

Abstract
The melanoma antigen (MAGE) proteins all contain a MAGE homology domain. MAGE genes are conserved in all eukaryotes and have expanded from a single gene in lower eukaryotes to ∼40 genes in humans and mice. Whereas some MAGEs are ubiquitously expressed in tissues, others are expressed in only germ cells with aberrant reactivation in multiple cancers. Much of the initial research on MAGEs focused on exploiting their antigenicity and restricted expression pattern to target them with cancer immunotherapy. Beyond their potential clinical application and role in tumorigenesis, recent studies have shown that MAGE proteins regulate diverse cellular and developmental pathways, implicating them in many diseases besides cancer, including lung, renal, and neurodevelopmental disorders. At the molecular level, many MAGEs bind to E3 RING ubiquitin ligases and, thus, regulate their substrate specificity, ligase activity, and subcellular localization. On a broader scale, the MAGE genes likely expanded in eutherian mammals to protect the germline from environmental stress and aid in stress adaptation, and this stress tolerance may explain why many cancers aberrantly express MAGEs. Here, we present an updated, comprehensive review on the MAGE family that highlights general characteristics, emphasizes recent comparative studies in mice, and describes the diverse functions exerted by individual MAGEs.
Keywords: cancer-testis antigen, spermatogenesis, cancer, stress adaptation, p53, metabolism, Fe-S cluster, Prader-Willi syndrome, Schaaf-Yang syndrome, stress granule, alternative polyadenylation, DNA damage response, ubiquitination, E3 ligase, melanoma antigen, MAGE, apoptosis, stress response, E3 ubiquitin ligase, ubiquitin, AMP-activated kinase (AMPK), cell metabolism
Introduction: A comparative view of the MAGE gene family
Discovery of MAGEs
Classic studies in the 1940s and 1950s provided experimental evidence for the concept that the immune system can recognize and reject tumor cells (1, 2) and opened the floodgates for identifying and characterizing tumor antigens, which could be targeted for cancer therapy. In addition to mutated, fused, overexpressed, and oncoviral proteins (2), male germ cell–specific proteins were added to the inventory in 1991 when melanoma antigen 1 (MAGE-1) was discovered in the melanoma cell line MZ2-MEL (3). MZ2-MEL cells were established from a patient (MZ-2) who had, for 10 years, presented with strong T-cell reactivity against autologous tumor cells in culture (4). This patient had stage IV amelanotic melanoma of an unknown primary tumor and never achieved complete remission despite multiple surgical interventions followed by chemotherapy. Remarkably, continued vaccination with autologous melanoma cell clones that had been mutagenized in vitro and lethally irradiated led to the patient surviving for more than 30 years without disease recurrence. To identify the tumor-associated antigens recognized by the cytotoxic T cells in this patient, Boon and his group (3) applied autologous typing and transfection of a cosmid library into the patient-derived MZ2-E cell line. Their efforts led to the discovery of MAGE-1, the first human tumor antigen, which was later renamed MAGE-A1 upon the identification of additional gene family members (3, 5, 6). Namely, subsequent studies (5, 7, 8) identified a whole family of MAGE genes, present in all placental mammals. Humans and mice have ∼40 MAGE genes, which include some designated as pseudogenes, that are further subdivided into two major categories based on their sequence homology, tissue expression pattern, and chromosomal location (Figs. 1 and 2) (5, 8–10). The type I MAGEs include the MAGE-A, -B, primate-specific -C, and mouse-specific Mage-a–like (-al and -k1) subfamily members. Type I MAGEs are also called cancer-testis antigens (CTAs) because they are primarily expressed in the testis but are normally silent in other tissues (Fig. 2A) (5, 11); however, they are often aberrantly reactivated during oncogenic transformation (Fig. 2B) and code for antigens recognized by cytolytic T lymphocytes (5). In contrast, the type II MAGEs, consisting of the MAGE-D, -E, -F, -G, -H, -L, and NECDIN genes, are more ubiquitously expressed in humans and mice and not typically associated with human cancer (5, 9, 11–13).
Figure 1.

Overview of the MAGE gene family in humans and mice. A, phylogenetic tree showing the relationship between human and mouse MAGE proteins. The tree was created by the neighbor-joining construction method using the Jukes–Cantor protein distance measurement from the CLC Main Workbench 20. B, chromosomal location of human and mouse MAGE genes. C, locations of MAGE genes on the human and mouse X chromosome based on the recent NCBI's genome assembly HRCh38.p13 and GRCm38.p6. For all figures, the type II MAGEs are represented in green, MAGE-A and -C subfamilies in red, and MAGE-B subfamily in blue. Light colors indicate mouse Mages and dark colors indicate human MAGEs.
Figure 2.

Expression of MAGEs in normal tissues and cancer. A, human and mouse MAGE expression during different life stages is indicated. Starting with the top part of the outer circle, the expression of MAGEs is depicted during spermatogenesis, in ES cells, in an embryo, and finally in adults. B, the heatmap displays the percentage of various tumors that express each type I MAGE. The results are based upon data generated by the TCGA Research Network (RRID:SCR_003193).
Since the discovery of MAGEs, a major research focus has been developing MAGE-targeted immunotherapies. Despite promising results from initial clinical trials (14, 15), MAGE-A3 vaccines ultimately failed in Phase III due to a lack of efficacy (16, 17), which suggested that activation of the T-cell response to MAGE-A3 antigen is not sufficient to inhibit disease progression (18). Furthermore, some patients treated with anti-MAGE therapies developed serious off-target effects, like neuro- and cardiotoxicity (19, 20). The neurotoxicity may have been caused by the anti-MAGE-A3-TCR–engineered T cells recognizing a similar MAGE that is expressed in the brain (i.e. MAGE-A12) (11), and the cardiotoxicity was attributed to vaccine recognition of an unrelated peptide (titin) in the heart (19, 20). Besides inefficacy and unexpected side effects, resistance has been another major roadblock. For example, MAGE-A expression correlates with poor response to the CTLA-4 checkpoint inhibitors in melanoma patients (21) and faster development of resistance to the epidermal growth factor receptor tyrosine kinase inhibitors and chemotherapy (22–24). Despite these setbacks, research is ongoing to improve clinical outcomes and limit off-target effects of MAGE-based immunotherapies (25, 26). Alternative methods to target MAGE-expressing cancers by utilizing combinations of conventional therapy and immunotherapy are also being investigated (27–30). To successfully and safely target the type I MAGEs, it is important to understand the mechanisms by which these proteins contribute to oncogenesis, how they are regulated, and what they normally do in physiological contexts. In this review, we dive into what is known about the diverse functions of individual MAGEs, as well as their roles in cancer and other diseases. Although MAGE proteins have diverse functions, emerging studies suggest that responding to stress is a unifying theme of MAGEs.
Genomic organization and structure of human and mouse MAGEs
Most of the MAGE genes are located in clusters that are preserved in diverse mammalian species; however, each cluster has undergone a different degree of expansion by duplication or retrotransposition, leading to a number of species-specific genes (13). As shown in Fig. 1, human and mouse genomes encompass different numbers of MAGE subfamily members. They also differ in that only humans possess MAGE-C genes, and mice possess additional Mage-a–like genes that form another subfamily (Fig. 1) (11). Consistent with their classification as CTAs, the type I MAGE subfamilies reside in syntenic regions on the X chromosome (Fig. 1, B and C) (2, 5, 11, 31, 32), where testis-expressed genes are overrepresented (33–35). The autosomal murine gene Mage-b3 is an exception, as it resides on chromosome 2. Another distinction between humans and mice is that Mage-a genes map to two different loci on the murine X chromosome, which could be the result of an interchromosomal recombination event during genome evolution in rodents (Fig. 1, B and C) (36). In contrast to the uniform genomic location of type I MAGEs, type II MAGE genes are located on both autosomes and the X chromosome (Fig. 1, B and C). The type II MAGEs also exhibit species-specific copy number variations. For example, the mouse genome has only three Mage-d genes and an additional Mage-g gene, Mage-g2 (11, 37). Intriguingly, Mage-f1 has a point mutation in rodents and is predicted to either be a pseudogene or code for a truncated protein (38); thus, Mage-g2 may be a rodent adaptation to this Mage-f1 mutation loss and may serve important functions during germ cell development (37).
Although most MAGE proteins (and their murine homologs) are encoded by a single exon, the coding regions of the MAGE-D subfamily span across more than 11 exons (5, 13). The MAGE-D subfamily is also the most conserved subfamily between species, with over 90% identity in the coding sequences (13), and the genomic structure of the murine Mage-d genes closely resembles that of humans (39). The majority of the type I genes acquired several 5′ noncoding exons, potentially allowing for differential regulation of expression (6, 40). Some mouse Mage-b genes that were originally thought to be pseudogenes (Mage-b7, -b8, and -b17) because they have the structure of a processed transcript (10) code for full proteins and are expressed on the transcriptional level in a cell-specific manner in the testis, suggesting a functional role in spermatogenesis (11). Furthermore, during primate evolution, human MAGE-A11 acquired three additional 5′ coding exons that are unique among the type I genes (41). Together, the genomic organization and structure of the MAGE gene family indicate that it has expanded by retrotransposition and local duplication events. After splitting from their phylogenetic ancestor, the MAGE genes independently evolved in each species, with the type I MAGEs evolving most rapidly (13).
Evolution of the MAGE gene family
The MAGE gene family is evolutionarily conserved across eukaryotes. Unlike the large multigene family found in placental mammals, earlier eukaryotes, from protozoa to nonplacental mammals like the platypus, possess a single MAGE gene (9, 13, 42, 43). The first expansion of the MAGE gene family possibly occurred in marsupials, but with the emergence of the placenta and LINE elements in eutherian mammals, the family rapidly expanded (13). During eutherian radiation, the subfamily ancestors were formed by retrotransposition and expanded by gene duplications (5, 12).
Although most of the MAGEs that exist today appear to derive from a single ancestral gene, the identity of the founder family member is still a matter of debate. The unique genomic architecture of the MAGE-D genes suggests that one of them is the founder (5, 12); however, functional studies of MAGE-G1 imply that it is most closely related to the ancestral MAGE (42, 44). Nevertheless, the type II MAGEs clearly appeared earlier, as evidenced by the high homology shared between the human and mouse orthologs (>80% nucleotide sequence identity) (13, 43). In contrast, the type I MAGE paralogs within species are more similar to their subfamily members than to their orthologs between species (Fig. 1A), suggesting that these duplications occurred after the separation of the species. Mice also lack MAGE-C genes, whereas humans lack Mage-a–like genes (11), further implying that the type I MAGE subfamilies underwent a more recent and rapid evolution.
Within type II MAGE proteins, the N- and C-terminal regions that flank the MAGE homology domain (MHD) are completely different in paralogs but are highly conserved between human and mouse orthologs. This pattern further indicates that the type II genes independently evolved before the phylogenetic separation of the two species, which is also evident by the branching of the human and mouse MAGE phylogenetic tree (Fig. 1A) (5). Integrative analysis of genomic structures and codon changes of MAGEs and their distinct evolution patterns indicates that negative or purifying selection maintained the established essential, nonredundant functions of type II MAGEs, whereas positive selection allowed the redundant type I MAGEs to diversify or acquire additional functions (13).
The MAGE gene family is unique among cancer-testis antigens and the multigenic families of the X chromosome. Although the X chromosome is generally considered to be the most evolutionarily stable chromosome in placental mammals, which is true of the single-copy genes (35, 45), its ampliconic regions are rapidly evolving (46–50). MAGE genes fall into both categories of genes, as single-copy type II genes are conserved among mammals, whereas several of the type I genes recently expanded (Fig. 1). The rapid expansion of multicopy/ampliconic genes on the X chromosome is thought to be driven by male X chromosome hemizygosity and the benefits these genes offer to male reproductive fitness (51). Due to rapid and selective evolution, these genes often lack murine counterparts, barring traditional in vivo genetic studies (52–54). Type I MAGEs are an intriguing exception, as they are present in all mammals, which enables investigation into their physiological function in animal models (11). Even though several type I genes have diversified in a species-specific manner, they expanded to the same extent in both mice and humans, resulting in a similar number of genes in both species, which suggests that they convergently evolved to serve similar functions.
Comparative MAGE expression
MAGEs in the adult tissues
Upon the initial discovery and characterization of MAGE genes, their expression was only detectable in tumor samples and could not be identified in the limited set of normal somatic tissues available to the Boon group (3). Later, mRNA of MAGE-A, -B, and -C subfamily members was discovered in the testis and, in some cases, the placenta, hence their classification as CTAs (5–7, 40, 55–59). Additional studies identified more distant family members that are broadly expressed in normal tissues and are now referred to as type II MAGEs (5, 39, 60, 61). Comparative anatomical and developmental gene expression profiling of the entire MAGE family revealed five distinct subgroups (Fig. 2A) that may predict the functional categories and tissue-specific activities of MAGE proteins (https://mage.stjude.org/) (11).
Type I MAGEs show expression restricted to either the testis only (referred to as type Ia MAGEs) or to the testis and placenta (type Ib) (Fig. 2A). In mice, several type Ib genes are also expressed in the ovary (57, 62). In contrast to the idea that expression of type I MAGEs is completely restricted to reproductive organs (5, 6, 8, 55, 63), several type I MAGEs (type Ic) are expressed in a variety of organs in both species, including bladder, brain, spleen, small intestine, skeletal muscle, heart, and esophagus (11). Besides indicating potential function(s) outside the gonads, this unexpected expression pattern may also explain some cancer immunotherapy side effects, such as the neurological toxicities observed in patients treated with genetically engineered anti-MAGE-A3 T cells (19, 20). This finding has important implications in cancer vaccine and immunotherapy development because MAGEs are one of the most frequently targeted proteins, and several clinical trials are under way (31, 64, 65).
Type II MAGEs display a more ubiquitous pattern of tissue expression and are expressed at higher absolute levels than the type I genes in both species (9, 11, 66–69). The type IIa genes are uniformly and highly expressed in the majority of tissues, and the type IIb MAGEs show enriched expression in the brain (Fig. 2A) (11, 66–68). Notably, some type IIa genes are also expressed at high levels in the brain, such as MAGE-D in the cerebral cortex, medulla, and hippocampus (9, 70). As a type IIb MAGE, MAGE-L2 is widely expressed in various human adult tissues and highly enriched in the brain, particularly in the hypothalamus (11, 67, 68). In mice, Mage-l2 expression is even more restricted to the brain, and enrichment in the hypothalamus is already detectable in the later embryonic stages (67, 68), suggesting a role for Mage-l2 during neural development and in the adult brain. Prominent Mage-l2–expressing neurons are located in regions (i.e. the arcuate nuclei, suprachiasmatic nuclei, paraventricular nuclei, and supraoptic nuclei) involved in appetite and feeding behaviors, underscoring the phenotypes seen in Prader–Willi (PWS) and Schaaf–Yang syndrome (SYS) patients, which will be explained in more detail in later sections of this review (71).
MAGE expression during embryonic development
The expression of type I and II MAGEs in placenta and several fetal tissues in human and mouse suggest developmental functions (6, 11, 62, 72, 73). Human placenta expresses several MAGE-A genes (6); in contrast, mouse Mage-a genes are restricted to expression in the testis, whereas the Mage-a–like genes (Mage-al2 and -al3) are highly enriched in the mouse placenta (11). This finding suggests that Mage-al genes may be the functional orthologs of human MAGE-A8, -A10, and -A11 in this tissue (11).
Unlike the adult tissues, expression of the type Ia MAGE genes is not restricted to the male gonad during embryonic development. Expression in the developing testis and ovary implicates a role for type I MAGEs in gametogenesis of both sexes (11, 58, 62, 73). Consistent with mouse expression, human MAGE-A1 and -A4 proteins have been detected in premeiotic germ cells (58) and in fetal ovary (62, 73), suggesting that human and mouse MAGE-A genes might share similar functions in premeiotic germ cell development of both species.
Type II MAGEs are broadly expressed during embryonic development in humans (69, 74) and mice (Fig. 2A) (9, 11, 69, 70). The high expression of type II genes in the brain suggests a role in the development and/or function of the central nervous system (11, 75–77). For example, MAGE-D1 is highly expressed in the neural tube during early human development and later in the ventricular zone, subplate, and cortical plate (76, 78). Interestingly, several type IIb brain-enriched genes, such as Ndn and Mage-l2, are more ubiquitously expressed during embryonic development, which implies involvement in a diverse array of biological functions during embryonic development and in pathogenesis of neurodevelopmental disorders (43). In later sections focusing on MAGE-D1, -D2, -G1, and -L2, we cover these roles in further detail.
Besides expression during late embryonic development, MAGEs are also expressed in human and mouse embryonic stem (ES) cells (Fig. 2A) (11, 72, 79, 80). Like in adult tissues, MAGE-D1 and -D2 are the most highly expressed MAGEs in human (76, 77) and mouse ES cells (11, 72, 79, 80), teratocarcinoma cells, and extraembryonic endoderm cells (77). Furthermore, expression of several type II MAGEs is increased by retinoic acid–induced differentiation (80, 81). Additional research is warranted to define the contribution of MAGEs in regulation of stemness, differentiation of pluripotent stem cells, and embryonic development.
MAGE expression during spermatogenesis and folliculogenesis
From the early mapping of the MAGE gene family (6), it was evident that the majority of type I MAGEs exhibit male germline-restricted expression in both humans and mice (7, 11, 56–58, 73, 82–85), which implicated that the potential physiological function of these proteins is related to spermatogenesis. Mammalian spermatogenesis is a highly coordinated and cyclic process of male germ cell generation entailing cell divisions and differentiation to ultimately yield a large number of haploid spermatozoa. Spermatogenesis takes place in the seminiferous tubules of the testis, where somatic Sertoli cells develop an epithelium to support male germ cell proliferation and differentiation (86). In the basal compartment (i.e. the gap between the basement membrane and the Sertoli cell tight junction), spermatogonial stem cells (SSCs) give rise to progenitors, also referred to as undifferentiated spermatogonia, which undergo a series of rapid transit-amplifying mitotic divisions. A surge in retinoic acid signals for progenitors to differentiate and go through a few more rounds of division to ultimately give rise to spermatocytes (87). Spermatocytes then enter meiotic division and cross the blood-testis barrier (BTB) to become pachytene spermatocytes. In the apical compartment of seminiferous tubules, spermatocytes then undergo two meiotic divisions to generate haploid round spermatids that undergo morphological changes to eventually mature into spermatozoa that are released into the lumen. This process takes ∼35 days in mice and ∼75 days in humans. Cyclic retinoic acid pulsation, which occurs every 8.6 days in the mouse testis, ensures continuity in spermatogenesis and a permanent supply of sperm throughout the life of a male (88–92).
The first round of mouse spermatogenesis is a distinctive program that provides a good model system to study gene expression during sperm development, as specific germ cell types (i.e. spermatogonia, spermatocytes, and spermatids) appear postnatally in a well-defined order (93, 94). Analysis of age-dependent MAGE expression patterns following initiation of spermatogenesis revealed that the majority of type I MAGEs are expressed at distinct stages in premeiotic, meiotic, and postmeiotic cells during sexual maturation (Fig. 2A) (11). Specifically, Mage-b4 and -b16 are expressed in spermatogonia, including SSCs (11, 57). Prepachytene spermatocytes exhibit peak expression of all Mage-a subfamily members, whose expression starts in spermatogonia and hits the highest point just before entry into meiosis and the BTB transition (11, 56). Interestingly, the non-X-chromosome–residing MAGE genes, Mage-g1, -g2, and -b3, are expressed in pachytene spermatocytes during meiosis (Fig. 2A) (11). The majority of MAGE genes expressed in haploid spermatids are the testis-restricted type Ia Mage genes, including Mage-b1, -b2, and -b5 (Fig. 2A) (7, 11, 56). Consistent with the broad expression of type II MAGEs in many tissues and somatic cell types, most type II Mages are expressed predominantly in the Sertoli cells (11, 95).
Besides the testis, several type I Mages are also expressed in the mouse ovary during follicle growth and maturation (Fig. 2A) (11). For example, Mage-b4 is expressed in the first 2 weeks after birth (11), when the rate of primordial follicle recruitment into the growth phase is the greatest, which is in line with immunohistochemistry analysis showing that female germ cells express Mage-b4 throughout meiosis and in dormant primary oocytes (57). Mage-a10, -b3, and -b7 are enriched later, during follicle maturation (Fig. 2A) (11). Intriguingly, the pseudogene Mage-a9ps, which is not expressed in any other tissue, is expressed during early ovary development, implying a potential regulatory function of this gene in oogenesis (11). All type II genes are expressed in the ovary, but only a few are regulated during ovary development, such as Mage-l2, which is enriched during early follicle growth (Fig. 2A). Taken together, these results indicate that MAGE genes are expressed in specific cell types and stages during spermatogenesis or folliculogenesis to perform unique and nonoverlapping functions during germ cell differentiation.
Epigenetic and transcriptional regulation of MAGE gene expression
Since first being identified in melanoma, MAGEs have been described in a myriad of tumors of various histological types and stages of progression (Fig. 2B) (3, 6, 82–84, 96–101). Given this widespread expression in different cancers, many studies have sought to identify and understand the underlying mechanisms that lead to the ectopic expression of MAGEs in cancer. Both the distinct stage-specific expression of MAGE CTAs in the male germline (Fig. 2A) (11) and the diverse pattern of activation in specific tumor types (Fig. 2B) (43, 101) suggest that a combination of epigenetic alterations with tissue-specific transcription factors is required to permit stable transcriptional activation of MAGE expression, although the precise regulatory mechanisms are still not fully understood.
DNA methylation
The discovery that a methyltransferase inhibitor, 5′-aza-2′-deoxycytidine (DAC) was capable of inducing MAGE-A1 expression indicated that DNA methylation status contributes to MAGE silencing in normal tissues and aberrant expression in cancer (96). Accordingly, the level of promoter methylation of various MAGEs inversely correlates with their expression in cancers (97, 100, 102–108). The predominant methyltransferase involved in the maintenance of CpG (5′-C-phosphate-G-3′) methylation of MAGE promoters is DNMT1 (109). In addition, methyl-CpG–binding domain proteins contribute to the silencing of MAGE-A genes (110, 111), further implying the important role of DNA methylation in transcriptional regulation of MAGE genes. Although the role of DNA methylation in physiological regulation of MAGEs is mostly unknown, the methylation reprograming pattern during gametogenesis (112) suggests that it could contribute to the cell type–specific MAGE expression pattern during spermatogenesis.
In line with this idea, BORIS (brother of the regulator of imprinted sites), a demethylation factor involved in regulation of the site specificity and timing of epigenetic reprogramming in germ cells, was recently found to promote aberrant activation of MAGEs in human tumors (113, 114). Thus, BORIS—itself a cancer-testis gene—highlights a possible overlap between the regulatory system for induction of MAGE genes in both normal male germ cells and cancer cells with respect to CpG methylation. The involvement of BORIS also suggests that aberrant activation of MAGEs might not just be a random consequence of genome-wide demethylation in cancer, as previously thought, but rather a process of targeted epigenetic modifications (113). Expression of BORIS in male germ cells overlaps with several MAGEs that are expressed from spermatogonia to spermatocytes and also coincides with erasure of the global methylation pattern (11, 113, 115). Furthermore, the illegitimate activation of BORIS also correlates with the up-regulation of several MAGEs in cancer (115). However, MAGE-A1 and other CTA genes are expressed in melanoma in the absence of BORIS activation, suggesting more complex activation of these genes (116).
Altogether, the importance of DNMTs and BORIS in spermatogenesis, their stage-specific expression during male germline development and their implication in cancer suggest that DNA methylation impacts MAGE expression in germ cells and cancer. Furthermore, the differential acquisition of methylation marks between male and female gametes (112) may also underlie differential expression of MAGE genes in male and female gonads, but further studies are required to provide experimental evidence and molecular details of such regulation.
Histone modifications
DNA methylation of MAGE promoters is intertwined with post-translational modification of histones, and both work together to enhance MAGE gene expression in cancer cells (96, 98, 104, 117). Tumor cells with high expression of MAGE-A1 and -A3 exhibit an enrichment in activation marks with a concomitant decrease in the repressive mark (118). Inhibition of DNA methyltransferases and histone deacetylases (HDACs) leads to MAGE-A11 expression, supporting the idea that DNA methylation and histone modifications play a synergistic role in regulating MAGE expression (119). In thyroid cancer and pituitary tumors, reactivation of the fibroblast growth factor receptor 2-IIIb (FGFR2-IIIb) led to repression of MAGE-A3 and -A6 by increasing histone deacetylation and histone methylation (120, 121). Histone deacetylation was also shown to be responsible for the silencing of MAGE-A1, -A2, -A3, and -A12 expression (122), whereas in female pituitary tumors, estradiol promoted H3 acetylation and MAGE-A3 expression (121). In addition to histone lysine acetylation, histone lysine methylation was also shown to affect MAGE gene expression in cancer cells (118). G9A, also known as euchromatic histone lysine N-methyltransferase 2 (EHMT2), methylates histone H3 Lys-9 in the MAGE-A2, -A6, and -A8 promoter regions, leading to the maintenance of their heterochromatic and silent state (123). Together, a growing body of evidence indicates that diverse epigenetic mechanisms regulate the expression and silencing of MAGE genes in cancer cells; however, how these epigenetic mechanisms contribute to their expression in the germline is still mostly unknown. Further studies into epigenetic regulation of MAGE gene expression are warranted, in particular, as epigenetic drugs are used in combination with immunotherapy to improve the response of cancer patients (118, 124).
Transcription factors and signal transduction pathways
Both the cell-specific MAGE expression during spermatogenesis and their distinct expression in diverse cancers raise questions about the specificity of the regulation of these genes and potential transcription factors involved. In contrast to epigenetic regulation of MAGE genes, the transcription factors and upstream activating pathways are still mostly undetermined. Mapping of type I MAGE promoter regions using deletional analysis and transcription factor–binding site analysis identified ETS- and SP1-binding elements, which were able to activate MAGE-A1 expression upon binding ETS transcription factors (125). Methylation of ETS- and SP1-binding sites in several MAGE-A promoters was subsequently shown to silence MAGE-A expression by preventing transcription factor binding and recruiting methyl-CpG–binding domain proteins (110, 126).
In several cancers, MAGE-A and -C expression is activated by deregulated proto-oncogenic KIT tyrosine kinase and concurrent promoter DNA demethylation (102, 104, 110, 127, 128). Upon treatment of KIT-dependent mast cells with the tyrosine kinase inhibitor imatinib, the expression of several MAGE-A and -C genes was inhibited (128). Intriguingly, Mage-a gene expression peaks in the seminiferous cycle after the retinoic acid surge that induces Kit signaling and activates differentiation of male gem cells (11, 129, 130). In line with this expression pattern, Mage-a protein expression is the highest in Stra8/Kit-positive spermatogonia and is induced in cultured primary spermatogonia after a retinoic acid spike (11), suggesting that Kit may also regulate expression of Mage-a genes during spermatogenesis.
Other signaling pathways may also regulate MAGE expression. For example, fibronectin and FGFR2 have been shown to induce expression of MAGE-A3 in some cancer types (131). MAGE-A3 expression is also inducible by carcinogens, such as Helicobacter pylori (132) or smoking (133), and is affected by miRNAs and lncRNAs (134–138). In all, current data suggest that the MAGE family of genes is repressed in somatic cells by many layers of epigenetic marks and the activities of transcription factors and signaling pathways that become coordinately dysregulated in cancer. However, the exact mechanisms and transcription factors involved in controlling MAGE gene expression in cancer and germ cells await discovery.
General characteristics of MAGE proteins
The defining feature of all MAGE proteins is an ∼180-amino acid domain known as the MAGE homology domain (MHD). The MHD is present in lower eukaryotes, such as Saccharomyces cerevisiae, Aspergillus spp., Drosophila melanogaster, and Arabidopsis thaliana (5, 9, 44, 139, 140). In the mammalian MAGE family, ∼40% of amino acids in the MHD are identical across all the MAGE subfamilies, but higher conservation is evident at the subfamily level, as the MAGE-D and MAGE-A subfamily members share 75 and 70% MHD residues, respectively (141). The MHD is generally positioned near the C terminus of MAGE family proteins and is flanked by short, poorly conserved N- and C termini in type I MAGEs (except human MAGE-C1 and mouse Mage-b4), as well as MAGE-F1, -G1, and NECDIN (Fig. 3A) (5, 9). In contrast, the remaining type II MAGEs contain extended N- and C-terminal sequences, but the biological importance of these MHD-flanking regions is unknown (9). Although MAGE family proteins typically have a single MHD, some MAGEs have a duplicated or a truncated MHD (Fig. 3A) (5, 9).
Figure 3.
