

Introduction
Acute respiratory distress syndrome (ARDS) is a pathophysiologic state of inflammatory response to lung injury, which encompasses a heterogeneous group of direct and indirect causes, but with similar terminal pathophysiologic characteristics: vascular endothelial and alveolar epithelial cell damage, production of inflammatory mediators, and accumulation of inflammatory cells, mainly neutrophils, in the lung (1, 2). The structural damage is translated clinically into a syndrome of acute respiratory failure that debuts with dyspnea, progressive arterial hypoxemia secondary to severely impaired gas exchange, pulmonary edema, intrapulmonary hemorrhage, and marked increase in ventilatory work (3–7).
Thomas L. Petty and colleagues first described ARDS in 1967 in a heterogeneous group of patients and demonstrated the beneficial effect of PEEP to treat of patients with acute respiratory failure (8). The definition, pathophysiology, and treatment of ARDS, however, has evolved constantly throughout. Therefore, a historical comparison of patients within the context of ARDS is difficult and may become imprecise (9). The current incidence of this disease is estimated as approximately 200,000 patients annually in the United States with mortality rates ranging from 25 to 60% (10, 11). Yet, the data are not homogenous for all cohorts studied; for example African American and Hispanic patients have a higher mortality in ARDS than Caucasians and the difference can not be solely explained based on socio-economic status and access to healthcare (12–14). Thus, beyond population statistics, there is a need for molecular and biochemical pathway tools to provide better individualized diagnostics and therapies in the care for ARDS.
ARDS is defined according to the Berlin Definition consensus as an acute inflammatory response present in patients within 1 week of an insult, with bilateral pulmonary infiltrates, non-cardiogenic pulmonary edema, and arterial hypoxemia, using the arterial oxygen to inhaled oxygen ratio as mild (PaO2/FiO2 = 200 to 300), moderate (PaO2/FiO2 = 100 to 200), and severe (PaO2/FiO2 = 200 to 300), with PEEP ≥ 5 cm H2O (15). The arterial hypoxemia is secondary to decreased blood gas exchange due to accumulation of edema and subsequent alveolar damage (16). In parallel, carbon dioxide elimination is also impaired, leading to higher respiratory rate, minute ventilation, and work of breathing.
Better survival rates due to improved supportive treatment with lung-protective ventilation, improved sepsis survival, and conservative fluid therapy are examples of physiologically based rational care based (9). However, we have no specific pharmacological therapy for ARDS approved to date (17, 18). We know that most patients who succumb to this syndrome do not die of hypoxemia or frank pulmonary failure, rather from multi-system organ failure that is likely mediated by the inflammatory mediators at play in this disease. Therefore, understanding these mediators and pathways is paramount to further development and hope for a pharmacologic treatment in the future.
In ARDS, an initial insult triggers massive liberation of inflammatory mediators, including alarmins, complement activation products, cytokines, chemokines, proteases, and oxidants (19–22). While sepsis and aspiration are frequent reasons for initiating ARDS, other etiologies are also observed (Table 1). Recent advances in the understanding of the molecular mechanisms responsible for the development of ARDS involving lung and systemic inflammation and tissue repair have opened a door to the development and improvement of new management strategies designed to increase survival. However, considerable work is needed to translate the basic research knowledge on lung injury to clinical practice.
Table 1.
Causes and predisposing factors for ARDS
| Factor | Association | |
|---|---|---|
| Individual | Age | Risk |
| Male gender | Risk | |
| Genetic | Genes | Risk |
| Polymorphisms | Risk and severity | |
| Anatomic | Malformations | Risk |
| Structural defects | Risk | |
| Comorbidities | Aspiration of gastric contents | Causative |
| Acute pancreatitis | Risk | |
| Chronic alcohol abuse | Risk and severity | |
| Coronary artery disease | Risk and severity | |
| Diabetes | Risk and severity | |
| Embolism (air, fat, amniotic fluid) | Causative | |
| History of smoking | Risk and severity | |
| Ischemia-reperfusion injury | Causative | |
| Multiple transfusions of blood products | Risk | |
| Pulmonary contusion | Causative | |
| Pulmonary infection | Causative | |
| Sepsis | Risk | |
| Severe trauma with shock | Risk | |
| Vasculitis | Causative | |
| Environmental | Burn (massive) | Risk |
| Inhalational injury (smoke, noxious gases) | Causative | |
| Medications (opioids, salicylates, amiodarone, tocolytics, chemotherapy) | Risk | |
| Near drowning | Causative | |
| Thoracic irradiation | Causative | |
To understand the molecular alterations observed during lung injury, one needs to know the normal molecular dynamic of the lung, which is a highly specialized organ with main functions in gas exchange, blood circulation regulation, and defense (23–26). Lungs are a primordial example of innate and adaptive immunity collaboration of the human body (Figure 1). In this complex organ, no single mechanism can fully account for all the molecular complexities observed. In this chapter, we focus on the molecular, structural, and physiological bases that contribute to ARDS. We describe the proteins, peptides, and other molecules that are and could be used as biomarkers for disease severity and therapy in ARDS. Molecular mechanisms of lung injury will undoubtedly provide better insights into the operative and perioperative management of critically ill patients.
Figure 1.
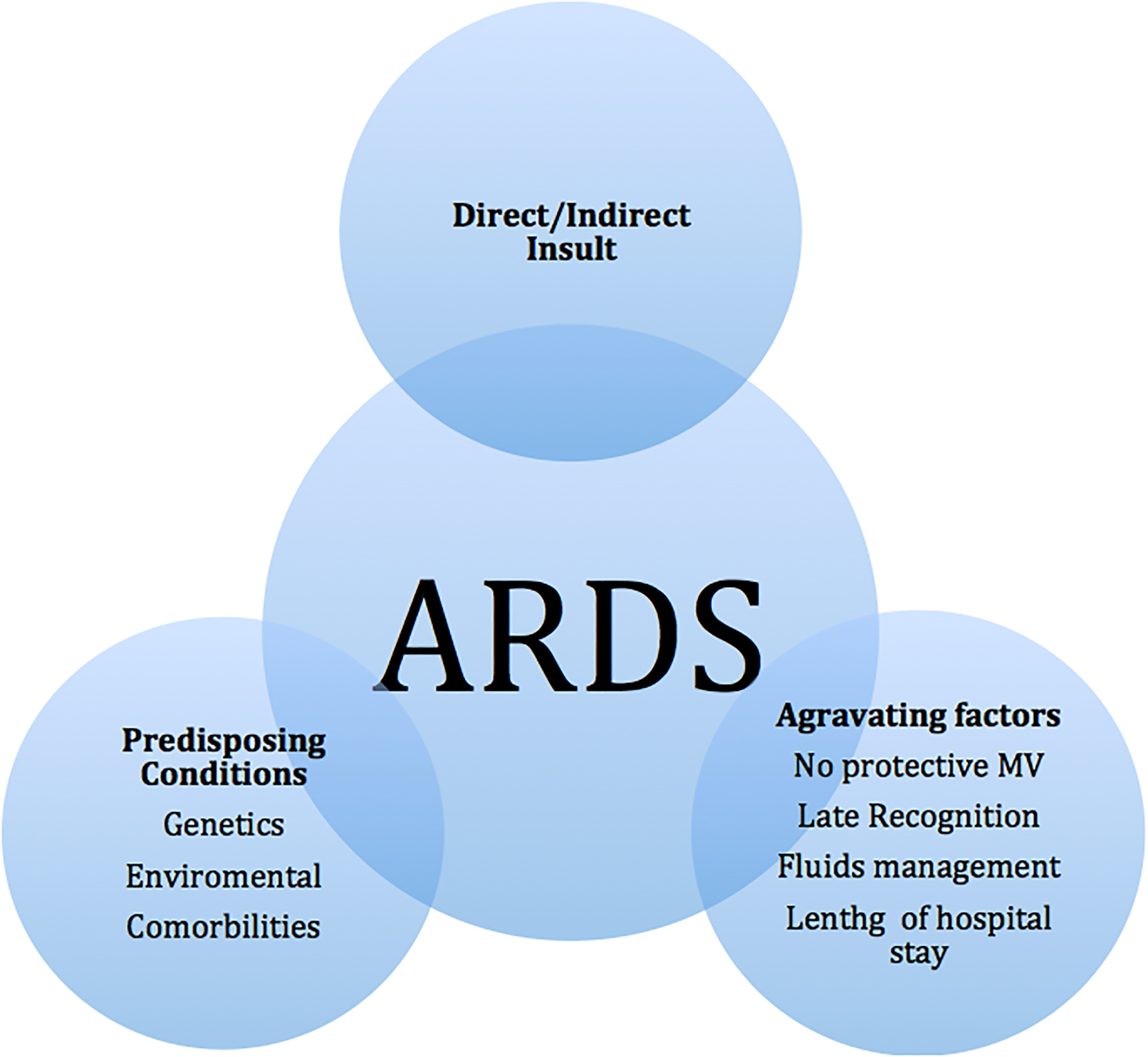
Associated factors to the development of ARDS
Acute Lung Injury in Mechanical Ventilation
An appropriate knowledge and analysis of mechanical ventilation (MV) is necessary for proper care of the ARDS patients (27). The objective of MV is to decrease the respiratory work, providing adequate gas exchange, while minimizing injury (28). However, since the first descriptions of the use of mechanical ventilation, ventilator-associated lung injury has been reported to either initiate lung injury or augment underlying lung injury, coining the term ventilator-induced lung injury (VILI) (29, 30).
