

Abstract
Malignant melanoma, the most aggressive form of skin cancer, is characterized by high prevalence of BRAF/NRAS mutations and hyperactivation of extracellular signal-regulated kinase 1 and 2 (ERK1/2), mitogen-activated protein kinases (MAPK), leading to uncontrolled melanoma growth. Efficacy of current targeted therapies against mutant BRAF or MEK1/2 have been hindered by existence of innate or development of acquired resistance. Therefore, a better understanding of the mechanisms controlled by MAPK pathway driving melanogenesis will help develop new treatment approaches targeting this oncogenic cascade. Here, we identify E3 ubiquitin ligase PARK2 as a direct target of ELK1, a known transcriptional effector of MAPK signaling in melanoma cells. We show that pharmacological inhibition of BRAF-V600E or ERK1/2 in melanoma cells increases PARK2 expression. PARK2 overexpression reduces melanoma cell growth in vitro and in vivo and induces apoptosis. Conversely, its genetic silencing increases melanoma cell proliferation and reduces cell death. Further, we demonstrate that ELK1 is required by the BRAF-ERK1/2 pathway to repress PARK2 expression and promoter activity in melanoma cells. Clinically, PARK2 is highly expressed in WT BRAF and NRAS melanomas, but it is expressed at low levels in melanomas carrying BRAF/NRAS mutations. Overall, our data provide new insights into the tumor suppressive role of PARK2 in malignant melanoma and uncover a novel mechanism for the negative regulation of PARK2 via the ERK1/2-ELK1 axis. These findings suggest that reactivation of PARK2 may be a promising therapeutic approach to counteract melanoma growth.
Keywords: melanoma, mitogen-activated protein kinase, MAPK, gene regulation, chromatin immunoprecipitation, ChIP, parkin, extracellular signal-regulated kinase, ERK, transcription factor, tumor suppressor gene
Malignant melanoma is the most aggressive form of skin cancer. The most prevalent genetic alterations in melanoma are mutually exclusive mutations in BRAF and NRAS, which occur in nearly 50 and 25% of melanoma patients, respectively (1). These mutations result in hyperactivation of the mitogen-activated protein kinases (MAPK) extracellular signal-regulated kinase 1 and 2 (ERK1/2), and consequent uncontrolled melanoma growth. These terminal kinases of the cascade catalyze the phosphorylation, mainly at Ser/Thr-Pro residues, of hundreds of cytoplasmic and nuclear substrates, including regulatory molecules and transcription factors (2). Among the ERK1/2 nuclear targets is the transcription factor ELK1, a member of ETS (E 26) oncogene family of transcription factors, which is directly phosphorylated by ERK1/2 on multi-sites in its transactivation domain (3–5). The aberrant activation of the RAS-RAF-MEK1/2-ERK1/2-ELK1 signaling pathway has provided the basis for efficient targeted therapy with specific inhibitors of mutant BRAF and MEK in melanoma. However, the presence of innate and development of acquired resistance have hindered the long-term clinical benefits of these treatments. Therefore, a better understanding of the mechanisms controlled by MAPK signaling will help develop efficient treatment approaches targeting this pathway in dismal skin cancer.
Here, we identified PARK2 as a novel target of the oncogenic ERK1/2-ELK1 pathway, and we provided insights into the role of PARK2 in melanoma. The E3 ubiquitin ligase PARK2 has been shown to act as a tumor suppressor in several contexts (6). PARK2 loss of heterozygosity and copy number loss have been observed in human cancers, including melanoma (7). In addition, PARK2 inactivating mutations are associated with increased risk of melanoma (8). Consistent with its tumor suppressive role, PARK2 ectopic expression has been shown to reduce cell proliferation in several types of cancer (9–18). Our findings define a novel mechanism through which the MAPK pathway controls melanoma cell growth through the suppression of PARK2 in an ELK1-dependent manner, and thus will contribute develop new treatment approaches targeting this oncogenic cascade.
Results
PARK2 is repressed by the RAS-RAF-MEK1/2-ERK1/2 signaling in melanoma cells
Western blotting (WB) and quantitative real-time PCR (qPCR) analyses in melanoma cell lines showed that PARK2 expression is lower in cells harboring BRAFV600E or NRASQ61R mutations (SK-Mel-2, SK-Mel-5, SK-Mel-28, A375, 501-Mel) compared with those with WT BRAF or NRAS (M51, SSM2c, SK-Mel-197). Only SK-Mel-197 cells express high levels of pERK1/2 and low levels of PARK2, although they do not carry mutations in BRAF or NRAS (Fig. 1, A and B). Further analysis showed reduced PARK2 mRNA expression in melanoma cell lines compared with normal human epidermal melanocytes (NHEM) (Fig. 1B). In support of the biological relevance of this finding, analysis of The Cancer Genome Atlas (TCGA) melanoma cohort shows higher PARK2 expression in WT compared with mutant BRAS/NRAS metastatic melanomas (p = 0.0027) (Fig. 1C). In agreement with these data, analysis of publicly available transcriptomic datasets (GDS1375) showed that PARK2 mRNA was expressed at higher level in human nevi (n = 18) compared with malignant melanomas (n = 45) (p < 0.05) (Fig. 1D). In addition, there is a trend toward improved overall survival with increased PARK2 expression in metastatic disease, although not statistically significant (Fig. S1). At the cellular level, Western blot analysis indicated a cytosolic localization of PARK2 in melanoma cells (Fig. 1E). Immunofluorescence confirmed these data and showed a colocalization of PARK2 with the protein COXIV, a mitochondrial marker (Fig. 1F). Altogether, our data indicate that PARK2 expression is down-regulated in human melanomas compared with nevi and that metastatic melanomas carrying WT BRAF/NRAS show higher expression of PARK2 compared with those with mutant BRAF/NRAS.
Figure 1.
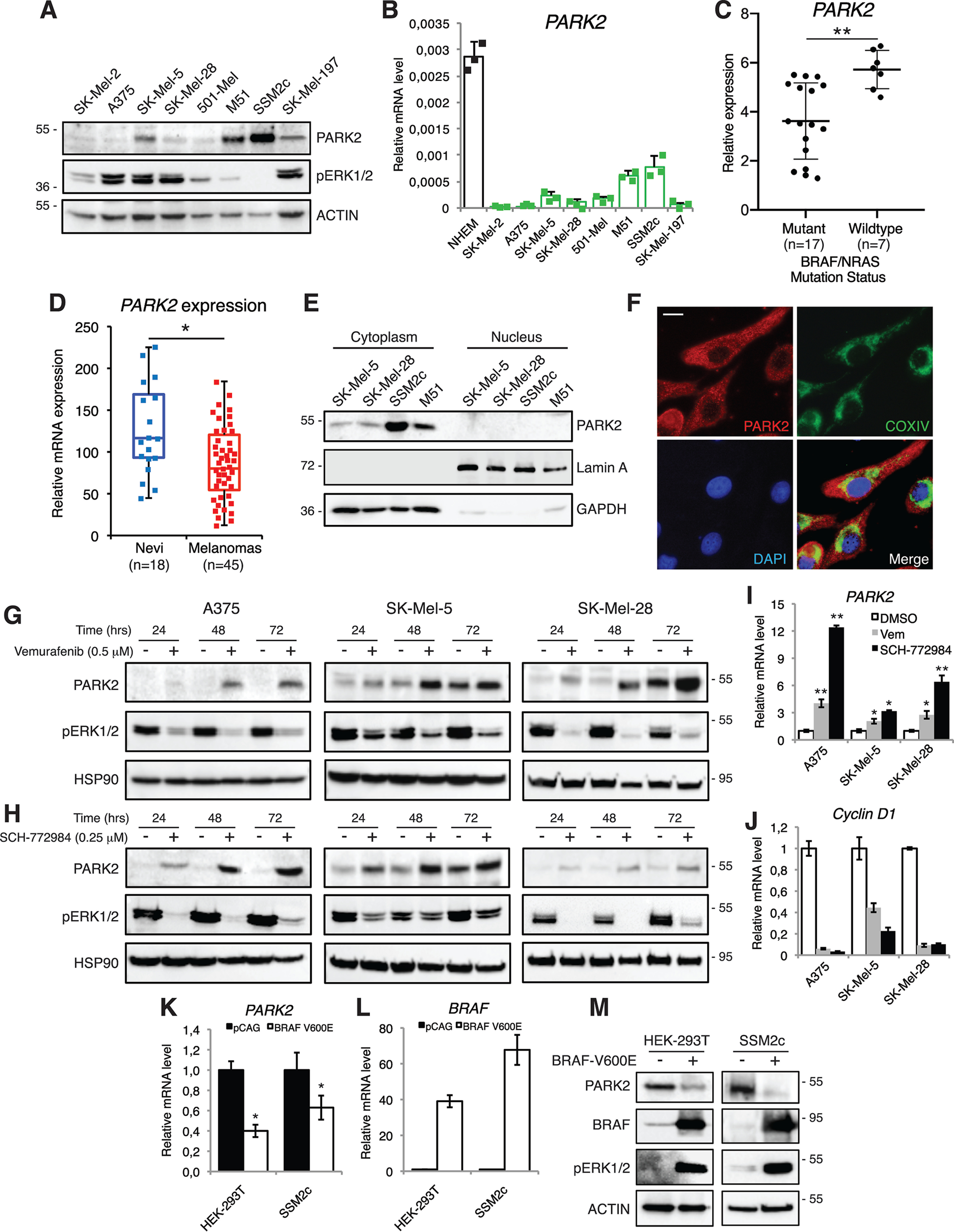
PARK2 is negatively modulated by the RAS-RAF-MEK1/2-ERK1/2 signaling in melanoma. A, Western blot analysis of cytosolic PARK2 and pERK1/2 in a panel of human melanoma cell lines. ACTIN was used as loading control. B, qPCR of PARK2 in a panel of human melanoma cell lines and in NHEM. The y axis represents expression ratio of gene/(GAPDH and β-ACTIN average). C, analysis of PARK2 expression by RNA-sequencing in mutant (n = 17) and WT (n = 7) BRAF/NRAS metastatic melanomas. D, expression of PARK2 mRNA in human nevi and in malignant melanoma samples, obtained from the analysis of the microarray dataset GDS1375. Median is represented as a line inside the box. Lines at the top and the bottom of the box represent, respectively, the 25th and 75th quartile, and lines above and below the box show the minimum and maximum. E, subcellular localization of endogenous PARK2 in four melanoma cell lines. Cell fractionation was performed and lysates were subjected to WB with anti-PARK2, anti-GAPDH (control for cytoplasmic proteins), and anti-Lamin A (control for nuclear proteins) antibodies. F, representative images of endogenous PARK2 in SK-Mel-5 melanoma cells. COXIV was used as a marker for mitochondria. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Scale bar = 10 μm. G and H, WB of PARK2 and pERK1/2 in A375, SK-Mel-5, and SK-Mel-28 melanoma cells treated with vemurafenib or SCH-772984 for the time indicated. HSP90 was used as loading control. I and J, expression of PARK2 (I) and Cyclin D1 (J) mRNA by qPCR after treatment with vemurafenib (Vem) or SCH-772984 in A375, SK-Mel-5, and SK-Mel-28 melanoma cells. The y axis represents expression ratio of gene/(GAPDH and β-ACTIN average), with the level of control equated to 1. K and L, expression of PARK2 (K) and BRAF (L) mRNA by qPCR in HEK-293T and SSM2c cells transiently transfected with BRAF-V600E. The y axis represents expression ratio of gene/(GAPDH and β-ACTIN average), with the level of control equated to 1. M, Western blot analysis of PARK2, BRAF, and pERK1/2 in HEK-293T and SSM2c cells transiently transfected with BRAF-V600E. ACTIN was used as loading control. Data shown are mean ± S.D. (B) or ± S.E. (I–L) of at least three biological replicates, each performed in triplicate. *, p < 0.05; **, p < 0.01. P values were calculated with two-tailed unpaired t test.
