EDTA, 1% Triton X-100) with protease and phosphatase inhibitors. Homogenates or lysates were equally loaded (15–30 μg) and electrophoresed on SDS polyacrylamide gels, transferred to nitrocellulose membrane, blocked with 5% milk-TBST for 1h, incubated with primary antibody (1:1,000–1:2,000) against: HSP70 Cell Signaling 4872S; Actin, Cell Signaling 3700S; alpha-tubulin, Sigma T0198; CPT2, Millipore ABS85; AKT and AKT(S473) Cell Signaling 2920S and 4060S; HSL, HSL(S563), and HSL(S660), Cell Signaling 4107S, 4139S, 4126S; eIF2A and eIF2A(S51), Cell Signaling 9722S and 3398S; acetylated lysine Cell Signaling 9441S; HMGCS2 Cell Signaling 40364S, washed, and incubated with secondary anti-mouse or anti-rabbit antibodies conjugated to IR dye 800CW or 680LT (LI-COR, Lincoln, NE). Proteins were visualized with Odyssey (LI-COR, Lincoln, NE) and quantified using Image Studio (LI-COR, Lincoln, NE).
Substrate oxidation assay
Freshly isolated muscle was minced and homogenized with a motor-driven Teflon pestle and glass mortar in ice-cold buffer oxidation buffer (100 mM KCl, 40 mM Tris·HCl, 10 mM Tris base, 5 mM MgCl2·6H2O, 1 mM EDTA, and 1 mM ATP, pH 7.4) at a 20-fold dilution (wt/vol) and the rate of [1-14C]palmitate, [1-14C]octanoate, or [2-14C]pyruvate, was measured, as previously described (Ellis et al., 2010; Pereyra et al., 2017). Briefly, carbon dioxide was trapped in a suspended well filled with sodium hydroxide-soaked filter paper. Acid soluble metabolites (ASM) were detected from unprecipitated incubation materials exposed to sulfuric acid.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data is presented as mean ± SEM, unless otherwise specified. Statistical analysis and figures were generated using GraphPad Prism version 8.0.0 for Windows (GraphPad Software, San Diego, California USA). Data was compared using either a paired (urinary acylcarnitines before-after high-fat diet feeding) or unpaired Student’s t test, or 2-way ANOVA followed by multiple comparison analysis. Significance level was set at p < 0.05. Statistical details for each dataset are described on figure legends. Heatmapper online tool was used to generate heatmaps (Wishart Research Group, University of Alberta, Edmonton, Canada). The EE ANCOVA analysis was done using the NIDDK Mouse Metabolic Phenotyping Centers (MMPC) Energy Expenditure Analysis page (http://www.mmpc.org/shared/regression.aspx).
Artwork
Graphical Abstract was created with BioRender.com
Supplementary Material
Highlights.
Long-chain acylcarnitine accumulation is not sufficient to drive insulin resistance
Loss of muscle CPT2 does not increase complex lipids but remodels membrane lipids
Loss of CPT2 in muscle prevents diet-induced obesity
L-carnitine supplementation improves insulin response independent of muscle CPT2
ACKNOWLEDGMENTS
We thank Michael J. Wolfgang for gifting the CPT2 floxed mice and Quin Waterbury for technical assistance. This work was supported by the Ralph and Grace Showalter Trust Young Investigator Program and U.S. Public Health Services grant R01 DK125812-01.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j. celrep.2020.108374.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, Hwang DH, Newman JW, and Garvey WT (2009). Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid β-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr 139, 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguer C, McCoin CS, Knotts TA, Thrush AB, Ono-Moore K, McPherson R, Dent R, Hwang DH, Adams SH, and Harper M-E (2015). Acylcarnitines: potential implications for skeletal muscle insulin resistance. FASEB J. 29, 336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn ML, Ono-Moore KD, Sobhi HF, and Adams SH (2020). Carnitine palmitoyltransferase 2 knockout potentiates palmitate-induced insulin resistance in C2C12 myotubes. Am. J. Physiol. Endocrinol. Metab 319, E265–E275. [DOI] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Bouchouirab F-Z, Fortin M, Noll C, Dubé J, and Carpentier AC (2018). Plasma Palmitoyl-Carnitine (AC16:0) is a Marker of Increased Postprandial Nonesterified Incomplete Fatty Acid Oxidation Rate in Adults with Type 2 Diabetes. Can. J. Diabetes 42, 382–388. [DOI] [PubMed] [Google Scholar]
