

Abstract
The octapeptins are lipopeptide antibiotics that are structurally similar to polymyxins yet retain activity against polymyxin-resistant Gram-negative pathogens, suggesting they might be used to treat recalcitrant infections. However, the basis of their unique activity is unclear because of the difficulty in generating high-resolution experimental data of the interaction of antimicrobial peptides with lipid membranes. To elucidate these structure–activity relationships, we employed all-atom molecular dynamics simulations with umbrella sampling to investigate the conformational and energetic landscape of octapeptins interacting with bacterial outer membrane (OM). Specifically, we examined the interaction of octapeptin C4 and FADDI-115, lacking a single hydroxyl group compared with octapeptin C4, with the lipid A–phosphoethanolamine modified OM of Acinetobacter baumannii. Octapeptin C4 and FADDI-115 both penetrated into the OM hydrophobic center but experienced different conformational transitions from an unfolded to a folded state that was highly dependent on the structural flexibility of their respective N-terminal fatty acyl groups. The additional hydroxyl group present in the fatty acyl group of octapeptin C4 resulted in the molecule becoming trapped in a semifolded state, leading to a higher free energy barrier for OM penetration. The free energy barrier for the translocation through the OM hydrophobic layer was ∼72 kcal/mol for octapeptin C4 and 62 kcal/mol for FADDI-115. Our results help to explain the lower antimicrobial activity previously observed for octapeptin C4 compared with FADDI-115 and more broadly improve our understanding of the structure–function relationships of octapeptins. These findings may facilitate the discovery of next-generation octapeptins against polymyxin-resistant Gram-negative 'superbugs.'
Keywords: octapeptin, polymyxin, antimicrobial resistance, Gram-negative bacteria, molecular dynamics, conformational transition, antibiotic action, antibiotic resistance, drug design, Gram-negative bacteria, conformational change, octapeptin
The World Health Organization has identified antimicrobial resistance as a serious global threat to human health (1). Of particular concern are the Gram-negative 'superbugs,' including Acinetobacter baumannii, that show resistance to almost all currently available antibiotics (2). Given that the development of new antibiotics has stagnated in recent years, polymyxins (i.e. polymyxin B and colistin) are increasingly used as a last-line therapy against these multidrug-resistant Gram-negative pathogens (3–5). Unfortunately, polymyxin resistance is increasing, such as the emergence and spread of plasmid-borne mcr genes that encode phosphoethanolamine transferases for the modification of lipid A (6–8). Novel antibiotic treatments that can target polymyxin resistance are therefore urgently required.
The octapeptins are lipopeptide antibiotics discovered over 40 years ago as natural products in the soil bacterium Bacillus circulans (9). Although structurally similar to the polymyxins (Table S1), there are major differences in the antibacterial activity between the two classes of lipopeptides. Whereas the polymyxins are only active against Gram-negative bacteria (3, 5), the octapeptins possess much broader antimicrobial activity against Gram-negative and Gram-positive bacteria, yeast, fungi, and even protozoa (9). Importantly, the octapeptins do not exhibit cross-resistance with polymyxins and retain activity against polymyxin-resistant Gram-negative bacteria (10). The development of resistance to the octapeptins in Gram-negative organisms also appears to be more difficult than occurs with the polymyxins (11). These unique features make the octapeptins promising candidates for the development of novel lipopeptide antibiotics (12).
Given the structural similarities to the polymyxins, the octapeptins are believed to have a similar mode of action that involves insertion into the OM of Gram-negative bacteria followed by membrane disorganization, lysis, and eventual cell death (3, 9, 13). Resistance to the polymyxins generally develops through modification of the lipid A component of lipopolysaccharide in the OM with cationic moieties such as ethanolamine and 4′-aminoarabinose (14, 15). Interestingly, lipid A–based NMR models revealed that the octapeptins can specifically bind to the ethanolamine- or 4′-aminoarabinose–modified lipid A molecule through strong hydrophobic contacts (10, 16). Minor structural modifications to the octapeptin core scaffold not involving the α-γ-diaminobutyric acid (Dab) residues have also been shown to dramatically hinder their bactericidal activity (9, 10, 17, 18). These findings suggest that the activity of the octapeptins depends upon their unique interaction pattern with the OM rather than simply acting as a cationic detergent. However, the interaction between the octapeptins and the bacterial OM has never been investigated at the atomic level, significantly limiting our understanding of their mechanism of activity and hindering the discovery of novel octapeptins.
In the present study, an asymmetric lipid A–phosphoethanolamine (lipid A–pEtN; Fig. S1)–modified OM model was constructed based on our quantitative lipidomics results of polymyxin-resistant A. baumannii (Fig. 1B) (19). For the first time, we employed all-atom molecular dynamics (MD) simulations and umbrella sampling technique to investigate the conformation and energetics governing the penetration of two octapeptins (octapeptin C4 and FADDI-115) into the lipid A–pEtN modified OM. Despite their close structural similarity, significant changes in their free energy profiles and conformational transitions during OM penetration were observed, revealing new insight into the structure–interaction relationship of octapeptins.
Figure 1.
