

Abstract
Methylenetetrahydrofolate reductase (MTHFR) links the folate cycle to the methionine cycle in one-carbon metabolism. The enzyme is known to be allosterically inhibited by SAM for decades, but the importance of this regulatory control to one-carbon metabolism has never been adequately understood. To shed light on this issue, we exchanged selected amino acid residues in a highly conserved stretch within the regulatory region of yeast MTHFR to create a series of feedback-insensitive, deregulated mutants. These were exploited to investigate the impact of defective allosteric regulation on one-carbon metabolism. We observed a strong growth defect in the presence of methionine. Biochemical and metabolite analysis revealed that both the folate and methionine cycles were affected in these mutants, as was the transsulfuration pathway, leading also to a disruption in redox homeostasis. The major consequences, however, appeared to be in the depletion of nucleotides. 13C isotope labeling and metabolic studies revealed that the deregulated MTHFR cells undergo continuous transmethylation of homocysteine by methyltetrahydrofolate (CH3THF) to form methionine. This reaction also drives SAM formation and further depletes ATP reserves. SAM was then cycled back to methionine, leading to futile cycles of SAM synthesis and recycling and explaining the necessity for MTHFR to be regulated by SAM. The study has yielded valuable new insights into the regulation of one-carbon metabolism, and the mutants appear as powerful new tools to further dissect out the intersection of one-carbon metabolism with various pathways both in yeasts and in humans.
Keywords: methionine, allosteric regulation, S-adenosylmethionine (SAM), Saccharomyces cerevisiae, AMP, feedback insensitive, futile cycle, MTHFR, nucleotide depletion
One-carbon metabolism is a universal metabolic process, in which single-carbon methyl groups are transferred to enable the synthesis of several essential metabolites. These include DNA synthesis (thymidine and purines), amino acid homeostasis (serine, glycine, and methionine), redox balance, and epigenetic maintenance. This carbon partitioning across various cellular outputs involves three different pathways, namely, the folate cycle, the methionine cycle, and the transsulfuration pathways (Fig. 1A) (1). Although the centrality of one-carbon metabolism has always been recognized, recent studies have revealed the immense importance of its homeostasis for health and disease (2).
Figure 1.

MTHFR, a critical branch point of one-carbon metabolism. A, one-carbon metabolism encompasses the folate cycle, methionine cycle, and transsulfuration pathway. The folate cycle plays a crucial role in nucleotide biosynthesis (purines and thymidine) and redox balance (by utilization and production of NADPH at various steps). Folate metabolism is linked to the methionine cycle via 5-CH3THF, a folate intermediate synthesized by the activity of the MTHFR enzyme. MS remethylates homocysteine to methionine by expending 5-CH3THF, which enters the methionine cycle. The methionine cycle, in addition to providing methionine for protein translation, is the major source of SAM, the cellular methylation currency. SAM also yields polyamines via the methionine salvage pathway or enters the transsulfuration pathway via hydrolysis of SAH to homocysteine. The transsulfuration pathway synthesizes GSH via a series of steps. GSH is the major anti-oxidant moiety in the cell. SHMT, serine hydroxyl methyltransferase; TYMS, thymidine synthase; GART, glycinamide ribonucleotide transformylase; AICART, aminoimidazolecarboxamide ribonucleotide formyltransferase. B, schematic representation of eukaryotic MTHFR. Shown is the domain organization of the yeast MTHFR, Met13p, with ∼300-amino acid-long catalytic domain at the N terminus and an equally large regulatory domain at the C terminus.
Methylenetetrahydrofolate reductase (MTHFR) is an enzyme that straddles the folate and methionine cycles of one-carbon metabolism. It is a flavoprotein that catalyzes the reduction of methylenetetrahydrofolate (CH2THF) to methyltetrahydrofolate using NADPH as the reducing equivalent. The methyl tetrahydrofolate that is formed is then utilized in the methylation of homocysteine to methionine. MTHFR is thus found at a critical branch point of one-carbon metabolism and is one of the key regulatory nodes being under allosteric feedback inhibition by SAM (Fig. 1A). Typical eukaryotic MTHFR enzymes have two domains: a catalytic domain of ∼300 amino acids and an equally large regulatory domain for feedback regulation by SAM (Fig. 1B). The catalytic domain of the enzyme has been extensively studied, and the catalytic mechanism as well as the NADPH, FAD, and substrate-binding sites have been clearly delineated (3–5). Several mutations in humans leading to decreased catalytic activity have also been identified. These mutants have been implicated in several disorders such as neural tube defects and pregnancy-related complications, motor and gait disturbances, seizures, psychiatric disturbances, and other neurological abnormalities and cardiovascular diseases and continue to be investigated (5–8). The role of the regulatory domain of MTHFR, however, in contrast, has not been rigorously investigated. This is surprising, considering that MTHFR essentially links the two cycles, and thus its regulation is likely to be important for the homeostasis of one-carbon metabolism. Indeed, the consequences of an absence of regulation by SAM of an otherwise functional MTHFR has been unclear till now.
In this article, we describe the isolation and characterization of feedback-insensitive, deregulated yeast MTHFR mutants that emerged from a rigorous search for such mutants. The observation that such mutants show a severe growth defect in methionine medium enabled their characterization and helped to define a region within the regulatory domain that is critical for regulation by SAM, which we established in vitro. Using targeted metabolite analysis, we found that although the folate, the methionine cycle and the transulfuration pathway were affected in these mutants, the major consequences appeared to be in the depletion of nucleotides. Further genetic analysis coupled with 13C isotope labeling and metabolic studies suggested that the cells undergo continuous methionine and SAM synthesis where the ATP and methionine were being continuously recycled. This led to futile cycles of SAM synthesis and recycling and consequently an accentuated nucleotide depletion in these cells.
Results
The regulatory region of the yeast MTHFR protein, Met13p: predicting potential residues critical for SAM binding
Toward the goal of creating a deregulated eukaryotic MTHFR protein in yeast that would be insensitive to allosteric inhibition by SAM (9), we initially carried out an analysis of the regulatory region of MTHFR. This region has not been subjected to any systematic investigations so far. The yeast MTHFR amino acid sequence was compared with other eukaryotic MTHFR enzymes. The catalytic domains of these proteins, expectedly, show high similarity (42% identity is seen with the human and yeast protein across the entire catalytic region). The eukaryotic MTHFRs also show a high sequence similarity in the regulatory region, with a 43% identity with the human and yeast proteins, across the entire regulatory region of the yeast MTHFR, residues 337–600 (Fig. S1). Further, within the large regulatory domain, one observes short blocks of high identity across all these organisms. We have defined these regions as conserved region 1 (CR1) corresponding to amino acids 340–361, conserved region 2 (CR2) corresponding to amino acids 440–445, and conserved region 3 (CR3) corresponding to amino acids 517–527 (Fig. 2A).
Figure 2.
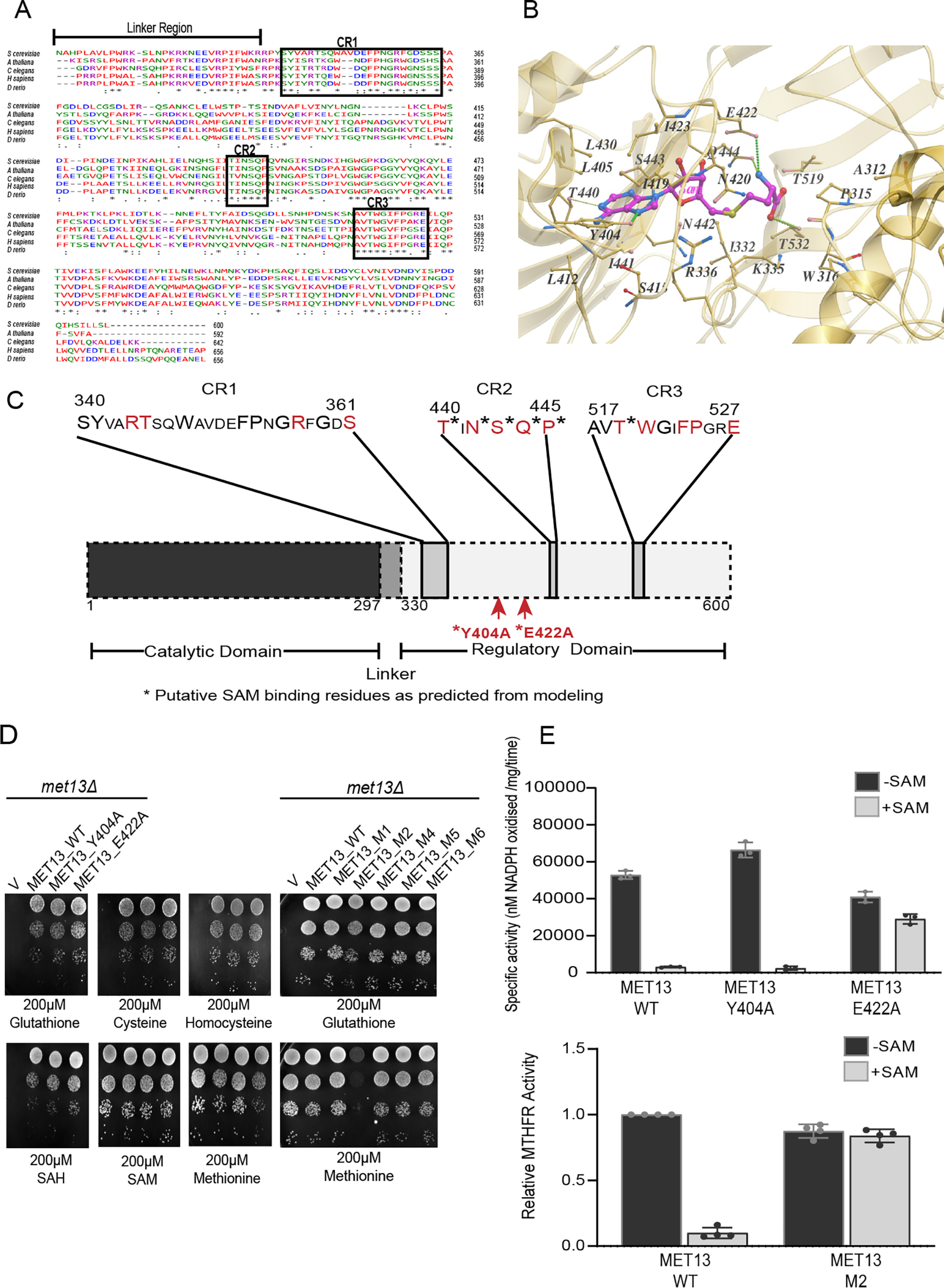
The regulatory region of the yeast MTHFR protein, Met13p: predicting potential residues critical for SAM binding and enzyme inhibition. A, sequence alignment of the regulatory domain of yeast (NP_011390.2), plant (NP_191556.1), roundworm (NP_741028.1), human (NP_005948.3), and zebrafish (NP_001121727.1) MTHFR proteins performed using Clustal omega. Asterisks indicate identical residues; periods and colons indicate varying degrees of conserved substitution; and dashes indicate gaps in the alignment. Sequence alignment analysis of MTHFR orthologs indicates three blocks of highly conserved regions (marked here as CR1, CR2, and CR3) within the regulatory domain. B, molecular docking and structural analysis of the modeled MET13 protein predicts several residues within the regulatory domain as the probable SAM-binding residues. SAM (represented in pink sticks) was docked with modeled Met13p using AutodockVina. Amino acid residues that might be involved in SAM binding are marked with yellow sticks. C, schematic illustration of residues selected for mutational analysis based on insights from structure and sequence analysis. Conserved residues are indicated in a bigger font, residues subjected to mutational analysis are marked in red, and an asterisk indicates the residues that were predicted to bind to SAM through modeling studies. D, S. cerevisiae met13Δ strains transformed with vector control (V), MET13_WT, structure-based mutants (MET13_Y404A and MET13_E422A), and conserved region mutants (MET13_M1, MET13_M2, MET13_M4, MET13_M5, and MET13_M6) were grown to exponential phase in minimal medium containing 200 μm GSH, harvested, washed, resuspended in water, and serially diluted to give 0.1, 0.01, 0.001, and 0.0001 A600 of cells. 10 μl of these dilutions were spotted on minimal medium containing 200 μm of different sulfur sources (GSH, cysteine, homocysteine, SAH, SAM, and methionine). The photographs were taken after 48 h of incubation at 30 °C. The experiment was repeated three times, and a representative data set is shown. E, measurement of catalytic activity of MET13_WT and its structural mutants (MET13_Y404A and MET13_E422A) and MET13_M2 without and with 200 μm SAM was performed using NADPH-menadione oxidoreductase assay as described under “Experimental procedures.” The activity was calculated as the rate of disappearance of NADPH by monitoring absorbance at 343 nm and using the extinction coefficient of NADPH as 6220 m−1 cm−1. The concentration of the protein samples measured using the Bradford assay was used for calculating the specific activities of all the purified protein samples. The experiment was done thrice, along with three technical replicates for each sample protein. The graph here corresponds to the representative data set plotted using the average of the three technical replicates along with ± S.D. values.
