

Abstract
SARS-COV-2, the novel coronavirus and root of global pandemic COVID-19 caused a severe health threat throughout the world. Lack of specific treatments raised an effort to find potential inhibitors for the viral proteins. The recently invented crystal structure of SARS-CoV-2 main protease (Mpro) and its key role in viral replication; non-resemblance to any human protease makes it a perfect target for inhibitor research. This article reports a computer-aided drug design (CADD) approach for the screening of 118 compounds with 16 distinct heterocyclic moieties in comparison with 5 natural products and 7 repurposed drugs. Molecular docking analysis against Mpro protein were performed finding isatin linked with a oxidiazoles (A2 and A4) derivatives to have the best docking scores of −11.22 kcal/mol and −11.15 kcal/mol respectively. Structure-activity relationship studies showed a good comparison with a known active Mpro inhibitor and repurposed drug ebselen with an IC50 value of −0.67 μM. Molecular Dynamics (MD) simulations for 50 ns were performed for A2 and A4 supporting the stability of the two compounds within the binding pocket, largely at the S1, S2 and S4 domains with high binding energy suggesting their suitability as potential inhibitors of Mpro for SARS-CoV-2.
Keywords: SARS-CoV-2, heterocyclic inhibitors, density functional theory, molecular docking, molecular dynamics
Graphical Abstract

Communicated by Ramaswamy H. Sarma
1. Introduction
During December 2019, a novel coronavirus named “Severe Acute Respiratory Syndrome Corona Virus 2 (SARS-CoV-2), caused an outbreak of a respiratory disease in the city of Wuhan, capital of the Hubei province in China and since has been spreading globally (Wu et al., 2020b; Zhou et al., 2020) and the pulmonary disease caused by SARS-CoV-2 is known as COVID-19. The virus was named SARS-CoV-2 (Gorbalenya et al., 2020) due to the resemblance of the RNA genome with the previous coronavirus in 2003, i.e. the SARS-CoV. Both viruses belong to the clade b of the genus Betacoronavirus (Wu et al., 2020b; Zhou et al., 2020). Since the outbreak, the incredible capability of the virus to spread from human-to-human led to an exponential growth in the number of patients with world-wide spreading of the virus. According to the World Health Organization (WHO), the virus infects most of the patients with mild symptoms such as fever, cough and difficulty in breathing that are cured with proper care. However, in older patients or in patients with underlying health conditions, the disease can progress into fatal pneumonia and acute respiratory failure (Chen et al., 2020a; Zhou et al., 2020). The progress of the virus was due to its novel transfer from human to human through cough, sneeze, touch from an infected person i.e. through the common droplet infection (Chen et al., 2020c; Wu et al., 2020b; Zhou et al., 2020). As of July 2nd, 11,000,000 cases and over 525,000 deaths worldwide exerting an enormous social impact and severely effecting world economic status, as previous pandemics (Keogh-Brown & Smith, 2008).
Currently, many researchers have been working to develop specific drugs through repurposing of drugs for diseases like HIV, Ebola, malaria etc. (Fischer et al., 2020; Wang et al., 2016; Xu et al., 2020b; Zhang et al., 2020a). Still these significant developments have yet to achieve prevention of disease spreading. A key strategy for treatment viral infection has been inhibiting its main protease, such inhibitors are in clinical use as effective treatments for human immunodeficiency virus (HIV) and hepatitis C (Ghosh et al., 2016; Yang et al., 2006).
The SARS-CoV-2 virion (Scheme 1) has four structural proteins (Wu et al., 2020a) spike proteins (S), Envelope (E), Membrane protein (M) and nucleocapsid proteins (N) which holds the RNA genome, similar to the other coronaviruses. The Spike protein and Mpro (also called as 3CLpro) are the two main sites for drug development for SARS-CoV-2. Mpro plays a key role in mediating viral replication and transcription (Liu & Wang, 2020; Xu et al., 2020b; Jin et al., 2020b; Xu et al., 2020b). Mpro operates at 11 cleavage sites on the large polyprotein 1ab (replicase 1ab, ∼790 kDa), inhibiting the activity of this enzyme directly blocks viral replication (Jin et al., 2020b). As there are no human proteases with a homolog of Mpro, it is an ideal target for drug design as inhibitors are less likely to be toxic for humans (Zhang et al., 2020c). Thus, several studies describing Mpro inhibitors have been recently published (Chen et al., 2020b; Fan et al., 2020; Jin et al., 2020b; Liu & Wang, 2020; Raugi et al., 2016; Xu et al., 2020b).
Scheme 1.

Schematic representation of the structure of SARS-CoV-2.
The crystal structure of SARS-CoV-2 Mpro was recently solved and deposited to the protein data bank (PDB-6LU7) enabling rational design of specific inhibitors (Bzówka et al., 2020). There is a strong resemblance between SARS-CoV-2 and SARS-CoV-1, thus, many researchers were motivated to develop drugs based on their activities of the SARS- CoV-1 (Dai et al., 2020; Xu et al., 2020b). While some of the Mpro inhibitors are in preclinical or early clinical stages, there have been no approved treatment yet (Li et al., 2020; Pillaiyar et al., 2016). Hence, we were encouraged to perform a virtual screen of inhibitors that have a wide structural diversity and include a variety of structures to generate a large pool of potential Mpro inhibitors.
