

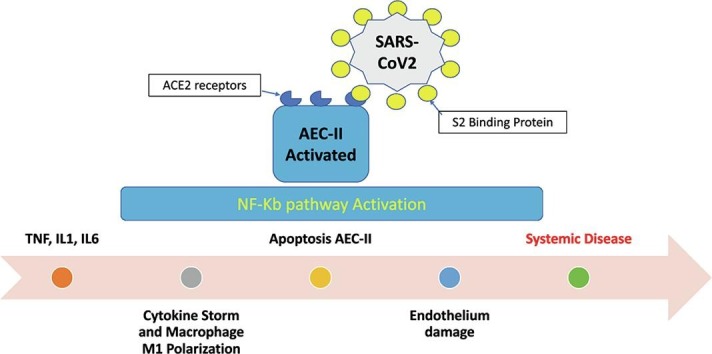
Graphical abstract
Keywords: COVID-19, SARS-CoV-2, COVID-19 pathogenesis, NF-kB pathway activation, Alveolar epithelial cell type 2, AEC-II, ARDS
Abstract
The Corona Virus Disease (COVID-19) pandemic caused by Severe Acute Respiratory Syndrome Corona Virus 2 (SARS-CoV-2) requires a rapid solution and global collaborative efforts in order to define preventive and treatment strategies. One of the major challenges of this disease is the high number of patients needing advanced respiratory support due to the Acute Respiratory Distress Syndrome (ARDS) as the lung is the major – although not exclusive – target of the virus. The molecular mechanisms, pathogenic drivers and the target cell type(s) in SARS-CoV-2 infection are still poorly understood, but the development of a “hyperactive” immune response is proposed to play a role in the evolution of the disease and it is envisioned as a major cause of morbidity and mortality. Here we propose a theory by which the main targets for SARS-CoV-2 are the Type II Alveolar Epithelial Cells and the clinical manifestations of the syndrome are a direct consequence of their involvement. We propose the existence of a vicious cycle by which once alveolar damage starts in AEC II cells, the inflammatory state is supported by macrophage pro-inflammatory polarization (M1), cytokines release and by the activation of the NF-κB pathway. If this theory is confirmed, future therapeutic efforts can be directed to target Type 2 alveolar cells and the molecular pathogenic drivers associated with their dysfunction with currently available therapeutic strategies.
Introduction
In the past twenty years there have been precedents for epidemic clusters sustained by viruses that have bats as their main hosts. The dynamic of the transmission from bat-borne viruses to humans is very complex and often involves the presence of additional intermediate hosts [1], [2], [3]. The human Coronavirus (hCoV) family belongs to a well-recognized bat-borne family of viruses that infects humans causing predominant damage to the respiratory system strictly connected with the development of ARDS [4], [5]. Previous Coronavirus-mediated diseases are Severe Acute Respiratory Syndrome Coronavirus (SARS-Co-V) or Middle East Respiratory Syndrome CoV (MERS CoV) [6], [7], [8] closely related to the new SARS-CoV-2 responsible for Coronavirus Disease 19 (COVID-19) [1], [9], [10]. ARDS is a manifestation of lung injury, therefore not a disease itself, rather a syndrome induced by different insults and characterized by substantial heterogeneity [4], [5]. We propose the hypothesis that the main target of the virus is the Alveolar Epithelial Cell Type II and that a dysregulation of the Nuclear Factor kappa-light-chain-enhancer of activated B cells pathway (NF-κB pathway) drives ARDS leading to Multiple Organ Failure (MOF), one of the most frequent causes of death. By correlating the clinical data and examining the pathophysiology of the various diseases present in COVID-19, we focused our attention on the intra and extra cellular signaling mechanisms that are activated following damage at the alveolar level and on how these signals are distantly propagated and amplified.
Physio-pathogenic theory
Physiological and pathological functions of AEC-II cells
When a pathogen like a virus infects humans, the immune system could clear the infection by limiting viral spread (effective antiviral response), or cause an excessive inflammation and tissue destruction by cytotoxic cells and consequent development of immuno-pathological damage. The epithelial cells are critical for balancing these two extreme situations [11]. The upper respiratory tract provides the first line of defense in that it activates a very complex system of signaling and recruitment of immune cells, in order to prevent pathogens from reaching the alveoli, the most important part of the respiratory system because responsible of gas exchange. Nevertheless, coronaviruses (especially SARS-CoV and MERS) are able to reduce or delay the expression of cytokines in human lung epithelial cell lines. This is an effective system to escape immune recognition by innate receptors in the infected cell [12], [13], which could therefore facilitate the progression of the virus into the lower respiratory airways (i.e. the alveolar space) (Fig. 1 ).
Fig. 1.

SARS-CoV2 immunoescape strategy to reach the alveolus.