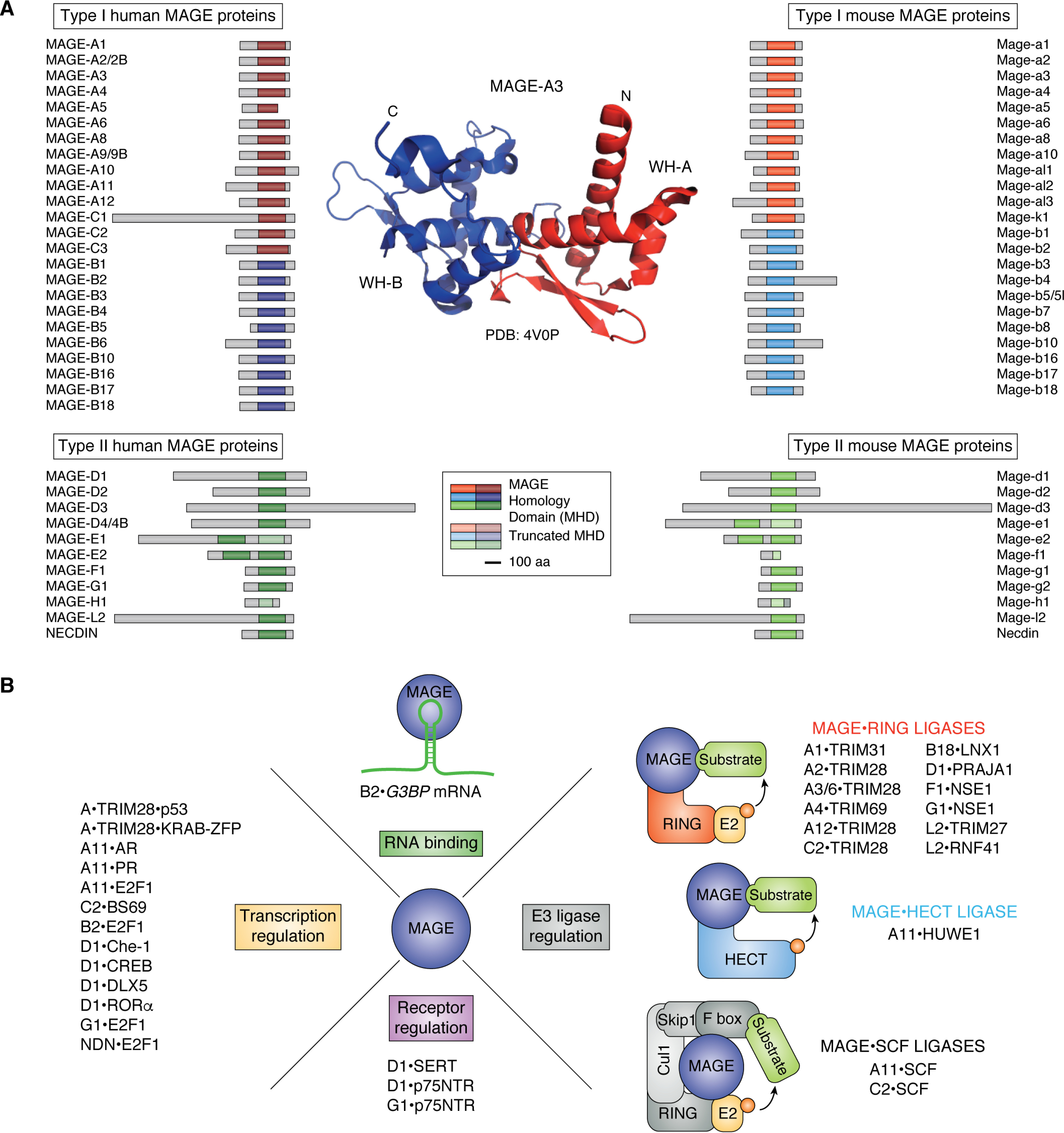
General structure of the MHD and biochemical function of MAGE proteins. A, schematic structure of human and mouse MAGE proteins. The MHD for each MAGE is indicated by a solid colored box, and the size corresponding to 100 amino acids (aa) is shown. The crystal structure of the double winged-helix motif of the MHD of MAGE-A3 (Protein Data Bank entry 4V0P) is shown. The N- and C termini are indicated, and the two WH motifs (WH-A and WH-B) are represented in red and blue, respectively. B, MAGEs bind to and regulate E3 ligases, receptors, transcription factors, and RNA (as an RNA-binding protein) to exert diverse molecular functions (General characteristics of MAGE proteins).
The MHD contains two tandem winged-helix (WH) motifs, referred to as WH-A and WH-B (Fig. 3A), the latter of which contains a dileucine motif that is important for MAGE biochemical function (141, 142). The crystal structures of MAGE-A3 and -A4 revealed tight binding of an extended peptide sequence within the tandem WH domain of the MHD, suggesting the MHD conveys binding capabilities that may be central to MAGE functionality (142). NMR and native MS of the MAGE-A4 MHD suggest that the domain encompasses compact folded structures and disordered regions with a broad charge state distribution, all of which are suggestive of a dynamic protein (143). This dynamic flexibility of the MHD may confer unique binding preferences and functions to individual MAGEs.
Despite long-lasting interest in MAGE proteins in cancer therapy, the diverse molecular functions of these proteins are just starting to be unraveled. In line with the dynamic nature of the MHD structure, MAGE proteins exert their function through interactions with diverse proteins (Fig. 3B). A growing body of evidence suggests that MAGEs assemble with different E3 ligases and, by doing so, modulate ubiquitination of target proteins (43, 141). E3 ligases recognize target substrates and mediate the transfer of activated ubiquitin from the E2 enzyme to a specific substrate (144). E3 ligases are categorized into four major classes: RING (really interesting new gene) finger, U-box, PHD finger, and HECT (145, 146). Through efforts to identify the function of MAGE proteins, we and others discovered that both type I and II MAGEs bind E3 ubiquitin ligases with RING domains and form MAGE-RING ligases (MRLs) (141, 147–154). Since their identification, several distinct MRLs have been described (Fig. 3B). MAGEs recognize their cognate E3 ligase partner through their MHD, and the dileucine motif in WH-B is particularly critical for this interaction (141). In contrast, the region on the RING protein that is recognized by a particular MAGE is variable (141). The disorder and flexibility of the MHD structure likely contributes to the specificity of each MAGE binding to its associated ligase (143). Indeed, the crystal structure of MAGE-G1 in complex with its NSE1 RING ligase demonstrated that the MHD undergoes extensive rearrangements for MRL formation (141).
MAGEs have been shown to regulate their respective E3 ligases through a diverse set of means; of particular interest is their ability to specify novel substrates for ubiquitination (141, 149, 151, 152, 155). For example, MAGE-A3 and -A6 bind the E3 ligase TRIM28 to specify 5′ AMP-activated protein kinase (AMPK) for ubiquitination and subsequent proteasomal degradation (151). AMPK ubiquitination by TRIM28 only in the presence of MAGE-A3 and -A6 implies that MAGEs reprogram ubiquitously expressed E3 ligases, like TRIM28, to serve cell type–specific functions in the germline or promote tumorigenesis in cancer cells (Fig. 4A) (151, 156). As another example of MAGEs specifying novel substrates, MAGE-F1 interacts with the E3 ligase NSE1 to target MMS19 for ubiquitination and degradation, which renders cells less competent in repairing DNA damage and predisposes them for oncogenic transformation (38). In another case, MAGE-L2-TRIM27–mediated ubiquitination of WASH protein leads to WASH activation, rather than proteasomal degradation, to facilitate transport of cargo proteins by retromer, a complex involved in endosomal protein sorting (149).
Figure 4.
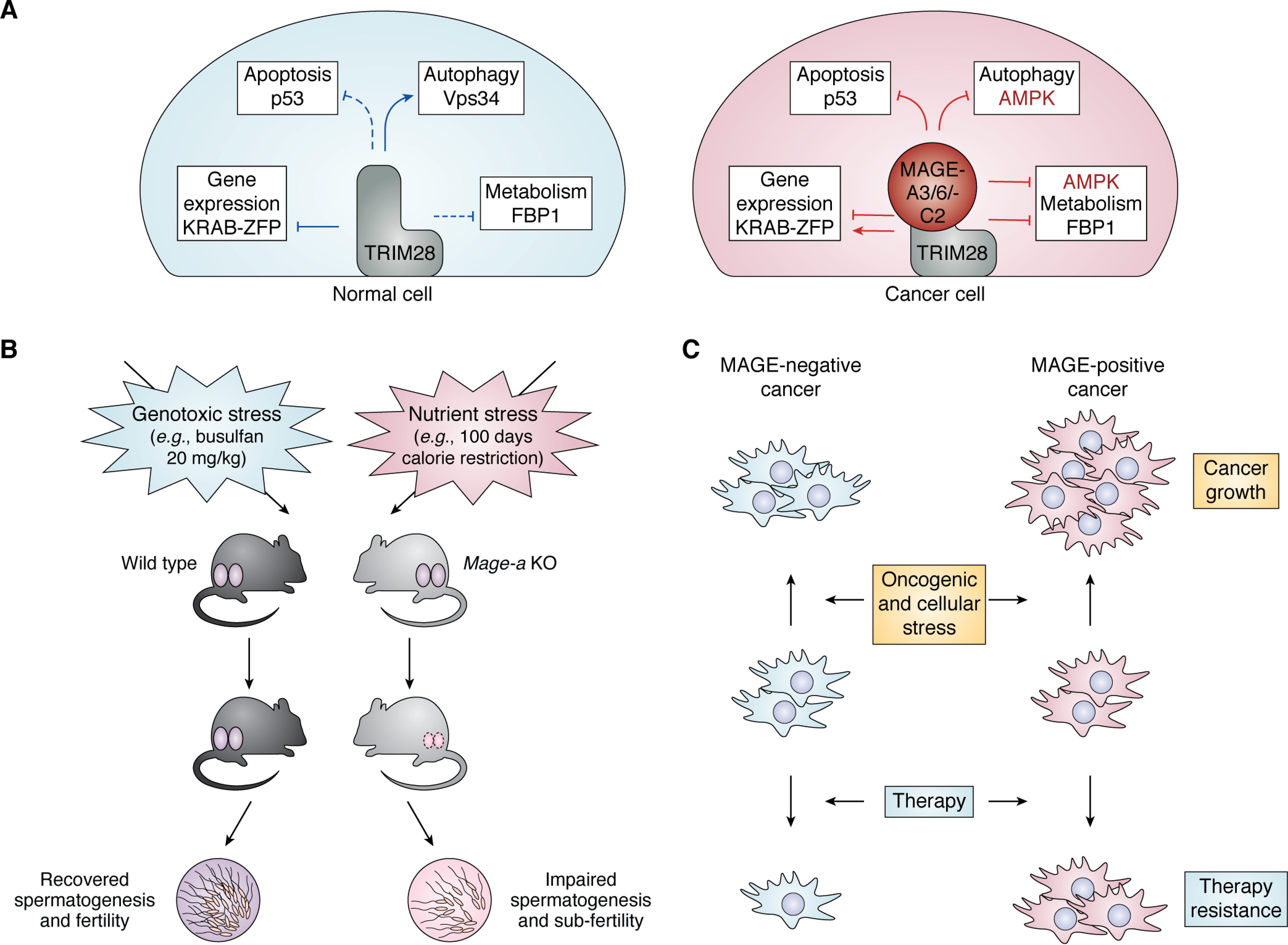
MAGE-A3/6 and -C2 are cell type–specific regulators of TRIM28 that confer stress resistance to male germline and cancer. A, MAGE-A3/6 and -C2 act as specific regulators of TRIM28 function in transcriptional regulation, apoptosis, autophagy, and cell metabolism (MAGE-A3/6 and MAGE-C2 are cancer cell-specific regulators of TRIM28). B, after genotoxic or nutritional stress, recovery of spermatogenesis in Mage-a KO mice is compromised compared with WT mice. C, MAGE-A3/6 and -C2 promote cancer growth and enable therapy resistance, likely by protecting cells against diverse stressors they encounter during tumorigenesis and treatment.
In addition to reprogramming E3 ligases and determining novel substrates, MAGEs often enhance ubiquitination of distinct ligase substrates. Since the initial discovery that MAGE-A3, -A2, -A6, and -C2 all bind TRIM28, subsequent biochemical analysis of the ubiquitin conjugation capacity of TRIM28 in vitro and in cells demonstrated that these MAGEs enhance ubiquitination of several TRIM28 substrates and autoubiquitination of TRIM28 (141, 148, 157, 158). The ability to enhance E3 ligase activity seems to be a conserved and shared function of both type I and II MAGEs, as MAGE-G1 also promotes the activity of NSE1 (141, 159). Additionally, MAGE proteins may enhance E3 ligase activity by recruiting E2 ubiquitin-conjugating enzymes to the E3 ligase or by stabilizing the E3-substrate complex (43, 141). Furthermore, MAGEs can also alter E3 ligase subcellular localization to bring the E3 ligase to the substrate. For example, besides specifying WASH for ubiquitination, MAGE-L2 also mediates the localization of TRIM27 to retromer-positive endosomes (149, 160).
Beyond RING ligases, MAGE-A11 and MAGE-C2 have been found to affect Skp1-Cullin-F-box (SCF) cullin-RING ligases (CRLs) (155, 161), but no evidence supports the existence of MAGE-SCF-CRL complexes. CRLs are the largest family of the multicomponent E3 ligases that consist of cullins, RING proteins, adaptor proteins, and substrate recognition proteins (Fig. 3B) (162). The cullin-RING module, which structurally resembles MRL, is the catalytic core that recruits E2 ubiquitin-conjugating enzyme and activates the transfer of ubiquitin from the E2 to the substrate (43). MAGE-C2 interacts with the RING protein Rbx1, a component of the SCF ligase (161). In contrast to an activating role in the MRL complex (141), MAGE-C2 inhibits SCF-mediated ubiquitination of cyclin E, preventing its degradation (161). MAGE-A11 binds and regulates the substrate specificity of Skp2, an F-box domain substrate recognition protein in the SCF ligase, and this interaction increases degradation of cyclin A and decreases degradation of the transcription factor E2F1 (155). In addition to MRLs and SCF-CRLs, MAGE-A11 binds HUWE1, a HECT (E6AP type) E3 ligase (Fig. 3B). MAGE-A11-HUWE1–dependent ubiquitination of PCF11 promotes alternative polyadenylation and 3′-UTR shortening in cancer cells (163).
In addition to regulation of ubiquitination, a number of MAGE proteins have been implicated in transcriptional regulation, either directly through binding transcription factors or indirectly through regulating their cognate E3 ligases (Fig. 3B). For example, MAGE-A2, -A3, and -C2-TRIM28 regulate Krüppel-associated box (KRAB) domain zinc finger protein (KRAB-ZFP) transcription factors and p53 (164, 165). Additionally, MAGE-A11 activates the androgen receptor, and both NECDIN and MAGE-G1 repress E2F1 (43). Interestingly, MAGE-B2 was recently shown to function as an RNA-binding protein (85), further expanding the range of MAGE protein molecular functions that are enabled by flexibility of the MHD.
MAGE proteins exert diverse biological functions
The discoveries over the last decades have provided evidence that MAGEs engage in diverse molecular and cellular functions, but how they contribute to normal physiology and the pathogenesis of cancer and diverse genetic diseases is only beginning to be understood (67, 166–170). In this section, we detail the current understanding of the biological functions of select MAGEs. Although both type I and II MAGEs engage in diverse functions, we are now starting to understand that they commonly converge in providing protection against diverse stressors, suggesting that ever-changing conditions drove MAGE evolution to confer faster adaptation to emerging stressors.
MAGE-A3/6 and MAGE-C2 are cancer cell–specific regulators of TRIM28
MAGE-A3, -A6, and -C2 are some of the most commonly expressed CTAs in human cancer (Fig. 2B). Given their restricted normal expression (11) and strong antigenic potential (171), these MAGEs attracted a lot of attention as targets for cancer immunotherapy (101). MAGE-A3 and MAGE-A6 are highly similar genes that encode proteins with 96% identity (156); we will refer to these as MAGEA3/6 herein. MAGEA3/6 are expressed in 30–80% of tumors from diverse types of cancer (Fig. 2B) (101), and expression of MAGEA3/6 significantly correlates with more aggressive disease progression, poor patient prognosis, and decreased overall survival (151, 172–177). Cancer cells that aberrantly activate MAGE-A3/6 become dependent on them, as depletion of MAGE-A3/6 leads to decreased cell viability and decreased clonogenicity (151, 178). Furthermore, expression of either MAGE-A3 or -A6 in MAGE-A3/6–negative cancer cells promotes several hallmarks of cancer, all of which suggests that these proteins have a pro-oncogenic function (131, 151, 158). Additionally, MAGE-A3 is enriched in the stem cell population of bladder cancer and in the stem cell-like population of multiple cancer cell lines (179, 180), further indicating a function in the progression of malignancies. MAGE-C2 was first discovered in melanoma cell lines (84, 181) and was then identified as an immunogenic molecule in hepatocellular carcinoma (HCC) (182). MAGE-C2 expression was subsequently associated with high tumor grade, reduced recurrence- and metastasis-free survival, and decreased overall survival in multiple tumor types (84, 133, 183–195).
Given their importance in cancer, many studies have sought to understand the molecular functions and tumorigenic role of MAGE-A3/6 and -C2. The first insight came with the discovery that MAGE-C2 and MAGE-A proteins bind to TRIM28 (43, 141, 151, 158, 196). TRIM28, also known as KAP1 or Tif1β, is a versatile protein that was first described as a cofactor for transcription factors from the KRAB-ZFP family. Now it is a well-established regulator of gene repression that is particularly important for the silencing of endogenous retroviruses (164, 165). In addition, TRIM28 regulates the activity of transcription factors without a KRAB domain (i.e. c-Myc and E2F1), promotes histone deacetylation and methylation, and recruits heterochromatin protein 1 (HP-1) (164, 165). Besides transcriptional and epigenetic regulation, TRIM28 also functions as a signaling scaffold protein and as a SUMO and ubiquitin E3 ligase (197). Furthermore, TRIM28 promotes p53 ubiquitination and degradation (198). Given its versatile molecular functions, TRIM28 is important for many biological processes, several of which are regulated by MAGE proteins (Fig. 4A) (199–205).
TRIM28 MRLs regulate transcription
By binding to TRIM28, MAGE-A3 and -C2 regulate KRAB-ZFP transcription factor–mediated gene repression (Fig. 4A) (148, 157). With some types of KRAB-ZFPs, MAGEs can relieve the TRIM28-KRAB-ZFP–mediated repression by promoting ubiquitination and degradation of distinct ZFPs (157). In contrast, MAGEs can also further enhance TRIM28-mediated repression without causing ubiquitination or degradation of the KRAB-ZFPs, whereas ZFPs with a Scan box motif KRAB domain are not affected by MAGEs (148, 157). By modifying TRIM28-KRAB-ZFP function, MAGEs alter the expression of several genes (148, 157), which suggests a potential molecular mechanism underlying the oncogenic role of MAGEs. For example, TRIM28 promotes gene silencing when at the ID1 promoter, but MAGE-A3 and -C2 are able to relieve the silencing via removal and subsequent proteasomal degradation of ZNF382, the KRAB-ZFP that acts as a tumor suppressor and normally inhibits ID1 (206). The resulting chromatin relaxation promotes ID1 expression and unleashes its pro-oncogenic functions (148, 206).
MAGE-C2 has also been implicated in transcriptional regulation, independent of TRIM28. MAGE-C2 interacts with the putative tumor suppressor BS69 (207), which negatively regulates the Epstein–Barr virus (EBV) protein, LMP1, and prevents LMP1-mediated NF-κB activation and IL-6 production (207). MAGE-C2 binding to BS69 mediates the ubiquitination and proteasomal degradation of BS69, thus promoting LMP1-induced IL-6 production, STAT3 signaling, and oncogenic transformation of EBV-infected cells (207, 208). In melanoma cells, MAGE-C2 also interacts with STAT3 and inhibits its degradation to promote amoeboid invasion of cells and potentially confers metastatic potential in tumors (209).
TRIM28 MRLs inhibit p53
TRIM28 regulates p53 protein stability in cells through E3 ligase MDM2 (198, 210); however, TRIM28 MRLs promote p53 degradation independently of MDM2 (Fig. 4A) (141, 158). In addition to promoting TRIM28-mediated p53 degradation, MAGE-A proteins directly bind p53 and inhibit its function by recruiting HDAC3 to the promoter binding sites of p53, thereby converting p53 from a trans-activator of CDKN1A (p21) into its repressor (211). MAGE-A proteins also interfere with the DNA-binding surface of the p53 core domain and prevent p53 from interacting with its cognate binding sites in chromatin, further inhibiting the expression of p53-responsive genes (212). In multiple myeloma, MAGE-A3 was predominantly detected in relapsed patients, where its expression correlated with higher proliferation status (213). Further investigation revealed that MAGE-A3 is required for the survival of proliferating myeloma cells through both p53-dependent and -independent mechanisms (213).
MAGE-C2 may also regulate tumor growth by enhancing cellular proliferation and DNA damage apart from TRIM28-mediated regulation of p53. Through binding to another RING domain protein Rbx1 (Fig. 3B), MAGE-C2 inhibits the E3 ligase activity of the SCF complex and prevents ubiquitination and proteasomal degradation of cyclin E; thus, MAGE-C2 increases the levels of cyclin E, which promotes G1-S transition and cell proliferation (161). In addition, binding of MAGE-C2 to TRIM28 increases ataxia-telangiectasia-mutated (ATM)-dependent phosphorylation of TRIM28 Ser-824, which facilitates the repair of DNA double-strand breaks (214).
Together, these findings indicate that MAGE-A3/6 and -C2 inhibit apoptosis, promote cell growth and tumor survival, and likely confer resistance to antitumor drugs, such as etoposide (Fig. 4C) (158, 198, 211, 213, 215). MAGE-mediated p53 inactivation might be particularly important in cancers with low mutation rates, like in melanomas and cervical carcinomas, where WT p53 is often present (216, 217). Interestingly, several cancer cell lines with p53 deletion are dependent on MAGEs for viability, and p53 mutation status does not correlate with MAGE-A3/6 or -C2 expression, which led to the finding that these MAGEs have p53-independent functions that contribute to their pro-oncogenic activity (151).
TRIM28 MRLs inhibit autophagy and rewire cancer metabolism
In addition to regulating p53 and cell viability, MAGE-A3/6 and -C2 also act as oncogenes by inhibiting autophagy and enabling metabolic flexibility in cancer cells (Fig. 4, A and C) (151, 196). MAGE-A3/6 and -C2 accomplish the metabolic rewiring by specifying two major metabolic proteins, AMPK and fructose-1,6-biphosphatase (FBP1), for TRIM28-mediated ubiquitination and subsequent proteasomal degradation (151). Unlike the targeting of p53 (141), AMPKα1 is targeted by TRIM28 only in the presence of MAGE-A3/6 or -C2 (151). Through binding to AMPKα1 and TRIM28, these MAGE proteins enable the ubiquitination and subsequent proteasomal degradation of AMPKα1 that leads to a reduction in overall AMPK protein levels (151). In line with MAGE-A3/6 regulating AMPK stability, MAGE-A3/6 mRNA expression inversely correlates with AMPK activity and protein levels in diverse cancer patient samples, including breast, lung, and colon cancer (134, 151). In colon cancer, MAGE-A3/6 mRNA levels inversely correlate with the expression of miR-1273g-3p, which silences MAGE-A3/6 expression and inhibits human colorectal cancer cell growth via AMPK activation (134). Likewise, in hepatocellular carcinoma, another miRNA, miR-448, was shown to inhibit expression of MAGE-A6, thereby activating AMPK signaling and inhibiting tumor growth, as well as inhibiting stemness maintenance and self-renewal of cancer stem cells (137). In glioma and renal cell carcinoma, MAGE-A6 was also shown to promote cell survival by targeting AMPKα1 (135, 138). Furthermore, MAGEA6-AMPK signaling was activated by lnc-THOR silencing, which inhibited human glioma cell survival (136), further corroborating an important role of AMPK regulation in the oncogenic function of MAGE-A3/6.
As a master sensor of cellular energy that is activated in response to energy stress, AMPK promotes catabolic processes, such as autophagy, while inhibiting anabolic processes and cell growth to restore energy balance (218–220). Accordingly, depletion of MAGE-A3/6 and -C2 or TRIM28 in several MAGE-positive cancer cells not only increases the levels of both total AMPK and the active form, but also leads to concomitant suppression of the mTOR signaling pathway (134–138, 151, 156). These results suggest that MAGE-A3/6 and MAGE-C2 can rewire cancer metabolism toward dependence on mTOR signaling for survival (221). Given that inhibition of MAGE-A3/6 expression and consequent activation of AMPK signaling inhibits cell growth in several cancer cell types (101, 151), MAGE-A3/6 may serve as biomarkers for effective use of AMPK agonists (e.g. metformin) and mTOR inhibitors (e.g. everolimus) that are already used in the clinic (134–138, 222, 223).
By inhibiting AMPK, MAGE-A3/6 and -C2 also act as molecular switches that convert TRIM28 from a pro-autophagic to an anti-autophagic factor (Fig. 4A) (151, 156). In the absence of MAGEs, TRIM28 functions as a pro-autophagic factor through its SUMO ligase activity, where it SUMOylates PIK3C3/VPS34 to promote formation of the PIK3C3-BECN1 complex and autophagy (156). However, in cells that express MAGE-A3/6 or MAGE-C2, TRIM28 MRLs target AMPK for degradation and, by doing so, inhibit autophagy and promote mTOR signaling, which may provide the optimal environment for early tumor formation and growth (151, 224).
In addition to regulating AMPK and mTOR signaling, MAGE-A3/6 and -C2-TRIM28 also impact glucose metabolism (151). In HCC, MAGE-A3 and -C2 enhanced TRIM28-mediated degradation of FBP1, a rate-limiting enzyme in gluconeogenesis (196). By promoting FBP1 degradation, TRIM28 MRLs increased glucose consumption and lactate production, promoted the Warburg effect, and reprogrammed cancer cell metabolism to support HCC progression (196). Thus, inhibiting MAGE-TRIM28–mediated degradation of substrates, such as FBP1, could be a therapeutic option for the treatment of advanced HCC. Intriguingly, the FBP1 substrate, fructose 1,6-bisphosphate (FBP), has been recently identified as a glucose sensor and inhibitor of AMPK activation (225), suggesting that TRIM28 MRLs may inhibit AMPK activity by several mechanisms. However, additional studies are necessary to understand the underlying mechanistic details and the specific contributions of MAGE-A3/6 suppression of AMPK through MAGE-mediated AMPKα1 ubiquitination compared with indirect regulation by degradation of FBP1 and subsequent increase in FBP.
Beyond controlling cell metabolism, MAGE-A3/6 themselves are also regulated in response to nutrient availability (226). Upon short-term cellular starvation, MAGE-A2, -A3/6, and -A12 are rapidly degraded by the ubiquitin–proteasome system, suggesting dynamic regulation of these proteins in different metabolic states (226). Starvation-induced degradation of these MAGE-A proteins is controlled by the CRL4-DCAF12 E3 ligase and depends on two glutamate residues (–EE*) at the extreme C terminus of the proteins that serve as a degron signal for CRL4-DCAF12 targets (226, 227). Intriguingly, CRL4-DCAF12 likely does not degrade MAGE-C2 because it lacks these C-terminal glutamates, which is indicative of diverse regulation and functional complementarity of seemingly redundant MAGE proteins.
The regulation of MAGE-A3/6 by CRL4-DCAF12 is important to achieve robust autophagy induction during nutrient starvation (226). Although inhibition of autophagy is often critical for tumor initiation, the reinstitution of autophagy promotes tumor progression (224). Thus, MAGE-A3/6 may suppress autophagy during early phases of tumor initiation, but as nutrient stress on tumors emerges, this brake may be relieved through CRL4-DCAF12–mediated down-regulation of MAGE-A3/6 to allow autophagy induction and tumor progression (226). Besides nutrient deficiency, acquisition of cancer-associated mutations in MAGE-A6 was also found to release autophagy inhibition in pancreatic cancer (228), in which MAGE-A6 is among the top 16 most commonly mutated genes (229). The identified MAGE-A6 mutations lead to its proteasomal degradation, suggesting that pancreatic cancer progression depends on the release of autophagy inhibition through degradation of MAGE-A6, induced either by nutrient deficiency or acquisition of mutations (228). Further studies will show how these processes are fine-tuned during tumorigenesis and the relevance to their physiological function in germ cells. Because glycolysis is important for stemness maintenance in SSCs, MAGE-A3/6 expression in spermatogonia implies a role in SSC biology and spermatogenesis regeneration (11, 83, 99, 230).
Mage-a proteins enable robust spermatogenesis under genotoxic and nutritional stress in mice
In primary cultures of undifferentiated spermatogonia, Mage-a proteins are important for maintaining the stemness of SSCs as knockdown of Mage-a proteins leads to the loss of ID4-positive cells and lower transplantation efficiency (11, 231). Intriguingly, the depletion of two (Mage-a4 and -a10), six (Mage-a1, -a2, -a3, -a5, -a6, and -a8), or all eight Mage-a genes does not affect male reproduction in mice (11, 232), suggesting that Mage-a proteins are dispensable for unperturbed male spermatogenesis when animals are kept under normal laboratory conditions.
Given the very recent evolutionary appearance of Mage-a genes and the astonishingly conserved basic processes of spermatogenesis, the lack of a phenotype in these Mage-a KO mice in an optimal environment suggests that Mage-a proteins may provide an advantage when animals endure stress (Fig. 4B) (11, 232). In support of this idea, short-term treatment of Mage-a KO mice with the genotoxic agent N-ethyl-N-nitrosourea results in higher germ cell apoptosis (11, 232). Interestingly, unchallenged Mage-a KO mice exhibit increased p53 protein levels, indicative of increased apoptosis and conservation of p53 regulation by human and mouse MAGE-A proteins within both germ cells and cancer (11, 141, 233). This result is consistent with increased survival of SSCs after irradiation in p53 KO mice (234). Furthermore, exposure to genotoxic stress by treatment with the chemotherapeutic busulfan, which completely ablates the germline, impairs the recovery of spermatogenesis in Mage-a KO mice, likely a result of both greater germ cell damage during the treatment and lower stem cell regenerative capacity after treatment (Fig. 4B) (11).
In accordance with MAGE-A3/6 regulation of AMPK and metabolism in human cancer cells and nutrient availability being one of the major drivers of evolution, mouse Mage-a proteins are also involved in metabolic adaptation of germ cells. Inducing nutrient stress with caloric restriction impairs spermatogenesis in Mage-a KO mice, indicating that Mage-a proteins provide an advantage to spermatogenesis when food is limited (Fig. 4B) (11). Interestingly, the peak expression of Mage-a genes just before male germ cells start transitioning through the BTB (Fig. 2A) suggests that Mage-a genes may have evolved to provide protection to the germline during metabolic stress, either arising from the environment or due to the metabolic switch during spermatogenesis. Accordingly, spermatogonial cells from Mage-a KO mice are more sensitive to 2-deoxy-d-glucose (2DG)-induced glycolysis inhibition than WT cells (11). Intriguingly, MAGE-A6–expressing human pancreatic cancer cells hijack this protective function when treated with 2DG, as they adapt faster to the induced metabolic stress and develop resistance against 2DG (Fig. 4C) (11).
MAGE-A11 regulates androgen receptor signaling and alternative polyadenylation
MAGE-A11 is a unique, primate-specific member of the MAGE-A subfamily that acts as a steroid hormone receptor transcriptional coregulator and proto-oncogenic protein implicated in prostate cancer (41). Another molecular function for MAGE-A11 in promoting mRNA alternative polyadenylation (APA) was recently discovered, defining an additional oncogenic function of this enigmatic member of the MAGE-A subfamily (163).
MAGE-A11 is normally expressed in the testis (Fig. 2A), in syncytiotrophoblasts of the placenta (11, 163), and cyclically in human endometrium (235). Like other type I MAGEs, MAGE-A11 is often aberrantly activated in human tumors (Fig. 2B) (43, 101, 105, 111, 163, 236–238). Not only is MAGE-A11 aberrantly expressed in cancer, but its expression is necessary and sufficient to drive tumorigenesis (163). Additionally, MAGE-A11 has been associated with worse disease progression (239–241) and therapy resistance (242, 243).