We initially built a homology model of the yeast MTHFR, Met13p, based on the crystal structure of human MTHFR, which has been recently solved (10). The modeled yeast MTHFR structure aligns well with human MTHFR protein with an root-mean-square-deviation value of 0.48 Å (Fig. S2), which exists as a homodimer with each monomer consisting of a catalytic domain and a regulatory domain (Fig. S3). The two monomers are held together by interactions of the regulatory domain, as has been reported for the human MTHFR (10).
To predict the SAM-binding residues of MET13, we docked the SAM molecule using a computational docking program, AutodockVina. Among the probable SAM-binding residues, two residues, Glu422 and Tyr404, appeared critical (Fig. 2B). Glu422 is the equivalent of the mammalian Glu463 that is predicted to be involved in binding to SAM (10). In the yeast MTHFR, Tyr404 also appeared to be critical because its benzyl ring can putatively interact with the aromatic ring of SAM around the hydrophobic region within the SAM-binding pocket.
These structural predictions, as well as the structural predictions made with the human enzyme (10), were not consistent with earlier experimental studies in which photoaffinity labeling of SAM was carried out to find out the SAM-binding regions of porcine and human MTHFR (7). These studies had indicated a completely different region of the MTHFR regulatory domain, i.e. within a stretch of ∼30 amino acids starting 30 residues after the junction between the two domains (7, 9, 11, 12). In particular, a 6-kDa region of the protein located at the N-terminal end of the regulatory domain was suggested in these studies to be important for SAM binding (7) that corresponds approximately to residues 340–390 in the yeast MTHFR. However, this region was quite distinct from the structural predictions.
Mutational analysis of different regions of the regulatory domain of MTHFR, reveals a region in CR1 critical for SAM regulation
To identify which regions of the regulatory region might be important for SAM-mediated repression of MTHFR, we targeted both the structure-based predictions of SAM-binding residues and residues within the conserved regions for mutational analysis. We generated independent single-point alanine mutants of both the structure predicted Tyr404 and Glu422, and also targeted multiple residues within the conserved regions (CR1, CR2, and CR3) using site-directed mutagenesis (Fig. 2C). In these regions, we created the mutants M1, M2, M4, M5, and M6. M1 (R344A and T345A) and M2 (R357A and S361A) are present in the CR1 region, M4 (T440A, N442A, S443A, Q444A, and P445A) is in the CR2 region, whereas M5 (T519A and W520A) and M6 (F523A, P524A, and E527A) are the mutations made in the CR3 region (Table S1).
These mutants in Met13p, the yeast MTHFR, were assayed for functionality by their ability to complement the methionine auxotrophy of the met13Δ strain. All the mutants were functional based on the utilization of GSH as sulfur source (Fig. 2D). However, when we examined their growth in minimal medium containing different sulfur sources, we observed that MET13_E422A showed a slight defect in growth on minimal medium containing methionine, whereas the M2 mutant (R357A and S361A), which was also functional, showed an even more serious growth defect, once again on methionine medium (Fig. 2D). Neither MET13_Y404A (predicted from the modeled structure) nor any of the other conserved region mutants (M1, M4, M5, and M6) exhibited any growth defect either on methionine or in any of the other tested sulfur sources (Fig. 2D).
Because it was possible that a deregulated MTHFR might show a growth defect, we examined this more rigorously by evaluating in vitro the inhibition of enzymatic activity by SAM at the protein level. Accordingly, these mutants were subcloned into an Escherichia coli expression vector, and the purified recombinant proteins (Fig. S4) were evaluated for enzymatic activity and SAM inhibition. We included in this analysis the WT protein MET13_WT, MET13_Y404A and the two mutants that showed a growth defect, MET13_E422A and MET13_R357AS361A (MET13_M2). All three mutants and the WT displayed MTHFR activity. The WT and MET13_Y404A mutant, which did not present any growth phenotype on the methionine plate, exhibited strong repression (∼95%) in the presence of 200 μm SAM (Fig. 2E). However, MET13_E422A, which displayed a mild growth defect on methionine medium, showed only mild repression (∼30%) in the presence of SAM (Fig. 2E). Moreover, in the case of the MET13_M2 mutant, which bears point mutations at two amino acid residues (R357A and S361A) within the CR1 region mutants, we observed an almost complete absence of allosteric inhibition by SAM (Fig. 2E).
A short stretch (residues from Phe353 to Gly359) within CR1 is critical for MTHFR regulation
The MET13_M2 mutant carried two mutations, R357A and S361A. To determine whether the mutation in both the residues together (R357A and S361A) contributed to this phenotype and also whether other amino acids within the CR1 region might be important, we carried out a more detailed analysis of the CR1 region. We made individual alanine mutants for 11 of the conserved residues within CR1 (S340A, Y341A, R344A, T345A, W348A, F353A, P354A, G356A, R357A, G359A, and S361A) and checked their growth on methionine medium. The mutants were functional, as seen by functional complementation of met13Δ strain on GSH supplemented medium. However, the methionine-specific growth defect was observed in the F353A, P354A, G356A, R357A, and G359A mutants of Met13p (Fig. 3A). This analysis of the CR1 region spanning 340–361 residues indicated that the stretch 353–359 seems critical for MTHFR regulation by SAM because residues outside this region did not exhibit any growth defect on methionine medium.
Figure 3.

A short stretch (residues from Phe353 to Gly359) within CR1 is critical for MTHFR regulation. A, S. cerevisiae met13Δ strains transformed with vector control (V), MET13_WT, and single point alanine mutants of conserved residues (S340A, Y341A, R344A, T345A, W348A, F353A, P354A, G356A, R357A, G359A, and S361A) within CR1 were selected on 200 μm GSH. These transformants were grown to exponential phase in minimal medium containing 200 μm GSH, harvested, washed, resuspended in water, and serially diluted to give 0.1, 0.01, 0.001, and 0.0001 A600 of cells. 10 μl of these dilutions were spotted on minimal medium containing either 200 μm GSH or 200 μm methionine as the sulfur source. The photographs were taken after 48 h of incubation at 30 °C. The experiment was repeated three times, and a representative data set is shown. B, measurement of catalytic activity of MET13_WT, MET13_P354A, and MET13_R357A, without and with 200 μm SAM, was performed using NADPH-menadione oxidoreductase assay as described under “Experimental procedures.” The experiment was done thrice, along with three technical replicates for each sample protein. The graph here corresponds to the representative data set plotted using the average of the three technical replicates along with ± S.D. values. C, the methionine-dependent growth defect of deregulated MET13 mutant (MET13_R357A) requires a functionally active enzyme. S. cerevisiae met13Δ strains transformed with vector control (V), MET13_WT, MET13_E422A (catalytically inactive mutant), MET13_R357A, and MET13_E22A.R357A were selected on 200 μm GSH. These transformants were grown to exponential phase in minimal medium containing 200 μm GSH, harvested, washed, resuspended in water, and serially diluted to give 0.1, 0.01, 0.001, and 0.0001 A600 of cells. 10 μl of these dilutions were spotted on minimal medium containing either 200 μm GSH or 200 μm methionine as the sulfur source. The photographs were taken after 48 h of incubation at 30 °C. The experiment was repeated three times, and a representative data set is shown.
MET13_P354A and MET13_R357A, two of the mutants within the CR1 region that display the most severe growth defect in methionine medium, were assayed for SAM insensitivity. The catalytic activity in the absence of SAM for both MET13_P354A and MET13_R357A was slightly higher than the WT protein (Fig. 3B). Additionally, both these mutants retained MTHFR activity even in the presence of the SAM (Fig. 3B). MET13_P354A was partially inhibited (repression, 50%), whereas MET13_R357A completely lacked inhibition (Fig. 3B).
The methionine-dependent growth defect of deregulated MET13 mutant (MET13_R357A) requires a functionally active enzyme
The deregulated mutants showed a severe growth defect on methionine plates. The possibility existed that the growth defect of these mutants was unrelated to its activity and was a consequence of the interference of these mutants with some other protein or an unrelated pathway. It was therefore important to establish whether the growth defect of these mutants was due to its unregulated activity. One way of establishing this link was to create a catalytically inactive mutation of the deregulated MTHFR. We therefore first created a mutation of the active-site residue of MTHFR, Glu22 (13). An E22A mutation led to an inactive MTHFR, as seen by its inability to complement the met13Δ defect (Fig. 3C). We then introduced this E22A mutation in the R357A deregulated mutant background. We observed that the R357A deregulated mutant carrying the E22A mutation no longer displayed the growth defect on methionine (Fig. 3C). This result clearly established that the deregulated mutants displayed the methionine-specific growth defect because of the deregulated property of MTHFR.
MTHFR deregulation disrupts the cellular folate and nucleotide pools
MTHFR is an enzyme that straddles two cycles: the folate cycle and the methionine cycle. It is also a key regulatory step in one-carbon metabolism. Therefore, it was possible that the deregulated enzyme might impact the metabolic homeostasis of both cycles, although which of the cycles might be primarily affected was difficult to predict. Because MTHFR-deregulated enzymes have not been isolated earlier, we were keen to understand the metabolic consequences. We exploit in particular the acute phenotype seen in the methionine medium for our analysis. We first examined the folate pools and whether cells might be facing folate deficiency caused by the deregulated MTHFR. Although plasma membrane folate transporters have not been reported in yeast, we nevertheless supplemented cells with increasing concentrations of folic acid in the medium. At higher concentrations of folic acid, we observed a significant rescue of growth (Fig. 4A). To further define which folate pools were deficient, we examined the steady-state levels of the different folate pools. Although folate pools are continuously cycling and are also difficult to measure reliably because of their lability, we nevertheless attempted to estimate these pools. Using LC–MS/MS analysis, we observed a decrease in the levels of the substrate of MTHFR, CH2THF in the deregulated mutant, MET13_R357A, whereas pools of the product of MTHFR, CH3THF, appeared to be higher in the mutant (Fig. S5). The levels of these two folate intermediate (CH2THF and CH3THF) indicated increased activity of MTHFR while suggesting that the cells could be facing some limitations in the pools of CH2THF. Interestingly, an increase in the THF pools for the deregulated mutant suggested that the activity of the downstream enzyme methionine synthase (MET6) involving the donation of the CH3 group to homocysteine could be increased (Fig. S5).
Figure 4.