These observations are coherent with some clinical evidence of CoVID-19 as well: 1) A high number of asymptomatic patients are positive at the nasopharyngeal swab for virus detection with real-time reverse transcription (RT)-PCR, which strongly suggests that despite viral load is present, no symptoms occur due to the lack of innate immune response; 2) On the other hand, in mild symptomatic patients –some time negative with nasopharyngeal swab-, early alveolar damage can still be detected with CT scan [14], thus demonstrating that the first and most important site of damage is the lung parenchyma.
The alveolar epithelium is composed by two different Alveolar Epithelial Cells (AEC): Alveolar Epithelial Cell Type I (AEC-I) that represents 85–90% of all alveolar cells and Alveolar Epithelial Cell Type II (AEC-II), 10–15% [15]. The latter, despite the small number, is fundamental for many functions, that we can summarize as follows: 1) production of the surfactant (S-protein SP); 2) stabilization of the airway epithelial barrier (AEB); 3) immune-defense; 4) airway regeneration during lung injury, a process carried out by progenitors for AEC-I cells [16]. Moreover AEC-II is the only lung cell that produces all of the components of the surfactant complex and aids in the biosynthesis, storage and release of the entire production cycle which is fundamental to lung integrity [17], [18] because responsible of lowering the superficial tension, thereby preventing alveolar collapse. Once the alveolar damage starts, in the absence of an effective response, the pathogenesis progresses to ARDS.
At least three different phases have been identified in ARDS: 1) The exudative phase; 2) The proliferative phase; 3) The fibrotic phase [4]. In the exudative phase the lung responds to insults by activating innate cell-mediated immunity that leads to an alteration of both the vascular endothelium and the alveolar epithelium. In this phase there is a ”cytokine storm” with an intense promotion of inflammation supported by the recruitment of neutrophils, monocytes and macrophages and by the activation of alveolar epithelial cells [19], [20]. The second proliferative phase is the most important for the recovery of normal lung function and for the restoration of normal alveolar-endothelial architecture. The third fibrotic phase is associated with an increase in mortality and the need for longer mechanical ventilation [4], [5], [21]. In the COVID-19 syndrome, the high mortality rate in critically ill patients suggests that there could be an imbalance between these phases. Furthermore, pulmonary fibrosis may be one of the most severe complications and a cause of permanent lung damage after patients recover from CoV-2 infection, thus prevention of this complication is an issue that urgently needs to be addressed.
SARS-CoV-2 is an enveloped virion containing one positive-strand RNA genome whose main sequence has been reported [22]. We also know that the Angiotensin Converting Enzyme 2 (ACE2) receptors are the main point of entry for coronaviruses through the binding with glycoprotein (GP) spikes proteins [23]. Relative to other coronaviruses, CoV-2 has a greater affinity (10–20 fold) for the receptor probably due to a different conformation of GP spikes within the subunit [24]. After binding the virus envelope is fused to target cells via a Trans Membrane Protease Serin 2, TMPRSS2 [25], [26]. Into the alveoli the ACE2 receptors are present on the AEC-II cell membrane and recent evidence suggests that one of the highest expression of both ACE2 and TMPRSS2 is in AEC-II [27], [28] and in transient secretory cell in more central bronchial branches [28], where the latter could act as a transporter to the more peripheral alveoli. This line of evidence lends strong support to our theory of the main role played by AEC-II.
Previous studies have shown that in the case of SARS-CoV and MERS the virus downregulates the expression of ACE2 [29], [30], and recently confirmed in SARS-CoV2 [31]. This is not a contradiction because Ace2 receptors counteract inflammation. Indeed ACE 2 plays a fundamental role in the Renin Angiotensin System (RAS), having a protective function through the conversion of Angiotensin II to Angiotensin 1–7, that counteracts the effects of Angiotensin II, promoting vasodilatory, anti-inflammatory, antioxidant activities. Moreover, in murine models, the ACE2 knockout displayed more severe symptoms of the disease compared to wild-type mice, while ACE2 overexpression appeared protective [30], [32], [33], [34]. ACE2 is also known to play a crucial role in the apoptosis of alveolar epithelial cells and is downregulated in lung fibrosis [35], and its actions on peptide signals balance and offset those of ACE [36]. Intriguingly, a decreased glomerular expression of ACE2 has been described in rodent models of diabetes [37], [38], [39]. These observations are consistent with some clinical aspects, also common to other coronavirus infections that correlate the severity of the disease with the presence of comorbidities such as diabetes, hypertension and cardiovascular diseases. This line of evidence supports the hypothesis that ACE2 is actually reduced as already suggested by some authors [31], [40] and plays a possible protective role, also explaining why angiotensin converting enzyme inhibitors (ACEI) and angiotensin receptor blocker (ARB) [32], [41], may also be protective.
The role of the endothelium and the interplay with macrophages and neutrophils.
A characteristic element of ARDS is the increase in capillary permeability resulting from the damage of the AEC cells and the endothelium which includes an accumulation of liquid rich in proteins in the alveoli and consequent release of proinflammatory cytokines: TNF, IL1 e IL6 [42]. This corresponds to the exudative phase mentioned above [4], during which resident macrophages, with the M1 phenotype, release additional proinflammatory cytokines, in a series of events also called “cytokine storm”, ultimately leading to the apoptosis of the AEC-II and AEC-I cells with further damage to the alveolus-capillary barrier, to the reduction of surfactant and to the formation of intravascular- and microvascular-clots.