- Bowman TA, O’Keeffe KR, D’Aquila T, Yan QW, Griffin JD, Killion EA, Salter DM, Mashek DG, Buhman KK, and Greenberg AS (2016). Acyl CoA synthetase 5 (ACSL5) ablation in mice increases energy expenditure and insulin sensitivity and delays fat absorption. Mol. Metab 5, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center SA, Warner KL, Randolph JF, Sunvold GD, and Vickers JR (2012). Influence of dietary supplementation with (L)-carnitine on metabolic rate, fatty acid oxidation, body condition, and weight loss in overweight cats. Am. J. Vet. Res 73, 1002–1015. [DOI] [PubMed] [Google Scholar]
- Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi H-S, Kang SG, Choi MJ, Lee SE, Jung S-B, et al. (2017). Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol 216, 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MN, Kjalarsdottir L, Thompson JW, Dubois LG, Stevens RD, Ilkayeva OR, Brosnan MJ, Rolph TP, Grimsrud PA, and Muoio DM (2016). The Acetyl Group Buffering Action of Carnitine Acetyltransferase Offsets Macronutrient-Induced Lysine Acetylation of Mitochondrial Proteins. Cell Rep. 14, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima CB, Ferreira CR, Milazzotto MP, Sobreira TJP, Vireque AA, and Cooks RG (2018). Comprehensive lipid profiling of early stage oocytes and embryos by MRM profiling. J. Mass Spectrom 53, 1247–1252. [DOI] [PubMed] [Google Scholar]
- Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, Watkins SM, Muoio DM, and Coleman RA (2010). Adipose acyl-CoA synthetase-1 directs fatty acids toward β-oxidation and is required for cold thermogenesis. Cell Metab. 12, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco J, Ferreira C, Paschoal Sobreira TJ, Sundberg JP, and HogenEsch H (2018). Profiling of epidermal lipids in a mouse model of dermatitis: Identification of potential biomarkers. PLoS ONE 13, e0196595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Wicks SE, Vandanmagsar B, Mendoza TM, Bayless DS, Salbaum JM, Dearth SP, Campagna SR, Mynatt RL, and Noland RC (2019). Extensive metabolic remodeling after limiting mitochondrial lipid burden is consistent with an improved metabolic health profile. J. Biol. Chem 294, 12313–12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harayama T, and Riezman H (2018). Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol 19, 281–296. [DOI] [PubMed] [Google Scholar]
- Kaiyala KJ, and Schwartz MW (2011). Toward a more complete (and less controversial) understanding of energy expenditure and its role in obesity pathogenesis. Diabetes 60, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keung W, Ussher JR, Jaswal JS, Raubenheimer M, Lam VHM, Wagg CS, and Lopaschuk GD (2013). Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes 62, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, He L, Johnson MS, Li Y, Zeng L, Ding Y, Long Q, Moore JF, Sharer JD, Nagy TR, et al. (2014). Carnitine Palmitoyltransferase 1b Deficiency Protects Mice from Diet-Induced Insulin Resistance. J. Diabetes Metab 5, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, and Mangelsdorf DJ (2019). A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 29, 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, et al. (2008). Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56. [DOI] [PubMed] [Google Scholar]
- Kraus D, Yang Q, and Kahn BB (2015). Lipid Extraction from Mouse Feces. Bio. Protoc 5, e1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ellis JM, and Wolfgang MJ (2015). Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell Rep. 10, 266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Choi J, Scafidi S, and Wolfgang MJ (2016). Hepatic Fatty Acid Oxidation Restrains Systemic Catabolism during Starvation. Cell Rep. 16, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LO, Grevengoed TJ, Paul DS, Ilkayeva O, Koves TR, Pascual F, Newgard CB, Muoio DM, and Coleman RA (2015). Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes 64, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepinsh E, Makrecka-Kuka M, Makarova E, Volska K, Svalbe B, Sevostjanovs E, Grinberga S, Kuka J, and Dambrova M (2016). Decreased acylcarnitine content improves insulin sensitivity in experimental mice models of insulin resistance. Pharmacol. Res 113 (Pt B), 788–795. [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Makrecka-Kuka M, Makarova E, Volska K, Vilks K, Sevostjanovs E, Antone U, Kuka J, Vilskersts R, Lola D, et al. (2017). Acute and long-term administration of palmitoylcarnitine induces muscle-specific insulin resistance in mice. BioFactors 43, 718–730. [DOI] [PubMed] [Google Scholar]