To investigate the effect of the RAS-RAF-MEK1/2-ERK1/2 signaling on PARK2 expression, melanoma cells harboring BRAFV600E (A375, SK-Mel-5, SK-Mel-28, and 501-Mel) were treated with specific inhibitors of the BRAF-MEK1/2-ERK1/2 cascade. Treatment with the BRAF-V600E inhibitor vemurafenib (19, 20) led to a time-dependent increase in PARK2 protein levels (Fig. 1G and Fig. S2A). Likewise, treatment with SCH-772984, an ERK1/2 inhibitor (21), consistently increased PARK2 protein levels in all four melanoma cell lines (Fig. 1H and Fig. S2B). To further clarify whether modulation of PARK2 by the BRAF-MEK1/2-ERK1/2 signaling was exerted also at transcriptional level, qPCR analysis of PARK2 mRNA was performed after inhibition of BRAF-V600E or ERK1/2. Expression of PARK2 mRNA was drastically increased upon treatment with vemurafenib or SCH-772984 (Fig. 1I). The efficiency of these inhibitors was confirmed by strong down-regulation of phosphorylated ERK1/2 level (Fig. 1, G and H and Fig. S2) and of Cyclin D1 (Fig. 1J), an established mitogenic target of mutant RAS signaling. Consistent with these results, transient overexpression of BRAF-V600E in SSM2c melanoma cells and in HEK-293T cells, which harbor WT BRAF and NRAS, led to a reduction of PARK2 both at mRNA and protein levels (Fig. 1, K–M). Furthermore, qPCR analysis in HEK-293T cells treated with EGF, which induces phosphorylation of ERK1/2, shows an increase in Cyclin D1 and c-Fos levels (Fig. S3, A and B) and a time-dependent decrease of PARK2 level (Fig. S3C). Altogether, these data indicate that in melanoma cells the RAS-RAF-MEK1/2-ERK1/2 pathway negatively regulates expression of PARK2.
The transcription factor ELK1 is required by RAS-RAF-MEK1/2-ERK1/2 pathway to repress PARK2 expression
Bioinformatic analysis identified putative ELK1 binding sites (BS) within the human PARK2 promoter (obtained from the UCSC Genome Browser assembly, ID: hg38) near the transcription start site (TSS) (Fig. 2A and Fig. S4). ELK1, a major downstream effector of ERK1/2, is a member of the ETS (E 26) oncogene family of transcription factors involved in many biological processes, such as cell growth, differentiation and survival, inflammation, and cancer (3–5, 22–24). Therefore, we investigated whether ELK1 might be a mediator of the RAS-RAF-MEK1/2-ERK1/2 cascade in modulating PARK2 expression. Treatment of A375 and SK-Mel-5 melanoma cells with the ERK1/2 inhibitor SCH-772984 drastically reduced ELK1 phosphorylation at serine 383 in both cell lines (Fig. 2B). To investigate whether ELK1 directly binds PARK2 promoter, we performed ChIP assay using two different ELK1-specific antibodies. Analysis of the immunoprecipitated DNA by qPCR showed that endogenous ELK1 binds to PARK2 promoter (Fig. 2C). This result was confirmed also upon ELK1 overexpression in HEK-293T cells (Fig. S5A) and upon ELK1 silencing in A375 melanoma cells (Fig. S5B). To confirm the ability of ELK1 to regulate PARK2 expression, the PARK2 promoter (−635 to +98 from the TSS) containing the putative ELK1 BS (CCGGAAA) was cloned into a luciferase reporter. Ectopic expression of ELK1 in A375 and SK-Mel-197 melanoma cells showed a strong reduction of luciferase activity (Fig. 2D). Mutation of the ELK1-binding element strongly reduces the inhibition of PARK2 transactivation by ELK1 overexpression in both cell lines (Fig. 2D). Consistent with the negative regulation of PARK2 by ELK1, silencing of ELK1 with two specific independent shRNAs (LV-shELK1-1, LV-shELK1-2) increased PARK2 mRNA and protein levels in A375 and SK-Mel-5 melanoma cells (Fig. S5, C–F), whereas overexpression of ELK1 in HEK-293T cells had the opposite effect (Fig. S5, G and H). In support of the relevance of the negative regulation of PARK2 expression by ELK1, a statistically significant negative correlation was found between ELK1 and PARK2 expression in the TCGA melanoma cohort (n = 481) (Fig. 2E).
Figure 2.
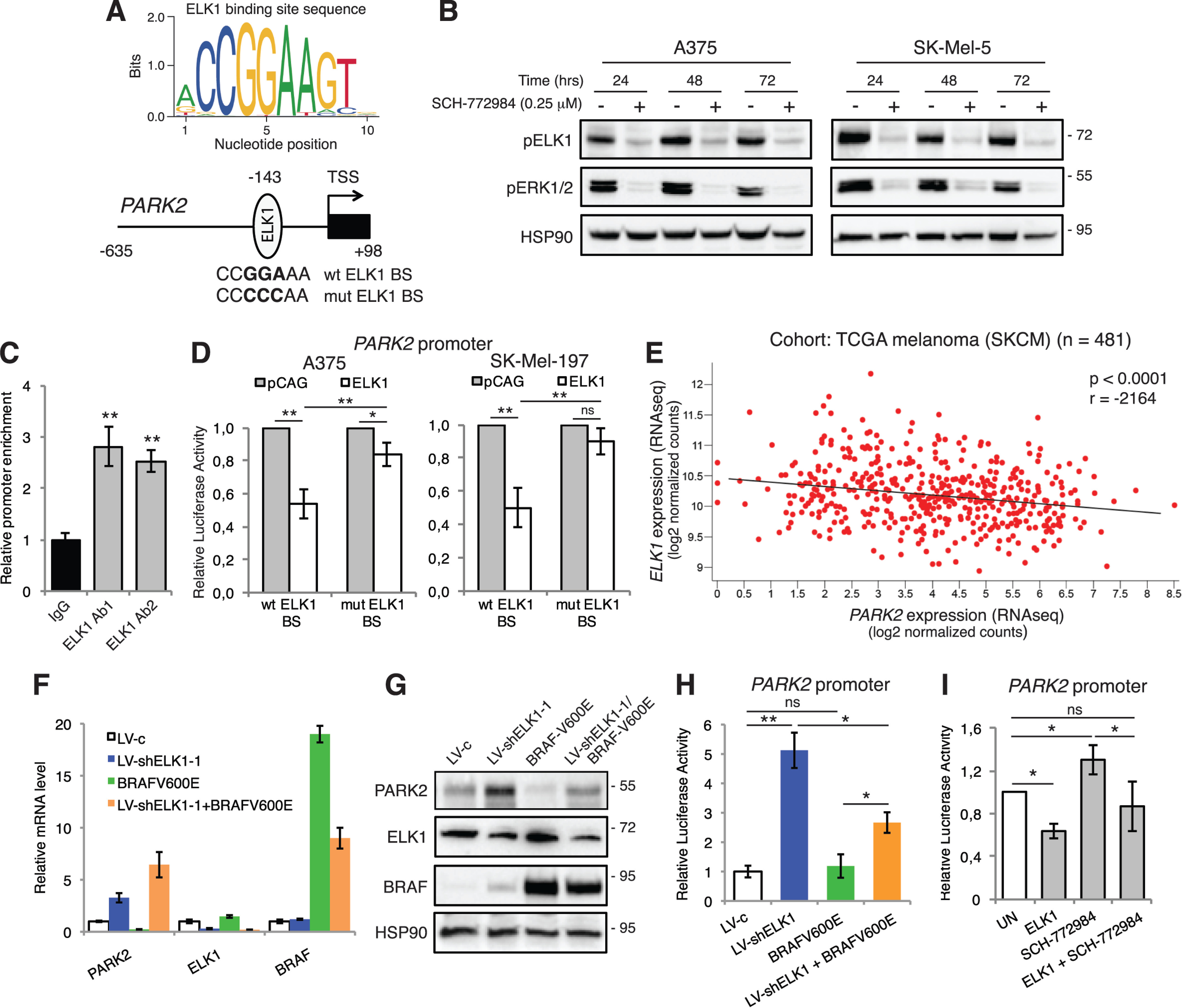
The transcription factor ELK1 is required by RAS-RAF-MEK1/2-ERK1/2 signaling to repress PARK2 expression. A, consensus ELK1 DNA-binding motif calculated using WebLogo3. Position of ELK1 DNA BS in the −635/+98 PARK2 promoter. Both WT (CCGGAAA) and mutagenized (mut, CCCCCAA) ELK1 BS are shown. B, Western blot analysis of pELK1 and pERK1/2 in A375 and SK-Mel-5 melanoma cells treated with SCH-772984 for the time indicated. HSP90 was used as loading control. C, ChIP assay showing that endogenous ELK1 binds to PARK2 promoter in HEK-293T cells. Two different specific anti-ELK1 antibodies were used. The y axis represents the relative promoter enrichment, normalized on the input material. IgG was used as negative control and set to 1. D, quantification of dual reporter luciferase assay in A375 and SK-Mel-197 cells transfected with the empty vector pCAG or ELK1 on PARK2 promoter carrying WT or mut ELK1 BS. Relative luciferase activities were firefly/Renilla ratios, with the level induced by control equated to 1. E, correlation between the expression of PARK2 and ELK1 using the TCGA melanoma cohort (n = 481). Pearson's correlation test. F, expression of PARK2, ELK1, and BRAF mRNA by qPCR in SK-Mel-28 melanoma cells transduced with LV-shELK1-1 and transiently transfected with BRAF-V600E. The y axis represents expression ratio of gene/(GAPDH and β-ACTIN average), with the level of control equated to 1. G, Western blot analysis of PARK2, ELK1, and BRAF in SK-Mel-28 cells transduced with LV-shELK1-1 and transiently transfected with BRAF-V600E. HSP90 was used as loading control. H, quantification of dual reporter luciferase assay in SK-Mel-28 cells showing that ELK1 silencing increases the transactivation of PARK2 promoter and partially prevents the inhibition by BRAF-V600E. Relative luciferase activities were firefly/Renilla ratios, with the level induced by control equated to 1. I, quantification of dual reporter luciferase assay in SK-Mel-28 cells showing that ELK1 decreases the transactivation of PARK2 promoter, whereas SCH-772984 treatment prevents the reduction of transactivation by ELK1. Relative luciferase activities were firefly/Renilla ratios, with the level induced by control equated to 1. Data shown are mean ± S.D. (C, D, H, I) or ± S.E. (F) of at least three biological replicates, each performed in triplicate. *, p < 0.05; **, p < 0.01.