The pulmonary endothelium plays a leading role in lung damage, so much so that some authors have called it “the orchestra conductor in respiratory diseases” [43]. In the lung at rest the endothelium assumes an inhibitory effect on inflammation and coagulation, whereas after cytokines stimulation it converts to a pro-inflammatory phenotype [44]. Neutrophil activation is fundamental in this process, because their accumulation enhances the progression of inflammation and alveolar damage. The NF-κB pathway is fundamental to regulate many important cellular behaviors, in particular, inflammatory responses, cellular growth and apoptosis, as well as in regulating the survival, activation and differentiation of innate immune cells and of inflammatory T cells [45]. Furthermore, NF-κB prevents the pro-inflammatory cytokine TNF-alpha from inducing the apoptosis of neutrophils in response to inflammation or viral-infections and exposes ligands for neutrophils by promoting the degradation of endothelial glycocalyx [44]. We envision that the result of this chain of events substantially leads to: 1) Apoptosis of AEC-II cells; 2) NF-κB pathway hyper-activation; 3) Enhanced survival of neutrophils and their accumulation, 4) Downregulation of ACE2, 5) Surfactant reduction due to apoptosis of AEC-II (Fig. 2 ). NF-κ B is activated by viruses, such as HIV-1, HTLV-1, hepatitis B virus (HBV), hepatitis C virus (HCV), Ebstein Barr Virus (EBV), and influenza virus [46]. The role of NF-Kb pathway in SARS-CoV infection has already been postulated by other authors in the past [47].
Fig. 2.

Endothelium-Macrophage interplay in COVID-19 development.
The reduction of the surfactant is a major driver of the collapse of the alveolus, therefore all the pathologies that involve its reduction aggravate the damage. It is known that diabetes for example involves a reduction in the production of surfactant as well as a reduction in ACE2 [48]; this explains why diabetes represents an aggravating condition for ARDS and for COVID-19 patients as well. The transition to the next restoration or proliferative phase is essential for tissue homeostasis after damage. This phase is governed by the phagocytosis of apoptotic neutrophils (efferocytosis), by resident macrophages that eventually switch towards the M2 phenotype [4]. However, we believe that this latter phase is delayed by SARS-CoV2 by NF-κB pathway amplification and inflammation sustained in a vicious circle manner (Fig. 3 ). Recently, in an Italian study on more than 40 autopsies, pathological alterations of the exudative phase and the early proliferative phase were found, where fibrotic phase is reported as focal and not widespread; therefore, the authors suggest that none of the patients had progressed to the fibrotic phase, possibly because of the short duration of the disease [49]. Otherwise they describe mural fibrosis in 65% of patients. It is important to underline that the three phases of ARDS do not behave exactly as described and that not every patient will develop fibrosis. These steps may overlap and occur in a heterogenous way regarding time and different lung region. Thus, inflammatory and repair mechanisms initiate in parallel and not in a serial way. This evidence is also supported by a recent review [50]. Furthermore, we believe that the early fibrotic pattern showed before the onset of symptoms could be COVID-19 related.
Fig. 3.

Inflammation state sustained by NF-Kb pathway and M1 macrophage phenotype.
Macrophages are key regulators of both inflammation and its resolution, and NF-κB plays a key role in macrophage responses and differentiation into pro-inflammatory (M1) or anti-inflammatory (M2) phenotype, which is strongly influenced by cytokines [51]. In the inactive form NF-κB is sequestered in the cytoplasm through the combined action of inhibitory protein IκB and its subunits (IκBα/β/γ). One possible mechanism that could play an important role in the inhibition of IkB protein family members and therefore upholds activation of NF-κB pathway, is the involvement of microRNA(s) that physiologically regulate the inflammatory response. Micro RNAs (miRNAs) are small, non-coding RNAs that regulate the expression of protein-coding genes [52]. Recently, miRNA levels have been used as a novel non-invasive biomarker for the diagnosis of various diseases [53], [54]. Expression of some miRNAs, such as miR 132 and miR 146a, have the capacity to dampen the inflammatory response in alveolar macrophages [55].
Corroborative evidence
During this pandemic, various therapies were tested, with some effects on improvement, where we find confirmation of our hypotheses on the dysregylation of the NF-κB pathway as a potential key driver of disease progression.