- Mai M, Tönjes A, Kovacs P, Stumvoll M, Fiedler GM, and Leichtle AB (2013). Serum levels of acylcarnitines are altered in prediabetic conditions. PLoS ONE 8, e82459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataki C, Magnier BC, Houten SM, Annicotte JS, Argmann C, Thomas C, Overmars H, Kulik W, Metzger D, Auwerx J, and Schoonjans K (2007). Compromised intestinal lipid absorption in mice with a liver-specific deficiency of liver receptor homolog 1. Mol. Cell. Biol 27, 8330–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoin CS, Knotts TA, and Adams SH (2015a). Acylcarnitines—old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol 11, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoin CS, Knotts TA, Ono-Moore KD, Oort PJ, and Adams SH (2015b). Long-chain acylcarnitines activate cell stress and myokine release in C2C12 myotubes: calcium-dependent and -independent effects. Am. J. Physiol. Endocrinol. Metab 308, E990–E1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morand R, Bouitbir J, Felser A, Hench J, Handschin C, Frank S, and Krähenbühl S (2014). Effect of carnitine, acetyl-, and propionylcarnitine supplementation on the body carnitine pool, skeletal muscle composition, and physical performance in mice. Eur. J. Nutr 53, 1313–1325. [DOI] [PubMed] [Google Scholar]
- Morrow RM, Picard M, Derbeneva O, Leipzig J, McManus MJ, Gouspillou G, Barbat-Artigas S, Dos Santos C, Hepple RT, Murdock DG, and Wallace DC (2017). Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc. Natl. Acad. Sci. USA 114, 2705–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh Scholle L, Lehmann D, Joshi PR, and Zierz S (2019). Normal FGF-21-Serum Levels in Patients with Carnitine Palmitoyltransferase II (CPT II) Deficiency. Int. J. Mol. Sci 20, 1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller DM, Seim H, Kiess W, Löster H, and Richter T (2002). Effects of oral L-carnitine supplementation on in vivo long-chain fatty acid oxidation in healthy adults. Metabolism 51, 1389–1391. [DOI] [PubMed] [Google Scholar]
- Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, Stevens RD, Hegardt FG, and Muoio DM (2009). Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J. Biol. Chem 284, 22840–22852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereyra AS, Hasek LY, Harris KL, Berman AG, Damen FW, Goergen CJ, and Ellis JM (2017). Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J. Biol. Chem 292, 18443–18456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power RA, Hulver MW, Zhang JY, Dubois J, Marchand RM, Ilkayeva O, Muoio DM, and Mynatt RL (2007). Carnitine revisited: potential use as adjunctive treatment in diabetes. Diabetologia 50, 824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradas I, Huynh K, Cabré R, Ayala V, Meikle PJ, Jové M, and Pamplona R (2018). Lipidomics Reveals a Tissue-Specific Fingerprint. Front. Physiol 9, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringseis R, Keller J, and Eder K (2012). Role of carnitine in the regulation of glucose homeostasis and insulin sensitivity: evidence from in vivo and in vitro studies with carnitine supplementation and carnitine deficiency. Eur. J. Nutr 51, 1–18. [DOI] [PubMed] [Google Scholar]
- Samimi M, Jamilian M, Ebrahimi FA, Rahimi M, Tajbakhsh B, and Asemi Z (2016). Oral carnitine supplementation reduces body weight and insulin resistance in women with polycystic ovary syndrome: a randomized, double-blind, placebo-controlled trial. Clin. Endocrinol. (Oxf.) 84, 851–857. [DOI] [PubMed] [Google Scholar]
- Sampey BP, Freemerman AJ, Zhang J, Kuan P-F, Galanko JA, O’Connell TM, Ilkayeva OR, Muehlbauer MJ, Stevens RD, Newgard CB, et al. (2012). Metabolomic profiling reveals mitochondrial-derived lipid biomarkers that drive obesity-associated inflammation. PLoS ONE 7, e38812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schooneman MG, Vaz FM, Houten SM, and Soeters MR (2013). Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes 62, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schooneman MG, Achterkamp N, Argmann CA, Soeters MR, and Houten SM (2014). Plasma acylcarnitines inadequately reflect tissue acylcarnitine metabolism. Biochim. Biophys. Acta 1841, 987–994. [DOI] [PubMed] [Google Scholar]
