

Abstract
Podocyte injury is a critical step toward the progression of renal disease and is often associated with a loss of slit diaphragm proteins, including Podocin. Although there is a possibility that the extracellular domain of these slit diaphragm proteins can be a target for a pathological proteolysis, the precise mechanism driving the phenomenon remains unknown. Here we show that Matriptase, a membrane-anchored protein, was activated at podocytes in CKD patients and mice, whereas Matriptase inhibitors slowed the progression of mouse kidney disease. The mechanism could be accounted for by an imbalance favoring Matriptase over its cognate inhibitor, hepatocyte growth factor activator inhibitor type 1 (HAI-1), because conditional depletion of HAI-1 in podocytes accelerated podocyte injury in mouse model. Matriptase was capable of cleaving Podocin, but such a reaction was blocked by either HAI-1 or dominant-negative Matriptase. Furthermore, the N terminus of Podocin, as a consequence of Matriptase cleavage of Podocin, translocated to nucleoli, suggesting that the N terminus of Podocin might be involved in the process of podocyte injury. Given these observations, we propose that the proteolytic cleavage of Podocin by Matriptase could potentially cause podocyte injury and that targeting Matriptase could be a novel therapeutic strategy for CKD patients.
Keywords: chronic kidney disease, Matriptase, hepatocyte growth factor activator inhibitor type 1, Podocin, podocyte, kidney, podocyte, protease, protease inhibitor, diabetic nephropathy
Chronic kidney disease (CKD) has come to be recognized as a global public health problem (1). An increasing number of CKD patients end up suffering from end-stage renal disease, requiring renal hemodialysis, and loading huge financial burden on the shoulder of their home countries. Developing specific treatments for curing CKD are warranted.
The glomerular filtration unit consists of three layers: fenestrated endothelial cells, the glomerular basement membrane, and podocytes (2). Podocytes, a major cell type of renal glomerulus, are terminally differentiated and interdigitate with adjacent podocytes to form glomerular filtration barrier (3). Interdigitated foot processes of podocytes form a 40-nm-wide cell junction that is composed of several proteins called slit diaphragm (SD) proteins. Podocyte injury is often associated with a loss of SD proteins, and the unique characteristic of several SD proteins with the extracellular domain might be a key to understand podocyte injury. However, the precise mechanism underlying this phenomenon remains unclear.
Proteolysis is a physiological process that is required for the maintenance of renal physiology at multiple levels, including gene transcription, cell trafficking, and extracellular secretion. Aberrant activation of proteolysis is likely involved in the pathogenesis of renal disease. In particular, a number of protease families likely play a key role for the development of renal fibrosis, including, matrix metalloproteinases (MMPs), serine proteases, and cathepsins (4, 5). Accumulating evidence demonstrates that intracellular proteolytic processing is required to maintain physiological function in podocytes, whereas aberrant activation causes cellular injury (6). Podocyte injury is often initiated by SD disruption, but its precise mechanism has yet to be clarified. Recently, it was shown that a nonspecific serine protease inhibitor, camostat mesilate, slowed the progression of renal disease caused by the protection of podocytes (7), suggesting that serine proteases should be involved. Nonetheless, the specific type of enzyme involved in this process also remains unknown.
Results
We established a model of adriamycin (ADR) nephropathy in mice developing albuminuria and podocyte injury with a reduction in mRNA expressions for several markers of podocytes in mouse glomerulus, including Podocin, Synaptopodin, and WT-1 (Fig. S1, A–D). Podocyte injury was also histologically confirmed by periodic acid–Schiff reagent staining and immunohistochemistry. Likewise, desmin, a marker of podocyte injury, was up-regulated along with glomerular injury (data not shown). Interestingly, the injured podocytes still expressed, to some extent, both Nephrin and Podocin in this model (Fig. S1E). A microarray analysis using glomeruli of ADR nephropathy mice identified 15 serine proteases, which were highly up-regulated (>2.0-fold) in glomeruli of mice with ADR nephropathy. This is compared with control mice at 6 weeks after administration (Fig. 1A and Table S1). Among these factors, only a type II transmembrane serine protease Matriptase (also known as suppressor of tumorigenicity 14 protein, or ST14) is expressed at the epithelial cell junctions in a wide variety of tissues (8). We examined the expressions of other type II transmembrane serine protease and found that Matriptase was induced at mRNA and protein levels in glomeruli of ADR mice at day 42 (Figs. 1, B and C, and 2, A–C). In particular, Matriptase was predominantly expressed in podocytes (Fig. 2C). Likewise, Matriptase expression was only detected in cultured podocytes but not in cultured endothelium and mesangial cells (data not shown). Glomerular Matriptase protein was also significantly higher in patients with clinical proteinuria because of diabetic nephropathy or membranous nephropathy but not in subjects with IgA nephropathy who had no clinical proteinuria (Fig. 2D).
Figure 1.

Microarray analysis. A, microarray was performed with glomerular RNA of control mice at 2 weeks (n = 3) and at 6 weeks (n = 3) and ADR nephropathy mice at 2 weeks (n = 3) and at 6 weeks (n = 3). Statistical significance (p < 0.05) was calculated using moderated Student's t test followed by the Benjamini–Hochberg false discovery rate correction on GeneSpring GX. Fold change cutoff of >2 led to extraction of 357 probes in ADR nephropathy mice at 6 weeks compared with control mice. Among those genes, 15 genes were categorized as serine protease, whereas only Matriptase is a type II membrane-anchored serine protease. The heat map shows that all serine proteases were up-regulated at 42 days in glomerulus of mice with ADR nephropathy. B, among seven probes, which are categorized as membrane-anchored serine proteinase and its cognate inhibitors, microarray analysis shows that only Matriptase and HAI-1 were significantly up-regulated in ADR-nephropathy mice compared with control mice (B–D). Hgfac, hepatocyte growth factor activator; Hpn, hepsin; Prss8, serine protease 8.
Figure 2.