The pressure to move air into the alveoli requires forces necessary to overcome the airway resistances, the gas acceleration resistance (inertia), and the elastic properties of the lung as well as the chest wall. Clinically, the transpulmonary pressure, a measurement of the distending force applied to the lung to inflate the lung and distend the chest wall (alveolar pressure minus pleural pressure) is difficult to calculate, but can be estimated through measuring the esophageal pressures (31). The plateau pressure (a surrogate measure of alveolar pressure) can be easily obtained during mechanical ventilation by applying an inspiratory pause on the ventilator. Variables having an effect on the plateau pressure measurement include respiratory efforts and chest wall stiffness. The differences of transpulmonary pressure, as a consequence of imbalance between lung stress and strain (barotrauma), have been identified as main drivers of VILI (32). Other factors attributed include volutrauma (injury secondary to regional overdistention), atelectrauma (cyclic collapse or re-opening of airway units), and biotrauma (cell signaling and direct release of cell mediators induced by physical forces) (33). With a heterogeneous distribution of transpulmonary pressures, it is a challenge to achieve optimal airway distension to prevent VILI. While it is tempting to achieve an open lung strategy by applying higher levels of positive-end expiratory pressures (PEEP), thus preventing atelectasis, recent work suggests that such an approach also increases the risk for volutrauma (33). We will attempt to focus on the molecular mechanisms of injury that result from the mechanical transduction of physical stress on the alveoli, i.e, biotrauma.
In all these scenarios, alveolar-capillary barrier dysfunction results in increased endothelial and epithelial permeability, alteration of surfactant function, and inflammation propagating the injury further (34). In general, the pathogenesis of ARDS is divided into three phases that frequently overlap temporally and spatially: exudative, proliferative and resolution (35, 36). Further, VILI can change the trajectory of ARDS, acting as an independent risk factor for patient outcomes.
Initial insult
Predisposing conditions
Predisposing conditions within and outside of the lung, also called “intrinsic” and “extrinsic” factors, are implicated in ARDS development (37, 38). Intrinsic factors include genetic predispositions and anatomic malformations, and extrinsic factors include comorbidities and environmental triggers (Table 1). Recently, it has been identified that some genes contribute to both susceptibility as well as severity of the disease (Figure 2) (39–45). Others genes associated with cell regulation, cell growth, and apoptosis have also been related to the pathogenesis of ARDS. Those genes include p53, p16INK4a, p15INK4b, WT1 (Wilms’ Tumor 1), and ATM (ataxia telangiectasia mutant) (46–48). P53 may be particularly important in ARDS, since overexpression of p53, at protein and transcription levels, occurs frequently in pulmonary fibrotic disorders. Poly(ADP-ribose) polymerase (PARP), a putative p53 regulating protein, is needed for the fibroplastic activity, since PARP deficient V79 fibroblasts are unable to induce p53 mRNA and are incapable of undergoing apoptosis (49). Independent mechanisms triggered via cell senescence, TGF-β and ROS (reactive oxygen species) have all been implicated in the mechanism of development of ARDS (50, 51).
Figure 2.
Genes implicated in direct ARDS
As described previously, extrinsic factors can trigger lung injury (Table 1), including chemical, biologic, and physical agents (52). Chemical agents include occupational particles (asbestos), and dust; biologic agents such as virus, bacteria, fungi and parasites; and finally, physical agents include inhalation of hot and harmful gases (53).
In addition to the complexity observed in ARDS, other previous co-morbidities can aggravate ARDS and worsen the prognosis. Moreover, patients with ARDS are commonly treated with MV; therefore the risk of VILI is inherently present. Thus, there is an interplay between the lungs suffering from ARDS and mechanical ventilation supporting the system with minimal injury.
Immune mediated Lung Injury
Early in ARDS, after an initial injury that triggers the liberation of alarmins, the exudative phase sets in. This early response is then amplified by the NLRP3 (NLR Family Pyrin Domain Containing 3) inflammasome, which is heavily regulated by Toll-like receptors (TLRs). NLRP3 is a major intracellular multiprotein inflammatory pathway of the innate immune system leading to IL-1β, IL-18, and caspase-1 activation and apoptosis as well as pyroptosis (54). Subsequently, elevated levels of both IL-1β and IL-18 are associated with poor long-term survival in ARDS (55, 56). The NLRP3 inflammasome triggers or alarmins are divided in endogenous factors (DAMPS –Damage Associated Molecular Patterns, ATP, uric acid crystals) or exogenous factors (bacterial hemolysins, pneumolysin, etc.), and are regulated by TLRs and NF-κB (57, 58). The regulatory effect of TLRs is established through the TLR-4 receptor signaling, which shows remarkable similarity to IL-1β signaling (57). This leads to subsequent cell activation of monocytes and macrophages, establishing a cascade of inflammatory cell recruitment and later release of active mediators. Polymorphonuclear neutrophils (PMN) and alveolar epithelial cells mediate this initial phase of intrapulmonary inflammation. This initial response also leads to a potent release of interleukin (IL)-12 and IL-23, promoting strong proinflammatory Th1 immune responses (59, 60). In addition, they trigger the release of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and other proinflammatory cytokines (e.g., TNF-α, IL-6) leading to apoptosis and necrosis. The apoptotic pathway most studied in ARDS, the Fas/FasL system, is mediated by the TNF-α death membrane receptor Fas (CD95) and its natural ligand (FasL) (61). Increased FasL concentrations in BAL from ARDS patients are also associated with an increase in mortality (46).
Lung injury mediated by mechanical forces
Ventilator-induced ARDS is the result of a complex interplay among diverse physical forces on lung anatomy during mechanical ventilation (62–65). Even the pattern and degree of stretch are relevant in determining cellular and molecular response, or biotrauma (66, 67). In addition to mechanical injury, stretch-induced trauma leads to the appearance of inflammatory mediators such as extracellular histones (68). Extracellular histones are proinflammatory mediators; they directly activate the NLRP3 inflammasome, resulting in neutrophil recruitment (69, 70). This process of conversion of mechanical stimuli into a biochemical signal (i.e. biotrauma) is called mechanotransduction. Cells act as mechanosensors after stimulation for physical forces, motion, osmotic forces, shear stress, and interactions between cells and/or cell matrix (71). The mechanical stretch induced by mechanical ventilation regulates ionic channels, including Na-K- ATPase and Ca2+ activity, factors that may play important roles in pulmonary edema development and clearance (72). Ca2+ also induces fos gene expression, as shown in pre-clinical models, propagating the lung injury. Interestingly, systemic inflammation increases the sensitivity of the lung epithelium to injury (73, 74). Additionally, mechanical injury induces translocation of NF-κB into the nucleus unleashing several proinflammatory cytokines and chemokines from macrophages upon mechanical stretch, such as IL-6, IL-8, IL-1β, and TNF-α, and this could be activated independent of TLR4 pathways (75). Follow-up human studies have demonstrated that patients who are ventilated with lower tidal volumes and lower airway pressures had a reduction in interleukin IL-8, −6, soluble TNF receptor 1, and neutrophils (76).
Moreover, mechanical forces can induce direct expression of immediate early response genes, such as c-fos, c-jun, c-myc, IL-1β, and Egr-1 (75). An initial release of pro-inflammatory cytokines and chemokines has long term effects, such as pulmonary fibrosis and thrombosis via platelet-derived growth factor-B, tissue plasminogen activator, intracellular adhesion molecule-1 (ICAM-1), and TGF-β in lung fibrosis (77, 78). Mechanical forces also regulate the production of ROS through the activity of nitric oxide synthase and cyclooxygenase-2 leading to endothelial and epithelial damage (79).
Transfusion-Related Acute Lung Injury
Transfusion-related ARDS is an uncommon complication during or shortly after transfusion. In recent years, the alterations of several inflammatory markers have been described in this immune mediated injury. Multiple factors, such as female donors, neutrophil alloantigens, platelets activation, sepsis and ischemia-reperfusion injury, have been implicated in this process (80–82). Vigilance in keeping TRALI in the differential diagnosis as well as restrictive use of blood has led to a relative decrease in its incidence, yet TRALI continues to be an independent risk factor for mortality (83). More information on this type of lung injury is available elsewhere in this volume.
Anatomic basis, pathophysiologic and molecular mechanism
Conducting airways and alveoli are both derived from the endoderm-derived bulbs (84). The alveoli are covered by a simple squamous epithelium composed of two types of cells, pneumocytes type I and II. This epithelium is a tight barrier impermeable to proteins and solutes. The flat type I pneumocyte covers around 90% of the alveolar surface; they have abundance in lipid-rich lamellar bodies, and high expression of MUC1, ABCA3, and surfactant proteins (SP) that decrease the alveolar surface tension, along with other biologic functions (85). The more cuboidal type II pneumocytes cover the remaining extension, and they have the critical role of surfactant synthesis/regulation, and as progenitors for type I pneumocytes. Mature airways are lined by an impermeable cylindrical pseudostratified epithelium with abundant superficial cilias. The permeability barrier is maintained by inter-cellular junctional proteins including claudins (Cld), connexins (especially Cnx43), paranexins, adhesins, and occludins (86–88). Gap junctions rapidly exchange the stimulus-induced responses among ciliated cells. The number of ciliated and non-ciliated cells varies along the airways, and their number is strongly influenced by toxic and inflammatory processes. The epithelial cells are interspersed with secretory goblet cells that produce abundant mucins, fluids and electrolytes (Cl– and HCO3–) together with the submucosal glands that produce mainly MUC5B and fluids. These mucins form a highly adhesive thin mucous aqueous subphase that creates a direct host defense barrier able to disrupt bacterial aggregation, adhesion and internalization into the epithelial surface. Furthermore, epithelial cells secrete host defense molecules and proteases such as human β-defensins, lysozyme, lactoferrin, cathelicidin LL37, and surfactant proteins. The joint work between the aforementioned mechanisms create an escalator apparatus that is able to mobilize harmful chemical, physical, and microbiologic agents from the surface of the airways (89). The crucial engine of this apparatus is the coordinated directional movement of cilia regulated by paracrine signals, including purinergic signaling. They are composed by microtubules associated with dynein arms to form axonemes that are powered by ATP (90).