To confirm the involvement of ELK1 downstream of MEK1/2-ERK1/2 in regulating PARK2 expression, we overexpressed BRAF-V600E in ELK1-silenced cells. ELK1 depletion enhanced PARK2 mRNA and protein levels, whereas BRAF-V600E overexpression reduced PARK2 expression, as expected. Interestingly, silencing of ELK1 in presence of BRAF-V600E overexpression rescued the reduction of PARK2 expression elicited by BRAFV-600E (Fig. 2, F and G). Consistent with these results, genetic silencing of ELK1 strongly increased PARK2 promoter activity, even when it was co-expressed with BRAF-V600E (Fig. 2H). In addition, PARK2 transactivation induced by the ERK1/2 inhibitor SCH-772984 was reverted by ELK1 overexpression (Fig. 2I). The negative modulation of PARK2 by ELK1 was also confirmed in glioblastoma (U87MG) and breast cancer (MCF7) cell lines. Indeed, in both cell types, PARK2 mRNA and protein levels were decreased upon ELK1 expression (Fig. S6). Altogether, our data indicate that in melanoma cells PARK2 expression is negatively regulated by the RAS-RAF-MEK1/2-ERK1/2 signaling through the transcription factor ELK1, a new repressor of PARK2 transcription. Further, our results suggest that the regulation of PARK2 by ELK1 may take place in other cancer types beyond melanoma, and it might be a general mechanism to restrain PARK2 function in cancer cells.
PARK2 reduces melanoma growth in vitro and in vivo
To evaluate the effect of restoring PARK2 expression in melanoma, we overexpressed it in four melanoma cell lines having low level of PARK2 (A375, SK-Mel-5, SK-Mel-28, and 501-Mel) using a retroviral vector encoding full-length PARK2. Stable overexpression of PARK2, confirmed at protein level (Fig. 3A and Fig. S7A), induced a decrease of melanoma cell growth in all four cell lines (Fig. 3, B and C and Fig. S7, B and C). Consistently, ectopic expression of PARK2 drastically decreased the number of adherent colonies in all four melanoma cell lines (Fig. 3, D and E and Fig. S7, D and E). Next, we tested whether ectopic PARK2 expression might affect the ability to form colonies in soft agar. We found that the number of colonies formed by PARK2-expressing melanoma cells was reduced compared with those in the control melanoma cells in A375 and SK-Mel-5 (Fig. 3, F and G). Further, we demonstrated that the reduced growth of melanoma cells expressing PARK2 was due to an increase in apoptosis. FACS-based Annexin V/7AAD analysis showed an increase of the fraction of apoptotic cells in PARK2 overexpressing cell lines (Fig. 3, H and I and Fig. S7, F and G). Further analysis showed reduced expression of the anti-apoptotic factors BCL-2 and BCL-XL at protein level upon PARK2 overexpression (Fig. 3J and Fig. S7H). qPCR showed also an increase of the pro-apoptotic factor PIG3 in PARK2-overexpressing cells (Fig. 3, K and L). PARK2 has been shown to negatively regulate the AKT pathway, a known survival pathway in melanoma (9, 25). Our data confirmed the reduction of AKT activation (phosphorylation of Ser-473) upon PARK2 overexpression in SK-Mel-5 and, to a lesser extent, in A375 and SK-Mel-28 melanoma cells (Fig. S7I).
Figure 3.
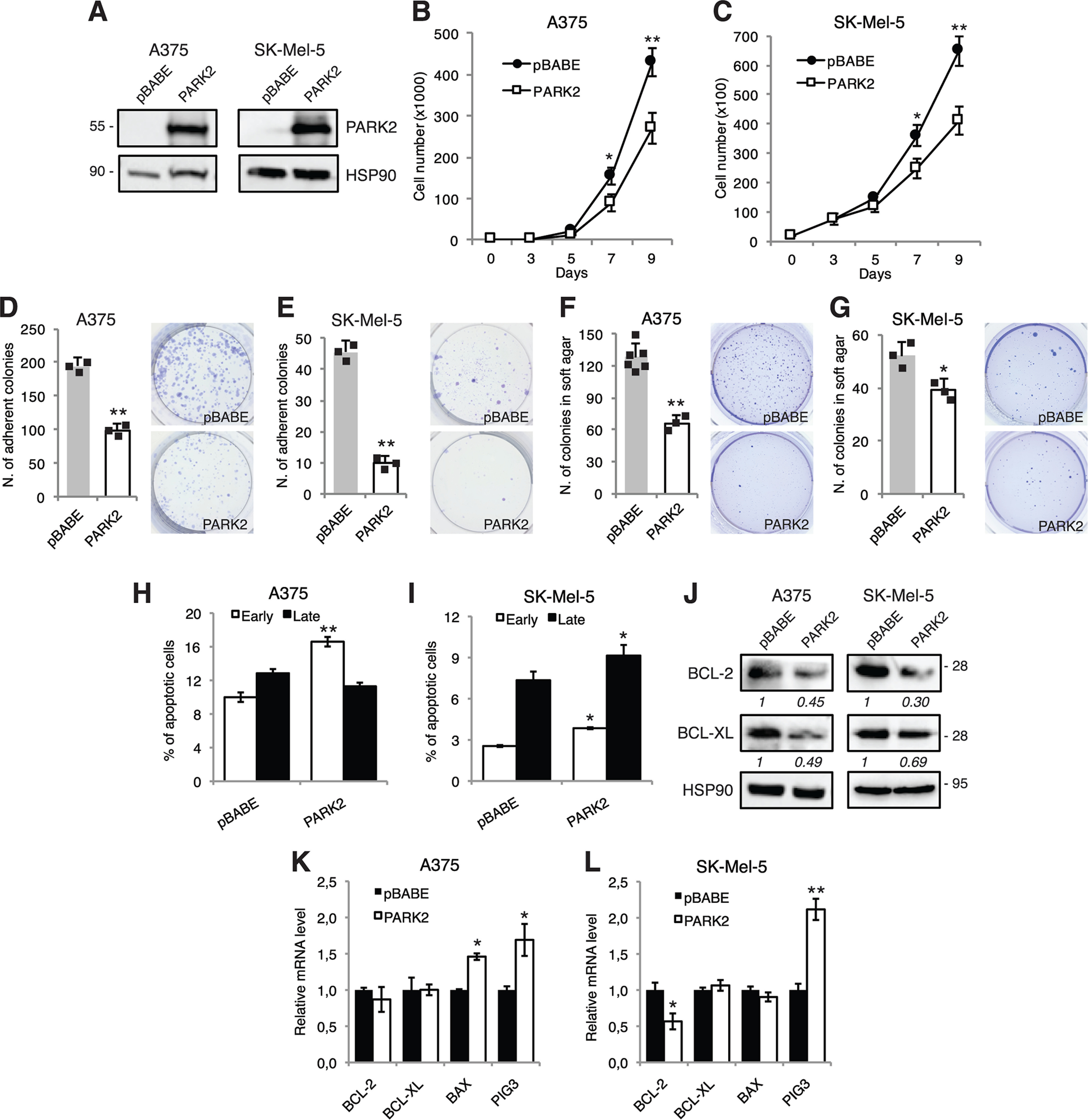
Ectopic expression of PARK2 reduces melanoma cell growth and increases apoptosis. A, Western blot analysis of PARK2 in A375 and SK-Mel-5 cells transduced with pBABE or PARK2. HSP90 was used as loading control. B and C, growth curves in A375 and SK-Mel-5 transduced as indicated. D–G, histogram of the quantification of colony (D and E) and soft agar (F and G) assays in A375 and SK-Mel-5 cells transduced as indicated. H and I, A375 and SK-Mel-5 melanoma cells, transduced as indicated, were subjected to cytometric analysis of apoptotic cells after staining with Annexin V/7-AAD (Annexin V+/7-AAD−: early apoptosis; Annexin V+/7-AAD+: late apoptosis). J, Western blot analysis of BCL-2 and BCL-XL in A375 and SK-Mel-5 transduced as described above. HSP90 was used as loading control. Protein quantifications are indicated in italics. K and L, qPCR analysis of BCL-2, BCL-XL, BAX, and PIG3 in A375 and SK-Mel-5 cells transduced as indicated. The y axis represents expression ratio of gene/(GAPDH and β-ACTIN average), with the control equated to 1. Data shown are mean ± S.D. (B–I) or mean ± S.E. (K and L) of at least three biological replicates, each performed in triplicate. *, p < 0.05; **, p < 0.01.
To further investigate the role of PARK2 in melanoma, PARK2 was silenced in two patient-derived melanoma cell lines (SSM2c and M51), which expressed the highest levels of PARK2, and in a commercial melanoma cell line (SK-Mel-197), which expressed low PARK2 levels, using two different short hairpin RNAs (shRNA) specific for PARK2 (LV-shPARK2-1, LV-shPARK2-2). Western blot analysis showed a strong reduction of PARK2 protein level in cells transduced with both shRNAs (Fig. 4A and Fig. S8A). PARK2 silencing induced an increase of melanoma cell viability over time (Fig. 4, B and C and Fig. S8B) and enhanced colony formation in soft agar in SSM2c and SK-Mel-197 PARK2-depleted cells compared with control, although only LV-shPARK2-1 led to a significant reduction of colony formation in SK-Mel-197 (Fig. 4, D and E). Cytometric analysis of Annexin V/7AAD staining revealed a decrease in the percentage of apoptotic cells in PARK2-silenced cells (Fig. 4, F and G and Fig. S8C). Consistently, Western blot analysis showed that PARK2 silencing increased expression level of the anti-apoptotic factor BCL-2 in all melanoma cell lines (Fig. 4H and Fig. S8D). Moreover, PARK2 silencing led to a consistent increase in the expression of pAKT in SK-Mel-197 and M51 cells (Fig. S8E). Altogether, our data indicate that PARK2 reduces melanoma cell growth promoting apoptosis.
Figure 4.
