

Abstract
Damage to the genome can accelerate aging. The percentage of aneuploid cells, that is, cells with an abnormal number of chromosomes, increases during aging; however, it is not clear whether increased aneuploidy accelerates aging. Here, we report an individual showing premature aging phenotypes of various organs including early hair loss, atrophic skin, and loss of hematopoietic stem cells; instability of chromosome numbers known as mosaic variegated aneuploidy (MVA); and spindle assembly checkpoint (SAC) failure. Exome sequencing identified a de novo heterozygous germline missense mutation of c.856C>A (p.R286S) in the mitotic activator CDC20. The mutant CDC20 showed lower binding affinity to BUBR1 during the formation of the mitotic checkpoint complex (MCC), but not during the interaction between MCC and the anaphase‐promoting complex/cyclosome (APC/C)–CDC20 complex. While heterozygous knockout of CDC20 did not induce SAC failure, knock‐in of the mutant CDC20 induced SAC failure and random aneuploidy in cultured cells, indicating that the particular missense mutation is pathogenic probably via the resultant imbalance between MCC and APC/C‐CDC20 complex. We postulate that accelerated chromosome number instability induces premature aging in humans, which may be associated with early loss of stem cells. These findings could form the basis of a novel disease model of the aging of the body and organs.
Keywords: aging, Cdc20 proteins, chromosomal instability, chromosome segregation, genomic instability, M phase cell cycle checkpoints, premature
We report an individual showing premature aging, instability of chromosome numbers, spindle assembly checkpoint failure, and a heterozygous missense mutation in CDC20. The CDC20 mutant showed low binding affinity to BUBR1 in the formation of the mitotic checkpoint complex and induced spindle assembly checkpoint failure in vitro. The case is postulated to be a novel syndrome showing that accelerated chromosome number instability induces premature aging.
1. INTRODUCTION
Premature aging syndromes are rare disorders with clinical features that mimic physiological aging at an early age. The known causative genes are related to the maintenance and repair of genomic DNA (Vijg & Suh, 2013). A failure of their genomic integrity is considered a primary driving force of human aging (Vijg & Suh, 2013).
Aneuploidy, the presence of an abnormal number of chromosomes, is an example of the failure of genomic integrity. The number of aneuploid cells increases with aging in various organs (Duncan et al., 2012; Jacobs et al., 1961; Rehen et al., 2005). However, it is not known whether an increased number of aneuploid cells directly drive aging in humans. To date, three causative genes, BUBR1, CEP57, and TRIP13, have been found to induce mosaic variegated aneuploidy (MVA) syndromes that show random aneuploidy in a significant percentage of somatic cells (Hanks et al., 2004; Snape et al., 2011; Yost et al., 2017). Increased risk for carcinogenesis, congenital abnormalities, including intrauterine growth retardation, and infantile death are major symptoms in those MVA syndromes. Only tissue‐specific premature aging phenotypes, including cataracts, have been reported in those MVA syndromes, while Bubr1 hypomorphic mice show MVA and systemic premature aging phenotypes, including kyphosis, muscle atrophy, dermal thinning, heart failure, and loss of fat tissues (Baker et al., 2004).
Here, we present a patient exhibiting premature aging of various organs, early stem cell loss, and MVA. We identified a missense variant of CDC20 that occurred de novo and characterized the variant, which induced spindle assembly checkpoint (SAC) failure and MVA. The findings of this case could provide the basis for a novel disease model of human aging.
2. RESULTS
The patient, a 54‐year‐old Japanese woman (II‐1 in Figure 1a), was born healthy from healthy parents but gradually developed growth retardation without mental retardation (Figure 1b); dry and atrophic skin with hyper‐ and hypo‐pigmented macules, atrophic uvula, and oral mucosa; and hair loss on the scalp and body before she reached 20 years of age (Figure 1c‐e and Figure S1). From age 20 to 50, she developed bilateral renal atrophy, bilateral cataracts, femur head necrosis, kyphosis, anemia, cardiac insufficiency, and thyroid atrophy (Tables S1 and S2; and Supplemental Text 1). She was cancer‐free until uterine endometrial carcinoma appeared at age 48, which was completely excised and without recurrence 5 years after the operation without adjuvant therapy. Bone marrow biopsy at age 50 revealed replacement of bone marrow by fat tissue and a marked decrease in colony‐forming efficiency of hematopoietic cells (Figure 1f, g). The patient showed no neurodevelopmental, cognitive, memory, or other neurological defects. No immunodeficiency or autoimmunity was identified.
Figure 1.
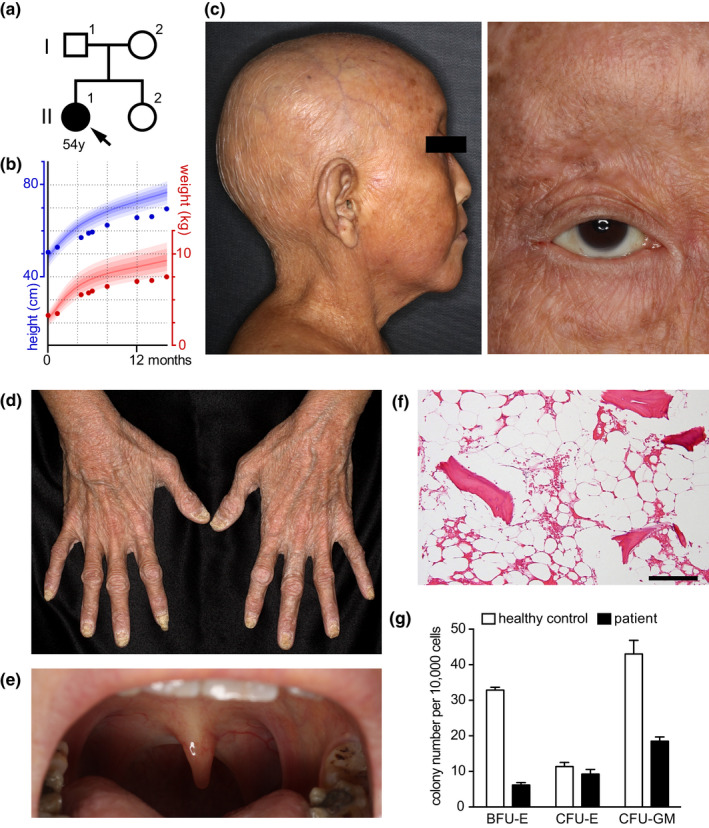
Clinical and cellular phenotypes of the patient. (a) The patient's family tree. (b) The patient's growth curve of height and weight plotted on a standard growth chart of a Japanese girl (Ministry of Health, 2010a, 2010b). Colored lines correspond to the 97th, 90th, 75th, 50th, 25th, 10th, and 3rd percentiles from top to bottom. (c–e) Clinical photographs of the patient showing the loss of scalp hair, eyelashes and eyebrows (c), dry, atrophic skin (d), and atrophic tonsils and uvula (e) at age 51. (f) Hematoxylin and eosin staining of a bone marrow biopsy section. Scale = 100 µm. (g) The numbers of colonies per 1 × 104 bone marrow mononuclear cells in the patient and an age‐matched control (means ± SEM from six technical replicates from each subject). BFU‐E, burst‐forming unit–erythroid; CFU‐E, colony‐forming unit–erythroid; CFU‐GM, colony‐forming unit–granulocytes/macrophages.
The patient showed complete female genitalia but had a history of surgical extrusion of streak gonads and renal failure, and thus, Frasier syndrome was first suspected (Barbaux et al., 1997). Karyotype analyses and genomic sequencing revealed the patient had XY chromosomes but no causative mutations of Frasier syndrome in WT1 (data not shown). Instead, the patient's PBMCs revealed MVA, a random gain and loss of various chromosomes, in ~15% of cells (38/250 cells, Figure 2a, b; Table S3) with no detectable chromosome breakage (0/250 cells). Premature chromatid separation (PCS) was observed in ~6% of cells (12/200 cells), while this was <2% in a normal cohort (Kajii et al., 1998). Micronuclei, the hallmark of chromosome missegregation, was observed in 2.5% of cells (Table S4, control: 0.7 ± 0.2% [mean ± SEM], n = 3). Treatment of the patient's PBMCs with nocodazole, a microtubule‐depolymerizing reagent, showed the accumulation of octoploid cells (Figure 2c), indicating aberrant cell cycle exit from metaphase due to SAC failure. Thus, we diagnosed the patient with MVA syndrome and decided not to administer adjuvant chemotherapy using taxanes after surgery for uterine endometrial carcinoma at the age of 48 years.