Both airway and alveoli epitheliums are anchored to the extracellular matrix (ECM) through a basal membrane. The ECM is composed of collagen and elastic fibers, surrounded by an amorphous substance rich in glycosaminoglicans such as hyaluronan. The space formed in the ECM is also known as interstitial space. The epithelial ciliated cells as well as the secretory goblet cells originate from epithelial stem cells that express α6β4 and are located in close proximity to the epithelial basal membrane. These epithelial stem cells play a critical role in airway regeneration after injury (91). Their differentiation is determined by the transcription factor SOX2 and requires SPDEF, a transcription factor that regulates genes encoding a network of molecules involved in mucus biosynthesis (92, 93). Such pluripotency will be important during the recovery phase in ARDS. In the interstitial space of the alveoli are macrophages with surfactant catabolism and the innate immune defense functions. Impairment of the surfactant regulatory effect of macrophages leads to acquired pulmonary alveolar proteinosis (Figure 3).
Figure 3.
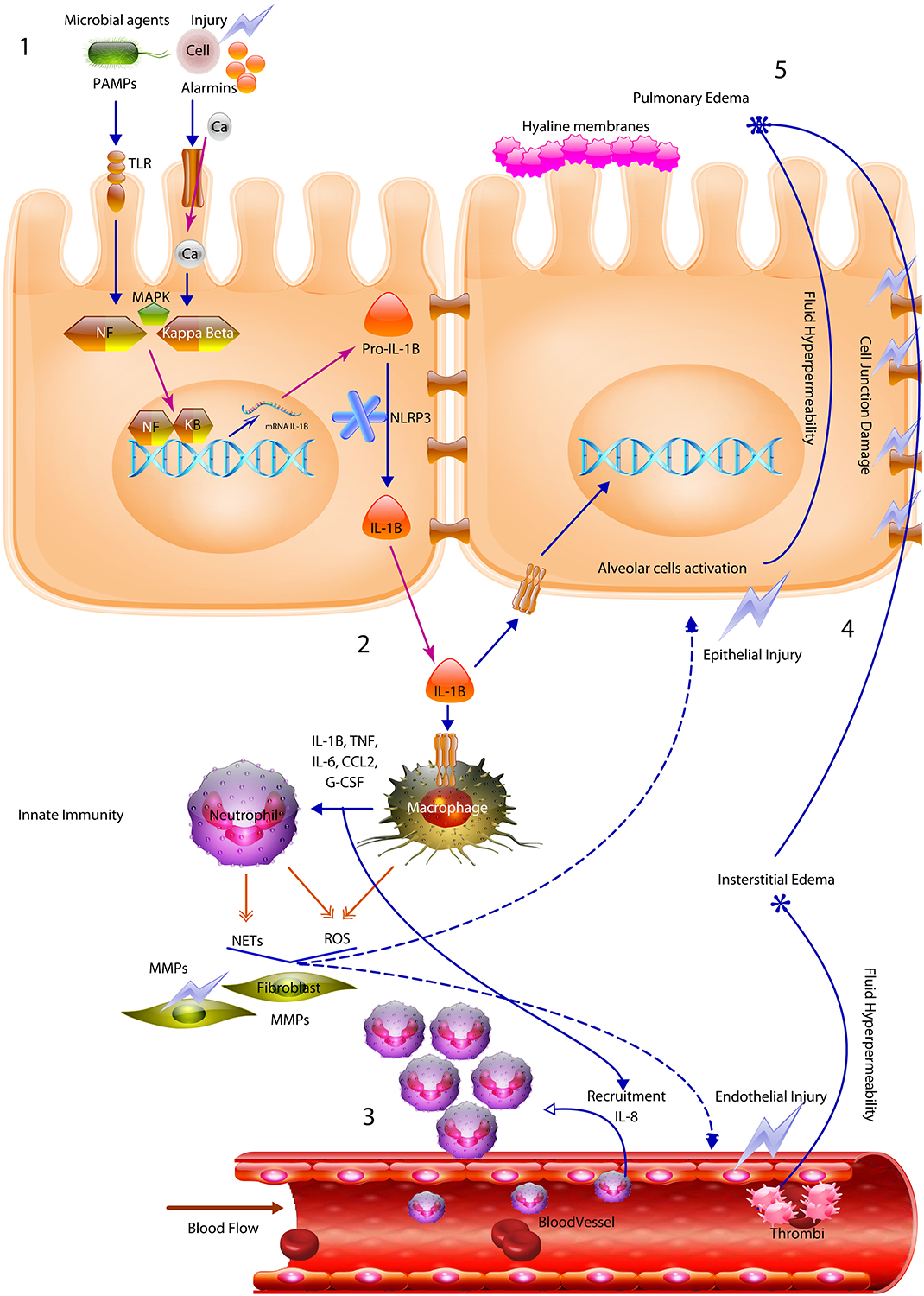
Pathogenesis of the ARDS is triggered after an initial injury that interacts through alarmin receptors and toll like receptors activating nuclear transcription factors including NF-Kappa-beta. Then a powerful acute immune response triggered mainly by IL-1B causes macrophage/neutrophil activation and recruitment. The cell mediated immune response produces tissue damage mediated by NOS and NETs allowing the development of edema and epithelial damage.
The pulmonary surfactant and alveolar liquid subphase keep the alveolus expanded. Pulmonary surfactant is composed mainly of lipids and limited amount of SPs (mainly SP-A and SP-D). Its main role is to reduce surface tension in the alveolus during the dynamic ventilation changes, but also actively participate in the opsonization and killing of pathogens, mediating in the regulation of macrophages, neutrophils and lymphocytes by pathogen-associated molecular patterns (PAMPs) (94–96). Additionally, the main surfactant lipids dipalmitoylphosphatidylcholine and phosphatidylglycerol have intrinsic antimicrobial activities. Type I pneumocytes act as alveolar mechanosensors, responding to the stretch of lung expansion by conveying surfactant-secreting calcium mediated signals to the type II pneumocytes.
Exudative Phase
Increased permeability and pulmonary edema
ARDS is characterized by an initial exudative phase, which presents with diffuse alveolar damage associated with severe accumulation of liquid forming pulmonary edema (97). This hydraulic imbalance governs together with the already described barometric alteration in the pulmonary microvasculature. A major microvascular pressure produces an enhanced filtration into the pulmonary interstitium. A higher conductance product of endothelial cell activation, mediated by P-selectin, also allows the high filtration and release of thromboxane A2 observed in ARDS (98). Thromboxane A2 activates endothelial thromboxane A2 receptors and induces de novo expression of ICAM-1, triggering firm PMN adherence to endothelium via β2-integrins (99). The alterations of both barometric and hydraulic forces induce the release of inflammatory mediators and gene transcription. The concentrations of the liberated inflammatory mediators are intimately linked with the grade of severity of the injury. Two relevant mediators are IL-1α and IL-6, which activate neutrophils and monocytes, inducing the liberation of ROS with damage of the endothelium (100). Additionally, angiopoetin-2 (Ang-2) also regulates vascular permeability, promoting cell death and vascular regression (101). The production of neutrophil extracellular traps (NETs) also damage the lung endothelium contributing to the edema (102).
Endothelial injury
The microvascular damage induces hyperpermeability after the formation of gaps between endothelial cells mediated by myosin light-chain kinase (MLCK) phosphorylation and subsequent contraction. Endothelial wall integrity and maintenance of the epithelial/endothelial barrier requires epithelial (E-cadherin)- and vascular endothelial cadherin (VE- cadherin)-mediated adherence of the junction bonds (103, 104). A proinflammatory state with elevation in vascular endothelial growth factor, TNF-α, or histamine induces Src-mediated phosphorylation of VE-cadherin at Tyr685 (p-Y685), which leads to endothelial activation and higher permeability (105).
In ARDS, high intravascular hydrostatic pressure and endothelial cell activation lead to massive leak of plasma, including large proteins such as albumin. Subsequently, the intravascular-interstitium balance of proteins is altered, and a disruption of the oncotic pressure results in worsening of the pulmonary edema, restriction of the effective gas exchange and thus hypoxemia.