It has been noted that disease severity is not only attributed to direct viral damage, but also by immune-mediated injury induced by SARS-CoV-2, where distinctive features have been noticed in severe and critical patients with COVID-19: progressive increase of inflammation and an unusual trend of hypercoagulation [56]. Indeed, thromboprophylaxis with Low Weight Molecular Heparin (LMWH) in COVID-19 patients has been implemented in several centers worldwide, where it has demonstrated a clear overall benefit in hospitalized patients on mechanical ventilation, reducing the death rate in this particular patient population [57], [58]. Currently, no study has methodically evaluated the benefit of LMWH at earlier stages of the disease. However, given that the endothelial damage and consequent micro-thrombi formation are likely key pathogenic triggers of downstream events [59], including of inflammation, studies in this direction could be very useful; It is further particularly interestingly to note that heparin also acts on macrophages reducing pro-inflammatory cytokine secretion and, most importantly, inhibits the NF-κB pathway in AEC-II cells reducing inflammation [60]. Failure to downregulate NF-κB was indeed observed in other, unrelated chronic inflammatory diseases, such as in rheumatoid arthritis [61]. Furthermore, one of the first drugs used in COVID-19 was Tocilizumab, a monoclonal antibody directed to the IL6 receptor that blocks the interaction with its ligand IL-6, one of the main pro-inflammatory cytokines involved in the “cytokines storm” [62], [63]. A reduction in IL-6 produce a negative feedback on NF-Kb pathway [64].
Recently some authors have reported on suspect Kawasaki Syndrome induced by SARS-CoV2 in children [65]. This finding, previously already reported for others coronavirus infections [66], further supports our hypothesis of the essential role of NF-Kb pathway. In fact, and strikingly, recent studies have highlighted the importance of the NF-κB pathway activation as a direct and cause-effect factor in the immunopathology underlying Kawasaki syndrome [67], and the role of the miRNAs was also hypothesized for this pathology [68]. Therefore, we suppose that Kawasaki Syndrome is likely to be secondarily triggered, rather than directly caused by SARS-CoV2 and could explain even other occasional reporting of this association in the past [66], [69], and may be our findings could be useful to better understand the Kawasaki Syndrome etiopathogenesis.
Summary of pathogenic mechanisms
In essence, we propose the following
-
1.
The main target of SARS-CoV-2 is the AEC-II cell and the NF-κB activation herein is a key driver of severe ARDS in Covid 19 patients. The first colonization of the upper airways is a necessary but not sufficient condition for the development of COVID-19 syndrome, because of an initial immuno-escape strategy of the virus from the Innate Immune response. We believe that the greater affinity of SARS-CoV-2 for ACE2 relatively to other coronaviruses enables it to more efficiently reach the main target, namely the AEC-II cells which though represent only 10% of all alveolar cells, play a fundamental role in the mediation of inflammatory responses and, ultimately in the remodeling of the lung and its homeostasis. The infection of these cells subsequently leads to the activation and amplification of the inflammatory process in other cell types, including AEC-I and the endothelium, during which the activation of the NF-κB pathway is amplified by an excessive transcription mediated by the virus with a mechanism to be ascertained.
Two different mechanisms could explain the NF-κB pathway involvement, namely the activation of Interferons during the course of the Innate Immune Response; alternatively, but not mutually exclusive, miRNAs could also be directly or indirectly involved, as shown in other pathological conditions. A similarly important question is whether the endothelium itself, which also expresses ACE2 receptors, is a direct port of entry for the virus and direct contributor to the pathogenesis of the coagulopathy, as speculated recently [56]. The direct endothelial damage theory could certainly explain the hyper-coagulation state observed in many Covid 19 patients and could also contribute to the enhanced pathology seen in hypertensive patients, given that hypertension promotes endothelial damage and a hyper-coagulative state.
-
2.
The early Alveolar Damage and delayed of the proliferative phase contributes to severe and permanent damage. We think that there is a protracted phase of exudation with destruction of the alveolus and the endothelium and Diffuse Alveolar Damage (DAD). All these features are the consequence of a pro-inflammatory state, in which the main role is played by the M1 polarized alveolar macrophages. Moreover, macrophages are identified as predominant cells in the intra-alveolar cavity of COVID-19 patients [50]. The endothelium is exposed to injury and therefore contributes to development of ARDS, by widespread and extensive deposition of extracellular matrix leading to early interstitial and intra-alveolar fibrosis, and non-pulmonary syndrome (e.g., Immune Thrombocytopenic Purpura) [70]. The timing of macrophage polarization changing from M1 to M2 is furthermore relevant and dependant from miRNAs expression [51], [71], [72] (Fig. 4 ), and could be justify the overlapping of epithelial vascular and fibrotic pattern of lung injury.
Fig. 4.

Early lung fibrosis development by impairment of Proliferative phase during Acute Respiratory Distress Syndrome.
After the ARDS started a Multi Organ Failure (MOF) could be develop, probably by endothelium infection via ACE2 receptors, as recently reported [73].
Some co-morbidities of patients may favor the entry mechanism of SARS-CoV2 into AEC-II cells, such as scarce presence of surfactant (like in diabetes). Another possible enhancing cause of viral infection is the reduced expression of ACE2 in diabetes, hypertension and cardiac pathologies. In addition, some gender differences can also be explained by the influence of hormonal factors in the production of surfactant: female hormones are known to have a function favoring the production of surfactant. This may explain why women have a less severe disease course. This observation is also supported by the fact that, regardless of the course of the disease, the incidence of the disease in women remains lower in all age groups up to menopause, from there onwards the incidence is comparable to that of men. Additionally, there could be a hormonal influence that could also justify the lower incidence in children under 10 years of age.