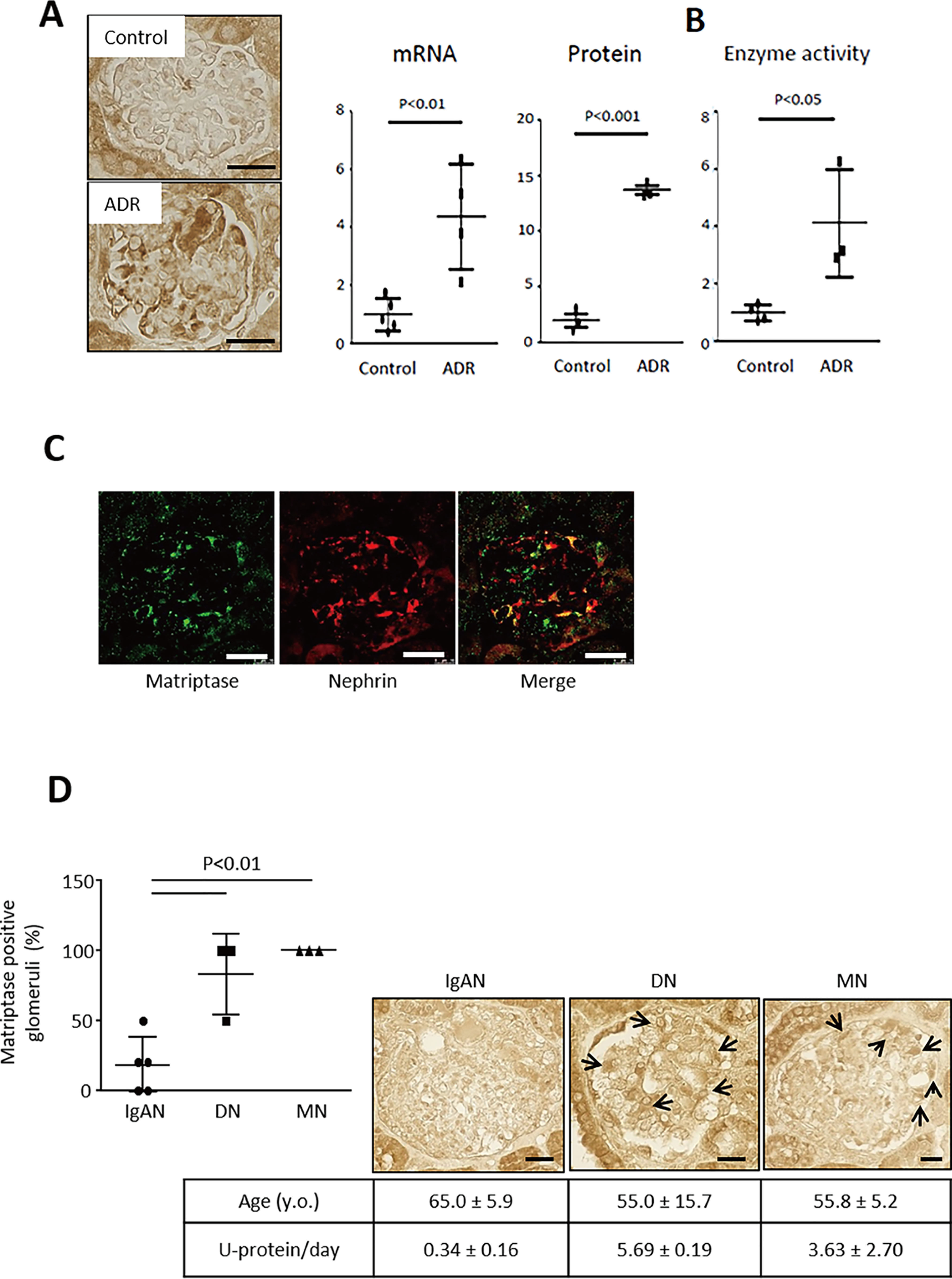
Matriptase is induced in podocytes of mice with ADR nephropathy and patients with clinical proteinuria. A, immunohistochemistry showed an increase in glomerular Matriptase protein expression. RT–quantitative PCR showed a significant increase in mRNA expression of glomerular Matriptase and quantitative analysis using immunohistochemistry confirmed the protein expression in mice with ADR nephropathy compared with control mice. B, glomerular serine protease activity was enhanced in ADR mice compared with control mice. C, Matriptase (green) was co-localized (yellow) with Nephrin (red) in glomerulus of mouse with ADR nephropathy. D, Matriptase protein expression was observed in human glomerulus of diabetic nephropathy (DN) and membranous nephropathy (MN), whereas it was negative in that of IgA nephropathy (IgAN). Bar, 20 μm.
Matriptase is tightly regulated by its cognate inhibitor HAI-1 (also known as SPINT1), a Kunitz-type serine protease inhibitor (9). HAI-1 mRNA expression was significantly induced in the glomeruli of ADR mice compared with that of WT mice (Fig. 1, B and D). Subsequently another type of mouse model with podocyte injury (10, 11), streptozotocin-induced diabetic endothelial nitric-oxide synthase knockout (diabetic eNOS-KO) mice, was examined using a microarray analysis by isolated glomeruli (Table S2). Among 13 candidate genes (Fig. S2A), HAI-1 level was significantly up-regulated in the podocytes (confirmed by immunohistochemistry) and in the serum of diabetic eNOS-KO mice (Fig. 3, A–C, and Fig. S2B). A microarray analysis showed that glomerular Matriptase mRNA expression was highly up-regulated by 5.3 times in diabetic eNOS-KO mice compared with eNOS-KO mice (Fig. S2, C and D). Analyses of these two independent mouse models suggest that the Matriptase/HAI-1 pathway plays a key role in podocyte injury and could therefore be a potential therapeutic target for kidney disease.
Figure 3.

Role of HAI-1 in diabetic eNOS-KO mice and mice with ADR nephropathy. A, the number of HAI-1 positive cells (arrows) increased in glomeruli of streptozotocin–induced diabetic mice and eNOS-KO mice, and further, it was higher in diabetic eNOS-KO mice than in any other groups. Bar, 20 μm. B, HAI-1 partially co-localized with Podocin. Bar, 20 μm. C, HAI-1 level in serum was also higher in diabetic (DM) WT and diabetic eNOS-KO mice compared with nondiabetic (NDM) WT and NDM eNOS-KO mice. D, 42 days after 10.5 mg/kg ADR was injected into HAI-1 wt/wt: Podocin-Cre (+) mice (n = 5), HAI-1 flox/flox: Podocin-Cre (−) mice (n = 8), and HAI-1 flox/flox: Podocin-Cre (+) mice (n = 6), urinary albumin/creatinine ratio (ACR) was examined. E, compared with foot process (white arrows) in HAI-1 wt/wt: Podocin-Cre (+) mice, the effacement (red arrows) of podocytes in HAI-1 flox/flox: Podocin-Cre (+) injected with ADR was observed under transmission electron microscopy. Bar, 500 nm. CL, capillary lumen; US, urinary space.