Initially identified through a yeast two-hybrid screen using the androgen receptor (AR) FXXLF motif as bait, MAGE-A11 was subsequently shown to act as a coregulator that stabilizes ligand-free AR (Fig. 3B) (244). The AR FXXLF motif interacts with a putative F-box motif located in the MHD of MAGE-A11, and this interaction is modulated by checkpoint kinase 1 and MAPK phosphorylation and ubiquitination (245, 246). MAGE-A11 promotes AR transcriptional activity though F-box–mediated interactions with coactivators, such as steroid receptor coactivator (SRC)/p160, transcriptional intermediary factor 2 (TIF2), and histone acetyltransferase p300 (244, 246, 247). MAGE-A11 expression has also been shown to increase during androgen deprivation therapy in prostate cancer, suggesting that the capacity of MAGE-A11 to activate AR in an androgen-independent fashion may play a critical role in the development of castration-recurrent prostate cancer (43, 105).
In addition to AR, MAGE-A11 interacts with the progesterone receptor (PR). MAGE-A11 specifically binds the PR-B isoform, which positively regulates the effects of progesterone and induces transcription via interactions with p300 during the cyclic development of the human endometrium (235). The highest levels of MAGE-A11 occur in the mid-secretory stage, coincident with the window of uterine receptivity to embryo implantation; however, the role of MAGE-A11 during decidualization and pregnancy establishment (248) has not been fully explored. The cellular functions of MAGE-A11 in female reproduction are almost completely unknown. Intriguingly, MAGE-A11 expression correlates with delayed endometrial decidualization and infertility in polycystic ovary syndrome, a common gynecological disorder that affects up to 12% of women of reproductive age (249). This association suggests that MAGE-A11 may also have an important role in female reproduction.
Besides interacting with steroid hormone receptors, AR, and PR, MAGE-A11 has also been shown to function as an E3 ligase substrate adapter to regulate protein ubiquitination and stability (Fig. 3B) (150). MAGE-A11 interacts with Skp2, the substrate recognition protein of the SCF E3 ligase, as well as with cyclin A, a target of Skp2; the presence of MAGE-A11 leads to enhanced E2F1 transcription activity by increasing Skp2-mediated degradation of cyclin A and decreasing degradation of E2F1 (155). MAGE-A11 also interacts with and stabilizes RB family proteins, including p107 and RB (237). Remarkably, MAGE-A11 binding to p107 flipped p107 from a transcriptional repressor to a transcriptional activator of AR and E2F1 to promote tumorigenesis in prostate cancer (237). Interestingly, the tumor suppressor p14-ARF has been demonstrated to target MAGE-A11 for degradation in a lysine-independent fashion and prevent MAGE-A11 activation of E2F1 (250).
Beyond the function of MAGE-A11 in conjunction with AR, PR, and E2F1, MAGE-A11 also regulates APA—the process by which the mRNA 3′-end processing complex utilizes one of several possible polyadenylation sites within the UTRs of a gene and, thus, regulates the length of the 3′-UTR of an mRNA transcript (Fig. 5A) (163). At least 70% of mammalian transcripts are alternatively polyadenylated (251, 252). APA-mediated 3′-UTR shortening (3′-US) can affect mRNA transcript stability, translation efficiency, nuclear export, and cellular localization (253). APA often occurs in a tissue– or developmental stage–specific manner and is associated with diverse biological processes, including T lymphocyte activation, brain development and function, and male germ cell differentiation (254–256).
Figure 5.

MAGE-A11 and -B2 affect transcription and translation, respectively. A, by binding to the E3 ligase HUWE1, MAGE-A11 specifies PCF11 for ubiquitination, which displaces CFIm25 from the mRNA 3′-end processing complex. The subsequent remodeling of the complex leads to 3′-UTR shortening, which leads to increased levels of oncogenes through loss of miRNA repression. Additionally, down-regulation of ceRNAs inhibits tumor suppressors. Thus, MAGE-A11 function in APA contributes to tumorigenesis. B, MAGE-B2 binds to the G3BP mRNA transcript to repress its translation and decrease G3BP protein concentration. As a result, MAGE-B2/-b4 inhibit stress granule formation and promote cellular stress tolerance, giving a growth advantage to cancer cells and heat tolerance to male germ cells.
Despite its integral function in normal biology, aberrant APA, including 3′-US, is often associated with cancer as a hallmark of most tumors (257, 258). An effort to uncover the molecular mechanisms of MAGE-A11 oncogenic activity identified PCF11, the poly(A) cleavage factor of the mRNA 3′-end processing complex, as a MAGE-A11 binding partner (163). The direct interaction of MAGE-A11 with PCF11 leads to the ubiquitination and degradation of PCF11 by recruiting the E3 ligase HUWE1 (Fig. 5A) (163). This regulation of PCF11 by MAGE-A11 drives APA and contributes to 3′-US in human tumors (163). Consistent with previous findings that depletion of CFIm25, another component of the mRNA 3′-end processing complex, induces 3′-US (259–261), MAGE-A11-HUWE1 ubiquitination of PCF11 inhibits the association of CFIm25 with RNA polymerase II, leading to remodeling of the mRNA 3′-end processing complex and inducing 3′-US (163).
Several bona fide oncogenes and tumor suppressor genes have altered 3′- UTRs associated with MAGE-A11-HUWE1–mediated ubiquitination of PCF11, which directly links MAGE-A11 function to tumorigenesis (Fig. 5A) (163). Analysis of ovarian and lung squamous carcinomas from TCGA data sets shows that MAGE-A11–expressing tumors have a significantly higher number of transcripts with 3′-US than MAGE-A11–negative tumors, implying that MAGE-A11 drives 3′-US in human cancer (163). MAGE-A11–induced 3′-US has both cis and trans effects on oncogenes (i.e. cyclin D2) and tumor suppressors (i.e. PTEN), either by increasing protein levels through loss of miRNA repression or down-regulating competing endogenous mRNAs (ceRNAs), respectively, to eventually stimulate key progrowth pathways (163).
Interestingly, polyadenylation site choice in germ cells differs from somatic cells and often results in 3′-US, which generates germ cell–specific transcripts and protein isoforms (255, 256, 262). The mechanism underlying widespread APA in germ cells leading to 3′-US is not well-understood, but changes in composition of the polyadenylation machinery have been proposed (263). Given their molecular function in cancer, MAGE-A11 and HUWE1 may be important factors in promoting APA in male germ cells. Consistently, HUWE1 has been shown to be important for spermatogonial differentiation and entry into meiosis (264).
MAGE-B2/Mage-b4 regulate stress tolerance in cancer and germ cells
Like other type I MAGE CTAs, MAGE-B2 is primarily expressed in the testis and is aberrantly expressed in various cancers, where it has been implicated in tumor growth and progression (Figs. 1 and 2). More specifically, MAGE-B2 overexpression promotes cell proliferation in transformed oral keratinocytes, whereas MAGE-B2 depletion reduces proliferation in osteosarcoma cell lines (106, 265). Moreover, subcutaneous injection of mouse melanoma cell lines expressing human MAGE-B2 enhances tumor xenograft growth in mice (265). Interestingly, MAGE-B2 is thought to be activated early during carcinogenesis and is also expressed in the cancer stem cell–like population derived from colon adenocarcinoma cells (180). Despite the mounting evidence indicative of MAGE-B2's oncogenic potential, little was known about its molecular function until a recent study revealed a theme analogous to MAGE-A function—stress tolerance (85).
MAGE-B2 enhances the cellular stress threshold by suppressing stress granule (SG) assembly (Fig. 5B) (85). SGs are conserved ribonucleoprotein membraneless organelles that form in response to a variety of stress stimuli (266, 267). Upon exposure to stress, translation stalls, polysomes disassemble, and a number of proteins and mRNAs condense into cytoplasmic SGs via liquid–liquid phase separation (LLPS) (267–270). Although the specific proteins and mRNAs that localize to SGs are stress-dependent, G3BP1 and its paralog G3BP2 (collectively referred to as G3BP) are uniquely critical for SG core assembly, as overexpression of G3BP induces spontaneous SG formation and deletion ablates SGs in response to sodium arsenite (271–279). Whereas the exact molecular features that drive SG formation are still being elucidated, G3BP is predicted to promote LLPS of SGs due to its ability to bind RNA and form higher-order oligomers (272).
MAGE-B2 regulates SG dynamics, such that depletion of MAGE-B2 leads to increased SG formation and overexpression of MAGE-B2 has the opposite effect (85). MAGE-B2 depletion in U2OS osteosarcoma cells was previously shown to decrease cell number and colony formation capacity without changing BrdU incorporation (265), suggesting that MAGE-B2 depletion does not affect cell proliferation (85). However, upon exposure to prolonged oxidative stress by a low dose of sodium arsenite, MAGE-B2 KO cells exhibit reduced cell viability that can be rescued by re-expression of MAGE-B2 (85, 265). Furthermore, low-dose treatment with the ribotoxic agent actinomycin D also leads to reduced proliferation of MAGE-B2 KO cells compared with WT MAGE-B2–expressing cells (265). Together, these findings suggest that MAGE-B2 increases cellular stress tolerance and provides a growth advantage in nonoptimal conditions.
Further investigation into the mechanism by which MAGE-B2 alters SG assembly revealed that MAGE-B2 reduces G3BP protein levels (85). Because in vitro LLPS is highly dependent on protein concentration (266), MAGE-B2-mediated regulation of G3BP protein levels alters SG formation and cell viability under prolonged stress (85). In a surprising deviation from the prototypical MAGE-RING ligase complex, MAGE-B2 works as an RNA-binding protein that directly binds the G3BP transcript to inhibit its translation and, thus, alters G3BP protein levels (85). In addition, MAGE-B2 binding to the 5′-UTR of G3BP displaces the translational activator DDX5, indicating that MAGE-B2 and DDX5 act in a competitive manner to fine-tune G3BP concentration and to regulate SG dynamics (Fig. 5B) (85).
In the context of normal physiology, the enriched expression of MAGE-B2 and its mouse orthologs, Mage-b4 and Mage-b10 (referred to herein as Mage-b4 due to highly similar sequence identity), in undifferentiated spermatogonia and SSCs (Fig. 2A) suggested that MAGE-B2 functions in stem cell maintenance and differentiation (11, 57, 85, 280–283). In support of this idea, Mage-b4 depletion in in vitro primary cultures of undifferentiated spermatogonia from Id4-eGFP reporter mice demonstrated that Mage-b4 plays a key role in SSC maintenance and promotes recovery after in vivo transplantation (85). Because male germ cells are very sensitive to heat (284–286), most mammals maintain testes in scrotum outside the body, thereby sustaining spermatogenesis at temperatures 5–7 °C lower than the core body temperature (287). In line with MAGE-B2 inhibition of SG formation being important for increasing the cellular threshold against stress, Mage-b4 provides thermotolerance for the male germline (85). Mage-b4 KO mice exposed to testicular heat stress exhibit increased SG formation in spermatogonia and severely impaired recovery of spermatogenesis with significantly reduced fertility and increased damage within the seminiferous tubules (Fig. 5B) (85). Together, these data suggest that, like Mage-a genes, Mage-b4 evolution was driven by protecting the male germline and preserving fertility in more extreme conditions.
MAGE-B2 also enhances the activity of E2Fs (265), which are transcriptional regulators of cell cycle progression (288). Elevated expression of E2F target genes in tumors is thought to induce aberrant cell proliferation and increase cell cycle–generated genomic errors (288). MAGE-B2 overexpression enhances E2F reporter activity, whereas depletion of MAGE-B2 reduces transcript levels of known cell cycle–associated E2F target genes, such as MCM6, CyclinD1, and CDK1 (265). Through binding the E2F1 repressor HDAC1, MAGE-B2 reduces the inhibitory E2F1-HDAC1 interaction and promotes E2F1 function (265). Interestingly, both MAGE-A11 and -B2 promote E2F transcriptional activity and cell growth, whereas the type II MAGE NECDIN inhibits E2F function and induces growth arrest (66, 155, 289). Whether MAGE-B2–mediated activation of E2F contributes to stress tolerance warrants further investigation.
Curiously, whereas MAGE function is typically assessed in the context of spermatogenesis or tumorigenesis, MAGE-B2 was originally identified in pediatric systemic lupus erythematosus (SLE) patients (290). SLE is a chronic autoimmune disease that causes widespread inflammation and tissue damage in affected organs (OMIM #152700). Because genome-wide methylation abnormalities are present in SLE patients (291, 292), it is plausible that disrupted DNA methylation allows for aberrant MAGE-B2 expression and the presentation of normally hidden antigens to provoke an autoimmune response and inflammation. Whereas the pathogenic role of MAGE-B2 in SLE remains unknown, the presence of MAGE-B2 protein and autoantibodies in patients with active lupus nephritis and the ability of MAGE-B2 to stimulate an immune response when it is presented by the MHC suggest a potential role in immune activation (290, 293–296).
Since its identification in SLE, MAGE-B2 autoantibodies have also been detected in patients with autoimmune polyendocrine syndrome type 1 (APS1), a monogenic autoimmune disorder that is caused by loss-of-function mutations in the autoimmune regulator (AIRE) gene (297, 298). AIRE encodes a transcription factor that plays an essential role in establishing self-tolerance in the thymus by driving promiscuous expression of tissue-restricted antigens (299, 300). This AIRE-driven antigen display allows naive T cells to be exposed to tissue-specific antigens and for the subsequent elimination of autoreactive T cells. In patients with APS1, defective AIRE allows autoreactive T cells to survive, thereby creating an autoimmune response. Intriguingly, infertility is a common manifestation of APS1 in both male and female patients. Whether MAGE-B2 antigens play a role in APS1 infertility remains unknown. Interestingly, two variant alleles of the rs1800522 AIRE SNP were shown to differently modulate MAGE-B2–specific T-cell survival and in vivo susceptibility to melanoma in mice (301); however, whether this finding translates to cancer predisposition in humans is yet to be determined.
MAGE-D1 fine-tunes apoptosis and differentiation during neurogenesis and oncogenesis
The MAGE-D subfamily, comprised of four genes in humans and three highly homologous orthologs in mice (Fig. 1), was identified through sequence homology with the initially discovered MAGE-A genes (39, 60). Among all of the MAGEs, MAGE-D1 (also referred to as NRAGE and Dlxin-1) is expressed at the absolute highest level across diverse tissues (11). In the central nervous system, MAGE-D1 is expressed throughout the neural tube during the early stage of neurogenesis and becomes restricted within the ventricular zone, subplate, and cortical plate during the later stage (69). High expression in the brain during development and in adults suggests a functional relevance in neurogenesis and brain physiology (70, 302–304)
In addition to the MHD, MAGE-D1 protein contains a unique WQXPXX hexapeptide repeat domain that confers specific interactions. Both the hexapeptide repeat domain and the MHD are important for MAGE-D1's molecular functions (75, 305–308). The cytosolic region of p75 neurotrophin receptor (p75NTR) was identified as the first binding partner of MAGE-D1 that promotes neuronal apoptosis during development (75, 308). Further studies not only uncovered additional MAGE-D1 interacting partners, but also expanded its diverse biological functions to include apoptosis, cell cycle progression, cell adhesion, angiogenesis, and developmental morphogenesis (75, 76, 78, 305, 309, 310).
MAGE-D1 is required for apoptosis during embryonic development and neurogenesis
MAGE-D1 regulates several apoptotic pathways, each caused by distinct input signals to ultimately drive neurogenesis (311). By binding to p75NTR, MAGE-D1 triggers cell cycle arrest and mediates neurotrophin-dependent apoptosis that requires JNK activation (75, 308). Following nerve growth factor treatment, MAGE-D1 accumulates at the plasma membrane to prevent p75NTR from binding to the receptor tyrosine kinase TrkA, which normally blocks p75NTR-dependent apoptotic signaling (75). In vitro, MAGE-D1 also promotes neurodifferentiation of PC12 cells by interacting with TrkA and by early activation of the MEK and Akt signaling pathways (312).
In addition, MAGE-D1 promotes apoptosis through interaction with the axon guidance receptor UNC5H1 (313) and different antiapoptotic proteins, like members of the inhibitors of apoptosis protein (IAP) family (314). In neural progenitor cells, MAGE-D1 interacts with the RING domain of X-linked IAP (XIAP), promoting its caspase-mediated cleavage and degradation (314) to transmit proapoptotic signals and NF-κB activation via the BMP alternative pathway (315). MAGE-D1 can also interact with CHE-1, an apoptosis-antagonizing transcription factor, which inhibits apoptotic signaling by binding RB and removing HDAC1 from E2F target promoters (78). MAGE-D1 sequesters CHE-1 in the cytoplasm, thereby promoting its ubiquitination and proteasomal degradation to further promote apoptosis (78).
Interestingly, MAGE-D1 itself is also regulated by ubiquitination. Bone morphogenetic protein 4 (BMP4) enhances Lys-63–linked polyubiquitination of MAGE-D1 by the SCFFBXO7 E3 ligase complex and facilitates formation of the MAGE-D1–TAK1–TAB1 complex, which up-regulates NF-κB and p38 and promotes caspase-dependent apoptosis (316, 317). BMP4-mediated activation also leads to formation of the MAGE-D1-TAK1-TAB1-XIAP complex, which has been shown to stimulate p38 in renal branching morphogenesis (310), IKK-α/β in macrophage migration inhibitory factor production (315), and G0/G1 arrest and apoptosis in dental pulp cells (318).
Together, the pleiotropic molecular functions of MAGE-D1 all converge on promoting apoptosis and cell cycle exit of neural progenitors to promote neuronal differentiation (316). To balance proliferation and differentiation during neurogenesis, MAGE-D1 protein level is controlled by PRAJA1, a RING E3 ligase that ubiquitinates MAGE-D1 and promotes its degradation by the proteasome (319).
Beyond neurogenesis, MAGE-D1 is also implicated in the differentiation of other cell types through regulation of the DLX/MSX family of homeodomain proteins, which MAGE-D1 binds via its hexapeptide repeat domain (320). MAGE-D1 binds to and enhances the activity of DLX5, a homeodomain-containing transcription factor that is expressed in the forebrain, limbs, and branchial arches during embryonic development and is important for digit formation in mice (306). Besides DLX5, MAGE-D1 also interacts with other homeodomain proteins, DLX4 and MSX2 (306), the latter of which is targeted for ubiquitination and proteasomal degradation by the MAGE-D1-PRAJA1 MRL (141, 320). Additionally, MAGE-D1 cooperates with the type II MAGE NECDIN in the developing brain and skeletal muscle, where MAGE-D1-NECDIN heterodimer inhibits MSX2 to promote terminal differentiation of postmitotic cells (320, 321). Besides PRAJA1, MAGE-D1 levels are also controlled by ROR2 receptor tyrosine kinase, which binds and sequesters MAGE-D1 at the cell membrane to prevent it from interacting with MSX2 (322).
In addition to expression and function during embryonic development, MAGE-D1 can be induced by muscle injury and is required for p21 induction and muscle regeneration (323). Furthermore, MAGE-D1 was implicated in regulation of the cytoskeleton and cell adhesion, in part by inhibiting hypoxia-induced HIF-1 activation (324, 325). However, further study is needed to understand the molecular mechanisms and the biological significance of MAGE-D1 functions in these processes.
Mage-d1 KO mice exhibit defects in neurogenesis and brain function
Based on its significance in facilitating neuronal apoptosis and neurogenesis during embryonic development, a Mage-d1 KO mouse model was generated to further explore MAGE-D1 functions (5). Although Mage-d1 KO mice are normal in terms of gross morphological and histopathological features, developmental apoptosis of the sympathetic neurons of the superior cervical ganglia is defective, similar to that observed in p75NTR KO mice (326). In line with in vitro data (75), primary cultures of sympathetic neurons derived from Mage-d1 KO animals are resistant to BDNF-p75NTR–mediated apoptosis. Further, Mage-d1 KO mice show defects in hair follicle catagen phase, another p75NTR-dependent apoptotic process, where hair follicles regress following morphogenesis (326). Mage-d1 KO mice also present with defects in motor neuron apoptosis that are not perturbed in p75NTR KO mice, supporting the role of Mage-d1 in p75NTR-independent apoptosis (76, 313, 326).
These KO mice also revealed the role of Mage-d1 in other brain functions, including regulation of mood, behavior, memory, body weight, and circadian rhythm (70, 302–304). Although not cyclic by itself, Mage-d1 regulates the circadian rhythm by binding to nuclear receptor RORα (303). Further, Mage-d1 promotes ubiquitination and proteasomal degradation of serotonin transporter (SERT), which prevents serotonin uptake, leading to lower serotonin levels in the prefrontal cortex, which, ultimately, results in symptoms of depression (70). Further analysis of KO mice implicated Mage-d1 in a complex behavioral syndrome that includes anxiety, decreased social interaction, memory loss, and obesity. These symptoms are explained by a reduction of mature oxytocin, a neuropeptide produced within the hypothalamus and released into the blood from the posterior pituitary that stimulates bonding and prosocial behavior (302), but the molecular mechanism underlying Mage-d1 regulation of oxytocin levels warrants further investigation. Mage-d1 KO mice also exhibit impaired cognitive functions, suggesting that Mage-d1 is involved in synaptic transmission and hippocampus-dependent learning and memory formation (304). By interacting with cAMP-response element–binding protein (CREB) transcription factor, Mage-d1 regulates the expression of BDNF, which is instrumental in hippocampus-dependent learning and memory formation (304).
By interacting with a range of proteins, from transcription factors, like CREB and RORα, to transmembrane receptors, like SERT and p75NTR, MAGE-D1 modulates diverse physiological pathways to fine-tune neurogenesis and brain functions. Intriguingly, despite these extensive functions, mice can survive without Mage-d1. Yet because Mage-d1 exhibits many diverse functions in the brain, we speculate that it enabled animals to better adapt to challenges and provided an evolutionary advantage.
MAGE-D2 in cellular stress response and kidney function
The function of MAGE-D2 remained mysterious for a long time, but discovering the cellular localization of MAGE-D2 provided the first insights into its biological role. MAGE-D2 may be found within the cytoplasm and the nucleolus; however, the location of MAGE-D2 changes throughout the cell cycle (327). Whereas MAGE-D2 is cytoplasmic and nucleolar in G1 phase, it becomes progressively more nucleoplasmic upon the entrance into S phase (327). In prophase before the disassembly of the nucleus, MAGE-D2 leaves the nucleolus and becomes completely cytoplasmic during mitosis, until it eventually re-enters the nucleolus in early G1 (327).
MAGE-D2 regulates DNA damage response
The main function of the nucleolus is the rapid production of small and large ribosome subunits, a process that must be highly regulated to achieve proper cellular proliferation and growth, as well as to respond quickly to stress (328). Interestingly, more than half of the nucleolar proteome represents proteins, including MAGE-D2, that are transiently stored in the nucleolus and can be rapidly released to control cellular stress responses (327, 328). Upon genotoxic and nucleolar stress, MAGE-D2 is shuttled from nucleoli to the nucleoplasm, leading to a G1/S block in cell cycle progression (327). DNA damage triggers MAGE-D2 phosphorylation by ATM/ATR, which is required to maintain proper levels of p21 and p27 to facilitate cell cycle arrest (329). Depletion of MAGE-D2 leads to reduced CHK2 phosphorylation and increased CHK1 phosphorylation, an indication of sustained ATR activation, suggesting a role for MAGE-D2 in maintaining genomic stability (329). Conversely, MAGE-D2 can also regulate p53 by decreasing p53 transcriptional activity and inhibiting TRAIL-induced apoptosis (330, 331). Altogether, the storage of MAGE-D2 in nucleoli appears to be a way to harbor MAGE-D2 until cellular stressors are encountered, allowing for rapid cell cycle arrest (327).
Antenatal Bartter's syndrome and MAGE-D2's role in embryonic kidney function
The discovery of another important MAGE-D2 function stemmed from the identification of MAGE-D2 mutations in patients with antenatal Bartter's syndrome (OMIM #300971), which confers an increased risk of adverse perinatal outcome, especially preterm delivery. The MAGE-D2 mutations identified in infants with transient antenatal Bartter's syndrome (332) account for 9% of all cases of antenatal Bartter's syndrome and explain 38% of patients who lack other characterized mutations (333). This syndrome is normally caused by loss-of-function mutations in the proteins that mediate renal salt reabsorption in the thick ascending limb of the loop of Henle, where MAGE-D2 is specifically expressed in fetal and adult kidneys (11, 332, 334).
Follow-up studies revealed that MAGE-D2 affects the expression and function of sodium chloride cotransporters, Na-K-Cl cotransporter (NKCC2) and Na-Cl cotransporter (NCC), which are important regulators of salt reabsorption (332). Rather than being properly embedded in the apical cell membrane of tubular epithelial cells, NKCC2 and NCC were predominantly cytoplasmic in patients and co-localized with endoplasmic reticulum markers (332). Loss-of-function and gain-of-function studies revealed that MAGE-D2 protects NKCC2 and NCC from Hsp40-mediated endoplasmic reticulum–associated degradation, indicating that MAGE-D2 is essential for fetal renal salt reabsorption, amniotic fluid homeostasis, and the maintenance of pregnancy (332). Given that MAGE-D2 mutant–associated antenatal Bartter's syndrome self-resolved postnatally, MAGE-D2 is likely less important for postnatal sodium homeostasis mechanisms (5, 332, 335). Intriguingly, patients with MAGE-D2 mutations also presented with lower plasma bicarbonate, implying that MAGE-D2 potentially also affects sodium bicarbonate transport (333).
In adult kidneys or renal cells, MAGE-D2 expression is inducible after acute kidney injury or stress caused by cisplatin, folic acid, the inflammatory cytokine TWEAK, or serum deprivation (336). Future studies are needed to elucidate the molecular mechanisms behind stress-induced MAGE-D2 expression and the role of MAGE-D2 in stress response within the kidney.
MAGE-F1 regulates the cytosolic iron-sulfur (Fe-S) assembly (CIA) pathway
Unlike all of the other MAGE genes, MAGE-F1 is uniquely located on chromosome 3 (Fig. 1B). As expected for a type II MAGE, MAGE-F1 expression is ubiquitous across human and murine normal tissues (Fig. 2A) and is found in many tumor types, such as ovarian, breast, cervical, melanoma, and leukemia (11, 337). Beyond initial characterization of the MAGE-F1 gene and its expression, MAGE-F1 had no known functions until a 2018 study from our laboratory reported a role in the CIA pathway (38).
Although MAGE-F1 is capable of interacting in vitro with two RING domain proteins, NSE1 and TRIM27, the interaction with NSE1 is stronger (141). Through binding to the NSE1 E3 ligase, MAGE-F1 specifically targets MMS19 for ubiquitination and subsequent degradation by the proteasome (Fig. 6A) (38). MMS19, together with CIA1 and CIA2B, is part of the CIA-targeting complex that transfers Fe-S clusters to client Fe-S proteins, many of which are involved in DNA repair processes (e.g. FANCJ, POLD1, XPD, and RTEL1) (338–340). The synthesis and insertion of Fe-S clusters into apoproteins is a highly coordinated process among several proteins that are members of the mitochondrial iron-sulfur cluster assembly and export system and the CIA machinery (341, 342). Following generation of iron- and sulfur-containing cofactors from a precursor product by the mitochondrial iron-sulfur cluster machinery, components of the CIA pathway assemble the Fe-S cluster and incorporate the Fe-S cluster into cytosolic and nuclear apoproteins (342, 343).
Figure 6.
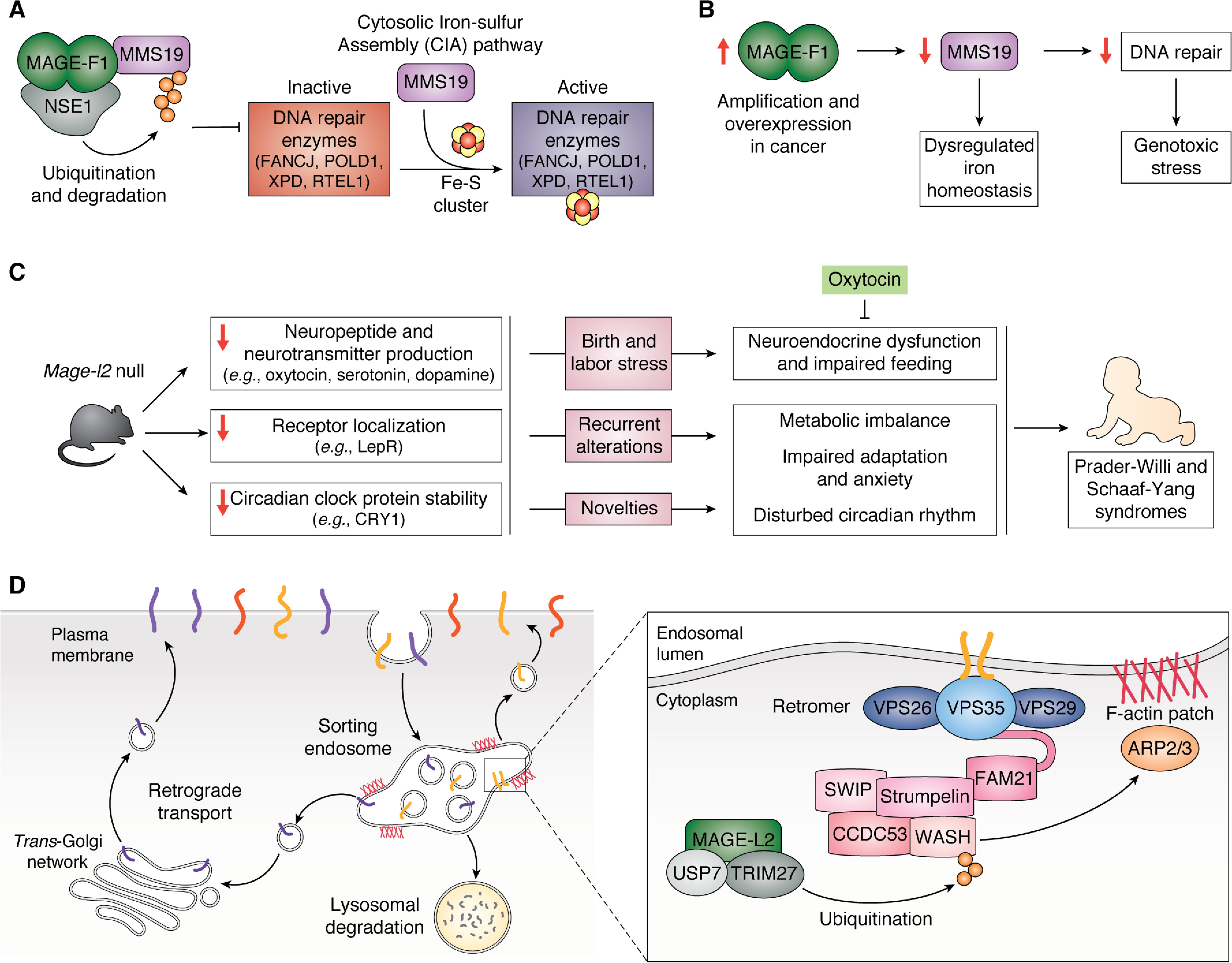
Functions and pathways of the type II MAGEs, MAGE-F1 and -L2. A, MAGE-F1 controls flux through the CIA pathway by regulating MMS19 protein levels. MAGE-F1 interacts with the E3 ligase NSE1 to form an MRL that ubiquitinates MMS19, promoting its degradation. Decreased MMS19 protein levels lead to decreased iron-sulfur (Fe-S) cluster incorporation into downstream targets, like DNA repair enzymes. B, increased levels of MAGE-F1 contribute to dysregulated iron homeostasis by degrading MMS19 and genotoxic stress by suppressing DNA repair pathways. C, MAGE-L2 contributes to the neurodevelopmental disorders PWS and SYS. Mage-l2–null mice exhibit decreased levels of mature neuropeptides, transmembrane receptors, like LepR, and circadian rhythm proteins, all of which may contribute to impaired adaptation to recurring and acute changes in the environment and contribute to the phenotypes seen in KO mice and PWS and SYS patients. Administration of oxytocin immediately after birth or during the first postnatal week rescues survival and normal adult social behavior of Mage-l2–null mice. D, MAGE-L2 is involved in endosomal protein trafficking. Cargo proteins on endosomes are trafficked to either the plasma membrane, the trans-Golgi network, or the lysosome for degradation. The retromer complex (blue) recognizes cargo proteins and, based on their destination, sorts them into endosomal tubules reshaped by localized F-actin patches. VPS35 interacts with the WASH regulatory complex (pink). The MUST complex (MAGE-L2-USP7-TRIM27) activates WASH by adding a Lys-63–linked polyubiquitin chain, which recruits ARP2/3 and promotes downstream F-actin nucleation.