According to our literature survey, the presence of heterocyclic moieties was found in most of the potential drugs designed inhibitors for SARS-CoV-2 Mpro. Here, we have screened a wide variety of heterocyclic compounds including 118 previously studied anticancer, anti-bacterial and anti-microbial compounds in comparison to the published Mpro inhibitor N3 along, 5 natural products and 7 repurposed drugs.
1.1. Selection of candidate drugs that were screened
The 7 repurposed drugs are presently used by various researchers and medical institutes for the treatment of COVID-19, like Chloroquine (Cortegiani et al., 2020; Mallikarjuna et al., 2020), Hydroxychloroquine (Gautret et al., 2020), Remdesivir (Scavone et al., 2020), Favipiravir (Shiraki & Daikoku, 2020), Niclosamide (Xu et al., 2020a), Ebeselen (Jin et al., 2020a) and Eidd-2801 (Sheahan et al., 2020). Five natural compounds like Ursodeoxychloric acid, Quercetin, Kaempferol, Pinocembrin and Rutin are also included in this study. Chloroquine and Hydroxychloroquine are known drugs for treatment of malaria (Ben-Zvi et al., 2012; Bhattacharjee, 2016; Cortegiani et al., 2020). Remdesivir and Favipiravir are antiviral drugs potentially against several viruses (Scavone et al., 2020; Shiraki & Daikoku, 2020).
The broad spectrum antiviral drug Remdesivir, developed by the Gilead Sciences and the anti malaria drug Chloroquine were recently studied in an in vitro research work by G. Xiao et al., to be effective inhibitors for the 2019-nCoV (Wang et al., 2020). Chloroquine was also previously predicted to be an inhibitor for the SARS-CoV-1 (De Clercq, 2006). The other malaria drug Hydrochloroquine was also found to be effective in inhibiting SARS-CoV-2 infection in vitro and attenuate inflammatory response as given in a recent study (Liu et al., 2020). An experimental antiviral drug developed for the treatment of influenza Eidd2801 was also repurposed for SARS, MERS and SARS-CoV-2, still in pre-clinical trials though very fruitful results are not expected (Sheahan et al., 2020). Niclosamide, a drug designed to treat tapeworm infestations, is known for its broad antiviral activity, recent studies are performed to understand its activity against SARS-CoV-2 (Xu et al., 2020a). The natural compound Ursodeoxychloric acid is a mammalian bile acid found in bear and is used in treatment of cholesterol (Einarsson, 1994). Quercetin, Kaempferol, Pinocembrin are natural flavonoids found in several fruits and vegetables and used as antioxidants (Wang et al., 2018). Pinocembrin is also found in honey (Rasul et al., 2013). Rutin is a bioflavonoid, found in many citrus food and have powerful antioxidant properties (Enogieru et al., 2018).
Many of the screened drug molecules for our docking studies were taken from previously studied drugs, which were designed for anticancer activities. Some of them were also known for their antimicrobial and antibacterial activities (Afifi et al., 2017; Agarwal et al., 2016; Beedie et al., 2015; Bhatt et al., 2020; Blair et al., 2007; Fischmann et al., 2008; Gerova et al., 2016; Ivanova et al., 2007; Kok et al., 2008; Lane et al., 2001; Mendel et al., 2003; Pessoa et al., 2010; Schelman et al., 2011; Yadagiri et al., 2015; Zhang et al., 2011). Many of these drugs are already FDA approved drugs like Sunitinib (Bhatt et al., 2020) and many are in their pre-clinical trials like Zibotentan (Schelman et al., 2011).
Compounds based on oxidiazoles and isatin moieties, well known for vigorous research for anticancer, antiparasitic activities and such type of compounds were also screened on the basis of their structure similarities (Bhatt et al., 2020; Ivanova et al., 2007; Lakshmithendral et al., 2019; Mendel et al., 2003; Serafim et al., 2017; Venkatarao et al., 2019; Zhang et al., 2011). Other heterocyclic moieties with azo based structures and antibacterial, antitumor or anticancer activities were also screened (Bhatt et al., 2018; Jha & Ramarao, 2018; Tamokou et al., 2016). Antimicrobial compounds based on benzylpyrrolidine groups have also been screened for our cause (Sreekanth & Jha, 2020). As for comparison our in-silico screen included the very recent virtually screened 11 compounds that showed inhibitors of SARS-CoV2 (Fischer et al., 2020). In addition we selected compounds with a new heterocyclic ring system, piperdin-4-one semicarbazones, that exhibits moderate antibacterial and antifungal activities (Anand et al., 2019). A recent study by Zhang et al. (2020b) showed that α-keto amide inhibitors are very important inhibitors for MERS and predicted it to be better for SARS-CoV-2 as well. We have chosen the best structure (A118) for our docking analysis.
Our docking analyses showed high interaction of 12 novel heterocyclic drug moieties with SARS-CoV-2 main protease Mpro that can serve as potential novel drug candidates. Further Molecular dynamic (MD) analyses of the two best docked compounds A2 and A4, both derivatives of isatin and oxadiazole, showed that the interactions for these compounds are mostly governed by the hydrophobic and electrostatics interactions. The electrostatic charges and Molecular electrostatic potentials were also calculated for these compounds using Density Functional Theory (DFT) to understand their charge electrostatic charge, shape and the surface of the ligands. These calculations were done to predict the electronic distribution of the ligand which is one of the most important factor for analysing the binding between the protein and the ligand. Drug like properties of the compounds, ADME profile analysis, were also performed for these compounds.