Figure 2.
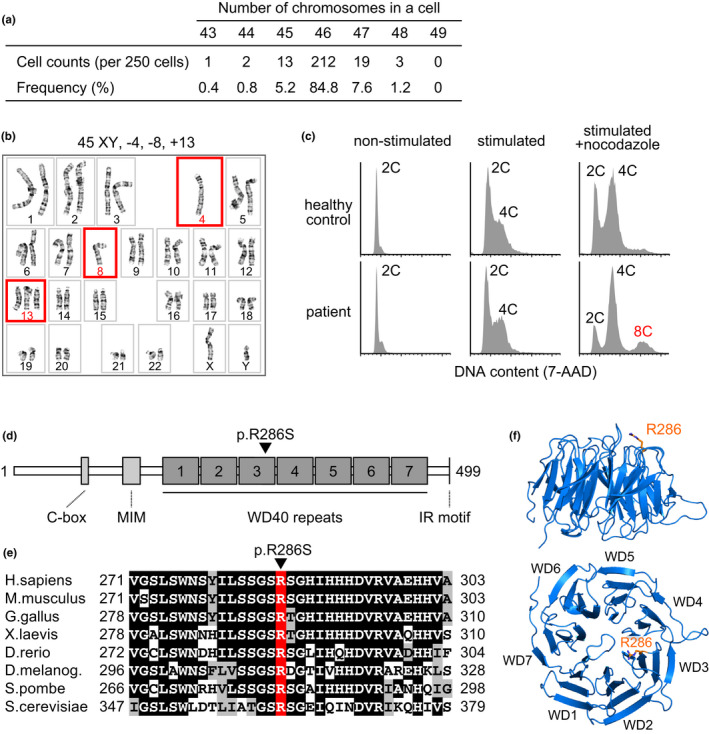
Chromosome number instability and CDC20 mutation in the patient's peripheral blood mononuclear cells. (a) Chromosome number instability observed in the patient's peripheral blood mononuclear cells (PBMCs). (b) A representative karyotype of the patient's PBMCs. Red squares indicate chromosome gain or loss. (c) Flow‐cytometric analyses of DNA contents in the PBMCs of the patient and a healthy control with or without mitotic stimulation and nocodazole treatment. X‐axes indicate nuclear DNA contents (2C, diploid; 4C, tetraploid; 8C, octoploid) stained with 7‐amino‐actinomycin D (7‐AAD) on a linear scale. (d) A schematic representation of the human CDC20 protein. The R286 amino acid residue is indicated with a black arrowhead. MIM, MAD2 interacting motif. (e) A cross‐species amino acid sequence comparison of CDC20 around the R286 residue (red). Identical and conserved amino acids are marked with black and gray boxes, respectively. (f) Side and top view of the WD40 repeat propeller domains (WD1–7) of the human CDC20 protein. The R286 residue (orange stick) is located at the top of WD3.
Typical symptoms of the known MVA syndromes caused by mutations in BUBR1, CEP57, and TRIP13 are intrauterine growth retardation, mental retardation, microcephaly, and childhood cancer (Hanks et al., 2004; Snape et al., 2011; Yost et al., 2017), none of which were observed in the patient (Table S1). Exome sequencing of genomic DNA from the patient revealed no rare variants in the causative genes of known MVA syndromes and no significant accumulation of somatic mutations. Screening for genes related to premature aging syndromes revealed that the patient was a heterozygous carrier of several rare variants for Rothmund–Thomson syndrome and Fanconi anemia (Table S5), but these are autosomal‐recessive diseases and their phenotypes differ from those of the patient. Exome sequencing revealed that the patient possessed nine homozygous, hemizygous, or compound heterozygous pairs of rare variants and three de novo germline monoallelic variants (Figure S2, Table S6). Among them, we focused on the de novo c.856C>A (p.R286S) variant in CDC20 (RefSeq: NM_001255.3/ NP_001246.2) and homozygous in‐frame deletion of c.862+3G>C (p.N235_N287del) in CENPT (RefSeq: NM_025082.4/ NP_079358.3, see Supplemental Text 2). CDC20 is a component of the mitotic checkpoint complex (MCC) and a cofactor of the anaphase‐promoting complex/cyclosome (APC/C) (Yu, 2007). CENPT is a kinetochore‐localizing protein important for kinetochore assembly and function (Nishino et al., 2013). The patient's younger sister was a heterozygous carrier of the CENPT variant and did not have the CDC20 variant (Figure S2). The CENPT variant correctly localized to the kinetochore in the Epstein–Barr virus‐transformed lymphoblastoid cell line of the patient (Figure S3). Because the biallelic defect of CENPT causes a syndrome characterized by severe growth failure and microcephaly without chromosome number instabilities (Hung et al., 2017), which differed from the patient's phenotypes (Table S7), we further investigated the CDC20 variant in this study.
CDC20 binds to BUBR1 in the formation of the MCC and in the inhibition of the APC/C via the MCC. The R286 residue of CDC20 locates at the binding site to BUBR1 and is conserved among eukaryotes (Figure 2d‐f); therefore, we examined whether the p.R286S variant affects SAC via knocking in the variation to HCT116 cells (Figure S4). Knocked‐in cells showed allele dosage‐dependent slippage from nocodazole‐induced metaphase arrest and accumulation of octoploid cells (Figure 3a and Figure S5a). By contrast, the induction of a monoallelic frameshift mutation to CDC20 did not induce SAC failure (Figure S6), suggesting that the p.R286S variant but not haploinsufficiency of CDC20 specifically induced SAC failure. Furthermore, a significant correlation was observed between the knocked‐in allele dosage of CDC20 p.R286S and aneuploid cell ratios in HCT116 clones (F(2,6) = 5.824, p = 0.0393 by one‐way ANOVA, Figure 3b and Figure S5b), although aneuploid cells were spontaneously observed in the parent clone of the HCT116 cells. These results indicate the CDC20 p.R286S variant induces SAC failure and MVA.
Figure 3.