Matriptase and HAI-1 are co-expressed in many epithelial cells (12), and the regulation of Matriptase by HAI-1 is required for epidermal integrity (13). In turn, an imbalance favoring Matriptase over HAI-1 contributes to various diseases (14–17). According to the Nephroseq database, HAI-1 is induced (1.5-fold), along with 2-fold increase in Matriptase expression, in the podocytes of diabetic nephropathy. This indicates that enhancing Matriptase activation over HAI-1 could be a potential mechanism for the progression of podocyte injury. Consistent with these observations, the podocyte-specific depletion of HAI-1 deteriorated podocyte injury in ADR nephropathy (Fig. 3, D and E, and Fig. S3, A–C). Podocyte injury could be accounted for by aberrant Matriptase activation caused by the absence of HAI-1 in the podocyte.
To develop a therapeutic approach to nephropathy, we subsequently examined whether the selective inhibition of Matriptase could protect kidneys from disease progression. We examined two types of serine protease inhibitors toward Matriptase activity: a selective peptide-mimetic inhibitor of Matriptase (IN-1) (18) and a synthetic nonspecific serine protease inhibitor nafamostat mesilate (NM), which ameliorates rat kidney disease (7). It was found that the two compounds inhibited Matriptase activity in a dose-response manner, and inhibitory effects were nearly equipotent, with an IC50 of IN-1 of 1.3 nm versus the IC50 of NM at 0.86 nm (Fig. 4A). However, inhibition assays toward thrombin activity showed that IC50 was 2.8 μm for IN-1, whereas it was 99 nm for NM (Fig. 4B), suggesting that IN-1 is more specific to Matriptase than NM. To examine the therapeutic effect of IN-1, this compound was applied to BALB/c mice with ADR nephropathy upon 15 days after ADR treatment. We observed that Matriptase inhibitor IN-1 slowed the progression of ADR nephropathy with blocking podocyte injury and ameliorated albuminuria (Fig. 4, C–F). Likewise, NM-1 also blocked the glomerular injury and suppressed urinary albumin excretion in mice with ADR nephropathy (Fig. S4). These results indicate that blocking Matriptase could be a potential therapeutic approach against chronic kidney diseases.
Figure 4.

Pharmacological inhibition of Matriptase slows the progression of ADR nephropathy in mice. A, IC50 of IN-1 for Matriptase activity is 1.3 nm in a protease activity assay, whereas that of NM was 0.86 nm. B, IC50 of IN-1 for thrombin activity was 2.8 μm, whereas that of NM was 99 nm. C, urinary albumin/creatinine ratio (ACR) was higher in mice with ADR nephropathy, but it was significantly suppressed by the injection of 10 mg/kg IN-1 three times a week as measured on days 10, 28 and 42. D, glomerular sclerosis was higher in mice with ADR nephropathy compared with normal control mice. However, it was significantly reduced by IN-1 treatment on day 42. E and F, Podocin-positive area and the number of WT positive cells in glomerulus were significantly lower by ADR treatment, but such a detrimental effect of ADR was slightly but significantly ameliorated by the chronic treatment with IN-1 on day 42.
Next, we sought to identify the substrates of Matriptase in podocytes. Interestingly, Matriptase was found to directly cleave mouse Podocin (mPodocin) (Fig. S5, A and I) but not with other slit membrane proteins, such as Claudin5, β-Catenin, or Nephrin (Fig. S5, B–D). Fig. 5A shows that enzymatic dead G827R Matriptase mutant failed to cleave mPodocin, and multiple bands in anti-His blot indicated that Matriptase was activated by self-cleavage at multiple sites. The mPodocin was cleaved at the membrane but not in the cytoplasm (Fig. 5B). A mutagenesis on mPodocin identified Arg50 as the specific cleavage site of Matriptase and not at Arg36, Arg45, or Arg54 (Fig. 5C and Fig. S5, H and I). Matriptase cleavage of mPodocin was blocked by co-expression of WT, as well as the extracellular domain of HAI-1 (Fig. 5D). We also identified the fragmented cleaved mPodocin in the urine of mice with ADR nephropathy (Fig. S5E), suggesting that Matriptase cleaves Podocin in in vivo.
Figure 5.

Matriptase cleaves Podocin at Arg50, which is blocked by HAI-1. A, although WT of Matriptase cleaved Podocin and produces its small fragment, the inactive form of Matriptase (G827R) failed to do it in HEK293 cells overexpressing mPodocin-FLAG and either WT-Matriptase or G827R Matriptase mutant. B, Matriptase-induced Podocin cleavage occurred at the membrane fraction, but not in the cytoplasm of HEK293 cells overexpressing Matriptase-His and mPodocin-FLAG. C, Matriptase cleavage of Podocin R50A mutant, as opposed to WT, R45A, and R54A mutants, was declined using in vitro cleavage assay. D, full-length of HAI-1 or secreted type of HAI-1 (deleted transmembrane region (dTM)) blocked the cleavage of Podocin in response to Matriptase in HEK293 cells. E, red signal indicates location of FLAG-tagged Podocin. Podocin-N (amino acids 2–50) was localized in the nucleus, whereas full-length Podocin and ΔN-Podocin (amino acids 51–385) remained in cytosol.
Exogenous expression of mPodocin N-terminal fragment (mPodocin-N; 2–50 amino acids) showed the mPodocin-N as being localized in the nucleus, whereas full-length mPodocin and △N-mPodocin (51–385 amino acids) remained at the plasma membrane (Fig. 5E). Human Podocin (hPodocin) was also cleaved by Matriptase (Fig. S5G). N-terminal fragment of hPodocin (hPodocin-N) appears to be co-localized with Fibrillarin, a marker of nucleoli, suggesting that hPodocin-N migrated to the nucleus and bound nucleoli (Fig. S6B).