This study suggests two compounds that bind Mpro extremely potently in-silico and may serve as the basis for potent inhibitors of COVID-19. Our calculations also shed light on the shape, electrostatic charge and surface of the binders showing adequate ADME properties.
2. Result and discussion
For our virtual screen of inhibitors of Mpro we selected structures of a library of newly designed compounds, A1-A118, in addition to natural products used as antiviral, anticancer, antibacterial drugs and COVID-19 drugs in clinical trials (Figure S1), as discussed in the introduction. All the structures were optimized with Gaussian 16 software at B3LYP/6-31 + G(d) level of theory (Gibbs free energy and the zero point energies are also given in Figure S1). Drugs used in clinical trials with specific crystal structures were taken directly without optimizing. All the structures were subjected to the docking analysis with the main protease Mpro of the SARS-CoV-2, PDB id: 6LU7. The free energy of binding (kcal/mol) and inhibitory constant (Ki) for these compounds are given in Table 1. The active site of Mpro is consists of five sub-sites (S1, S2, S1’, S2’, S4). S1 site consists of residues (Phe140, Leu141, Gln166, His163, Met165 and His172). The bulky hydrophobic S2 subsite (Met49 and Asp187, Glu189) (Wang et al., 2016), while S1’ (Thr25, Leu27, Cys38, Pro-39, Val42, and Cys145) is essential for catalysis (Xue et al., 2008), S2’ (Thr-26, Asn-28, Tyr-118, Asn-119, and Gly-143) and flexible S4 site (Leu167, Gln192) in nature (Mittal et al., 2020; Wang et al., 2016).
Table 1.
The calculated Ki, Free binding energy and bond length of hydrogen bonds and π-π interaction of selected FDA approved drugs, natural products containing heterocyclic moieties compared with the top 12 compounds with the best binding affinity to SARS-Co-V-2 Mpro.
| S. No. |
Compound (Pubchem ID) |
Inhibitory Constant (Ki) |
Free binding energy (kcal/mol) |
H-bond and π-π interactions (bond length in Å) |
|---|---|---|---|---|
| 1 | N3 (native drug) | 307.96 nm | −8.88 | Cys145 (1.91 Å) |
| 2 | Ebselen (3194) | 18.66 μm | −6.45 | Glu166 (2.18 Å), Cys145 |
| 3 | Favipiravir (492405) | 378.76 μm | −4.67 | – |
| 4 | Remdesivir (121304016) | 676.24 nm | −8.42 | Glu166-NH (2.09 Å), Glu166-OH (1.92 Å) |
| 5 | Chloroquine (2719) | 7.27 μm | −7.01 | Glu166 (2.04 Å), Arg188 (1.78 Å) |
| 6 | Hydroxychloroquine (3652 ) | 3.80 μm | −7.39 | His164 (1.88 Å), Ser144 (2.21 Å), Leu141 (1.69 Å) |
| 7 | EIDD-2801 (145996610) | 8.29 μm | −6.93 | His41 (2.02 Å), Gly143 (2.21 Å), Ser144 (2.02 Å), Cys145 (2.11 Å), His 163 (2.14 Å), |
| 8 | Niclosamide (4477) | 2.66 μm | −7.60 | Gly143(1.76 Å), Cys145(2.16 Å), Glu166 (2.068 Å), |
| 9 | Ursodeoxycholic acid (31401) | 123.64 nm | −9.42 | Leu141 (2.22 Å), Met163 (2.16 Å), His164 (2.20 Å), Gln192 (2.09 Å) |
| 10 | Quercetin (5280343) | 1.50 μm | −7.94 | His164 (2.23 Å), Glu166 (2.12 Å), Asp187 (2.02 Å), |
| 11 | Kaempferol (5280863) | 1.47 μm | −7.96 | Glu166 (2.07 Å), Asp187 (2.05 Å), Thr190 (1.92 Å), Gln192 (2.026 Å) |
| 12 | Pinocembrin (68071) | 2.07 μm | −7.76 | Thr190(1.73 Å) |
| 13 | Rutin (6728944) | 1.11 μm | −8.12 | Asn142 (2.10 Å), His164(2.15 Å), Glu166 (1.93 Å), Thr190 (2.06 Å), Gln189-NH (1.79 Å), Gln189 C = O (2.19 Å), Gln189 C = O (2.08 Å). |
| 14 | A1 | 15.17 nm | −10.67 | His163 (2.13 Å), Glu166 (2.03 Å) (π-π interactions with Phe140 and Tyr54) |
| 15 | A2 | 5.94 nm | −11.22 | Glu166 (2.14 Å), Ser144, OH, (1.89 Å), Ser144, NH (1.99 Å) |
| 16 | A4 | 6.66 nm | −11.15 | Cys145 (2.16 Å), His164 (2.21 Å), (π-π interaction with His172) |
| 17 | A5 | 30.51 nm | −10.25 | His163 (1.73 Å), Thr190 (1.87 Å), Glu192 (1.94 Å) |
| 18 | A7 | 13.08 nm | −10.75 | Glu166 (2.08 Å), His164 (2.13 Å), His163 (2.01 Å), (π-π interaction with Phe140) |
| 19 | A8 | 9.10 nm | −10.97 | His163 (1.66 Å), Glu166 (2.02 Å), Glu192 (2.8 Å), Thr190 (1.87 Å) |
| 20 | A9 | 22.20 nm | −10.44 | Glu166 (2.09 Å), Gln192 (1.85 Å) |
| 21 | A11 | 22.64 nm | −10.43 | Gly13 (2.24 Å), Thr190 (1.86 Å), Glu192 (2.14 Å) |
| 22 | A12 | 10.84 nm | −10.87 | Gln189 (2.27 Å), Glu166 (2.15 Å), Glu192 (1.68 Å) |
| 23 | A20 | 11.33 nm | −10.84 | Gly143 (1.74 Å) |