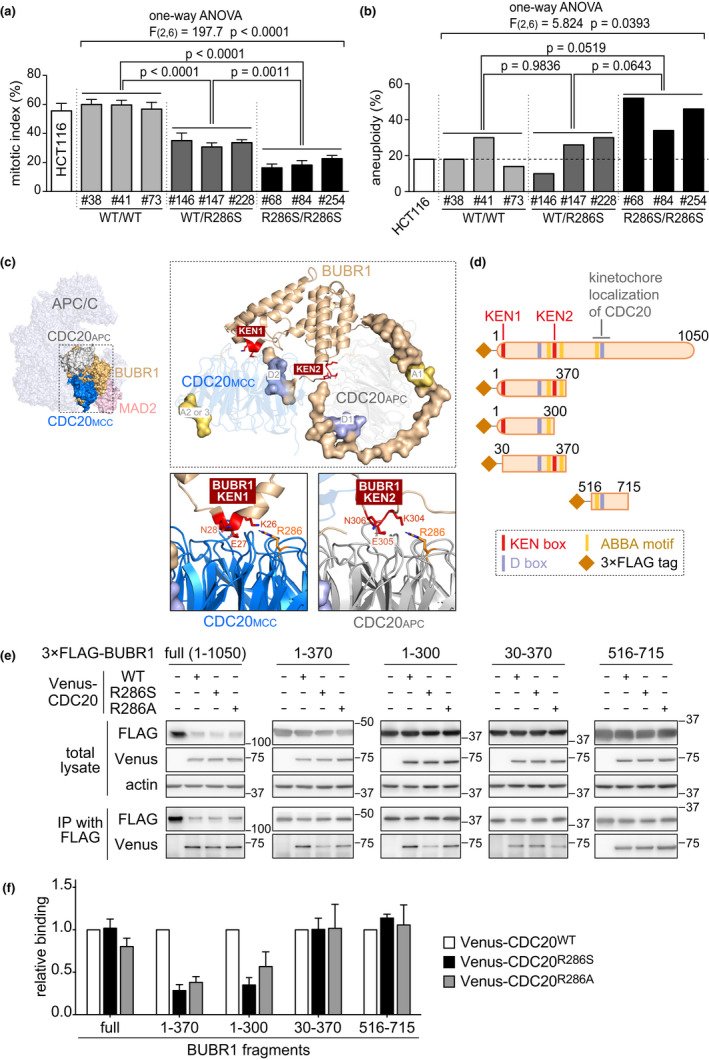
The impact of the CDC20 variant on spindle assembly checkpoint and interaction with BUBR1. (a) The mitotic indices after 12‐h nocodazole treatment of an HCT116 parent clone and its CRISPR‐mediated mutant clones harboring either the wild‐type (WT) silence mutation or the p.R286S mutation in CDC20 (means ± SEM from three independent experiments). The numbers are the code numbers of the mutant clones. (b) The percentage of aneuploid cells in the parent and mutant clones (50 metaphase spreads were counted per clone). (c) The 3D structural model of the mitotic checkpoint complex (MCC) interacting with anaphase‐promoting complex/cyclosome (APC/C) (left panel) through the binding of BUBR1 with CDC20 s consisting of the APC/C‐CDC20 complex and MCC (CDC20APC and CDC20MCC, respectively, right upper panel). KEN boxes (KEN1 and KEN2), destruction boxes (D1 and D2), and ABBA motifs (A1, A2, and A3) on BUBR1 are shown with red, blue, and yellow, respectively. The detailed spatial relationships of KEN boxes with the R286 residue of CDC20MCC and CDC20APC are shown in right lower panels. (d) Schematics of the BUBR1 deletion constructs used in immunoprecipitation. Colored lines indicate KEN1, KEN2, destruction boxes (D box; R224STL and R555RPL), and ABBA motifs (I272TVFDE, F340TPYVE, and F528SIFDE). (e) Immunoprecipitation of Venus‐tagged wild type (WT), p.R286S, and p.R286A variants of CDC20 precipitated by 3×FLAG‐tagged BUBR1 deletion constructs shown in d using anti‐FLAG antibody. Upper three rows show Western blotting of the total cell lysates, and lower two rows show Western blotting of the immunoprecipitants. Tags used for the detection and molecular weight indicators (kilodaltons) are shown on the left and right sides of the blot, respectively. The uncropped images are shown in Figure S9. (f) The relative ratios of the band densities for the p.R286S and p.R286A variants of CDC20 compared with WT CDC20 in e (means ± SEM from three independent experiments). p‐values, ANOVA/Tukey's test.
To determine how the p.R286S variant induces SAC failure, we investigated the interaction between CDC20 and BUBR1. One BUBR1 molecule binds to two CDC20 molecules, namely CDC20MCC and CDC20APC; the former is included in the MCC, which consists of BUBR1, CDC20MCC, BUB3, and MAD2. The latter is included in the APC/C‐CDC20 complex (APC/CCDC20) (Izawa & Pines, 2015). MCC binding to CDC20APC via BUBR1 inhibits the ubiquitination activity of APC/CCDC20 and the degradation of APC/C substrates, such as cyclin B1, and therefore blocks cell cycle exit from mitosis (Izawa & Pines, 2015).
The interactions between CDC20 and BUBR1 are mediated by the K26EN and K304EN boxes (KEN1 and KEN2, respectively), destruction (D) boxes, and ABBA motifs of BUBR1 (Figure 3c,d, RefSeq: NM_001211.5/ NP_001202.4) (Alfieri et al., 2016; Di Fiore et al., 2016). Among them, the predicted structural interaction model suggests KEN1 and KEN2 of BUBR1 are faced to the R286 residues of CDC20MCC and CDC20APC, respectively (Alfieri et al., 2016) (Figure 3c). An immunoprecipitation assay using FLAG‐tagged BUBR1 fragments and Venus‐tagged CDC20 (Figure 3d‐f) revealed that the N‐terminal BUBR11–370, including both KEN1 and KEN2 showed significant lower binding affinity to the p.R286S variant of CDC20. The binding affinity of the CDC20 variant to full‐length BUBR1 and the BUBR1516–715 fragment, a middle part of BUBR1 responsible for CDC20 recruitment to the kinetochores (Lischetti et al., 2014), showed no differences (Figure 3e, f), consistent with the results of the p.R286S variant showing normal kinetochore localization in vitro (Figure S7). Remarkably, BUBR11–300 including only KEN1 and BUBR11–370 including both KEN1 and KEN2, but not BUBR130–370 including only KEN2, showed reduced immunoprecipitated CDC20 p.R286S variant when compared with the immunoprecipitation of wild‐type CDC20 (Figure 3e, f). Therefore, the KEN1‐mediated binding to CDC20, but not the KEN2‐mediated binding to another CDC20, was affected in the interaction between BUBR1 and the CDC20 p.R286S variant. The CDC20 p.R286A variant, which had been reported to show lower binding affinity to BUBR1 (Tian et al., 2012) and was used as a control, showed similar specific reduction in the KEN1‐mediated binding between BUBR1 and CDC20 (Figure 3e, f). These results indicate that the R286 residue of CDC20 is specifically responsible for the KEN1‐mediated binding of BUBR1 to CDC20MCC, but not KEN2‐mediated binding to CDC20APC. The p.R286S CDC20 variant was suggested to induce aberrant activation of the APC/CCDC20, possibly via insufficient formation of MCC to inhibit APC/CCDC20, resulting in SAC failure and MVA in the patient (Figure 4).
Figure 4.
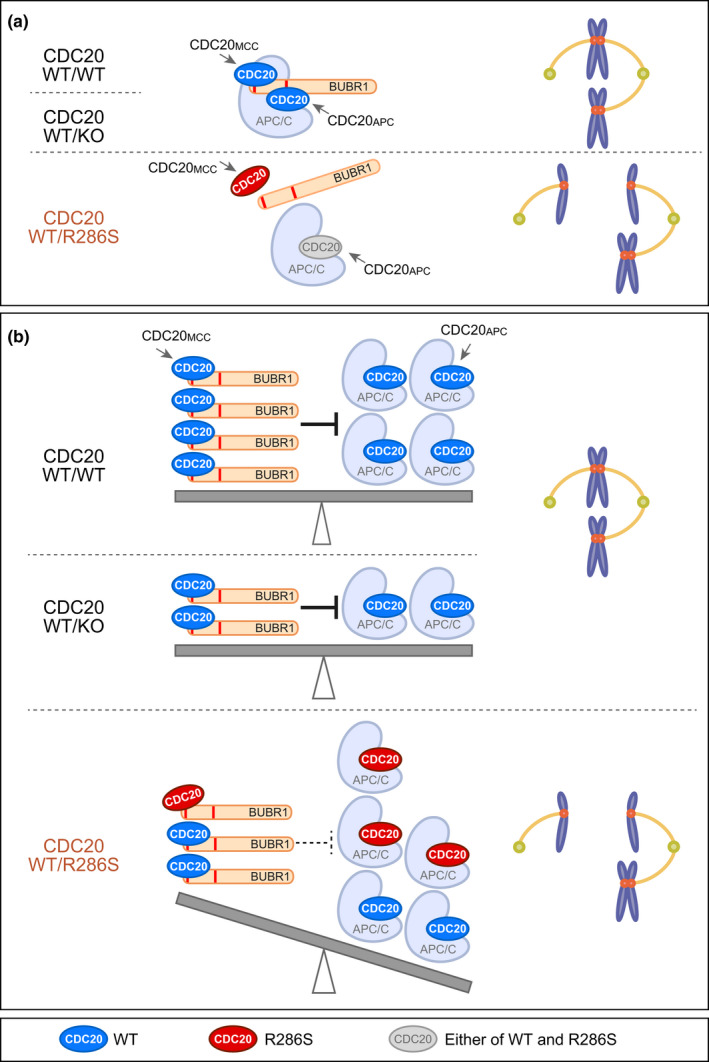
Hypothetical models of the aberrant activation of the APC/C‐CDC20 complex. Hypothetical models of APC/C inhibition by MCC focusing on the interaction of BUBR1 with wild type (blue) and p.R286S variant (red) of CDC20. BUB3 and MAD2 in MCC are not shown for simplification. (a) The increased frequency of stochastic dissociation of the p.R286S variant of CDC20MCC from BUBR1 induces aberrant dissociation of MCC from the APC/C‐CDC20 complex (APC/CCDC20), resulting in aberrant entry into anaphase and unequal chromosome segregation (WT/R286S). Haploinsufficiency of CDC20 (WT/KO) does not induce this aberrant dissociation of MCC. (b) Balanced inhibition of the APC/CCDC20 by MCC prohibits aberrant entry into anaphase in wild type (WT/WT) and in haploinsufficiency of CDC20 (WT/KO). Reduction in the binding affinity of the CDC20 p.R286S variant to BUBR1 specifically in the formation of MCC but not of the APC/CCDC20 induces a molecular imbalance between MCC and the APC/CCDC20, resulting in aberrant entry into anaphase (WT/R286S).