Discussion
Podocytes have been found to produce other types of proteases, matrix metalloproteases MMP-2 and -9, all of which are believed to have significant roles within the glomerulus because they degrade type IV collagen, a major protein of the glomerular basement membrane (19). It has also been observed that altered MMPs/tissue inhibitors of MMPs (TIMPs) balance leads to increased extracellular matrix deposition or excessive degradation activity, a finding with critical implications for glomerular diseases (2). The up-regulation of MMP-9 expression can be found in several glomerular diseases accompanied by proteinuria in human patients (20, 21) and diabetic nephropathy murine model (22). Podocytes express CD40 and CD154, the ligand of CD40, to induce MMP-9 expression in an autocrine fashion (23). In various diseases, coagulation proteases also contribute to tissue injuries, including cancer progression and cardiovascular diseases. These injuries are mediated by protease-activated receptors (PARs) (24, 25), a family of G protein–coupled receptors consisting of four members (PAR1–PAR4). Consistent with findings from an earlier study suggesting that PARs play a role in kidney disease progression (26), PAR-1 has indeed been found to contribute to the development of podocyte and glomerular injuries (27).
It is important to realize that individual proteases induce divergent signaling pathways that lead to differential functional consequences (28). These studies imply that the strategy to target all the proteases via nonspecific protease inhibitor may not always be beneficial; the targeting of specific protease(s) by specific inhibitor(s) may be a better approach. Although proteases play key roles in the maintenance of renal filtration barriers, and there are indications to suggest that serine protease inhibition is involved in kidney disease progression (7), the precise mechanism(s) underlying the progression of kidney disease, in which serine proteases are involved, as well as the target serine protease(s), remain unknown and to be identified.
In this study, we showed that HAI-1 was essential to the protection of podocytes from nephropathy by blocking of Matriptase-mediated cleavage of Podocin. We also demonstrated that a transmembrane serine protease Matriptase acted as a potential therapeutic target, findings that imply a new class of selective peptide-mimetic inhibitor of Matriptase can be applied as a potential drug for the treatment of chronic kidney diseases. Although careful preclinical studies are required before applying the Matriptase inhibitor in clinical practice, our finding that NM, which is commonly used for the treatment of pancreatitis and disseminated intravascular coagulation (29), also protects the kidney from disease progression might give some reassurance that the inhibition of Matriptase is a safe and promising approach.
In conclusion, we identified Matriptase as a potential cause for podocyte injury. The aberrantly activated enzyme beyond the suppression by HAI-1 potently cleaves Podocin and disturbs podocyte integrity, leading to renal injury. Most surprisingly, Podocin-N migrates to the nucleus and binds nucleoli, indicating its roles in physiological and active cell death process.
Experimental procedures
Animal study
All animal experiments were performed in accordance with either the Animal Experimentation Committee of Kyoto University, Tanabe R&D Service Co., Ltd. (Osaka, Japan), or both. The protocol was approved by the animal care and use committee. Male C57BL/6J-Nos3tm1nc (eNOS-KO) mice at 7 weeks of age were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The mice with diabetic nephropathy were developed as described previously (10). Diabetes was induced with intraperitoneal injection of 50 mg/kg/day streptozotocin for 5 consecutive days. Diabetes was defined as nonfasting blood glucose of >250 mg/dl using a blood glucose meter. The mice were fed a standard laboratory chow ad libitum. Mean blood pressure was measured using the CODA multichannel, computerized, noninvasive blood pressure system (Kent Scientific Corporation, Torrington, CT, USA), whereas the blood glucose level was determined by GLUCOCARD MyDIA (Arkray, Edina, MN, USA). Urine was collected overnight using metabolic cages. At 14 weeks of age, all mice were sacrificed.
For ADR nephropathy model, ADR at a dose of 10.5 mg/kg body weight was injected via the tail vein of male BALB/c mice at 8 weeks of age. Matriptase inhibitor (IN-1) at a dose of 10 mg/kg body weight was intraperitoneally injected to mice with ADR nephropathy three times a week for 4 weeks. Urine was collected overnight using metabolic cages. At 14 weeks of age, all mice were sacrificed (30). Matriptase inhibitor (IN-1) was synthesized (WuXi AppTec Co., Ltd., Shanghai, China), and the purity of this compound was 97.23% as previously described (18). HAI-1 flox/flox mice were kindly provided from Dr. Hiroaki Kataoka (31). Establishment of Podocin-CreERT2 (Podocin-Cre (+)) mice was reported previously (32). HAI-1 flox/flox:Podocin-Cre (+) mice were produced by crossing HAI-1 flox/flox mice and Podocin-Cre mice. The primers for genotyping Podocin-Cre were TTTGCCTGCATTACCGGTCGATGCAAC and TGCCCCTGTTTCACTATCCAGGTTACGGA. The primers used for genotyping HAI-1 flox were ACCACTGGCTCATTTGGTGTTGGC and TGAAGCCTGGCCACTTCCTGATG. To induce functional Cre protein, 150 mg/kg tamoxifen was injected intraperitoneally for 3 consecutive days at the age of 8 weeks. ADR at a dose of 10.5 mg/kg body weight was injected into the tail veins of male mice at 10 weeks of age. At 16 weeks of age, all mice were sacrificed.
Molecular analysis
Urinary albumin concentration was measured using Albuwell M (Exocell, Philadelphia, PA, USA) or microfluoral microalbumin test (Progen, Heidelberg, Germany). Urine creatinine was measured using LabAssay creatinine (Wako, Osaka, Japan). Serum HAI-1 concentration was measured using mouse HAI-1 ELISA kit (R&D Systems, Minneapolis, MN, USA).
Isolation of glomeruli
The mice were perfused with 8 × 107 Dynabeads (Life Technologies, Inc.) diluted in 40 ml of Hanks' balanced salt solution through the heart under anesthesia. The kidneys were isolated and digested in collagenase solution (1 mg/ml collagenase A and 100 units/ml DNase I) for 30 min at 37 °C, and then glomeruli containing Dynabeads were gathered by a magnetic particle concentrator. During the procedure, the kidney tissues were kept at 4 °C except for the collagenase digestion. Finally, glomerular RNA was purified with RNeasy mini kit (Qiagen). For measuring protease activity in glomeruli, the glomeruli were lysed with RIPA buffer (Santa Cruz Biotechnology Inc., Dallas, TX, USA).
Microarray analysis
Glomerular RNA was extracted and purified with RNeasy mini kit (Qiagen). RNA quality was assessed with Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). In accordance with the Agilent Technologies protocol, all samples were processed and hybridized to SurePrint G3 mouse gene expression 8 × 60 K (Agilent Technologies). Fluorescence was detected using an Agilent DNA microarray scanner. The data were analyzed with GeneSpring GX (Agilent Technologies).