| 24 | A38 | 46.88 nm | −10.00 | Glu166 (1.99 Å), His163 (1.93 Å) |
| 25 | A40 | 23.39 nm | −10.41 | Glu166 (2.12 Å) |
| 26 | A68 | 11.67 nm | −10.82 | Glu166 (2.23 Å) |
Our obtained results were also compared with the recently published inhibitors for possible interaction with 6LU7-the main protease (Mpro) of the SARS-CoV-2. The docking analyses were also performed for the native drug N3 (−8.88 kcal/mol) and ebselen (−6.45 kcal/mol) as given in Table 1. Furthermore, we compared these to the FDA approved drugs Remdesivir (−8.42 kcal/mol), Chloroquine (−7.01 kcal/mol); Hydroxychloroquine (−7.39 kcal/mol); Niclossamide (−7.60 kcal/mol) and EIDD-2801 (−6.93 kcal/mol). The natural flavonoids Quercetin (−7.94 kcal/mol), Kaempferol (−7.96 kcal/mol), Pinocembrin (−7.76 kcal/mol) and Rutin (−8.12 kcal/mol) also showed very similar scores like the FDA approved drugs (Table 1). Ursodeoxychloric acid (−9.42 kcal/mol) showed a higher docking score than all the other natural products and the FDA approved drugs featured (Table 1). Our calculations (−8.42 kcal/mol) with AutoDock4.2 showed comparable interactions with the main protease of the SARS-CoV-2 with the Remdesivir drug by Ji et al., using Schrodinger docking suites (−7.215 kcal/mol) (Hall Jr & Ji, 2020).
2.1. Orientation and binding interaction of A2 and A4
The two best inhibitors found from our docking studies were A2 (−11.22 kcal/mol) and A4 (−11.15 kcal/mol) with calculated Ki of 5.94 nM and 6.66 nM, respectively, see structures in Table S1. The oxodiazole moiety of A2, oriented toward the S1 site and establishes a hydrogen bond with residue Glu166, and weakens the salt bridge between positively charged N-terminal Ser1 (one monomer) of the negatively charged Glu166 (another monomer), thus destablilizes the Mpro pocket (Huynh et al., 2020) this may be the reason for increasing the potency of compound A2. The Ebselen a Mpro inhibitor FDA approved drugs Remdesivir, Chloroquine and Niclosamide were also formed a similar type of hydrogen bonding with Glu166. The the Phenol segment of A2 has formed two H-bond with Ser144 by fully occupying S1' subsite. The chlorobenzene segment orients toward the S4 pocket, while the isatin segment is oriented towards the bulky hydrophobic S2 site (Figure 1). Compound A4 showed a different picture with its benzo[d]oxazol-2(3H)-1 segment, the benzyl moiety orienting towards the S2’ subsite while the oxazole moiety has accommodated well in S1’subsite by forming a strong hydrogen bond between the ring oxygen with the catalytic Cys145 residue; The chlorobenzene segment near oxazole moiety orients toward the S1 site and form a π-π interaction with the residue His172. The connecting 2° amine segment of the A4 is oriented towards the bulky hydrophobic S1 site and forms a hydrogen bond with the residue His164. The chlorobenzene segment near oxadiazole moiety orients towards the S4 pocket (Figure 1). In Figure 1, the other higher docked compounds like A2, A4, A8 and A20 are also represented. The best docked compounds A2 and A4 have been selected for further analysis with MD simulations using GROMACS.
Figure 1.

The binding mode of the four best docked compounds in the active site of the SARS-CoV-2 virus Mpro (PDB ID: 6LU7). The interacted amino acid residues and the distances in Å are given in yellow. Top, the ligands are shown together in the binding pocket. At bottom, 4 panels, individual compounds docked into the binding site of SARS CoV-2 virus Mpro. A2: Saffron; A4: Sky; A8: Ocean blue; A20: Dark brown.
2.2. Structural activity relationship
In this article, we performed screening of 16 distinct heterocyclic skeletons (Table 2) with a total of 118 compounds, 5 natural products and 8 repurposed drugs (including Ebselen, which has a similar structure to isatin (Jin et al., 2020b)) (Figure 2). The docking analysis of several isatin compounds A2, A4, A8, A20, showed the best interaction with the Mpro (above −10.84 kcal/mol) as presented in Table 1 and Figure 1. In addition, highly potent compounds with Ki under 10 nM, A2, A4 and A8 were comprised of three separate ring systems, branched from a central carbon enabling multiple binding interactions with Mpro.
Table 2.