3. DISCUSSION
To the best of our knowledge, this is the first reported case of systemic premature aging associated with SAC failure and MVA in humans. The de novo‐occurring monoallelic p.R286S missense variant of CDC20 is considered pathogenic. The patient data suggest that aging could be accelerated by increased numbers of aneuploid cells in humans.
Several nonsense mutations of CDC20 have been registered in human exome databases (dbSNP, 2019). No SAC failure was observed in heterozygous CDC20 knockout human cells (Figure S6), suggesting that heterozygous CDC20‐knockout humans likely exist in the general population without showing SAC failure and MVA phenotype. Cdc20‐null mice were embryonic lethal at the two‐cell stage (Li et al., 2007; Manchado et al., 2010). Tamoxifen‐induced conditional knockout of Cdc20 in mice during the developmental stage revealed severe developmental defects with widespread metaphase arrest in proliferating cells caused by anaphase onset failure (Manchado et al., 2010). In contrast, patient cells investigated in this study showed SAC failure, which accelerate cells to exit metaphase and enter anaphase. Therefore, the cellular phenotypes induced by the monoallelic p.R286S missense mutation of CDC20 are different from the phenotypes induced by CDC20 haploinsufficiency or CDC20 deletion. Our results strongly suggest that a specific missense mutation of the R286 residue of CDC20 induced SAC failure and MVA by affecting the KEN1‐mediated binding of BUBR1 to CDC20MCC, but not affecting the KEN2‐mediated binding to CDC20APC. This may explain the extreme rarity of the disease because only specific missense mutations on a specific residue may cause the disease.
It is likely that instability of the MCC caused by the lower binding affinity of BUBR1 to the CDC20 p.R286S mutant resulted in insufficient inhibition of the APC/CCDC20 by MCC in the patient, for example, via aberrant early dissociation of the MCC from the APC/CCDC20 or an imbalance between the MCC and APC/CCDC20 ratio (Figure 4). Because the patient has one normal CDC20 allele, aberrant activation of the APC/CCDC20 is supposed to be less frequent in the patient than in the MVA syndrome patients caused by biallelic BUBR1 mutations. This hypothesis likely explains why the frequency and extent of MVA and PCS were relatively low in the patient compared with BUBR1‐deficient MVA syndrome patients (Table S1), and why the patient did not show severe anomalies, childhood cancer, and early death.
The phenotype of systemic premature aging and being XY female were the major clinical differences in our patient compared with known MVA syndromes (Table S1). Exome sequencing revealed no causative genetic defect related to sex determination in the patient. As XO/XY mosaicism can result in the XY female phenotype (Johansen et al., 2012), accidental loss of the Y chromosome due to MVA in the early development of the gonads may have caused the XY female phenotype in our patient. It remains unclear whether systemic premature aging observed in the patient was solely induced by the CDC20 mutation or by the combination with other genetic factors, for example, the homozygous in‐frame deletion in CENPT encoding kinetochore‐localizing protein. The biallelic defect of CENPT in humans has been reported to cause severe growth failure and microcephaly but not to cause chromosome number instabilities (Hung et al., 2017), which differed from the phenotypes of our patient. Based on our in vitro study, it is likely that SAC failure and MVA in the patient are solely caused by the CDC20 mutation. If systemic premature aging was also solely induced by the CDC20 mutation, the disease is considered a novel premature aging syndrome distinct from known MVA syndromes and premature aging syndromes. However, if the patient's phenotype is induced by the combination of CDC20 mutation and homozygous in‐frame deletion mutations of CENPT, the phenotype is considered unique to the patient. Other patients are required to determine whether the systemic premature aging phenotype is solely induced by the CDC20 mutation.
Several SAC failure mouse models, including Bubr1 hypomorphic mice, develop systemic premature aging without severe congenital anomalies and pediatric cancer (Baker et al., 2004, 2006; Wijshake et al., 2012). Various cellular stresses, including proteotoxic and metabolic stresses, are known to be induced by aneuploidy in in vitro cultured cells (Santaguida & Amon, 2015). Early exhaustion of stem cells has been observed in tissue‐specific knockout mice of Mad2, a component to form MCC with Bubr1 and Cdc20MCC (Foijer et al., 2013; Kollu et al., 2015). Bubr1 hypomorphic mice showed exhaustion of hematopoietic stem cells after serial transplantation of bone marrow cells (Pfau et al., 2016). The various premature aging phenotypes observed in the patient are likely associated with exhaustion of stem cells in highly proliferating tissues, for example, early hair loss and bone marrow hypoplasia with decreased colony‐forming efficiency of hematopoietic cells, and are associated with cellular stresses in slowly proliferating tissues, for example, kidney and thyroid failure, and cataracts.
The reason why only partial premature aging phenotypes but not systemic premature aging have been observed in the known human MVA syndromes including BUBR1 deficiency remains unclear. One possible explanation is that the premature aging phenotypes in MVA syndromes are difficult to observe in humans because they have a long lifespan but develop lethal phenotypes including childhood cancer much earlier than the development of premature aging phenotypes. In this context, it is possible that the systemic premature aging phenotypes observed in our patient were induced by MVA and could be observed because the patient did not develop cancer until age 48. Another possible explanation is that the systemic premature aging phenotype is related to the non‐mitotic function of the APC/CCDC20 associated with the maintenance of stem cells, which is shown in a model of cancer stem cells where APC/CCDC20 is associated with self‐renewal activity of stem cells by interacting with pluripotency‐related transcription factor SOX2 (Mao et al., 2015). Further studies are required to evaluate the non‐mitotic function of the APC/CCDC20 in the context of stem cell maintenance.
The present case suggests that SAC failure and chromosome number instabilities can accelerate aging in humans and is suggestive of a vicious cycle between increased aneuploid cells and aging. Further studies on the CDC20 p.R286S variant and other MVA syndrome model animals could form the basis of a novel disease model to investigate aging of the body and organs.
4. EXPERIMENTAL PROCEDURES
4.1. Ethics statement
This study was approved by the Medicine Ethics Committee of the Keio University School of Medicine. Written informed consent was obtained from the patient, the parents, and the younger sister according to the guidelines of the Institutional Review Board of Keio University School of Medicine. The patient gave written informed consent to publish the clinical images.
4.2. Extraction of genomic DNA
Genomic DNA in blood samples was isolated using the Wizard® Genomic DNA Purification Kit (Promega). Genomic DNA from patient saliva was isolated using the Oragene DNA Collection Kit (Kyodo International).
4.3. Exome sequencing
Exome sequencing was performed as described previously (Kubo et al., 2013; Takenouchi et al., 2015, 2016). Exome sequencing produced ~90,000,000 paired reads per sample, of which ~99.76% were mapped to the hs37d5 exon region of the human genome sequence assembly. The average coverage of the targeted exonic region was 97.22×, with more than 98.94% of targeted bases covered at over 10× reads. Candidate variants were screened using the autosomal dominant model (de novo mutations identified in the patient, but not in the parents) with an MAF <0.001 and the autosomal‐recessive model (homozygous, hemizygous, or compound heterozygous rare variants identified in the patient) with an MAF <0.03 in public databases including the Human Genetic Variation Database (Higasa et al., 2016), the Tohoku Medical Megabank Organization 3.5KJPNv2 (Tadaka et al., 2018), Japanese Genotype‐phenotype Archive (Kodama et al., 2015), 1000 Genomes Project (Genomes Project et al., 2015), and Exome Aggregation Consortium (Lek et al., 2016).
4.4. Sanger sequencing
The target DNA fragments were amplified by PCR using HotStarTaq DNA Polymerase (Qiagen) and subjected to direct Sanger sequencing. The sequences of PCR primer pairs are listed in Table S8.