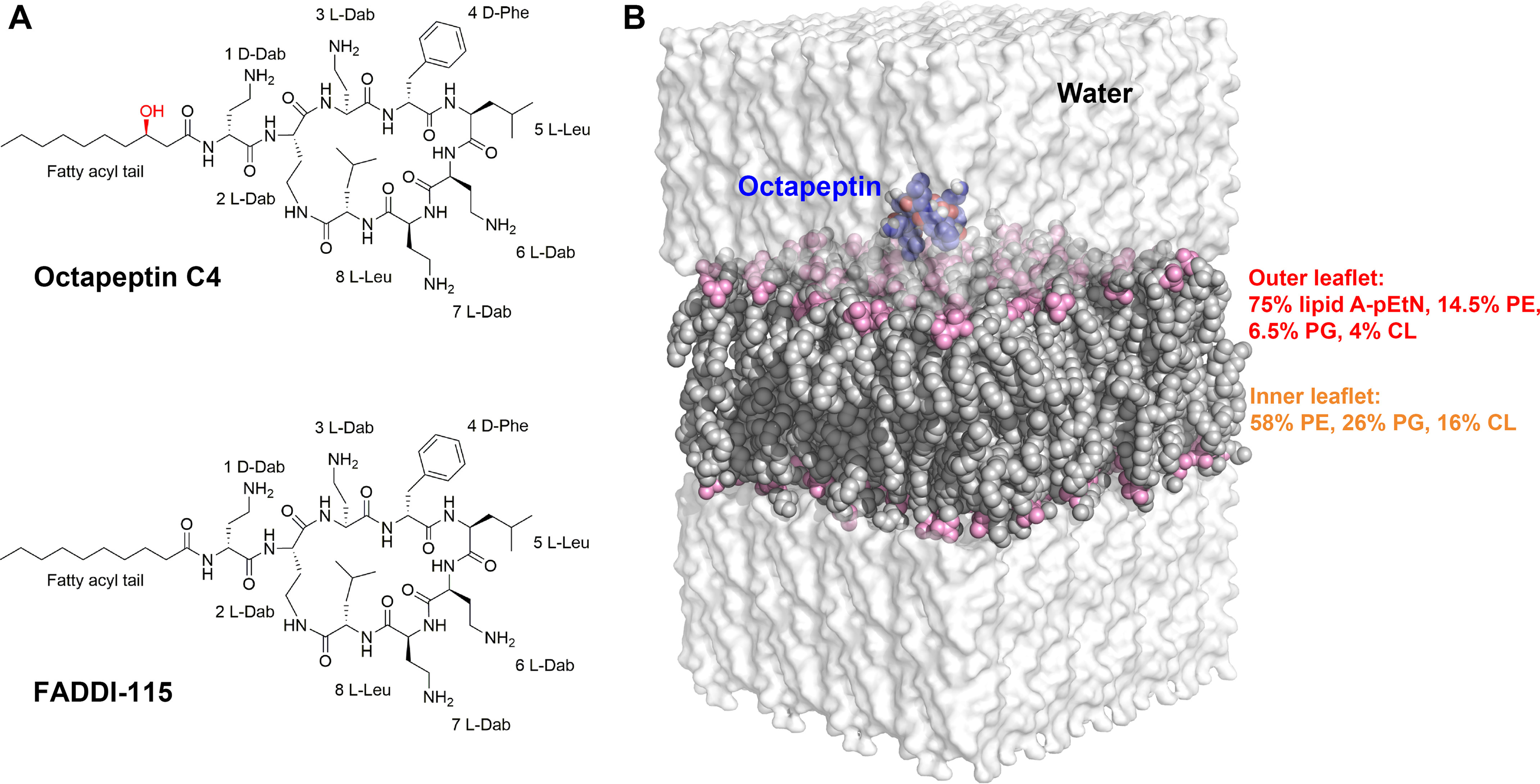
Chemical structures and description of molecular dynamics simulation systems. A, chemical structures of octapeptin C4 and the octapeptin analog FADDI-115. The additional hydroxyl group in octapeptin C4 is shown in red. B, the equilibrated simulation box used for molecular dynamics simulations. The water shell is shown as transparent surface model, the octapeptin molecule is displayed as blue spheres, and the bacterial outer membrane is shown as gray spheres with the phosphate groups in pink. The lipid composition of the outer membrane is shown for each membrane leaflet. Lipid A–pEtN, phosphoethanolamine-modified lipid A; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; CL, cardiolipin.
Results
Interaction of octapeptin C4 and FADDI-115 with the bacterial OM
The motion trajectories of the octapeptin molecules in the steered simulations were tracked relative to the center of the OM (Fig. 2A). After ∼5 ns, both the octapeptin C4 and FADDI-115 molecules approached the surface of the outer leaflet of the OM, traversing the polar headgroup region after ∼12 ns. After a short plateau phase (∼2 ns for octapeptin C4 and 4 ns for FADDI-115), the octapeptin molecule completely penetrated into the hydrophobic region of the OM after roughly 36 ns. We investigated the interactions between the octapeptin molecules and the OM at different stages of penetration. At the surface-binding stage, the positively charged side chains of d-Dab1 and Dab6 of octapeptin C4 closely interacted with the negatively charged phosphate groups of adjacent lipid A–pEtN molecules (Fig. 2B, panel i). Similarly, the side chains of d-Dab1 and Dab3 in FADDI-115 bound to the phosphate groups of lipid A–pEtN molecules (Fig. 2C, panel i). These results indicate that the initial binding of the octapeptins to the bacterial OM was mainly driven by the long-range electrostatic interactions. Subsequently, both octapeptin C4 and FADDI-115 inserted their fatty acyl groups into the hydrocarbon region of the OM, although their cyclic heptapeptide segments formed multiple hydrogen bonds with the headgroups of lipid A–pEtN molecules (Fig. 2, B and C, panels ii). After this, both octapeptins formed a folded conformation within the hydrophobic center of the OM. For octapeptin C4, its N-terminal fatty acyl group and the d-Phe4 oriented toward the OM center and formed hydrophobic contacts with the hydrocarbon chains of lipid A–pEtN and phospholipid molecules, whereas the polar heptapeptide ring made intensive interactions with the phosphate groups of lipid A–pEtN molecules (Fig. 2B, panel iii). The interaction was somewhat different for FADDI-115; the fatty acyl chain and Leu8 were embedded in the hydrocarbon region of the OM, whereas the cyclic head interacted with the phosphate groups of lipid A–pEtN molecules (Fig. 2C, panel iii). During the OM penetration process, the positively charged Dab residues of both octapeptins formed strong interactions with the headgroups of lipid A–pEtN molecules, which triggered ∼150° rotation of the headgroups of the adjacent lipid A–pEtN molecules (Fig. 2D) and the local structural disorganization of the OM (Fig. S2).
Figure 2.

Octapeptin interaction with the bacterial outer membrane. A, distance of the center of mass of the octapeptin molecule from the center of the outer membrane. The polar headgroup region of the outer membrane is labeled by the two orange dashed lines. B and C show the structural snapshots of the penetration of octapeptin C4 and FADDI-115 in different regions of the outer membrane, respectively. The octapeptin molecule is shown as blue sticks, the lipid A–pEtN molecules are shown as teal lines, and the phospholipid molecules are shown as gray lines. The hydrogen bonds between the octapeptin molecule and the lipid A–pEtN molecules in the outer membrane are shown as purple dashed lines. D, the orientation of lipid A–pEtN molecule relative to the normal of the outer membrane.