By regulating MMS19 protein levels, MAGE-F1 controls flux through the CIA pathway (38). MAGE-F1-NSE1–mediated ubiquitination and degradation of MMS19 leads to decreased Fe-S incorporation into downstream targets, such as DNA repair enzymes, and sensitizes cells to DNA-damaging agents due to their reduced DNA repair capacity (Fig. 6B) (38). Given that the efficiency of cytosolic Fe-S cluster assembly regulates intracellular iron homeostasis, as iron regulatory protein 1 (IRP1) itself contains an Fe-S cluster that is lost in iron-replete conditions, we speculate that MAGE-F1-NSE1 ubiquitination and degradation of MMS19 also alters iron homeostasis regulation (38, 342).
MAGE-F1 expression in several tumors suggested that it may play a role in cancer. In line with this idea, MAGE-F1 overexpression in lung squamous carcinoma cell lines is necessary and sufficient to drive xenograft tumor growth (38). Furthermore, MAGE-F1 is amplified along with known oncogenes on chromosome 3q (e.g. PIK3CA, SOX2, and TP63) in several cancers (38). Consistent with MAGE-F1 down-regulating the MMS19 CIA pathway and reducing cellular DNA repair capacity, patient tumors with MAGE-F1 amplification have greater mutation burdens (38). In head and neck squamous carcinoma tumors, increased expression of MAGE-F1 and NSE1 correlates with decreased patient survival (38), further implying clinical relevance of MAGE-F1-NSE1 MRL.
Interestingly, MAGE-F1 amplification is found in cancers that are frequently associated with smoking, like lung squamous cell carcinomas (344, 345). Given that MMS19 is important for nucleotide excision repair, which is involved in the repair of smoking-induced lesions, the down-regulation of MMS19 by increased MAGE-F1 levels may foster tumorigenesis through promoting replication stress and increasing mutational burden (38, 339). Thus, by promoting replicative stress and suppressing DNA repair pathways, MAGE-F1 amplification and down-regulation of the CIA pathway may contribute to genomic instability and oncogenesis.
Intriguingly, MAGE-F1 is only conserved in placental mammals, and many mammalian species, including rodents, have acquired insertions, deletions, or mutations in the MAGE-F1 gene that lead to in-frame stop codons (12, 13, 335). Although the reason for MAGE-F1 pseudogenization in specific mammalian lineages awaits future determination, it is possible that MAGE-F1 pseudogenization prevented excessive genomic instability in distinct species or is a consequence of differential regulation of iron and oxygen homeostasis.
MAGE-G1 is a component of the SMC5/6 complex involved in promoting genome stability
MAGE-G1, also referred to as NDNL2 or NSMCE3, is the most closely related to MAGE-F1 with 59% identical amino acids within the MHD (43, 337). Human MAGE-G1 and its murine ortholog Mage-g1 are broadly expressed (Fig. 2A), with the highest levels found in the testis, ovary, and brain (11, 66, 67). Although MAGE-G1 is closely located to the imprinted region on chromosome 15 that is implicated in neurodevelopmental disorders and autism susceptibility, including PWS (OMIM #176270) and Angelman syndrome (OMIM #105830), MAGE-G1 is not imprinted and is not known to be involved in these disorders (67, 346, 347).
MAGE-G1 role in maintaining genome stability
A proteomic approach identified MAGE-G1 as a subunit of the SMC5/6 complex (139, 140) that is critical for chromosome replication and DNA repair in somatic cells (348, 349) and meiotic recombination in yeast and mammals (350–352). The structural maintenance of chromosome (SMC) heterodimers, in association with non-SMC element (NSE) subunits, perform important roles in the organization, packaging, and repair of chromosomes. SMC5/6 associates with six NSE subunits, NSE1–6, among which MAGE-G1 represents NSE3 (139, 140, 353–357). The SMC5/6 complex is crucial for maintenance of genomic stability, as it regulates DNA replication, checkpoint responses, and DNA repair processes, including telomere maintenance and repair pathways for double-strand breaks and homologous recombination (349, 358–362). Accordingly, cells with mutations in SMC5/6 complex subunits are sensitive to genotoxic stressors (363). The role of MAGE-G1 in SMC5/6 function and DNA damage repair is still enigmatic; however, depletion of MAGE-G1 by siRNA results in degradation of all of the other complex subunits, suggesting that it is a critical component of the complex like the other members (140). Namely, NSE1, NSE3, and NSE4 form a trimeric complex that bridges the globular heads between SMC5 and SMC6 (364, 365). In both S. pombe and human cells, MAGE-G1 strongly interacts with NSE1 not only within the SMC5/6 complex, but also independently (141), which indicates that MAGE-G1-NSE1 MRL may have additional functions besides those mediated by the SMC5/6 complex (140, 357).
Given their expression in meiotic male germ cells during the pachytene stage (Fig. 2A) (11, 351) and their crucial role in yeast meiosis (139), MAGE-G1 and other components of the SMC5/6 complex are predicted to have an important function during mammalian meiosis. MAGE-G1 was also suggested to be the closest ortholog of the first autosomal MAGE gene in marsupials, implying that the autosomal transposition of the ancestral MAGE was driven by its important function in meiosis and the appearance of sex chromosome inactivation (366). Interestingly, although current data suggest that the SMC5/6 complex is not acutely required for premeiotic DNA replication and meiotic progression during mouse spermatogenesis, the SMC5/6 complex ensures genome integrity and, thus, fertility when germ cells are challenged by exogenous DNA damage (367).
Notably, the mouse genome contains the additional, related Mage-g2 gene, which also exhibits increased expression at the pachytene spermatocyte stage (Fig. 2A) (11, 37). Proteomic and genomic analyses suggest that Mage-g2 does not engage with the Smc5/6 complex but, rather, binds independently to the testicular germ cell-specific serine/threonine-protein kinase 31 (Stk31) and heat shock protein 9 (Hspa9) (37). Although further analysis showed that Mage-g2 reduced the kinase activity of Stk31, the functional relevance of binding to Hspa9 is currently unknown (37). Because various stressors up-regulate Hspa9 to suppress the engagement of apoptosis and regulate the functions of p53 (368), Mage-g2 binding to Hspa9 may be related to stress response in the testis, which further supports the emerging concept that MAGE genes evolved to provide an advantage in stressful circumstances.
MAGE-G1 in lung disease immunodeficiency and chromosome breakage syndrome
Interestingly, missense mutations in MAGE-G1 have been identified in children with lung disease immunodeficiency and chromosome breakage syndrome (LICS; OMIM #617241) (369). All four affected children manifested clinical features similar to those seen in Nijmegen breakage and ataxia-telangiectasia chromosomal breakage syndromes, yet LICS patients were uniquely predisposed to severe and ultimately fatal pneumonia during early childhood (369).
The mutations in Pro-209 and Leu-264 abolish formation of the trimeric MAGE-G1-NSE1-NSE4 complex, which facilitates the bridging of SMC5 and SMC6 (365, 370), thereby disrupting the whole SMC5/6 complex (369). Furthermore, patient-derived fibroblasts containing the homozygous L264F MAGE-G1 mutation showed multiple chromosomal rearrangements and increased levels of micronuclei (369), indicative of genome instability during mitosis, similar to those seen in cells with mutations in NSE2 (371). Also similar to cells with NSE2 mutations, these fibroblasts displayed an increased sensitivity to genotoxic treatment, defects in the homologous recombination repair pathway, and delays in recovery from hydroxyurea-induced replication stress (369). These results imply that the MAGE-G1 mutant proteins destabilize the SMC5/6 complex, resulting in incoherent homologous recombination and impaired recovery from replication stress. The impact of the identified MAGE-G1 mutations in LICS further underscores the importance of MAGE-G1 in maintaining chromosome stability.
MAGE-dependent switching of NSE1 function
As mentioned earlier, NSE1 also functions outside the SMC5/6 complex through binding to MAGE-F1 to regulate the CIA pathway during DNA damage response (38). Although similar MAGEs have been reported to bind the same RING protein with redundant activity, like MAGE-A3/6 and -C2 binding TRIM28, the interaction of MAGE-F1 or MAGE-G1 with NSE1 is novel because these MRLs exhibit nonoverlapping functions (38, 140, 141, 151, 156, 357). Whereas it is not known how the binding of MAGE-F1 or MAGE-G1 to NSE1 may mechanistically switch its function, we have a few possible explanations. Because the N and C termini of these two MAGEs differ, it is plausible that these regions impart different substrate specificity on NSE1. Another possible explanation is that the binding of MAGE-F1 or MAGE-G1 leads to different NSE1 conformations that promote different activities. Additionally, the binding of MAGE-G1-NSE1 to the SMC5/6 complex or other proteins may hinder interaction with MMS19. Yet another possibility is that MAGE-G1-NSE1 and MAGE-F1-NSE1 localize to distinct subcellular compartments.
Whether one or more of these proposed mechanisms is responsible for switching the function of NSE1 warrants further investigation. However, it is clear that MAGE proteins enable diversification of E3 ligase function by expanding their regulatory potential and allowing contextual changes in multiple pathways through altering formation of distinct MRLs. The differential binding of MAGE-G1 and MAGE-F1 to NSE1 not only diversifies function but also promotes opposing activities, as MAGE-G1-NSE1 promotes DNA repair through the SMC5/6 complex (369), whereas MAGE-F1-NSE1 inhibits DNA repair through degradation of MMS19 (38).
MAGE-L2 regulates protein recycling and hypothalamic functions
MAGE-L2 is one of the largest MAGE proteins (Figs. 3A) with an N-terminal region that is highly proline-rich (>30%) (372), but the functional significance of this domain is still unknown. The human MAGE-L2 gene is one of ∼150 imprinted genes with monoallelic expression (373) located within the imprinted domain on the chromosome 15 that is critical for the manifestation of two distinct neurodevelopmental disorders, the PWS and Angelman syndrome (372). In addition to MAGE-L2, the PWS region contains five other protein-coding genes, including NECDIN, also a MAGE gene, and six small nucleolar RNA genes.
MAGE-L2 in PWS, SYS, and other genetic disorders
PWS is a multigenic disorder that affects one in 15,000 children and results either from the deletion of paternal 15q11-q13 (65–75% of cases), from maternal uniparental disomy (20–30%), or from imprinting defects (1–3%) (374). The major clinical features of PWS include intellectual and physical disabilities, endocrine dysfunctions, obesity, and maladaptive behaviors (170, 374–377), all implying hypothalamic dysfunction. At birth, PWS is characterized by neonatal hypogonadism, hypotonia with feeding difficulties, and failure to thrive (374). Later, PWS children develop intellectual disability, hypothyroidism, short stature, maladaptive social behaviors, and, most notably, hyperphagia leading to childhood obesity and type II diabetes (374).
Besides PWS, MAGE-L2 has also been implicated in the PWS-like neurodevelopmental disorder Schaaf–Yang syndrome (SYS, OMIM #615547) (372), where patients harbor diverse truncating pathogenic variants of the paternal MAGE-L2 gene (170, 376, 378). The most pathogenic variant, c.1996delC, resulted in lethal arthrogryposis in several patients (376, 378, 379). Although many SYS symptoms overlap with those of PWS, the lack of hyperphagia in SYS distinguishes it from PWS (374). In addition, SYS patients have a higher prevalence of autism spectrum disorder and display joint contractures that range in severity from mild contractures of the distal phalanges to lethal arthrogryposis (170, 376, 378–380). The severity of many SYS features varies based on mutation location, yet it is interesting that deletion of the entire MAGE-L2 gene in PWS results in a milder phenotype than the MAGE-L2 truncating mutations in SYS. However, this genotype-to-phenotype correlation needs further investigation to discover the molecular underpinnings that drive these differences.
MAGE-L2 has also been implicated in other developmental syndromes. Truncating mutations of MAGE-L2 have been identified in patients with arthrogryposis multiplex congenita (OMIM #208100) (379) and Chitayat–Hall syndrome (OMIM #208080) (381), which are characterized by multiple severe joint contractures that result in minimum fetal movement, global developmental delays, and growth hormone deficiency. Although the genetic background for most patients with Opitz trigonocephaly C syndrome (OTCS, OMIM #211750) is still enigmatic, the de novo nonsense MAGE-L2 mutation c.1912C > T (p.Q638) was identified in one of 10 OTCS patients (382). Given the phenotypic overlap between SYS and OTCS, the promising candidates for OTCS causative genes include those encoding MAGE-L2 molecular partners that form the MUST complex (MAGE-L2-TRIM27-USP7) (43, 68, 149, 372), as well as proteins implicated in MUST-regulated retrograde transport (43, 68, 372, 382). Analogously, components of the MUST-regulated WASH-retromer protein recycling pathways have been implicated in various neurological disorders, including SWIP mutations in intellectual disability (383), strumpellin in hereditary sporadic paraplegia (384), and USP7 variants in pediatric neurodevelopmental disorders like hypotonia, seizures, and autism spectrum disorder (68). These disorder associations suggest an important role for MAGE-L2–regulated molecular pathways, which are described in the following paragraphs, in neurodevelopment and central nervous system functions. All studies so far are based on nonsense frameshifting mutations that result in truncated MAGE-L2 (170, 376, 378, 379, 382, 385), yet the pathogenic mechanism of missense MAGE-L2 mutations remains completely unstudied.
Mage-l2–null mice yield insights into PWS and SYS
Two mouse models with targeted deletion of Mage-l2 recapitulate several fundamental aspects of PWS and SYS (372). Similar to the phenotypes seen in children with PWS and SYS, these animals display growth retardation at early stages of life, followed by weight gain after weaning, increased adiposity, and unbalanced metabolism (Fig. 6C) (168, 169, 386, 387). Additionally, Mage-l2–null mice exhibit abnormal circadian rhythm and feeding behaviors, similar to the hyperphagia present in PWS patients (168, 169, 386–388). In line with predicted hypothalamic defects and disturbed endocrine phenotypes seen in patients, Mage-l2–null mice show diverse neuroendocrine dysfunctions, including decreased levels of the neuropeptides and biogenic amines, which may account for maladaptive behaviors, like increased anxiety and deficit in preference for social novelty (386, 389).
The mouse model developed by Muscatelli and her group (390), where the Mage-l2 promoter and most of the coding region was deleted—in contrast to the lacZ insertion that replaces the C-terminal domain of Mage-l2 in the first model developed by Wevrick's group—but leaving intact the last 1165 base pairs that code for the MHD, also showed a significant reduction in oxytocin level, and, impressively, a single bolus of oxytocin immediately after birth rescued the suckling deficit and neonatal survival (Fig. 6C) (390). Moreover, daily administration of oxytocin in the first postnatal week restored normal social behavior and learning abilities in adult animals (391), further implicating oxytocin as a critical mediator of phenotype development. Accordingly, Mage-l2–null mice have a significantly decreased level of mature oxytocin in the hypothalamus (391), which is similar to the orexin maturation defects observed in the Wevrick model (168, 169). Although the mechanism leading to neuropeptide reduction in PWS patients and Mage-l2-null mice is still not clear, increased pro-hormone levels suggest defective processing into bioactive circulating hormones (386).
MAGE-L2 regulates endosomal protein recycling
Consistent with MAGEs regulating E3 ligases (43), MAGE-L2 directly binds RING E3 ligase TRIM27 (also known as Ret finger protein, or RFP) through its MHD (68, 149). In addition to TRIM27, MAGE-L2 binds the deubiquitinating enzyme USP7 to form the MUST complex that controls WASH activity to facilitate endosomal protein recycling (Fig. 6D). USP7 stabilizes this complex by interacting with both MAGE-L2 and TRIM27 (68, 141, 149). MAGE-L2-TRIM27 mediates Lys-63–linked ubiquitination of the protein WASH (149), which, unlike most ubiquitin linkage forms that target proteins for degradation, activates WASH and enables WASH-mediated endosomal protein recycling (Fig. 6D) (149). USP7-mediated deubiquitination prevents WASH overactivation and also counteracts TRIM27 autoubiquitination to prevent its proteolytic degradation, thereby stabilizing the MUST complex (68).
Endosomal protein recycling is an essential process that enables proper sorting of membrane proteins from endosomes to either the plasma membrane (e.g. diverse transporters, signaling receptors, and cell adhesion molecules (392–398)) or the trans-Golgi network (e.g. intracellular sorting receptors, transmembrane peptidases, and SNAREs (399–402)). Given the diversity of cargo proteins dependent on the endosomal protein recycling pathway for their proper localization and function, dysfunction of MAGE-L2 or the MUST complex can impact a range of physiological and pathophysiological processes.
On a mechanistic level, MUST-dependent WASH activation enables F-actin nucleation to provide the initial force needed for membrane invagination and subsequent vesicle scission that triggers retrograde transport in the early endosome (160, 384, 403–406). In the early endosomes, cargo proteins originating either from the plasma membrane or the biosynthetic pathway are sorted and then dissociated through the interconnected network of endosomal membranes. The master regulator of cargo protein recognition and sorting is the retromer complex (composed of VPS35, -26, and -29 (407, 408)), which also recruits the WASH regulatory complex (SHRC; composed of WASH, FAM21, CCDC53, SWIP, and strumpellin) to start the transport after cargos are selected. MAGE-L2 directly interacts with VPS35 to recruit the MUST complex to endosomes for WASH activation (Fig. 6D) (149).
Upon its activation, WASH then activates the ARP2/3 actin-nucleating complex and triggers F-actin nucleation at the endosomal surface, a necessary step for proper protein trafficking (160, 384, 403). WASH, like all WASP family members, contains a C-terminal VCA (verprolin homologous or WH2, central hydrophobic, and acidic) domain that binds both actin and the ARP2/3 complex to trigger actin filament nucleation (160). Because the VCA domain is autoinhibited through intra- and intermolecular interactions, activating signals expose the motifs to enable F-actin nucleation (160, 372, 384, 403, 404). Whereas the exact mechanistic role of the Lys-63–linked polyubiquitin chains on WASH is unclear, this modification may physically disrupt the autoinhibitory contact between WASH and other components of the SHRC, thus exposing the WASH VCA domain for interactions with ARP2/3 (149, 409). Alternatively, the Lys-63–linked polyubiquitin chain on WASH may also recruit additional factors to promote ARP2/3 complex-mediated actin nucleation on endosomes. Given that MAGE-L2 is mammalian-specific and highly enriched in the hypothalamus (11, 68), whereas WASH and retromer are conserved in all eukaryotes (407, 408), MAGE-L2 may represent a tissue-specific regulator of WASH that is critical for protein trafficking, localization, and function in the mammalian hypothalamus.
Interestingly, MAGE-L2 also engages another E3 ligase, RNF41, and deubiquitinating enzyme, USP8, to regulate endosomal protein recycling of the leptin receptor (LepR) (154). LepR activity in the hypothalamus is vital for the regulation of appetite and energy balance (410, 411). Compared with WT littermates, Mage-l2–null mice display reduced levels of LepR and Rnf41, indicating a defect in endosomal recycling, which is rescued by Mage-l2 expression (154). Concurrently, these mice exhibit increased levels of Usp8 and Stam1, a component of another sorting complex called ESCRT (endosomal sorting complexes required for transport) (154). Stam1 was previously shown to stabilize the ESCRT complex and push trafficked cargo proteins like LepR into multivesicular bodies for subsequent lysosomal degradation (412). Thus, the altered abundances of Usp8 and Rnf41 may increase Stam1 and promote LepR degradation, ultimately contributing to the obesity phenotype observed in PWS (154). This study suggests an additional MAGE-L2 complex, which resembles MUST, that plays a role in hypothalamic protein recycling; however, further studies are required to determine the exact molecular mechanisms involved.
The day-night routine disruptions commonly present in PWS and SYS patients and the circadian rhythm defects observed in Mage-l2-null mice implicated another role for MAGE-L2 in the circadian clock (Fig. 6C) (168, 372). Indeed, MAGE-L2 manipulates the ubiquitination and stability of proteins that regulate circadian rhythm, in particular the key circadian rhythm protein cryptochrome 1 (CRY1) (388, 413). Furthermore, Mage-l2 has circadian expression and is highly expressed in the SCN, the area of the brain that establishes and controls circadian rhythm (71, 168). Through anticipating recurring changes in the environment to appropriately adjust behavior and physiology, the internal circadian clock confers an evolutionary advantage to most species on Earth.
Taken together, the brain- and hypothalamus-enriched MAGE-L2 regulates different E3 ligases to control sorting and proper localization of proteins, as well as the stability of neuropeptides and circadian proteins. Identifying which cargo proteins are aberrantly trafficked due to the loss of MAGE-L2 will be imperative for teasing out its contribution to the behavior, feeding, circadian rhythm, and endocrine homeostasis anomalies seen in PWS and SYS, which will lead to developing better therapeutic strategies. Although the exact molecular mechanisms underlying MAGE-L2 function need further investigation, current data suggest that MAGE-L2 evolved to enable better adaptation to recurrent and unanticipated changes in the environment.
Conclusions and future perspectives
Investigation of human and mouse MAGE proteins has uncovered many of their diverse cellular functions, but we are just starting to understand how these functions contribute to normal physiological processes and the pathogenesis of cancer and other genetic diseases. At the molecular level, the dynamic nature of the MHD structure enables individual MAGE proteins to preferentially bind different proteins, including E3 ligases, to exert their functions. MAGEs regulate their respective E3 ligases through enhancing ligase activity, specifying novel substrates for ubiquitination, and altering ligase subcellular localization. As a result, MAGEs regulate many biological pathways, including metabolism and autophagy, DNA repair, cell cycle progression, apoptosis, mRNA polyadenylation and stability, stress granule formation, and membrane protein recycling.
Through in-depth examination of the diverse functions exhibited by individual MAGEs, the overarching theme of stress tolerance has emerged as a unifying thread that further ties the MAGE family together. Based on thorough characterization of Mage expression in spermatogenesis and the location of many MAGE genes on the X chromosome, where the rapid expansion of multicopy/ampliconic genes is likely driven by their beneficial roles in male reproductive fitness, the type I MAGE genes presumably expanded in eutherian mammals to protect the germline from environmental stress and aid in stress adaptation to preserve fertility. This theory is supported by the detrimental effects on spermatogenesis reported in Mage-a and -b4 KO mice exposed to genotoxic, metabolic, and heat stress. Many type II MAGE proteins, like MAGE-G1 and Mage-g2, also play a role in protecting the genomic integrity and fitness of the germline. Additionally, ubiquitously expressed MAGE proteins respond to stressors in other tissues, like MAGE-D2 after kidney injury, and may have provided an evolutionary advantage, as is likely the case for MAGE-D1 and MAGE-L2 in the brain. Interestingly, analysis of novel eutherian genes, many of which reside on the X chromosome like MAGEs, suggests extensive genetic modification to pathways involved in testis and brain function during eutherian mammal evolution, indicating that these genes provided an evolutionary advantage (414). In all, our findings evoke the conclusion that MAGEs provide a selective advantage by enabling better stress adaptation, either on a cellular or an organismal level.
Intriguingly, the protective responses of MAGEs to stress in the germline are likely hijacked by cancer cells that aberrantly express MAGEs, to foster a tumorigenic environment. For example, MAGE-A11's ability to induce APA and result in 3′-US, typical for germ cell–specific transcripts, appears to be co-opted by cancer cells to promote tumor growth. Furthermore, MAGE-A3/6 confer resistance to metabolic stress, and MAGE-B2 increases cellular stress tolerance in cancer cells by suppressing stress granule formation. Given that MAGE expression often correlates with resistance to checkpoint inhibitors, targeted therapy, and chemotherapy (21–24), we speculate that MAGEs may contribute to therapy resistance by activating diverse stress-adaptation pathways.
As highlighted throughout this review, many aspects related to MAGE family expression and function remain enigmatic. Although the expression patterns of MAGE CTAs in male germ cells and various cancers indicate that epigenetic alterations work in concert with tissue-specific transcription factors to transcriptionally activate MAGEs, we do not yet fully grasp which mechanisms control expression of MAGEs within germ, somatic, and cancer cells. Beyond these epigenetic mechanisms, defining the contribution of MAGEs to regulation of stemness, differentiation of pluripotent stem cells, and embryonic development is also warranted. Similarly, the possible roles of MAGEs in female reproduction have not been studied, despite the fact that some MAGEs, like MAGE-A11, are expressed in the placenta and uterus. Ongoing efforts to successfully and safely target the type I MAGEs will greatly benefit from understanding the mechanisms by which these proteins contribute to oncogenesis, their regulation, and their normal physiological functions. Furthermore, the recently published expression analysis of MAGEs (11) in various tissues and life stages will aid in predicting and avoiding potential off-target effects of immunotherapies that target a single MAGE. In addition, elucidating which molecular mechanisms are responsible for poor response to immunotherapy and chemotherapy resistance development in MAGE-positive tumors will be critical.
In summary, the MAGE family comprises an exciting group of proteins with diverse functions that contribute to stress tolerance in germ and cancer cells. Addressing the open questions mentioned in this review will deepen our understanding of MAGEs and reveal strategies for treating cancer and other diseases.
Acknowledgments
We thank members of the Potts laboratory for advice and critical discussions. We thank M. Tacer for help with the TCGA data analysis. We apologize for our inability to discuss and reference all work in the field due to space limitations.
Funding and additional information—This work was supported by NIGMS, National Institutes of Health, Grant R01GM111332, Worldwide Cancer Research Grant 15-0177, Foundation for Prader–Willi Syndrome Research Grant 342604, and American Cancer Society Research Scholar Award 181691010 (to P. R. P.) and Cancer Prevention and Research Institute of Texas Scholar Award RR200059 (to K. F. T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—P. R. P. is a paid consultant of Levo Therapeutics, Inc. and Amgen, Inc.
- MAGE
- melanoma antigen
- CTA
- cancer-testis antigen
- ceRNA
- competing endogenous mRNA
- BDNF
- brain-derived neurotrophic factor
- MHD
- MAGE homology domain
- PWS
- Prader–Willi syndrome
- SYS
- Schaaf–Yang syndrome
- ES cells
- embryonic stem cells
- SSC
- spermatogonial stem cell
- BTB
- blood-testis barrier
- DAC
- 5′-aza-2′-deoxycytidine
- HDAC
- histone deacetylase
- MRL
- MAGE-RING ligase
- AMPK
- AMP-activated kinase
- SCF
- Skp1-Cullin-F-box
- CRL
- cullin-RING ligase
- KRAB
- Krüppel-associated box
- ZFP
- zinc finger protein
- HCC
- hepatocellular carcinoma
- SUMO
- small ubiquitin-like modifier
- EBV
- Epstein–Barr virus
- ATM
- ataxia-telangiectasia-mutated
- FBP1
- fructose-1,6-biphosphatase
- FBP
- fructose 1,6-bisphosphate
- 2DG
- 2-deoxy-d-glucose
- AR
- androgen receptor
- PR
- progesterone receptor
- SG
- stress granule
- LLPS
- liquid–liquid phase separation
- SLE
- systemic lupus erythematosus
- APS1
- autoimmune polyendocrine syndrome type 1
- IAP
- inhibitors of apoptosis protein
- XIAP
- X-linked IAP
- SERT
- serotonin transporter
- LepR
- leptin receptor
- CREB
- cAMP-response element–binding protein
- CIA
- cytosolic iron-sulfur (Fe-S) assembly
- SMC
- structural maintenance of chromosome
- NSE
- non-SMC element
- LICS
- lung disease immunodeficiency and chromosome breakage syndrome
- OTCS
- Opitz trigonocephaly C syndrome
- SHRC
- WASH regulatory complex
- KO
- knockout.