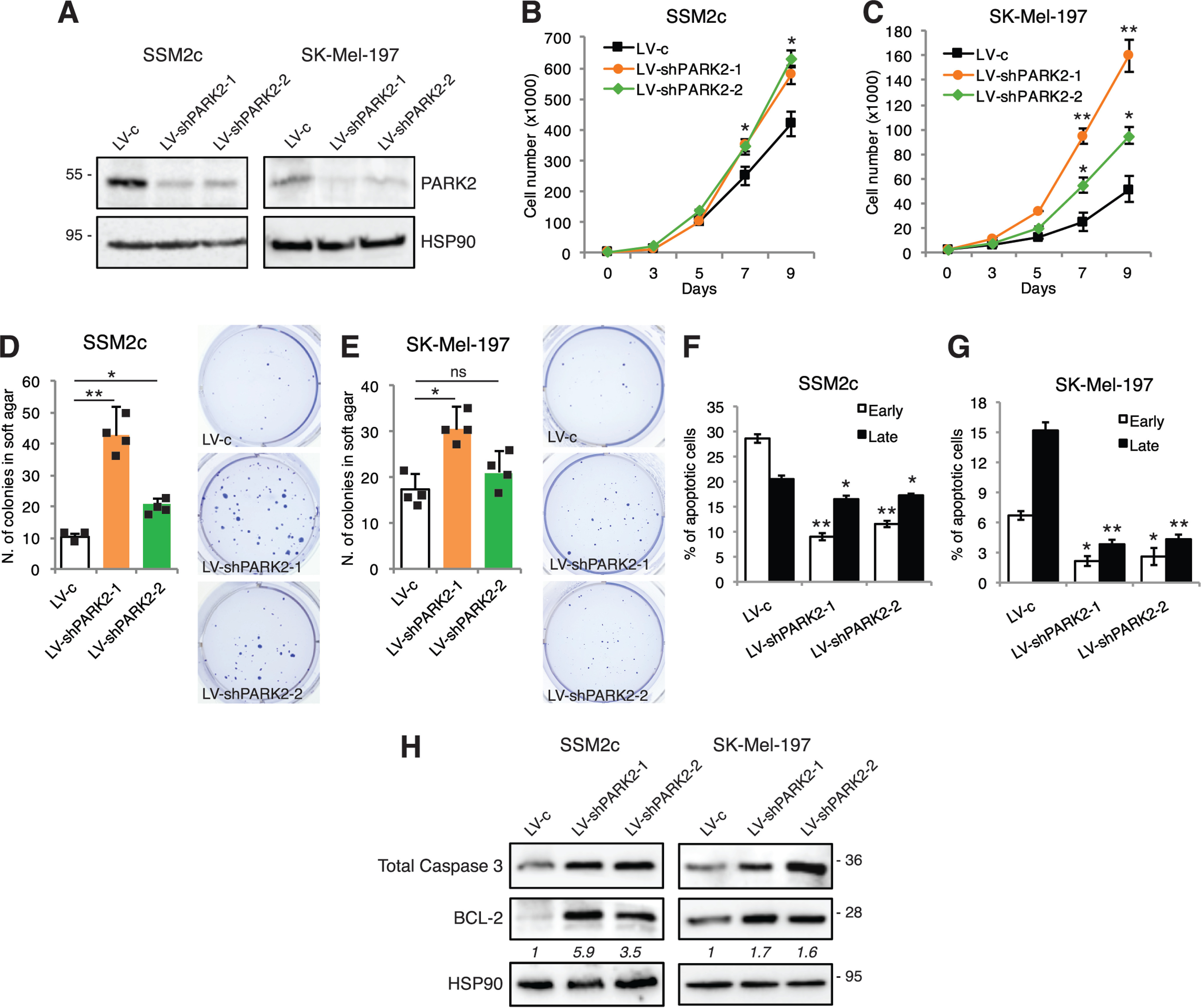
PARK2 silencing increases melanoma cell growth and reduces apoptosis. A, Western blot analysis of PARK2 in SSM2c and SK-Mel-197 cells transduced with LV-c, LV-shPARK2-1, or LV-shPARK2-2. HSP90 was used as loading control. B and C, growth curves in SSM2c and SK-Mel-197 cells transduced as indicated. D and E, histogram of the quantification of colonies in soft agar in SSM2c and SK-Mel-197 cells transduced as indicated. F and G, SSM2c and SK-Mel-197 melanoma cells transduced as indicated, were subjected to cytometric analysis of apoptotic cells after staining with Annexin V/7-AAD (Annexin V+/7-AAD−: early apoptosis; Annexin V+/7-AAD+: late apoptosis). H, Western blot analysis of caspase-3 and BCL-2 in SSM2c and SK-Mel-197 cells transduced as indicated. HSP90 was used as loading control. Protein quantifications are indicated in italics. Data shown are mean ± S.D. of at least three biological replicates, each performed in triplicate. *, p < 0.05; **, p < 0.01.
To investigate whether PARK2 might affect melanoma xenograft growth in vivo, A375 cells stably transduced with pBABE or PARK2 were subcutaneously injected into the flanks of athymic nude mice and tumor growth was monitored over time. Ectopic expression of PARK2 reduced by 60% the size of melanoma xenografts compared with pBABE control (Fig. 5A and B). Western blotting in dissected tumors confirmed PARK2 overexpression with a 20- to 30-fold increase compared with controls (Fig. 5C) and a drastic decrease of BCL-2 protein level (Fig. 5C), consistent with in vitro tumor cell growth experiments. Induction of apoptosis was confirmed at the molecular level in PARK2-overexpressing xenografts by increased BAX/BCL-2 ratio (Fig. 5D), an indicator of apoptosis (26, 27). PARK2 overexpression in xenografts was also confirmed by immunohistochemistry (Fig. 5E). The degree of reduction of melanoma growth upon PARK2 overexpression in vivo was greater than the decrease of melanoma cell growth observed in vitro, suggesting a potential role of the tumor microenvironment. Altogether, these results indicate that PARK2 represses melanoma cell growth in vitro and in vivo, further confirming the tumor suppressive role of PARK2 in melanoma.
Figure 5.
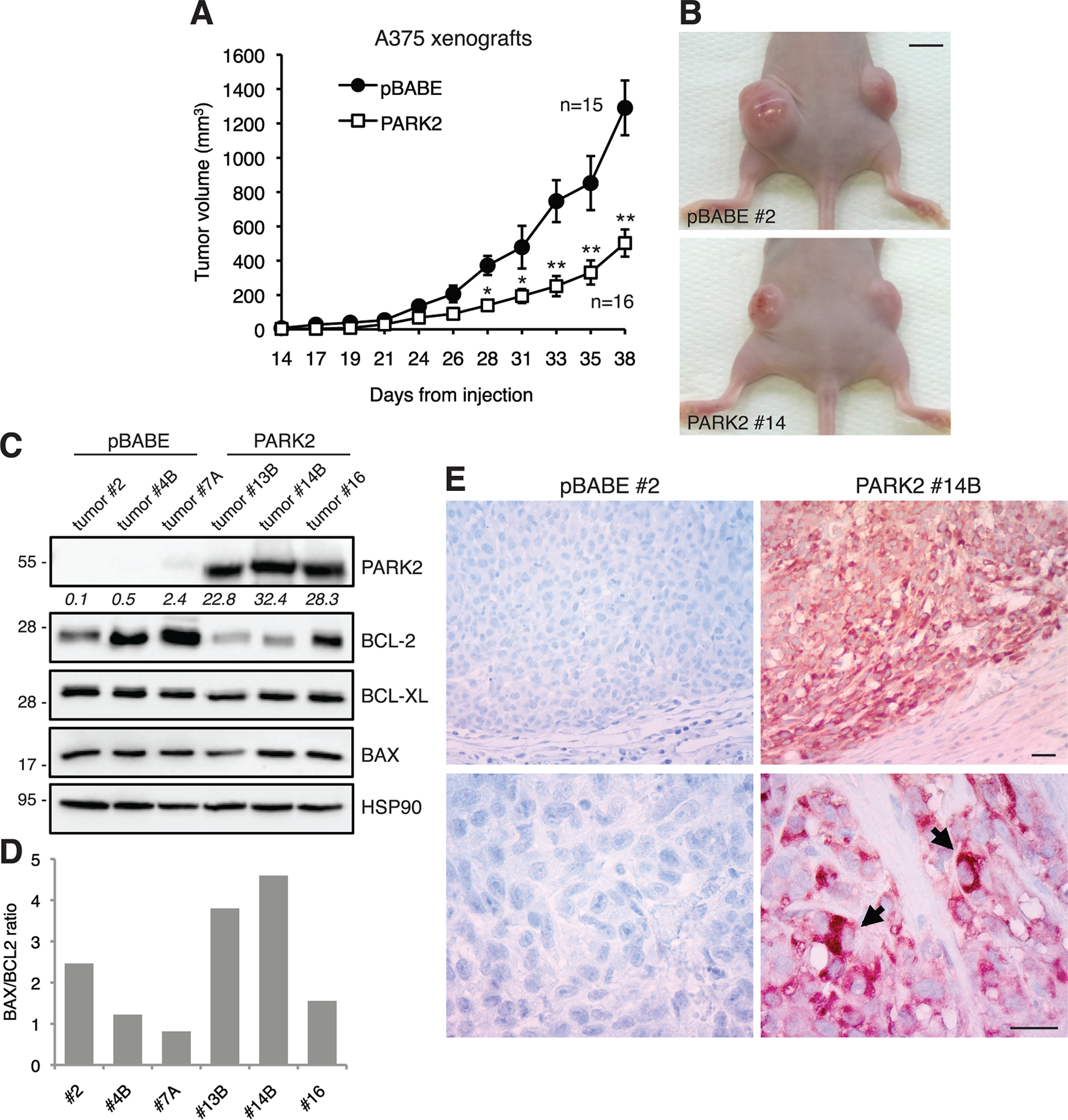
PARK2 suppresses melanoma cell growth in vivo. A, A375 cells transduced with pBABE or PARK2 were injected subcutaneously in athymic nude mice (n = 8 per group). Quantification of tumor volume (n = 15 in pBABE group; n = 16 in PARK2 group), showing that PARK2 drastically decreases tumor growth. Data shown are mean ± S.E. B, representative images of A375 xenografts, as indicated. Scale bar = 0.7 cm. C, Western blot analysis of PARK2, BCL-2, BCL-XL, and BAX in tumors derived from A375 xenografts. HSP90 was used as loading control. PARK2 protein quantification is indicated in italics. D, densitometric quantification of BAX/BCL2 ratio in xenografts. E, immunohistochemical analysis of PARK2 in paraffin sections of pBABE and PARK2 xenografts. Nuclei were counterstained with hematoxylin. Representative cytoplasmic localization of PARK2 is indicated (arrows). Scale bar = 30 μm. *, p < 0.05; **, p < 0.01.