Histological analysis
Formalin-fixed, paraffin-embedded sections (2 μm) were stained with periodic acid–Schiff reagent for the light microscopy. Glomeruli (50–100 per kidney) were examined on coronal sections to evaluate the degrees of glomerulosclerosis. Glomerulosclerosis was defined as obstruction of the capillary lumen caused by mesangial expansion or collapsed capillaries.
Immunohistochemistry and immunofluorescence
Following deparaffinization, the formalin-fixed, paraffin-embedded section samples were incubated with 3% H2O2 for 20 min to inactivate endogenous peroxidase activity. The samples were incubated with citrate buffer (pH 6.0) for retrievals of antigens for Podocin, Nephrin, and WT-1. The sections were subsequently incubated with primary antibodies. The following antibodies were used: anti-Nephrin antibody (R&D Systems AF3159), anti–HAI-1 antibody (R&D Systems AF1141), anti–WT-1 antibody (Santa Cruz Biotechnology Inc.; SC192), and anti-Podocin antibody (Sigma–Aldrich; P0372). For Matriptase antibody, anti-serum was collected from rabbits immunized with peptide corresponding mouse and human Matriptase sequence (single-letter code, CAQRNKPGVYTRLP). A validation of the antibody specificity using Basic Local Alignment Search Tool (BLAST) analysis showed that a number of proteins contained significant identity with CAQRNKPGVYTRLP, including Klk5, urokinase, and tryptase β2. However, the microarray analysis showed these proteins were not up-regulated in our model, indicating that our antibody was likely specific to Matriptase in this experiment. After reaction with primary antibody, the sections were incubated with ImmPRESS reagent kit (Vector Laboratory, Burlingame, CA, USA) for immunohistochemistry or incubated with Alexa Fluor–conjugated secondary antibodies (Invitrogen) for immunofluorescence. Positive areas were measured using MetaMorph (Molecular Devices, Sunnyvale, CA, USA). The percentage of positive area in the glomerular tuft was determined in 30 glomeruli/section. After permeabilization for 10 min, fixed U2OS was incubated in blocking solution (5% normal goat serum + 0.1% Triton X-100 in PBS) and then incubated with primary antibodies diluted in blocking solution overnight at 4 °C. The cells were incubated with Alexa Fluor–conjugated secondary antibodies diluted in the blocking solution for 1 h at room temperature and mounted on slide glass with VECTASHIELD mounting medium with 4′,6-diamino-2-phenylindole (Vector Laboratories). The following antibodies were used: anti-FLAG M2 monoclonal antibody (Sigma–Aldrich; F1804) and anti-fibrillarin (Cell Signaling Technology). Samples were analyzed using Olympus confocal microscope Fluoview FV-1000 under a 100× objective lens.
Quantitative PCR
cDNA was synthesized from purified RNA using ReverTra Ace quantitative PCR RT kit (TOYOBO, Osaka, Japan). Real-time PCR was performed using a Step One Plus thermal cycler (Applied Biosystems, Waltham, MA, USA). The amount of PCR products was normalized with 18S rRNA mRNA. The following primers were used: 18S rRNA, AGGGGAGAGCGGGTAAGAGA (forward) and GGACAGGACTAGGCGGAACA (reverse); Matriptase, TCCCTCAGAGCCAGAAGTGT (forward) and ACAGTCCGTCTTCCCATCAC (reverse); Podocin, GTGTCCAAAGCCATCCAGTT (forward) and GTCTTTGTGCCTCAGCTTCC (reverse); Synaptopodin, TTCCGAGTGGCATCCTTAAGTC (forward) and GCTGCTGCTTGGTAGGTTCA (reverse).
Cell culture
HEK293 was used for transfection of plasmids with FuGENE HD transfection reagent (Promega, Tokyo, Japan). Madin–Darby canine kidney cells were grown in RPMI 1640 and infected with adenovirus expressing LacZ or FLAG-mPodocin or FLAG-mPodocin(2–50) or FLAG-Podocin(51–385). After incubation for 3 days, the cells were lysed with RIPA buffer or fixed with 4% paraformaldehyde. For primary podocytes, primary glomerular cells isolated from isolated glomeruli with the Dynabeads methods were seeded in culture in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (33). After podocytes formed the colonies, the cells were harvested with trypsin and used for RNA analysis. Synaptopodin was used as a marker to confirm the characteristics of primary podocytes. RIPA buffer was used for obtaining proteins from whole cells. For separating cytoplasm and membrane proteins, the subcellular protein fractionation kit (Thermo Fisher Scientific) was used. U2OS was seeded in cover glass on 6-well plate (Falcon) with Dulbecco's modified Eagle's complete medium. Transfection was performed the following day using FuGENE HD transfection reagent. After 24 h, the cells were fixed with 4% paraformaldehyde.
Plasmid constructs
Full-length cDNA of mouse Podocin, mouse Claudin5, and mouse β-Catenin were cloned into pFLAG-CMV-6a (Sigma–Aldrich). FLAG-mPodocin or FLAG-mPodocin(2–50) or FLAG-Podocin(51–385) were cloned into pAd-CMV-V5 vector (Invitrogen) using the Gateway system. LacZ gene was used as a control included in the kit. The recombinant adenovirus backbones were linearized by PacI digestion and transfected into 293A cells for packaging. Viral titers were estimated using a horseradish peroxidase–conjugated polyclonal antibody to adenovirus hexon (Thermo Fisher Scientific). cDNA of mutant Podocin (R45A, R50A, and R54A) was generated by PCR and cloned into pFLAG-CMV-6a. The full-length cDNA of Matriptase constructed in pcDNA3.1-V5-His (Thermo Fisher Scientific) was provided by Dr. Hiroaki Kataoka. Matriptase mutant (G827R) was generated by PCR and cloned into pcDNA3.1-V5-His. The full-length of cDNA of HAI-1 and the deletion type of transmembrane region cloned into pCIneo were provided by Dr. Kataoka. A human Podocin-N fragment cDNA with 3× FLAG was amplified by PCR using a human kidney cDNA in human MTC panel I (TaKaRa) as a template. A 3× FLAG Podocin-N was cloned into pcDNA3.1(+). The adenovirus system was used to express mouse Nephrin-DsRed.