Parallel to the development of edema, the initial proinflamatory stimuli leads to the recruitment and accumulation of neutrophils in the pulmonary microcirculation, where they are activated, producing NETs and ROS with subsequent direct endothelial wall damage (106). Non-activated neutrophils perform endothelial cell adhesion rolling, chemotaxis, sequestration and activation in the lung interstitium due to the activity of IL-8, epithelial neutrophil-activating peptide (ENA-78), and β1 and β2 integrins (107). Integrins mediate cell adhesion by interaction with adhesion molecules (for example, intercellular adhesion molecule [ICAM]-1, ICAM-2, vascular cell adhesion molecule [VCAM]-1). During ARDS, neutrophils adhere to the endothelium in an integrin–dependent or in an integrin–independent mechanisms that are activated after highly antigenic or other mediators (99, 108).
Epithelial injury
As previously described epithelial cells or pneumocytes line the alveolar wall. They play critical roles including the functions of protective barrier, regulating surfactant production, and fluid homeostasis. In ARDS, alveolar epithelial cells undergo severe damage following insults of neutrophils, proinflammatory cytokines, hypoxia, and physical forces. All of these insults induce cell damage by autocrine or paracrine apoptotic mechanisms (109, 110). Mitochondrial-mediated apoptotic pathways also contribute to this increased apoptotic rates (110). In addition, the activation of the Fas/FasL pathway is important in mediating apoptosis in ARDS. Apoptosis is induced when membrane-bound or FasL binds to Fas-bearing cells.
In the airways multiple epithelial changes are observed during ARDS. Alterations in the number of ciliated cells, goblet cell metaplasia and excessive mucus production are all observed. The development of these changes are linked to inflammatory signaling via TLRs and transcription factors of the IRF and NF-κB families and cytokine signaling via the Jak kinase–STAT transcription factor pathway in respiratory epithelial cells. Airway Clara cells are altered during ARDS, which are normally devoted to the protection of the respiratory tract against toxic inhaled agents, the repair of damaged epithelium, xenobiotics detoxification, and the secretion of proteins with important biological activities. The Clara cell protein (CC16) is highly expressed after injury and possibly increasing the product by modulating the production and/or activity of phospholipase- A2, interferon-γ, and TNF-α.
Lung extracellular matrix alteration
The extracellular matrix (ECM) forms the region of the lung situated between the alveolar wall and the blood vessels; it is also known as lung interstitium. ECM is a complex and dynamic meshwork that regulates cellular and extracellular functions including development, nutrition, proliferation, repair, and migration. ECM is composed of protein fibers and amorphous substance. The protein fibers are mainly collagen and elastin, and the amorphous substance is composed of water, glycoproteins, and proteoglycans including hyaluronic acid. In ARDS, lung ECM undergoes significant remodeling because of physical damage as well as degradation of its components, including hyaluronan fragments (111–113). If the damage is not controlled, it can lead to irreversible structural changes as well as progressive inflammation with long-term sequelae, including lung fibrosis (114, 115). An adequate clearance of hyaluronan fragments is necessary to allow adequate healing, a process dependent on the hyaluronan receptor CD44 (113). Unquenched hyaluronan degradation products on the other hand can induce chemokines, cytokines, growth factors, signal transduction molecules, and adhesion molecules propagating inflammation and injury (116).
The family of zinc-dependent enzymes, matrix metalloproteinases (MMPs), are responsible for ECM remodeling and these enzymes are produced by a variety of cell types, including macrophages, monocytes, and PMNs (117, 118). MMPs are known to exert a myriad of regulatory actions critical in tissue repair and remodeling, including epithelial cell migration, proliferation, differentiation, and apoptosis, as well as release of latent or bound growth factors from the ECM. In ARDS, MMP-2 (gelatinase A), MMP-8 (collagenase 2), and MMP-9 (gelatinase B), are extensively studied and their concentrations correlate with clinical severity (119, 120). In addition to degrading the epithelial and endothelial basal membrane allowing migration of inflammatory cells, MMPs modulate inflammation by either activating or inactivating cytokines and chemokines (121, 122).
Neutrophil-Mediated Injury
Excessive recruitment and activation of neutrophils with subsequent recruitment of monocytic cells is observed in early stages of ARDS. This leads to release of toxic enzymes, damage of alveolar epithelium, vascular leakage, pulmonary edema, and hypoxemia. Phagocytic cells, both neutrophils and macrophages, respond to several potent chemoattractants expressed during lung injury, including IL-17, macrophage inflammatory protein-2 (MIP-2), CCL3, and CXCL2. These phagocytic cells are terminally differentiated effector cells with primary roles in innate immunity. They are able to perform phagocytosis of pathogens, production of reactive oxygen intermediates and toxic enzymes such as elastase, and formation of neutrophil extracellular traps (NETs) (99, 123).
NETs have been recently described as an innate immune mechanism, which increases the local capture of pathogens by the DNA. When neutrophils are activated, they emit their DNA fibers, which carry nuclear and cytoplasmic proteins, including histones, elastase, myeloperoxidase (MPO), pentraxin, cytokines, MMPs, and bactericidal peptides (124). The DNA fibers rich in proteolytic and pro-inflammatory molecules are interwoven to create web-like structures able to capture and destroy pathogens. While effective, this massive response may lead to tissue damage because its effect is non-specific, injuring the endothelium (vasculitis), alveoli, terminal bronchioles, and extracellular fibers allowing the formation of abscesses and damage of the alveolar–capillary barrier (102). Interestingly, bacteria such as Streptococcus pneumoniae and Staphylococcus aureus can produce endonucleases able to digest NETs (125). ROS and MPO together with neutrophil elastase are known to mediate NETs induction (126).
Chemokines, cytokines, and lipid signaling molecules-mediated inflammation and injury
The role of cytokines, cytokine binding proteins, and growth factors in regulating ARDS pathology has been extensively studied. The ARDS inflammatory cascade involves release of these mediators, where alveolar macrophages and PMNs are considered the primary source of these molecules, but they are also produced by type II pneumocytes (127, 128). Yet, both proinflammatory cytokines (IL-1 −6, −8, −18, TNF-α, and TGF-β) and anti-inflammatory cytokines (IL-10, −13) are altered in ARDS, making it a balancing act (129). Other mediators implicated in lung injury include omega-3 fatty acid derived lipid mediators released from cell injury during the early phases of ARDS (130).
Oxidant-Mediated Injury
Oxidative-mediated mediators are implicated in the development of lung injury. Toxic reactive oxygen species (ROS) are generated from normal cellular respiration and aerobic metabolism, and increased significantly under cellular stress and damage. In injury, several free radicals, such as hydrogen peroxide (H2O2), superoxide anion (O2−) and the hydroxyl radical (HO−), and reactive nitrogen species (RNS), produce deleterious effects (131, 132). These oxidative mediators are regulated at both cellular and mitochondrial levels.
The most frequent pathophysiologic basis of noninfectious ARDS is hyperoxia-induced injury secondary to free oxygen radicals (133). The hyperoxia effect killing type II pneumocytes appears to be mediated by p53 as well as p21 dependent mechanisms, which also play a role in long-term fibrosis (134–136). TLRs contribute to increased inflammation during hyperoxia in the absence of infection via NF-κB activation (137, 138). Tyrosine kinase (TK), protein kinase C (PKC), and mitogen activated protein kinase (MAPK) signaling cascades also activate NF-κB pathways.
The second mechanism implicates the release of ROS, especially O2− and H2O2, upon activation of inflammatory cells (139). In endotoxin mediated ARDS, radical oxygen species are formed by inducible cyclo-oxygenase-2 and inducible nitric oxide synthase (iNOS) in alveolar macrophages (140). The activation of these enzymes also causes increased pulmonary microvascular permeability especially in burn and smoke inhalation mediated injuries, and activation of response genes (c-fos and c-jun), and pro-apoptotic FAS and FAS-ligand in alveolar macrophages and alveolar epithelial cells suggests that ROS play a critical role in ARDS mediated apoptosis and cellular injury (141).
A third mechanism is the liberation of ROS from the mitochondria (142). Mitochondrial superoxide dismutase converts the superoxide to H2O2, which diffuses out of the mitochondria to activate cytosolic targets such as Weibel-Palade bodies, causing endothelial expression of P-selectin, and activating the NF-κB pathway. The mitochondrial participation depends on Ca2+-induced activation of mitochondrial electron transport.
Other mechanisms include restitution by hydrogen donation, nucleotide excision, and recombination, such as nicotinamide adenine dehydrogenase (NADPH) oxidase, xanthine/ xanthine oxidase, and/or phospholipase C pathways, also activated during ROS production in lung injury (143).
Counter-regulatory mediators including both non-enzymatic and enzymatic antioxidant defenses are activated to protect the lung tissue from the deleterious effects of ROS. Non-enzymatic defenses include vitamins, bilirubin, sulfhydryl-containing glutathione, albumin, ceruloplasmin, hemosiderin and transferrin/lactoferrin. In addition, enzymatic antioxidants counter balance superoxide dismutase. Such enzymes are Superoxide Dismutase (SOD), Myeloperoxidase (MPO), catalase and glutathione-S-transferases, and peroxidases. These enzymes also regulate NETs, suggesting a complex balance between the protective and destructive effects of ROS in lung injury (144).
Coagulation and fibrinolysis
ARDS is characterized by alterations in both systemic and intra-alveolar coagulation and fibrinolysis. Blood coagulation pathways are involved in ARDS. The inflammatory endothelial damage has an effect inducing the formation of thrombin creating a hypercoagulable state (145). In addition, inflammation activates the endothelium and reduces blood flow with subsequent higher propensity for thrombosis. Thus, the formation of thrombi and vasculopathy is a common event in ARDS.