Finally, we addressed whether silencing of PARK2 would revert the reduction of melanoma cell proliferation induced by inhibition of BRAF-V600E. Vemurafenib treatment reduced melanoma cell growth in a dose-dependent manner in SK-Mel-28 cells transduced with LV-c, as expected. On the other hand, vemurafenib had a very minor effect in reducing melanoma cell growth in absence of PARK2 (Fig. 6, A and B). For instance, treatment with vemurafenib at 50 nm reduced growth of LV-c–transduced cells by 26%, but only by 3.6% in PARK2-silenced cells (Fig. 6C). These results indicate that PARK2 depletion partially rescues the effect of BRAF-V600E inhibition, suggesting that PARK2 is important to mediate the effects of BRAF-ERK1/2 activation in melanoma.
Figure 6.
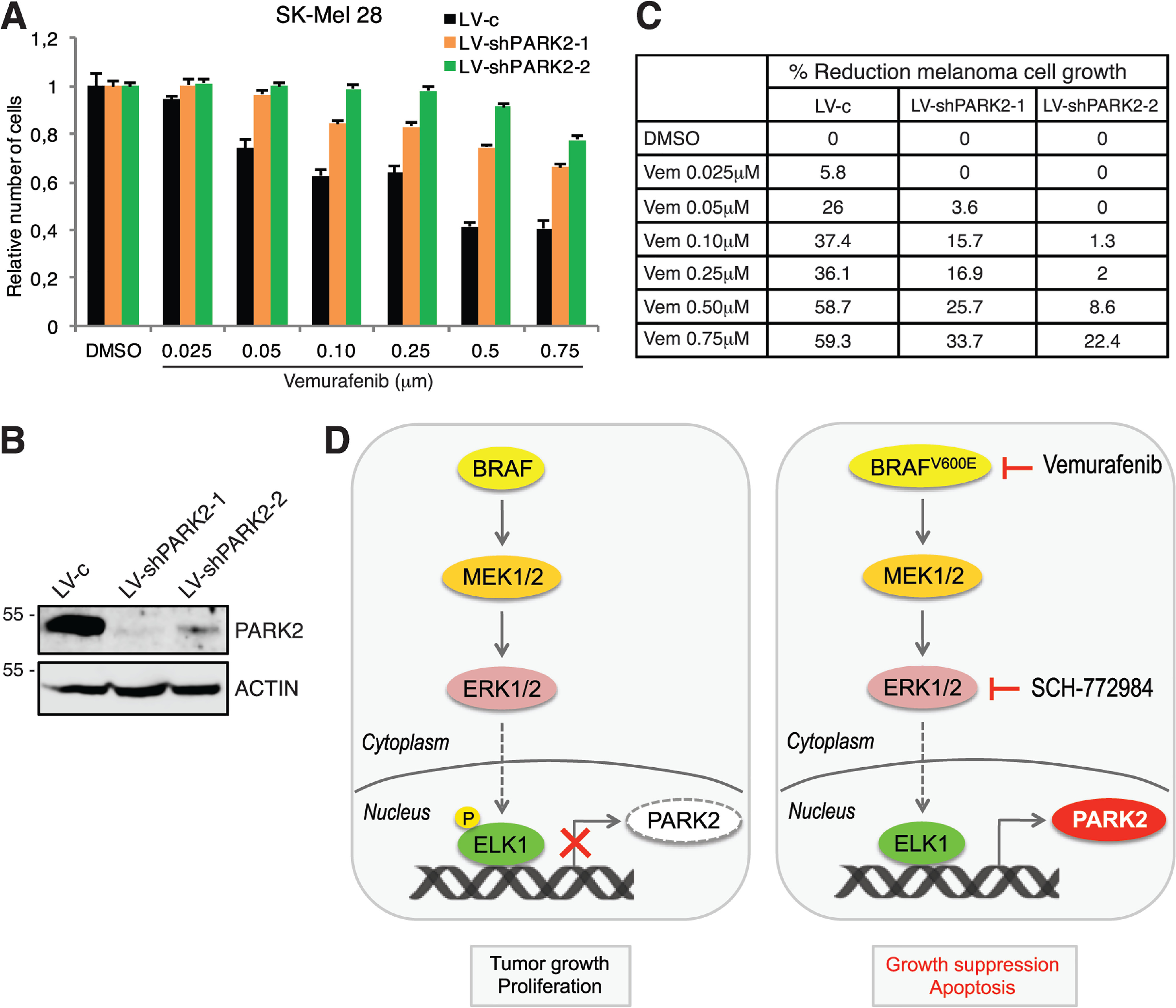
Schematic representation of the negative regulation of PARK2 by the oncogenic ERK1/2-ELK1 transcriptional axis. A, SK-Mel-28 cell growth after transduction with LV-c, LV-shPARK2-1, or LV-shPARK2-2 and treatment with the vehicle (DMSO) or increasing doses of vemurafenib for 72 h. Data are shown as mean ± S.D. of at least three biological replicates, each performed in triplicate. B, WB of PARK2 in SK-Mel-28 cells transduced as indicated. ACTIN was used as loading control. C, table summarizes % reduction of melanoma cell growth as shown in A. D, schematic model. On the left, BRAF-MEK1/2-ERK1/2 signaling induces phosphorylation and nuclear translocation of ELK1, which binds PARK2 proximal promoter acting as negative transcriptional regulator of PARK2. On the right, inhibition of BRAF-V600E or ERK1/2 reduces ELK1 phosphorylation with consequent induction of PARK2 transcription and suppression of tumor growth. Genetic silencing of ELK1 resembles the effect of BRAF-V600E or ERK1/2 inhibition on PARK2 expression. Vemurafenib is a BRAF-V600E inhibitor, whereas SCH-772984 is an ERK1/2 inhibitor.
Discussion
The RAS-RAF-MEK1/2-ERK1/2 pathway is a complex signaling network that integrates numerous upstream stimuli to modulate several cellular processes, including cell growth, proliferation, and survival. Aberrant activation of this signaling pathway occurs in the majority of malignant melanomas (1). In this study, we provide the evidence that PARK2 is negatively modulated by the RAS-RAF-MEK1/2-ERK1/2 signaling via the transcription factor ELK1. In addition, we provide new evidence of the tumor-suppressive role of PARK2 in melanoma, where PARK2 restrains melanoma cell growth downstream of ERK1/2 through the induction of cellular apoptosis.
Our data highlight a previously unexplored mechanism of PARK2 regulation by the RAS-RAF-MEK1/2-ERK1/2 signaling through the transcription factor ELK1, which belongs to the ETS family. ELK1 is the best studied ETS member, and it is directly phosphorylated and activated by ERK1/2 (3, 4, 24), functioning as both activator and repressor of transcription (28). Like all ETS proteins, ELK1 binds the conserved core motif (GGAA/T) embedded in a larger 10-bp consensus sequence that determines the specific recognition of target sites in different genes (29, 30). Our findings indicate that ELK1 represses PARK2 expression through binding to a consensus sequence (CCGGAAA) within the proximal promoter of PARK2. The biological relevance of this regulation is supported by the negative correlation between the expression of PARK2 and ELK1 in a cohort of 481 human melanoma samples. Ultimately, the negative regulation of PARK2 by the ERK1/2-ELK1 axis leads to an increase of proliferation and tumor growth. Consistently, our findings indicate that inhibition of BRAF-V600E or ERK1/2 reduces ELK1 phosphorylation and, as such, ELK1 cannot longer repress PARK2 transcription, with consequent tumor growth arrest and increased cellular apoptosis (Fig. 6D).
PARK2 is an RBR type E3 ubiquitin ligase that mediates degradation of several substrates through the ubiquitin-proteasome system (9, 25, 31). A number of studies in the last few years have shown that PARK2 is involved in protein turnover, stress response, mitochondria homeostasis, genomic stability, metabolism, and many other cellular processes regulating cell growth and survival (6). Mutations in PARK2 gene have been originally associated with the pathogenesis of autosomal recessive juvenile Parkinson's disease (32, 33) and a wide spectrum of brain disorders (34–36). Although the link between PARK2 and cancer susceptibility is not clear, PARK2 deletion, copy number alteration, mutations, and altered mRNA/protein expression have been found in several types of cancer, such as glioblastoma, breast, ovarian, lung, and colorectal. In particular, in glioblastoma PARK2 activation correlates inversely with disease progression and patient survival (12). Interestingly, recent evidence shows a link between PARK2 somatic mutations in melanoma and Parkinson's disease (37, 38).
A recent study proposed that PARK2-inactivating mutations increase the risk of melanoma and that restoration of PARK2 expression in PARK2-deficient melanoma cell lines reduces colony formation (8). However, another report suggested an oncogenic role of PARK2 in melanoma (39). Our findings provide several lines of evidence supporting the tumor suppressive role of PARK2 in human melanoma. First, re-expression of PARK2 in different melanoma cell lines expressing mutated BRAF strongly reduces proliferation in vitro and melanoma xenograft growth in vivo. Second, PARK2 genetic silencing enhances melanoma cell growth and colony formation. Third, PARK2 expression is down-regulated in human melanomas compared with nevi or normal melanocytes. Altogether our data suggest that PARK2 loss-of-function may cooperate with BRAF mutations/amplifications during the early phase of melanoma progression. Following this hypothesis, a previous report suggested an association between alterations in BRAF gene (BRAF-V600E mutation and BRAF amplification) and PARK2 copy loss in primary melanomas (7).
The molecular mechanism by which PARK2 exerts its tumor suppressive function in melanoma is, in part, through the induction of cellular apoptosis, as revealed by the strong up-regulation of the anti-apoptotic factors BCL-2 and BCL-XL in PARK2-silenced melanoma cells and their down-regulation upon PARK2 overexpression in vitro and in vivo. Our findings are consistent with a recent report showing that PARK2 directly binds to and ubiquitinates BCL-XL (40) and other members of the BCL-2 family, such as MCL1 (41). There is evidence in other cancer types that PARK2 targets both Cyclin D1 and E1 for degradation (42), and it interacts with both β-catenin and EGFR to promote their ubiquitination in glioblastoma (9). In addition, PARK2 negatively regulates the PI3K/AKT pathway and PARK2 depletion promotes PTEN inactivation by S-nitrosylation and ubiquitination (25). However, in melanoma cells we did not observe any effect of PARK2 on Cyclin D and E, β-catenin, or PTEN. Nevertheless, our results confirm a role of PARK2 in the negative modulation of AKT pathway (9, 25, 43), evident upon PARK2 depletion. These data suggest that in melanoma PARK2 explicates anti-proliferative effects mainly by regulating programmed cell death, unlike in glioblastoma and colon cancer where PARK2 is mostly involved in controlling cell cycle progression.