References
- 1. Dunn G. P., Old L. J., and Schreiber R. D. (2004) The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 10.1146/annurev.immunol.22.012703.104803 [DOI] [PubMed] [Google Scholar]
- 2. Whitehurst A. W. (2014) Cause and consequence of cancer/testis antigen activation in cancer. Annu. Rev. Pharmacol. Toxicol. 54, 251–272 10.1146/annurev-pharmtox-011112-140326 [DOI] [PubMed] [Google Scholar]
- 3. van der Bruggen P., Traversari C., Chomez P., Lurquin C., De Plaen E., Van den Eynde B., Knuth A., and Boon T. (1991) A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647 10.1126/science.1840703 [DOI] [PubMed] [Google Scholar]
- 4. Jager E., and Knuth A. (2012) The discovery of cancer/testis antigens by autologous typing with t cell clones and the evolution of cancer vaccines. Cancer Immun. 12, 6 [PMC free article] [PubMed] [Google Scholar]
- 5. Chomez P., De Backer O., Bertrand M., De Plaen E., Boon T., and Lucas S. (2001) An overview of the mage gene family with the identification of all human members of the family. Cancer Res. 61, 5544–5551 [PubMed] [Google Scholar]
- 6. De Plaen E., Traversari C., Gaforio J. J., Szikora J. P., De Smet C., Brasseur F., van der Bruggen P., Lethé B., Lurquin C., Chomez P., and De Backer O. (1994) Structure, chromosomal localization, and expression of 12 genes of the mage family. Immunogenetics 40, 360–369 10.1007/BF01246677 [DOI] [PubMed] [Google Scholar]
- 7. Chomez P., Williams R., De Backer O., Boon T., and Vennström B. (1996) The smage gene family is expressed in post-meiotic spermatids during mouse germ cell differentiation. Immunogenetics 43, 97–100 10.1007/BF00186613 [DOI] [PubMed] [Google Scholar]
- 8. De Plaen E., De Backer O., Arnaud D., Bonjean B., Chomez P., Martelange V., Avner P., Baldacci P., Babinet C., Hwang S. Y., Knowles B., and Boon T. (1999) A new family of mouse genes homologous to the human MAGE genes. Genomics 55, 176–184 10.1006/geno.1998.5638 [DOI] [PubMed] [Google Scholar]
- 9. Barker P. A., and Salehi A. (2002) The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J. Neurosci. Res. 67, 705–712 10.1002/jnr.10160 [DOI] [PubMed] [Google Scholar]
- 10. Forslund K. Ö., and Nordqvist K. (2001) The melanoma antigen genes—any clues to their functions in normal tissues? Exp. Cell Res. 265, 185–194 10.1006/excr.2001.5173 [DOI] [PubMed] [Google Scholar]
- 11. Fon Tacer K., Montoya M. C., Oatley M. J., Lord T., Oatley J. M., Klein J., Ravichandran R., Tillman H., Kim M., Connelly J. P., Pruett-Miller S. M., Bookout A. L., Binshtock E., Kamiński M. M., and Potts P. R. (2019) MAGE cancer-testis antigens protect the mammalian germline under environmental stress. Sci. Adv. 5, eaav4832 10.1126/sciadv.aav4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katsura Y., and Satta Y. (2011) Evolutionary history of the cancer immunity antigen mage gene family. PLoS ONE 6, e20365 10.1371/journal.pone.0020365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao Q., Caballero O. L., Simpson A. J. G., and Strausberg R. L. (2012) Differential evolution of mage genes based on expression pattern and selection pressure. PLoS ONE 7, e48240 10.1371/journal.pone.0048240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kruit W. H. J., Suciu S., Dreno B., Mortier L., Robert C., Chiarion-Sileni V., Maio M., Testori A., Dorval T., Grob J.-J., Becker J. C., Spatz A., Eggermont A. M. M., Louahed J., Lehmann F. F., et al. (2013) Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J. Clin. Oncol. 31, 2413–2420 10.1200/JCO.2012.43.7111 [DOI] [PubMed] [Google Scholar]
- 15. Vansteenkiste J., Zielinski M., Linder A., Dahabreh J., Gonzalez E. E., Malinowski W., Lopez-Brea M., Vanakesa T., Jassem J., Kalofonos H., Perdeus J., Bonnet R., Basko J., Janilionis R., Passlick B., et al. (2013) Adjuvant MAGE-A3 immunotherapy in resected non–small-cell lung cancer: phase II randomized study results. J. Clin. Oncol. 31, 2396–2403 10.1200/JCO.2012.43.7103 [DOI] [PubMed] [Google Scholar]
- 16. Dreno B., Thompson J. F., Smithers B. M., Santinami M., Jouary T., Gutzmer R., Levchenko E., Rutkowski P., Grob J.-J., Korovin S., Drucis K., Grange F., Machet L., Hersey P., Krajsova I., et al. (2018) MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (derma): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 19, 916–929 10.1016/S1470-2045(18)30254-7 [DOI] [PubMed] [Google Scholar]
- 17. Vansteenkiste J. F., Cho B. C., Vanakesa T., De Pas T., Zielinski M., Kim M. S., Jassem J., Yoshimura M., Dahabreh J., Nakayama H., Havel L., Kondo H., Mitsudomi T., Zarogoulidis K., Gladkov O. A., et al. (2016) Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 17, 822–835 10.1016/S1470-2045(16)00099-1 [DOI] [PubMed] [Google Scholar]
- 18. Daud A. I. (2018) Negative but not futile: MAGE-A3 immunotherapeutic for melanoma. Lancet Oncol. 19, 852–853 10.1016/S1470-2045(18)30353-X [DOI] [PubMed] [Google Scholar]
- 19. Linette G. P., Stadtmauer E. A., Maus M. V., Rapoport A. P., Levine B. L., Emery L., Litzky L., Bagg A., Carreno B. M., Cimino P. J., Binder-Scholl G. K., Smethurst D. P., Gerry A. B., Pumphrey N. J., Bennett A. D., et al. (2013) Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced t cells in myeloma and melanoma. Blood 122, 863–871 10.1182/blood-2013-03-490565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morgan R. A., Chinnasamy N., Abate-Daga D., Gros A., Robbins P. F., Zheng Z., Dudley M. E., Feldman S. A., Yang J. C., Sherry R. M., Phan G. Q., Hughes M. S., Kammula U. S., Miller A. D., Hessman C. J., et al. (2013) Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 36, 133–151 10.1097/CJI.0b013e3182829903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shukla S. A., Bachireddy P., Schilling B., Galonska C., Zhan Q., Bango C., Langer R., Lee P. C., Gusenleitner D., Keskin D. B., Babadi M., Mohammad A., Gnirke A., Clement K., Cartun Z. J., et al. (2018) Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell 173, 624–633.e8 10.1016/j.cell.2018.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jin J., Liu B.-Z., and Wu Z.-M. (2015) Evaluation of melanoma antigen gene A3 expression in drug resistance of epidermal growth factor receptor-tyrosine kinase inhibitors in advanced nonsmall cell lung cancer treatment. J. Cancer Res. Ther. 11, 271–274 10.4103/0973-1482.170549 [DOI] [PubMed] [Google Scholar]
- 23. Chen Y., Zhao H., Li H., Feng X., Tang H., Qiu C., Zhang J., and Fu B. (2020) LINC01234/MicroRNA-31-5p/MAGEA3 axis mediates the proliferation and chemoresistance of hepatocellular carcinoma cells. Mol. Ther. Nucleic Acids 19, 168–178 10.1016/j.omtn.2019.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24. Xie C., Subhash V. V., Datta A., Liem N., Tan S. H., Yeo M. S., Tan W. L., Koh V., Yan F. L., Wong F. Y., Wong W. K., So J., Tan I. B., Padmanabhan N., Yap C. T., et al. (2016) Melanoma associated antigen (MAGE)-A3 promotes cell proliferation and chemotherapeutic drug resistance in gastric cancer. Cell. Oncol. 39, 175–186 10.1007/s13402-015-0261-5 [DOI] [PubMed] [Google Scholar]
- 25. Sun Q., Zhang X., Wang L., Gao X., Xiong Y., Liu L., Wei F., Yang L., and Ren X. (2019) T-cell receptor gene therapy targeting melanoma-associated antigen-A4 by silencing of endogenous TCR inhibits tumor growth in mice and human. Cell Death Dis. 10, 475 10.1038/s41419-019-1717-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khalaf W. S., Garg M., Mohamed Y. S., Stover C. M., and Browning M. J. (2019) In vitro generation of cytotoxic T cells with potential for adoptive tumor immunotherapy of multiple myeloma. Front. Immunol. 10, 1792–1792 10.3389/fimmu.2019.01792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bao L., Dunham K., and Lucas K. (2011) MAGE-A1, MAGE-A3, and NY-ESO-1 can be upregulated on neuroblastoma cells to facilitate cytotoxic t lymphocyte-mediated tumor cell killing. Cancer Immunol. Immunother. 60, 1299–1307 10.1007/s00262-011-1037-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Serrano A., Tanzarella S., Lionello I., Mendez R., Traversari C., Ruiz-Cabello F., and Garrido F. (2001) Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2'-deoxycytidine treatment. Int. J. Cancer 94, 243–251 10.1002/ijc.1452 [DOI] [PubMed] [Google Scholar]
- 29. Sigalotti L., Altomonte M., Colizzi F., Degan M., Rupolo M., Zagonel V., Pinto A., Gattei V., and Maio M. (2003) 5-Aza-2'-deoxycytidine (decitabine) treatment of hematopoietic malignancies: a multimechanism therapeutic approach? Blood 101, 4644–4646 10.1182/blood-2002-11-3458 [DOI] [PubMed] [Google Scholar]
- 30. Krishnadas D. K., Shusterman S., Bai F., Diller L., Sullivan J. E., Cheerva A. C., George R. E., and Lucas K. G. (2015) A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol. Immunother. 64, 1251–1260 10.1007/s00262-015-1731-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scanlan M. J., Gure A. O., Jungbluth A. A., Old L. J., and Chen Y.-T. (2002) Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol. Rev. 188, 22–32 10.1034/j.1600-065X.2002.18803.x [DOI] [PubMed] [Google Scholar]
- 32. Caballero O. L., and Chen Y.-T. (2009) Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 100, 2014–2021 10.1111/j.1349-7006.2009.01303.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warburton P. E., Giordano J., Cheung F., Gelfand Y., and Benson G. (2004) Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 14, 1861–1869 10.1101/gr.2542904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Skaletsky H., Kuroda-Kawaguchi T., Minx P. J., Cordum H. S., Hillier L., Brown L. G., Repping S., Pyntikova T., Ali J., Bieri T., Chinwalla A., Delehaunty A., Delehaunty K., Du H., Fewell G., et al. (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 10.1038/nature01722 [DOI] [PubMed] [Google Scholar]
- 35. Mueller J. L., Skaletsky H., Brown L. G., Zaghlul S., Rock S., Graves T., Auger K., Warren W. C., Wilson R. K., and Page D. C. (2013) Independent specialization of the human and mouse X chromosomes for the male germ line. Nat. Genet. 45, 1083–1087 10.1038/ng.2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ross M. T., Grafham D. V., Coffey A. J., Scherer S., McLay K., Muzny D., Platzer M., Howell G. R., Burrows C., Bird C. P., Frankish A., Lovell F. L., Howe K. L., Ashurst J. L., Fulton R. S., et al. (2005) The DNA sequence of the human X chromosome. Nature 434, 325–337 10.1038/nature03440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeong J., Jin S., Choi H., Kwon J., Kim J., Kim J., Park Z., and Cho C. (2017) Characterization of MAGEG2 with testis-specific expression in mice. Asian J. Androl. 19, 659–665 10.4103/1008-682X.192033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weon J. L., Yang S. W., and Potts P. R. (2018) Cytosolic iron-sulfur assembly is evolutionarily tuned by a cancer-amplified ubiquitin ligase. Mol. Cell 69, 113–125.e6 10.1016/j.molcel.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 39. Lucas S., Brasseur F., and Boon T. (1999) A new mage gene with ubiquitous expression does not code for known mage antigens recognized by T cells. Cancer Res. 59, 4100–4103 [PubMed] [Google Scholar]
- 40. Rogner U. C., Wilke K., Steck E., Korn B., and Poustka A. (1995) The melanoma antigen gene (MAGE) family is clustered in the chromosomal band xq28. Genomics 29, 725–731 10.1006/geno.1995.9945 [DOI] [PubMed] [Google Scholar]
- 41. Willett C. S., and Wilson E. M. (2018) Evolution of melanoma antigen-A11 (MAGEA11) during primate phylogeny. J. Mol. Evol. 86, 240–253 10.1007/s00239-018-9838-8 [DOI] [PubMed] [Google Scholar]
- 42. López-Sánchez N., González-Fernández Z., Niinobe M., Yoshikawa K., and Frade J. M. (2007) Single mage gene in the chicken genome encodes CMage, a protein with functional similarities to mammalian type II mage proteins. Physiol. Genomics 30, 156–171 10.1152/physiolgenomics.00249.2006 [DOI] [PubMed] [Google Scholar]
- 43. Lee A. K., and Potts P. R. (2017) A comprehensive guide to the mage family of ubiquitin ligases. J. Mol. Biol. 429, 1114–1142 10.1016/j.jmb.2017.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nishimura I., Shimizu S., Sakoda J-y., and Yoshikawa K. (2007) Expression of Drosophila MAGE gene encoding a necdin homologous protein in postembryonic neurogenesis. Gene Expr. Patterns 7, 244–251 10.1016/j.modgep.2006.09.008 [DOI] [PubMed] [Google Scholar]
- 45. Ohno S. (1967) Sex chromosomes and sex-linked genes, pp. 46–73, Springer, Berlin [Google Scholar]
- 46. Wang P. J., McCarrey J. R., Yang F., and Page D. C. (2001) An abundance of X-linked genes expressed in spermatogonia. Nat. Genet. 27, 422–426 10.1038/86927 [DOI] [PubMed] [Google Scholar]
- 47. Mueller J. L., Mahadevaiah S. K., Park P. J., Warburton P. E., Page D. C., and Turner J. M. A. (2008) The mouse X chromosome is enriched for multicopy testis genes showing postmeiotic expression. Nat. Genet. 40, 794–799 10.1038/ng.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu W.-S. (2019) Mammalian sex chromosome structure, gene content, and function in male fertility. Annu. Rev. Anim. Biosci. 7, 103–124 10.1146/annurev-animal-020518-115332 [DOI] [PubMed] [Google Scholar]
- 49. Vicoso B., and Charlesworth B. (2006) Evolution on the X chromosome: unusual patterns and processes. Nat. Rev. Genet. 7, 645–653 10.1038/nrg1914 [DOI] [PubMed] [Google Scholar]
- 50. Khil P. P., Smirnova N. A., Romanienko P. J., and Camerini-Otero R. D. (2004) The mouse X chromosome is enriched for sex-biased genes not subject to selection by meiotic sex chromosome inactivation. Nat. Genet. 36, 642–646 10.1038/ng1368 [DOI] [PubMed] [Google Scholar]
- 51. Coyne J. A. (1992) Genetics and speciation. Nature 355, 511–515 10.1038/355511a0 [DOI] [PubMed] [Google Scholar]
- 52. Stevenson B. J., Iseli C., Panji S., Zahn-Zabal M., Hide W., Old L. J., Simpson A. J., and Jongeneel C. V. (2007) Rapid evolution of cancer/testis genes on the X chromosome. BMC Genomics 8, 129 10.1186/1471-2164-8-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wyckoff G. J., Wang W., and Wu C.-I. (2000) Rapid evolution of male reproductive genes in the descent of man. Nature 403, 304–309 10.1038/35002070 [DOI] [PubMed] [Google Scholar]
- 54. Gibbs Z. A., and Whitehurst A. W. (2018) Emerging contributions of cancer/testis antigens to neoplastic behaviors. Trends Cancer 4, 701–712 10.1016/j.trecan.2018.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. De Backer O., Verheyden A. M., Martin B., Godelaine D., De Plaen E., Brasseur R., Avner P., and Boon T. (1995) Structure, chromosomal location, and expression pattern of three mouse genes homologous to the human MAGE genes. Genomics 28, 74–83 10.1006/geno.1995.1108 [DOI] [PubMed] [Google Scholar]
- 56. Clotman F., De Backer O., De Plaen E., Boon T., and Picard J. (2000) Cell- and stage-specific expression of mage genes during mouse spermatogenesis. Mamm. Genome 11, 696–699 10.1007/s003350010116 [DOI] [PubMed] [Google Scholar]
- 57. Osterlund C., Töhönen V., Forslund K. O., and Nordqvist K. (2000) Mage-b4, a novel melanoma antigen (MAGE) gene specifically expressed during germ cell differentiation. Cancer Res. 60, 1054–1061 [PubMed] [Google Scholar]
- 58. Takahashi K., Shichijo S., Noguchi M., Hirohata M., and Itoh K. (1995) Identification of MAGE-1 and MAGE-4 proteins in spermatogonia and primary spermatocytes of testis. Cancer Res. 55, 3478–3482 [PubMed] [Google Scholar]
- 59. Jurk M., Kremmer E., Schwarz U., Förster R., and Winnacker E. L. (1998) MAGE-11 protein is highly conserved in higher organisms and located predominantly in the nucleus. Int. J. Cancer 75, 762–766 [DOI] [PubMed] [Google Scholar]
- 60. Põld M., Zhou J., Chen G. L., Hall J. M., Vescio R. A., and Berenson J. R. (1999) Identification of a new, unorthodox member of the MAGE gene family. Genomics 59, 161–167 10.1006/geno.1999.5870 [DOI] [PubMed] [Google Scholar]
- 61. Boccaccio I., Glatt-Deeley H., Watrin F., Roëckel N., Lalande M., and Muscatelli F. (1999) The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader-Willi region. Hum. Mol. Genet. 8, 2497–2505 10.1093/hmg/8.13.2497 [DOI] [PubMed] [Google Scholar]
- 62. Nelson P. T., Zhang P. J., Spagnoli G. C., Tomaszewski J. E., Pasha T. L., Frosina D., Caballero O. L., Simpson A. J. G., Old L. J., and Jungbluth A. A. (2007) Cancer/testis (CT) antigens are expressed in fetal ovary. Cancer Immun. 7, 1 [PMC free article] [PubMed] [Google Scholar]
- 63. Gaugler B., Van den Eynde B., van der Bruggen P., Romero P., Gaforio J. J., De Plaen E., Lethé B., Brasseur F., and Boon T. (1994) Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 179, 921–930 10.1084/jem.179.3.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Goldman B., and DeFrancesco L. (2009) The cancer vaccine roller coaster. Nat. Biotechnol. 27, 129–139 10.1038/nbt0209-129 [DOI] [PubMed] [Google Scholar]
- 65. Brichard V. G., and Lejeune D. (2007) GSK's antigen-specific cancer immunotherapy programme: pilot results leading to phase III clinical development. Vaccine 25, B61–B71 10.1016/j.vaccine.2007.06.038 [DOI] [PubMed] [Google Scholar]
- 66. Kuwako K-I., Taniura H., and Yoshikawa K. (2004) Necdin-related mage proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J. Biol. Chem. 279, 1703–1712 10.1074/jbc.M308454200 [DOI] [PubMed] [Google Scholar]
- 67. Lee S., Kozlov S., Hernandez L., Chamberlain S. J., Brannan C. I., Stewart C. L., and Wevrick R. (2000) Expression and imprinting of MAGEL2 suggest a role in Prader-Willi syndrome and the homologous murine imprinting phenotype. Hum. Mol. Genet. 9, 1813–1819 10.1093/hmg/9.12.1813 [DOI] [PubMed] [Google Scholar]
- 68. Hao Y. H., Fountain M. D., Fon Tacer K., Xia F., Bi W., Kang S. H. L., Patel A., Rosenfeld J. A., Le Caignec C. D., Isidor B., Krantz I. D., Noon S. E., Pfotenhauer J. P., Morgan T. M., Moran R., et al. (2015) USP7 acts as a molecular rheostat to promote wash-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol. Cell 59, 956–969 10.1016/j.molcel.2015.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bertrand M., Huijbers I., Chomez P., and De Backer O. (2004) Comparative expression analysis of the MAGED genes during embryogenesis and brain development. Dev. Dyn. 230, 325–334 10.1002/dvdy.20026 [DOI] [PubMed] [Google Scholar]
- 70. Mouri A., Sasaki A., Watanabe K., Sogawa C., Kitayama S., Mamiya T., Miyamoto Y., Yamada K., Noda Y., and Nabeshima T. (2012) MAGE-D1 regulates expression of depression-like behavior through serotonin transporter ubiquitylation. J. Neurosci. 32, 4562–4580 10.1523/JNEUROSCI.6458-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Maillard J., Park S., Croizier S., Vanacker C., Cook J. H., Prevot V., Tauber M., and Bouret S. G. (2016) Loss of MAGEL2 impairs the development of hypothalamic anorexigenic circuits. Hum. Mol. Genet. 25, 3208–3215 10.1093/hmg/ddw169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gjerstorff M. F., Harkness L., Kassem M., Frandsen U., Nielsen O., Lutterodt M., Møllgård K., and Ditzel H. J. (2008) Distinct GAGE and MAGE-A expression during early human development indicate specific roles in lineage differentiation. Hum. Reprod. 23, 2194–2201 10.1093/humrep/den262 [DOI] [PubMed] [Google Scholar]
- 73. Gjerstorff M. F., Kock K., Nielsen O., and Ditzel H. J. (2007) MAGE-A1, GAGE and NY-ESO-1 cancer/testis antigen expression during human gonadal development. Hum. Reprod. 22, 953–960 10.1093/humrep/del494 [DOI] [PubMed] [Google Scholar]
- 74. Langnaese K., Kloos D. U., Wehnert M., Seidel B., and Wieacker P. (2001) Expression pattern and further characterization of human maged2 and identification of rodent orthologues. Cytogenet. Cell Genet. 94, 233–240 10.1159/000048822 [DOI] [PubMed] [Google Scholar]
- 75. Salehi A. H., Roux P. P., Kubu C. J., Zeindler C., Bhakar A., Tannis L. L., Verdi J. M., and Barker P. A. (2000) NRAGE, a novel mage protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron 27, 279–288 10.1016/S0896-6273(00)00036-2 [DOI] [PubMed] [Google Scholar]
- 76. Kendall S. E., Goldhawk D. E., Kubu C., Barker P. A., and Verdi J. M. (2002) Expression analysis of a novel p75ntr signaling protein, which regulates cell cycle progression and apoptosis. Mech. Dev. 117, 187–200 10.1016/S0925-4773(02)00204-6 [DOI] [PubMed] [Google Scholar]
- 77. Lifantseva N., Koltsova A., Krylova T., Yakovleva T., Poljanskaya G., and Gordeeva O. (2011) Expression patterns of cancer-testis antigens in human embryonic stem cells and their cell derivatives indicate lineage tracks. Stem Cells Int. 2011, 795239 10.4061/2011/795239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Di Certo M. G., Corbi N., Bruno T., Iezzi S., De Nicola F., Desantis A., Ciotti M. T., Mattei E., Floridi A., Fanciulli M., and Passananti C. (2007) NRAGE associates with the anti-apoptotic factor Che-1 and regulates its degradation to induce cell death. J. Cell Sci. 120, 1852–1858 10.1242/jcs.03454 [DOI] [PubMed] [Google Scholar]
- 79. Gaspar J. A., Srinivasan S. P., Sureshkumar P., Doss M. X., Hescheler J., Papadopoulos S., and Sachinidis A. (2017) Depletion of Mageb16 induces differentiation of pluripotent stem cells predominantly into mesodermal derivatives. Sci. Rep. 7, 14285 10.1038/s41598-017-14561-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gordeeva O., Gordeev A., and Khaydukov S. (2019) Expression dynamics of Mage family genes during self-renewal and differentiation of mouse pluripotent stem and teratocarcinoma cells. Oncotarget 10, 3248–3266 10.18632/oncotarget.26933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu Y., Yang S., Yang J., Que H., and Liu S. (2012) Relative expression of type ii mage genes during retinoic acid-induced neural differentiation of mouse embryonic carcinoma p19 cells: a comparative real-time PCR analysis. Cell. Mol. Neurobiol. 32, 1059–1068 10.1007/s10571-012-9826-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lurquin C., De Smet C., Brasseur F., Muscatelli F., Martelange V., De Plaen E., Brasseur R., Monaco A. P., and Boon T. (1997) Two members of the human MAGEB gene family located in xp21.3 are expressed in tumors of various histological origins. Genomics 46, 397–408 10.1006/geno.1997.5052 [DOI] [PubMed] [Google Scholar]
- 83. Jungbluth A. A., Busam K. J., Kolb D., Iversen K., Coplan K., Chen Y. T., Spagnoli G. C., and Old L. J. (2000) Expression of MAGE-antigens in normal tissues and cancer. Int. J. Cancer 85, 460–465 [DOI] [PubMed] [Google Scholar]
- 84. Lucas S., De Plaen E., and Boon T. (2000) MAGE-B5, MAGE-B6, MAGE-C2, and MAGE-C3: Four new members of the mage family with tumor-specific expression. Int. J. Cancer 87, 55–60 [DOI] [PubMed] [Google Scholar]
- 85. Lee A. K., Klein J., Fon Tacer K., Lord T., Oatley M. J., Oatley J. M., Porter S. N., Pruett-Miller S. M., Tikhonova E. B., Karamyshev A. L., Wang Y. D., Yang P., Korff A., Kim H. J., Taylor J. P., et al. (2020) Translational repression of G3BP in cancer and germ cells suppresses stress granules and enhances stress tolerance. Mol. Cell 79, 645–659.e9 10.1016/j.molcel.2020.06.037 [DOI] [PubMed] [Google Scholar]
- 86. Lord T., and Oatley J. M. (2018) Spermatogonial response to somatic cell interactions. in Encyclopedia of Reproduction, 2nd Ed. (Skinner M. K., ed) pp. 53–58, Academic Press, Oxford [Google Scholar]
- 87. Tagelenbosch R. A. J., and de Rooij D. G. (1993) A quantitative study of spermatogonial multiplication and stem cell renewal in the C3H/101 F1 hybrid mouse. Mutat. Res. 290, 193–200 10.1016/0027-5107(93)90159-D [DOI] [PubMed] [Google Scholar]
- 88. de Rooij D. G. (2017) The nature and dynamics of spermatogonial stem cells. Development 144, 3022–3030 10.1242/dev.146571 [DOI] [PubMed] [Google Scholar]
- 89. Griswold M. D. (2016) Spermatogenesis: the commitment to meiosis. Physiol. Rev. 96, 1–17 10.1152/physrev.00013.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Oatley J. M., and Brinster R. L. (2008) Regulation of spermatogonial stem cell self-renewal in mammals. Annu. Rev. Cell Dev. Biol. 24, 263–286 10.1146/annurev.cellbio.24.110707.175355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hogarth C. A., Arnold S., Kent T., Mitchell D., Isoherranen N., and Griswold M. D. (2015) Processive pulses of retinoic acid propel asynchronous and continuous murine sperm production. Biol. Reprod. 92, 37 10.1095/biolreprod.114.126326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schlatt S., and Ehmcke J. (2014) Regulation of spermatogenesis: an evolutionary biologist's perspective. Semin. Cell Dev. Biol. 29, 2–16 10.1016/j.semcdb.2014.03.007 [DOI] [PubMed] [Google Scholar]
- 93. Bellvé A. R., Cavicchia J. C., Millette C. F., O'Brien D. A., Bhatnagar Y. M., and Dym M. (1977) Spermatogenic cells of the prepuberal mouse: isolation and morphological characterization. J. Cell Biol. 74, 68–85 10.1083/jcb.74.1.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Schultz N., Hamra F. K., and Garbers D. L. (2003) A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc. Natl. Acad. Sci. U. S. A. 100, 12201–12206 10.1073/pnas.1635054100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hennuy B., Reiter E., Cornet A., Bruyninx M., Daukandt M., Houssa P., N'Guyen V. H., Closset J., and Hennen G. (2000) A novel messenger ribonucleic acid homologous to human MAGE-D is strongly expressed in rat Sertoli cells and weakly in Leydig cells and is regulated by follitropin, lutropin, and prolactin. Endocrinology 141, 3821–3831 10.1210/endo.141.10.7705 [DOI] [PubMed] [Google Scholar]
- 96. Weber J., Salgaller M., Samid D., Johnson B., Herlyn M., Lassam N., Treisman J., and Rosenberg S. A. (1994) Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2'-deoxycytidine. Cancer Res. 54, 1766–1771 [PubMed] [Google Scholar]
- 97. Sigalotti L., Coral S., Nardi G., Spessotto A., Cortini E., Cattarossi I., Colizzi F., Altomonte M., and Maio M. (2002) Promoter methylation controls the expression of MAGE2, 3 and 4 genes in human cutaneous melanoma. J. Immunother. 25, 16–26 10.1097/00002371-200201000-00002 [DOI] [PubMed] [Google Scholar]
- 98. Suyama T., Ohashi H., Nagai H., Hatano S., Asano H., Murate T., Saito H., and Kinoshita T. (2002) The MAGE-A1 gene expression is not determined solely by methylation status of the promoter region in hematological malignancies. Leuk. Res. 26, 1113–1118 10.1016/S0145-2126(02)00048-6 [DOI] [PubMed] [Google Scholar]
- 99. Li B., Qian X. P., Pang X. W., Zou W. Z., Wang Y. P., Wu H. Y., and Chen W. F. (2003) Hca587 antigen expression in normal tissues and cancers: correlation with tumor differentiation in hepatocellular carcinoma. Lab. Invest. 83, 1185–1192 10.1097/01.lab.0000080605.73839.96 [DOI] [PubMed] [Google Scholar]
- 100. Honda T., Tamura G., Waki T., Kawata S., Terashima M., Nishizuka S., and Motoyama T. (2004) Demethylation of MAGE promoters during gastric cancer progression. Br. J. Cancer 90, 838–843 10.1038/sj.bjc.6601600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Weon J. L., and Potts P. R. (2015) The mage protein family and cancer. Curr. Opin. Cell Biol. 37, 1–8 10.1016/j.ceb.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. De Smet C., De Backer O., Faraoni I., Lurquin C., Brasseur F., and Boon T. (1996) The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc. Natl. Acad. Sci. U. S. A. 93, 7149–7153 10.1073/pnas.93.14.7149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. De Smet C., Loriot A., and Boon T. (2004) Promoter-dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE-A1 in tumor cells. Mol. Cell Biol. 24, 4781–4790 10.1128/MCB.24.11.4781-4790.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. De Smet C., Lurquin C., Lethé B., Martelange V., and Boon T. (1999) DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell Biol. 19, 7327–7335 10.1128/mcb.19.11.7327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Karpf A. R., Bai S., James S. R., Mohler J. L., and Wilson E. M. (2009) Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic amp. Mol. Cancer Res. 7, 523–535 10.1158/1541-7786.MCR-08-0400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pattani K. M., Soudry E., Glazer C. A., Ochs M. F., Wang H., Schussel J., Sun W., Hennessey P., Mydlarz W., Loyo M., Demokan S., Smith I. M., and Califano J. A. (2012) MAGEB2 is activated by promoter demethylation in head and neck squamous cell carcinoma. PLoS ONE 7, e45534 10.1371/journal.pone.0045534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Furuta J., Umebayashi Y., Miyamoto K., Kikuchi K., Otsuka F., Sugimura T., and Ushijima T. (2004) Promoter methylation profiling of 30 genes in human malignant melanoma. Cancer Sci. 95, 962–968 10.1111/j.1349-7006.2004.tb03184.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jones P. A., and Baylin S. B. (2002) The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 3, 415–428 10.1038/nrg816 [DOI] [PubMed] [Google Scholar]