In vitro cleavage assay
Cell lysate from HEK293 transfected with FLAG-Podocin (WT, R45A, R50A, and R54A) was immobilized on anti-FLAG beads (Sigma–Aldrich). The beads were washed with IP buffer (1–2% Triton-X, 150 mm NaCl, 50 mm Tris-HCl, pH 7.5). The samples eluted with FLAG peptide were used as substrate for Matriptase. 15 ng/ml recombinant mouse Matriptase (R&D Systems) and the substrate were incubated for 2 h in assay buffer including 50 mm Tris-HCl (pH 8.5), 50 mm NaCl, and 0.01% Tween 20.
Western blotting
Cell lysate was separated by SDS-PAGE and electrotransferred onto polyvinylidene fluoride membranes. After overnight incubation with primary antibody at 4 °C, the membranes were incubated with horseradish peroxidase–conjugated anti-mouse (GE Healthcare; NXA931) or anti-rabbit (GE Healthcare; NA934) secondary antibody for 1 h at room temperature, followed by the addition of ECL prime (GE Healthcare) to detect bands using Image Quant LAS4000mini (GE Healthcare). The following primary antibodies were used: anti-FLAG antibody (Sigma–Aldrich; F1804), anti-His antibody (MBL, Nagoya, Japan; D291-3), anti-actin antibody (Sigma–Aldrich; A2228), anti-NA-K ATPase antibody (Cell Signaling, Danvers, MA, USA; 3010), and anti-HAI-1 antibody (R&D Systems; AF1141). All blots were run under the same experimental conditions. Each image was obtained at a single time point and was not combined into a single image.
Protease activity assay
Matriptase or thrombin activity was assessed by measuring 7-amino-4-methylcoumarin release from synthetic substrates. For Matriptase activity, 25 μm substrate (Boc-QAC–7-amino-4-methylcoumarin) (Enzo Life Sciences, Farmingdale, NY, USA), 100 pg/μl recombinant mouse Matriptase (R&D Systems) in assay buffer including 50 mm Tris-HCl (pH 8.5), 50 mm NaCl, and 0.01% Tween 20 was used. For thrombin activity, 60 mU thrombin (Sigma–Aldrich) was used instead of Matriptase. Nafamostat mesilate (Sigma–Aldrich) or Matriptase inhibitor (IN-1) was added 10 min before adding substrate. For both assays, the released fluorescence (excitation, 380 nm; emission, 460 nm) was measured using an EnVision (PerkinElmer) at a certain time.
Transmission EM
The kidneys were fixed with 4% PFA and 2% glutaraldehyde with 0.1 m phosphate buffer at 4 °C overnight. After postfixation with 1% osmium tetroxide in 0.1 m phosphate buffer for 2 h, the samples were dehydrated in ethanol and propylene oxide. Then the samples were penetrated in propylene oxide and Epon after polymerization in pure Epon. Ultrathin sections were cut with an ultramicrotome. The sections were stained in uranyl acetate and lead citrate. The grids were examined with a transmission electron microscope (H-7650; Hitachi).
Analysis of human kidney specimens
All of the human specimens were procured and analyzed after informed consent was obtained and with the approval of the Ethics Committee of Ikeda City Hospital and Kyoto University. Tissue samples were obtained from diagnostic renal biopsies performed at Ikeda City Hospital. We investigated samples from patients who had been diagnosed with IgA nephropathy (n = 5), diabetic nephropathy (n = 3), and membranous nephropathy (n = 3).
Nephroseq database analysis
The kidney transcriptomics data repository Nephroseq (University of Michigan, RRID:SCR_019050) was used to analyze various deposited data sets. The data set used in the current study is available under accession number GSE30122 in Gene Expression Omnibus (RRID:SCR_005012). The expression of HAI-1, HAI-2, and Matriptase in the diabetic nephropathy data set was checked against that of healthy living with diabetic nephropathy (34). We checked the gene expressions of glomeruli (threshold; p < 0.05, fold change > 1.5) and tubulointerstitium (threshold; none).
Statistical analysis
All values are presented as means ± S.D. Statistical analysis of variance was performed, and Tukey's method was used to compare groups or two-tailed t test. A level of p < 0.05 was considered statistically significant.
Data availability
The raw data sets from the microarray analysis generated in this study have been deposited and available under accession numbers E-GEAD-374 and E-GEAD-387 in the DNA Data Bank of Japan (RRID:SCR_002359). The rest of the data are contained within the article.
Supplementary Material
Acknowledgments
We thank Prof. Pierre Chambon for the CreERT2 plasmid, E. Keren-Happuch for the critical reading of the manuscript, and Hideyo Mukai, Masaru Kawai, Mariko Iwaki and Takayuki Kanemoto for technical assistance.
This article contains supporting information.
Author contributions—T. N. conceptualization; S. O., M. M., H. N., and T. N. data curation; S. O., E. M., and T. N. formal analysis; S. O., E. M., and T. N. writing-original draft; M. Y., K. M., K. A., N. K., K. H., H. O., M. K., H. Y., and H. K. investigation; E. M. and T. N. project administration; E. M. and T. N. writing-review and editing.
Funding and additional information—This work was supported by Grants from the TMK Project; JSPS (Japan Society for the Promotion of Science) KAKENHI JP26460274 (to T. N.), JP17H07031 (to E. M.), JP20H03199 (to E. M.), JP18H06202 (to H. N.), JP19K21306 (to H. N.), and JP20K16583 (to H. N.); AMED (Japan Agency for Medical Research and Development) Brain/MINDS Beyond Grant JP20dm0307032 (to E. M.); funds from Suzuken Co., Ltd. (to T. N.); funds from the MSD Lifescience Foundation (to T. N. and E. M.); Novartis Research Grants (to T. N. and E. M.); funds from Fuji Yakuhin Co., Ltd. (to T. N. and E. M.), the Takeda Science Foundation (to E. M.), the Kanzawa Medical Research Foundation (to E. M.), the Uehara Memorial Foundation (to E. M.), the Nakatomi Foundation (to E. M.), the Konica Minolta Science and Technology Foundation (to E. M.), the Naito Foundation (to E. M.), the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to E. M.), the Senshin Medical Research Foundation (to E. M.), the Terumo Foundation for Life Sciences and Arts (to E. M.), and the Nara Kidney Disease Research Foundation (to E. M.); and unrestricted funds (to E. M.) from Dr. Taichi Noda (KTX Corp., Aichi, Japan) and Dr. Yasuhiro Horii (Koseikai, Nara, Japan).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- CKD
- chronic kidney disease
- HAI-1
- hepatocyte growth factor activator inhibitor type 1
- Podocin-N
- N terminus of Podocin
- SD
- slit diaphragm
- MMP
- matrix metalloproteinase
- ADR
- adriamycin
- eNOS-KO
- endothelial nitric-oxide synthase knockout
- NM
- nafamostat mesylate
- PAR
- protease-activated receptor
- ADR
- adriamycin
- m
- mouse
- h
- human
- RIPA
- radioimmune precipitation assay.