Potential therapeutic interventions
If this hypothesis were to be confirmed by clinical and molecular finding, the following interventions could be attempted (Fig. 5 ):
-
1.
NF-kB Inhibitors: the NF-κB signaling pathway has become a potential target for pharmacological intervention for a wide range of pathologies, such as cancer (angiogenesis and metastases) and chronic inflammation. Numerous inhibitors of NF-κB have been identified that act on different mechanism of NF-kB activation. In a review by Gilmore and Hersovitch reported over 750 inhibitors of NF-κB activation, and summarized their activity essentially in three mechanisms: (1) blockage of the incoming stimulating signal at an early stage; (2) interference with a cytoplasmic step in the NF-κB activation pathway by blockage of a specific component of the cascade (e.g., the activation of the IKK complex, or degradation of IκB); or (3) blockage of NF-κB nuclear activity [74]. Two examples of these inhibitors are: Denosumab, a humanized monoclonal antibody that binds Receptor Activator of Nuclear factor Kappa B Ligand (RANKL), used in metastatic bone cancer and in Rheumatoid Arthritis, a well-known and tolerate drug with a potent NF-Kb pathway inhibition activity; as to Bortezonib, a drug used in multiple myeloma, that inhibits the 26S subunit of the proteasome which leads to the inability of IκB degradation and NF-κB activation [75].
-
2.
Macrophage control of polarization to promote the switching from proinflammatory (M1) to anti-inflammatory (M2) phenotype [51], [71], [76].
-
3.Surfactant (Replacement/Improvement)
-
a.Replacement by Liquid Ventilation (Partial/Total): is a technique of mechanical ventilation in which the lungs are insufflated with an oxygenated perfluorochemical liquid (PFC). Some studies suggest that PFC reduced neutrophil adhesion, activation, and migration [77]; moreover, inhibit inflammatory cytokine expression and NF-kB pathway activation [78]. However, although fascinating and promising, to date it is not clear which patients with ARDS can benefit from this type of treatment, which still requires mechanical ventilation (at least in the Total Liquid Ventilation technique) [79]. Furthermore, is to establish the potential virus diffusion risk with this technique by aerosol of PFC elimination.
-
b.Improvement: ACE2 Human Recombinant use demonstrates in preliminary study that Surfactant protein D concentrations were increased, and interleukin-6 concentrations were decreased [80], [81], [82]. Moreover, in a recent study, human recombinant soluble ACE2 (hrsACE2) are used to blocks growth of SARS-CoV-2 in VERO cells [82], [83].
-
c.Special consideration for diabetic patient: Recently a study demonstrated the potential role of Liraglutide, a drug used in diabetes, to enhance ACE2 expression in the lung and to reduce the lung and cardiovascular impairment induced by diabetes, moreover improving the surfactant production (SP-A and SP-B)[84].
-
a.
-
4.
Endothelium protection: Low Weight Molecular Heparin, already widely used, but with early administration before the onset of clinical symptoms and at therapeutic dosage.
Fig. 5.

Possible therapeutic interventions in different steps of COVID-19 development.
Funding
No funding received.
Contributors
Maurizio Carcaterra had the idea of this physio-pathological Theory, Cristina Caruso contributed to collecting data and writing of the report.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song H.D., Tu C.C., Zhang G.W., Wang S.Y., Zheng K., Lei L.C. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci USA. 2005;102(7):2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang T., Wu Q., Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. 2020 doi: 10.1016/j.cub.2020.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson B.T., Chambers R.C., Liu K.D. Acute respiratory distress syndrome. N Engl J Med. 2017;377:562–572. doi: 10.1056/NEJMra1608077. [DOI] [PubMed] [Google Scholar]
- 5.Ware L.B., Matthay M.A. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 6.Drosten C., Günther S., Preiser W., van der Werf S., Brodt H.-R., Becker S. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 7.Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev. 2005;69:635. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Channappanavar R., Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39:529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shereen M.A., Khan S., Kazmi A., Bashir N., Siddique R. COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J Adv Res. 2020 doi: 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng S.C., Chang Y.C., Fan Chiang Y.L., Chien Y.C., Cheng M., Yang C.H. First case of coronavirus disease 2019 (COVID-19) pneumonia in Taiwan. J Formos Med Assoc. 2020 doi: 10.1016/j.jfma.2020.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denney L., Ho L.-P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed J. 2018;41:218–233. doi: 10.1016/j.bj.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau S.K.P., Lau C.C.Y., Chan K.H., Li C.P.Y., Chen H., Jin D.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J General Virol. 2013;94:2679–2690. doi: 10.1099/vir.0.055533-0. [DOI] [PubMed] [Google Scholar]