Structural activity relationship of designed compounds.
| 1 | A1 to A3 | 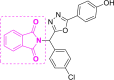 |
A2 (−11.22) |
| 2 | A4 to A6 | 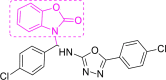 |
A4 (−11.15) |
| 3 | A7 to A12 | 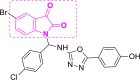 |
A8 (−10.97) |
| 4 | A15 to A29 |  |
A20 (−10.84) |
| 5 | A30 to A37 and A38 to A40 | 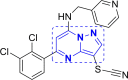 |
A39 (−10.41) |
| 6 | A42 to A45 and A88 to A92 | 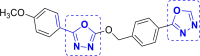 |
A91 (−9.60) |
| 7 | A94 to A101 | 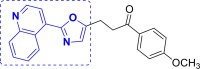 |
A95 (−9.58) |
| 8 | A75 to A87 | 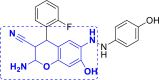 |
A86 (−9.47) |
| 9 | A57 to A63 |  |
A58 (−9.44) |
| 10 | A102 |  |
A102 (−9.44) |
| 11 |  |
Ursodeoxycholic acid (−9.42) | |
| 12 | A50 to A56 |  |
A51 (−9.23) |
| 13 | A103 to A105 | 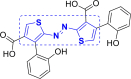 |
A105 (−8.96) |
| 14 | A106 to A112 | A107 (−8.94) | |
| 15 | A47 | 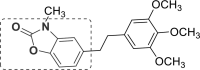 |
A47 (−8.07) |
| 16 | A48 and A49 | 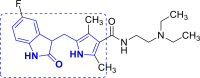 |
A48 (−7.98) |
| 17 | A113 to A117 | 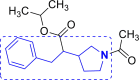 |
A117 (−7.30) |
Figure 2.

IC50 and binding energies of potent compounds similar to Ebselen - a potent published SARS-CoV-2 Mpro inhibitor (Jin et al., 2020b). The top 3 were identified for SARS-CoV1 (Chen et al., 2005; Zhou et al., 2006).
According to a literature survey, isatin based compounds are well known for their anticancer (Bhatt et al., 2020; Popp, 1969) and antibacterial activities (Pandeya et al., 2000; Varma & Nobles, 1967). They have also been known for their antiviral activities for a various pathogen virus (Bauer & Sadler, 1960; Burger, 1979)89,90 including HIV (Pauwels et al., 1988; Teitz et al., 1994) and SARS-CoV-1 (Selvam et al., 2008). Oxadiazole, based derivatives have also been found very important for potent biological functions. Especially antiviral, anti-HIV, anti-HCV, anti-HBV, anti-HSV activities, etc (Dong et al., 2016; Li et al., 2011). Triazole derivatives also occupy a pivotal position in modern medicinal chemistry and several derivatives are well known for their applications in medicine especially anticancer and antiviral drugs (Kharb et al., 2011; Xu et al., 2019). The specific reason behind the isatin; oxadiazole and triazole scaffold acting as potential anticancer, antiviral drugs are their strong tendency to accept multiple hydrogen bonds from their target protein. The Oxidiazole based compounds A1, A2, A4, A5, A7-A9, A11 & A12 and triazole based compound A20 showed good binding energies and Inhibition constants. Previously designed Cyclin-dependent kinases inhibitors known for their anticancer activity, A38 and A40 also showed high binding interaction with the Mpro of the SARS CoV-2, though their inhibition constants were higher than the isatin oxidiazoles/triazoles based compounds (Table 1). Compound A68, also showed high scores in our calculation (−10.82 kcal/mol), also comprised of a three separate ring system. A68 inhibitor was already reported by Fischer et al.10. Other compounds with very efficient docked scores that were subjected to further analysis, from our docking calculations are A1 (−10.67 kcal/mol); A5 (−10.25 kcal/mol); A7 (−10.75 kcal/mol); A8 (−10.97 kcal/mol); A9 (−10.44 kcal/mol); A11 (−10.43 kcal/mol); A12 (−10.87 kcal/mol); A20 (−10.84 kcal/mol); A38 (−10.00 kcal/mol) and A40 (−10.41 kcal/mol). All other compounds with lower docked scores are given in the supporting information (Table S2). A2 (−11.22 kcal/mol) and A4 (−11.15 kcal/mol) compounds showed the highest docked scores and were subjected to further analysis.
2.3. Chelpg interaction
Molecular recognition between a protein and small molecules (ligand) occurs at their surfaces. The extent of binding forces in the protein-ligand complex largely depends on the electronic distribution of the ligand which can be predicted by the electrostatic charges of the ligand molecule. The electrostatic charges of the electronegative atoms of the A2 and A4 ligands are given in Figure 3. Such molecular details of the ligands helps predicting the binding probabilities of the ligands in the S1, S2, S1’, S2’ and S4 pockets as discussed above. Our charge analysis shown that more electronegative atoms are present in A2 predicting it to interact better with the protein than A4. The ChelPG charges of the electronegative atoms for the other potential ligands are given in the supporting information (Figure S1). As observed from the charge analysis, A1-an analogue of A2, showed less interaction to Mpro due to the presence of chlorine atom in place of the hydroxide. A5, an analogue of A4, showed low interactions despite of the presence of hydroxide ion. This may be due to the steric hindrance between the isatin and the chlorobenzene occurring, owing to the flexibility of bonds. A7, A8, A9, A11 and A12 having the similar structures showed very comparable interactions. The ChelPG charge analysis could not be performed for A7, A8 and A9 ligands due to restrictions in the Gaussian format with presence of bromine atom. The ligand A20, having a distinct structure with a hydrophobic end, showed higher interaction to the protein. The ChelPG charges of the other important ligands (A38, A40 and A68) with good interactions to the Mpro are also given in the Figure S1 in the supporting information.
Figure 3.