MTHFR deregulation disrupts the cellular folate and nucleotide pools. A, yeast met13Δ strains transformed with vector control (V), MET13_WT, and MET13_R357A were grown to the exponential phase in minimal medium containing 200 μm GSH, harvested, washed, resuspended in water, and serially diluted to give 0.1, 0.01, 0.001, and 0.0001 A600 of cells. 10 μl of these dilutions were spotted on minimal medium containing either 200 μm GSH, methionine, and methionine along with the increasing concentrations of folic acid (10–1000 μm). The photographs were taken after 72 h of incubation at 30 °C. The experiment was repeated three times, and a representative data set is shown. B, the relative abundance of nucleotide pools in transformants bearing the WT MET13 or deregulated mutant of yeast MTHFR, MET13_R357A. Transformants were grown overnight in SD minimal medium along with GSH (200 μm) as a sulfur source and reinoculated to medium containing methionine (200 μm), and different nucleotide intermediates were determined by LC–MS/MS. The graphs show a representative data set of three biological replicates. C, S. cerevisiae met13Δ strains transformed with vector control (V), and MET13_WT and MET13_R357A were grown to exponential phase in minimal medium containing 200 μm GSH, harvested, washed, resuspended in water, and serially diluted to give 0.1, 0.01, 0.001, and 0.0001 A600 of cells. 10 μl of these dilutions were spotted on minimal medium containing 200 μm GSH, 200 μm methionine, and different concentrations of either adenine or guanine (50–1000 μm) in methionine-supplemented plates. The photographs were taken after 48 h of incubation at 30 °C. The experiment was repeated three times, and a representative data set is shown.
Because CH2THF, the substrate of MTHFR is essential for the synthesis of nucleotides (purines and thymidine), depleted CH2THF pools would be reflected in depleted nucleotide pools. Therefore, we determined the nucleotide levels in the MTHFR-deregulated mutant strains as a readout of an impaired folate cycle. We observed a striking 10-fold depletion of the cellular AMP, GMP, and CMP pools in the mutant-bearing cells (Fig. 4B). To examine whether external supplementation might rescue the growth, we performed a growth assay by supplementing mutant cells with either adenine or guanine (0.05–1 mm). We observed that adenine supplementation as little as 0.05 mm rescued the growth defect of the deregulated MTHFR mutant (Fig. 4C). However, guanine at even 1 mm did not restore the growth of the deregulated mutant (Fig. 4C). A likely explanation for the inability of guanine to complement the growth defect is the inability of guanine to interconvert to adenine, whereas adenine is able to interconvert to guanine intracellularly (14). Hence, the restoration of both would be required to restore cell growth. Although these observations are consistent with the depletion of CH2THF, which is a precursor in nucleotide biosynthesis, the much steeper decreases in the nucleotide levels suggested that folate depletion might be only one of the causative factors.
Cells bearing deregulated MTHFR display depleted cofactor pools
The MTHFR enzyme uses NADPH as the reducing equivalent for the catalytic activity. Additionally, it has a FAD moiety noncovalently bound to it, which is also essential for the oxidoreductase activity. Both NADPH/NADP and FAD/FADH2 derive from adenine pools. Furthermore, recent studies have shown that in addition to the pentose phosphate pathway, the folate pathway is also an important contributor to the NADPH pools in the cell (15). Thus, with lower folate and adenine pools in the cells, the pools of the cofactors would also be expectedly lower. Furthermore, with unregulated MTHFR activity, the reduced NADPH pools that are required for numerous anabolic reactions would be further depleted, whereas the specific oxidized FAD pools that are cofactors in many essential enzymes would also be similarly depleted. We measured total NADPH/NADP pools in cellular extracts of transformants bearing either MET13_WT or MET13_R357A plasmid. We observed an approximate 2–3-fold reduction in total NADPH/NADP pools in cells expressing the deregulated MET13 mutant when compared with the WT cells upon growth minimal medium containing 200 μm methionine (Fig. 5A). Deficiency in the nucleotide pools could be responsible for this significant decrease. Further, because NADPH represents ∼95% (ratio of NADPH/NADP varies from 15 to 60) of the total content of NADPH and NADP pools, it indicates primarily a depletion in the reduced NADPH pools (16). If this is the case, then merely replenishing the reduced pools might in itself provide some benefit to the cells. We examined this by replenishing the NADPH pools by overexpression of the ZWF1 gene, which encodes the glucose-6-phosphate dehydrogenase (G6PDH) enzyme. G6PDH, the first step in the pentose phosphate pathway, is the major enzyme converting NADP to NADPH, and this is the main NADPH-generating enzyme in living cells (17). We observed that the coexpression of ZWF1 with the deregulated mutant, MET13_R357A, led to a significant growth rescue (Fig. 5B). These findings confirm that the unregulated MTHFR activity leads to the depletion of the cellular reserves of NADPH, which can be partly restored by the reductive biosynthetic activity of G6PDH.
Figure 5.

Cells bearing deregulated MTHFR display lack of desired cofactor pools. A, intracellular levels of NADPH in transformants bearing the WT MET13 or deregulated mutant of yeast MTHFR, MET13_R357A. Transformants were grown overnight in SD minimal medium along with GSH (200 μm) as a sulfur source. These were then reinoculated at 0.15 A600 in fresh SD medium without any sulfur source. Methionine was added to the transformants after 3 h of secondary inoculation, and samples were collected for NADPH estimation as the cells reached 0.6-0.8 OD (WT ∼ 10 h, MET13_R357A ∼22 h). NADPH was estimated using NADP/NADPH-GloTM assay kit. The graph shows a representative data set of three biological replicates. Error bars indicate S.D. (n = 3). *, p < 0.05; **, p < 0.01. B and C, S. cerevisiae met13Δ strains were cotransformed with MET13_R357A either with vector control (V) and ZWF1 (B) or FRD1 (C). The transformants were grown to exponential phase in minimal medium containing 200 μm GSH, harvested, washed, resuspended in water, and serially diluted to give 0.1, 0.01, 0.001, and 0.0001 A600 of cells. 10 μl of these dilutions were spotted on minimal medium containing either 200 μm GSH or methionine. The photographs were taken after 72 h of incubation at 30 °C. The experiment was repeated three times, and a representative data set is shown.
The cofactor FAD that is bound to MTHFR plays a critical role in many other oxidoreductase reactions as well, where it aids in transferring electrons in these redox reactions. MTHFR is a flavin-dependent oxidoreductase that requires oxidized FAD for transferring the electrons and reducing equivalents to its substrate, CH2THF. Because FAD is also derived from adenine, the deregulated cells would have lower total pools of FAD/FADH2 and a higher demand for oxidized FAD. We investigated this possibility by restoring the cellular reserves of oxidized FAD. Fumarate reductase is a flavo-protein and catalyzes the reduction of fumarate to succinate along with the synthesis of oxidized FAD (18). Yeast have two fumarate reductase: FRD1, a cytosolic fumarate reductase, and OSM1, a mitochondrial fumarate reductase (19). We coexpressed the cytoplasmic fumarate reductase FRD1 gene along with a deregulated Met13p and examined the growth on methionine plates. FRD1 overexpression significantly rescues the defective growth of the deregulated mutant on methionine medium (Fig. 5C). These results confirm that NADPH and oxidized FAD, both of which are required for the catalytic activity of MTHFR, becomes limiting in a deregulated mutant of MTHFR.
The impact of the deregulated MTHFR on amino acids pools, SAM, and GSH
MTHFR links the folate cycle to the methionine cycle that leads to the synthesis of methionine, SAM, cysteine, and GSH. In addition to the sulfur amino acids, cysteine and methionine, which are required for protein biosynthesis, SAM is required for methylation in a host of reactions with lipids, proteins, and DNA. At the same time, GSH is critical for redox homeostasis. We were, therefore, interested in understanding the consequences of MTHFR deregulation on the levels of these different sulfur metabolites, their intermediates that fall in the reverse transsulfuration pathway and amino acid pools in general.
We therefore carried out the analysis of the sulfur metabolites in a met15Δmet13Δ background. MET15 deletions are deficient in inorganic sulfur assimilation; thus, all the sulfur in such cells is derived from the organic sulfur supplemented in the medium (methionine or GSH). The cells carried a deletion in the MET13 gene and thus lacked the endogenous MTHFR (met13Δ). The strains thus only carried on the plasmids, either the WT or mutant MTHFR. The transformants exposed to methionine were harvested at two different cell densities, i.e. A600 0.5 and 1.0, corresponding to the early and midexponential phase of growth, respectively, and these samples were used to extract metabolites for targeted LC–MS/MS–based metabolite estimations.
Even though the cells were deregulated for MTHFR and were also grown in medium containing exogenous methionine, the methionine levels surprisingly remained almost unchanged; instead, they showed a minor reduction (1.2-fold) in the mutants (Fig. 6A). SAM levels were, however, increased, whereas SAH showed decreased levels in the deregulated mutant (Fig. 6A). The metabolites of the reverse transsulfuration pathway, homocysteine, cystathionine, and cysteine showed decreased levels, the most striking decrease being in the levels of cystathionine (Fig. 6A). We also observed lower levels of both the oxidized and reduced forms of glutathione (GSH and GSSG). To confirm whether the cells were indeed facing a GSH depletion, we examined their sensitivity to methylglyoxal (MG) because GSH is required for detoxification of MG (20). Indeed, cells transformed with MET13_R357A showed enhanced sensitivity to MG compared with the WT Met13p, suggesting that GSH was depleted in these strains (Fig. S6).
Figure 6.

The impact of the deregulated MTHFR on amino acids, SAM, and GSH. A, relative intracellular levels of different intermediates in the methionine cycle and transsulfuration pathway positioned in the context of the sulfur amino acid pathway, from the S. cerevisiae (ABC2613) transformants with MET13_WT or MET13_R357A that were grown overnight in minimal medium with amino acid supplements and GSH. These were then reinoculated at 0.15 A600 in fresh SD medium without any sulfur source. Methionine was added to the transformants after 3 h of secondary inoculation, and the samples were collected for metabolite extraction at 0.5 and 1.0 OD. The graph here corresponds to the representative data set plotted using the average of four biological replicates along with ± S.D. values. Error bars indicate S.D. (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001. B, relative intracellular levels of the amino acid pools estimated from yeast transformants of MET13_WT or MET13_R357A in met13Δmet15Δ (ABC2613) that were grown overnight in minimal medium with amino acid supplements and GSH. These were then reinoculated at 0.15 A600 in fresh SD medium without any sulfur source. Methionine was added to the transformants after 3 h of secondary inoculation, and samples were collected in triplicate at 0.5 or 1.0 OD. The graph here corresponds to the representative data set of 1.0 OD cells plotted using the average of three biological replicates along with ± S.D. values. Error bars indicate S.D. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant.
Estimations of the pools of the other nonsulfur amino acids were also carried out, and although the pools of the majority of amino acids were lower, a few of the amino acids revealed sharper differences (Fig. 6B). This included aspartic acid, threonine, and arginine. The trends for all the metabolites were similar at 0.5 (Fig. S7, A and B) and 1.0 OD cells; however, the differences were more striking for sulfur metabolites and arginine at the midexponential phase of growth.
Methionine biosynthesis continues in the deregulated MTHFR even in the presence of exogenous methionine
The surprisingly unchanged levels of methionine suggested that in the presence of exogenous methionine, the homeostasis of cellular methionine might be maintained by repression of synthesis. To determine the relative importance of endogenous biosynthesis of methionine and direct uptake of exogenous methionine in the observed phenotype, we initially adopted a genetic strategy to investigate the phenomenon. MET6 encodes methionine synthase enzyme that accepts the methyl group from CH3THF to convert homocysteine to methionine. When we deleted this gene, we observed that the strain, met6Δ lacked methionine biosynthetic capacity. This was evident from its inability to grow on GSH as the sulfur source because the strain had now become strictly auxotrophic for methionine. Importantly, the strain also showed a reduced growth defect when transformed with the deregulated MTHFR (Fig. 7A). We then evaluated the contributions of methionine uptake, where we deleted MUP1, a high-affinity transporter of methionine. Interestingly, the mup1Δ strains also did not show the growth defect on methionine in the presence of the deregulated MTHFR (Fig. 7B). These experiments seemed to indicate that both endogenous biosynthesis and the transport of exogenous methionine were essential factors in the growth defect on methionine.