- 13.Josset L., Menachery V.D., Gralinski L.E., Agnihothram S., Sova P., Carter V.S. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. mBio. 2013;4 doi: 10.1128/mBio.00165-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu G., Yang Y., Du Y., Peng F., Hu P., Wang R. Clinical pathway for early diagnosis of COVID-19: updates from experience to evidence-based practice. Clinic Rev Allerg Immunol. 2020 doi: 10.1007/s12016-020-08792-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ochs M., Nyengaard J.R., Jung A., Knudsen L., Voigt M., Wahlers T. The number of alveoli in the human lung. Am J Respir Crit Care Med. 2004;169:120–124. doi: 10.1164/rccm.200308-1107oc. [DOI] [PubMed] [Google Scholar]
- 16.Barkauskas C.E., Cronce M.J., Rackley C.R., Bowie E.J., Keene D.R., Stripp B.R. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuo Y.Y., Veldhuizen R.A.W., Neumann A.W., Petersen N.O., Possmayer F. Current perspectives in pulmonary surfactant – inhibition, enhancement and evaluation. Biochim Biophys Acta (BBA) – Biomembr. 2008 doi: 10.1016/j.bbamem.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 18.Gong M.N., Wei Z., Xu L.-L., Miller D.P., Thompson B.T., Christiani D.C. Polymorphism in the surfactant protein-B gene, gender, and the risk of direct pulmonary injury and ARDS. Chest. 2004;125:203–211. doi: 10.1378/chest.125.1.203. [DOI] [PubMed] [Google Scholar]
- 19.Aggarwal N.R., King L.S., D'Alessio F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol. 2014;306:L709–L725. doi: 10.1152/ajplung.00341.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaughan A.E., Brumwell A.N., Xi Y., Gotts J.E., Brownfield D.G., Treutlein B. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cochi S.E., Kempker J.A., Annangi S., Kramer M.R., Martin G.S. Mortality trends of acute respiratory distress syndrome in the United States from 1999 to 2013. Ann Am Thoracic Soc. 2016;13:1742–1751. doi: 10.1513/AnnalsATS.201512-841OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y. Epidemiological and clinical characteristics of 99 cases of 2019-novel coronavirus (2019-nCoV) pneumonia in Wuhan China. SSRN Electron J. 2020 doi: 10.2139/ssrn.3523861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge X.Y., Li J.L., Yang Lou X., Chmura A.A., Zhu G., Epstein J.H. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.aax0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heurich A., Hofmann-Winkler H., Gierer S., Liepold T., Jahn O., Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181 doi: 10.1016/j.cell.2020.02.052. 271–80.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziegler C.G.K., Allon S.J., Nyquist S.K., Mbano I.M., Miao V.N., Tzouanas C.N. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181 doi: 10.1016/j.cell.2020.04.035. 1016–35.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lukassen S., Lorenz Chua R., Trefzer T., Kahn N.C., Schneider M.A., Muley T. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020 doi: 10.15252/embj.20105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glowacka I., Bertram S., Herzog P., Pfefferle S., Steffen I., Muench M.O. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol. 2010;84:1198–1205. doi: 10.1128/jvi.01248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuba K., Imai Y., Rao S., Jiang C., Penninger J.M. Lessons from SARS: control of acute lung failure by the SARS receptor ACE2. J Mol Med. 2006;84:814–820. doi: 10.1007/S00109-006-0094-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verdecchia P., Cavallini C., Spanevello A., Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14. doi: 10.1016/j.ejim.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuba K., Zhang L., Imai Y., Arab S., Chen M., Maekawa Y. Impaired heart contractility in Apelin gene–deficient mice associated with aging and pressure overload. Circ Res. 2007;101 doi: 10.1161/CIRCRESAHA.107.158659. [DOI] [PubMed] [Google Scholar]
- 34.Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uhal B.D., Li X., Xue A., Gao X.u., Abdul-Hafez A. Regulation of alveolar epithelial cell survival by the ACE-2/angiotensin 1–7/Mas axis. Am J Physiol Lung Cell Mol Physiol. 2011;301 doi: 10.1152/ajplung.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tikellis C., Thomas M.C. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Peptides. 2012;2012:1–8. doi: 10.1155/2012/256294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soler M.J., Wysocki J., Ye M., Lloveras J., Kanwar Y., Batlle D. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007;72:614–623. doi: 10.1038/sj.ki.5002373. [DOI] [PubMed] [Google Scholar]