Increased levels of coagulation factor III (tissue factor) and plasminogen activator inhibitor type 1 mediated by IL-1 and −6 have been reported during ARDS (145, 146). These two mediators lead to the activation of the extrinsic pathway of coagulation and inhibition of fibrinolysis, respectively. They also mediate the degradation of fibrin and induce platelet activation. Additionally, inflammatory mediators induce plasma prekallikrein (KLKB1) production, leading to a potent vascular permeability stimulus (147).
Thrombosis and fibrin deposition is a beneficial counter regulatory mechanism able to quickly repair the disrupted vascular and alveolar wall. This effect is mediated by von Willebrand Factor (vWF). However, massive fibrin deposition and thrombosis can lead to activation of neutrophils and fibroblasts, endothelial injury, loss of surfactant activity favoring alveolar collapse, and impaired alveolar fluid clearance.
Fibroproliferative phase
When a persistent or massive injury occurs and counter regulatory mechanisms cannot resolve the damage, persistent inflammation results in a fibroproliferative response. Accumulation of macrophages, fibrocytes, fibroblasts, and myofibroblasts in the alveolar and interstitial compartment contribute to excessive deposition of ECM (50). Macrophages contribute to the fibroproliferative phase in ARDS, producing numerous proinflammatory mediators (IL-1, - 4, and −13) and growth factors (transforming growth factor [TGF]-α, TNF- α, platelet derived growth factor [PDGF], fibroblast growth factor [FGF], and insulin-like growth factor [IGF]-I) (148).
While alveolar wall repair is mediated by keratinocyte growth factor (KGF), hepatocyte growth factor (HGF), FGF, and TGF- α, endothelial wall repair is mediated by VEGF and HIF-1α (149, 150). Proliferating cells are regulated and eliminated through apoptosis, resolving the massive cell infiltrate and pneumocyte type II cell hyperplasia; fibrosis develops when these counter regulatory mechanisms are arrested or fail.
TGF-β production is the main mediator implicated in lung fibrosis development. Other soluble factors such as endothelin and thrombin may mediate similar effects, although their relative roles in fibrotic diseases of the lung are unclear (51, 151). Multiple cell types synthesize TGF- β such as alveolar macrophages, lymphocytes, fibroblasts, and pneumocytes. This cytokine mediates its activity through p53 and cyclin E encoding for connective tissue matrix accumulation, pulmonary mesenchyme cell growth and chemotaxis, and arrest of counter fibrotic mechanisms (152).
Leukotrienes (leukotriene D4) and prostaglandins (in particular PGE2), oppose fibrotic changes in ARDS. Potential antifibrotic mechanisms of action of PGE2 include inhibition of fibroblast proliferation, migration, contractility, and myofibroblast differentiation (153).
Resolution phase
Despite the severe clinical and molecular manifestations of ARDS, the majority of patients survive. This illustrates clearly that repair mechanisms can reestablish normal or near-normal endothelial and epithelial barriers and normalize the structure and function of the injured alveoli. An effective lung repair in ARDS requires correction of both micro-vascular hyper-permeability and edema. The alveolo-capillary barrier repair depends on effective and rapid adherents junctions being re-established. A rapid re-absorption and fluid clearance from inside the alveoli is also required. Several catecholamine-independent pathways, including glucocorticoids and thyroid hormone, increase the rate of alveolar fluid clearance (154, 155). Na, K-ATPase pumps and the CFTR (cystic fibrosis transmembrane regulator) play a key role regulating the alveolar fluid transport (156, 157). The first step toward resolution of ARDS is to remove the alveolar edema fluid to the lung interstitium, where net clearance can occur through lung lymphatics, the pulmonary microcirculation and even bulk flow into the pleural space. Removal of alveolar edema fluid from the air spaces requires vector transport of sodium and chloride across the apical and basolateral membranes of alveolar epithelial type I and II cells, which creates an osmotic gradient for the reabsorption of water. Thus, sodium is actively absorbed across the apical surface of alveolar epithelial type I and II cells, predominantly by the epithelial sodium channel; the sodium is then extruded from the cell by the Na/K-ATPase pumps on the basolateral membrane. Chloride is transported by the transcellular and perhaps the paracellular route, although the molecular pathways are not completely understood.
Conclusion
ARDS continues to be a clinical challenge, yet there have been significant improvements in the acute mortality in the last 20 years. Lung protective approaches to mechanical ventilation (low Tv and appropriate use of PEEP) along with a multi-disciplinary approach to the patient have led to this overall improvement, presumably through less iatrogenic lung injury (VILI). However, we are still faced with the long-term challenges, including the reality that most patients dying from ARDS perish from unmitigated multisystem organ dysfunction rather than the direct lung injury itself. Therefore, as we continue to improve our understanding of the inflammatory pathways and mediators, we hope to develop further treatments and strategies to improve the complications and management of this syndrome to further improve the survival (158).
III. References
- 1.Esteban A, Fernandez-Segoviano P, Frutos-Vivar F, Aramburu JA, Najera L, Ferguson ND, et al. Comparison of clinical criteria for the acute respiratory distress syndrome with autopsy findings. Ann Intern Med. 2004;141(6):440–5. [DOI] [PubMed] [Google Scholar]
- 2.Thille AW, Esteban A, Fernandez-Segoviano P, Rodriguez JM, Aramburu JA, Penuelas O, et al. Comparison of the Berlin definition for acute respiratory distress syndrome with autopsy. Am J Respir Crit Care Med. 2013;187(7):761–7. [DOI] [PubMed] [Google Scholar]
- 3.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122(8):2731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thille AW, Esteban A, Fernandez-Segoviano P, Rodriguez JM, Aramburu JA, Vargas-Errazuriz P, et al. Chronology of histological lesions in acute respiratory distress syndrome with diffuse alveolar damage: a prospective cohort study of clinical autopsies. Lancet Respir Med. 2013;1(5):395–401. [DOI] [PubMed] [Google Scholar]
- 5.Bachofen M, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis. 1977;116(4):589–615. [DOI] [PubMed] [Google Scholar]
- 6.Papazian L, Doddoli C, Chetaille B, Gernez Y, Thirion X, Roch A, et al. A contributive result of open-lung biopsy improves survival in acute respiratory distress syndrome patients. Crit Care Med. 2007;35(3):755–62. [DOI] [PubMed] [Google Scholar]
- 7.Ichikado K, Suga M, Muranaka H, Gushima Y, Miyakawa H, Tsubamoto M, et al. Prediction of prognosis for acute respiratory distress syndrome with thin-section CT: validation in 44 cases. Radiology. 2006;238(1):321–9. [DOI] [PubMed] [Google Scholar]
- 8.Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2(7511):319–23. [DOI] [PubMed] [Google Scholar]
- 9.Kallet RH, Jasmer RM, Pittet JF, Tang JF, Campbell AR, Dicker R, et al. Clinical implementation of the ARDS network protocol is associated with reduced hospital mortality compared with historical controls. Crit Care Med. 2005;33(5):925–9. [DOI] [PubMed] [Google Scholar]
- 10.Fahr M, Jones G, O’Neal H, Duchesne J, Tatum D. Acute Respiratory Distress Syndrome Incidence, But Not Mortality, Has Decreased Nationwide: A National Trauma Data Bank Study. Am Surg. 2017;83(4):323–31. [PubMed] [Google Scholar]
- 11.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA. 2016;315(8):788–800. [DOI] [PubMed] [Google Scholar]
- 12.Ryb GE, Cooper C. Race/ethnicity and acute respiratory distress syndrome: a National Trauma Data Bank study. J Natl Med Assoc. 2010;102(10):865–9. [DOI] [PubMed] [Google Scholar]
- 13.Soto GJ, Martin GS, Gong MN. Healthcare disparities in critical illness. Crit Care Med. 2013;41(12):2784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erickson SE, Shlipak MG, Martin GS, Wheeler AP, Ancukiewicz M, Matthay MA, et al. Racial and ethnic disparities in mortality from acute lung injury. Crit Care Med. 2009;37(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–33. [DOI] [PubMed] [Google Scholar]
- 16.Martin TR. Interactions between mechanical and biological processes in acute lung injury. Proc Am Thorac Soc. 2008;5(3):291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McAuley DF, Cross LM, Hamid U, Gardner E, Elborn JS, Cullen KM, et al. Keratinocyte growth factor for the treatment of the acute respiratory distress syndrome (KARE): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Respir Med. 2017;5(6):484–91. [DOI] [PubMed] [Google Scholar]
- 18.Zeiher BG, Artigas A, Vincent JL, Dmitrienko A, Jackson K, Thompson BT, et al. Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Crit Care Med. 2004;32(8):1695–702. [DOI] [PubMed] [Google Scholar]
- 19.Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185(11):1225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robbins RA, Russ WD, Rasmussen JK, Clayton MM. Activation of the complement system in the adult respiratory distress syndrome. Am Rev Respir Dis. 1987;135(3):651–8. [DOI] [PubMed] [Google Scholar]