- 109. Loriot A., De Plaen E., Boon T., and De Smet C. (2006) Transient down-regulation of DNMT1 methyltransferase leads to activation and stable hypomethylation of MAGE-A1 in melanoma cells. J. Biol. Chem. 281, 10118–10126 10.1074/jbc.M510469200 [DOI] [PubMed] [Google Scholar]
- 110. Wischnewski F., Friese O., Pantel K., and Schwarzenbach H. (2007) Methyl-CpG binding domain proteins and their involvement in the regulation of the MAGE-A1, MAGE-A2, MAGE-A3, and MAGE-A12 gene promoters. Mol. Cancer Res. 5, 749–759 10.1158/1541-7786.MCR-06-0364 [DOI] [PubMed] [Google Scholar]
- 111. Liu S., Liu F., Huang W., Gu L., Meng L., Ju Y., Wu Y., Li J., Liu L., and Sang M. (2018) MAGE-A11 is activated through TFCP2/ZEB1 binding sites de-methylation as well as histone modification and facilitates escc tumor growth. Oncotarget 9, 3365–3378 10.18632/oncotarget.22973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Trasler J. M. (2009) Epigenetics in spermatogenesis. Mol. Cell. Endocrinol. 306, 33–36 10.1016/j.mce.2008.12.018 [DOI] [PubMed] [Google Scholar]
- 113. Vatolin S., Abdullaev Z., Pack S. D., Flanagan P. T., Custer M., Loukinov D. I., Pugacheva E., Hong J. A., Morse H. 3rd, Schrump D. S., Risinger J. I., Barrett J. C., and Lobanenkov V. V. (2005) Conditional expression of the CTCF-paralogous transcriptional factor BORIS in normal cells results in demethylation and derepression of MAGE-A1 and reactivation of other cancer-testis genes. Cancer Res. 65, 7751–7762 10.1158/0008-5472.CAN-05-0858 [DOI] [PubMed] [Google Scholar]
- 114. Schwarzenbach H., Eichelser C., Steinbach B., Tadewaldt J., Pantel K., Lobanenkov V., and Loukinov D. (2014) Differential regulation of MAGE-A1 promoter activity by BORIS and Sp1, both interacting with the TATA binding protein. BMC Cancer 14, 796 10.1186/1471-2407-14-796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Martin-Kleiner I. (2012) Boris in human cancers—a review. Eur. J. Cancer 48, 929–935 10.1016/j.ejca.2011.09.009 [DOI] [PubMed] [Google Scholar]
- 116. Kholmanskikh O., Loriot A., Brasseur F., De Plaen E., and De Smet C. (2008) Expression of BORIS in melanoma: lack of association with MAGE-A1 activation. Int. J. Cancer 122, 777–784 10.1002/ijc.23140 [DOI] [PubMed] [Google Scholar]
- 117. Karpf A. R., Lasek A. W., Ririe T. O., Hanks A. N., Grossman D., and Jones D. A. (2004) Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2'-deoxycytidine. Mol. Pharmacol. 65, 18–27 10.1124/mol.65.1.18 [DOI] [PubMed] [Google Scholar]
- 118. Rao M., Chinnasamy N., Hong J. A., Zhang Y., Zhang M., Xi S., Liu F., Marquez V. E., Morgan R. A., and Schrump D. S. (2011) Inhibition of histone lysine methylation enhances cancer–testis antigen expression in lung cancer cells: implications for adoptive immunotherapy of cancer. Cancer Res. 71, 4192–4204 10.1158/0008-5472.CAN-10-2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. James S. R., Cedeno C. D., Sharma A., Zhang W., Mohler J. L., Odunsi K., Wilson E. M., and Karpf A. R. (2013) DNA methylation and nucleosome occupancy regulate the cancer germline antigen gene magea11. Epigenetics 8, 849–863 10.4161/epi.25500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kondo T., Zhu X., Asa S. L., and Ezzat S. (2007) The cancer/testis antigen melanoma-associated antigen-A3/A6 is a novel target of fibroblast growth factor receptor 2-IIIB through histone H3 modifications in thyroid cancer. Clin. Cancer Res. 13, 4713–4720 10.1158/1078-0432.CCR-07-0618 [DOI] [PubMed] [Google Scholar]
- 121. Zhu X., Asa S. L., and Ezzat S. (2008) Fibroblast growth factor 2 and estrogen control the balance of histone 3 modifications targeting MAGE-A3 in pituitary neoplasia. Clin. Cancer Res. 14, 1984–1996 10.1158/1078-0432.CCR-07-2003 [DOI] [PubMed] [Google Scholar]
- 122. Wischnewski F., Pantel K., and Schwarzenbach H. (2006) Promoter demethylation and histone acetylation mediate gene expression of MAGE-A1, -A2, -A3, and -A12 in human cancer cells. Mol. Cancer Res. 4, 339–349 10.1158/1541-7786.MCR-05-0229 [DOI] [PubMed] [Google Scholar]
- 123. Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H., and Shinkai Y. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 10.1101/gad.989402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Chiappinelli K. B., Zahnow C. A., Ahuja N., and Baylin S. B. (2016) Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 76, 1683–1689 10.1158/0008-5472.CAN-15-2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. De Smet C., Courtois S. J., Faraoni I., Lurquin C., Szikora J. P., De Backer O., and Boon T. (1995) Involvement of two Ets binding sites in the transcriptional activation of the MAGE1 gene. Immunogenetics 42, 282–290 10.1007/BF00176446 [DOI] [PubMed] [Google Scholar]
- 126. Serrano A., García A., Abril E., Garrido F., and Ruiz-Cabello F. (1996) Methylated CpG points identified within MAGE-1 promoter are involved in gene repression. Int. J. Cancer 68, 464–470 [DOI] [PubMed] [Google Scholar]
- 127. Miranda E. I. (2010) MAGE, biological functions and potential clinical applications. Leuk. Res. 34, 1121–1122 10.1016/j.leukres.2010.03.045 [DOI] [PubMed] [Google Scholar]
- 128. Yang B., Wu J., Maddodi N., Ma Y., Setaluri V., and Jack Longley B. (2007) Epigenetic control of MAGE gene expression by the kit tyrosine kinase. J. Invest. Dermatol. 127, 2123–2128 10.1038/sj.jid.5700836 [DOI] [PubMed] [Google Scholar]
- 129. Endo T., Freinkman E., de Rooij D. G., and Page D. C. (2017) Periodic production of retinoic acid by meiotic and somatic cells coordinates four transitions in mouse spermatogenesis. Proc. Natl. Acad. Sci. U. S. A. 114, E10132–E10141 10.1073/pnas.1710837114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ahmed E. A., and de Rooij D. G. (2009) Staging of mouse seminiferous tubule cross-sections. in Meiosis: Volume 2, Cytological Methods (Keeney S., ed) pp. 263–277, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 131. Liu W., Cheng S., Asa S. L., and Ezzat S. (2008) The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis. Cancer Res. 68, 8104–8112 10.1158/0008-5472.CAN-08-2132 [DOI] [PubMed] [Google Scholar]
- 132. Fukuyama T., Yamazaki T., Fujita T., Uematsu T., Ichiki Y., Kaneko H., Suzuki T., and Kobayashi N. (2012) Helicobacter pylori, a carcinogen, induces the expression of melanoma antigen-encoding gene (MAGE)-A3, a cancer/testis antigen. Tumor Biol. 33, 1881–1887 10.1007/s13277-012-0448-6 [DOI] [PubMed] [Google Scholar]
- 133. Chen X., Wang L., Liu J., Huang L., Yang L., Gao Q., Shi X., Li J., Li F., Zhang Z., Zhao S., Zhang B., Van der Bruggen P., and Zhang Y. (2017) Expression and prognostic relevance of MAGE-A3 and MAGE-C2 in non-small cell lung cancer. Oncol. Lett. 13, 1609–1618 10.3892/ol.2017.5665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wu F., Liu F., Dong L., Yang H., He X., Li L., Zhao L., Jin S., and Li G. (2018) miR-1273g silences MAGEA3/6 to inhibit human colorectal cancer cell growth via activation of AMPK signaling. Cancer Lett. 435, 1–9 10.1016/j.canlet.2018.07.031 [DOI] [PubMed] [Google Scholar]
- 135. Ye X., Xie J., Huang H., and Deng Z. (2018) Knockdown of MAGEA6 activates AMP-activated protein kinase (AMPK) signaling to inhibit human renal cell carcinoma cells. Cell. Physiol. Biochem. 45, 1205–1218 10.1159/000487452 [DOI] [PubMed] [Google Scholar]
- 136. Xue J., Zhong S., Sun B.-M., Sun Q.-F., Hu L.-Y., and Pan S.-J. (2019) Lnc-THOR silencing inhibits human glioma cell survival by activating MAGEA6-AMPK signaling. Cell Death Dis. 10, 866–866 10.1038/s41419-019-2093-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Guo J.-C., Yang Y.-J., Zhang J.-Q., Guo M., Xiang L., Yu S.-F., Ping H., and Zhuo L. (2019) MicroRNA-448 inhibits stemness maintenance and self-renewal of hepatocellular carcinoma stem cells through the MAGEA6-mediated AMPK signaling pathway. J. Cell. Physiol. 234, 23461–23474 10.1002/jcp.28915 [DOI] [PubMed] [Google Scholar]
- 138. Pan S.-J., Ren J., Jiang H., Liu W., Hu L.-Y., Pan Y.-X., Sun B., Sun Q.-F., and Bian L.-G. (2018) MAGEA6 promotes human glioma cell survival via targeting AMPKα1. Cancer Lett. 412, 21–29 10.1016/j.canlet.2017.09.051 [DOI] [PubMed] [Google Scholar]
- 139. Pebernard S., McDonald W. H., Pavlova Y., Yates J. R., and Boddy M. N. (2004) Nse1, Nse2, and a novel subunit of the Smc5-Smc6 complex, Nse3, play a crucial role in meiosis. Mol. Biol. Cell 15, 4866–4876 10.1091/mbc.e04-05-0436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Taylor E. M., Copsey A. C., Hudson J. J. R., Vidot S., and Lehmann A. R. (2008) Identification of the proteins, including MAGEG1, that make up the human SMC5-6 protein complex. Mol. Cell Biol. 28, 1197–1206 10.1128/MCB.00767-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Doyle J. M., Gao J., Wang J., Yang M., and Potts P. R. (2010) Mage-ring protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 39, 963–974 10.1016/j.molcel.2010.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Newman J. A., Cooper C. D., Roos A. K., Aitkenhead H., Oppermann U. C., Cho H. J., Osman R., and Gileadi O. (2016) Structures of two melanoma-associated antigens suggest allosteric regulation of effector binding. PLoS ONE 11, e0148762 10.1371/journal.pone.0148762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Hagiwara Y., Sieverling L., Hanif F., Anton J., Dickinson E. R., Bui T. T. T., Andreeva A., Barran P. E., Cota E., and Nikolova P. V. (2016) Consequences of point mutations in melanoma-associated antigen 4 (MAGE-A4) protein: insights from structural and biophysical studies. Sci. Rep. 6, 25182–25182 10.1038/srep25182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Werner A., Manford A. G., and Rape M. (2017) Ubiquitin-dependent regulation of stem cell biology. Trends Cell Biol. 27, 568–579 10.1016/j.tcb.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Deshaies R. J., and Joazeiro C. A. P. (2009) Ring domain e3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 10.1146/annurev.biochem.78.101807.093809 [DOI] [PubMed] [Google Scholar]
- 146. Rotin D., and Kumar S. (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409 10.1038/nrm2690 [DOI] [PubMed] [Google Scholar]
- 147. Feng Y., Gao J., and Yang M. (2011) When MAGE meets RING: insights into biological functions of mage proteins. Protein Cell 2, 7–12 10.1007/s13238-011-1002-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Xiao T. Z., Bhatia N., Urrutia R., Lomberk G. A., Simpson A., and Longley B. J. (2011) MAGE I transcription factors regulate KAP1 and KRAB domain zinc finger transcription factor mediated gene repression. PLoS ONE 6, e23747 10.1371/journal.pone.0023747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hao Y. H., Doyle J. M., Ramanathan S., Gomez T. S., Jia D., Xu M., Chen Z. J., Billadeau D. D., Rosen M. K., and Potts P. R. (2013) Regulation of wash-dependent actin polymerization and protein trafficking by ubiquitination. Cell 152, 1051–1064 10.1016/j.cell.2013.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kozakova L., Vondrova L., Stejskal K., Charalabous P., Kolesar P., Lehmann A. R., Uldrijan S., Sanderson C. M., Zdrahal Z., and Palecek J. J. (2015) The melanoma-associated antigen 1 (MAGEA1) protein stimulates the E3 ubiquitin-ligase activity of TRIM31 within a TRIM31-MAGEA1-NSE4 complex. Cell Cycle 14, 920–930 10.1080/15384101.2014.1000112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Pineda C. T., Ramanathan S., Fon Tacer K., Weon J. L., Potts M. B., Ou Y.-H., White M. A., and Potts P. R. (2015) Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728 10.1016/j.cell.2015.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Gao Y., Mutter-Rottmayer E., Greenwalt A. M., Goldfarb D., Yan F., Yang Y., Martinez-Chacin R. C., Pearce K. H., Tateishi S., Major M. B., and Vaziri C. (2016) A neomorphic cancer cell-specific role of MAGE-A4 in trans-lesion synthesis. Nat. Commun. 7, 12105 10.1038/ncomms12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Rual J.-F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., et al. (2005) Towards a proteome-scale map of the human protein–protein interaction network. Nature 437, 1173–1178 10.1038/nature04209 [DOI] [PubMed] [Google Scholar]
- 154. Wijesuriya T. M., De Ceuninck L., Masschaele D., Sanderson M. R., Carias K. V., Tavernier J., and Wevrick R. (2017) The Prader-Willi syndrome proteins MAGEL2 and NECDIN regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum. Mol. Genet. 26, 4215–4230 10.1093/hmg/ddx311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Su S., Chen X., Geng J., Minges J. T., Grossman G., and Wilson E. M. (2017) Melanoma antigen-A11 regulates substrate-specificity of SKP2-mediated protein degradation. Mol. Cell. Endocrinol. 439, 1–9 10.1016/j.mce.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Pineda C. T., and Potts P. R. (2015) Oncogenic MAGEA-TRIM28 ubiquitin ligase downregulates autophagy by ubiquitinating and degrading AMPK in cancer. Autophagy 11, 844–846 10.1080/15548627.2015.1034420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Xiao T. Z., Suh Y., and Longley B. J. (2014) Mage proteins regulate KRAB zinc finger transcription factors and KAP1 E3 ligase activity. Arch. Biochem. Biophys. 563, 136–144 10.1016/j.abb.2014.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Yang B., Herrin S. M., Wu J., Reagan-Shaw S., Ma Y., Bhat K. M. R., Gravekamp C., Setaluri V., Peters N., Hoffmann F. M., Peng H., Ivanov A. V., Simpson A. J. G., and Longley B. J. (2007) MAGE-A, mMAGE-B, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in mage-positive cell lines. Cancer Res. 67, 9954–9962 10.1158/0008-5472.CAN-07-1478 [DOI] [PubMed] [Google Scholar]
- 159. Pebernard S., Perry J. J. P., Tainer J. A., and Boddy M. N. (2008) Nse1 ring-like domain supports functions of the Smc5-Smc6 holocomplex in genome stability. Mol. Biol. Cell 19, 4099–4109 10.1091/mbc.e08-02-0226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Derivery E., Sousa C., Gautier J. J., Lombard B., Loew D., and Gautreau A. (2009) The Arp2/3 activator wash controls the fission of endosomes through a large multiprotein complex. Dev. Cell 17, 712–723 10.1016/j.devcel.2009.09.010 [DOI] [PubMed] [Google Scholar]
- 161. Hao J., Song X., Wang J., Guo C., Li Y., Li B., Zhang Y., and Yin Y. (2015) Cancer-testis antigen MAGE-C2 binds Rbx1 and inhibits ubiquitin ligase-mediated turnover of cyclin E. Oncotarget 6, 42028–42039 10.18632/oncotarget.5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Petroski M. D., and Deshaies R. J. (2005) Function and regulation of cullin–ring ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 10.1038/nrm1547 [DOI] [PubMed] [Google Scholar]
- 163. Yang S. W., Li L., Connelly J. P., Porter S. N., Kodali K., Gan H., Park J. M., Tacer K. F., Tillman H., Peng J., Pruett-Miller S. M., Li W., and Potts P. R. (2020) A cancer-specific ubiquitin ligase drives mRNA alternative polyadenylation by ubiquitinating the mRNA 3′ end processing complex. Mol. Cell 77, 1206–1221.e7 10.1016/j.molcel.2019.12.022 [DOI] [PubMed] [Google Scholar]
- 164. Friedman J. R., Fredericks W. J., Jensen D. E., Speicher D. W., Huang X. P., Neilson E. G., and Rauscher F. J. (1996) KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 10, 2067–2078 10.1101/gad.10.16.2067 [DOI] [PubMed] [Google Scholar]
- 165. Cheng C.-T., Kuo C.-Y., and Ann D. K. (2014) Kaptain in charge of multiple missions: emerging roles of KAP1. World J. Biol. Chem. 5, 308–320 10.4331/wjbc.v5.i3.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Jay P., Rougeulle C., Massacrier A., Moncla A., Mattei M. G., Malzac P., Roëckel N., Taviaux S., Lefranc J. L., Cau P., Berta P., Lalande M., and Muscatelli F. (1997) The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat. Genet. 17, 357–361 10.1038/ng1197-357 [DOI] [PubMed] [Google Scholar]
- 167. MacDonald H. R., and Wevrick R. (1997) The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum. Mol. Genet. 6, 1873–1878 10.1093/hmg/6.11.1873 [DOI] [PubMed] [Google Scholar]
- 168. Kozlov S. V., Bogenpohl J. W., Howell M. P., Wevrick R., Panda S., Hogenesch J. B., Muglia L. J., Van Gelder R. N., Herzog E. D., and Stewart C. L. (2007) The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 39, 1266–1272 10.1038/ng2114 [DOI] [PubMed] [Google Scholar]
- 169. Bischof J. M., Stewart C. L., and Wevrick R. (2007) Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Genet. 16, 2713–2719 10.1093/hmg/ddm225 [DOI] [PubMed] [Google Scholar]
- 170. Schaaf C. P., Gonzalez-Garay M. L., Xia F., Potocki L., Gripp K. W., Zhang B., Peters B. A., McElwain M. A., Drmanac R., Beaudet A. L., Caskey C. T., and Yang Y. (2013) Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 45, 1405–1408 10.1038/ng.2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.CTDatabase. ( RRID:SCR_007614)
- 172. Ayyoub M., Scarlata C.-M., Hamaï A., Pignon P., and Valmori D. (2014) Expression of MAGE-A3/6 in primary breast cancer is associated with hormone receptor negative status, high histologic grade, and poor survival. J. Immunother. 37, 73–76 10.1097/CJI.0000000000000013 [DOI] [PubMed] [Google Scholar]
- 173. Lausenmeyer E. M., Braun K., Breyer J., Gierth M., Denzinger S., Burger M., Voelker H. U., and Otto W. (2019) Strong expression of cancertestis antigens CTAG1B and MAGEA3 is correlated with unfavourable histopathological features and MAGEA3 is associated with worse progression-free survival in urothelial bladder cancer. Urol. Int. 102, 77–82 10.1159/000493577 [DOI] [PubMed] [Google Scholar]
- 174. Abikhair M., Roudiani N., Mitsui H., Krueger J. G., Pavlick A., Lee J., Therrien J.-P., Meehan S. A., Felsen D., and Carucci J. A. (2017) MAGEA3 expression in cutaneous squamous cell carcinoma is associated with advanced tumor stage and poor prognosis. J. Invest. Dermatol. 137, 775–778 10.1016/j.jid.2016.10.036 [DOI] [PubMed] [Google Scholar]
- 175. Gure A. O., Chua R., Williamson B., Gonen M., Ferrera C. A., Gnjatic S., Ritter G., Simpson A. J. G., Chen Y.-T., Old L. J., and Altorki N. K. (2005) Cancer-testis genes are coordinately expressed and are markers of poor outcome in non–small cell lung cancer. Clin. Cancer Res. 11, 8055–8062 10.1158/1078-0432.CCR-05-1203 [DOI] [PubMed] [Google Scholar]
- 176. Trippel A., Halling F., Heymann P., Ayna M., Al-Nawas B., and Ziebart T. (2019) The expression of melanoma-associated antigen a (MAGE-A) in oral squamous cell carcinoma: an evaluation of the significance for tumor prognosis. Oral Maxillofac. Surg. 23, 343–352 10.1007/s10006-019-00778-x [DOI] [PubMed] [Google Scholar]
- 177. Conley P. A., Wang W.-L., Livingston A. J., Ravi V., Tsai J.-W., Ali A., Ingram R. D., Lowery D. C., Roland L. C., Somaiah N., Hwu P., Yee C., Subbiah V., Futreal A., Lazar J. A., et al. (2019) MAGE-A3 is a clinically relevant target in undifferentiated pleomorphic sarcoma/myxofibrosarcoma. Cancers (Basel) 11, 677 10.3390/cancers11050677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Gao X., Chen G., Cai H., Wang X., Song K., Liu L., Qiu T., and He Y. (2020) Aberrantly enhanced melanoma-associated antigen (MAGE)-A3 expression facilitates cervical cancer cell proliferation and metastasis via actuating wnt signaling pathway. Biomed. Pharmacother. 122, 109710 10.1016/j.biopha.2019.109710 [DOI] [PubMed] [Google Scholar]
- 179. Yin B., Zeng Y., Liu G., Wang X., Wang P., and Song Y. (2014) MAGE-A3 is highly expressed in a cancer stem cell-like side population of bladder cancer cells. Int. J. Clin. Exp. Pathol. 7, 2934–2941 [PMC free article] [PubMed] [Google Scholar]
- 180. Yamada R., Takahashi A., Torigoe T., Morita R., Tamura Y., Tsukahara T., Kanaseki T., Kubo T., Watarai K., Kondo T., Hirohashi Y., and Sato N. (2013) Preferential expression of cancer/testis genes in cancer stem-like cells: proposal of a novel sub-category, cancer/testis/stem gene. Tissue Antigens 81, 428–434 10.1111/tan.12113 [DOI] [PubMed] [Google Scholar]
- 181. Güre A. O., Stockert E., Arden K. C., Boyer A. D., Viars C. S., Scanlan M. J., Old L. J., and Chen Y.-T. (2000) CT10: a new cancer-testis (CT) antigen homologous to CT7 and the MAGE family, identified by representational-difference analysis. Int. J. Cancer 85, 726–732 [DOI] [PubMed] [Google Scholar]
- 182. Wang Y., Han K. J., Pang X. W., Vaughan H. A., Qu W., Dong X. Y., Peng J. R., Zhao H. T., Rui J. A., Leng X. S., Cebon J., Burgess A. W., and Chen W. F. (2002) Large scale identification of human hepatocellular carcinoma-associated antigens by autoantibodies. J. Immunol. 169, 1102–1109 10.4049/jimmunol.169.2.1102 [DOI] [PubMed] [Google Scholar]
- 183. Hou S., Sang M., Zhao L., Hou R., and Shan B. (2016) The expression of MAGE-C1 and MAGE-C2 in breast cancer and their clinical significance. Am. J. Surg. 211, 142–151 10.1016/j.amjsurg.2015.05.028 [DOI] [PubMed] [Google Scholar]
- 184. Pabst C., Zustin J., Jacobsen F., Luetkens T., Kröger N., Schilling G., Bokemeyer C., Sauter G., Atanackovic D., and Marx A. (2010) Expression and prognostic relevance of MAGE-C1/CT7 and MAGE-C2/CT10 in osteolytic lesions of patients with multiple myeloma. Exp. Mol. Pathol. 89, 175–181 10.1016/j.yexmp.2010.06.011 [DOI] [PubMed] [Google Scholar]
- 185. Liu Y., Wen L., Ma L., Kang Y., Liu K. Y., Huang X. J., Ruan G. R., and Lu J. (2019) MAGE genes: prognostic indicators in AL amyloidosis patients. J. Cell. Mol. Med. 23, 5672–5678 10.1111/jcmm.14475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Curioni-Fontecedro A., Nuber N., Mihic-Probst D., Seifert B., Soldini D., Dummer R., Knuth A., van den Broek M., and Moch H. (2011) Expression of MAGE-C1/CT7 and MAGE-C2/CT10 predicts lymph node metastasis in melanoma patients. PLoS ONE 6, e21418 10.1371/journal.pone.0021418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. von Boehmer L., Keller L., Mortezavi A., Provenzano M., Sais G., Hermanns T., Sulser T., Jungbluth A. A., Old L. J., Kristiansen G., van den Broek M., Moch H., Knuth A., and Wild P. J. (2011) MAGE-C2/CT10 protein expression is an independent predictor of recurrence in prostate cancer. PLoS ONE 6, e21366 10.1371/journal.pone.0021366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Ghadban T., Perez D. R., Vashist Y. K., Bockhorn M., Koenig A. M., El Gammal A. T., Izbicki J. R., Metzger U., Hauswirth F., Frosina D., and Jungbluth A. A. (2014) Expression of cancer testis antigens CT10 (MAGE-C2) and GAGE in gastrointestinal stromal tumors. Eur. J. Surg. Oncol. 40, 1307–1312 10.1016/j.ejso.2014.03.011 [DOI] [PubMed] [Google Scholar]
- 189. Hodgson A., Jungbluth A. A., Katabi N., Xu B., and Downes M. R. (2020) Evaluation of cancer testis antigen (CT10, PRAME) and MHC I expression in high-grade urothelial carcinoma of the bladder. Virchows Arch. 476, 535–542 10.1007/s00428-019-02661-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Zhao Q., Xu W. T., and Shalieer T. (2016) Pilot study on MAGE-C2 as a potential biomarker for triple-negative breast cancer. Dis. Markers 2016, 1–8 10.1155/2016/2325987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Yang F., Zhou X., Miao X., Zhang T., Hang X., Tie R., Liu N., Tian F., Wang F., and Yuan J. (2014) MAGEC2, an epithelial-mesenchymal transition inducer, is associated with breast cancer metastasis. Breast Cancer Res. Treat. 145, 23–32 10.1007/s10549-014-2915-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Riener M. O., Wild P. J., Soll C., Knuth A., Jin B., Jungbluth A., Hellerbrand C., Clavien P. A., Moch H., and Jochum W. (2009) Frequent expression of the novel cancer testis antigen MAGE-C2/CT-10 in hepatocellular carcinoma. Int. J. Cancer 124, 352–357 10.1002/ijc.23966 [DOI] [PubMed] [Google Scholar]
- 193. Gu X., Mao Y., Shi C., Ye W., Hou N., Xu L., Chen Y., and Zhao W. (2019) MAGEC2 correlates with unfavorable prognosis and promotes tumor development in HCC via epithelial-mesenchymal transition. Onco Targets Ther. 12, 7843–7855 10.2147/OTT.S213164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Jiang S., Liu X., Li D., Yan M., Ju C., Sun J., and Jiang F. (2018) Study on attenuating angiogenesis and epithelial-mesenchymal transition (EMT) of non-small cell lung carcinoma (NSCLC) by regulating MAGEC2. Technol. Cancer Res. Treat. 17, 153303381879758 10.1177/1533033818797587 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 195. Chen X., Wang L., Yue D., Liu J., Huang L., Yang L., Cao L., Qin G., Li A., Wang D., Wang M., Qi Y., Zhang B., van der Bruggen P., and Zhang Y. (2017) Correlation between the high expression levels of cancer-germline genes with clinical characteristics in esophageal squamous cell carcinoma. Histol. Histopathol. 32, 793–803 10.14670/hh-11-847 [DOI] [PubMed] [Google Scholar]
- 196. Jin X., Pan Y., Wang L., Zhang L., Ravichandran R., Potts P. R., Jiang J., Wu H., and Huang H. (2017) MAGE-TRIM28 complex promotes the Warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis 6, e312 10.1038/oncsis.2017.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Iyengar S., and Farnham P. J. (2011) Kap1 protein: an enigmatic master regulator of the genome. J. Biol. Chem. 286, 26267–26276 10.1074/jbc.R111.252569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Wang C., Ivanov A., Chen L., Fredericks W. J., Seto E., Rauscher F. J., and Chen J. (2005) MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 24, 3279–3290 10.1038/sj.emboj.7600791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. White D. E., Negorev D., Peng H., Ivanov A. V., Maul G. G., and Rauscher F. J. (2006) KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 66, 11594–11599 10.1158/0008-5472.CAN-06-4138 [DOI] [PubMed] [Google Scholar]
- 200. Czerwińska P., Mazurek S., and Wiznerowicz M. (2017) The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 24, 63–63 10.1186/s12929-017-0374-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Rowe H. M., Jakobsson J., Mesnard D., Rougemont J., Reynard S., Aktas T., Maillard P. V., Layard-Liesching H., Verp S., Marquis J., Spitz F., Constam D. B., and Trono D. (2010) KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240 10.1038/nature08674 [DOI] [PubMed] [Google Scholar]
- 202. Matsui T., Leung D., Miyashita H., Maksakova I. A., Miyachi H., Kimura H., Tachibana M., Lorincz M. C., and Shinkai Y. (2010) Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464, 927–931 10.1038/nature08858 [DOI] [PubMed] [Google Scholar]
- 203. Tao Y., Yen M.-R., Chitiashvili T., Nakano H., Kim R., Hosohama L., Tan Y. C., Nakano A., Chen P.-Y., and Clark A. T. (2018) TRIM28-regulated transposon repression is required for human germline competency and not primed or naive human pluripotency. Stem Cell Rep. 10, 243–256 10.1016/j.stemcr.2017.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Brattås P. L., Jönsson M. E., Fasching L., Nelander Wahlestedt J., Shahsavani M., Falk R., Falk A., Jern P., Parmar M., and Jakobsson J. (2017) TRIM28 controls a gene regulatory network based on endogenous retroviruses in human neural progenitor cells. Cell Rep. 18, 1–11 10.1016/j.celrep.2016.12.010 [DOI] [PubMed] [Google Scholar]
- 205. Leseva M., Knowles B. B., Messerschmidt D. M., and Solter D. (2015) Erase–maintain–establish: natural reprogramming of the mammalian epigenome. Cold Spring Harb. Symp. Quant. Biol. 80, 155–163 10.1101/sqb.2015.80.027441 [DOI] [PubMed] [Google Scholar]