- 38.Liu C.X., Hu Q., Wang Y., Zhang W., Ma Z.Y., Feng J.B. Angiotensin-converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: a comparison with ACE inhibition. Mol Med. 2011;17:59–69. doi: 10.2119/molmed.2010.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tikellis C., Johnston C.I., Forbes J.M., Burns W.C., Burrell L.M., Risvanis J. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41:392–397. doi: 10.1161/01.HYP.0000060689.38912.CB. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y., Yang Y., Zhang C., Huang F., Wang F., Yuan J. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Science China Life Sci. 2020;63:364–374. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan R., Zhang Y., Li Y., Xia L.u., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020 doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pierrakos C., Karanikolas M., Scolletta S., Karamouzos V., Velissaris D. Acute respiratory distress syndrome: pathophysiology and therapeutic options. J Clin Med Res. 2012;4:7. doi: 10.4021/JOCMR761W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huertas A., Guignabert C., Barberà J.A., Bärtsch P., Bhattacharya J., Bhattacharya S. Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases: highlights from basic research to therapy. Eur Respir J. 2018;51 doi: 10.1183/13993003.00745-2017. [DOI] [PubMed] [Google Scholar]
- 44.Millar F.R., Summers C., Griffiths M.J., Toshner M.R., Proudfoot A.G. The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax. 2016;71(5):462–473. doi: 10.1136/THORAXJNL-2015-207461. [DOI] [PubMed] [Google Scholar]
- 45.Liu T., Zhang L., Joo D., Sun S.-C. NF-κB signaling in inflammation. Sig Transduct Target Ther. 2017;2 doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hiscott J., Kwon H., Génin P. Hostile takeovers: Viral appropriation of the NF-κB pathway. J Clin Invest. 2001;107:143–151. doi: 10.1172/JCI11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeDiego M.L., Nieto-Torres J.L., Regla-Nava J.A., Jimenez-Guardeño J.M., Fernandez-Delgado R., Fett C. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J Virol. 2014;88:913–924. doi: 10.1128/JVI.02576-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foster D.J., Ravikumar P., Bellotto D.J., Unger R.H., Hsia C.C.W. Fatty diabetic lung: altered alveolar structure and surfactant protein expression. Am J Physiol Lung Cell Mol Physiol. 2010;298:392–403. doi: 10.1152/ajplung.00041.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carsana L., Sonzogni A., Nasr A., Rossi R.S., Pellegrinelli A., Zerbi P. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. 2020 doi: 10.1016/S1473-3099(20)30434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Polak S.B., Van Gool I.C., Cohen D., von der Thüsen J.H., van Paassen J. A systematic review of pathological findings in COVID-19: a pathophysiological timeline and possible mechanisms of disease progression. Mod Pathol. 2020:1–11. doi: 10.1038/s41379-020-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Essandoh K., Li Y., Huo J., Fan G.C. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock. 2016;46:122–131. doi: 10.1097/SHK.0000000000000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bartel D.P. MicroRNAs. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 53.Cortez M.A., Calin G.A. MicroRNA identification in plasma and serum: a new tool to diagnose and monitor diseases. Expert Opin Biol Ther. 2009;9:703–711. doi: 10.1517/14712590902932889. [DOI] [PubMed] [Google Scholar]
- 54.Fan J., Fan Y., Wang X., Xie H., Gao H., Zhang Y. miR-429 is involved in regulation of NF-κBactivity by targeting IKKβ and suppresses oncogenic activity in cervical cancer cells. FEBS Lett. 2017;591:118–128. doi: 10.1002/1873-3468.12502. [DOI] [PubMed] [Google Scholar]
- 55.Vergadi E., Vaporidi K., Theodorakis E.E., Doxaki C., Lagoudaki E., Ieronymaki E. Akt2 deficiency protects from acute lung injury via alternative macrophage activation and miR-146a induction in mice. J Immunol. 2014 doi: 10.4049/jimmunol.1300959. [DOI] [PubMed] [Google Scholar]
- 56.Cao W., Li T. COVID-19: towards understanding of pathogenesis. Cell Res. 2020;30:367–369. doi: 10.1038/s41422-020-0327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spyropoulos A.C., Ageno W., Barnathan E.S. Hospital-based use of thromboprophylaxis in patients with COVID-19. Lancet. 2020;395:e75. doi: 10.1016/S0140-6736(20)30926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang N., Li D., Wang X., Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–847. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teuwen L.-A., Geldhof V., Pasut A., Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. 2020 doi: 10.1038/s41577-020-0343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Camprubí-Rimblas M., Guillamat-Prats R., Lebouvier T., Bringué J., Chimenti L., Iglesias M. Role of heparin in pulmonary cell populations in an in-vitro model of acute lung injury. Respir Res. 2017;18:1–12. doi: 10.1186/s12931-017-0572-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mussbacher M., Salzmann M., Brostjan C., Hoesel B., Schoergenhofer C., Datler H. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front Immunol. 2019;10 doi: 10.3389/FIMMU.2019.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. ChinaXiv 2020. [DOI] [PMC free article] [PubMed]