- 22.Bos LD, Schouten LR, van Vught LA, Wiewel MA, Ong DSY, Cremer O, et al. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gattinoni L, Caironi P, Cressoni M, Chiumello D, Ranieri VM, Quintel M, et al. Lung recruitment in patients with the acute respiratory distress syndrome. N Engl J Med. 2006;354(17):1775–86. [DOI] [PubMed] [Google Scholar]
- 24.Martin GS, Brigham KL. Fluid flux and clearance in acute lung injury. Compr Physiol. 2012;2(4):2471–80. [DOI] [PubMed] [Google Scholar]
- 25.Sawheny E, Ellis AL, Kinasewitz GT. Iloprost improves gas exchange in patients with pulmonary hypertension and ARDS. Chest. 2013;144(1):55–62. [DOI] [PubMed] [Google Scholar]
- 26.Herlihy JP, Vermeulen MW, Joseph PM, Hales CA. Impaired alveolar macrophage function in smoke inhalation injury. J Cell Physiol. 1995;163(1):1–8. [DOI] [PubMed] [Google Scholar]
- 27.Silversides JA, Ferguson ND. Clinical review: Acute respiratory distress syndrome - clinical ventilator management and adjunct therapy. Crit Care. 2013;17(2):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kallet RH, Hemphill JC 3rd, Dicker RA, Alonso JA, Campbell AR, Mackersie RC, et al. The spontaneous breathing pattern and work of breathing of patients with acute respiratory distress syndrome and acute lung injury. Respir Care. 2007;52(8):989–95. [PubMed] [Google Scholar]
- 29.Bendixen HH, Hedley-Whyte J, Laver MB. Impaired Oxygenation in Surgical Patients during General Anesthesia with Controlled Ventilation. A Concept of Atelectasis. N Engl J Med. 1963;269:991–6. [DOI] [PubMed] [Google Scholar]
- 30.Woo SW, Hedley-Whyte J. Macrophage accumulation and pulmonary edema due to thoracotomy and lung over inflation. J Appl Physiol. 1972;33(1):14–21. [DOI] [PubMed] [Google Scholar]
- 31.Talmor D, Sarge T, O’Donnell CR, Ritz R, Malhotra A, Lisbon A, et al. Esophageal and transpulmonary pressures in acute respiratory failure. Crit Care Med. 2006;34(5):1389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amato MB, Meade MO, Slutsky AS, Brochard L, Costa EL, Schoenfeld DA, et al. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med. 2015;372(8):747–55. [DOI] [PubMed] [Google Scholar]
- 33.Cressoni M, Chiumello D, Algieri I, Brioni M, Chiurazzi C, Colombo A, et al. Opening pressures and atelectrauma in acute respiratory distress syndrome. Intensive Care Med. 2017;43(5):603–11. [DOI] [PubMed] [Google Scholar]
- 34.Andrews PL, Sadowitz B, Kollisch-Singule M, Satalin J, Roy S, Snyder K, et al. Alveolar instability (atelectrauma) is not identified by arterial oxygenation predisposing the development of an occult ventilator-induced lung injury. Intensive Care Med Exp. 2015;3(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greer R The temporal evolution of acute respiratory distress syndrome following shock. Eur J Anaesthesiol. 2010;27(3):226–32. [DOI] [PubMed] [Google Scholar]
- 36.Sekiguchi H, Schenck LA, Horie R, Suzuki J, Lee EH, McMenomy BP, et al. Critical care ultrasonography differentiates ARDS, pulmonary edema, and other causes in the early course of acute hypoxemic respiratory failure. Chest. 2015;148(4):912–8. [DOI] [PubMed] [Google Scholar]
- 37.Szilagyi KL, Liu C, Zhang X, Wang T, Fortman JD, Zhang W, et al. Epigenetic contribution of the myosin light chain kinase gene to the risk for acute respiratory distress syndrome. Transl Res. 2017;180:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown SM, Grissom CK, Rondina MT, Hoidal JR, Scholand MB, Wolff RK, et al. Polymorphisms in key pulmonary inflammatory pathways and the development of acute respiratory distress syndrome. Exp Lung Res. 2015;41(3):155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Acosta-Herrera M, Pino-Yanes M, Blanco J, Ballesteros JC, Ambros A, Corrales A, et al. Common variants of NFE2L2 gene predisposes to acute respiratory distress syndrome in patients with severe sepsis. Crit Care. 2015;19:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tejera P, O’Mahony DS, Owen CA, Wei Y, Wang Z, Gupta K, et al. Functional characterization of polymorphisms in the peptidase inhibitor 3 (elafin) gene and validation of their contribution to risk of acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2014;51(2):262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer NJ, Feng R, Li M, Zhao Y, Sheu CC, Tejera P, et al. IL1RN coding variant is associated with lower risk of acute respiratory distress syndrome and increased plasma IL-1 receptor antagonist. Am J Respir Crit Care Med. 2013;187(9):950–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Azevedo ZM, Moore DB, Lima FC, Cardoso CC, Bougleux R, Matos GI, et al. Tumor necrosis factor (TNF) and lymphotoxin-alpha (LTA) single nucleotide polymorphisms: importance in ARDS in septic pediatric critically ill patients. Hum Immunol. 2012;73(6):661–7. [DOI] [PubMed] [Google Scholar]
- 43.Bajwa EK, Cremer PC, Gong MN, Zhai R, Su L, Thompson BT, et al. An NFKB1 promoter insertion/deletion polymorphism influences risk and outcome in acute respiratory distress syndrome among Caucasians. PLoS One. 2011;6(5):e19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheu CC, Zhai R, Wang Z, Gong MN, Tejera P, Chen F, et al. Heme oxygenase-1 microsatellite polymorphism and haplotypes are associated with the development of acute respiratory distress syndrome. Intensive Care Med. 2009;35(8):1343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong MN, Zhou W, Williams PL, Thompson BT, Pothier L, Boyce P, et al. −308GA and TNFB polymorphisms in acute respiratory distress syndrome. Eur Respir J. 2005;26(3):382–9. [DOI] [PubMed] [Google Scholar]
- 46.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, et al. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol. 2002;161(5):1783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A. 2009;106(35):14978–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eickmeier O, Kim SY, Herrmann E, Doring C, Duecker R, Voss S, et al. Altered mucosal immune response after acute lung injury in a murine model of Ataxia Telangiectasia. BMC Pulm Med. 2014;14:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, Hall J, et al. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008;36(13):4454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keshari RS, Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Chaaban H, et al. Acute lung injury and fibrosis in a baboon model of Escherichia coli sepsis. Am J Respir Cell Mol Biol. 2014;50(2):439–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dhainaut JF, Charpentier J, Chiche JD. Transforming growth factor-beta: a mediator of cell regulation in acute respiratory distress syndrome. Crit Care Med. 2003;31(4 Suppl):S258–64. [DOI] [PubMed] [Google Scholar]
- 52.Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol. 2015;194(3):855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moazed F, Calfee CS. Environmental risk factors for acute respiratory distress syndrome. Clin Chest Med. 2014;35(4):625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He X, Qian Y, Li Z, Fan EK, Li Y, Wu L, et al. TLR4-Upregulated IL-1beta and IL-1RI Promote Alveolar Macrophage Pyroptosis and Lung Inflammation through an Autocrine Mechanism. Sci Rep. 2016;6:31663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107(4):1062–73. [DOI] [PubMed] [Google Scholar]
- 56.Guo J, Huang F, Liu J, Chen Y, Wang W, Cao B, et al. The Serum Profile of Hypercytokinemia Factors Identified in H7N9-Infected Patients can Predict Fatal Outcomes. Sci Rep. 2015;5:10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113(10):2324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marchant A, Amedei A, Azzurri A, Vekemans J, Benagiano M, Tamburini C, et al. Polarization of PPD-specific T-cell response of patients with tuberculosis from Th0 to Th1 profile after successful antimycobacterial therapy or in vitro conditioning with interferon-alpha or interleukin-12. Am J Respir Cell Mol Biol. 2001;24(2):187–94. [DOI] [PubMed] [Google Scholar]
- 60.Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015;21(7):719–29. [DOI] [PubMed] [Google Scholar]
- 61.Lopez AD, Avasarala S, Grewal S, Murali AK, London L. Differential role of the Fas/Fas ligand apoptotic pathway in inflammation and lung fibrosis associated with reovirus 1/L-induced bronchiolitis obliterans organizing pneumonia and acute respiratory distress syndrome. J Immunol. 2009;183(12):8244–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol. 1970;28(5):596–608. [DOI] [PubMed] [Google Scholar]
- 63.Meade MO, Cook DJ, Guyatt GH, Slutsky AS, Arabi YM, Cooper DJ, et al. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2008;299(6):637–45. [DOI] [PubMed] [Google Scholar]
- 64.Mercat A, Richard JC, Vielle B, Jaber S, Osman D, Diehl JL, et al. Positive end-expiratory pressure setting in adults with acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2008;299(6):646–55. [DOI] [PubMed] [Google Scholar]
- 65.Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974;110(5):556–65. [DOI] [PubMed] [Google Scholar]
- 66.Chen ZL, Song YL, Hu ZY, Zhang S, Chen YZ. An estimation of mechanical stress on alveolar walls during repetitive alveolar reopening and closure. J Appl Physiol (1985). 2015;119(3):190–201. [DOI] [PubMed] [Google Scholar]