References
- 1. Jha V., Garcia-Garcia G., Iseki K., Li Z., Naicker S., Plattner B., Saran R., Wang A. Y., and Yang C. W. (2013) Chronic kidney disease: global dimension and perspectives. Lancet 382, 260–272 10.1016/S0140-6736(13)60687-X [DOI] [PubMed] [Google Scholar]
- 2. Lenz O., Elliot S. J., and Stetler-Stevenson W. G. (2000) Matrix metalloproteinases in renal development and disease. J. Am. Soc. Nephrol. 11, 574–581 [DOI] [PubMed] [Google Scholar]
- 3. Brinkkoetter P. T., Ising C., and Benzing T. (2013) The role of the podocyte in albumin filtration. Nat. Rev. Nephrol. 9, 328–336 10.1038/nrneph.2013.78 [DOI] [PubMed] [Google Scholar]
- 4. Eddy A. A. (2009) Serine proteases, inhibitors and receptors in renal fibrosis. Thromb. Haemost. 101, 656–664 [PMC free article] [PubMed] [Google Scholar]
- 5. Fox C., Cocchiaro P., Oakley F., Howarth R., Callaghan K., Leslie J., Luli S., Wood K. M., Genovese F., Sheerin N. S., and Moles A. (2016) Inhibition of lysosomal protease cathepsin D reduces renal fibrosis in murine chronic kidney disease. Sci. Rep. 6, 20101 10.1038/srep20101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yaddanapudi S., Altintas M. M., Kistler A. D., Fernandez I., Möller C. C., Wei C., Peev V., Flesche J. B., Forst A.-L., Li J., Patrakka J., Xiao Z., Grahammer F., Schiffer M., Lohmüller T., et al. (2011) CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J. Clin. Invest. 121, 3965–3980 10.1172/JCI58552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hayata M., Kakizoe Y., Uchimura K., Morinaga J., Yamazoe R., Mizumoto T., Onoue T., Ueda M., Shiraishi N., Adachi M., Miyoshi T., Sakai Y., Tomita K., and Kitamura K. (2012) Effect of a serine protease inhibitor on the progression of chronic renal failure. Am. J. Physiol. Renal Physiol. 303, F1126–F1135 10.1152/ajprenal.00706.2011 [DOI] [PubMed] [Google Scholar]
- 8. Tanabe L. M., and List K. (2017) The role of type II transmembrane serine protease-mediated signaling in cancer. FEBS J. 284, 1421–1436 10.1111/febs.13971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimomura T., Denda K., Kitamura A., Kawaguchi T., Kito M., Kondo J., Kagaya S., Qin L., Takata H., Miyazawa K., and Kitamura N. (1997) Hepatocyte growth factor activator inhibitor, a novel Kunitz-type serine protease inhibitor. J. Biol. Chem. 272, 6370–6376 10.1074/jbc.272.10.6370 [DOI] [PubMed] [Google Scholar]
- 10. Nakagawa T., Sato W., Glushakova O., Heinig M., Clarke T., Campbell-Thompson M., Yuzawa Y., Atkinson M. A., Johnson R. J., and Croker B. (2007) Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J. Am. Soc. Nephrol. 18, 539–550 10.1681/ASN.2006050459 [DOI] [PubMed] [Google Scholar]
- 11. Tanabe K., Lanaspa M. A., Kitagawa W., Rivard C. J., Miyazaki M., Klawitter J., Schreiner G. F., Saleem M. A., Mathieson P. W., Makino H., Johnson R. J., and Nakagawa T. (2012) Nicorandil as a novel therapy for advanced diabetic nephropathy in the eNOS-deficient mouse. Am. J. Physiol. Renal Physiol. 302, F1151–F1160 10.1152/ajprenal.00596.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oberst M., Anders J., Xie B., Singh B., Ossandon M., Johnson M., Dickson R. B., and Lin C.-Y. (2001) Matriptase and HAI-1 are expressed by normal and malignant epithelial cells in vitro and in vivo. Am. J. Pathol. 158, 1301–1311 10.1016/S0002-9440(10)64081-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carney T. J., Von Der Hardt S., Sonntag C., Amsterdam A., Topczewski J., Hopkins N., and Hammerschmidt M. (2007) Inactivation of serine protease Matriptase1a by its inhibitor Hai1 is required for epithelial integrity of the zebrafish epidermis. Development 134, 3461–3471 10.1242/dev.004556 [DOI] [PubMed] [Google Scholar]
- 14. Saleem M., Adhami V. M., Zhong W., Longley B. J., Lin C.-Y., Dickson R. B., Reagan-Shaw S., Jarrard D. F., and Mukhtar H. (2006) A novel biomarker for staging human prostate adenocarcinoma: overexpression of Matriptase with concomitant loss of its inhibitor, hepatocyte growth factor activator inhibitor-1. Cancer Epidemiol. Biomarkers Prev. 15, 217–227 10.1158/1055-9965.EPI-05-0737 . [DOI] [PubMed] [Google Scholar]
- 15. Vogel L. K., Saebø M., Skjelbred C. F., Abell K., Pedersen E. D., Vogel U., and Kure E. H. (2006) The ratio of Matriptase/HAI-1mRNA is higher in colorectal cancer adenomas and carcinomas than corresponding tissue from control individuals. BMC Cancer 6, 176 10.1186/1471-2407-6-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sales K. U., Masedunskas A., Bey A. L., Rasmussen A. L., Weigert R., List K., Szabo R., Overbeek P. A., and Bugge T. H. (2010) Matriptase initiates activation of epidermal pro-kallikrein and disease onset in a mouse model of Netherton syndrome. Nat. Genet. 42, 676–683 10.1038/ng.629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bardou O., Menou A., François C., Duitman J. W., Von Der Thüsen J. H., Borie R., Sales K. U., Mutze K., Castier Y., Sage E., Liu L., Bugge T. H., Fairlie D. P., Königshoff M., Crestani B., et al. (2016) Membrane-anchored serine protease Matriptase is a trigger of pulmonary fibrogenesis. Am. J. Resp. Crit. Care Med. 193, 847–860 10.1164/rccm.201502-0299OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colombo É., Désilets A., Duchêne D., Chagnon F., Najmanovich R., Leduc R., and Marsault E. (2012) Design and synthesis of potent, selective inhibitors of Matriptase. ACS Med. Chem. Lett. 3, 530–534 10.1021/ml3000534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lelongt B., Legallicier B., Piedagnel R., and Ronco P. M. (2001) Do matrix metalloproteinases MMP-2 and MMP-9 (gelatinases) play a role in renal development, physiology and glomerular diseases? Curr. Opin. Nephrol. Hypertens. 10, 7–12 10.1097/00041552-200101000-00002 [DOI] [PubMed] [Google Scholar]