- 63.Fu B., Xu X., Wei H. Why tocilizumab could be an effective treatment for severe COVID-19? J Transl Med. 2020 doi: 10.1186/s12967-020-02339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee J., Nakagiri T., Oto T., Harada M., Morii E., Shintani Y. IL-6 Amplifier, NF-κB–triggered positive feedback for IL-6 signaling, in grafts is involved in allogeneic rejection responses. J Immunol. 2012;189:1928–1936. doi: 10.4049/jimmunol.1103613. [DOI] [PubMed] [Google Scholar]
- 65.Jones V.G., Mills M., Suarez D., Hogan C.A., Yeh D., Segal J.B., Nguyen E.L., Barsh G.R., Maskatia S., Mathew R. COVID-19 and Kawasaki disease: novel virus and novel case. Hospital Pediatr. 2020;10(6):537–540. doi: 10.1542/hpeds.2020-0123. [DOI] [PubMed] [Google Scholar]
- 66.Esper F., Shapiro E., Weibel C., Ferguson D., Landry M., Kahn J. Association between a novel human coronavirus and Kawasaki disease. J Infect Dis. 2005;191:499–502. doi: 10.1086/428291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim K.Y., Kim D.S. Recent advances in Kawasaki disease. Yonsei Med J. 2016;57:15–21. doi: 10.3349/ymj.2016.57.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yun K.W., Lee J.Y., Yun S.W., Lim I.S., Choi E.S. Elevated serum level of microRNA (miRNA)-200c and miRNA-371-5p in children with Kawasaki disease. Pediatr Cardiol. 2014;35:745–752. doi: 10.1007/s00246-013-0846-6. [DOI] [PubMed] [Google Scholar]
- 69.Shimizu C., Shike H., Baker S.C., Garcia F., van der Hoek L., Kuijpers T.W. Human coronavirus NL63 is not detected in the respiratory tracts of children with acute Kawasaki disease. J Infect Dis. 2005;192:1767–1771. doi: 10.1086/497170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zulfiqar A.-A., Lorenzo-Villalba N., Hassler P., Andrès E. Immune thrombocytopenic purpura in a patient with covid-19. N Engl J Med. 2020:1–2. doi: 10.1056/NEJMc2010472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vergadi E., Ieronymaki E., Lyroni K., Vaporidi K., Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198:1006–1014. doi: 10.4049/JIMMUNOL.1601515. [DOI] [PubMed] [Google Scholar]
- 72.Ponomarev E.D., Veremeyko T., Weiner H.L. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia. 2013;61:91–103. doi: 10.1002/glia.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Varga Z., Flammer A.J., Steiger P., Haberecker M., Andermatt R., Zinkernagel A.S. Endothelial cell infection and endotheliitis in COVID-19. Lancet (London, England) 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gilmore T.D., Herscovitch M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene. 2006;25:6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 75.Pascal L., Gay J., Willekens C., Wemeau M., Balkaran S., Robu D. Bortezomib and Waldenstrom’s macroglobulinemia. Expert Opin Pharmacother. 2009;10:909–916. doi: 10.1517/14656560902800160. [DOI] [PubMed] [Google Scholar]
- 76.Garash R., Bajpai A., Marcinkiewicz B.M., Spiller K.L. Drug delivery strategies to control macrophages for tissue repair and regeneration. Exp Biol Med (Maywood) 2016;241(10):1054–1063. doi: 10.1177/1535370216649444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Varani J., Hirschl R.B., Dame M., Johnson K. Perfluorocarbon protects lung epithelial cells from neutrophil-mediated injury in an in vitro model of liquid ventilation therapy. Shock (Augusta, Ga) 1996;6:339–344. doi: 10.1097/00024382-199611000-00007. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Z., Liang Z., Li H., Li C., Yang Z., Li Y. Perfluorocarbon reduces cell damage from blast injury by inhibiting signal paths of NF-κB, MAPK and Bcl-2/Bax signaling pathway in A549 cells. PLoS One. 2017;12 doi: 10.1371/journal.pone.0173884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaisers U., Kelly K.P., Busch T. Liquid ventilation. Br J Anaesth. 2003;91:143–151. doi: 10.1093/bja/aeg147. [DOI] [PubMed] [Google Scholar]
- 80.Khan A., Benthin C., Zeno B., Albertson T.E., Boyd J., Christie J.D. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21:234. doi: 10.1186/s13054-017-1823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oudit G.Y., Liu G.C., Zhong J., Basu R., Chow F.L., Zhou J. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2010;59:529–538. doi: 10.2337/db09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Monteil V., Kwon H., Prado P., Hagelkrüys A., Wimmer R.A., Stahl M. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181 doi: 10.1016/J.CELL.2020.04.004. 905–913.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pang X., Cui Y., Zhu Y. Recombinant human ACE2: potential therapeutics of SARS-CoV-2 infection and its complication. Acta Pharmacol Sin. 2020:1–3. doi: 10.1038/s41401-020-0430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Romaní-Pérez M., Outeiriño-Iglesias V., Moya C.M., Santisteban P., González-Matías L.C., Vigo E. Activation of the GLP-1 receptor by liraglutide increases ACE2 expression, reversing right ventricle hypertrophy, and improving the production of SP-A and SP-B in the lungs of type 1 diabetes rats. Endocrinology. 2015;156:3559–3569. doi: 10.1210/en.2014-1685. [DOI] [PubMed] [Google Scholar]