- 67.Halter JM, Steinberg JM, Gatto LA, DiRocco JD, Pavone LA, Schiller HJ, et al. Effect of positive end-expiratory pressure and tidal volume on lung injury induced by alveolar instability. Crit Care. 2007;11(1):R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol. 2004;172(11):6692–700. [DOI] [PubMed] [Google Scholar]
- 69.Grailer JJ, Ward PA. Lung inflammation and damage induced by extracellular histones. Inflamm Cell Signal. 2014;1(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol. 2014;192(12):5974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, et al. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol. 2005;167(2):609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fisher JL, Margulies SS. Na(+)-K(+)-ATPase activity in alveolar epithelial cells increases with cyclic stretch. Am J Physiol Lung Cell Mol Physiol. 2002;283(4):L737–46. [DOI] [PubMed] [Google Scholar]
- 73.Barlow CA, Shukla A, Mossman BT, Lounsbury KM. Oxidant-mediated cAMP response element binding protein activation: calcium regulation and role in apoptosis of lung epithelial cells. Am J Respir Cell Mol Biol. 2006;34(1):7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DebRoy A, Vogel SM, Soni D, Sundivakkam PC, Malik AB, Tiruppathi C. Cooperative signaling via transcription factors NF-kappaB and AP1/c-Fos mediates endothelial cell STIM1 expression and hyperpermeability in response to endotoxin. J Biol Chem. 2014;289(35):24188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Held HD, Boettcher S, Hamann L, Uhlig S. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med. 2001;163(3 Pt 1):711–6. [DOI] [PubMed] [Google Scholar]
- 76.Talmor D, Sarge T, Malhotra A, O’Donnell CR, Ritz R, Lisbon A, et al. Mechanical ventilation guided by esophageal pressure in acute lung injury. N Engl J Med. 2008;359(20):2095–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quesnel C, Nardelli L, Piednoir P, Lecon V, Marchal-Somme J, Lasocki S, et al. Alveolar fibroblasts in acute lung injury: biological behaviour and clinical relevance. Eur Respir J. 2010;35(6):1312–21. [DOI] [PubMed] [Google Scholar]
- 78.Briassoulis G, Papassotiriou I, Mavrikiou M, Lazaropoulou C, Margeli A. Longitudinal course and clinical significance of TGF-beta1, sL- and sE-Selectins and sICAM-1 levels during severe acute stress in children. Clin Biochem. 2007;40(5–6):299–304. [DOI] [PubMed] [Google Scholar]
- 79.Frank JA, Wray CM, McAuley DF, Schwendener R, Matthay MA. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291(6):L1191–8. [DOI] [PubMed] [Google Scholar]
- 80.Peters AL, Van Stein D, Vlaar AP. Antibody-mediated transfusion-related acute lung injury; from discovery to prevention. Br J Haematol. 2015;170(5):597–614. [DOI] [PubMed] [Google Scholar]
- 81.Greinacher A, Wesche J, Hammer E, Furll B, Volker U, Reil A, et al. Characterization of the human neutrophil alloantigen-3a. Nat Med. 2010;16(1):45–8. [DOI] [PubMed] [Google Scholar]
- 82.Qing DY, Conegliano D, Shashaty MG, Seo J, Reilly JP, Worthen GS, et al. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am J Respir Crit Care Med. 2014;190(11):1243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li G, Kojicic M, Reriani MK, Fernandez Perez ER, Thakur L, Kashyap R, et al. Long-term survival and quality of life after transfusion-associated pulmonary edema in critically ill medical patients. Chest. 2010;137(4):783–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Del Moral PM, Warburton D. Explant culture of mouse embryonic whole lung, isolated epithelium, or mesenchyme under chemically defined conditions as a system to evaluate the molecular mechanism of branching morphogenesis and cellular differentiation. Methods Mol Biol. 2010;633:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baker CS, Evans TW, Randle BJ, Haslam PL. Damage to surfactant-specific protein in acute respiratory distress syndrome. Lancet. 1999;353(9160):1232–7. [DOI] [PubMed] [Google Scholar]
- 86.Overgaard CE, Schlingmann B, Dorsainvil White S, Ward C, Fan X, Swarnakar S, et al. The relative balance of GM-CSF and TGF-beta1 regulates lung epithelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2015;308(12):L1212–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ward C, Schlingmann B, Stecenko AA, Guidot DM, Koval M. NF-kappaB inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers. 2015;3(1–2):e982424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sugamata R, Dobashi H, Nagao T, Yamamoto K, Nakajima N, Sato Y, et al. Contribution of neutrophil-derived myeloperoxidase in the early phase of fulminant acute respiratory distress syndrome induced by influenza virus infection. Microbiol Immunol. 2012;56(3):171–82. [DOI] [PubMed] [Google Scholar]
- 89.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163(6):1376–83. [DOI] [PubMed] [Google Scholar]
- 90.Antonov A, Snead C, Gorshkov B, Antonova GN, Verin AD, Catravas JD. Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am J Respir Cell Mol Biol. 2008;39(5):551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, et al. Integrin alpha6beta4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest. 2011;121(7):2855–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park KS, Wells JM, Zorn AM, Wert SE, Laubach VE, Fernandez LG, et al. Transdifferentiation of ciliated cells during repair of the respiratory epithelium. Am J Respir Cell Mol Biol. 2006;34(2):151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tompkins DH, Besnard V, Lange AW, Wert SE, Keiser AR, Smith AN, et al. Sox2 is required for maintenance and differentiation of bronchiolar Clara, ciliated, and goblet cells. PLoS One. 2009;4(12):e8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sever-Chroneos Z, Krupa A, Davis J, Hasan M, Yang CH, Szeliga J, et al. Surfactant protein A (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J Biol Chem. 2011;286(6):4854–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hartshorn KL, Crouch E, White MR, Colamussi ML, Kakkanatt A, Tauber B, et al. Pulmonary surfactant proteins A and D enhance neutrophil uptake of bacteria. Am J Physiol. 1998;274(6 Pt 1):L958–69. [DOI] [PubMed] [Google Scholar]
- 96.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16(1):27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li Y, Huang J, Foley NM, Xu Y, Li YP, Pan J, et al. B7H3 ameliorates LPS-induced acute lung injury via attenuation of neutrophil migration and infiltration. Sci Rep. 2016;6:31284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rossaint J, Kuhne K, Skupski J, Van Aken H, Looney MR, Hidalgo A, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun. 2016;7:13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaynar AM, Houghton AM, Lum EH, Pitt BR, Shapiro SD. Neutrophil elastase is needed for neutrophil emigration into lungs in ventilator-induced lung injury. Am J Respir Cell Mol Biol. 2008;39(1):53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Matuschak GM, Munoz CF, Johanns CA, Rahman R, Lechner AJ. Upregulation of postbacteremic TNF-alpha and IL-1alpha gene expression by alveolar hypoxia/reoxygenation in perfused rat lungs. Am J Respir Crit Care Med. 1998;157(2):629–37. [DOI] [PubMed] [Google Scholar]
- 101.Lomas-Neira J, Venet F, Chung CS, Thakkar R, Heffernan D, Ayala A. Neutrophil-endothelial interactions mediate angiopoietin-2-associated pulmonary endothelial cell dysfunction in indirect acute lung injury in mice. Am J Respir Cell Mol Biol. 2014;50(1):193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. 2011;179(1):199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Herwig MC, Tsokos M, Hermanns MI, Kirkpatrick CJ, Muller AM. Vascular endothelial cadherin expression in lung specimens of patients with sepsis-induced acute respiratory distress syndrome and endothelial cell cultures. Pathobiology. 2013;80(5):245–51. [DOI] [PubMed] [Google Scholar]
- 104.Shah D, Romero F, Duong M, Wang N, Paudyal B, Suratt BT, et al. Obesity-induced adipokine imbalance impairs mouse pulmonary vascular endothelial function and primes the lung for injury. Sci Rep. 2015;5:11362. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 105.Adam AP, Sharenko AL, Pumiglia K, Vincent PA. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem. 2010;285(10):7045–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H, et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep. 2016;6:37252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Arndt PG, Young SK, Worthen GS. Regulation of lipopolysaccharide-induced lung inflammation by plasminogen activator Inhibitor-1 through a JNK-mediated pathway. J Immunol. 2005;175(6):4049–59. [DOI] [PubMed] [Google Scholar]
- 108.Mulligan MS, Polley MJ, Bayer RJ, Nunn MF, Paulson JC, Ward PA. Neutrophil-dependent acute lung injury. Requirement for P-selectin (GMP-140). J Clin Invest. 1992;90(4):1600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peteranderl C, Morales-Nebreda L, Selvakumar B, Lecuona E, Vadasz I, Morty RE, et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J Clin Invest. 2016;126(4):1566–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, et al. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am J Respir Crit Care Med. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Morales MM, Pires-Neto RC, Inforsato N, Lancas T, da Silva LF, Saldiva PH, et al. Small airway remodeling in acute respiratory distress syndrome: a study in autopsy lung tissue. Crit Care. 2011;15(1):R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Noble PW, Jiang D. Matrix regulation of lung injury, inflammation, and repair: the role of innate immunity. Proc Am Thorac Soc. 2006;3(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11(11):1173–9. [DOI] [PubMed] [Google Scholar]