In conclusion, our study uncovers a novel mechanism of negative regulation of PARK2 by the ERK1/2-ELK1 transcriptional axis, suggesting that reactivation of PARK2 in cancer cells might have a potential therapeutic effect. Our data suggest that inhibitors of the BRAF-MEK1/2-ERK1/2 cascade could be useful to induce PARK2 expression. In addition, X-ray data combined with computational modeling have contributed to establish a complete structural model of human PARK2 (44, 45) providing the basis for targeted drug design to identify small-molecule activators of this E3 ubiquitin ligase. The complete understanding of PARK2's activation and the ability to improve target specificity will be key determinants in future drug discovery efforts to reactivate PARK2 function.
Experimental procedures
Cell lines
HEK-293T (CRL-3216) cells were purchased from ATCC (Manassas, VA, USA) and NHEM cells from PromoCell (Heidelberg, Germany). A375, SK-Mel-2, SK-Mel-5, SK-Mel-28, SK-Mel-197, and 501-Mel melanoma cells were kindly provided by Dr. Laura Poliseno (Core Research Laboratory, ISPRO, Pisa, Italy). Patient-derived SSM2c and M51 melanoma cells were described already (Table 1) (46, 47). HEK-293T and all melanoma cell lines were grown in DMEM (Euroclone, Milan, Italy) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 2 mm l-glutamine (Lonza, Basel, Switzerland). Human melanoma samples were obtained after approved protocols by the Ethics Committee. Mycoplasma was periodically tested by PCR upon thawing of a new batch of cells and cultures were renewed every month. Transduced cells were selected with puromycin (Invivogen, San Diego, CA, USA) at 1-2 μg/ml for 72 h.
Table 1.
List of cell lines used in this study and their genetic alterations
| Cell line | BRAF/NRAS mutational status | Characteristics |
|---|---|---|
| NHEM | WT BRAF/WT NRAS | Normal human epidermal melanocytes |
| A375 | BRAF V600E | Metastatic melanoma (skin) |
| SK-Mel-2 | NRAS Q61R | Metastatic melanoma (skin) |
| SK-Mel-5 | BRAF V600E | Metastatic melanoma (lymph node) |
| SK-Mel-28 | BRAF V600E | Metastatic melanoma (skin) |
| 501-Mel | BRAF V600E | Metastatic melanoma |
| SK-Mel-197 | WT BRAF/WT NRAS | Metastatic melanoma |
| SSM2c | WT BRAF/WT NRAS | Primary cell line, obtained from a metastatic melanoma |
| M51 | WT BRAF/WT NRAS | Primary cell line, obtained from a metastatic melanoma |
| U87MG | WT BRAF/WT NRAS | Glioblastoma cell line |
| MCF7 | WT BRAF/WT NRAS | Breast cancer cell line |
U87MG and MCF7 harbor CDKN2A-del471; U87MG carries homozygous PTEN-Ins; MCF7 harbors PI3KCA-E545K.
Drug treatments
Vemurafenib (BRAF inhibitor, Selleck Chemicals, Houston, TX, USA) and SCH-772984 (ERK1/2 inhibitor, Sigma) were used at 0.5 μm and 0.25 μm, respectively. Each treatment was performed at low serum condition (1% FBS) and cells were serum-starved for 24 h before treatment.
Plasmid construction, mutagenesis, retroviral and lentiviral vectors
The coding sequence of PARK2 was amplified by PCR with KOD polymerase (Merck Millipore) and cloned into the retroviral vector pBABE using BamHI and EcoRI sites. Primers used were the following: Forward, 5′-ATGATAGTGTTTGTCAGGTTCAACTC-3′, reverse, 5′-CTACACGTCGAACCAGTGGT-3′.
Lentiviral vectors used were pLKO.1-puro (LV-c); pLKO.1-puro-shPARK2-1 (LV-shPARK2-1, targeting sequence 5′-CGTGAACATAACTGAGGGCAT-3′); pLKO.1-puro-shPARK2-2 (LV-shPARK2-2, targeting sequence 5′-CGCAACAAATAGTCGGAACAT-3′); pLKO.1-puro-shELK1-1 (LV-shELK1-1, targeting sequence 5′-CCCAAGAGTAACTCTCATTAT-3′); pLKO.1-puro-shELK1-2 (LV-shELK1-2, targeting sequence 5′-TGAAATCGGAAGAGCTTAATG-3′). Each couple of oligos were annealed, phosphorylated, and then cloned into pLKO.1-puro vector (Addgene) to generate the shRNAs. Retroviruses and lentiviruses were produced in HEK-293T cells.
pcDNA 3.1-BRAFV600E was a kind gift from Dr. Laura Poliseno and pCMV-HA-ELK1 was kindly provided by Martyn Bullock and Roderick Clifton-Bligh (Northern Clinical School, University of Sydney, Sydney, Australia). PARK2 promoter was amplified by PCR with KOD polymerase and cloned into pGL3Basic vector (Promega, Madison, WI, USA) using NheI and XhoI sites, to generate PARK2 promoter-luciferase reporter (−635 bp from TSS to +98bp). Primers used were forward, 5′-GATCACTTACGACTGAGTTT-3′, reverse, 5′-GGTCACTGGGTAGGTGGC-3′. Mutation of the ELK1 binding site was introduced using QuikChange II (Agilent Technologies, Santa Clara, CA, USA) with the following oligos: PARK2prom forward mut1 (5′-CTGGGCCTGAAGCCCCCAAGGGCGGCGGTGG-3′) and PARK2prom reverse mut1 (5′-CCACCGCCGCCCTTGGGGGCTTCAGGCCCAG-3′) (Fig. S4). All transfections were performed in OptiMEM (Life Technologies, Carlsbad, CA, USA) using X-tremeGENE transfection reagent (Roche Diagnostic) or polyethylenimine (Sigma-Aldrich).
Growth curve, colony assay, and soft agar assay
For growth curve, cells were plated in 24-well plates (1500 cells/well for A375 and SSM2c; 2000 cells/well for SK-Mel-5 and SK-Mel-197) and counted on days 3, 5, and 7. For colony formation assay, cells were plated at low density (500 cells/well) in 6-well plate. After 2 weeks, cells were fixed with methanol for 30 min at −20°C and stained with Crystal Violet 0.1%. For soft agar assay, cells were suspended in 0.5% agarose supplemented with DMEM 10% FBS and overlaid on 1% agarose in 6-well plates at density of 2000 cells/well. After 15-20 days, colonies were fixed and stained with Crystal Violet 0.01%. Colonies were counted using ImageJ software. Crystal violet staining was used to measure cell proliferation in SK-Mel-28 cells treated with increasing doses of vemurafenib for 72 h using a plate reader (Victor × 5, Perkin Elmer). Each experimental condition was plated in triplicate.
Immunofluorescence
Cells were plated after coating with poly-lysine and allowed to grow. Cells were fixed with cold methanol and incubated with mouse anti-PARK2 (no. 4211) and rabbit anti-COXIV (no. 4850) (Cell Signaling Technology, Danvers, MA, USA) antibodies. Secondary antibodies were anti-rabbit FITC-conjugated and anti-mouse rhodamine-conjugated (Life Technologies). Cells were counterstained with 4′,6-diamidino-2-phenylindole. Immunofluorescence was visualized with a Zeiss Observer.z1.
Western blotting and cell fractionation
For total protein extraction, cells were harvested, lysed in RIPA buffer (1% Nonidet P-40, 150 mm NaCl, 5 mm EDTA, 0.25% NaDOC, 50 mm Tris-HCl, pH 7.5, SDS 0.1%) supplemented with protease and phosphatase inhibitors, for 20 min in ice and centrifuged 20 min at 14,000 rpm, as described previously (48). Supernatant were quantified using Coomassie Protein Assay Kit (Thermo Fisher Scientific) and separated on SDS-PAGE and transferred into nitrocellulose membranes (Bio-Rad). For cytosolic/nuclear fractionation, cells were lysed first in Buffer A (20 mm Hepes buffer, 10 mm KCl, 1 mm EDTA, 0.2% Nonidet P-40, 10% glycerol), centrifuged 2 min at 14,000 rpm at 4°C, and supernatants containing cytosolic fraction were collected. Pellets (nuclei and membranes) were dissolved in RIPA buffer and processed as described above. The supernatant contained the nuclear protein extract. Antibodies used for Western blotting were mouse anti-PARK2 (no. 4211), rabbit anti-pERK1/2 (no. 9101), mouse anti-BCL-2 (no. 15071), rabbit anti-BCL-XL (no. 2764), rabbit anti-BAX (no. 2772), rabbit anti-p-ELK1 Ser-383 (no. 9181), rabbit anti-ELK1 (no. 9182), rabbit anti-pAKT Ser-473 (no. 4060) (Cell Signaling Technology), mouse anti-ACTIN (sc-47778), mouse anti-LAMIN A (sc-293162), goat anti-GAPDH (sc-20357), mouse anti-HSP90 (sc-13119), mouse anti-CASPASE-3 (sc-7272), mouse anti-BRAF (sc-5284) (Santa Cruz Biotechnology, Dallas, TX, USA). ChemiDoc XRS (Bio-Rad) was used for chemiluminescent detection. Images were recorded as TIFF files for quantification with ImageJ software.
RNA isolation and quantitative real-time PCR
Total RNA was isolated from cells using TRIzol Reagent (Thermo Fisher Scientific) and treated with DNase I (Sigma-Aldrich) to remove genomic contamination. cDNA was obtained using the High-Capacity RNA-to-cDNATM Kit (Thermo Fisher Scientific). qPCR was carried out at 60°C using Sso Advanced Universal SYBR Green Supermix (Bio-Rad) in a Rotorgene-Q (Qiagen, Hilden, Germany). Primer sequences are listed in Table 2.
Table 2.
List of primers used for qPCR
FW, forward; RV, reverse.
| Gene name | Sequences (5′ to 3′) |
|---|---|
| PARK2 | FW: CCAAACCGGATGAGTGGTGA RV: GACGTCTGTGCACGTAATGC |
| ACTIN | FW: GAAAATCTGGCACCACACC RV: TAGCACAGCCTGGATAGCAA |
| GAPDH | FW: TAGCACAGCCTGGATAGCAA RV GCTGGTGGTCCAGGGGTC |
| BAX | FW: CGGGTTGTCGCCCTTTTCTA RV: AGGAGTCTCACCCAACCACC |
| BCl-XL | FW: GGTAAACTGGGGTCGCATTG RV: GCTGCTGCATTGTTCCCATAG |
| BCl-2 | FW: CTTTGAGTTCGGTGGGGTCA RV: GGGCCGTACAGTTCCACAAA |
| PIG3 | FW: CTGAACCGGGCGGACTTAAT RV: GTGTCCCCGATCTTCCAGTG |
| CycD1 | FW: CGTGGCCTCTAAGATGAAGG RV: GTGTTCAATGAAATCGTGCGG |
| BRAF | FW: TGGCAGAGTGCCTCAAAAAGA RV: ACTCATTTGTTTCAGTGGACAGG |
| ELK1 | FW: TTCTGGAGCACCCTGAGTC RV: GAGCATGGATGGAGTGACC |
| c-Fos | FW: GGGGCAAGGTGGAACAGTTA RV: AGGTTGGCAATCTCGGTCTG |
ChIP
ChIP experiments were performed using EZ-Magna ChIP A/G Kit (Millipore, Burlington, MA, USA, cat. no. 17-10086) according to manufacturer's instructions. Briefly, for each assay, 2 × 106 cells were crosslinked with 1% formaldehyde (Sigma) at room temperature for 10 min followed by quenching with 125 mm glycine for 5 min. Cells were harvested and lysed in Cell Lysis Buffer supplemented with a protease inhibitor mixture (Millipore). Nuclei were collected and lysed in nuclear lysis buffer added with protease inhibitors. Chromatin was sonicated and sheared to an average size of 200–500 bp using Bioruptor NGS sonicator (Diagenode, Liege, Belgium). The sheared chromatin was immunoprecipitated at 4°C overnight using anti-ELK1 (Ab1, Santa Cruz Biotechnology, sc-365876) or anti-ELK1 (Ab2, Cell Signaling Technology, no. 9182) antibodies. Normal mouse IgG (Millipore) were used as negative control. qPCR was performed as described above. Primers used for ChIP-qPCR are PARK2chip-F2: 5′-GCTAAGCGACTGGTCAACAC-3′; PARK2chip-R2: 5′-AACGCGTAGTTTCTCCTCACG-3′.