Figure 7.

Methionine biosynthesis continues in the deregulated MTHFR even in the presence of exogenous methionine. Yeast met13Δ and met6Δmet13Δ (A) or mup1Δmet13Δ (B) strains were transformed with vector control (V), MET13_WT, MET13_F353A, MET13_P354A, MET13_G356A, and MET13_R357A were grown to exponential phase in minimal medium containing 200 μm GSH, harvested, washed, resuspended in water, and serially diluted to give 0.1, 0.01, 0.001, and 0.0001 A600 of cells. 10 μl of these dilutions were spotted on minimal medium containing 200 μm GSH or different concentrations of methionine. The photographs were taken after 48 h of incubation at 30 °C. The experiment was repeated three times, and a representative data set is shown. C, 13C label redistribution, fate of externally fed, histidine, lysine, leucine, and methionine is depicted. The proportional MIDs of protein-derived amino acid fragments retrobiosynthetically report on the labeling of precursors. These amino acid fragments were obtained by TBDMS derivatization and GC-MS ionization and are presented by their m/z and carbon backbone.
Despite the suggestions from the genetic experiments, we needed more rigorous confirmation of whether methionine biosynthesis by CH3THF was indeed taking place. Although methionine biosynthesis appeared to be required (based on the met6Δ observations), this normally does not occur in a strain that is supplemented by methionine, because in the presence of exogenous methionine, methionine biosynthesis is repressed. To evaluate this aspect, we resorted to 13C metabolic studies.
We first examined the 13C incorporation in the four externally fed amino acids: leucine, lysine, histidine, and methionine that were added to fulfill the nutritional requirement. When external amino acids are supplemented, the corresponding biosynthetic pathway is strongly repressed. Thus, as expected, in the case of leucine, lysine, and histidine de novo biosynthesis was not observed, and thus these amino acids are principally used from the external medium (Table S3 and Fig. 7C). We see only an exception with methionine. The [1-13C]-fed cells indicated significantly higher m + 1 isotopomer for all these fragments of methionine in the case of deregulated MTHFR bearing cells compared with the WT (Fig. 7C). The first four-carbon backbone of methionine are derived from aspartate, and the last carbon, which is 13C-labeled, is derived from one-carbon metabolism. The presence of higher m + 1 isotopomer in fragment ions of methionine, Met292[C2-5] lacking C1 and Met320[C1-5] (Fig. 7C) confirms CH3THF (a one-carbon intermediate of folate cycle) as the precursor for the increased label of methionine in mutant. These results reveal a continuous biosynthesis of methionine in yeast (with significantly higher rates in the mutants) through the substrates of 13C-labeled CH3 and homocysteine. The mass isotopomer distributions of methionine also shows that the homocysteine, one of the substrates of methionine synthesis, is primarily derived from SAM via methionine cycle rather than from aspartate, which would have otherwise led to m + 2 mass isotopomers in methionine. This finding confirms the significance of externally fed methionine for the defective growth phenotype of the deregulated mutant.
Cells with the deregulated MTHFR show minor readjustments in relative metabolic fluxes
The continuous synthesis of methionine even in the presence of exogenous methionine and its specific effect on growth retardation required a better understanding of the impact of methionine over the metabolic fluxes and the cellular metabolism of the deregulated mutant. Interestingly, one observes a dramatic difference in growth seen on plates; however, when the growth was analyzed in the liquid medium, the generation time of the mutant-bearing cells was only ∼2.3 times the WT (generation time of 10.5 h for the mutant as compared with 4.5 h for the WT) (Fig. 8A).
Figure 8.

Cells bearing the deregulated MTHFR do not show major changes in relative metabolic fluxes. A, yeast transformants of MET13_WT or MET13_R357A in met13Δmet15Δ (ABC2613) that were grown overnight in minimal medium with amino acid supplements and GSH were analyzed for growth. These were then reinoculated at 0.15 A600 in fresh SD medium without any sulfur source. Methionine was added to the transformants after 3 h of secondary inoculation. The experiment was repeated three times, and a representative data set is shown. B, the 13C label redistribution via glycolysis, pentose phosphate pathway, TCA cycle, one-carbon metabolism (represented along with folate cycle, methionine cycle, methionine salvage pathway, polyamine synthesis, transsulfuration pathway, and amino acid biosynthesis are highlighted. Also, the fate of externally fed, histidine, lysine, leucine, and methionine is depicted. The proportional MIDs of protein-derived amino acid fragments retrobiosynthetically report on the labeling of precursors. These amino acid fragments were obtained by TBDMS derivatization and GC-MS ionization and are presented by their m/z and carbon backbone. The 13C redistribution in the carbon atoms of central metabolites is derived from our analysis and highlighted via filled light-gray circles. The open circles represent unlabeled carbon, whereas the circles with close vertical lines represents either unlabeled 12C or labeled 13C incorporation. In the methionine cycle and methionine salvage pathway, additional carbons from ATP (adenine and ribose unit) are depicted in circles with close vertical lines. An asterisk represents significance at 99% significance level analyzed via t test. Filled gray circles, 13C; white circles, labeled carbon; circles with close vertical lines, unlabeled 12C or labeled 13C; PPP, pentose phosphate pathway; G6P, glucose-6-phosphate; 3PG, 3-phosphoglycerate; DHF, dihydrofolate; THF, tetrahydrofolate; 5,10-CH2THF, 5,10-methylene tetrahydrofolate; 5-CH3THF, 5-methyl tetrahydrofolate; 5-HCO-THF, 5- formyltetrahydrofolate; 10-HCOTHF, 10-formyltetrahydrofolate; 5,10-CH*THF, 5,10-methenyl tetrahydrofolate; dcSAM, decarboxylated SAM; MTA, methylthioadenosine; MTR, methylthioribose; SHMT, serine hydroxymethyl transferase; TYMS, thymidylate synthase; GART, glycinamide ribonucleotide transformylase; AICART, aminoimidazolecarboxamide ribonucleotide formyltransferase.
To examine alterations in pathway preferences, we further analyzed the 13C isotope labeling data to analyze the fluxes of these pathways under pseudo–steady-state conditions, as mentioned in the previous section. The label incorporation in all the amino acids except leucine, lysine, histidine (discussed in the previous section), and glycine were observed as indicated by the relative mass isotopomer distributions (Table S3 and Fig. 7C). The 13C redistribution in the amino acids retrobiosynthetically reporting on the central precursors of different pathways of central metabolism shows that the relative fluxes in the WT and mutant are predominantly stable with only minor but appreciably significant readjustments as supported by the quantitative analysis of the extent of 13C proportions in the mass isotopomers.
Glycolysis is active in both WT and mutant cells based on 13C incorporation in serine and alanine that retrobiosynthetically report on the label incorporations in the glycolytic intermediates 3-phosphoglycerate and pyruvate, respectively (Fig. 8B).13C label incorporation detected in phenylalanine MIDs indicates an active pentose phosphate pathway for both the MET13_WT and MET13_R357A transformed cells (Table S2). Likewise, the label incorporation in aspartate, glutamate, and proline confirms an active TCA cycle (Fig. 8B and Table S3). However, minor differences in the relative abundances of mass isotopomers were observed between the WT and the mutant, suggesting minor readjustments of central metabolism.
From the [1-13C]glucose-fed cells, two amino acids serine and alanine displayed minor but significant differences in 13C incorporation between WT and mutant highlighting readjustment in glycolysis. The relative proportion of m + 1 in Ser362[C2-3] fragment (lacking C1) compared with the Ser390[C1-3] confirms that C3 was labeled via glycolysis (Fig. 8B). The relative levels of m + 1 in the mutant for both serine and alanine (that are derived from glycolysis) were marginally yet significantly lower than the WT, implying a slightly lesser rate of glucose oxidation (significant based on t test) (Fig. 8B and Table S3).The data provide strong evidence that relative glucose oxidation via glycolysis was marginally lower in the mutant as compared with the WT, and this could be a consequence of the depleted ATP levels in the cell. With lower glycolysis, it can be speculated that the relative activities via a pentose phosphate pathway would be higher. The conversion of serine to glycine releases the labeled C3 into one-carbon pools (CH2THF), which was 13C-enriched (Fig. 8B). However, regardless of these minor differences, our data suggest that the deregulated mutant, which has a slower growth rate, is undergoing minor metabolic readjustments to behave metabolically like the WT without any major changes in the relative fluxes.
In the deregulated MTHFR mutant cells, SAM cycles in futile salvage pathways wherein both methionine and adenine are continuously utilized and recycled
The absence of any major metabolic readjustments in the deregulated mutant as compared with the WT suggested that pathways downstream of methionine were responsible for the methionine-dependent growth defect in the MTHFR-deregulated mutants. Methionine formation drives the ATP-dependent enzymatic synthesis of SAM. The metabolite analysis had indicated an increase in SAM, which can meet two distinct fates. In the first case, SAM is involved in methylation of a variety of cellular substrates such as nucleic acids, proteins, and lipids. Recent studies in yeast have reported phosphatidylethanolamine (PE) methylation as the major SAM consumer (21). Therefore, we quantified the relative abundance of phospholipids (PE and phosphatidylcholine (PC)) in the deregulated MTHFR mutant. Mutant harboring cells show a noticeable increase (≈1.6-fold) in the total PC levels along with a marginal decline in cellular abundance of PE (Fig. 9). An increase in SAH, a side product of PE methylation and other methylation reactions, is generally observed with increased methylation. However, SAH, in turn, is hydrolyzed to homocysteine and adenosine, both of which are in increased requirements in the mutant. Adenosine is required because of the depleted adenine pools, whereas homocysteine is required because of the continuous unregulated formation of CH3THF. The CH3 group of CH3THF is accepted by homocysteine to form methionine again, and subsequently, the methionine formed drives the ATP-dependent formation of SAM.
Figure 9.