- 20. McMillan J. I., Riordan J. W., Couser W. G., Pollock A. S., and Lovett D. H. (1996) Characterization of a glomerular epithelial cell metalloproteinase as matrix metalloproteinase-9 with enhanced expression in a model of membranous nephropathy. J. Clin. Invest. 97, 1094–1101 10.1172/JCI118502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ebihara I., Nakamura T., Shimada N., and Koide H. (1998) Increased plasma metalloproteinase-9 concentrations precede development of microalbuminuria in non-insulin-dependent diabetes mellitus. Am. J. Kidney Dis. 32, 544–550 10.1016/S0272-6386(98)70015-0 [DOI] [PubMed] [Google Scholar]
- 22. Li S.-Y., Huang P.-H., Yang A.-H., Tarng D.-C., Yang W.-C., Lin C.-C., Chen J.-W., Schmid-Schönbein G., and Lin S.-J. (2014) Matrix metalloproteinase-9 deficiency attenuates diabetic nephropathy by modulation of podocyte functions and dedifferentiation. Kidney Int. 86, 358–369 10.1038/ki.2014.67 [DOI] [PubMed] [Google Scholar]
- 23. Rigothier C., Daculsi R., Lepreux S., Auguste P., Villeneuve J., Dewitte A., Doudnikoff E., Saleem M., Bourget C., Combe C., and Ripoche J. (2016) CD154 induces matrix metalloproteinase-9 secretion in human podocytes. J. Cell. Biochem. 117, 2737–2747 10.1002/jcb.25571 [DOI] [PubMed] [Google Scholar]
- 24. Schaffner F., and Ruf W. (2008) Tissue factor and protease-activated receptor signaling in cancer. Semin. Thromb. Hemost. 34, 147–153 10.1055/s-2008-1079254 [DOI] [PubMed] [Google Scholar]
- 25. Badeanlou L., Furlan-Freguia C., Yang G., Ruf W., and Samad F. (2011) Tissue factor–protease-activated receptor 2 signaling promotes diet-induced obesity and adipose inflammation. Nat. Med. 17, 1490–1497 10.1038/nm.2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harris J. J., McCarthy H. J., Ni L., Wherlock M., Kang H., Wetzels J. F., Welsh G. I., and Saleem M. A. (2013) Active proteases in nephrotic plasma lead to a Podocin-dependent phosphorylation of VASP in podocytes via protease activated receptor-1. J. Pathol. 229, 660–671 10.1002/path.4149 [DOI] [PubMed] [Google Scholar]
- 27. Guan Y., Nakano D., Zhang Y., Li L., Liu W., Nishida M., Kuwabara T., Morishita A., Hitomi H., Mori K., Mukoyama M., Masaki T., Hirano K., and Nishiyama A. (2017) A protease-activated receptor-1 antagonist protects against podocyte injury in a mouse model of nephropathy. J. Pharmacol. Sci. 135, 81–88 10.1016/j.jphs.2017.09.002 [DOI] [PubMed] [Google Scholar]
- 28. Jiang Y., Yau M. K., Kok W. M., Lim J., Wu K. C., Liu L., Hill T. A., Suen J. Y., and Fairlie D. P. (2017) Biased signaling by agonists of protease activated receptor 2. ACS Chem. Biol. 12, 1217–1226 10.1021/acschembio.6b01088 [DOI] [PubMed] [Google Scholar]
- 29. Murakawa M., Okamura T., Shibuya T., Harada M., Otsuka T., and Niho Y. (1992) Use of a synthetic protease inhibitor for the treatment of L-asparaginase-induced acute pancreatitis complicated by disseminated intravascular coagulation. Ann. Hematol. 64, 249–252 10.1007/BF01738305 [DOI] [PubMed] [Google Scholar]
- 30. Wang Y., Wang Y. P., Tay Y. C., and Harris D. C. (2000) Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 58, 1797–1804 10.1046/j.1523-1755.2000.00342.x [DOI] [PubMed] [Google Scholar]
- 31. Kawaguchi M., Takeda N., Hoshiko S., Yorita K., Baba T., Sawaguchi A., Nezu Y., Yoshikawa T., Fukushima T., and Kataoka H. (2011) Membrane-bound serine protease inhibitor HAI-1 is required for maintenance of intestinal epithelial integrity. Am. J. Pathol. 179, 1815–1826 10.1016/j.ajpath.2011.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yokoi H., Kasahara M., Mukoyama M., Mori K., Kuwahara K., Fujikura J., Arai Y., Saito Y., Ogawa Y., Kuwabara T., Sugawara A., and Nakao K. (2010) Podocyte-specific expression of tamoxifen-inducible Cre recombinase in mice. Nephrol. Dial. Transplant 25, 2120–2124 10.1093/ndt/gfq029 [DOI] [PubMed] [Google Scholar]
- 33. Ueda S., Ozawa S., Mori K., Asanuma K., Yanagita M., Uchida S., and Nakagawa T. (2015) ENOS deficiency causes podocyte injury with mitochondrial abnormality. Free Radic. Biol. Med. 87, 181–192 10.1016/j.freeradbiomed.2015.06.028 [DOI] [PubMed] [Google Scholar]
- 34. Woroniecka K. I., Park A. S., Mohtat D., Thomas D. B., Pullman J. M., and Susztak K. (2011) Transcriptome analysis of human diabetic kidney disease. Diabetes 60, 2354–2369 10.2337/db10-1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data sets from the microarray analysis generated in this study have been deposited and available under accession numbers E-GEAD-374 and E-GEAD-387 in the DNA Data Bank of Japan (RRID:SCR_002359). The rest of the data are contained within the article.