Luciferase reporter assay
PARK2 promoter-luciferase reporter was used in combination with Renilla luciferase pRL-TK reporter vector (Promega) in a ratio 10:1, to normalize luciferase activities; pGL3Basic vector (Promega) was used to equal DNA amounts. Luminescence was measured using the Dual-Glo Luciferase Assay System (Promega) and the GloMax 20/20 Luminometer (Promega).
Flow cytometry analysis
For analysis of apoptosis, Annexin V-PE/7-AAD staining was used to detect cells in early or late apoptosis (Becton Dickinson, Franklin Lakes, NJ, USA) after exposure to serum-deprived conditions for 48 h. Cytometric analysis was performed with CytoFLEX S (Beckman Coulter, Brea, CA, USA).
Xenograft experiments
A375 melanoma cells transduced with pBABE or PARK2 retroviruses were resuspended in Matrigel (BD Biosciences)/DMEM (1/1 ratio) and subcutaneously injected (10,000 cells/injection) into both lateral flanks of adult female athymic nude mice (n = 8 per group) (CD-1 nude mice) (Charles River Laboratories Italy, Milan, Italy). Subcutaneous tumor size was measured three times a week by a caliper, and tumor volumes were calculated using the formula V = W2 × L × 0.5, where W represents the tumor width and L the length. Animals were monitored daily, housed in specific pathogen-free conditions and the experiment was approved by the Italian Ministry of Health in accordance with the Italian guidelines and regulations.
Immunohistochemistry
Immunohistochemistry was performed on 5-μm thick formalin-fixed paraffin-embedded sections of A375 xenografts. After deparaffinization, hydration, and citrate buffer antigen retrieval, slides were incubated with mouse monoclonal anti-PARK2 (Santa Cruz Biotechnology, sc-32282), followed by detection with UltraVision Large Volume Detection System anti-Polyvalent HRP (no. TP-060-HL, Thermo Fisher Scientific) according to manufacturer's instructions. AEC (3-amino-9-ethylcarbazole) (Dako, Copenhagen, Denmark, no. K3461) was used as chromogen. Sections were counterstained with hematoxylin.
Bioinformatic analysis
PARK2 expression in nevi and melanoma samples was analyzed using publicly available microarray dataset (GDS1375) (49), from Gene Expression Omnibus profiled on Affymetrix U133 platforms. The University of California Santa Cruz Xena platform was used to analyze correlation, transcriptomic, and survival data from TCGA melanoma (SKCM) cohort of 17 datasets (50). This curated survival data from the pan-cancer Atlas manuscript highlighting four types of curated survival endpoints of recommended use including overall survival (51). Gene expression profile was measured using the Illumina HiSeq 2000 RNA-Seq platform by the University of North Carolina TCGA genome characterization center. Level 3 data were downloaded from TCGA data coordination center and gene-level transcription estimates were shown as log2(+1) transformed RSEM normalized count. Genes were mapped onto the human genome coordinates using UCSC Xena HUGO probeMap.
Statistical analysis
Data are presented as mean ± S.D. or ± S.E. from at least three independent experiments. P values were calculated using two-tailed Student's t test (two groups) or analysis of variance (more than two groups; multiple comparison using Bonferroni's correction). Value of p < 0.05 was considered statistically significant. Correlation between the expression of PARK2 and ELK1 using the TCGA melanoma cohort (n = 481). Pearson's correlation test was used to analyze the correlation between PARK2 and ELK1 expression.
Data availability
All the described data are contained within this manuscript.
Supplementary Material
Acknowledgments
We thank Silvia Pietrobono, Laura Carrassa, and Giulia Anichini (ISPRO, Florence, Italy) for helpful comments on the paper and discussion. We are grateful to Laura Poliseno (ISPRO, Pisa, Italy) for providing SK-Mel-2, SK-Mel-5, SK-Mel-28, and 501-Mel melanoma cell lines and BRAF-V600E plasmid; Martyn Bullock and Roderick Clifton-Bligh (Northern Clinical School, University of Sydney, Sydney, Australia) for sharing pCMV-HA-ELK1 plasmid; Michela Sica (ISPRO, Florence, Italy) for assistance with flow cytometry; and Sinforosa Gagliardi (ISPRO, Florence, Italy) for assistance with in vivo experiments.
Author contributions—V. M., L. M., A. A., R. M. C., and M. E. F.-Z. data curation; V. M., L. M., A. A., S. P., and B. S. formal analysis; V. M., A. A., S. P., and R. M. C. methodology; V. M., M. E. F.-Z., and B. S. writing-original draft; V. M., L. M., M. E. F.-Z., and B. S. writing-review and editing; R. M. C. investigation; M. E. F.-Z., and B. S. supervision; B. S. conceptualization; B. S. funding acquisition.
Funding and additional information—This work was supported by Institute for Cancer Research, Prevention and Clinical Network (ISPRO) funding and by postdoctoral fellowships from Italian Association for Cancer Research (AIRC) Projects 19580 (to V. M.) and 22644 (to L. M.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- MAPK
- mitogen-activated protein kinases
- ERK
- extracellular signal-regulated kinase
- WB
- Western blotting
- qPCR
- quantitative real-time PCR
- NHEM
- normal human epidermal melanocytes
- TCGA
- The Cancer Genome Atlas
- BS
- binding sites
- TSS
- transcription start site.
References
- 1. Cancer Genome Atlas Network. (2015) Genomic classification of cutaneous melanoma. Cell 161, 1681–1696 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roskoski R., Jr. (2012) ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 66, 105–143 10.1016/j.phrs.2012.04.005 [DOI] [PubMed] [Google Scholar]
- 3. Gille H., Kortenjann M., Thomae O., Moomaw C., Slaughter C., Cobb M. H., and Shaw P. E. (1995) ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 14, 951–962 10.1002/j.1460-2075.1995.tb07076.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marais R., Wynne J., and Treisman R. (1993) The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 73, 381–393 10.1016/0092-8674(93)90237-K [DOI] [PubMed] [Google Scholar]
- 5. Mylona A., Theillet F. X., Foster C., Cheng T. M., Miralles F., Bates P. A., Selenko P., and Treisman R. (2016) Opposing effects of Elk-1 multisite phosphorylation shape its response to ERK activation. Science 354, 233–237 10.1126/science.aad1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu L., Lin D. C., Yin D., and Koeffler H. P. (2014) An emerging role of PARK2 in cancer. J. Mol. Med. 92, 31–42 10.1007/s00109-013-1107-0 [DOI] [PubMed] [Google Scholar]
- 7. Montagnani V., Benelli M., Apollo A., Pescucci C., Licastro D., Urso C., Gerlini G., Borgognoni L., Luzzatto L., and Stecca B. (2016) Thin and thick primary cutaneous melanomas reveal distinct patterns of somatic copy number alterations. Oncotarget 7, 30365–30378 10.18632/oncotarget.8758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu H. H., Kannengiesser C., Lesage S., André J., Mourah S., Michel L., Descamps V., Basset-Seguin N., Bagot M., Bensussan A., Lebbé C., Deschamps L., Saiag P., Leccia M. T., Bressac-de-Paillerets B., et al. (2015) PARKIN inactivation links Parkinson's disease to melanoma. J. Natl. Cancer Inst. 108 10.1093/jnci/djv340 [DOI] [PubMed] [Google Scholar]
- 9. Lin D. C., Xu L., Chen Y., Yan H., Hazawa M., Doan N., Said J. W., Ding L. W., Liu L. Z., Yang H., Yu S., Kahn M., Yin D., and Koeffler H. P. (2015) Genomic and functional analysis of the E3 ligase PARK2 in glioma. Cancer Res. 75, 1815–1827 10.1158/0008-5472.CAN-14-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poulogiannis G., McIntyre R. E., Dimitriadi M., Apps J. R., Wilson C. H., Ichimura K., Luo F., Cantley L. C., Wyllie A. H., Adams D. J., and Arends M. J. (2010) PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. U.S.A. 107, 15145–15150 10.1073/pnas.1009941107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Veeriah S., Taylor B. S., Meng S., Fang F., Yilmaz E., Vivanco I., Janakiraman M., Schultz N., Hanrahan A. J., Pao W., Ladanyi M., Sander C., Heguy A., Holland E. C., Paty P. B., et al. (2010) Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 42, 77–82 10.1038/ng.491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yeo C. W., Ng F. S., Chai C., Tan J. M., Koh G. R., Chong Y. K., Koh L. W., Foong C. S., Sandanaraj E., Holbrook J. D., Ang B. T., Takahashi R., Tang C., and Lim K. L. (2012) Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res. 72, 2543–2553 10.1158/0008-5472.CAN-11-3060 [DOI] [PubMed] [Google Scholar]
- 13. Tay S. P., Yeo C. W., Chai C., Chua P. J., Tan H. M., Ang A. X., Yip D. L., Sung J. X., Tan P. H., Bay B. H., Wong S. H., Tang C., Tan J. M., and Lim K. L. (2010) Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J. Biol. Chem. 285, 29231–29238 10.1074/jbc.M110.108241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun X., Liu M., Hao J., Li D., Luo Y., Wang X., Yang Y., Li F., Shui W., Chen Q., and Zhou J. (2013) Parkin deficiency contributes to pancreatic tumorigenesis by inducing spindle multipolarity and misorientation. Cell Cycle 12, 1133–1141 10.4161/cc.24215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li C., Zhang Y., Cheng X., Yuan H., Zhu S., Liu J., Wen Q., Xie Y., Liu J., Kroemer G., Klionsky D. J., Lotze M. T., Zeh H. J., Kang R., and Tang D. (2018) PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev. Cell 46, 441–455 10.1016/j.devcel.2018.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Picchio M. C., Martin E. S., Cesari R., Calin G. A., Yendamuri S., Kuroki T., Pentimalli F., Sarti M., Yoder K., Kaiser L. R., Fishel R., and Croce C. M. (2004) Alterations of the tumor suppressor gene Parkin in non-small cell lung cancer. Clin. Cancer Res. 10, 2720–2724 10.1158/1078-0432.ccr-03-0086 [DOI] [PubMed] [Google Scholar]