4.5. Culture of cell lines
The 293 T cell line, HCT116 cell line, and its derived clones were cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; Bovogen, Keilor East, Australia), 100 units ml−1 penicillin, and 100 μg mL−1 streptomycin (Wako Pure Chemical). Epstein–Barr virus‐transformed lymphoblastoid cell lines (LCLs) were established from the peripheral blood of the patient and the healthy control by SRL Inc. and cultured in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% FBS, 100 units ml−1 penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B (Sigma). Each cell line was maintained at 95% humidified air, 5% CO2, and 37°C.
4.6. Plasmid construction
For knock‐in of the CDC20 p.R286S missense mutation into HCT116 cells by CRISPR/Cas9, oligonucleotides encoding single‐guide RNAs (sgRNAs) against CDC20 were inserted into the pSpCas9(BB)‐2A‐Puro (px459) vector (Addgene). The sequences of sgRNAs were as follows: sgRNA‐E2 target sequence: 5′‐TCCTCCAGTGGTTCACGTTC‐3′; sgRNA‐E3 target sequence: 5′‐GAACATCATGGTGGTGGATG‐3′. pSpCas9(BB)‐2A‐Puro (PX459) V2.0 was a gift from Feng Zhang (Addgene plasmid # 62988; http://n2t.net/addgene:62988; RRID:Addgene_62988) (Ran et al., 2013).
To construct the pCAGGS‐Venus‐hCDC20 plasmid encoding, the fusion protein of Venus (Nagai et al., 2002) and human CDC20, the human CDC20 open reading frame (ORF) was isolated from the pCMV7.1–3×FLAG‐CDC20 plasmid (Miyamoto et al., 2011) by EcoRI digestion and subcloned into the pCAGGS vector (Niwa et al., 1991) with N‐terminal Venus. The p.R286S mutation was inserted into this plasmid using the CDC20‐QC‐T/B primer pair and the QuikChange Site‐Directed Mutagenesis Kit (Stratagene). The p.R286A mutation was inserted using the CDC20 (KOD‐R286A)‐F/R primer pair and the KOD‐Plus Mutagenesis Kit (Toyobo).
To construct the pCMV7.1–3×FLAG‐hBUBR1 plasmid encoding 3×FLAG‐tagged human BUBR1, human BUBR1 ORF was PCR‐amplified from the pEGFP‐hBUBR1 plasmid (Miyamoto et al., 2011) using a primer set with a Kpn1 site at the 5′‐end and a BamHI site at the 3’‐end and then was subcloned into the pCMV7.1–3×FLAG vector (Sigma). To construct the plasmids encoding the 3×FLAG‐tagged truncated human BUBR1 proteins, BUBR1 fragments were PCR‐amplified from pCMV7.1–3×FLAG‐hBUBR1 using primer sets with a BamHI site at the 5′‐end and a NotI site at the 3′‐end. These fragments were subcloned into a modified pCMV7.1–3×FLAG vector, which had a HindIII‐BamHI‐NotI‐SmaI multi‐cloning site.
All constructs used in this study were verified by Sanger sequencing. The primer sequences are listed in Table S8. The molecular weight and antibody reactivity of the product proteins were verified by Western blotting (Figure S8).
4.7. CRISPR/Cas9‐mediated knock‐in of the mutation
A single‐cell suspension of 1 × 106 HCT116 cells was electroporated with 2.5 µg px459 vector containing the sgRNA against CDC20 and 2 µl 100 μM ssODN by Nucleofector II (Lonza). To prevent further digestion of the knock‐in allele by Cas9, four silent mutations were inserted into each ssODNs. These ssODNs included BamHI and BspEI sites for genotyping (Figure S4). The sequences used were as follows: CDC20 WT ssODN for sgRNA‐E2: 5’‐GCTCTGGCTTGCTTGCATTTGGTGCTGCCACAGAACCTGATTCCCTTCTTTCCTCCTCCAGTGGATCCCGTTCCGGACACATCCACCACCATGATGTTCGGGTAGCAGAACACCATGTGGCCACACTGAGTGGCCACAGCCAGGAAGTGT‐3’; CDC20 p.R286S mutant ssODN for sgRNA‐E2: 5’‐GCTCTGGCTTGCTTGCATTTGGTGCTGCCACAGAACCTGATTCCCTTCTTTCCTCCTCCAGTGGATCCAGTTCCGGACACATCCACCACCATGATGTTCGGGTAGCAGAACACCATGTGGCCACACTGAGTGGCCACAGCCAGGAAGTGT‐3’; CDC20 WT ssODN for sgRNA‐E3: 5’‐CTGGCTTGCTTGCATTTGGTGCTGCCACAGAACCTGATTCCCTTCTTTCCTCCTCCAGTGGATCCCGTTCTGGTCATATCCACCACCATGATGTTCGGGTAGCAGAACACCATGTGGCCACACTGAGTGGCCACAGCCAGGAAGTGTGTG‐3’; CDC20 p.R286S mutant ssODN for sgRNA‐E3: 5’‐CTGGCTTGCTTGCATTTGGTGCTGCCACAGAACCTGATTCCCTTCTTTCCTCCTCCAGTGGATCCAGTTCTGGTCATATCCACCACCATGATGTTCGGGTAGCAGAACACCATGTGGCCACACTGAGTGGCCACAGCCAGGAAGTGTGTG‐3.’
The transfected cells were plated onto two 100‐mm dishes. After 24‐h incubation, the cells were treated with puromycin (0.75 μg/ml) for 48 h and then further incubated with puromycin‐free media for 2 weeks, after which the colonies were picked. To screen for positive clones, genomic DNA was extracted from each clone by the phenol/chloroform method, and the target locus was amplified by PCR using HotStarTaq and the CDC20 ex8F/R primer pair. PCR products were further digested with BamHI or BspEI restriction enzymes and electrophoresed on agarose gels. Positive clones were further confirmed by direct Sanger sequencing.
4.8. Chromosome analyses
Karyotype analyses and premature chromatid separation analyses of the patient blood sample were performed at the LSI Medience Corporation. For chromosome count analyses of the HCT116 clones, the cell cycle was arrested with 0.12 μg/ml colcemid for 1 h and then treated with 8 ml 0.075 M KCl at room temperature for 20 min. We added 2 ml Carnoy's solution, and the cells were incubated for 10 min and washed two or three times with Carnoy's solution. The chromosomes were spread onto glass slides using a HANABI metaphase spreader (ADSTEC).
4.9. Cytokinesis‐block micronucleus (CBMN) assay
The CBMN assay was performed as described previously (Fenech, 2007). Briefly, PBMCs were isolated from the peripheral blood of the patient and healthy controls by gradient centrifugation using Lymphoprep (Axis‐Shield) and cultured in RPMI 1640 supplemented with 10% FBS, 100 units ml penicillin, 100 μg/ml streptomycin, and 30 μg/ml phytohemagglutinin (Wako Pure Chemical) at 95% humidified air, 5% CO2, and 37°C. After 44 h, cytochalasin B (Sigma) was added, and the cells were further cultured for 22–26 h. The cells were smeared onto glass slides, stained with May–Grünwald–Giemsa, and mounted with DPX mounting medium (Sigma). More than 1000 binucleated cells were examined under a BIOREVO microscope BZ‐9000 (Keyence) at ×1000 magnification to calculate the frequency of micronucleus formation.
4.10. Colony‐forming unit assay of bone marrow cells
The patient bone marrow cells were obtained from the remainder of the bone marrow aspiration sample, and mononuclear cells were isolated by gradient centrifugation using Lymphoprep (Axis‐Shield). An age‐matched normal control bone marrow mononuclear cell sample was obtained from AllCells. Cells were cultured in MethoCult H4034 Optimum (STEMCELL Technologies) at 1 × 104 or 2 × 104 cells per 35 mm dish at 95% humidified air, 5% CO2, and 37°C for 14 days, after which the colony numbers were counted.
4.11. Mitotic checkpoint analyses
For the mitotic index analyses of the HCT116 cell clones, cells were grown on coverslips (Matsunami Glass), coated with collagen (Cellmatrix; Nitta Gelatin), and treated with 200 ng/ml nocodazole (Sigma). Following incubation for 0 or 12 h, the cells were fixed with 4% paraformaldehyde (Muto Pure Chemicals, Tokyo, Japan) for 15 min at room temperature, and the DNA was stained with Hoechst 33258 or 33342 (Thermo Fisher Scientific), before being mounted with Mowiol (Merck). At least 200 cells were analyzed under a confocal laser scanning microscope LSM710 (Carl Zeiss) at ×400 magnification.