The ChelpG charge of the electronegative atoms for both the compounds A2 and A4 are given here. The circle in coloured lines and the Sj (j = 1,1',2,2',4) term denotes the pockets where these sections of the compounds may interact in the Mpro protein. Carbon: Ash; Oxygen: Red; Chlorine: Green; Nitrogen: Blue; Hydrogen: White.
2.4. Molecular electrostatic potential analysis
Molecular electrostatic potential calculations (MESP) were performed for A2 and A4 including the shape and surface of the compound since electronic distribution in the compound is one of the most important factor to predict the binding to a protein. In the MESP surface below, the electronegative atoms are shown in red and they act as regions for H-bond acceptor, while the electron poor atoms are designated in blue, which act as H-bond donors. The green colour portions are neutral in nature and are regions where π- and other types of π-staking interactions are important (Figure 4). The MESP analysis shows A2 can form at least three hydrogen bonds, while A4 can form two (red patches). The electron density of the A2 was found to be higher than the A4 (green to yellow). The incorporation of an OH group in A2, rather than a Cl as in A4, is the reason behind the increasing electron density of A2, which should increases its binding interactions. Such enhancement of the electron density is favourable for the π-staking interaction. The Cl atom for both compounds also occupies a large electron density surface, showed in green, with negative values which is also suitable for multiple π-staking interactions. The small red, yellow and blue patches on the large green surface of compounds are balancing the hydrophilic and hydrophobic parts which are essential for good binding to the protein. The MESP analysis of the other ligands as given in Table 1 are also calculated and are given in the Figure S2, in the supporting information. The red, yellow and blue patches on the large green surface of compounds in these ligands are observed and the interactions patterns are clear from the MESP.
Figure 4.

The calculated Molecular Electrostatic Potential surface for the A2 and A4 compounds showing the electron density surface. The red patches hydrogen bonding donor and blue hydrogen bond acceptors sites. π-interaction sites are given as green color and yellow patches showed higher electron density than the green sites. The surfaces shown correspond to an isosurface value of 0.03 electrons/a.u.3.
2.5. Molecular dynamics simulation
Further calculations using MD simulation tools were performed for A2 and A4 to check the interaction analysis and binding ability with the Mpro protein of the SARS-CoV-2 to evaluate the results of the docking and the DFT charge analyses.
2.5.1. System and ligand stability inside the Mpro active site
The stability of each simulated model was determined by the calculations of backbone atom RMSD for ligand − Mpro complex as shown in Figure 5. The RMSD of A2 and A4 with- Mpro rises till 20 ns and stabilizes after 10 ns at 2 Å. This clearly indicates that ligands are stabilized inside the pocket and don’t change their orientation in the active site of Mpro. Similarly protein backbone atoms for A2 showed stability around 2.5 Å. However, A4 showed higher RMSD and maintained at 3 Å, which directly resembels to the charge analyses. Furthermore, A2 and A4 hydrogen bond with Mpro are plotted along the 50-ns MD simulation, shown in Figure 6. These simulations indicated that on an average A2 and A4 show approximately two hydrogen bonds and are in accordance with our docking results and further suggested the stability of the ligand in active site.
Figure 5.

The root mean square deviation (RMSD) plots for the A2 and A4 compounds interacting with the Mpro protein for the entire 50 ns MD simulation. Top, RMSD of A2 and A4, and bottom of the RMSD of the backbone atoms of Mpro during binding of the A2 and A4. The green line represents A2 and blue line represents A4.
Figure 6.

Number of Hydrogen bond of A2 and A4 with Mpro plotted along the 50-ns MD simulation.
Other than the Hydrogen bond, hydrophobic interactions were established among non-polar amino acids and provide the stability of proteins in solution by shielding the non-polar amino acids in hydrophobic cores, away from the aqueous environment. Solvation plays an important role in ligand-protein association and has a strong impact on comparisons of binding energies for dissimilar molecules.
Change in Solvent Accessible Surface Area (SASA) directly indicates the change of hydrophobic core caused by changes in tertiary structure. The calculation of the SASA of A2 and A4 do not show any drastic change that indicate the ligand were stable during the simulation run, as shown in Figure 7, and showed very similar hydrophobic area. This indicates that A2 and A4 are less exposed to water molecules and buried in Mpro active site.
Figure 7.

Hydrophobic, Hydrophilic, and solvent accessible surface area for A2 and A4 with Mpro plotted along the 50-ns MD simulation. Blue colour showed ΔGsolv, the free energy of ligand desolvation, based on the solvent accessible surface plotted along the 50-ns. Red line showed the hydrophilic SASA; black line, the hydrophobic SASA and the green line showed the combination of hydrophilic and hydrophobic SASA.
2.5.2. Binding energy analysis
The calculated binding energy using the mmpbsa approach indicates that A2 has higher energy (−48.33 kcal/mol) in comparison to A4 (-33.10 kcal/mol), Table 3, similar to the finding in the docking analysis, Table 1. Individual components of energy for each system indicate that A2 and A4 both show higher van der Waals energies and very comparable SASA energies and polar salvation energies. However a significant difference was observed in the electrostatic energies. As explained in the ChelpG charge analysis (Figure 3), the electrostatic interactions for the A2 showed much higher interactions than the A4 ligand, which effects the binding energy difference of the two systems. Thus, the binding energies are governed by the van der Waals energy and electrostatic energy.
Table 3.