- 17. Duan H., Lei Z., Xu F., Pan T., Lu D., Ding P., Zhu C., Pan C., and Zhang S. (2019) PARK2 suppresses proliferation and tumorigenicity in non-small cell lung cancer. Front Oncol. 9, 790 10.3389/fonc.2019.00790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lei Z., Duan H., Zhao T., Zhang Y., Li G., Meng J., Zhang S., and Yan W. (2018) PARK2 inhibits osteosarcoma cell growth through the JAK2/STAT3/VEGF signaling pathway. Cell Death Dis. 9, 375 10.1038/s41419-018-0401-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Søndergaard J. N., Nazarian R., Wang Q., Guo D., Hsueh T., Mok S., Sazegar H., MacConaill L. E., Barretina J. G., Kehoe S. M., Attar N., von Euw E., Zuckerman J. E., Chmielowski B., Comin-Anduix B., et al. (2010) Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J. Transl. Med. 8, 39 10.1186/1479-5876-8-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flaherty K. T., Puzanov I., Kim K. B., Ribas A., McArthur G. A., Sosman J. A., O'Dwyer P. J., Lee R. J., Grippo J. F., Nolop K., and Chapman P. B. (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819 10.1056/NEJMoa1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morris E. J., Jha S., Restaino C. R., Dayananth P., Zhu H., Cooper A., Carr D., Deng Y., Jin W., Black S., Long B., Liu J., Dinunzio E., Windsor W., Zhang R., et al. (2013) Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 3, 742–750 10.1158/2159-8290.CD-13-0070 [DOI] [PubMed] [Google Scholar]
- 22. Sharrocks A. D. (2001) The ETS-domain transcription factor family. Nat. Rev. Mol. Cell Biol. 2, 827–837 10.1038/35099076 [DOI] [PubMed] [Google Scholar]
- 23. Besnard A., Galan-Rodriguez B., Vanhoutte P., and Caboche J. (2011) Elk-1 a transcription factor with multiple facets in the brain. Front. Neurosci. 5, 35 10.3389/fnins.2011.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo X., Yang L., Xiao L., Xia X., Dong X., Zhong J., Liu Y., Li N., Chen L., Li H., Li W., Liu W., Yu X., Chen H., Tang M., et al. (2015) Grifolin directly targets ERK1/2 to epigenetically suppress cancer cell metastasis. Oncotarget 6, 42704–42716 10.18632/oncotarget.5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gupta A., Anjomani-Virmouni S., Koundouros N., Dimitriadi M., Choo-Wing R., Valle A., Zheng Y., Chiu Y. H., Agnihotri S., Zadeh G., Asara J. M., Anastasiou D., Arends M. J., Cantley L. C., and Poulogiannis G. (2017) PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol. Cell 65, 999–1013 10.1016/j.molcel.2017.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oltval Z. N., Milliman C. L., and Korsmeyer S. J. (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74, 609–619 10.1016/0092-8674(93)90509-O [DOI] [PubMed] [Google Scholar]
- 27. Pietrobono S., Santini R., Gagliardi S., Dapporto F., Colecchia D., Chiariello M., Leone C., Valoti M., Manetti F., Petricci E., Taddei M., and Stecca B. (2018) Targeted inhibition of Hedgehog-GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing replication stress and mitotic catastrophe. Cell Death Dis. 9, 142 10.1038/s41419-017-0142-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Göke J., Chan Y. S., Yan J., Vingron M., and Ng H. H. (2013) Genome-wide kinase-chromatin interactions reveal the regulatory network of ERK signaling in human embryonic stem cells. Mol. Cell 50, 844–855 10.1016/j.molcel.2013.04.030 [DOI] [PubMed] [Google Scholar]
- 29. Boros J., Donaldson I. J., O'Donnell A., Odrowaz Z. A., Zeef L., Lupien M., Meyer C. A., Liu X. S., Brown M., and Sharrocks A. D. (2009) Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res. 19, 1963–1973 10.1101/gr.093047.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Odrowaz Z., and Sharrocks A. D. (2012) ELK1 uses different DNA binding modes to regulate functionally distinct classes of target genes. PLoS Genet. 8, e1002694 10.1371/journal.pgen.1002694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu J., Zhang C., Zhao Y., Yue X., Wu H., Huang S., Chen J., Tomsky K., Xie H., Khella C. A., Gatza M. L., Xia D., Gao J., White E., Haffty B. G., et al. (2017) Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 8, 1823 10.1038/s41467-017-01947-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., and Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 10.1038/33416 [DOI] [PubMed] [Google Scholar]
- 33. Lücking C. B., Dürr A., Bonifati V., Vaughan J., De Michele G., Gasser T., Harhangi B. S., Meco G., Denèfle P., Wood N. W., Agid Y., Nicholl D., Breteler M. M. B., Oostra B. A., De Mari M., et al. (2000) Association between early-onset Parkinson's disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567 10.1056/NEJM200005253422103 [DOI] [PubMed] [Google Scholar]
- 34. Glessner J. T., Wang K., Cai G., Korvatska O., Kim C. E., Wood S., Zhang H., Estes A., Brune C. W., Bradfield J. P., Imielinski M., Frackelton E. C., Reichert J., Crawford E. L., Munson J., et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573 10.1038/nature07953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burns M. P., Zhang L., Rebeck G. W., Querfurth H. W., and Moussa C. E. (2009) Parkin promotes intracellular Aβ1-42 clearance. Hum. Mol. Genet. 18, 3206–3216 10.1093/hmg/ddp258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Periquet M., Latouche M., Lohmann E., Rawal N., De Michele G., Ricard S., Teive H., Fraix V., Vidailhet M., Nicholl D., Barone P., Wood N. W., Raskin S., Deleuze J. F., Agid Y., et al. (2003) Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 126, 1271–1278 10.1093/brain/awg136 [DOI] [PubMed] [Google Scholar]
- 37. Inzelberg R., Samuels Y., Azizi E., Qutob N., Inzelberg L., Domany E., Schechtman E., and Friedman E. (2016) Parkinson disease (PARK) genes are somatically mutated in cutaneous melanoma. Neurol. Genet. 2, e70 10.1212/NXG.0000000000000070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levin L., Srour S., Gartner J., Kapitansky O., Qutob N., Dror S., Golan T., Dayan R., Brener R., Ziv T., Khaled M., Schueler-Furman O., Samuels Y., and Levy C. (2016) Parkin somatic mutations link melanoma and Parkinson's disease. J. Genet. Genomics 43, 369–379 10.1016/j.jgg.2016.05.005 [DOI] [PubMed] [Google Scholar]
- 39. Lee Y. S., Jung Y. Y., Park M. H., Yeo I. J., Im H. S., Nam K. T., Kim H. D., Kang S. K., Song J. K., Kim Y. R., Choi D. Y., Park P. H., Han S. B., Yun J. S., and Hong J. T. (2018) Deficiency of parkin suppresses melanoma tumor development and metastasis through inhibition of MFN2 ubiquitination. Cancer Lett. 433, 156–164 10.1016/j.canlet.2018.07.007 [DOI] [PubMed] [Google Scholar]
- 40. Gong Y., Schumacher S. E., Wu W. H., Tang F., Beroukhim R., and Chan T. A. (2017) Pan-cancer analysis links PARK2 to BCL-XL-dependent control of apoptosis. Neoplasia 19, 75–83 10.1016/j.neo.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carroll R. G., Hollville E., and Martin S. J. (2014) Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 9, 1538–1553 10.1016/j.celrep.2014.10.046 [DOI] [PubMed] [Google Scholar]
- 42. Gong Y., Zack T. I., Morris L. G., Lin K., Hukkelhoven E., Raheja R., Tan I. L., Turcan S., Veeriah S., Meng S., Viale A., Schumacher S. E., Palmedo P., Beroukhim R., and Chan T. A. (2014) Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet. 46, 588–594 10.1038/ng.2981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fallon L., Bélanger C. M., Corera A. T., Kontogiannea M., Regan-Klapisz E., Moreau F., Voortman J., Haber M., Rouleau G., Thorarinsdottir T., Brice A., van Bergen En Henegouwen P. M., and Fon E. A. (2006) A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat. Cell Biol. 8, 834–842 10.1038/ncb1441 [DOI] [PubMed] [Google Scholar]
- 44. Caulfield T. R., Fiesel F. C., Moussaud-Lamodière E. L., Dourado D. F., Flores S. C., and Springer W. (2014) Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput. Biol. 10, e1003935 10.1371/journal.pcbi.1003935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Caulfield T. R., Fiesel F. C., and Springer W. (2015) Activation of the E3 ubiquitin ligase Parkin. Biochem. Soc. Trans. 43, 269–274 10.1042/BST20140321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Santini R., Vinci M. C., Pandolfi S., Penachioni J. Y., Montagnani V., Olivito B., Gattai R., Pimpinelli N., Gerlini G., Borgognoni L., and Stecca B. (2012) Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 30, 1808–1818 10.1002/stem.1160 [DOI] [PubMed] [Google Scholar]
- 47. Riverso M., Montagnani V., and Stecca B. (2017) KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene 36, 3322–3333 10.1038/onc.2016.481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pandolfi S., Montagnani V., Lapucci A., and Stecca B. (2015) HEDGEHOG/GLI-E2F1 axis modulates iASPP expression and function and regulates melanoma cell growth. Cell Death Differ. 22, 2006–2019 10.1038/cdd.2015.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Talantov D., Mazumder A., Yu J. X., Briggs T., Jiang Y., Backus J., Atkins D., and Wang Y. (2005) Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin. Cancer Res. 11, 7234–7242 10.1158/1078-0432.CCR-05-0683 [DOI] [PubMed] [Google Scholar]
- 50. Goldman M., Craft B., Hastie M., Repečka K., McDade F., Kamath A., Banerjee A., Luo Y., Rogers D., Brooks A. N., Zhu J., and Haussler D. (2019) The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 326470 10.1101/326470 [DOI]
- 51. Liu J., Lichtenberg T., Hoadley K. A., Poisson L. M., Lazar A. J., Cherniack A. D., Kovatich A. J., Benz C. C., Levine D. A., Lee A. V., Omberg L., Wolf D. M., Shriver C. D., Thorsson V., and Hu H., Cancer Genome Atlas Research Network. (2018) An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416 10.1016/j.cell.2018.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the described data are contained within this manuscript.