To analyze the DNA content of the PBMCs, mononuclear cells were obtained from the peripheral blood by gradient centrifugation using Lymphoprep (Axis‐Shield) and suspended in RPMI 1640 medium supplemented with 10% FBS, 100 units ml penicillin, and 100 μg/ml streptomycin. The cells were stimulated with 50 ng/ml phorbol 12‐myristate 13‐acetate (Sigma) and 1 μg/ml ionomycin (Wako Pure Chemical) at 95% humidified air, 5% CO2, and 37°C for 4 days in 96‐well round‐bottom plates. Next, 25 ng/ml nocodazole was added, and the cells were cultured for another 4 days. Harvested cells were stained with Live/Dead Fixable Aqua Dead Cell Stains (Thermo Fisher Scientific) and fixed with 4% paraformaldehyde (Muto Pure Chemicals) for 15 min on ice. Then the cells were permeabilized with 0.1% Triton X‐100 in PBS for 5 min, washed with PBS, and stained with 15 μg/ml 7‐AAD for 3 h at room temperature. The DNA contents were analyzed using a FACSCanto (Becton Dickinson) flow cytometer. For DNA content analyses of the HCT116 cell clones, the cells were treated with 200 ng/ml nocodazole (Sigma) for 36 h. The harvested cells were processed and analyzed in the same way as the PBMCs.
4.12. Immunoprecipitation analyses
The 293 T cells were plated onto a 35‐mm dish and transiently transfected with plasmids encoding 3×FLAG‐tagged truncated BUBR1 fragments and Venus‐CDC20 fusion proteins by Lipofectamine LTX with PLUS reagent (Thermo Fisher Scientific). One day after transfection, the cells were lysed with Triton X‐100‐based lysis buffer (0.5% Triton X‐100, 150 mM NaCl, 20 mM Tris‐HCl (pH 7.5), 1 mM EDTA with a 1/100 concentration of protease inhibitor cocktail (Sigma)). The lysates were sheared with a 23 G needle, incubated on ice for 30 min, and centrifuged at 20,000 × g for 10 min at 4°C. The supernatants were incubated with anti‐FLAG M2 Affinity Gel (P8340, Sigma) for 90 min at 4°C with constant rotation. The beads were washed three times with lysis buffer, boiled in SDS‐sample buffer for 5 min, and subjected to SDS‐PAGE and immunoblotting. All images were acquired with an LAS‐4000 mini (Fujifilm, Tokyo, Japan/GE Healthcare) and displayed without altering the brightness or contrast. Band densities were calculated with ImageQuant TL software (version 8.1, GE Healthcare). The following antibodies were used at the indicated dilutions: mouse anti‐DDDDK‐tag (M‐185–3, Medical and Biological Laboratories [MBL]) 1:10,000; mouse anti‐GFP (04363–66, Nacalai Tesque, Kyoto, Japan) 1:2000; rabbit anti‐CDC20 p55 CDC (H‐175, Santa Cruz Biotechnology, Dallas, TX, USA) 1:200; mouse anti‐BUBR1 (K0169–3, MBL) 1:1000; mouse anti‐beta‐actin (M177–3, MBL) 1:1000; goat anti‐rabbit IgG‐peroxidase conjugate (A0545, Sigma) 1:50,000; and goat anti‐mouse IgG‐peroxidase conjugate (P0447, Dako, Glostrup, Denmark) 1:1500. The uncropped images are shown in Figure S9.
4.13. Fluorescence imaging analyses of cultured cells
To analyze the localization of the Venus‐CDC20 fusion protein in the HCT116 cell line, cells were seeded onto coverslips (Matsunami Glass) coated with poly‐L‐lysine (Sigma) and transiently transfected with plasmids encoding WT or p.R286S mutant CDC20 protein fused to Venus fluorescent protein (Nagai et al., 2002) by Lipofectamine LTX with PLUS reagent (Thermo Fisher Scientific). At 24–48 h after transfection, the cells were fixed with 4% paraformaldehyde (Muto Pure Chemicals) for 15 min at room temperature. The DNA was stained with Hoechst 33258 (Thermo Fisher Scientific) for 30 min at room temperature, and the samples were mounted with Mowiol (Merck).
For the immunofluorescent staining of the CENPT protein, patient and healthy control LCLs were fixed with 2% paraformaldehyde (Muto Pure Chemicals) for 15 min at 37°C and smeared on glass slides. For membrane permeabilization, the slides were soaked in 0.1% Triton X‐100 in PBS for 15 min at room temperature, and nonspecific binding was blocked with 10% newborn calf serum (Thermo Fisher Scientific) and 5% normal goat serum (Dako) in PBS for >1 h at room temperature. Then, the slides were incubated with the primary antibodies for 1 h at room temperature, washed in PBS, and reacted with fluorescence‐conjugated secondary antibodies and Hoechst 33342 for 1 h at room temperature. Then, the slides were washed in PBS and mounted with Mowiol (Merck).
The images were acquired on a confocal laser scanning microscope LSM710 (Carl Zeiss) using a Plan‐Apochromat 63×/1.40 oil DIC M27 objective with a working distance of 0.19 mm, a 405 nm diode laser (30 mW), a 488 nm Argon laser (25/35 mW), and a 543 nm He‐Ne laser (1 mW). All images were processed with Imaris software (version 8.2.0; Bitplane, Zurich, Switzerland), Photoshop CS4 (Adobe), and Illustrator CS6 (Adobe). The following antibodies and reagents were used at the indicated dilutions: rat anti‐CENP‐T (D286–3, MBL) 1:200; rabbit anti‐alpha tubulin (ab52866, Abcam) 1:500; goat anti‐rat IgG‐Alexa Fluor 568 conjugate (ab175710, Abcam) 1:200; goat anti‐rabbit IgG‐Alexa Fluor 488 conjugate (A‐11034, Thermo Fisher Scientific) 1:200; and Hoechst 33258 or Hoechst 33342 (Thermo Fisher Scientific) 1:1000.
4.14. RT‐PCR
Total RNA was extracted from the patient and healthy control LCLs with TRIzol reagent (Thermo Fisher Scientific) and an RNeasy Mini Kit (Qiagen), and then subjected to DNase treatment (RNase‐Free DNase Set; Qiagen). Complementary DNA synthesis was performed with ReverTra Ace (Toyobo), and a subsequent PCR was performed with HotStarTaq DNA Polymerase (Qiagen). The primer sequences are described in Table S8.
4.15. Statistics
Statistical significance of the differences in the mitotic indices and the percentages of aneuploid cells between CRISPR‐mediated HCT116 mutant clones shown in Figure 3 were analyzed with a one‐way analysis of variance (ANOVA) and the post hoc Tukey's multiple comparison test using Prism software (ver.6; GraphPad Software).
4.16. Protein sequence alignment
Cross‐species CDC20 protein sequence alignment was generated using the ClustalW program (https://www.genome.jp/tools‐bin/clustalw) (Li, 2003) and the BoxShade 3.21 program (https://embnet.vital‐it.ch/software/BOX_form.html). We used the following protein sequences for comparison: Homo sapiens CDC20 (NP_001246.2), Mus musculus CDC20 (NP_075712.2), Gallus gallus CDC20 (NP_001006536.1), Xenopus laevis Cdc20.s (NP_001079443.1), Danio rerio Cdc20 (NP_998245.2), Drosophila melanogaster Fizzy/Cdc20 (NP_477501.1), Schizosaccaromyces pombe Slp1/Cdc20 (NP_593161.1), and Saccharomyces cerevisiae Cdc20 (NP_011399.1).
4.17. 3D modeling of the protein structure
All 3D images of the protein structures and their interactions were generated using the PyMOL Molecular Graphics System, Version 2.0.7 (Schrödinger LLC) (https://pymol.org). For Figure 2f, the 3D structural model of CDC20 was reconstituted from the published Protein Data Bank (PDB) file ID: 4GGD (Tian et al., 2012). For Figure 3c, the 3D structural model of the APC/C‐CDC20‐MCC interaction was reconstituted from the published PDB file ID: 5LCW (Alfieri et al., 2016).