MM/PBSA binding free energy of the selected compounds compared with the known inhibitors of the SARSCoV-2 Mpro.
| Complex |
Van der Waal energy kcal/mol |
Electrostatic energy kcal/mol |
Polar solvation energy kcal/mol |
SASA energy kcal/mol |
Binding energy kcal/mol |
|---|---|---|---|---|---|
| A2 | −58.38+/− 0.26 | −10.08+/−0.23 | 24.74+/−0.15 | −4.61+/−0.02 | −48.33+/−0.36 |
| A4 | −49.89+/− 0.27 | −2.43+/−0.19 | 23.61+/−0.24 | −4.39+/−0.02 | −33.10+/−0.20 |
2.5.3. Energy contribution of each residue to binding
To identify important interacting residues with A2 and A4 the contribution of each residues to the binding energy was calculated and plotted (Figure 8) indicating three important regions: amino acids 26 to 56, amino acids 136 to 148, and amino acids 162 to 194. These regions showed significant energy contribution to the binding energy. Leu27 Asn51 and Pro52 showed negative energy in both systems while His41 showed positive energy in A2. In the second region (amino acid 136 to 148) A4 showed higher energy values for residues Leu141, Asn142, Gly143, Ser144, Cys145. In the third region (amino acid residue 162 to 194) A2 had higher binding energy values for interactions with residues Met165, Leu167, Pro168, Phe185, Asp187, Thr190, Ala191 and Gln192. While A2 interacted with amino acid from region 162 to 192 and residue 44, A4 showed good interaction with region 140 to 146.
Figure 8.

The contribution energy of each residue to Mpro. Top box is a general energy plot of all residues. Bottom three boxes: the Y axis presents Energy [kcal/mol] and the X axis indicated the energy contribution of each residue by number in Mpro, green triangles represent A2 and blue triangle A4. Clear perturbations are detected at amino acids 26 to 56, amino acids 136 to 148 and amino acid 162 to 194. The respective structures of the ligand-protein complexes are given above.
The Mpro complex interacts with the ligands A2 and A4 with the different amino acid residues between energy −2.4 kcal/mol to −0.24 kcal/mol. A2 formed more interaction with high binding energy (up to 2.4 kcal/mol) in the active site, Figure 9. Cys44, Met165, Gln189 and Ala191 are major residues that highly contributing to binding with Mpro, this indicates the ligand fits well into the active site of Mpro. However, residues participating in A4 binding are somewhat different, very few residues bind with more than −0.95 kcal/mol, the major binding residue were found to be Gln189, Met165, Cys145 and Pro168. Several amino acids were common to binding both A2 and A4: Leu27, Cys44, Met49, His164, Met165, Leu167, Asp187, Gln189, Thr190, Ala191 and Gln192 these also contribute to the binding energy. Overall this analysis supports the better binding interactions of the A2 on the active site of Mpro than A4, Figure 10.
Figure 9.

Major residues contributing to the binding of A2 and A4 compounds are plotted. The Y axis represents the energy in kcal/mol for the energy contribution and X axis showed amino acid position for each residue.
Figure 10.

The different interactions the compounds A2 and A4 at the active site of the Mpro protein of the SARS-CoV-2 calculated using the Molecular dynamics simulations. Carbon: Ash; Oxygen: Red; Chlorine: Green; Nitrogen: Blue; Hydrogen: White. The different types of interactions are also given in the figure for both the compounds.
2.5.4. Flexible region and catalytic dyad of Mpro
The RMSF captures, for each residue, the fluctuation from its average position, gives insight into the flexibility of regions of the Mpro. The fluctuations per residue of COVID-19 Mpro indicated that after the binding, three regions, amino acid residues from 40 to 54, 124 to 144 and 180 to 200, and in addition 272 to 306 showed higher fluctuations, Figure 11. Moreover, these residues are also participating in ligand binding. These might suggest that these flexible regions are important in substrate or ligand binding.
Figure 11.

The RMSF plot for each residue out of the four highly fluctuating regions, is show over the 50 ns MD simulation. A2 (green) and A4 (blue); (A): showing fluctuations in region 40 to 54; (B): Showing fluctuation in region 124 to 144 & 180 to 200; (C): showing fluctuation in region 272 to 306 during Mpro-Ligands complex.
2.6. ADME parameters prediction
In order to screen the pharmacokinetic behaviour, the in-silico ADME parameters of the designed ligands with free energy of binding higher than −10 kcal/mol were calculated using the SwissADME software (http://www.swissadme.ch). Lipinski’s rule of five is a prerequisite to ensure drug-like properties when using rational drug design. The designed derivatives follow the four properties of Lipinski’s rule of five (mol. wt. ≤ 500 Da; log P o/w ≤ 5; HBD ≤ 5; HBA ≤10). All the ligands have showed ADME parameters in an acceptable range and possess significant drug-like characteristics based on Lipinski’s rule of 5, Table 4.
Table 4.