- 206. Ling F., Kang B., and Sun X.-H. (2014) Id proteins: small molecules, mighty regulators. in Curr. Top. Dev. Biol. 110, 189–216 10.1016/b978-0-12-405943-6.00005-1 [DOI] [PubMed] [Google Scholar]
- 207. Hao J., Shen R., Li Y., Zhang Y., and Yin Y. (2014) Cancer-testis antigen HCA587/MAGE-C2 interacts with BS69 and promotes its degradation in the ubiquitin-proteasome pathway. Biochem. Biophys. Res. Commun. 449, 386–391 10.1016/j.bbrc.2014.05.078 [DOI] [PubMed] [Google Scholar]
- 208. Zhang G., Tsang C. M., Deng W., Yip Y. L., Lui V. W., Wong S. C., Cheung A. L., Hau P. M., Zeng M., Lung M. L., Chen H., Lo K. W., Takada K., and Tsao S. W. (2013) Enhanced IL-6/IL-6R signaling promotes growth and malignant properties in EBV-infected premalignant and cancerous nasopharyngeal epithelial cells. PLoS ONE 8, e62284 10.1371/journal.pone.0062284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Song X., Hao J., Wang J., Guo C., Wang Y., He Q., Tang H., Qin X., Li Y., Zhang Y., and Yin Y. (2017) The cancer/testis antigen MAGEC2 promotes amoeboid invasion of tumor cells by enhancing STAT3 signaling. Oncogene 36, 1476–1486 10.1038/onc.2016.314 [DOI] [PubMed] [Google Scholar]
- 210. Okamoto K., Kitabayashi I., and Taya Y. (2006) KAP1 dictates p53 response induced by chemotherapeutic agents via Mdm2 interaction. Biochem. Biophys. Res. Commun. 351, 216–222 10.1016/j.bbrc.2006.10.022 [DOI] [PubMed] [Google Scholar]
- 211. Monte M., Simonatto M., Peche L. Y., Bublik D. R., Gobessi S., Pierotti M. A., Rodolfo M., and Schneider C. (2006) MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc. Natl. Acad. Sci. U. S. A. 103, 11160–11165 10.1073/pnas.0510834103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Marcar L., MacLaine N. J., Hupp T. R., and Meek D. W. (2010) Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin. Cancer Res. 70, 10362–10370 10.1158/0008-5472.CAN-10-1341 [DOI] [PubMed] [Google Scholar]
- 213. Nardiello T., Jungbluth A. A., Mei A., DiLiberto M., Huang X., Dabrowski A., Andrade V. C. C., Wasserstrum R., Ely S., Niesvizky R., Pearse R., Coleman M., Jayabalan D. S., Bhardwaj N., Old L. J., et al. (2011) MAGE-A inhibits apoptosis in proliferating myeloma cells through repression of Bax and maintenance of survivin. Clin. Cancer Res. 17, 4309–4319 10.1158/1078-0432.CCR-10-1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Bhatia N., Xiao T. Z., Rosenthal K. A., Siddiqui I. A., Thiyagarajan S., Smart B., Meng Q., Zuleger C. L., Mukhtar H., Kenney S. C., Albertini M. R., and Jack Longley B. (2013) MAGE-C2 promotes growth and tumorigenicity of melanoma cells, phosphorylation of KAP1, and DNA damage repair. J. Invest. Dermatol. 133, 759–767 10.1038/jid.2012.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Yang B., O'Herrin S., Wu J., Reagan-Shaw S., Ma Y., Nihal M., and Longley B. J. (2007) Select cancer testes antigens of the MAGE-A, -B, and -C families are expressed in mast cell lines and promote cell viability in vitro and in vivo. J. Invest. Dermatol. 127, 267–275 10.1038/sj.jid.5700548 [DOI] [PubMed] [Google Scholar]
- 216. Flørenes V. A., Øyjord T., Holm R., Skrede M., Børresen A. L., Nesland J. M., and Fodstad Ø. (1994) TP53 allele loss, mutations and expression in malignant melanoma. Br. J. Cancer 69, 253–259 10.1038/bjc.1994.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Olivier M., Hollstein M., and Hainaut P. (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 10.1101/cshperspect.a001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Hardie D. G. (2015) Molecular pathways: is AMPK a friend or a foe in cancer? Clin. Cancer Res. 21, 3836–3840 10.1158/1078-0432.CCR-14-3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Garcia D., and Shaw R. J. (2017) AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800 10.1016/j.molcel.2017.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M. J., and Thompson C. B. (2005) AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 10.1016/j.molcel.2005.03.027 [DOI] [PubMed] [Google Scholar]
- 221. Shaw R. J. (2015) Tumor metabolism: MAGE-A proteins help trim turn over AMPK. Curr. Biol. 25, R418–R420 10.1016/j.cub.2015.03.019 [DOI] [PubMed] [Google Scholar]
- 222. Heckman-Stoddard B. M., Gandini S., Puntoni M., Dunn B. K., DeCensi A., and Szabo E. (2016) Repurposing old drugs to chemoprevention: the case of metformin. Semin. Oncol. 43, 123–133 10.1053/j.seminoncol.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Baselga J., Campone M., Piccart M., Burris H. A., Rugo H. S., Sahmoud T., Noguchi S., Gnant M., Pritchard K. I., Lebrun F., Beck J. T., Ito Y., Yardley D., Deleu I., Perez A., et al. (2012) Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N. Engl. J. Med. 366, 520–529 10.1056/NEJMoa1109653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. White E. (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Zhang C.-S., Hawley S. A., Zong Y., Li M., Wang Z., Gray A., Ma T., Cui J., Feng J.-W., Zhu M., Wu Y.-Q., Li T. Y., Ye Z., Lin S.-Y., Yin H., et al. (2017) Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116 10.1038/nature23275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Ravichandran R., Kodali K., Peng J., and Potts P. R. (2019) Regulation of MAGE-A3/6 by the CRL4-DCAF12 ubiquitin ligase and nutrient availability. EMBO Rep. 20, e47352 10.15252/embr.201847352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Koren I., Timms R. T., Kula T., Xu Q., Li M. Z., and Elledge S. J. (2018) The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons. Cell 173, 1622–1635.e14 10.1016/j.cell.2018.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Tsang Y. H., Wang Y., Kong K., Grzeskowiak C., Zagorodna O., Dogruluk T., Lu H., Villafane N., Bhavana V. H., Moreno D., Elsea S. H., Liang H., Mills G. B., and Scott K. L. (2020) Differential expression of MAGEA6 toggles autophagy to promote pancreatic cancer progression. eLife 9, e48963 10.7554/eLife.48963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Biankin A. V., Waddell N., Kassahn K. S., Gingras M.-C., Muthuswamy L. B., Johns A. L., Miller D. K., Wilson P. J., Patch A.-M., Wu J., Chang D. K., Cowley M. J., Gardiner B. B., Song S., Harliwong I., et al. (2012) Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 10.1038/nature11547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Helsel A. R., Oatley M. J., and Oatley J. M. (2017) Glycolysis-optimized conditions enhance maintenance of regenerative integrity in mouse spermatogonial stem cells during long-term culture. Stem Cell Rep. 8, 1430–1441 10.1016/j.stemcr.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Chan F., Oatley M. J., Kaucher A. V., Yang Q.-E., Bieberich C. J., Shashikant C. S., and Oatley J. M. (2014) Functional and molecular features of the Id4+ germline stem cell population in mouse testes. Genes Dev. 28, 1351–1362 10.1101/gad.240465.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Hou S., Xian L., Shi P., Li C., Lin Z., and Gao X. (2016) The Magea gene cluster regulates male germ cell apoptosis without affecting the fertility in mice. Sci. Rep. 6, 26735 10.1038/srep26735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Ladelfa M. F., Peche L. Y., Toledo M. F., Laiseca J. E., Schneider C., and Monte M. (2012) Tumor-specific MAGE proteins as regulators of p53 function. Cancer Lett. 325, 11–17 10.1016/j.canlet.2012.05.031 [DOI] [PubMed] [Google Scholar]
- 234. Ishii K., Ishiai M., Morimoto H., Kanatsu-Shinohara M., Niwa O., Takata M., and Shinohara T. (2014) The Trp53-Trp53inp1-Tnfrsf10b pathway regulates the radiation response of mouse spermatogonial stem cells. Stem Cell Rep. 3, 676–689 10.1016/j.stemcr.2014.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Su S., Blackwelder A. J., Grossman G., Minges J. T., Yuan L., Young S. L., and Wilson E. M. (2012) Primate-specific melanoma antigen-A11 regulates isoform-specific human progesterone receptor-B transactivation. J. Biol. Chem. 287, 34809–34824 10.1074/jbc.M112.372797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Lian Y., Sang M., Ding C., Zhou X., Fan X., Xu Y., Lu W., and Shan B. (2012) Expressions of MAGE-A10 and MAGE-A11 in breast cancers and their prognostic significance: a retrospective clinical study. J. Cancer Res. Clin. 138, 519–527 10.1007/s00432-011-1122-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Su S., Minges J. T., Grossman G., Blackwelder A. J., Mohler J. L., and Wilson E. M. (2013) Proto-oncogene activity of melanoma antigen-A11 (MAGE-A11) regulates retinoblastoma-related p107 and E2F1 proteins. J. Biol. Chem. 288, 24809–24824 10.1074/jbc.M113.468579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Xia L. P., Xu M., Chen Y., and Shao W. W. (2013) Expression of MAGE-A11 in breast cancer tissues and its effects on the proliferation of breast cancer cells. Mol. Med. Rep. 7, 254–258 10.3892/mmr.2012.1126 [DOI] [PubMed] [Google Scholar]
- 239. Jia S., Zhang M., Li Y., Zhang L., and Dai W. (2020) MAGE-A11 expression predicts patient prognosis in head and neck squamous cell carcinoma. Cancer Manag. Res. 12, 1427–1435 10.2147/CMAR.S237867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Su S., Gu Q., Xu A., Shen S., Liu S., Zhang C., Miao C., Qin C., Liu B., and Wang Z. (2019) Genetic variations in MAGE-A11 predict the risk and survival of renal cell cancer. J. Cancer 10, 4860–4865 10.7150/jca.32675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Gu L., Sang M., Li J., Liu F., Wu Y., Liu S., and Shan B. (2019) Demethylation-mediated upregulation of melanoma-associated antigen-A11 correlates with malignant progression of esophageal squamous cell carcinoma. Dig. Liver Dis. 51, 1475–1482 10.1016/j.dld.2019.04.018 [DOI] [PubMed] [Google Scholar]
- 242. Hartmann S., Zwick L., Maurus K., Fuchs A. R., Brands R. C., Seher A., Kübler A. C., and Müller-Richter U. D. A. (2018) Melanoma-associated antigen A11 reduces erlotinib and afatinib efficacy in head and neck cancer. J. Craniomaxillofac. Surg. 46, 492–497 10.1016/j.jcms.2017.12.014 [DOI] [PubMed] [Google Scholar]
- 243. Hartmann S., Zwick L., Scheurer M. J. J., Fuchs A. R., Brands R. C., Seher A., Böhm H., Kübler A. C., and Müller-Richter U. D. A. (2018) Mage-a11 expression contributes to cisplatin resistance in head and neck cancer. Clin. Oral Investig. 22, 1477–1486 10.1007/s00784-017-2242-8 [DOI] [PubMed] [Google Scholar]
- 244. Bai S., He B., and Wilson E. M. (2005) Melanoma antigen gene protein mage-11 regulates androgen receptor function by modulating the interdomain interaction. Mol. Cell Biol. 25, 1238–1257 10.1128/MCB.25.4.1238-1257.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Bai S., and Wilson E. M. (2008) Epidermal-growth-factor-dependent phosphorylation and ubiquitinylation of MAGE-11 regulates its interaction with the androgen receptor. Mol. Cell Biol. 28, 1947–1963 10.1128/MCB.01672-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Askew E. B., Bai S., Hnat A. T., Minges J. T., and Wilson E. M. (2009) Melanoma antigen gene protein-A11 (MAGE-11) F-box links the androgen receptor NH2-terminal transactivation domain to p160 coactivators. J. Biol. Chem. 284, 34793–34808 10.1074/jbc.M109.065979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Askew E. B., Bai S., Blackwelder A. J., and Wilson E. M. (2010) Transcriptional synergy between melanoma antigen gene protein-A11 (MAGE-11) and p300 in androgen receptor signaling. J. Biol. Chem. 285, 21824–21836 10.1074/jbc.M110.120600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Douglas A. G., Ioannis S., and Philippa T. K. S. (2016) Regulation of androgen action during establishment of pregnancy. J. Mol. Endocrinol. 57, R35–R47 10.1530/JME-16-0027 [DOI] [PubMed] [Google Scholar]
- 249. Younas K., Quintela M., Thomas S., Garcia-Parra J., Blake L., Whiteland H., Bunkheila A., Francis L. W., Margarit L., Gonzalez D., and Conlan R. S. (2019) Delayed endometrial decidualisation in polycystic ovary syndrome; the role of AR-MAGEA11. J. Mol. Med. (Berl.) 97, 1315–1327 10.1007/s00109-019-01809-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Minges J. T., Grossman G., Zhang P., Kafri T., and Wilson E. M. (2015) Post-translational down-regulation of melanoma antigen-A11 (MAGE-A11) by human p14-ARF tumor suppressor. J. Biol. Chem. 290, 25174–25187 10.1074/jbc.M115.663641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Derti A., Garrett-Engele P., Macisaac K. D., Stevens R. C., Sriram S., Chen R., Rohl C. A., Johnson J. M., and Babak T. (2012) A quantitative atlas of polyadenylation in five mammals. Genome Res. 22, 1173–1183 10.1101/gr.132563.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Hoque M., Ji Z., Zheng D., Luo W., Li W., You B., Park J. Y., Yehia G., and Tian B. (2013) Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 10, 133–139 10.1038/nmeth.2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Tian B., and Manley J. L. (2017) Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 18, 18–30 10.1038/nrm.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Sandberg R., Neilson J. R., Sarma A., Sharp P. A., and Burge C. B. (2008) Proliferating cells express mRNAs with shortened 3′-untranslated regions and fewer microrna target sites. Science 320, 1643–1647 10.1126/science.1155390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. MacDonald C. C., and Redondo J.-L. (2002) Reexamining the polyadenylation signal: were we wrong about AAUAAA? Mol. Cell. Endocrinol. 190, 1–8 10.1016/S0303-7207(02)00044-8 [DOI] [PubMed] [Google Scholar]
- 256. MacDonald C. C. (2019) Tissue-specific mechanisms of alternative polyadenylation: testis, brain, and beyond (2018 update). Wiley Interdiscip. Rev. RNA 10, e1526 10.1002/wrna.1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Fu Y., Sun Y., Li Y., Li J., Rao X., Chen C., and Xu A. (2011) Differential genome-wide profiling of tandem 3' UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 21, 741–747 10.1101/gr.115295.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Mayr C., and Bartel D. P. (2009) Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684 10.1016/j.cell.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Li W., You B., Hoque M., Zheng D., Luo W., Ji Z., Park J. Y., Gunderson S. I., Kalsotra A., Manley J. L., and Tian B. (2015) Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 11, e1005166 10.1371/journal.pgen.1005166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Masamha C. P., Xia Z., Yang J., Albrecht T. R., Li M., Shyu A.-B., Li W., and Wagner E. J. (2014) Cfim25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510, 412–416 10.1038/nature13261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Ogorodnikov A., Levin M., Tattikota S., Tokalov S., Hoque M., Scherzinger D., Marini F., Poetsch A., Binder H., Macher-Göppinger S., Probst H. C., Tian B., Schaefer M., Lackner K. J., Westermann F., et al. (2018) Transcriptome 3′end organization by pcf11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 9, 5331 10.1038/s41467-018-07580-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Li W., Park J. Y., Zheng D., Hoque M., Yehia G., and Tian B. (2016) Alternative cleavage and polyadenylation in spermatogenesis connects chromatin regulation with post-transcriptional control. BMC Biol. 14, 6 10.1186/s12915-016-0229-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. McMahon K. W., Hirsch B. A., and MacDonald C. C. (2006) Differences in polyadenylation site choice between somatic and male germ cells. BMC Mol. Biol. 7, 35 10.1186/1471-2199-7-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Bose R., Sheng K., Moawad A. R., Manku G., O'Flaherty C., Taketo T., Culty M., Fok K. L., and Wing S. S. (2017) Ubiquitin ligase Huwe1 modulates spermatogenesis by regulating spermatogonial differentiation and entry into meiosis. Sci. Rep. 7, 17759 10.1038/s41598-017-17902-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Peche L. Y., Ladelfa M. F., Toledo M. F., Mano M., Laiseca J. E., Schneider C., and Monte M. (2015) Human MageB2 protein expression enhances E2F transcriptional activity, cell proliferation, and resistance to ribotoxic stress. J. Biol. Chem. 290, 29652–29662 10.1074/jbc.M115.671982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Protter D. S. W., and Parker R. (2016) Principles and properties of stress granules. Trends Cell Biol. 26, 668–679 10.1016/j.tcb.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Van Treeck B., and Parker R. (2018) Emerging roles for intermolecular RNA-RNA interactions in RNP assemblies. Cell 174, 791–802 10.1016/j.cell.2018.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Anderson P., Kedersha N., and Ivanov P. (2015) Stress granules, P-bodies and cancer. Biochim. Biophys. Acta 1849, 861–870 10.1016/j.bbagrm.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Kedersha N., and Anderson P. (2002) Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 30, 963–969 10.1042/bst0300963 [DOI] [PubMed] [Google Scholar]
- 270. Banani S. F., Rice A. M., Peeples W. B., Lin Y., Jain S., Parker R., and Rosen M. K. (2016) Compositional control of phase-separated cellular bodies. Cell 166, 651–663 10.1016/j.cell.2016.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Jain S., Wheeler J. R., Walters R. W., Agrawal A., Barsic A., and Parker R. (2016) ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498 10.1016/j.cell.2015.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Kedersha N., Panas M. D., Achorn C. A., Lyons S., Tisdale S., Hickman T., Thomas M., Lieberman J., McInerney G. M., Ivanov P., and Anderson P. (2016) G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 212, 845–860 10.1083/jcb.201508028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Khong A., Matheny T., Jain S., Mitchell S. F., Wheeler J. R., and Parker R. (2017) The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol. Cell 68, 808–820.e5 10.1016/j.molcel.2017.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Markmiller S., Soltanieh S., Server K. L., Mak R., Jin W., Fang M. Y., Luo E. C., Krach F., Yang D., Sen A., Fulzele A., Wozniak J. M., Gonzalez D. J., Kankel M. W., Gao F. B., et al. (2018) Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–604.e13 10.1016/j.cell.2017.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Namkoong S., Ho A., Woo Y. M., Kwak H., and Lee J. H. (2018) Systematic characterization of stress-induced RNA granulation. Mol. Cell 70, 175–187.e8 10.1016/j.molcel.2018.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Reineke L. C., Dougherty J. D., Pierre P., and Lloyd R. E. (2012) Large G3BP-induced granules trigger eIF2α phosphorylation. Mol. Biol. Cell 23, 3499–3510 10.1091/mbc.E12-05-0385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Souquere S., Mollet S., Kress M., Dautry F., Pierron G., and Weil D. (2009) Unravelling the ultrastructure of stress granules and associated P-bodies in human cells. J. Cell Sci. 122, 3619–3626 10.1242/jcs.054437 [DOI] [PubMed] [Google Scholar]
- 278. Wheeler J. R., Matheny T., Jain S., Abrisch R., and Parker R. (2016) Distinct stages in stress granule assembly and disassembly. eLife 5, e18413 10.7554/eLife.18413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Zhang P., Fan B., Yang P., Temirov J., Messing J., Kim H. J., and Taylor J. P. (2019) Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. eLife 8, e39578 10.7554/eLife.39578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Guo J., Grow E. J., Mlcochova H., Maher G. J., Lindskog C., Nie X., Guo Y., Takei Y., Yun J., Cai L., Kim R., Carrell D. T., Goriely A., Hotaling J. M., and Cairns B. R. (2018) The adult human testis transcriptional cell atlas. Cell Res. 28, 1141–1157 10.1038/s41422-018-0099-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Jung M., Wells D., Rusch J., Ahmad S., Marchini J., Myers S. R., and Conrad D. F. (2019) Unified single-cell analysis of testis gene regulation and pathology in five mouse strains. eLife 8, e43966 10.7554/eLife.43966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Sohni A., Tan K., Song H. W., Burow D., de Rooij D. G., Laurent L., Hsieh T. C., Rabah R., Hammoud S. S., Vicini E., and Wilkinson M. F. (2019) The neonatal and adult human testis defined at the single-cell level. Cell Rep. 26, 1501–1517.e4 10.1016/j.celrep.2019.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Xia B., Yan Y., Baron M., Wagner F., Barkley D., Chiodin M., Kim S. Y., Keefe D. L., Alukal J. P., Boeke J. D., and Yanai I. (2020) Widespread transcriptional scanning in the testis modulates gene evolution rates. Cell 180, 248–262.e21 10.1016/j.cell.2019.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Reid B. O., Mason K. A., Withers H. R., and West J. (1981) Effects of hyperthermia and radiation on mouse testis stem cells. Cancer Res. 41, 4453–4457 [PubMed] [Google Scholar]
- 285. Rockett J. C., Mapp F. L., Garges J. B., Luft J. C., Mori C., and Dix D. J. (2001) Effects of hyperthermia on spermatogenesis, apoptosis, gene expression, and fertility in adult male mice. Biol. Reprod. 65, 229–239 10.1095/biolreprod65.1.229 [DOI] [PubMed] [Google Scholar]
- 286. Yin Y., Hawkins K. L., DeWolf W. C., and Morgentaler A. (1997) Heat stress causes testicular germ cell apoptosis in adult mice. J. Androl. 18, 159–165 [PubMed] [Google Scholar]
- 287. Widlak W., and Vydra N. (2017) The role of heat shock factors in mammalian spermatogenesis. Adv. Anat. Embryol. Cell Biol. 222, 45–65 10.1007/978-3-319-51409-3_3 [DOI] [PubMed] [Google Scholar]
- 288. Kent L. N., and Leone G. (2019) The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 19, 326–338 10.1038/s41568-019-0143-7 [DOI] [PubMed] [Google Scholar]
- 289. Taniura H., Taniguchi N., Hara M., and Yoshikawa K. (1998) Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J. Biol. Chem. 273, 720–728 10.1074/jbc.273.2.720 [DOI] [PubMed] [Google Scholar]
- 290. Hoftman A. D., Tai L. Q., Tze S., Seligson D., Gatti R. A., and McCurdy D. K. (2008) MAGE-B2 autoantibody: a new biomarker for pediatric systemic lupus erythematosus. J. Rheumatol. 35, 2430–2438 10.3899/jrheum.080333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Ballestar E., Esteller M., and Richardson B. C. (2006) The epigenetic face of systemic lupus erythematosus. J. Immunol. 176, 7143–7147 10.4049/jimmunol.176.12.7143 [DOI] [PubMed] [Google Scholar]
- 292. Richardson B. (2003) DNA methylation and autoimmune disease. J. Clin. Immunol. 109, 72–79 10.1016/S1521-6616(03)00206-7 [DOI] [PubMed] [Google Scholar]
- 293. Barnea E., Beer I., Patoka R., Ziv T., Kessler O., Tzehoval E., Eisenbach L., Zavazava N., and Admon A. (2002) Analysis of endogenous peptides bound by soluble MHC class I molecules: a novel approach for identifying tumor-specific antigens. Eur. J. Immunol. 32, 213–222 [DOI] [PubMed] [Google Scholar]
- 294. Fleischhauer K., Gattinoni L., Dalerba P., Lauvau G., Zanaria E., Dabovic B., van Endert P. M., Bordignon C., and Traversari C. (1998) The DAM gene family encodes a new group of tumor-specific antigens recognized by human leukocyte antigen A2-restricted cytotoxic T lymphocytes. Cancer Res. 58, 2969–2972 [PubMed] [Google Scholar]
- 295. Novellino L., Castelli C., and Parmiani G. (2005) A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol. Immunother. 54, 187–207 10.1007/s00262-004-0560-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Mizukami M., Hanagiri T., Baba T., Fukuyama T., Nagata Y., So T., Ichiki Y., Sugaya M., Yasuda M., Takenoyama M., Sugio K., and Yasumoto K. (2005) Identification of tumor associated antigens recognized by IgG from tumor-infiltrating B cells of lung cancer: correlation between Ab titer of the patient's sera and the clinical course. Cancer Sci. 96, 882–888 10.1111/j.1349-7006.2005.00119.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Landegren N., Sharon D., Freyhult E., Hallgren A., Eriksson D., Edqvist P. H., Bensing S., Wahlberg J., Nelson L. M., Gustafsson J., Husebye E. S., Anderson M. S., Snyder M., and Kampe O. (2016) Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine syndrome type 1. Sci. Rep. 6, 20104 10.1038/srep20104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Nagamine K., Peterson P., Scott H. S., Kudoh J., Minoshima S., Heino M., Krohn K. J., Lalioti M. D., Mullis P. E., Antonarakis S. E., Kawasaki K., Asakawa S., Ito F., and Shimizu N. (1997) Positional cloning of the APECED gene. Nat. Genet. 17, 393–398 10.1038/ng1297-393 [DOI] [PubMed] [Google Scholar]
- 299. Anderson M. S., Venanzi E. S., Klein L., Chen Z., Berzins S. P., Turley S. J., von Boehmer H., Bronson R., Dierich A., Benoist C., and Mathis D. (2002) Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 10.1126/science.1075958 [DOI] [PubMed] [Google Scholar]
- 300. Liston A., Lesage S., Wilson J., Peltonen L., and Goodnow C. C. (2003) Aire regulates negative selection of organ-specific T cells. Nat. Immunol. 4, 350–354 10.1038/ni906 [DOI] [PubMed] [Google Scholar]
- 301. Conteduca G., Fenoglio D., Parodi A., Battaglia F., Kalli F., Negrini S., Tardito S., Ferrera F., Salis A., Millo E., Pasquale G., Barra G., Damonte G., Indiveri F., Ferrone S., et al. (2016) AIRE polymorphism, melanoma antigen-specific T cell immunity, and susceptibility to melanoma. Oncotarget 7, 60872–60884 10.18632/oncotarget.11506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Dombret C., Nguyen T., Schakman O., Michaud J. L., Hardin-Pouzet H., Bertrand M. J., and De Backer O. (2012) Loss of Maged1 results in obesity, deficits of social interactions, impaired sexual behavior and severe alteration of mature oxytocin production in the hypothalamus. Hum. Mol. Genet. 21, 4703–4717 10.1093/hmg/dds310 [DOI] [PubMed] [Google Scholar]
- 303. Wang X., Tang J., Xing L., Shi G., Ruan H., Gu X., Liu Z., Wu X., Gao X., and Xu Y. (2010) Interaction of MAGED1 with nuclear receptors affects circadian clock function. EMBO J. 29, 1389–1400 10.1038/emboj.2010.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Yang J., Lai B., Xu A., Liu Y., Li X., Zhao Y., Li W., Ji M., Hu G., Gao X., and Gao J. (2015) Maged1 co-interacting with CREB through a hexapeptide repeat domain regulates learning and memory in mice. Mol. Neurobiol. 51, 8–18 10.1007/s12035-014-8677-x [DOI] [PubMed] [Google Scholar]
- 305. Rochira J. A., Cowling R. A., Himmelfarb J. S., Adams T. L., and Verdi J. M. (2010) Mapping of NRAGE domains reveals clues to cell viability in BMP signaling. Apoptosis 15, 63–70 10.1007/s10495-009-0427-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Masuda Y., Sasaki A., Shibuya H., Ueno N., Ikeda K., and Watanabe K. (2001) Dlxin-1, a novel protein that binds Dlx5 and regulates its transcriptional function. J. Biol. Chem. 276, 5331–5338 10.1074/jbc.M008590200 [DOI] [PubMed] [Google Scholar]
- 307. Sullivan A. E., Peet D. J., and Whitelaw M. L. (2016) MaGED1 is a novel regulator of a select subset of bHLH PAS transcription factors. FEBS J 283, 3488–3502 10.1111/febs.13824 [DOI] [PubMed] [Google Scholar]
- 308. Salehi A. H., Xanthoudakis S., and Barker P. A. (2002) NRAGE, a p75 neurotrophin receptor-interacting protein, induces caspase activation and cell death through a JNK-dependent mitochondrial pathway. J. Biol. Chem. 277, 48043–48050 10.1074/jbc.M205324200 [DOI] [PubMed] [Google Scholar]
- 309. Bragason B. T., and Palsdottir A. (2005) Interaction of PRP with NRAGE, a protein involved in neuronal apoptosis. Mol. Cell. Neurosci. 29, 232–244 10.1016/j.mcn.2005.02.013 [DOI] [PubMed] [Google Scholar]
- 310. Nikopoulos G. N., Martins J. F., Adams T. L., Karaczyn A., Adams D., Vary C., Oxburgh L., and Verdi J. M. (2009) NRAGE: a potential rheostat during branching morphogenesis. Mech. Dev. 126, 337–349 10.1016/j.mod.2009.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Akihiro M., Yukihiro N., Ken W., and Toshitaka N. (2013) The roles of MAGE-D1 in the neuronal functions and pathology of the central nervous system. Rev. Neurosci. 24, 61–70 10.1515/revneuro-2012-0069 [DOI] [PubMed] [Google Scholar]