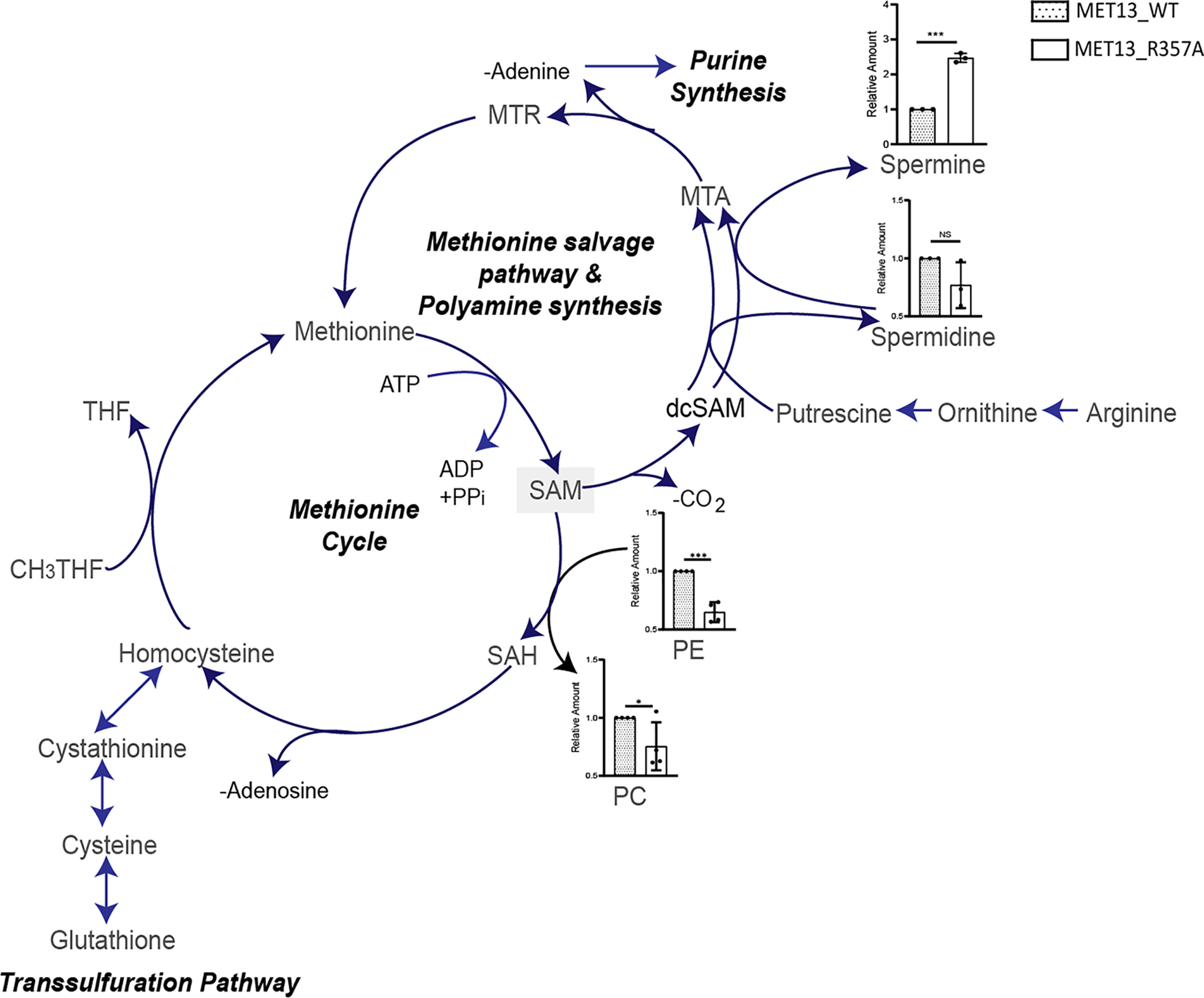
In deregulated MTHFR mutant cells, SAM cycles in futile salvage pathways wherein both methionine and adenine are continuously utilized and recycled. A schematic for the metabolism of SAM in budding yeast. Major catabolism of SAM to SAH occurs by methylation of PE to PC, or SAM may also be consumed for the production of spermidine, spermine, and methylthioadenosine (MTA) via polyamine synthesis, the latter then enters a futile methionine salvage pathway producing adenine as a side-product. Relative intracellular levels of the two phospholipids intermediates involved in PE methylation and polyamines (spermidine and spermine) estimated from yeast transformants of MET13_WT or MET13_R357A in met13Δmet15Δ (ABC2613) that were grown overnight in minimal medium with amino acid supplements and GSH. These transformants were then reinoculated at 0.15 A600 in fresh SD medium without any sulfur source. Methionine was added to the transformants after 3 h of secondary inoculation, and the samples were collected in triplicate at 1.0 OD. The graph here corresponds to the representative data set plotted using the average of three biological replicates and three technical replicates of each of these biological replicates along with ± S.D. values. Error bars indicate S.D. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant.
The second route of SAM metabolism is through the methionine salvage pathway. In this pathway, the methionine that converts to SAM is converted back to methionine with the release of adenine and polyamine following the decarboxylation of SAM. In the polyamine pathway, the decarboxylated SAM can be used in the synthesis of spermidine from putrescine or in the formation of spermine, which eventually releases into methylthioadenosine, subsequently yielding methionine and adenine (22). We measured the relative abundance of polyamines (spermidine and spermine) in the WT and mutant-bearing cells. Although there was no significant change observed in the relative abundance of spermidine in the WT and mutant transformed cells, we observed a significant increase of 2.5-fold in the amount of spermine for the deregulated mutant (MET13_R357A) (Fig. 9). This clearly suggested a significant conversion of SAM back to methionine and adenine, with the concomitant formation of spermine. The methionine so formed would again drive the ATP-dependent formation of SAM.
Thus, based on the metabolite data, it is clear that the methionine and ATP-driven formation of SAM is recycled back through both pathways of SAM metabolism. The first is via SAH and homocysteine, whereas the other is through decarboxylated SAM and the polyamines. As a consequence of this, with continuous methionine formation that is leading again to the formation of SAM, one observes futile cycles of ATP-dependent SAM formation and its recycling to methionine and adenine/adenosine. The activities of both the pathways in yeast with higher rates in the mutant in particular is supported by the higher proportion of mass isotopomer m + 1 in methionine mainly contributed by the incorporation of 13C of CH3THF caused by enhanced activities of either methionine cycle and/or methionine salvage pathway via SAM.
Discussion
In this study we have been able to show the critical role of SAM-mediated allosteric control of MTHFR in the maintenance of overall metabolic homeostasis in the cell. SAM-mediated inhibition of MTHFR has been considered to be a key regulatory feature in one-carbon metabolism ever since the discovery of allosteric inhibition of mammalian MTHFR by SAM (23, 24). However, there was neither direct evidence for the presence of this regulation in vivo nor clear demonstrations of the regions in the regulatory region that were important for this control. The current studies, therefore, although providing this evidence for a regulation in vivo and delineating the residues in MTHFR critical for this feedback inhibition, also provide a clear explanation as to why MTHFR is specifically regulated by SAM. We have been able to demonstrate this by isolating, for the first time, mutants in the regulatory region of MTHFR that show a lack of inhibition by SAM.
Although peptide mapping and photoaffinity labeling of the mammalian enzymes that were carried out more than two decades ago had suggested that the SAM-binding domain lay within a 6-kDa region of the regulatory region close to the junction of the catalytic and regulatory domains of MTHFR, the exact residues were never delineated (7). Recent crystal structural studies, however, suggested residues that conflicted with the earlier mapping data (10). In these studies the human MTHFR mutants E463D and E463Q were shown not to bind SAM based on size-exclusion chromatography experiments, although enzymatic inhibition studies were not carried out. We therefore carried out a rigorous investigation toward identifying these residues, and this led us to pinpoint a 7-amino acid stretch, 353FPNGRFG359, within the CR1 region of the regulatory domain, as being critical for the regulation by SAM. This region falls within the 6-kDa region that was earlier suggested from the photoaffinity labeling studies (7, 25), surprisingly completely outside the CR1 region (Fig. S9). The structural model does not indicate any proximity of Glu422 with the CR1 region, so the participation and involvement of Glu422 is unclear at present. One possibility is that at some early point of the conformational transitions, when SAM first binds to the region in CR1, it may also be in proximity to the Glu422. However, this awaits more dynamic insights into the process. Importantly, the CR1 region mapped to the interfacial region between the two monomers on the structure, and thus it would have been difficult to predict the involvement of these regions based on the modeled structure. Therefore, combining both structural approaches and targeted mutagenic approaches yielded valuable insights into the regions of the protein that are likely to determine regulation by SAM.
The growth defect seen in the feedback-insensitive deregulated MTHFR mutant on methionine medium provided an opportunity to understand the metabolic reasons behind the regulation of MTHFR by SAM. In the deregulated MTHFR mutant, we observed a drastic reduction in the nucleotide pools (which are derived from folate pools). However, growth could only be partly recovered by supplementation with external folate. Thus, although depleted folate pools appeared to be part of the explanation for the growth defects, additional mechanisms seemed to be important. Our investigations further revealed the presence of ongoing methionine synthesis driven by CH3THF formation that occurred even in the presence of biosynthetically repressive levels of methionine in the medium. Using stable isotope labeling studies, we could demonstrate that the 13C label seen in methionine arises from CH3THF, and quantitative estimations indicated a 31% increased consumption of CH2THF by the deregulated MTHFR mutant. Methionine, when it is in excess of its requirement in protein biosynthesis, is activated by ATP to the universal methyl donor, SAM. Because continuous generation of SAM would exacerbate nucleotide depletion, it suggests, therefore, that severe nucleotide depletion is a consequence of sustained SAM synthesis in a deregulated, feedback-insensitive MTHFR mutant.
In addition to the folate cycle, we also observed consequences on both the methionine cycle and the transulfuration pathway. Methionine biosynthesis occurs in the methionine cycle through the transmethylation of homocysteine (26). However, homocysteine also stands at the junction of another competing pathway, the reverse transsulfuration pathway, toward the formation of cystathionine and cysteine (27, 28). As a consequence of the deregulated MTHFR, its product, CH3THF, is formed continuously, and these higher CH3THF pools push the transmethylation of homocysteine toward methionine biosynthesis despite the presence of adequate external methionine. This depletes the homocysteine pools and limits its availability for reverse transsulfuration pathway. That this phenomenon is indeed occurring is reflected not only by the lower levels of homocysteine but also by the significantly lower levels of cystathionine and cysteine observed in these mutants. With lower cysteine there is consequently decreased GSH, and thus redox homeostasis is also partially disrupted as seen by the sensitivity to methylglyoxal. In contrast to the transulfuration pathway, the methionine cycle is overactive with the consequence that the accumulating methionine (through both de novo biosynthesis and external uptake) drives the formation of SAM despite the depleted ATP levels. This also explains the methionine dependence of the phenotype.
SAM is a critical methyl donor for various substrate-specific methyltransferases that enables the methylation of DNA, RNA, lipids, and proteins, and this can have very profound metabolic consequences in the cell. In yeasts, excess SAM can also be trapped in the methylation of phosphatidyl ethanolamine to form phosphatidyl choline, and this has been reported in yeast to function as a “methyl” sink (21). Irrespective of the substrates, however, the methytransferases generate SAH. SAH, in turn is hydrolyzed to adenosine and homocysteine through the action of the enzyme, SAH hydrolase. The homocysteine so generated is normally diverted to the synthesis of cysteine and GSH through the reverse transsulfuration pathway. However, this is prevented, as we have seen earlier, because of the continuous transmethylation of homocysteine by CH3THF and its diversion toward the formation of methionine in the deregulated MTHFR.
SAM is also metabolized through the methionine salvage pathway, wherein SAM is first decarboxylated and then recycled with the release of methionine, adenosine, and polyamines (29). In the deregulated MTHFR mutant, we observed a 2.5-fold increase in the amount of spermine, suggesting that the pathway is indeed active in the mutant. This possibly also explains why arginine, which is a precursor for spermine, is also observed at significantly lower levels in the deregulated mutant. Thus, in effect, the methionine- and ATP-driven formation of SAM enters both the pathways of SAM metabolism. It is difficult to conclude at this point which of the two pathways might be the predominant one in the regeneration of adenosine. However, irrespective of whether one or other is a major contributor to the recycling process, one observes futile cycles of SAM formation and its recycling to methionine and adenosine. This unregulated, “wasteful” synthesis of SAM and recycling further accentuates the nucleotide depletions seen in the cell and consequently also affects the cell growth. Spermine levels are also dependent on their catabolism. In yeast, spermine is catabolized by a FAD-utilizing enzyme, FMS1 (30). Because the SAM-insensitive mutants face depletion of FAD pools, it is possible that the activity of this FAD-utilizing enzyme might also be compromised. Therefore, inefficient catabolism of spermine might also be contributing to the increased spermine levels in the deregulated mutant.
Even though both yeast and human MTHFR enzymes require NADPH for their activity and are feedback-regulated by SAM, it is interesting to note that the plant enzyme is naturally feedback-insensitive while also requiring NADH for the activity in place of NADPH. In an effort to investigate the importance of SAM insensitivity in yeast, a previous study made use of Arabidopsis enzymes and yeast–Arabidopsis chimeric enzymes (9). However, the enzyme had quite different cofactor requirements compared with the native enzyme, and thus these studies, although yielding interesting results, could not be generalized to the actual situations in vivo and not surprisingly led to significantly different metabolic consequences than what was observed in the current study.