Before the penetration of the octapeptins, water molecules could only reach the polar headgroup region of the OM and were largely excluded from the hydrophobic region. However, following the penetration of the octapeptin molecule into the center of the OM, a large number of water molecules entered into the hydrophobic layer of the OM through the penetration site of the octapeptin molecules; the number of water molecules within the membrane–water interface increased from ∼350 to 650 in the octapeptin C4 system (Fig. 3A) and to 550 in the FADDI-115 system (Fig. 3B). This increase in the water permeability of the bacterial OM promoted by the octapeptins may lead to further destabilization of the bacterial OM and help explain the antimicrobial activity of the octapeptins.
Figure 3.

Water permeation of the outer membrane caused by octapeptin C4 (A) and FADDI-115 (B). Z = 0 indicates the membrane center. The structural snapshots show the different stages of the penetration of octapeptins into the outer membrane. The octapeptin molecule is shown as blue sticks. The water molecules are shown as spheres with red oxygen atoms and white hydrogen atoms. The reaction coordinates along z axis represent the positions relative to the center of the outer membrane.
Free energy profiles of the penetration of octapeptin C4 and FADDI-115 in the bacterial OM
Free energy profiles are useful to evaluate the membrane-penetrating ability of antimicrobials and closely relates to their efficacy (20–22). Through a series of umbrella sampling simulations, we characterized the free energy profiles for the octapeptins moving from the bulk water to the hydrophobic center of the OM (Fig. 4). The whole free energy profile was divided into three components. The ΔGbind describes the binding of the octapeptin to the surface of the OM outer leaflet (2.5 nm < Z < 3.0 nm), whereas the ΔGtranslocate-1 and ΔGtranslocate-2 values describe the translocations of the octapeptin molecule through the headgroup region (1.8 nm < Z < 2.5 nm) and the hydrocarbon region (0 nm < Z < 1.8 nm) of the OM outer leaflet, respectively. The ΔGbind was approximately −3.5 kcal/mol (favorable) for both octapeptin C4 and FADDI-115. The ΔGtranslocate-1 was comparatively unfavorable (∼8 kcal/mol) because the octapeptin C4 and FADDI-115 passed through the headgroup region of the OM. Curiously, the free energy profile diverged when traversing the hydrocarbon region of the OM for each octapeptin, especially in the range Z < 1.2 nm. To pass through this hydrophobic region, the ΔGtranslocate-2 was ∼72 kcal/mol for octapeptin C4 and 62 kcal/mol for FADDI-115 (energy-unfavorable). Interestingly, the free energy profiles of octapeptin C4 and FADDI-115 significantly diverged when they penetrated through the inner leaflet of the OM. The free energy curve reached the plateau at Z = −1 nm in the system with FADDI-115, and the free energy for FADDI-115 was ∼50 kcal/mol lower than that for octapeptin C4 at Z = −3 nm (Fig. S3). Because the only structural difference between these two octapeptins is an additional hydroxyl group on the fatty acyl chain of octapeptin C4 (Fig. 1A), these results show that this structural variation did not affect their binding affinity with the bacterial OM and penetrating ability through the polar headgroup region but significantly changed their ability to traverse the hydrophobic region of the OM.
Figure 4.

Free energy profiles for the penetration of octapeptin C4 and FADDI-115 into the outer membrane. The free energy barriers for octapeptin binding to the outer membrane, passing through the headgroup and hydrocarbon regions of the outer leaflet are depicted by ΔGbind, ΔGtranslocate-1, and ΔGtranslocate-2, respectively. The reaction coordinates along z axis represent the positions relative to the center of the outer membrane, with Z = 0 indicating the hydrophobic center of the outer membrane.
Conformational transition of octapeptin C4 and FADDI-11 in the bacterial OM
As shown in Fig. 2 (B and C, panels iii), both octapeptin C4 and FADDI-115 formed a folded conformation in the hydrophobic center of the OM, consistent with the previous NMR findings (10). To examine the mechanism of the conformational transition, we characterized the conformational ensembles of the octapeptin C4 and FADDI-115 molecules by measuring their radius of gyration (Rg) around the z axis during the penetration process. The octapeptin C4 molecule displayed three major conformational clusters during the entire penetration process (Fig. 5A). It adopted an unfolded state (Rg > 0.6 nm) in the polar headgroup region of the OM (Z > 1.8 nm), transformed into an intermediate, semifolded state (Rg = ∼0.54 nm) in the headgroup–hydrocarbon interface region of the OM (1.0 nm < Z < 1.5 nm), and finally stayed in a more compact, folded state (Rg = ∼0.45 nm) near the OM hydrophobic center (Z < 0.5 nm). Similarly, the FADDI-115 molecule also maintained an unfolded state in the polar headgroup region of the OM and formed a folded state near the OM center (Fig. 5B). However, the transition between these two states was achieved through an intermediate aisle (0.5 nm < Z < 1 nm), rather than the intermediate conformational cluster observed in the octapeptin C4 system.
Figure 5.