- 312. Reddy E. M., Chettiar S. T., Kaur N., Shepal V., and Shiras A. (2010) Dlxin-1, a mage family protein, induces accelerated neurite outgrowth and cell survival by enhanced and early activation of MEK and Akt signalling pathways in pc12 cells. Exp. Cell Res. 316, 2220–2236 10.1016/j.yexcr.2010.05.030 [DOI] [PubMed] [Google Scholar]
- 313. Williams M. E., Strickland P., Watanabe K., and Hinck L. (2003) UNC5H1 induces apoptosis via its juxtamembrane region through an interaction with NRAGE. J. Biol. Chem. 278, 17483–17490 10.1074/jbc.M300415200 [DOI] [PubMed] [Google Scholar]
- 314. Jordan B. W., Dinev D., LeMellay V., Troppmair J., Gotz R., Wixler L., Sendtner M., Ludwig S., and Rapp U. R. (2001) Neurotrophin receptor-interacting mage homologue is an inducible inhibitor of apoptosis protein-interacting protein that augments cell death. J. Biol. Chem. 276, 39985–39989 10.1074/jbc.C100171200 [DOI] [PubMed] [Google Scholar]
- 315. Matluk N., Rochira J. A., Karaczyn A., Adams T., and Verdi J. M. (2010) A role for NRAGE in NF-κB activation through the non-canonical BMP pathway. BMC Biol. 8, 7 10.1186/1741-7007-8-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Kendall S. E., Battelli C., Irwin S., Mitchell J. G., Glackin C. A., and Verdi J. M. (2005) NRAGE mediates p38 activation and neural progenitor apoptosis via the bone morphogenetic protein signaling cascade. Mol. Cell Biol. 25, 7711–7724 10.1128/MCB.25.17.7711-7724.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Kang J., and Chung K. C. (2015) The F-box protein FBXO7 positively regulates bone morphogenetic protein-mediated signaling through Lys-63-specific ubiquitination of neurotrophin receptor-interacting mage (NRAGE). Cell. Mol. Life Sci. 72, 181–195 10.1007/s00018-014-1665-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Wu Q., Qi S., Ma J., Chen F., Chen J., Li J., Zhang X., Xu Y., Pan Q., and Wang R. (2015) The effect of NRAGE on cell cycle and apoptosis of human dental pulp cells and MDPC-23. Int. J. Clin. Exp. Med. 8, 10657–10667 [PMC free article] [PubMed] [Google Scholar]
- 319. Teuber J., Mueller B., Fukabori R., Lang D., Albrecht A., and Stork O. (2013) The ubiquitin ligase PRAJA1 reduces NRAGE expression and inhibits neuronal differentiation of PC12 cells. PLoS ONE 8, e63067 10.1371/journal.pone.0063067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Sasaki A., Masuda Y., Iwai K., Ikeda K., and Watanabe K. (2002) A RING finger protein Praja1 regulates Dlx5-dependent transcription through its ubiquitin ligase activity for the Dlx/Msx-interacting MAGE/Necdin family protein, Dlxin-1. J. Biol. Chem. 277, 22541–22546 10.1074/jbc.M109728200 [DOI] [PubMed] [Google Scholar]
- 321. Kuwajima T., Taniura H., Nishimura I., and Yoshikawa K. (2004) Necdin interacts with the Msx2 homeodomain protein via MAGE-D1 to promote myogenic differentiation of C2C12 cells. J. Biol. Chem. 279, 40484–40493 10.1074/jbc.M404143200 [DOI] [PubMed] [Google Scholar]
- 322. Matsuda T., Suzuki H., Oishi I., Kani S., Kuroda Y., Komori T., Sasaki A., Watanabe K., and Minami Y. (2003) The receptor tyrosine kinase Ror2 associates with the melanoma-associated antigen (MAGE) family protein Dlxin-1 and regulates its intracellular distribution. J. Biol. Chem. 278, 29057–29064 10.1074/jbc.M302199200 [DOI] [PubMed] [Google Scholar]
- 323. Nguyen T. H., Bertrand M. J., Sterpin C., Achouri Y., and De Backer O. R. (2010) Maged1, a new regulator of skeletal myogenic differentiation and muscle regeneration. BMC Cell Biol. 11, 57 10.1186/1471-2121-11-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Xue B., Wen C., Shi Y., Zhao D., and Li C. (2005) Human NRAGE disrupts E-cadherin/β-catenin regulated homotypic cell-cell adhesion. Biochem. Biophys. Res. Commun. 336, 247–251 10.1016/j.bbrc.2005.08.069 [DOI] [PubMed] [Google Scholar]
- 325. Shen W. G., Xue Q. Y., Wu Y. D., Hu B. S., Zhu J., Zhang Y., and Su Q. (2007) Melanoma-associated antigen family protein-D1 regulation of tumor cell migration, adhesion to endothelium, and actin structures reorganization in response to hypoxic stress. Cell Commun. Adhes. 14, 21–31 10.1080/15419060701224948 [DOI] [PubMed] [Google Scholar]
- 326. Bertrand M. J., Kenchappa R. S., Andrieu D., Leclercq-Smekens M., Nguyen H. N., Carter B. D., Muscatelli F., Barker P. A., and De Backer O. (2008) NRAGE, a p75NTR adaptor protein, is required for developmental apoptosis in vivo. Cell Death Differ. 15, 1921–1929 10.1038/cdd.2008.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Pirlot C., Thiry M., Trussart C., Di Valentin E., Piette J., and Habraken Y. (2016) Melanoma antigen-D2: a nucleolar protein undergoing delocalization during cell cycle and after cellular stress. Biochim. Biophys. Acta 1863, 581–595 10.1016/j.bbamcr.2015.12.010 [DOI] [PubMed] [Google Scholar]
- 328. Boulon S., Westman B. J., Hutten S., Boisvert F.-M., and Lamond A. I. (2010) The nucleolus under stress. Mol. Cell 40, 216–227 10.1016/j.molcel.2010.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Trussart C., Pirlot C., Di Valentin E., Piette J., and Habraken Y. (2018) Melanoma antigen-D2 controls cell cycle progression and modulates the DNA damage response. Biochem. Pharmacol. 153, 217–229 10.1016/j.bcp.2018.01.035 [DOI] [PubMed] [Google Scholar]
- 330. Papageorgio C., Brachmann R., Zeng J., Culverhouse R., Zhang W., and McLeod H. (2007) Maged2: a novel p53-dissociator. Int. J. Oncol. 31, 1205–1211 [PubMed] [Google Scholar]
- 331. Tseng H.-Y., Chen L. H., Ye Y., Tay K. H., Jiang C. C., Guo S. T., Jin L., Hersey P., and Zhang X. D. (2012) The melanoma-associated antigen MAGE-D2 suppresses TRAIL receptor 2 and protects against TRAIL-induced apoptosis in human melanoma cells. Carcinogenesis 33, 1871–1881 10.1093/carcin/bgs236 [DOI] [PubMed] [Google Scholar]
- 332. Laghmani K., Beck B. B., Yang S. S., Seaayfan E., Wenzel A., Reusch B., Vitzthum H., Priem D., Demaretz S., Bergmann K., Duin L. K., Gobel H., Mache C., Thiele H., Bartram M. P., et al. (2016) Polyhydramnios, transient antenatal Bartter's syndrome, and MAGED2 mutations. N. Engl. J. Med. 374, 1853–1863 10.1056/NEJMoa1507629 [DOI] [PubMed] [Google Scholar]
- 333. Legrand A., Treard C., Roncelin I., Dreux S., Bertholet-Thomas A., Broux F., Bruno D., Decramer S., Deschenes G., Djeddi D., Guigonis V., Jay N., Khalifeh T., Llanas B., Morin D., et al. (2018) Prevalence of novel MAGED2 mutations in antenatal Bartter syndrome. Clin. J. Am. Soc. Nephrol. 13, 242–250 10.2215/CJN.05670517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Brochard K., Boyer O., Blanchard A., Loirat C., Niaudet P., Macher M.-A., Deschenes G., Bensman A., Decramer S., Cochat P., Morin D., Broux F., Caillez M., Guyot C., Novo R., et al. (2009) Phenotype–genotype correlation in antenatal and neonatal variants of Bartter syndrome. Nephrol. Dial. Transpl. 24, 1455–1464 10.1093/ndt/gfn689 [DOI] [PubMed] [Google Scholar]
- 335. De Donato M., Peters S. O., Hussain T., Rodulfo H., Thomas B. N., Babar M. E., and Imumorin I. G. (2017) Molecular evolution of type II MAGE genes from ancestral MAGED2 gene and their phylogenetic resolution of basal mammalian clades. Mamm. Genome 28, 443–454 10.1007/s00335-017-9695-6 [DOI] [PubMed] [Google Scholar]
- 336. Valiño-Rivas L., Cuarental L., Agustin M., Husi H., Cannata-Ortiz P., Sanz A. B., Mischak H., Ortiz A., and Sanchez-Niño M. D. (2019) MAGE genes in the kidney: identification of MAGED2 as upregulated during kidney injury and in stressed tubular cells. Nephrol. Dial. Transpl. 34, 1498–1507 10.1093/ndt/gfy367 [DOI] [PubMed] [Google Scholar]
- 337. Stone B., Schummer M., Paley P. J., Crawford M., Ford M., Urban N., and Nelson B. H. (2001) MAGE-F1, a novel ubiquitously expressed member of the MAGE superfamily. Gene 267, 173–182 10.1016/S0378-1119(01)00406-1 [DOI] [PubMed] [Google Scholar]
- 338. Stehling O., Mascarenhas J., Vashisht A. A., Sheftel A. D., Niggemeyer B., Rösser R., Pierik A. J., Wohlschlegel J. A., and Lill R. (2013) Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell Metab. 18, 187–198 10.1016/j.cmet.2013.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Stehling O., Vashisht A. A., Mascarenhas J., Jonsson Z. O., Sharma T., Netz D. J. A., Pierik A. J., Wohlschlegel J. A., and Lill R. (2012) Mms19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 337, 195–199 10.1126/science.1219723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Gari K., León Ortiz A. M., Borel V., Flynn H., Skehel J. M., and Boulton S. J. (2012) Mms19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 337, 243–245 10.1126/science.1219664 [DOI] [PubMed] [Google Scholar]
- 341. Saha P. P., Vishwanathan V., Bankapalli K., and D'Silva P. (2018) Iron-sulfur protein assembly in human cells. Rev. Physiol. Biochem. Pharmacol. 174, 25–65 10.1007/112_2017_5 [DOI] [PubMed] [Google Scholar]
- 342. Paul V. D., and Lill R. (2015) Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochim. Biophys. Acta 1853, 1528–1539 10.1016/j.bbamcr.2014.12.018 [DOI] [PubMed] [Google Scholar]
- 343. Lill R., Dutkiewicz R., Freibert S. A., Heidenreich T., Mascarenhas J., Netz D. J., Paul V. D., Pierik A. J., Richter N., Stümpfig M., Srinivasan V., Stehling O., and Mühlenhoff U. (2015) The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell Biol. 94, 280–291 10.1016/j.ejcb.2015.05.002 [DOI] [PubMed] [Google Scholar]
- 344. Mashberg A., Boffetta P., Winkelman R., and Garfinkel L. (1993) Tobacco smoking, alcohol drinking, and cancer of the oral cavity and oropharynx among U.S. veterans. Cancer 72, 1369–1375 [DOI] [PubMed] [Google Scholar]
- 345. Cook J. D., Kondapalli K. C., Rawat S., Childs W. C., Murugesan Y., Dancis A., and Stemmler T. L. (2010) Molecular details of the yeast frataxin-Isu1 interaction during mitochondrial FE-S cluster assembly. Biochemistry 49, 8756–8765 10.1021/bi1008613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Chibuk T. K., Bischof J. M., and Wevrick R. (2001) A necdin/MAGE-like gene in the chromosome 15 autism susceptibility region: expression, imprinting, and mapping of the human and mouse orthologues. BMC Genet. 2, 22 10.1186/1471-2156-2-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Glenn C. C., Driscoll D. J., Yang T. P., and Nicholls R. D. (1997) Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol. Hum. Reprod. 3, 321–332 10.1093/molehr/3.4.321 [DOI] [PubMed] [Google Scholar]
- 348. Potts P. R. (2009) The Yin and Yang of the MMS21–SMC5/6 SUMO ligase complex in homologous recombination. DNA Repair (Amst.) 8, 499–506 10.1016/j.dnarep.2009.01.009 [DOI] [PubMed] [Google Scholar]
- 349. Potts P. R., Porteus M. H., and Yu H. (2006) Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 25, 3377–3388 10.1038/sj.emboj.7601218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Copsey A., Tang S., Jordan P. W., Blitzblau H. G., Newcombe S., Chan A. C-h., Newnham L., Li Z., Gray S., Herbert A. D., Arumugam P., Hochwagen A., Hunter N., and Hoffmann E. (2013) Smc5/6 coordinates formation and resolution of joint molecules with chromosome morphology to ensure meiotic divisions. PLoS Genet. 9, e1004071 10.1371/journal.pgen.1004071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Gómez R., Jordan P. W., Viera A., Alsheimer M., Fukuda T., Jessberger R., Llano E., Pendás A. M., Handel M. A., and Suja J. A. (2013) Dynamic localization of SMC5/6 complex proteins during mammalian meiosis and mitosis suggests functions in distinct chromosome processes. J. Cell Sci. 126, 4239–4252 10.1242/jcs.130195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Lilienthal I., Kanno T., and Sjögren C. (2013) Inhibition of the Smc5/6 complex during meiosis perturbs joint molecule formation and resolution without significantly changing crossover or non-crossover levels. PLoS Genet. 9, e1003898 10.1371/journal.pgen.1003898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Fujioka Y., Kimata Y., Nomaguchi K., Watanabe K., and Kohno K. (2002) Identification of a novel non-structural maintenance of chromosomes (SMC) component of the SMC5-SMC6 complex involved in DNA repair. J. Biol. Chem. 277, 21585–21591 10.1074/jbc.M201523200 [DOI] [PubMed] [Google Scholar]
- 354. Harvey S. H., Sheedy D. M., Cuddihy A. R., and O'Connell M. J. (2004) Coordination of DNA damage responses via the SMC5/SMC6 complex. Mol. Cell Biol. 24, 662–674 10.1128/mcb.24.2.662-674.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. McDonald W. H., Pavlova Y., Yates J. R., and Boddy M. N. (2003) Novel essential DNA repair proteins Nse1 and Nse2 are subunits of the fission yeast Smc5-Smc6 complex. J. Biol. Chem. 278, 45460–45467 10.1074/jbc.M308828200 [DOI] [PubMed] [Google Scholar]
- 356. Pebernard S., Wohlschlegel J., McDonald W. H., Yates J. R., and Boddy M. N. (2006) The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol. Cell Biol. 26, 1617–1630 10.1128/MCB.26.5.1617-1630.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Sergeant J., Taylor E., Palecek J., Fousteri M., Andrews E. A., Sweeney S., Shinagawa H., Watts F. Z., and Lehmann A. R. (2005) Composition and architecture of the Schizosaccharomyces pombe Rad18 (Smc5-6) complex. Mol. Cell Biol. 25, 172–184 10.1128/MCB.25.1.172-184.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Andrews E. A., Palecek J., Sergeant J., Taylor E., Lehmann A. R., and Watts F. Z. (2005) Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol. Cell Biol. 25, 185–196 10.1128/MCB.25.1.185-196.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Lehmann A. R., Walicka M., Griffiths D. J., Murray J. M., Watts F. Z., McCready S., and Carr A. M. (1995) The rad18 gene of Schizosaccharomyces pombe defines a new subgroup of the SMC superfamily involved in DNA repair. Mol. Cell Biol. 15, 7067–7080 10.1128/mcb.15.12.7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Potts P. R., and Yu H. (2005) Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol. Cell Biol. 25, 7021–7032 10.1128/MCB.25.16.7021-7032.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Potts P. R., and Yu H. (2007) The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 14, 581–590 10.1038/nsmb1259 [DOI] [PubMed] [Google Scholar]
- 362. Verkade H. M., Bugg S. J., Lindsay H. D., Carr A. M., and O'Connell M. J. (1999) Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol. Biol. Cell 10, 2905–2918 10.1091/mbc.10.9.2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Piccoli G. D., Torres-Rosell J., and Aragón L. (2009) The unnamed complex: what do we know about Smc5-Smc6? Chromosome Res. 17, 251–263 10.1007/s10577-008-9016-8 [DOI] [PubMed] [Google Scholar]
- 364. Duan X., Sarangi P., Liu X., Rangi G. K., Zhao X., and Ye H. (2009) Structural and functional insights into the roles of the Mms21 subunit of the Smc5/6 complex. Mol. Cell 35, 657–668 10.1016/j.molcel.2009.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Palecek J., Vidot S., Feng M., Doherty A. J., and Lehmann A. R. (2006) The Smc5-Smc6 DNA repair complex: bridging of the Smc5-Smc6 heads by the KLEISIN, Nse4, and non-Kleisin subunits. J. Biol. Chem. 281, 36952–36959 10.1074/jbc.M608004200 [DOI] [PubMed] [Google Scholar]
- 366. Turner J. M. A. (2007) Meiotic sex chromosome inactivation. Development 134, 1823–1831 10.1242/dev.000018 [DOI] [PubMed] [Google Scholar]
- 367. Hwang G., Verver D. E., Handel M. A., Hamer G., and Jordan P. W. (2018) Depletion of SMC5/6 sensitizes male germ cells to DNA damage. Mol. Biol. Cell 29, 3003–3016 10.1091/mbc.E18-07-0459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Kaul S. C., Deocaris C. C., and Wadhwa R. (2007) Three faces of mortalin: a housekeeper, guardian and killer. Exp. Gerontol. 42, 263–274 10.1016/j.exger.2006.10.020 [DOI] [PubMed] [Google Scholar]
- 369. Van Der Crabben S. N., Hennus M. P., McGregor G. A., Ritter D. I., Nagamani S. C. S., Wells O. S., Harakalova M., Chinn I. K., Alt A., Vondrova L., Hochstenbach R., Van Montfrans J. M., Terheggen-Lagro S. W., Van Lieshout S., Van Roosmalen M. J., et al. (2016) Destabilized SMC5/6 complex leads to chromosome breakage syndrome with severe lung disease. J. Clin. Invest. 126, 2881–2892 10.1172/JCI82890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Guerineau M., Kriz Z., Kozakova L., Bednarova K., Janos P., and Palecek J. (2012) Analysis of the Nse3/MAGE-binding domain of the Nse4/EID family proteins. PLoS ONE 7, e35813 10.1371/journal.pone.0035813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Payne F., Colnaghi R., Rocha N., Seth A., Harris J., Carpenter G., Bottomley W. E., Wheeler E., Wong S., Saudek V., Savage D., O'Rahilly S., Carel J.-C., Barroso I., O'Driscoll M., et al. (2014) Hypomorphism in human NSMCE2 linked to primordial dwarfism and insulin resistance. J. Clin. Invest. 124, 4028–4038 10.1172/JCI73264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Fon Tacer K., and Potts P. R. (2017) Cellular and disease functions of the Prader–Willi syndrome gene MAGEL2. Biochem. J. 474, 2177–2190 10.1042/BCJ20160616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Matarazzo V., and Muscatelli F. (2013) Natural breaking of the maternal silence at the mouse and human imprinted Prader-Willi locus: a whisper with functional consequences. Rare Dis. 1, e27228 10.4161/rdis.27228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Holm V. A., Cassidy S. B., Butler M. G., Hanchett J. M., Greenswag L. R., Whitman B. Y., and Greenberg F. (1993) Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 91, 398–402 [PMC free article] [PubMed] [Google Scholar]
- 375. Cassidy S. B., and Driscoll D. J. (2009) Prader-Willi syndrome. Eur. J. Hum. Genet. 17, 3–13 10.1038/ejhg.2008.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Fountain M. D., Aten E., Cho M. T., Juusola J., Walkiewicz M. A., Ray J. W., Xia F., Yang Y., Graham B. H., Bacino C. A., Potocki L., van Haeringen A., Ruivenkamp C. A. L., Mancias P., Northrup H., et al. (2017) The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet. Med. 19, 45–52 10.1038/gim.2016.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Greaves N., Prince E., Evans D. W., and Charman T. (2006) Repetitive and ritualistic behaviour in children with Prader-Willi syndrome and children with autism. J. Intellect. Disabil. Res. 50, 92–100 10.1111/j.1365-2788.2005.00726.x [DOI] [PubMed] [Google Scholar]
- 378. McCarthy J., Lupo P. J., Kovar E., Rech M., Bostwick B., Scott D., Kraft K., Roscioli T., Charrow J., Schrier Vergano S. A., Lose E., Smiegel R., Lacassie Y., and Schaaf C. P. (2018) Schaaf-Yang syndrome overview: report of 78 individuals. Am. J. Med. Genet. A 176, 2564–2574 10.1002/ajmg.a.40650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Mejlachowicz D., Nolent F., Maluenda J., Ranjatoelina-Randrianaivo H., Giuliano F., Gut I., Sternberg D., Laquerrière A., and Melki J. (2015) Truncating mutations of MAGEL2, a gene within the Prader-Willi locus, are responsible for severe arthrogryposis. Am. J. Hum. Genet. 97, 616–620 10.1016/j.ajhg.2015.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Bennett J. A., Germani T., Haqq A. M., and Zwaigenbaum L. (2015) Autism spectrum disorder in Prader-Willi syndrome: a systematic review. Am. J. Med. Genet. A 167, 2936–2944 10.1002/ajmg.a.37286 [DOI] [PubMed] [Google Scholar]
- 381. Jobling R., Stavropoulos D. J., Marshall C. R., Cytrynbaum C., Axford M. M., Londero V., Moalem S., Orr J., Rossignol F., Lopes F. D., Gauthier J., Alos N., Rupps R., McKinnon M., Adam S., et al. (2018) Chitayat-Hall and Schaaf-Yang Syndromes: a common aetiology: expanding the phenotype of MAGEL2-related disorders. J. Med. Genet. 55, 316–321 10.1136/jmedgenet-2017-105222 [DOI] [PubMed] [Google Scholar]
- 382. Urreizti R., Cueto-Gonzalez A. M., Franco-Valls H., Mort-Farre S., Roca-Ayats N., Ponomarenko J., Cozzuto L., Company C., Bosio M., Ossowski S., Montfort M., Hecht J., Tizzano E. F., Cormand B., Vilageliu L., et al. (2017) A de novo nonsense mutation in MAGEL2 in a patient initially diagnosed as Opitz-C: similarities between Schaaf-Yang and Opitz-C syndromes. Sci. Rep. 7, 44138–44138 10.1038/srep44138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Ropers F., Derivery E., Hu H., Garshasbi M., Karbasiyan M., Herold M., Nürnberg G., Ullmann R., Gautreau A., Sperling K., Varon R., and Rajab A. (2011) Identification of a novel candidate gene for non-syndromic autosomal recessive intellectual disability: the wash complex member swip. Hum. Mol. Genet. 20, 2585–2590 10.1093/hmg/ddr158 [DOI] [PubMed] [Google Scholar]
- 384. Jia D., Gomez T. S., Metlagel Z., Umetani J., Otwinowski Z., Rosen M. K., and Billadeau D. D. (2010) WASH and WAVE actin regulators of the Wiskott-Aldrich syndrome protein (WASP) family are controlled by analogous structurally related complexes. Proc. Natl. Acad. Sci. U. S. A. 107, 10442–10447 10.1073/pnas.0913293107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Hidalgo-Santos A. D., DeMingo-Alemany M. D C., Moreno-Macián F., Roselló M., Orellana C., Martínez F., Caro-Llopis A., León-Cariñena S., and Tomás-Vila M. (2018) A novel mutation of MAGEL2 in a patient with Schaaf-Yang syndrome and hypopituitarism. Int. J. Endocrinol. Metab. 16, e67329 10.5812/ijem.67329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Mercer R. E., and Wevrick R. (2009) Loss of magel2, a candidate gene for features of Prader-Willi syndrome, impairs reproductive function in mice. PLoS ONE 4, e4291 10.1371/journal.pone.0004291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Tennese A. A., and Wevrick R. (2011) Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2-null mice. Endocrinology 152, 967–978 10.1210/en.2010-0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Devos J., Weselake S. V., and Wevrick R. (2011) Magel2, a Prader-Willi syndrome candidate gene, modulates the activities of circadian rhythm proteins in cultured cells. J. Circadian Rhythms 9, 12 10.1186/1740-3391-9-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Luck C., Vitaterna M. H., and Wevrick R. (2016) Dopamine pathway imbalance in mice lacking Magel2, a Prader-Willi syndrome candidate gene. Behav. Neurosci. 130, 448–459 10.1037/bne0000150 [DOI] [PubMed] [Google Scholar]
- 390. Schaller F., Watrin F., Sturny R., Massacrier A., Szepetowski P., and Muscatelli F. (2010) A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 19, 4895–4905 10.1093/hmg/ddq424 [DOI] [PubMed] [Google Scholar]
- 391. Meziane H., Schaller F., Bauer S., Villard C., Matarazzo V., Riet F., Guillon G., Lafitte D., Desarmenien M. G., Tauber M., and Muscatelli F. (2015) An early postnatal oxytocin treatment prevents social and learning deficits in adult mice deficient for Magel2, a gene involved in Prader-Willi syndrome and autism. Biol. Psychiatry 78, 85–94 10.1016/j.biopsych.2014.11.010 [DOI] [PubMed] [Google Scholar]
- 392. Caswell P., and Biology J. N. (2008) Endocytic transport of integrins during cell migration and invasion. Trends Cell Biol. 18, 257–263 10.1016/j.tcb.2008.03.004 [DOI] [PubMed] [Google Scholar]
- 393. Goldenring J. R. (2015) Recycling endosomes. Curr. Opin. Cell Biol. 35, 117–122 10.1016/j.ceb.2015.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Hsu V. W., Bai M., and Li J. (2012) Getting active: protein sorting in endocytic recycling. Nat. Rev. Mol. Cell Biol. 13, 323–328 10.1038/nrm3332 [DOI] [PubMed] [Google Scholar]
- 395. Ivaska J., and Heino J. (2011) Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 27, 291–320 10.1146/annurev-cellbio-092910-154017 [DOI] [PubMed] [Google Scholar]
- 396. Maxfield F. R., and McGraw T. E. (2004) Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5, 121–132 10.1038/nrm1315 [DOI] [PubMed] [Google Scholar]
- 397. Mellman I., and Nelson W. J. (2008) Coordinated protein sorting, targeting and distribution in polarized cells. Nat. Rev. Mol. Cell Biol. 9, 833–845 10.1038/nrm2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Sorkin A., and von Zastrow M. (2009) Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 10, 609–622 10.1038/nrm2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Ghosh P., Dahms N. M., and Kornfield S. (2003) Mannose 6-phosphate receptors: new twists in the tale. Nat. Rev. Mol. Cell Biol. 4, 201–212 10.1038/nrm1050 [DOI] [PubMed] [Google Scholar]
- 400. Molloy S. S., Anderson E. D., Jean F., and Thomas G. (1999) Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 9, 28–35 10.1016/s0962-8924(98)01382-8 [DOI] [PubMed] [Google Scholar]
- 401. Hettema E. H., Lewis M. J., Black M. W., and Pelham H. R. B. (2003) Retromer and the sorting nexins Snx4/41/42 mediate distinct retrieval pathways from yeast endosomes. EMBO J. 22, 548–557 10.1093/emboj/cdg062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Lewis M. J., Nichols B. J., Prescianotto-Baschong C., Riezman H., and Pelham H. R. B. (2000) Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol. Biol. Cell 11, 23–38 10.1091/mbc.11.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Gomez T. S., and Billadeau D. D. (2009) A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev. Cell 17, 699–711 10.1016/j.devcel.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Leung D. W., and Rosen M. K. (2005) The nucleotide switch in Cdc42 modulates coupling between the GTPase-binding and allosteric equilibria of Wiskott-Aldrich syndrome protein. Proc. Natl. Acad. Sci. U. S. A. 102, 5685–5690 10.1073/pnas.0406472102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Verboon J. M., Rahe T. K., Rodriguez-Mesa E., and Parkhurst S. M. (2015) Wash functions downstream of Rho1 GTPase in a subset of Drosophila immune cell developmental migrations. Mol. Biol. Cell 26, 1665–1674 10.1091/mbc.E14-08-1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Liu J., Sun Y., Drubin D. G., and Oster G. F. (2009) The mechanochemistry of endocytosis. PLoS Biol. 7, e1000204 10.1371/journal.pbio.1000204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Seaman M. N. J. (2012) The retromer complex—endosomal protein recycling and beyond. J. Cell Sci. 125, 4693–4702 10.1242/jcs.103440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Seaman M. N. J., Gautreau A., and Billadeau D. D. (2013) Retromer-mediated endosomal protein sorting: all WASHed up! Trends Cell Biol. 23, 522–528 10.1016/j.tcb.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Jia D., Gomez T. S., Billadeau D. D., and Rosen M. K. (2012) Multiple repeat elements within the FAM21 tail link the WASH actin regulatory complex to the retromer. Mol. Biol. Cell 23, 2352–2361 10.1091/mbc.E11-12-1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Hill J. W., Williams K. W., Ye C., Luo J., Balthasar N., Coppari R., Cowley M. A., Cantley L. C., Lowell B. B., and Elmquist J. K. (2008) Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Invest. 118, 1796–1805 10.1172/JCI32964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Wauman J., De Smet A.-S., Catteeuw D., Belsham D., and Tavernier J. (2008) Insulin receptor substrate 4 couples the leptin receptor to multiple signaling pathways. Mol. Endocrinol. 22, 965–977 10.1210/me.2007-0414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. De Ceuninck L., Wauman J., Masschaele D., Peelman F., and Tavernier J. (2013) Reciprocal cross-regulation between RNF41 and USP8 controls cytokine receptor sorting and processing. J. Cell Sci. 126, 3770–3781 10.1242/jcs.131250 [DOI] [PubMed] [Google Scholar]
- 413. Carias K. V., Zoeteman M., Seewald A., Sanderson M. R., Bischof J. M., and Wevrick R. (2020) A MAGEL2-deubiquitinase complex modulates the ubiquitination of circadian rhythm protein CRY1. PLoS ONE 15, e0230874 10.1371/journal.pone.0230874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Dunwell T. L., Paps J., and Holland P. W. H. (2017) Novel and divergent genes in the evolution of placental mammals. Proc. Biol. Sci. 284, 20171357 10.1098/rspb.2017.1357 [DOI] [PMC free article] [PubMed] [Google Scholar]