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
AK conceptualized the study. TS and KN Curated the data. TS, KN, KH, and HS involved in formal analysis. HF, KK, YM, MA, and AK acquired funding. HF, TMi, SNA, TMo, and AK investigated the study. HF, TMi, and SNA designed the methodology. MA and AK administered the project. HF, TMi, SS, TMo, and AK provided resources. MA and AK supervised the study. HF and AK validated and visualized the data, and wrote the original draft. TMi, SM, MA, and AK reviewed and edited the manuscript.
Supporting information
ACKNOWLEDGMENTS
We are grateful to the patient and her family for their participation and kind contribution to this study. We thank for Nobuyo Nishimura, Satomi Aoki, and the Collaborative Research Resources of Keio University for their technical support, and Mariko Okajima for laboratory management. We also thank Aiko Shiohama and Takeshi Matsui for providing advice and John Stanley and Mayumi Ito for helpful discussions. This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant Number 18K16040) and by research grants from the Japan Agency for Medical Research and Development under “Practical Research Project for Rare/Intractable Diseases Program” (Grant Number 18ek0109301s0201) and “Program for an Integrated Database of Clinical and Genomic Information” (Grant Number JP18kk0205002).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Alfieri, C. , Chang, L. , Zhang, Z. , Yang, J. , Maslen, S. , Skehel, M. , & Barford, D. (2016). Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature, 536(7617), 431–436. 10.1038/nature19083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, D. J. , Jeganathan, K. B. , Cameron, J. D. , Thompson, M. , Juneja, S. , Kopecka, A. , Kumar, R. , Jenkins, R. B. , de Groen, P. C. , Roche, P. , & van Deursen, J. M. (2004). BubR1 insufficiency causes early onset of aging‐associated phenotypes and infertility in mice. Nature Genetics, 36(7), 744–749. 10.1038/ng1382 [DOI] [PubMed] [Google Scholar]
- Baker, D. J. , Jeganathan, K. B. , Malureanu, L. , Perez‐Terzic, C. , Terzic, A. , & van Deursen, J. M. (2006). Early aging‐associated phenotypes in Bub3/Rae1 haploinsufficient mice. Journal of Cell Biology, 172(4), 529–540. 10.1083/jcb.200507081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaux, S. , Niaudet, P. , Gubler, M.‐C. , Grünfeld, J.‐P. , Jaubert, F. , Kuttenn, F. , Fékété, C. N. , Souleyreau‐Therville, N. , Thibaud, E. , Fellous, M. , & McElreavey, K. (1997). Donor splice‐site mutations in WT1 are responsible for Frasier syndrome. Nature Genetics, 17(4), 467–470. 10.1038/ng1297-467 [DOI] [PubMed] [Google Scholar]
- Database of Single Nucleotide Polymorphisms (dbSNP) (2019). Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine. dbSNP gene ID:991, (dbSNP Build ID: 153 (Aug 8, 2019)). Retrieved from https://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=991
- Di Fiore, B. , Wurzenberger, C. , Davey, N. E. , & Pines, J. (2016). The mitotic checkpoint complex requires an evolutionary conserved cassette to bind and inhibit active APC/C. Molecular Cell, 64(6), 1144–1153. 10.1016/j.molcel.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, A. W. , Hanlon Newell, A. E. , Smith, L. , Wilson, E. M. , Olson, S. B. , Thayer, M. J. , Strom, S. C. , & Grompe, M. (2012). Frequent aneuploidy among normal human hepatocytes. Gastroenterology, 142(1), 25–28. 10.1053/j.gastro.2011.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech, M. (2007). Cytokinesis‐block micronucleus cytome assay. Nature Protocols, 2(5), 1084–1104. 10.1038/nprot.2007.77 [DOI] [PubMed] [Google Scholar]
- Foijer, F. , DiTommaso, T. , Donati, G. , Hautaviita, K. , Xie, S. Z. , Heath, E. , Smyth, I. , Watt, F. M. , Sorger, P. K. , & Bradley, A. (2013). Spindle checkpoint deficiency is tolerated by murine epidermal cells but not hair follicle stem cells. Proceedings of the National Academy of Sciences United States of America, 110(8), 2928–2933. 10.1073/pnas.1217388110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C. , Auton, A. , Brooks, L. D. , Durbin, R. M. , Garrison, E. P. , Kang, H. M. , & Abecasis, G. R. (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks, S. , Coleman, K. , Reid, S. , Plaja, A. , Firth, H. , FitzPatrick, D. , Kidd, A. , Méhes, K. , Nash, R. , Robin, N. , Shannon, N. , Tolmie, J. , Swansbury, J. , Irrthum, A. , Douglas, J. , & Rahman, N. (2004). Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nature Genetics, 36(11), 1159–1161. 10.1038/ng1449 [DOI] [PubMed] [Google Scholar]
- Higasa, K. , Miyake, N. , Yoshimura, J. , Okamura, K. , Niihori, T. , Saitsu, H. , Doi, K. , Shimizu, M. , Nakabayashi, K. , Aoki, Y. , Tsurusaki, Y. , Morishita, S. , Kawaguchi, T. , Migita, O. , Nakayama, K. , Nakashima, M. , Mitsui, J. , Narahara, M. , Hayashi, K. , … Matsuda, F. (2016). Human genetic variation database, a reference database of genetic variations in the Japanese population. Journal of Human Genetics, 61(6), 547–553. 10.1038/jhg.2016.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung, C. Y. , Volkmar, B. , Baker, J. D. , Bauer, J. W. , Gussoni, E. , Hainzl, S. , Klausegger, A. , Lorenzo, J. , Mihalek, I. , Rittinger, O. , Tekin, M. , Dallman, J. E. , & Bodamer, O. A. (2017). A defect in the inner kinetochore protein CENPT causes a new syndrome of severe growth failure. PLoS One, 12(12), e0189324 10.1371/journal.pone.0189324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa, D. , & Pines, J. (2015). The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature, 517(7536), 631–634. 10.1038/nature13911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, P. A. , Court Brown, W. M. , & Doll, R. (1961). Distribution of human chromosome counts in relation to age. Nature, 191, 1178–1180. [DOI] [PubMed] [Google Scholar]
- Kajii, T. , Kawai, T. , Takumi, T. , Misu, H. , Mabuchi, O. , Takahashi, Y. , Tachino, M. , Nihei, F. , & Ikeuchi, T. (1998). Mosaic variegated aneuploidy with multiple congenital abnormalities: homozygosity for total premature chromatid separation trait. American Journal of Medical Genetics, 78(3), 245–249. [DOI] [PubMed] [Google Scholar]
- Kodama, Y. , Mashima, J. , Kosuge, T. , Katayama, T. , Fujisawa, T. , Kaminuma, E. , Ogasawara, O. , Okubo, K. , Takagi, T. , & Nakamura, Y. (2015). The DDBJ Japanese Genotype‐phenotype Archive for genetic and phenotypic human data. Nucleic Acids Research, 43(Database issue), D18–22. 10.1093/nar/gku1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollu, S. , Abou‐Khalil, R. , Shen, C. , & Brack, A. S. (2015). The spindle assembly checkpoint safeguards genomic integrity of skeletal muscle satellite cells. Stem Cell Reports, 4(6), 1061–1074. 10.1016/j.stemcr.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo, A. , Shiohama, A. , Sasaki, T. , Nakabayashi, K. , Kawasaki, H. , Atsugi, T. , Sato, S. , Shimizu, A. , Mikami, S. , Tanizaki, H. , Uchiyama, M. , Maeda, T. , Ito, T. , Sakabe, J.‐I. , Heike, T. , Okuyama, T. , Kosaki, R. , Kosaki, K. , Kudoh, J. , … Amagai, M. (2013). Mutations in SERPINB7, encoding a member of the serine protease inhibitor superfamily, cause Nagashima‐type palmoplantar keratosis. American Journal of Human Genetics, 93(5), 945–956. 10.1016/j.ajhg.2013.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , O’Donnell‐Luria, A. H. , Ware, J. S. , Hill, A. J. , Cummings, B. B. , Tukiainen, T. , Birnbaum, D. P. , Kosmicki, J. A. , Duncan, L. E. , Estrada, K. , Zhao, F. , Zou, J. , Pierce‐Hoffman, E. , Berghout, J. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. B. (2003). ClustalW‐MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics, 19(12), 1585–1586. 10.1093/bioinformatics/btg192 [DOI] [PubMed] [Google Scholar]