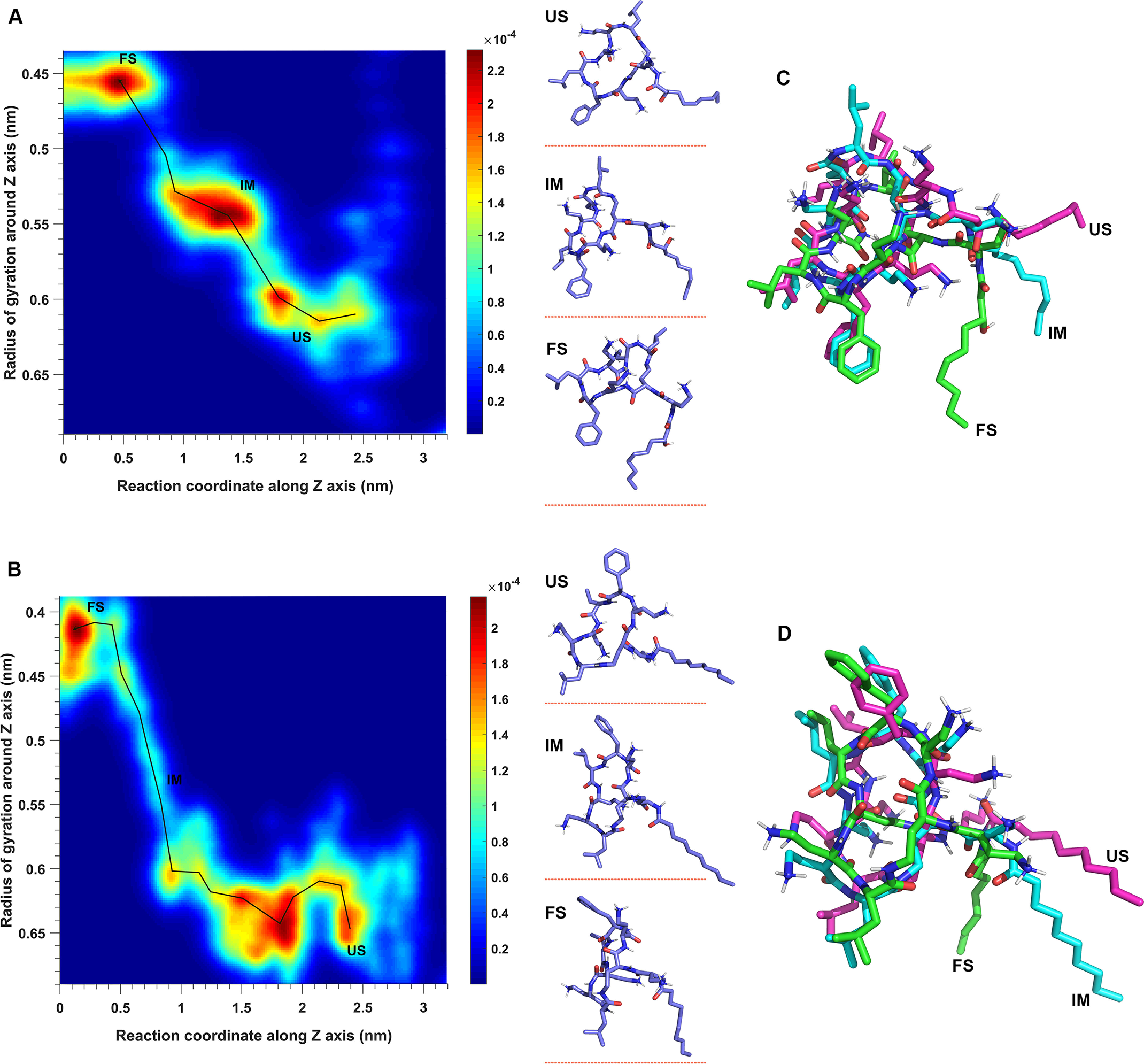
Conformational dynamics of octapeptin C4 (A) and FADDI-115 (B) in the outer membrane, with the superimposition of the octapeptin molecule at different conformational states shown for octapeptin C4 (C) and FADDI-115 (D). The conformational landscape is defined by sampling the radius of gyration of the octapeptin molecule around the z axis and the reaction coordinates of the penetration. The conformations of the octapeptin molecule are represented by points, and the color spectrum represents the density of points. The pathway of conformational transition from the unfolded state (US) to intermediate state (IM) and folded state (FS) is shown by a black line.
Interestingly, structural superimposition showed that the cyclic heptapeptide component of the octapeptin C4 and FADDI-115 molecules was well-aligned among their unfolded, intermediate, and folded states. The variation in the radius of gyration observed was due to conformational swing of their respective fatty acyl groups (Fig. 5, C and D). This may suggest that the hydroxyl group on the fatty acyl group of octapeptin C4 is a key factor for their observed different conformational transitions in the OM.
Structure–interaction relationships of octapeptins
The all-atom simulation results were utilized to examine the interactions between the OM and the octapeptin molecule in its folded state. A single octapeptin C4 molecule was observed to bind to three lipid A–pEtN molecules, and the complex was stabilized by a combination of electrostatic and hydrophobic interactions (Fig. 6A). The positively charged side chains of Dab7 interacted with the negatively charged phosphate group of the lipid A–pEtN molecule; the side chain of d-Dab1 made a hydrogen bond with the sugar of the lipid A–pEtN molecule; the main chain of the residues on the cyclic segment contributed five hydrogen bonds with the lipid A–pEtN molecules, and the N-terminal fatty acyl group and d-Phe4 formed hydrophobic contacts with the hydrocarbon chains of the adjacent lipid A–pEtN molecules. Notably, the side chains of Dab3 and Dab6 made few interactions with the lipid A–pEtN molecules, likely because of the shielding from the hydrophobic d-Phe4, Leu5 and Leu8. Similarly, the FADDI-115 molecule bound to three lipid A–pEtN molecules (Fig. 6B). The side chains of d-Dab1, Dab3, and Dab7 interacted with the phosphate groups of the lipid A–pEtN molecules; the main chains of Dab3 and Dab6 made three hydrogen bonds with the lipid A–pEtN molecules; and the N-terminal fatty acyl group and Leu8 formed hydrophobic contacts with the hydrocarbon tails of the adjacent lipid A–pEtN molecules.
Figure 6.

Interactions between the octapeptin and lipid A–pEtN molecules in the outer membrane. A, octapeptin C4. B, FADDI-115. C, the hydrogen bonds formed between the hydroxyl group on the fatty acyl chain of octapeptin C4 and the outer membrane lipids.
Additionally, we examined the interactions associated with the hydroxyl group on the fatty acyl group of octapeptin C4 (Fig. 6C). The hydroxyl group formed up to two hydrogen bonds with the OM lipids during the entire penetration process and likely contributed two hydrogen bonds when the octapeptin C4 molecule reached the headgroup–hydrocarbon interface region of the OM (1.0 nm < Z < 1.5 nm). These intermolecular hydrogen bonds with octapeptin C4 appear to change the conformational dynamics of the fatty acyl group, which stabilized a large intermediate, semifolded conformational cluster at this interface region (Fig. 5A).