Although the findings described in this report are restricted to yeasts, they have implications for human MTHFR. Human MTHFR shares 43% identity with the yeast protein. This similarity extends to both the catalytic and regulatory domains of the protein. The 7-amino acid region in CR1 important for regulation by SAM that we have shown here is also conserved in the human protein. Therefore, it is likely that mutations or polymorphisms in these regions of the human MTHFR might also exhibit a deregulated phenotype and exhibit similar metabolic consequences in humans. Innumerable studies on SNPs of human MTHFR have been carried out over the years because of their relevance to many diseases, but these studies have focused on SNPs exhibiting a loss of function (4, 31–33). Mutations or SNPs that might result in a gain of function have not been identified or even recognized so far. This study with the feedback-insensitive, deregulated MTHFR reveals that a single polymorphism in the critical region could significantly disrupt cellular folate and nucleotide homeostasis while also impacting homocysteine levels and affecting redox homeostasis. Investigations of patient MTHFR polymorphisms that affect MTHFR regulation are therefore likely to be important but are likely to manifest quite different phenotypes than what has been reported so far for the loss of function/decreased function SNPs. One of the possible phenotypes that might be predicted would be an increased sensitivity of cancer patients to drugs targeting the folate pathway, as has been recently observed with polymorphisms in the histidine catabolic pathway (34). In conclusion, these studies with the isolation and investigation of deregulated mutants of MTHFR have yielded valuable new insights into SAM cycle and one-carbon metabolism, and future investigations with these mutants, both in yeasts and humans, will throw more light on the interaction of these two pathways.
Experimental procedures
Chemicals and reagents
All chemicals used in the present study were of either analytical or molecular biology grades and were obtained from commercial sources. Media components were purchased from Difco. Oligonucleotides were purchased from Sigma and IDT. Restriction enzymes, vent DNA polymerase, and other DNA-modifying enzymes were obtained from New England Biolabs. Gel extraction kits and plasmid miniprep columns were obtained from Bioneer Inc. (Daejeon, South Korea) or Promega, and the nickel–nitrilotriacetic acid–agarose resin was obtained from Qiagen. NADP/NADPH-Glo™ assay kit for NADPH estimation was procured from Promega. The labeling substrates and derivatization agents N-methyl-N-(t-butyldimethylsilyl) trifluoroacetamide (TBDMS) + 1% t-butyl-dimethylchlorosilane were purchased from Sigma–Aldrich. The software used includes Agilent ChemStation, MassHunter, IsoCorr, Metalign, MATLAB, and Amdis (National Institute of Standards and Technology, Gaithersburg, MD, USA).
Strains and growth conditions
The yeast strains used in the study are described in Table S4. The yeast strains were regularly maintained on a nonselective YPD medium, which is composed of yeast extract (1%), peptone (2%), and dextrose (2%) medium. For yeast transformation experiments, synthetically defined minimal medium containing yeast nitrogen base (0.17%), ammonium sulfate (0.5%), and dextrose (2%) supplemented with histidine, leucine, and lysine (when not used as an auxotrophic marker) at 80 mg/liter were used. Growth, handling of bacteria and yeast, and all the molecular techniques used in the study were according to the standard protocols (35, 36). Yeast transformation was carried out by the lithium acetate method (37). E. coli strain DH5-α and RosettaTM were used as a cloning host and expression host, respectively.
Modeling of yeast MTHFR and docking with ligands
Yeast MTHFR sequence was extracted from National Center for Biotechnology Information (RRID:SCR_006472). The crystal structure of human MTHFR was downloaded from Protein Data Bank (entry 6FCX) and used as template structure. The missing coordinates for residues 161–171 in the human template structure were modeled using Modeller9v20. The refined human structure was used to generate a collection of 5000 homology models, and the best representative structure was selected based on the evaluation of discrete optimized protein energy potential. The model was further subjected to loop refinement module of Modeller9v20 to refine the connecting loops, and the energetically favorable model was selected among the 1000 generated models based on the discrete optimized protein energy score.
The selected model was then used as receptor structure for docking. For SAM, the coordinates were obtained from the PubChem database in sdf format and docked using the computational docking program AutodockVina. The template human structure consists of a SAH-binding site, and comparative analysis of both human and modeled yeast structure showed the presence of a similar binding pocket for yeast MTHFR as well. Therefore, SAM molecule was docked within the box volumes of 65 Å × 65 Å × 65 Å, enclosing the SAH-binding pocket. The residues within the binding pocket were defined as flexible residues to accommodate SAM molecule. AutodockVina predicted favorable docking poses for SAM, and the most favorable docked conformation was used to identify critical residues for further experimental evaluation.
MET13 cloning and construction of site-directed mutants
MET13 was cloned with a hexahistidine tag at the C terminus downstream of the TEF promoter at XbaI and ClaI sites of p416TEF vector, resulting in plasmid p416TEF-MET13-His. This construct was further used as a template for the creation of the different mutants of MET13 by splice overlap extension strategy. The primers used in this study are listed in the (Table S5). The PCR products were cloned back into the p416TEF-MET13-His vector. Clones were confirmed by sequencing for the presence of the desired changes.
Growth studies by dilution spotting
For growth assays, the different strains were grown overnight in minimal medium with amino acid supplements and GSH as a sulfur source without uracil. They were reinoculated in fresh medium containing methionine at an A600 of 0.15 and grown until the cells attained an A600 of 0.6–0.8. The exponential-phase cells were harvested, washed with water, and resuspended in water to an A600 of 0.2. These suspensions were serially diluted to 1:10, 1:100, and 1:1000. Of these cell resuspensions, 10 μl were spotted on the desired minimal medium plates. The plates were incubated at 30 °C for 2–3 days, and then the images were taken using the Bio-Rad Gel DocTM XR+ imaging system.
Cloning, expression, and purification of yeast WT and mutant MET13
For protein expression studies using the nickel-based affinity chromatography method, the WT and mutant MET13 genes were first cloned with a His tag at the C terminus in the pET21d vector. His tag was incorporated in the WT MET13 gene with the help of PCR-based approach using MET13_NheI-F and MET13_HIS SacI-R primers. The amplified 1.8-kb gene was digested with NheI and SacI restriction enzymes and cloned into the NheI and SacI sites in the pET21d vector. The in-frame fusions were confirmed by sequencing. The MET13 mutant genes were subcloned from their respective yeast expression clones, generated in 416TEF vector, into the bacterial expression plasmid pET21d using BamHI and SalI restriction sites.
All the proteins were expressed as C-terminal His6-tagged fusion proteins in E. coli RosettaTM strains. RosettaTM strains harboring either WT or the mutant MET13 clones were inoculated in LB broth, with 25 μg/ml chloramphenicol and 100 μg/ml ampicillin, to an A600 of 0.05 and grown aerobically at 37 °C to an A600 of ∼0.6. These cultures were then induced with 1 mm isopropyl β-d-thiogalactopyranoside and allowed to grow for 16 h at 18 °C. The cells were harvested after induction by centrifugation at 5000 [times] g for 20 min at room temperature. At this stage, the pellet was stored at −80 °C until further analysis.
The pellet was resuspended in 20 mm KPi buffer (pH 7.2) containing 10% glycerol, 500 mm NaCl, 0.3 mm EDTA, 20 mm imidazole, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture. The cells were lysed by sonication at 20 amplitude, and 10-s sonication cycles were alternated with 20-s recovery periods. The sonicate was centrifuged at 10,000 × g for 45 min at 4 °C, and the supernatant was collected. The cleared lysate was loaded on to a nickel–nitrilotriacetic acid column equilibrated with 20 mm KPi containing 10% glycerol, 500 mm NaCl, and 0.3 mm EDTA and subsequently washed with 20 mm KPi, 10% glycerol, 0.3 mm EDTA, and 50 mm imidazole. Elution was performed in the presence of elution buffer carrying 300 mm imidazole. MET13 being a flavoprotein can be visually identified by its intense yellow color. The purified proteins were analyzed by SDS-PAGE (12% gel). Protein concentration estimations were done by the Bradford method (38) with BSA as standard after the removal of imidazole by means of dialysis.
MTHFR activity and inhibition studies: NADPH-menadione oxidoreductase assay
MTHFR activity and inhibition studies were performed with several modifications in the NADPH-menadione oxidoreductase assay (39). In a total volume of 800 μl, the reaction mixture consisted of 400 μl of 100 mm potassium phosphate buffer (pH 7.2) with 0.6 mm EDTA, 80 μl of menadione from a stock concentration of 2 mm (menadione stock was prepared freshly in methanol) and 25–100 nm of enzyme. This mixture was incubated for 5 min at 25 °C. To initiate the enzyme activity, NADPH at a final concentration of 200 μm was added to the reaction mix. For inhibition studies, in the same reaction mix 200 μm of SAM was added just before adding the NADPH. In both cases, the rate of reaction was monitored as a decrease in the absorbance of NADPH at 343 nm. Activities are presented as the initial rate of NADPH oxidation observed, using an extinction coefficient of 6220 m−1 cm−1. Activity units represent nanomoles of NADPH oxidized per min per milligram of protein.
Targeted metabolite analysis: extractions and estimations
For metabolite analysis (amino acids, sulfur intermediates, and nucleotides), the Saccharomyces cerevisiae (ABC2613) transformants with MET13_WT or MET13_R357A were grown overnight in minimal medium with amino acid supplements and GSH. These were then reinoculated at 0.15 A600 in fresh SD medium without any sulfur source. Methionine was added to the transformants after 3 h of secondary inoculation, and the samples were collected in duplicate at 0.5 and 1.0 OD. The samples were rapidly quenched in a quenching solution, following which metabolite extraction was done. Metabolite quenching and extraction was performed as described previously (40). Metabolite extracts were dried down in a speed vacuum (3–4 h) and stored at −80 °C until analyzed by mass spectrometer.
These metabolites were estimated by LC–MS/MS, using a HPLC coupled to a triple-quadrupole mass spectrometer (ABSciex 6500). Details of the analysis are described in Ref. 40. In brief, metabolites were extracted, resolved, and estimated as described. For each metabolite, parameters for quantitation of the two most abundant daughter ions (that is, two Multiple Reaction Monitoring per metabolite) were included. To quantify metabolites, the area under each peak was quantitated by using Analyst software (ABSciex), inspected for accuracy, and normalized against total ion count, after which relative amounts were quantified.
Polyamines and folate intermediates were measured in QTRAP 6500+. Metabolites were extracted from the cells using chilled methanol method. Briefly 20 OD cells were quenched with chilled methanol (kept at −80 °C) and followed by bead beating using acid-washed glass beads. The cell suspension was transferred to a fresh tube and centrifuged at 15,000 × g for 15 min at 4 °C temperature. The supernatant was vacuum-dried and reconstituted in 100 μl of 50% methanol. 5 μl was injected for LC–MS/MS analysis. The data were acquired using a Sciex Exion LCTM analytical UHPLC system coupled with a triple quadrupole hybrid ion trap mass spectrometer (QTRAP 6500+) in a positive-ion mode. To quantify these metabolites, the area under each peak was measured using the Multiquant software (ABSciex), inspected for accuracy, and normalized against total ion count, after which relative amounts were quantified.
For PE, PC, and phosphatidylcholine estimation, a modified Bligh and Dyer method (41) was used for extraction. Briefly, 1.8 OD cells were taken and 2.0 ml of methanol was added with 1 ml of dichloromethane, vortexed, and made sure to have a mono-phase. The mixture was allowed to be incubated for 30 min at room temperature. After incubation, 500 μl of water and 1 ml of dichloromethane was added to the solution and vortexed for 5 s. The mixture was centrifuged at 1200 rpm for 10 min. The lower layer was collected into a fresh glass tube. 2 ml of dichloromethane was added to the remaining mixture in an extraction tube and centrifuged, and the above step was repeated once again. Solvent was evaporated in a vacuum dryer, and the lipids were suspended in 100 μl of 100% ethanol, vortexed for 5 min followed by sonication for 10 min, and again vortexed for 5 min. There suspension was transferred to LC vials and subjected to an LC–MS run.