- Li, M. , York, J. P. , & Zhang, P. (2007). Loss of Cdc20 causes a securin‐dependent metaphase arrest in two‐cell mouse embryos. Molecular and Cellular Biology, 27(9), 3481–3488. 10.1128/MCB.02088-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhardt Johansen, M. , Hagen, C. P. , Rajpert‐De Meyts, E. , Kjærgaard, S. , Petersen, B. L. , Skakkebæk, N. E. , Main, K. M. , & Juul, A. (2012). 45, X/46, XY mosaicism: Phenotypic characteristics, growth, and reproductive function–a retrospective longitudinal study. Journal of Clinical Endocrinology and Metabolism, 97(8), E1540–1549. 10.1210/jc.2012-1388 [DOI] [PubMed] [Google Scholar]
- Lischetti, T. , Zhang, G. , Sedgwick, G. G. , Bolanos‐Garcia, V. M. , & Nilsson, J. (2014). The internal Cdc20 binding site in BubR1 facilitates both spindle assembly checkpoint signalling and silencing. Nature Communications, 5, 5563 10.1038/ncomms6563 [DOI] [PubMed] [Google Scholar]
- Manchado, E. , Guillamot, M. , de Carcer, G. , Eguren, M. , Trickey, M. , Garcia‐Higuera, I. , & Malumbres, M. (2010). Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55alpha, delta phosphatase. Cancer Cell, 18(6), 641–654. 10.1016/j.ccr.2010.10.028 [DOI] [PubMed] [Google Scholar]
- Mao, D. D. , Gujar, A. D. , Mahlokozera, T. , Chen, I. , Pan, Y. , Luo, J. , Brost, T. , Thompson, E. A. , Turski, A. , Leuthardt, E. C. , Dunn, G. P. , Chicoine, M. R. , Rich, K. M. , Dowling, J. L. , Zipfel, G. J. , Dacey, R. G. , Achilefu, S. , Tran, D. D. , Yano, H. , & Kim, A. H. (2015). A CDC20‐APC/SOX2 signaling axis regulates human glioblastoma stem‐like cells. Cell Reports, 11(11), 1809–1821. 10.1016/j.celrep.2015.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ministry of Health (2010a). Standard growth chart of a Japanese child (height, 2010). Labour and Welfare; http://www.mhlw.go.jp/file/04‐Houdouhappyou‐11901000‐Koyoukintoujidoukateikyoku‐Soumuka/hyou2.xls [Google Scholar]
- Ministry of Health (2010b). Standard growth chart of a Japanese child (weight, 2010). Labour and Welfare; http://www.mhlw.go.jp/file/04‐Houdouhappyou‐11901000‐Koyoukintoujidoukateikyoku‐Soumuka/hyou1_1.xls [Google Scholar]
- Miyamoto, T. , Porazinski, S. , Wang, H. , Borovina, A. , Ciruna, B. , Shimizu, A. , Kajii, T. , Kikuchi, A. , Furutani‐Seiki, M. , & Matsuura, S. (2011). Insufficiency of BUBR1, a mitotic spindle checkpoint regulator, causes impaired ciliogenesis in vertebrates. Human Molecular Genetics, 20(10), 2058–2070. 10.1093/hmg/ddr090 [DOI] [PubMed] [Google Scholar]
- Nagai, T. , Ibata, K. , Park, E. S. , Kubota, M. , Mikoshiba, K. , & Miyawaki, A. (2002). A variant of yellow fluorescent protein with fast and efficient maturation for cell‐biological applications. Nature Biotechnology, 20(1), 87–90. 10.1038/nbt0102-87 [DOI] [PubMed] [Google Scholar]
- Nishino, T. , Rago, F. , Hori, T. , Tomii, K. , Cheeseman, I. M. , & Fukagawa, T. (2013). CENP‐T provides a structural platform for outer kinetochore assembly. EMBO Journal, 32(3), 424–436. 10.1038/emboj.2012.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa, H. , Yamamura, K. , & Miyazaki, J. (1991). Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene, 108(2), 193–199. [DOI] [PubMed] [Google Scholar]
- Pfau, S. J. , Silberman, R. E. , Knouse, K. A. , & Amon, A. (2016). Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes & Development, 30(12), 1395–1408. 10.1101/gad.278820.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, F. A. , Hsu, P. D. , Wright, J. , Agarwala, V. , Scott, D. A. , & Zhang, F. (2013). Genome engineering using the CRISPR‐Cas9 system. Nature Protocols, 8(11), 2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehen, S. K. , Yung, Y. C. , McCreight, M. P. , Kaushal, D. , Yang, A. H. , Almeida, B. S. , & Chun, J. (2005). Constitutional aneuploidy in the normal human brain. Journal of Neuroscience, 25(9), 2176–2180. 10.1523/JNEUROSCI.4560-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida, S. , & Amon, A. (2015). Short‐ and long‐term effects of chromosome mis‐segregation and aneuploidy. Nature Reviews Molecular Cell Biology, 16(8), 473–485. 10.1038/nrm4025 [DOI] [PubMed] [Google Scholar]
- Snape, K. , Hanks, S. , Ruark, E. , Barros‐Núñez, P. , Elliott, A. , Murray, A. , Lane, A. H. , Shannon, N. , Callier, P. , Chitayat, D. , Clayton‐Smith, J. , FitzPatrick, D. R. , Gisselsson, D. , Jacquemont, S. , Asakura‐Hay, K. , Micale, M. A. , Tolmie, J. , Turnpenny, P. D. , Wright, M. , … Rahman, N. (2011). Mutations in CEP57 cause mosaic variegated aneuploidy syndrome. Nature Genetics, 43(6), 527–529. 10.1038/ng.822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadaka, S. , Saigusa, D. , Motoike, I. N. , Inoue, J. , Aoki, Y. , Shirota, M. , Koshiba, S. , Yamamoto, M. , & Kinoshita, K. (2018). jMorp: Japanese multi omics reference panel. Nucleic Acids Research, 46(D1), D551–D557. 10.1093/nar/gkx978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenouchi, T. , Yamaguchi, Y. , Tanikawa, A. , Kosaki, R. , Okano, H. , & Kosaki, K. (2015). Novel overgrowth syndrome phenotype due to recurrent de novo PDGFRB mutation. Journal of Pediatrics, 166(2), 483–486. 10.1016/j.jpeds.2014.10.015 [DOI] [PubMed] [Google Scholar]
- Takenouchi, T. , Yoshihashi, H. , Sakaguchi, Y. , Uehara, T. , Honda, M. , Takahashi, T. , & Miyama, S. (2016). Hirschsprung disease as a yet undescribed phenotype in a patient with ARID1B mutation. American Journal of Medical Genetics Part A, 170(12), 3249–3252. 10.1002/ajmg.a.37861 [DOI] [PubMed] [Google Scholar]
- Tian, W. , Li, B. , Warrington, R. , Tomchick, D. R. , Yu, H. , & Luo, X. (2012). Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proceedings of the National Academy of Sciences of the United States of America, 109(45), 18419–18424. 10.1073/pnas.1213438109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg, J. , & Suh, Y. (2013). Genome instability and aging. Annual Review of Physiology, 75, 645–668. 10.1146/annurev-physiol-030212-183715 [DOI] [PubMed] [Google Scholar]
- Wijshake, T. , Malureanu, L. A. , Baker, D. J. , Jeganathan, K. B. , van de Sluis, B. , & van Deursen, J. M. (2012). Reduced life‐ and healthspan in mice carrying a mono‐allelic BubR1 MVA mutation. PLoS Genetics, 8(12), e1003138 10.1371/journal.pgen.1003138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost, S. , de Wolf, B. , Hanks, S. , Zachariou, A. , Marcozzi, C. , Clarke, M. , de Voer, R. M. , Etemad, B. , Uijttewaal, E. , Ramsay, E. , Wylie, H. , Elliott, A. , Picton, S. , Smith, A. , Smithson, S. , Seal, S. , Ruark, E. , Houge, G. , Pines, J. , … Rahman, N. (2017). Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nature Genetics, 49(7), 1148–1151. 10.1038/ng.3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. (2007). Cdc20: a WD40 activator for a cell cycle degradation machine. Molecular Cell, 27(1), 3–16. 10.1016/j.molcel.2007.06.009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.