Discussion
The octapeptins are structurally similar to the polymyxins, with both possessing an N-terminal fatty acyl group and three cationic Dab residues located at the same positions in a heptapeptide ring. However, despite these similarities, the octapeptins retain activity against many polymyxin-resistant Gram-negative bacteria (9, 10, 12). As a class of membrane-targeting antibiotics, the interaction between the octapeptins and the bacterial OM has not been investigated at the atomic level to date. In this study, all-atom MD simulations were employed to investigate how the octapeptins interact with the lipid A–pEtN modified OM that renders many Gram-negative bacteria polymyxin-resistant. Unlike previous simulation studies in which the bacterial OM was simplified into a symmetric or asymmetric lipopolysaccharide bilayer with model phospholipid compositions (23–26), we utilized the quantitative membrane lipidomics results from polymyxin-resistant A. baumannii 5075R to construct a more realistic OM model that allowed for better precision when characterizing interactions with the octapeptins (19). Analysis of the simulations revealed that a minor structural variation between octapeptin C4 and FADDI-115, namely, the presence of a hydroxyl group on the fatty acyl group of octapeptin C4 (Fig. 1A), was sufficient to impact their conformational transitions and resulted in different free energy profiles for OM penetration. These results provided novel atomic-scale insights into the structure–activity relationship of the octapeptins against polymyxin-resistant Gram-negative bacteria.
Free energy profiles quantitatively evaluated the energetics governing the penetration of the octapeptin molecules into the bacterial OM (Fig. 4). The free energy barrier for the translocation through the hydrophobic layer of the OM (ΔGtranslocate-2 = ∼72 and 62 kcal/mol for octapeptin C4 and FADDI-115, respectively; Fig. 4) was much lower than that of colistin A (∼82 kcal/mol) (27). The high free energy barrier in this region suggests that the hydrophobic layer of the OM constitutes the major defense to the action of both octapeptins and polymyxins. Lacking the hydroxyl group found on the fatty acyl tail of octapeptin C4 (Fig. 1A), FADDI-115 is slightly more hydrophobic than octapeptin C4. The lower free energy required for FADDI-115 to penetrate the membrane indicates that the additional hydroxyl group of octapeptin C4 negatively impacted OM penetration. This may explain the greater bacterial killing of FADDI-115 compared with octapeptin C4 against polymyxin-resistant A. baumannii strains (10). Importantly, the lower free energy barrier for the octapeptins compared with the polymyxins illustrates that their superior antimicrobial activity against polymyxin-resistant Gram-negative bacteria is very likely caused by their enhanced ability to traverse the hydrophobic region of the OM.
Our conformational analysis suggests that free energy differences between octapeptin C4 and FADDI-115 were linked to their different conformational dynamics within the OM. Specifically, a large conformational cluster populating an intermediate state was observed for the octapeptin C4 at the headgroup–hydrocarbon interface region of the OM and not for FADDI-115 (Fig. 5). This may indicate that during penetration into the membrane, octapeptin C4 is readily trapped in its intermediate state, making the transition to the folded state more difficult to achieve compared with FADDI-115. That octapeptin C4 formed the intermediate conformation close to the headgroup–hydrocarbon interface region of the OM (the region where the free energy profiles of octapeptin C4 and FADDI-115 diverged) suggests that the formation of the folded conformation plays a key role in regulating their penetration into the bacterial OM. With the folded conformation, the hydrophobic residues and charged Dab residues were separated into two domains, conferring structural amphipathicity (Fig. 6). This special structural arrangement suits the interactions with both the hydrocarbon tails and phosphate groups of lipid A–pEtN molecules.
A similar folded conformation was also found in an NMR-based interaction model of octapeptin C4 with a single lipid A molecule (10). Importantly, the specific interaction pairs derived from the NMR-based model and our simulation-based model are quite similar. In both models, the fatty acyl group and d-Phe4 formed hydrophobic contacts with the carbon tails of lipid A; the d-Dab1 residue interacted with the sugar of lipid A; and Dab3, Dab6, and Dab8 residues interacted with the phosphate groups of the lipid A (Fig. 6A). However, the octapeptin C4 molecule adopted a more compact conformation in the NMR-based model (10) compared with our simulation-based model, very likely because the octapeptin C4 interacted with only a single lipid A molecule in the NMR model but with three lipid A molecules in the membrane condition. Moreover, our recent studies discovered that the polymyxins also adopted a similar folded conformation in WT lipid A OM but not in lipid A–pEtN modified OM (27). Collectively, the reported NMR study and our simulation results reveal that the folding is critical for the OM penetration of both octapeptins and polymyxins. Once in the folded conformation, octapeptin and polymyxin molecules are able to fully penetrate the membrane, which leads to membrane destabilization. Given the bactericidal activity of the polymyxins involves the disorganization of the OM (3, 28), the ability of the octapeptins (but not the polymyxins) to form the folded conformation in the OM of polymyxin-resistant bacteria may explain their activity against these polymyxin-resistant organisms. Furthermore, we also found that the octapeptins formed the folded conformation within the WT, nonmodified lipid A OM of A. baumannii and interacted with the surrounding lipid A molecules via both electrostatic interaction (between Dab residues and phosphate groups of lipid A) and hydrophobic interaction (between hydrophobic regions of octapeptin and hydrocarbon tails of lipid A) (Fig. S4). These results support the similar activity of octapeptins against polymyxin-susceptible and polymyxin-resistant Gram-negative bacteria (10).