Cell growth for 13C labeling experiments
WT yeast (MET13_WT) and deregulated MTHFR (MET13_R357A) mutant strain were grown in SD medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate; 80 mg/liter leucine, 80 mg/liter histidine, methionine or GSH as per experimental requirement (200 μm), and 2% glucose). For 13C feeding experiments, the strains were grown in parallel in SD minimal medium with either 99% [1-13C]glucose, or unlabeled glucose. The cells were harvested during the midexponential phase of the growth (at ∼13 h for MET13_WT and 26 h for MET13_R357A mutant) that represent pseudo–steady-state condition. Cell pellets were quenched in liquid nitrogen, lyophilized, and stored at −80 °C until further analysis.
Acid hydrolysis of cell pellets and derivatization of amino acids
Lyophilized yeast cell pellets (∼2 mg each) were resuspended in 600 μl of 6 n HCl. The samples were incubated at 100 °C for 18 h to hydrolyze the protein and get the amino acid released (42). The hydrolysates were centrifuged, and 50 μl supernatant was transferred to fresh vial and subjected to vacuum drying in a speed vacuum (Thermo Scientific) to ensure the complete removal of moisture. The dried pellets were derivatized using TBDMS for the detection of amino acids using GC-MS (43). This is achieved by first dissolving the dried extracts in 30 μl of pyridine (Sigma–Aldrich) and incubated at 37 °C at 900 rpm for 30 min. The resuspended extracts were topped up with 50 μl of MtBSTFA + 1% BDMCS and incubated at 60 °C on a thermoshaker set at 900 rpm for 30 min. The derivatized samples were centrifuged for 10 min at 13,000 rpm, and the supernatant was transferred to new GC-MS vials followed by sealing with a septum screw cap.
GC-MS–based analysis of amino acids
Samples were analyzed by GC-MS (Agilent 7890B GC, electron impact ionization; 70 eV). Agilent HP 5-ms ultra-inert chromatography column (Agilent 19091S-433UI, 30 m × 250 μm × 0.25 μm) with 1 μl of injection volume, splitless mode, with a 1.3 ml/min helium carrier gas flow was used to separate the TBDMS-derivatized amino acids (44). The initial oven temperature was constant at 120 °C for 5 min, followed by ramp of 4 °C per min to 270 °C, hold for 3 min, then a ramp of 20 °C per min to 320 °C, and hold for 1 min. After 30 min the run temperature was reduced to 70 °C with a ramp of 100 °C per min. Solvent delay was set for ∼10 min. MassHunter (Agilent Technologies) was used to control the data acquisition parameters (both GC separation and MS) during all the sample runs. The raw GC-MS spectra of each sample obtained was baseline-corrected using MetAlign with default parameters for accurate assessment of MIDs of the metabolites. The National Institute of Standards and Technology database and authentic standards were used for amino acid peak and fragment identification. The intensity of amino acid fragment mass ions ranging from 40 to 600 were obtained using Agilent ChemStation software. Once the MIDs of all amino acid fragments were obtained from the averaged scans, the values were corrected for the presence of naturally occurring heavy isotopes attached to carbon backbone of derivatives using IsoCor (45). The average 13C abundance of each fragment was calculated for all mass corrected MIDs (46). The MIDs of each amino acid fragment can retrobiosynthetically be correlated to the label incorporation from the central precursors. The analysis further gives us a quantitative and qualitative understanding about the incorporation of glucose into amino acid via various metabolic pathways in the WT and deregulated mutant strain.
The WT and mutant-bearing cells grown in minimal medium supplemented with leucine, lysine, histidine, and methionine were fed either [12C]glucose or [1-13C]glucose. The derived mass isotopomer fragments were successfully validated by comparing the corrected MID with expected theoretical proportions in unlabeled fragments (Fig. S8A and Table S2). Among these fragments, the reliable mass ions (m/z) of fragments [M-85] or [M-57] were considered for comparison of label incorporation and distribution (47). In the case of the fragments derived from unlabeled amino acids (obtained from 12C tracer feeding), the fractions of 13C in these fragments were observed to be less than the natural abundance (i.e. <1.13%) (Table S3). In parallel, the cells fed with [1-13C]glucose confirmed the label redistribution of 15 amino acids under pseudo–steady-state conditions via different pathways of the central metabolism in MET13_WT, as well as in MET13_R357A (Fig. S8B and Table S3).
Generation of MET13 disruptions in different deletion strains
The MET13 gene was disrupted in the mup1Δ and met6Δ strain backgrounds using the one-step PCR-mediated gene disruption (48). The met13Δ::LEU2 disruption cassette was generated using the primer pair MET13Δ-LEU2F and MET13Δ-LEU2R and the plasmid pRS315TEF (ABE 3488, laboratory stock) as a template. The 1.6-kb PCR product obtained was transformed into the strains mentioned above, and the transformants were selected on minimal medium without leucine but containing methionine because the successful disruptants would be methionine auxotrophs. For met13Δ::LEU2 disruption in mup1Δ strain background, transformants were confirmed by lack of growth on SD plates without methionine. The transformants in met6Δ strain were confirmed for the disruption by diagnostic PCR using the primer pair MET13promF and MET13Δ-LEU2-R.
Disc diffusion assay for sensitivity methylglyoxal sensitivity
Disc diffusion assay has been performed after several modifications in the method (adapted from Ref. 49). Transformants were grown overnight in SD minimal medium along with reduced GSH (200 μm) as sulfur source and other amino acid supplements. These were then reinoculated in fresh SD medium to 0.15 A600 after washing them twice with sterile distilled water. Methionine (200 μm) was added to the culture 3 h after the secondary inoculation. At the exponential phase (1.0–1.5 OD), 5 OD of cells were plated onto SD plates supplemented with methionine and GSH. Grade 1 filter paper 1 cm in diameter (Whatman) was placed onto the SD plate, and 25 μl of MG (6.49 m) was soaked into the filter paper. The cells were grown at 30 °C for 48 h.
Total NADPH/NADP measurement by luminescence-based kit
For measurement of total NADPH/NADP pools of yeast cells, transformant expressing either WT or mutated MTHFR protein were grown overnight in SD minimal medium along with reduced GSH (200 μm) as sulfur source and other amino acid supplements at 30 °C for 12 h and reinoculated in fresh SD medium lacking a sulfur source at an initial A600 of 0.15. After 3 h of incubation, methionine was added to these transformants and allowed to grow at 30 °C till the early exponential phase of growth at 0.6–0.8 A600 with shaking at 220 rpm. Details of the treatment given to cells was as described in Ref. 50. Equal number of cells (A600 = ∼1.0) were harvested at 5000 rpm and washed with sterile MilliQ water followed by resuspension of the cells in lysis buffer (100 mm KH2PO4, 1.2 m sorbitol). Spheroplasts were prepared by adding the zymolase at the final concentration of 0.3 mg/ml and a subsequent incubation at 30 °C with shaking at 100 rpm for 1 h. A 100-μl aliquot of these spheroplasts were mixed with an equal volume of the NADP/NADPH GloTM detection reagent from NADP/NADPH-GloTM assay kit (Promega). The reaction mixture was incubated in dark at room temperature for 45 min, and readings were taken using luminescence spectrometer. The data were analyzed using GraphPad Prism 5.0.
Data availability
The LC–MS and GC/MS data used to support the findings of this study are available upon request from the corresponding author.
Supplementary Material
Acknowledgments
We acknowledge Swati Dhiman for help with the cloning work of the site-directed MET13 mutants of the CR1 region.
This article contains supporting information.
Author contributions—M. B. and A. K. B. conceptualization; M. B. data curation; M. B., S. Sengupta, S. L., S. K. M., and A. K. B. formal analysis; M. B., J. T., S. Suyal, R. O., R. C., and S. P. investigation; M. B. methodology; M. B. and A. K. B. writing-original draft; M. B., J. T., S. Sengupta, S. L., S. K. M., and A. K. B. writing-review and editing; A. K. B. supervision; A. K. B. project administration; J. T. 13C metabolic feeding experiments, image preparation for 13C metabolite experiments; S. Suyal fRD1 cloning and growth experiments; R. O. investigation and analysis of targeted metabolites; R. C. investigation and analysis of targeted metabolite analysis; S. P. investigation and analysis of targeted metabolites (phospholipid); M. S. analysis, interpretation, and image preparation of homology modeling and docking studies; S. Sengupta and S. L. design, plan, analysis, and interpretation of metabolite experiments; S. K. M. design, plan, analysis and interpretation of 13C metabolite feeding experiments image preparation for 13C metabolite data.
Funding and additional information—M. B. is a recipient of Indian Council of Medical Research, India (ICMR) Fellowship JRF-2013/HRD-64 (33138), and S. S. acknowledges fellowship support from Indian Institute of Science Education and Research Mohali for her doctoral work. J. T. acknowledges fellowship support from Ministry of Human Resource Development (MHRD) and IMPRINT. This work was supported by Grant-in-Aid CRG/2018/000190 from the Department of Science and Technology of the Government of India (to A. K. B.) S. K. M acknowledges the support of MHRD-IMPRINT Project 7801 IITM/IMPRINT/AD/169. S. L. acknowledges support from Department of Biotechnology, Ministry of Science and Technology, India (DBT) Grant BT/PR13446/COE/34/30/2015. M. S. acknowledges Department of Science and Technology for an INSPIRE Award and Research Grant IFA14-CH-165.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- MTHFR
- methylene tetrahydrofolate reductase
- CR1/2/3
- conserved region 1/2/3
- MG
- methylglyoxal
- MID
- mass isotopomer distribution
- PE
- phosphatidylethanolamine
- PC
- phosphatidylcholine
- G6PDH
- glucose-6-phosphate dehydrogenase
- MtBSTFA
- N-methyl-N-(t-butyldimethylsilyl) trifluoroacetamide
- TBDMS
- t-butyl-dimethylchlorosilane.