- Schutz Y, and Ravussin E (1980). Respiratory quotients lower than 0.70 in ketogenic diets. Am. J. Clin. Nutr 33, 1317–1319. [DOI] [PubMed] [Google Scholar]
- Soeters MR, Sauerwein HP, Duran M, Wanders RJ, Ackermans MT, Fliers E, Houten SM, and Serlie MJ (2009). Muscle acylcarnitines during short-term fasting in lean healthy men. Clin. Sci. (Lond.) 116, 585–592. [DOI] [PubMed] [Google Scholar]
- Su X, Han X, Mancuso DJ, Abendschein DR, and Gross RW (2005). Accumulation of long-chain acylcarnitine and 3-hydroxy acylcarnitine molecular species in diabetic myocardium: identification of alterations in mitochondrial fatty acid processing in diabetic myocardium by shotgun lipidomics. Biochemistry 44, 5234–5245. [DOI] [PubMed] [Google Scholar]
- Timmers S, Nabben M, Bosma M, van Bree B, Lenaers E, van Beurden D, Schaart G, Westerterp-Plantenga MS, Langhans W, Hesselink MKC, et al. (2012). Augmenting muscle diacylglycerol and triacylglycerol content by blocking fatty acid oxidation does not impede insulin sensitivity. Proc. Natl. Acad. Sci. USA 109, 11711–11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschöp MH, Speakman JR, Arch JR, Auwerx J, Brüning JC, Chan L, Eckel RH, Farese RV Jr., Galgani JE, Hambly C, et al. (2011). A guide to analysis of mouse energy metabolism. Nat. Methods 9, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Warfel JD, Wicks SE, Ghosh S, Salbaum JM, Burk D, Dubuisson OS, Mendoza TM, Zhang J, Noland RC, and Mynatt RL (2016). Impaired Mitochondrial Fat Oxidation Induces FGF21 in Muscle. Cell Rep. 15, 1686–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavrova E, Lenoir V, Alves-Guerra M-C, Denis RG, Castel J, Esnous C, Dyck JRB, Luquet S, Metzger D, Bouillaud F, and Prip-Buus C (2016). Muscle expression of a malonyl-CoA-insensitive carnitine palmitoyltransferase-1 protects mice against high-fat/high-sucrose diet-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab 311, E649–E660. [DOI] [PubMed] [Google Scholar]
- Vissing CR, Dunø M, Wibrand F, Christensen M, and Vissing J (2019). Hydroxylated Long-Chain Acylcarnitines are Biomarkers of Mitochondrial Myopathy. J. Clin. Endocrinol. Metab 104, 5968–5976. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Komen J, and Kemp S (2011). Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 278, 182–194. [DOI] [PubMed] [Google Scholar]
- Warfel JD, Bermudez EM, Mendoza TM, Ghosh S, Zhang J, Elks CM, Mynatt R, and Vandanmagsar B (2016). Mitochondrial fat oxidation is essential for lipid-induced inflammation in skeletal muscle in mice. Sci. Rep 6, 37941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfel JD, Vandanmagsar B, Wicks SE, Zhang J, Noland RC, and Mynatt RL (2017). A low fat diet ameliorates pathology but retains beneficial effects associated with CPT1b knockout in skeletal muscle. PLoS ONE 12, e0188850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicks SE, Vandanmagsar B, Haynie KR, Fuller SE, Warfel JD, Stephens JM, Wang M, Han X, Zhang J, Noland RC, and Mynatt RL (2015). Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc. Natl. Acad. Sci. USA 112, E3300–E3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Hansen JS, Zhao XJ, Chen S, Hoene M, Wang XL, Clemmesen JO, Secher NH, Häring HU, Pedersen BK, et al. (2016). Liver and Muscle Contribute Differently to the Plasma Acylcarnitine Pool During Fasting and Exercise in Humans. J. Clin. Endocrinol. Metab 101, 5044–5052. [DOI] [PubMed] [Google Scholar]
- Zhang D, Christianson J, Liu Z-X, Tian L, Choi CS, Neschen S, Dong J, Wood PA, and Shulman GI (2010). Resistance to high-fat diet-induced obesity and insulin resistance in mice with very long-chain acyl-CoA dehydrogenase deficiency. Cell Metab. 11, 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Light AR, Hoppel CL, Campbell C, Chandler CJ, Burnett DJ, Souza EC, Casazza GA, Hughen RW, Keim NL, et al. (2017). Acyl carnitines as markers of exercise-associated fuel partitioning, xenometabolism, and potential signals to muscle afferent neurons. Exp. Physiol 102, 48–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Pascual F, Bacudio L, Suchanek AL, Young PA, Li LO, Martin SA, Camporez J-P, Perry RJ, Shulman GI, et al. (2019). Defective fatty acid oxidation in mice with muscle-specific acyl-CoA synthetase 1 deficiency increases amino acid use and impairs muscle function. J. Biol. Chem 294, 8819–8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.