In the interaction model of the two octapeptins (Fig. 6), both the hydrophobic interactions (involving the fatty acyl group and d-Phe4 in octapeptin C4; or fatty acyl group and Leu8 in FADDI-115) and polar interactions (involving the Dab residues) stabilized the interaction complex with multiple lipid A–pEtN molecules. Our previous NMR results emphasized the importance of the hydrophobic interactions for octapeptin activity (10). In our simulations, we found that the main chain of the heptapeptide ring of octapeptins formed three to five hydrogen bonds with the phosphate groups of lipid A–pEtN molecules (Fig. 6). In contrast, although similar hydrophobic interactions were still present with polymyxin B, only the side chains of its Dab residues interacted with the phosphate groups of lipid A (29). These results indicate that the interactions with the bacterial OM of polymyxins, but not octapeptins, are more dependent on the electrostatic interactions between the positively charged side chains of Dab residues and the negatively charged phosphate groups of lipid A (3, 30). This explains why the addition of positively charged moieties (e.g. ethanolamine or 4′-aminoarabinose) to the phosphate of lipid A significantly attenuates the interaction with polymyxins, but not octapeptins. In the lipid A–pEtN modified OM, the octapeptins strongly interacted with the lipid A–pEtN molecules through a combination of main chain–mediated polar interactions and hydrophobic contacts (Fig. 6), thus retaining the strong interaction with the outer membrane and subsequent membrane penetration. As noted above, this may explain why the octapeptins maintain activity against polymyxin-resistant bacteria (10, 16). That the interaction of octapeptins with the bacterial OM is not highly dependent on electrostatic interactions with the lipid A phosphate groups may also explain their broad antimicrobial spectrum against Gram-positive bacteria, yeast, fungi, and protozoa that do not contain lipid A in their membranes (9).
Another interesting finding of our study was that the hydroxyl group on the fatty acyl group of octapeptin C4 formed multiple hydrogen bonds with the OM lipids (Fig. 6C), inhibiting the conformational transition of the molecule and increasing the free energy barrier for its penetration into the OM. A minor structural variation causing such a dramatic impact as observed in the present study (i.e. trapping the molecule in a semifolded state and creating a higher free energy barrier for OM penetration) indicates that the fatty acyl group of octapeptins is highly sensitive to structural modifications. This has important implications for antimicrobial activity. Indeed, previous studies with the octapeptins have shown that changes in the length of the fatty acyl group, as well as certain other structural modifications, significantly affect activity against Gram-negative bacteria (10, 17, 18). Although the present study is the first to examine the effect on membrane penetration of altering the hydroxyl group on the fatty acyl group of the octapeptins, the effects of other structural modifications at the remaining positions on membrane penetration and antimicrobial activity are warranted, with the aim of better understanding the structure–activity relationship of the octapeptins.
To the best of our knowledge, this is the first mechanistic study on the interaction between the octapeptins and the bacterial OM at the atomic level. The thermodynamics governing the penetration of the octapeptins into the OM and their conformational transitions within the OM environment were investigated. Importantly, our results help to explain why the octapeptins retain antimicrobial activity against polymyxin-resistant bacteria despite sharing a similar structural scaffold with the polymyxins. These novel findings enhance our understanding of the mechanism underlying the antimicrobial activity of the octapeptins and will facilitate the development of novel antibiotics for the treatment of infections caused by life-threatening Gram-negative 'superbugs.'
Experimental procedures
System preparation
A representative natural octapeptin (octapeptin C4) and a novel lead (FADDI-115) from our in-house library of octapeptin analogs were employed as the models (10). The only difference between these two octapeptins is that the hydroxyl group on the fatty acyl group of octapeptin C4 is absent in FADDI-115 (Fig. 1A). Importantly, both octapeptins show excellent activity against polymyxin-resistant Gram-negative bacteria (10). The structural models of the octapeptins were constructed in Chem3D. Energy minimization was performed based on the MM2 force field to relieve any intramolecular steric clashes (31). The topology parameters of the octapeptins were generated in the SwissParam server and are compatible with CHARMM force field (32). Consistent with the NMR experimental results (10), the octapeptin molecule formed a folded conformation in the simulations, which verified the topologies of the octapeptins. An asymmetric OM model was constructed using the CHARMM-GUI membrane builder (33). Notably, the accurate lipid composition of the OM (Fig. 1B) was based on our quantitative lipidomics results of polymyxin-resistant A. baumannii 5075R (19). Initially, the octapeptin molecules were placed above the outer leaflet of the OM using the gmx insert-molecules tool by replacing the overlapping water molecules (34). TIP3P water molecules and counter ions (CaCl2) were employed to hydrate and neutralize the simulation systems, respectively.
Steered MD
Because of the limitation of conventional MD simulations, it is not feasible to observe the spontaneous penetration of antimicrobials into the bacterial membrane within the time scale of hundreds to thousands of nanoseconds (23, 35). Therefore, the steered simulations (20–22) were conducted to construct the reaction coordinates of the penetration of the octapeptins into the lipid A–pEtN modified OM. A harmonic potential with a force constant of 500 KJ mol−1 nm−2 was applied on the centers of mass of both the octapeptin molecule and the OM along the z axis and changed adaptively according to the interaction between the octapeptin molecule and the OM. For each system, three independent steered simulations were performed.
Umbrella sampling
To characterize the equilibrated thermodynamics governing the penetration of the octapeptins into the OM, umbrella sampling simulations were performed (20–22). The system configurations from the steered simulation trajectory were used to construct a series of umbrella sampling windows in which the octapeptin molecule was harmonically restrained at various fixed positions at the z axis. The position interval between neighboring windows was 0.2 nm, and 17 simulation windows were constructed for each of the octapeptin C4 and FADDI-115 systems. Each window was simulated for 50 ns to enhance the conformational sampling; hence, a total of 850 ns of simulations were performed for each octapeptin system to obtain the free energy profile that quantitatively described the penetration of the octapeptin from bulk water to the hydrophobic center of the OM. The free energy profiles calculated using 10-ns time blocks and the overlap between position distributions of octapeptins in simulation windows were used to check the convergence of free energy calculations (Fig. S5). The WHAM integration method was used to calculate the free energy profile (36). The reaction coordinates were set based on the position of the octapeptin molecule relative to the OM center at z axis. Z > 0 indicated the bulk water shell and the outer leaflet of the OM; Z = 0 corresponded to the hydrophobic center of the OM.