References
- 1. Locasale J. W. (2013) Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583 10.1038/nrc3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ducker G. S., and Rabinowitz J. D. (2017) One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 10.1016/j.cmet.2016.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trimmer E. E. (2013) Methylenetetrahydrofolate reductase: biochemical characterization and medical significance. Curr. Pharm. Des. 19, 2574–2593 10.2174/1381612811319140008 [DOI] [PubMed] [Google Scholar]
- 4. Burda P., Schäfer A., Suormala T., Rummel T., Bürer C., Heuberger D., Frapolli M., Giunta C., Sokolová J., Vlásková H., Kozich V., Koch H. G., Fowler B., Froese D. S., and Baumgartner M. R. (2015) Insights into severe 5,10-methylenetetrahydrofolate reductase deficiency: molecular genetic and enzymatic characterization of 76 patients. Hum. Mutat. 36, 611–621 10.1002/humu.22779 [DOI] [PubMed] [Google Scholar]
- 5. Guenther B. D., Sheppard C. A., Tran P., Rozen R., Matthews R. G., and Ludwig M. L. (1999) The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat. Struct. Biol. 6, 359–365 10.1038/7594 [DOI] [PubMed] [Google Scholar]
- 6. Frosst P., Blom H. J., Milos R., Goyette P., Sheppard C. A., Matthews R. G., Boers G. J., den Heijer M., Kluijtmans L. A., and van den Heuvel L. P. (1995) A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 10, 111–113 10.1038/ng0595-111 [DOI] [PubMed] [Google Scholar]
- 7. Goyette P., Sumner J. S., Milos R., Duncan A. M., Rosenblatt D. S., Matthews R. G., and Rozen R. (1994) Human methylenetetrahydrofolate reductase: isolation of cDNA mapping and mutation identification. Nat. Genet. 7, 551 10.1038/ng0694-195 [DOI] [PubMed] [Google Scholar]
- 8. Rosenblatt D. S. (1995) Inherited disorders of folate transport and metabolism. In The Metabolic and Molecular Bases of Inherited Diseases (Scriver C. R., Beaudet A. L., Sly W. S., and Valle D., eds) 7th ed., pp. 3111–3128, McGraw-Hill Book Co., New York [Google Scholar]
- 9. Roje S., Chan S. Y., Kaplan F., Raymond R. K., Horne D. W., Appling D. R., and Hanson A. D. (2002) Metabolic engineering in yeast demonstrates that S-adenosylmethionine controls flux through the methylenetetrahydrofolate reductase reaction in vivo. J. Biol. Chem. 277, 4056–4061 10.1074/jbc.M110651200 [DOI] [PubMed] [Google Scholar]
- 10. Froese D. S., Kopec J., Rembeza E., Bezerra G. A., Oberholzer A. E., Suormala T., Lutz S., Chalk R., Borkowska O., Baumgartner M. R., and Yue W. W. (2018) Structural basis for the regulation of human 5,10-methylenetetrahydrofolate reductase by phosphorylation and S-adenosylmethionine inhibition. Nat. Commun. 9, 2261 10.1038/s41467-018-04735-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sumner J., Jencks D. A., Khani S., and Matthews R. G. (1986) Photoaffinity labeling of methylenetetrahydrofolate reductase with 8-azido-S-adenosylmethionine. J. Biol. Chem. 261, 7697–7700 [PubMed] [Google Scholar]
- 12. Raymond R. K., Kastanos E. K., and Appling D. R. (1999) Saccharomyces cerevisiae expresses two genes encoding isozymes of methylenetetrahydrofolate reductase. Arch. Biochem. Biophys. 372, 300–308 10.1006/abbi.1999.1498 [DOI] [PubMed] [Google Scholar]
- 13. Trimmer E. E., Ballou D. P., Ludwig M. L., and Matthews R. G. (2001) Folate activation and catalysis in methylenetetrahydrofolate reductase from Escherichia coli: roles for aspartate 120 and glutamate 28. Biochemistry 40, 6216–6226 10.1021/bi002790v [DOI] [PubMed] [Google Scholar]
- 14. Rébora K., Desmoucelles C., Borne F., Pinson B., and Daignan-Fornier B. (2001) Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate. Mol. Cell Biol. 21, 7901–7912 10.1128/MCB.21.23.7901-7912.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fan J., Ye J., Kamphorst J. J., Shlomi T., Thompson C. B., and Rabinowitz J. D. (2014) Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302 10.1038/nature13236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang J., Sonnenschein N., Pihl T. P., Pedersen K. R., Jensen M. K., and Keasling J. D. (2016) Engineering an NADPH/NADP+ redox biosensor in yeast. ACS Synth. Biol. 5, 1546–1556 10.1021/acssynbio.6b00135 [DOI] [PubMed] [Google Scholar]
- 17. Thomas D., Cherest H., and Surdin-Kerjan Y. (1991) Identification of the structural gene for glucose-6-phosphate dehydrogenase in yeast. Inactivation leads to a nutritional requirement for organic sulfur. EMBO J. 10, 547–553 10.1002/j.1460-2075.1991.tb07981.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Camarasa C., Faucet V., and Dequin S. (2007) Role in anaerobiosis of the isoenzymes for Saccharomyces cerevisiae fumarate reductase encoded by OSM1 and FRDS1. Yeast 24, 391–401 10.1002/yea.1467 [DOI] [PubMed] [Google Scholar]
- 19. Enomoto K., Arikawa Y., and Muratsubaki H. (2002) Physiological role of soluble fumarate reductase in redox balancing during anaerobiosis in Saccharomyces cerevisiae. FEMS Microbiol. Lett. 215, 103–108 10.1111/j.1574-6968.2002.tb11377.x [DOI] [PubMed] [Google Scholar]
- 20. Jain M., Nagar P., Sharma A., Batth R., Aggarwal S., Kumari S., and Mustafiz A. (2018) GLYI and D-LDH play key role in methylglyoxal detoxification and abiotic stress tolerance. Sci. Rep. 8, 5451 10.1038/s41598-018-23806-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ye C., Sutter B. M., Wang Y., Kuang Z., and Tu B. P. (2017) A metabolic function for phospholipid and histone methylation. Mol. Cell 66, 180–193.e8 10.1016/j.molcel.2017.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan S. Y., and Appling D. R. (2003) Regulation of S-adenosylmethionine levels in Saccharomyces cerevisiae. J. Biol. Chem. 278, 43051–43059 10.1074/jbc.M308696200 [DOI] [PubMed] [Google Scholar]
- 23. Kutzbach C., and Stokstad E. L. (1967) Feedback inhibition of methylene-tetrahydrofolate reductase in rat liver by S-adenosylmethionine. Biochim. Biophys. Acta 139, 217–220 10.1016/0005-2744(67)90140-4 [DOI] [PubMed] [Google Scholar]
- 24. Kutzbach C., and Stokstad E. L. (1971) Mammalian methylenetetrahydrofolate reductase: partial purification, properties, and inhibition by S-adenosylmethionine. Biochim. Biophys. Acta 250, 459–477 10.1016/0005-2744(71)90247-6 [DOI] [PubMed] [Google Scholar]
- 25. Matthews R. G., Sheppard C., and Goulding C. (1998) Methylenetetrahydrofolate reductase and methionine synthase: biochemistry and molecular biology. Eur. J. Pediatr. 157, S54–S59 10.1007/pl00014305 [DOI] [PubMed] [Google Scholar]
- 26. Finkelstein J. D. (1990) Methionine metabolism in mammals. J. Nutr. Biochem. 1, 228–237 10.1016/0955-2863(90)90070-2 [DOI] [PubMed] [Google Scholar]
- 27. Selhub J., and Miller J. W. (1992) The pathogenesis of homocysteinemia: interruption of the coordinate regulation by S-adenosylmethionine of the remethylation and transsulfuration of homocysteine. Am. J. Clin. Nutr. 55, 131–138 10.1093/ajcn/55.1.131 [DOI] [PubMed] [Google Scholar]
- 28. Thomas D., and Surdin-Kerjan Y. (1997) Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 61, 503–532 10.1128/.61.4.503-532.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murín R., Vidomanová E., Kowtharapu B. S., Hatok J., and Dobrota D. (2017) Role of S-adenosylmethionine cycle in carcinogenesis. Gen. Physiol. Biophys. 36, 513–520 10.4149/gpb_2017031 [DOI] [PubMed] [Google Scholar]
- 30. White W. H., Gunyuzlu P. L., and Toyn J. H. (2001) Saccharomyces cerevisiae is capable of de novo pantothenic acid biosynthesis involving a novel pathway of β-alanine production from spermine. J. Biol. Chem. 276, 10794–10800 10.1074/jbc.M009804200 [DOI] [PubMed] [Google Scholar]
- 31. Rummel T., Suormala T., Häberle J., Koch H. G., Berning C., Perrett D., and Fowler B. (2007) Intermediate hyperhomocysteinaemia and compound heterozygosity for the common variant c.677C>T and a MTHFR gene mutation. J. Inherit. Metab. Dis. 30, 401 10.1007/s10545-007-0445-x [DOI] [PubMed] [Google Scholar]
- 32. Lopez-Lopez E., Martin-Guerrero I., Ballesteros J., and Garcia-Orad A. (2013) A systematic review and meta-analysis of MTHFR polymorphisms in methotrexate toxicity prediction in pediatric acute lymphoblastic leukemia. Pharmacogenomics J. 13, 498–506 10.1038/tpj.2012.44 [DOI] [PubMed] [Google Scholar]
- 33. Wang H. L., Sun L., Zhou S., and Wang F. (2018) Association between 5,10-methylenetetrahydrofolate, gene polymorphism and congenital heart disease. J. Biol. Regul. Homeost. Agents 32, 1255–1260 [PubMed] [Google Scholar]
- 34. Kanarek N., Keys H. R., Cantor J. R., Lewis C. A., Chan S. H., Kunchok T., Abu-Remaileh M., Freinkman E., Schweitzer L. D., and Sabatini D. M. (2018) Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 559, 632–636 10.1038/s41586-018-0316-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sambrook J., Fritsch E. F., and Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 36. Guthrie C., and Fink G. R. (1991) Guide to Yeast Genetics and Molecular Biology, p. 194 Academic Press, Cambridge [Google Scholar]
- 37. Gietz R. D., Schiestl R. H., Willems A. R., and Woods R. A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11, 355–360 10.1002/yea.320110408 [DOI] [PubMed] [Google Scholar]
- 38. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 39. Matthews R. G., and Haywood B. J. (1979) Inhibition of pig liver methylenetetrahydrofolate reductase by dihydrofolate: some mechanistic and regulatory implications. Biochemistry 18, 4845–4851 10.1021/bi00589a012 [DOI] [PubMed] [Google Scholar]
- 40. Walvekar A., Rashida Z., Maddali H., and Laxman S. (2018) A versatile LC-MS/MS approach for comprehensive, quantitative analysis of central metabolic pathways. Wellcome Open Res. 3, 122 10.12688/wellcomeopenres.14832.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 42. Hayakawa K., Matsuda F., and Shimizu H. (2018) C-metabolic flux analysis of ethanol-assimilating Saccharomyces cerevisiae for S-adenosyl-l-methionine production. Microb. Cell Fact. 17, 82 10.1186/s12934-018-0935-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jyoti P., Shree M., Joshi C., Prakash T., Ray S. K., Satapathy S. S., and Masakapalli S. K. (2020) The Entner–Doudoroff and nonoxidative pentose phosphate pathways bypass glycolysis and the oxidative pentose phosphate pathway in Ralstonia solanacearum. mSystems 5, e00091–20 10.1128/mSystems.00091-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Masakapalli S. K., Bryant F. M., Kruger N. J., and Ratcliffe R. G. (2014) The metabolic flux phenotype of heterotrophic Arabidopsis cells reveals a flexible balance between the cytosolic and plastidic contributions to carbohydrate oxidation in response to phosphate limitation. Plant J. 78, 964–977 10.1111/tpj.12522 [DOI] [PubMed] [Google Scholar]
- 45. Millard P., Letisse F., Sokol S., and Portais J. C. (2012) IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics 28, 1294–1296 10.1093/bioinformatics/bts127 [DOI] [PubMed] [Google Scholar]
- 46. Shree M., and S K. M. (2018) Intracellular fate of universally labelled 13C isotopic tracers of glucose and xylose in central metabolic pathways of Xanthomonas oryzae. Metabolites 8, 66 10.3390/metabo8040066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Antoniewicz M. R., Kraynie D. F., Laffend L. A., González-Lergier J., Kelleher J. K., and Stephanopoulos G. (2007) Metabolic flux analysis in a nonstationary system: fed-batch fermentation of a high yielding strain of E. coli producing 1,3-propanediol. Metab. Eng. 9, 277–292 10.1016/j.ymben.2007.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baudin A., Ozier-Kalogeropoulos O., Denouel A., Lacroute F., and Cullin C. (1993) A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 21, 3329–3330 10.1093/nar/21.14.3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kato M., Yang Y. S., Sutter B. M., Wang Y., McKnight S. L., and Tu B. P. (2019) Redox state controls phase separation of the yeast ataxin-2 protein via reversible oxidation of its methionine-rich low-complexity domain. Cell 177, 711–721.e8 10.1016/j.cell.2019.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yadav S., Mody T. A., Sharma A., and Bachhawat A. K. (2020) A genetic screen to identify genes influencing the secondary redox couple NADPH/NADP+ in the yeast Saccharomyces cerevisiae. G3 (Bethesda) 10, 371–378 10.1534/g3.119.400606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The LC–MS and GC/MS data used to support the findings of this study are available upon request from the corresponding author.