- 114.Martin C, Papazian L, Payan MJ, Saux P, Gouin F. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest. 1995;107(1):196–200. [DOI] [PubMed] [Google Scholar]
- 115.Madtes DK, Rubenfeld G, Klima LD, Milberg JA, Steinberg KP, Martin TR, et al. Elevated transforming growth factor-alpha levels in bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 1998;158(2):424–30. [DOI] [PubMed] [Google Scholar]
- 116.Yamawaki H, Hirohata S, Miyoshi T, Takahashi K, Ogawa H, Shinohata R, et al. Hyaluronan receptors involved in cytokine induction in monocytes. Glycobiology. 2009;19(1):83–92. [DOI] [PubMed] [Google Scholar]
- 117.Lepidi S, Kenagy RD, Raines EW, Chiu ES, Chait A, Ross R, et al. MMP9 production by human monocyte-derived macrophages is decreased on polymerized type I collagen. J Vasc Surg. 2001;34(6):1111–8. [DOI] [PubMed] [Google Scholar]
- 118.Shapiro SD, Campbell EJ, Welgus HG, Senior RM. Elastin degradation by mononuclear phagocytes. Ann N Y Acad Sci. 1991;624:69–80. [DOI] [PubMed] [Google Scholar]
- 119.Rella JM, Jilma B, Fabry A, Kaynar AM, Mayr FB. MMP-8 genotypes influence the inflammatory response in human endotoxemia. Inflammation. 2014;37(2):451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hsu AT, Barrett CD, DeBusk GM, Ellson CD, Gautam S, Talmor DS, et al. Kinetics and Role of Plasma Matrix Metalloproteinase-9 Expression in Acute Lung Injury and the Acute Respiratory Distress Syndrome. Shock. 2015;44(2):128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA. Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol. 2010;184(3):1575–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, et al. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286(5437):113–7. [DOI] [PubMed] [Google Scholar]
- 123.Yildiz C, Palaniyar N, Otulakowski G, Khan MA, Post M, Kuebler WM, et al. Mechanical ventilation induces neutrophil extracellular trap formation. Anesthesiology. 2015;122(4):864–75. [DOI] [PubMed] [Google Scholar]
- 124.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. [DOI] [PubMed] [Google Scholar]
- 125.Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Koh Y, Hybertson BM, Jepson EK, Repine JE. Tumor necrosis factor induced acute lung leak in rats: less than with interleukin-1. Inflammation. 1996;20(5):461–9. [DOI] [PubMed] [Google Scholar]
- 128.Jacobs RF, Tabor DR, Burks AW, Campbell GD. Elevated interleukin-1 release by human alveolar macrophages during the adult respiratory distress syndrome. Am Rev Respir Dis. 1989;140(6):1686–92. [DOI] [PubMed] [Google Scholar]
- 129.Makabe H, Kojika M, Takahashi G, Matsumoto N, Shibata S, Suzuki Y, et al. Interleukin-18 levels reflect the long-term prognosis of acute lung injury and acute respiratory distress syndrome. J Anesth. 2012;26(5):658–63. [DOI] [PubMed] [Google Scholar]
- 130.Eickmeier O, Seki H, Haworth O, Hilberath JN, Gao F, Uddin M, et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol. 2013;6(2):256–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ghio AJ, Carter JD, Richards JH, Richer LD, Grissom CK, Elstad MR. Iron and iron-related proteins in the lower respiratory tract of patients with acute respiratory distress syndrome. Crit Care Med. 2003;31(2):395–400. [DOI] [PubMed] [Google Scholar]
- 132.Lamb NJ, Gutteridge JM, Baker C, Evans TW, Quinlan GJ. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med. 1999;27(9):1738–44. [DOI] [PubMed] [Google Scholar]
- 133.Lee SM, McLaughlin JN, Frederick DR, Zhu L, Thambiayya K, Wasserloos KJ, et al. Metallothionein-induced zinc partitioning exacerbates hyperoxic acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.O’Reilly MA, Staversky RJ, Stripp BR, Finkelstein JN. Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am J Respir Cell Mol Biol. 1998;18(1):43–50. [DOI] [PubMed] [Google Scholar]
- 135.O’Reilly MA, Staversky RJ, Watkins RH, Maniscalco WM. Accumulation of p21(Cip1/WAF1) during hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 1998;19(5):777–85. [DOI] [PubMed] [Google Scholar]
- 136.Staversky RJ, Watkins RH, Wright TW, Hernady E, LoMonaco MB, D’Angio CT, et al. Normal remodeling of the oxygen-injured lung requires the cyclin-dependent kinase inhibitor p21(Cip1/WAF1/Sdi1). Am J Pathol. 2002;161(4):1383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Murray LA, Knight DA, McAlonan L, Argentieri R, Joshi A, Shaheen F, et al. Deleterious role of TLR3 during hyperoxia-induced acute lung injury. Am J Respir Crit Care Med. 2008;178(12):1227–37. [DOI] [PubMed] [Google Scholar]
- 138.Ogawa Y, Tasaka S, Yamada W, Saito F, Hasegawa N, Miyasho T, et al. Role of Toll-like receptor 4 in hyperoxia-induced lung inflammation in mice. Inflamm Res. 2007;56(8):334–8. [DOI] [PubMed] [Google Scholar]
- 139.Johnson KJ, Fantone JC 3rd, Kaplan J, Ward PA. In vivo damage of rat lungs by oxygen metabolites. J Clin Invest. 1981;67(4):983–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Patel JD, Krupka T, Anderson JM. iNOS-mediated generation of reactive oxygen and nitrogen species by biomaterial-adherent neutrophils. J Biomed Mater Res A. 2007;80(2):381–90. [DOI] [PubMed] [Google Scholar]
- 141.Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang BH, et al. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol. 2008;180(5):3072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lee I, Dodia C, Chatterjee S, Feinstein SI, Fisher AB. Protection against LPS-induced acute lung injury by a mechanism-based inhibitor of NADPH oxidase (type 2). Am J Physiol Lung Cell Mol Physiol. 2014;306(7):L635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bjornsdottir H, Welin A, Michaelsson E, Osla V, Berg S, Christenson K, et al. Neutrophil NET formation is regulated from the inside by myeloperoxidase-processed reactive oxygen species. Free Radic Biol Med. 2015;89:1024–35. [DOI] [PubMed] [Google Scholar]
- 145.Bastarache JA, Wang L, Geiser T, Wang Z, Albertine KH, Matthay MA, et al. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007;62(7):608–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gando S, Kameue T, Matsuda N, Hayakawa M, Morimoto Y, Ishitani T, et al. Imbalances between the levels of tissue factor and tissue factor pathway inhibitor in ARDS patients. Thromb Res. 2003;109(2–3):119–24. [DOI] [PubMed] [Google Scholar]
- 147.Carvalho AC, DeMarinis S, Scott CF, Silver LD, Schmaier AH, Colman RW. Activation of the contact system of plasma proteolysis in the adult respiratory distress syndrome. J Lab Clin Med. 1988;112(2):270–7. [PubMed] [Google Scholar]
- 148.Deng X, Jin K, Li Y, Gu W, Liu M, Zhou L. Platelet-Derived Growth Factor and Transforming Growth Factor beta1 Regulate ARDS-Associated Lung Fibrosis Through Distinct Signaling Pathways. Cell Physiol Biochem. 2015;36(3):937–46. [DOI] [PubMed] [Google Scholar]
- 149.McClendon J, Jansing NL, Redente EF, Gandjeva A, Ito Y, Colgan SP, et al. Hypoxia-Inducible Factor 1alpha Signaling Promotes Repair of the Alveolar Epithelium after Acute Lung Injury. Am J Pathol. 2017;187(8):1772–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zeng H, He X, Tuo QH, Liao DF, Zhang GQ, Chen JX. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2alpha/Notch3 pathways. Sci Rep. 2016;6:20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wesselkamper SC, Case LM, Henning LN, Borchers MT, Tichelaar JW, Mason JM, et al. Gene expression changes during the development of acute lung injury: role of transforming growth factor beta. Am J Respir Crit Care Med. 2005;172(11):1399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Andre PA, Prele CM, Vierkotten S, Carnesecchi S, Donati Y, Chambers RC, et al. BARD1 mediates TGF-beta signaling in pulmonary fibrosis. Respir Res. 2015;16:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dackor RT, Cheng J, Voltz JW, Card JW, Ferguson CD, Garrett RC, et al. Prostaglandin E(2) protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am J Physiol Lung Cell Mol Physiol. 2011;301(5):L645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Finley N, Norlin A, Baines DL, Folkesson HG. Alveolar epithelial fluid clearance is mediated by endogenous catecholamines at birth in guinea pigs. J Clin Invest. 1998;101(5):972–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bhargava M, Runyon MR, Smirnov D, Lei J, Groppoli TJ, Mariash CN, et al. Triiodo-L-thyronine rapidly stimulates alveolar fluid clearance in normal and hyperoxia-injured lungs. Am J Respir Crit Care Med. 2008;178(5):506–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Factor P, Dumasius V, Saldias F, Brown LA, Sznajder JI. Adenovirus-mediated transfer of an Na+/K+-ATPase beta1 subunit gene improves alveolar fluid clearance and survival in hyperoxic rats. Hum Gene Ther. 2000;11(16):2231–42. [DOI] [PubMed] [Google Scholar]
- 157.Solymosi EA, Kaestle-Gembardt SM, Vadasz I, Wang L, Neye N, Chupin CJ, et al. Chloride transport-driven alveolar fluid secretion is a major contributor to cardiogenic lung edema. Proc Natl Acad Sci U S A. 2013;110(25):E2308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011;364(14):1293–304. [DOI] [PubMed] [Google Scholar]