MD simulations
GROMACS 5.1.2 was used to conduct all molecular simulations with the CHARMM36 all-atom force field (34, 37). Energy minimizations were performed using the steepest descent method with the maximum force tolerance of 1,000 KJ mol−1 nm−1. To fully equilibrate the simulation system, six equilibration cycles were carried out by gradually turning off the position restraints on the lipids. Periodic boundary conditions were considered. All production simulations were conducted at constant temperature and pressure (NPT ensemble). The temperature and pressure were maintained at 313 K using the Nose–Hoover algorithm and 1 bar using the semi-isotropic pressure coupling method with Parrinello–Rhaman barostat (38–40). The electrostatic and Van der Waals interactions were calculated using the particle mesh Ewald method and Lennard–Jones potential algorithm, respectively (41). The time step in production simulations was 2 fs.
Data availability
All data are contained in the article.
Supplementary Material
This article contains supporting information.
Author contributions—X. J. data curation; X. J. formal analysis; K. Y., B. Y., and F. S. methodology; B. G., L. Wan, N. A. P., K. D. R., and L. Wang resources; J. D. S., K. D. R., T. V., and J. L. writing-review and editing; T. V. and J. L. funding acquisition; J. L. conceptualization; J. L. supervision.
Funding and additional information—This work was supported by NIAID, National Institutes of Health Grant R01 AI132154 (to J. L. and T. V.). X. J. is recipient of the 2019 Faculty Bridging Fellowship from Monash University. J. L. is an Australia National Health Medical Research Council Principal Research Fellow. The simulations were performed on the supercomputer at Monash University (Australia) and the HPC Cloud Platform (National Key Research and Development Project 2016YFB0201702) at Shandong University (China). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- OM
- outer membrane
- Dab
- α-γ-diaminobutyric acid
- pEtN
- phosphoethanolamine.
References
- 1. Tacconelli E., Carrara E., Savoldi A., Harbarth S., Mendelson M., Monnet D. L., Pulcini C., Kahlmeter G., Kluytmans J., Carmeli Y., Ouellette M., Outterson K., Patel J., Cavaleri M., Cox E. M., et al. (2018) Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 10.1016/S1473-3099(17)30753-3 [DOI] [PubMed] [Google Scholar]
- 2. MacLean R. C., and San Millan A. (2019) The evolution of antibiotic resistance. Science 365, 1082–1083 10.1126/science.aax3879 [DOI] [PubMed] [Google Scholar]
- 3. Velkov T., Thompson P. E., Nation R. L., and Li J. (2010) Structure–activity relationships of polymyxin antibiotics. J. Med. Chem. 53, 1898–1916 10.1021/jm900999h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Landman D., Georgescu C., Martin D. A., and Quale J. (2008) Polymyxins revisited. Clin. Microbiol. Rev. 21, 449–465 10.1128/CMR.00006-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Velkov T., Roberts K. D., Thompson P. E., and Li J. (2016) Polymyxins: a new hope in combating Gram-negative superbugs? Future Med. Chem. 8, 1017–1025 10.4155/fmc-2016-0091 [DOI] [PubMed] [Google Scholar]
- 6. Srinivas P., and Rivard K. (2017) Polymyxin resistance in Gram-negative pathogens. Curr. Infect. Dis. Rep. 19, 38 10.1007/s11908-017-0596-3 [DOI] [PubMed] [Google Scholar]
- 7. Baron S., Hadjadj L., Rolain J. M., and Olaitan A. O. (2016) Molecular mechanisms of polymyxin resistance: knowns and unknowns. Int. J. Antimicrob. Agents 48, 583–591 10.1016/j.ijantimicag.2016.06.023 [DOI] [PubMed] [Google Scholar]
- 8. Liu Y.-Y., Wang Y., Walsh T. R., Yi L.-X., Zhang R., Spencer J., Doi Y., Tian G., Dong B., Huang X., Yu L.-F., Gu D., Ren H., Chen X., Lv L., et al. (2016) Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168 10.1016/S1473-3099(15)00424-7 [DOI] [PubMed] [Google Scholar]
- 9. Velkov T., Roberts K., and Li J. (2017) Rediscovering the octapeptins. Nat. Prod. Rep. 34, 295–309 10.1039/c6np00113k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Velkov T., Gallardo-Godoy A., Swarbrick J. D., Blaskovich M. A. T., Elliott A. G., Han M., Thompson P. E., Roberts K. D., Huang J. X., Becker B., Butler M. S., Lash L. H., Henriques S. T., Nation R. L., Sivanesan S., et al. (2018) Structure, function, and biosynthetic origin of octapeptin antibiotics active against extensively drug-resistant Gram-negative bacteria. Cell Chem. Biol. 25, 380–391 10.1016/j.chembiol.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meyers E., Pansy F. E., Basch H. I., Mcripley R. J., Slusarchyk D. S., Graham S. F., and Trejo W. H. (1973) EM49, a new peptide antibiotic: iii. biological characterization in vitro and in vivo. J. Antibiot. 26, 457–462 10.7164/antibiotics.26.457 [DOI] [PubMed] [Google Scholar]
- 12. Fayad A. A., Herrmann J., and Müller R. (2018) Octapeptins: lipopeptide antibiotics against multidrug-resistant superbugs. Cell Chem. Biol. 25, 351–353 10.1016/j.chembiol.2018.04.003 [DOI] [PubMed] [Google Scholar]
- 13. Hancock R. E. (1997) Peptide antibiotics. Lancet 349, 418–422 10.1016/S0140-6736(97)80051-7 [DOI] [PubMed] [Google Scholar]
- 14. Jeannot K., Bolard A., and Plésiat P. (2017) Resistance to polymyxins in Gram-negative organisms. Int. J. Antimicrob. Agents 49, 526–535 10.1016/j.ijantimicag.2016.11.029 [DOI] [PubMed] [Google Scholar]
- 15. Olaitan A. O., Morand S., and Rolain J. M. (2014) Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front. Microbiol. 5, 643 10.3389/fmicb.2014.00643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peterson A., Fesik S., and McGroarty E. (1987) Decreased binding of antibiotics to lipopolysaccharides from polymyxin-resistant strains of Escherichia coli and Salmonella typhimurium. Antimicrob. Agents Chemother. 31, 230–237 10.1128/aac.31.2.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swanson P. E., Paddy M. R., Dahlquist F. W., and Storm D. R. (1980) Characterization of octapeptin-membrane interactions using spin-labeled octapeptin. Biochemistry 19, 3307–3314 10.1021/bi00555a032 [DOI] [PubMed] [Google Scholar]
- 18. De Zoysa G. H., Cameron A. J., Hegde V. V., Raghothama S., and Sarojini V. (2015) Antimicrobial peptides with potential for biofilm eradication: synthesis and structure activity relationship studies of battacin peptides. J. Med. Chem. 58, 625–639 10.1021/jm501084q [DOI] [PubMed] [Google Scholar]
- 19. Zhu Y., Lu J., Han M., Jiang X., Azad M. A. K., Patil N. A., Lin Y., Zhao J., Hu Y., Yu H. H., Chen K., Boyce J. D., Dunstan R. A., Lithgow T., Barlow C. K., et al. (2020) Polymyxins bind to the cell surface of unculturable Acinetobacter baumannii and cause unique dependent resistance. Adv. Sci. 7, 2000704 10.1002/advs.202000704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yesylevskyy S., Marrink S. J., and Mark A. E. (2009) Alternative mechanisms for the interaction of the cell-penetrating peptides penetratin and the TAT peptide with lipid bilayers. Biophys. J. 97, 40–49 10.1016/j.bpj.2009.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao J., Zhao C., Liang G., Zhang M., and Zheng J. (2013) Engineering antimicrobial peptides with improved antimicrobial and hemolytic activities. J. Chem. Inf. Model. 53, 3280–3296 10.1021/ci400477e [DOI] [PubMed] [Google Scholar]
- 22. Sun D., Forsman J., Lund M., and Woodward C. E. (2014) Effect of arginine-rich cell penetrating peptides on membrane pore formation and life-times: a molecular simulation study. Phys. Chem. Chem. Phys. 16, 20785–20795 10.1039/c4cp02211d [DOI] [PubMed] [Google Scholar]
- 23. Santos D. E. S., Pol-Fachin L., Lins R. D., and Soares T. A. (2017) Polymyxin binding to the bacterial outer membrane reveals cation displacement and increasing membrane curvature in susceptible but not in resistant lipopolysaccharide chemotypes. J. Chem. Inf. Model. 57, 2181–2193 10.1021/acs.jcim.7b00271 [DOI] [PubMed] [Google Scholar]
- 24. Rice A., and Wereszczynski J. (2018) Atomistic scale effects of lipopolysaccharide modifications on bacterial outer membrane defenses. Biophys. J. 114, 1389–1399 10.1016/j.bpj.2018.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jefferies D., Hsu P. C., and Khalid S. (2017) Through the lipopolysaccharide glass: a potent antimicrobial peptide induces phase changes in membranes. Biochemistry 56, 1672–1679 10.1021/acs.biochem.6b01063 [DOI] [PubMed] [Google Scholar]
- 26. Fu L., Wan M., Zhang S., Gao L., and Fang W. (2020) Polymyxin B loosens lipopolysaccharide bilayer but stiffens phospholipid bilayer. Biophys. J. 118, 138–150 10.1016/j.bpj.2019.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang X., Yang K., Han M., Yuan B., Li J., Gong B., Velkov T., Schreiber F., Wang L., and Li J. (2020) Outer membranes of polymyxin-resistant Acinetobacter baumannii with phosphoethanolamine-modified lipid a and lipopolysaccharide loss display different atomic-scale interactions with polymyxins. ACS Infect. Dis. DOI: 10.1021/acsinfecdis.0c00330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hancock R. E. (1984) Alterations in outer membrane permeability. Annu. Rev. Microbiol. 38, 237–264 10.1146/annurev.mi.38.100184.001321 [DOI] [PubMed] [Google Scholar]
- 29. Jiang X., Yang K., Yuan B., Han M., Zhu Y., Roberts K. D., Patil N. A., Li J., Gong B., Hancock R. E. W., Velkov T., Schreiber F., Wang L., and Li J. (2020) Molecular dynamics simulations informed by membrane lipidomics reveal structure–interaction relationship of polymyxins with lipid A–based outer membrane of Acinetobacter baumannii. J. Antimicrob. Chemother. dkaa376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Velkov T., Roberts K. D., Nation R. L., Wang J., Thompson P. E., and Li J. (2014) Teaching 'old' polymyxins new tricks: new-generation lipopeptides targeting Gram-negative “superbugs.” ACS Chem. Biol. 9, 1172–1177 10.1021/cb500080r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Allinger N. (1977) MM2: a hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 99, 8127–8134 10.1021/ja00467a001 [DOI] [Google Scholar]
- 32. Zoete V., Cuendet M. A., Grosdidier A., and Michielin O. (2011) SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 2359–2368 10.1002/jcc.21816 [DOI] [PubMed] [Google Scholar]
- 33. Lee J., Patel D. S., Ståhle J., Park S.-J., Kern N. R., Kim S., Lee J., Cheng X., Valvano M. A., Holst O., Knirel Y. A., Qi Y., Jo S., Klauda J. B., Widmalm G., et al. (2019) CHARMM-GUI membrane builder for complex biological membrane simulations with glycolipids and lipoglycans. J. Chem. Theory Comput. 15, 775–786 10.1021/acs.jctc.8b01066 [DOI] [PubMed] [Google Scholar]
- 34. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- 35. Berglund N. A., Piggot T. J., Jefferies D., Sessions R. B., Bond P. J., and Khalid S. (2015) Interaction of the antimicrobial peptide polymyxin B1 with both membranes of E. coli: a molecular dynamics study. PLoS Comput. Biol. 11, e1004180 10.1371/journal.pcbi.1004180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hub J. S., De Groot B. L., and Van Der Spoel D. (2010) g_wham: a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput. 6, 3713–3720 10.1021/ct100494z [DOI] [Google Scholar]
- 37. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D., and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 10.1021/jp101759q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 10.1063/1.328693 [DOI] [Google Scholar]
- 39. Nosé S. (1984) A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268 10.1080/00268978400101201 [DOI] [Google Scholar]
- 40. Hoover W. G. (1985) Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 10.1103/physreva.31.1695 [DOI] [PubMed] [Google Scholar]
- 41. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained in the article.