In-silico predicted ADME properties of the designed active compounds.
| Compound | Mol. Wt. | Number of HBD | Number of HBA | MR | Log P o/w |
Log S (ESOL) |
Solubility (mg/ml) |
Lipinski Rule |
|---|---|---|---|---|---|---|---|---|
| A1 | 450.27 | 0 | 5 | 119.1 | 4.53 | −5.98 | 4.66e-04 | Yes; 0 violation |
| A2 | 431.83 | 1 | 6 | 116.02 | 3.52 | −5.25 | 2.41e-03 | Yes; 0 violation |
| A4 | 453.28 | 1 | 5 | 118.36 | 4.80 | −6.54 | 1.31e-04 | Yes; 0 violation |
| A5 | 434.9 | 2 | 6 | 115.37 | 3.66 | −5.80 | 6.89e-04 | Yes; 0 violation |
| A7 | 544.18 | 1 | 5 | 131.38 | 4.16 | −7.17 | 3.70e-05 | Yes; 1 violation: MW > 500 |
| A8 | 525.74 | 2 | 6 | 128.40 | 3.17 | −6.43 | 1.96e-04 | Yes; 1 violation: MW > 500 |
| A9 | 554.74 | 1 | 7 | 135.20 | 3.92 | −6.63 | 1.29e-04 | Yes; 1 violation: MW > 500 |
| A11 | 446.84 | 2 | 6 | 120.70 | 2.86 | −5.52 | 1.35e-03 | Yes; 0 violation |
| A12 | 475.84 | 1 | 7 | 127.50 | 2.26 | −5.72 | 8.99e-04 | Yes; 0 violation |
| A20 | 432.47 | 0 | 4 | 131.48 | 3.18 | −5.78 | 7.14e-04 | Yes; 0 violation |
| A38 | 341.41 | 1 | 2 | 102.48 | 3.18 | −4.52 | 1.04e-02 | Yes; 0 violation |
| A40 | 427.31 | 1 | 4 | 111.50 | 3.89 | −5.58 | 1.12e-03 | Yes; 0 violation |
Mol. Wt.: Molecular weight, HBD: Hydrogen bond donor, HBA: Hydrogen bond acceptor, MR: Molar Refractivity. Log Po/w: Octanol/water partition coefficient, Log S: Aqueous solubility.
3. Conclusion
In conclusion, we have studied the binding of a series of heterocyclic moieties to the active site of Mpro of the SARS-CoV2. Molecular docking analysis show that the isatin based derivatives possess excessive interactions to Mpro, with binding energies of over −10 kcal/mol. The best docked ligands A2 and A4 consisted of an Isatin moiety along with an oxidiazole ring, and both moieties are highly used in anticancer, antiviral, antibacterial drug development. Further, the binding interactions were more vividly discussed with a comprehensive MD simulation analysis using GROMACS, and binding energies were calculated using the MMPBSA method on the active site of the protein. These analyses show hydrogen bonds and π-π interactions with the residues His41, Cys145, Val42, Cys44, Thr45, Ser46, Glu47, Asp48, Met49 and Glu166, Met165, Leu167 and Pro168. Our analysis also shows that hydrophobic and electrostatic interactions are most important to predict the binding energies of the ligands to the protein and that A2 interacts more strongly than the A4. This finding was reinforced by the electrostatic charge analysis and MESP analysis through DFT calculations. All the ligands have showed ADME parameters in an acceptable range and possess significant drug-like characteristics based on Lipinski’s rule of 5. Our findings indicate that both isatin and oxidiazoles based drugs are indeed very good candidates as potential inhibitor for Mpro of SARS-CoV-2 and A2 and A4 compounds are the best candidates with extremely high potency in silico and further in vitro analysis will be performed as future studies.
4. Computational section
4.1. Quantum chemical calculations
All the ligands were initially optimized using Density functional theory with Gaussian 16 suite of programs to obtain the most stable structure of each system (Ogliaro et al., 2016) except the repurposed drugs with crystal structures available. All the compounds were optimized at B3LYP/6-31 + G(d) (Becke, 1993; Lee et al., 1988) level of theory in gas phase. Positive vibrational frequencies confirmed the structures to be minimum. PDB structures were saved using Gaussview and used for the Molecular docking studies. Electrostatic charges were calculated for the specific compounds using the ChelpG charge analysis (Martin & Zipse, 2005; Rozas, 1997; Tsuzuki et al., 1996) implemented in Gaussian software. The Molecular Electrostatic Potential (MESP) was generated for both the ligands at the same level of theory.
4.2. Molecular docking studies
The crystal structure of SARS-CoV-2 has been retrieved from the rcsb.org (PDB ID: 6LU7) (Jin et al., 2020b) and used to generate initial 3D coordinates. Co-crystallized water molecules were deleted along with addition of polar Hydrogen and compute gasteiger charges. The structures of selected inhibitors were superimposed against pre-docked ligand in the PDB, and the latter was then removed to generate initial conformation of inhibitor at the active site of SARS-CoV-2. Grid box was then determined by the native ligand position on the binding site (Thr26, Tyr54, Phe140, Asn142, Gly143, Ser144, Cys145, His163, His164, Glu166, and His172). Finally both the Auto-grid and Auto-dock were run with the default parameters (Badavath et al., 2016a). All molecular docking analysis were performed by AutoDock4.2 using earlier reported protocol (Badavath et al., 2016b; Badavath et al., 2015; Badavath et al., 2017; Badavath et al., 2016c; Gangireddy et al., 2019; Munusamy et al., 2019; Nath et al., 2018; Nayak et al., 2015; Vishnu Nayak et al., 2013) software version 4.2 (Forli et al., 2012). After completing the molecular docking process, the generated DLG (.dlg) file was analyzed using AutoDock v4.2 tools for all the possible interaction and binding free energies following the methodology (Huey et al., 2007). Top scoring molecules from largest cluster were considered for the interactions.
4.3. Molecular dynamics simulation
Gromacs 4.6.2 (Hess et al., 2008; Van Der Spoel et al., 2005) with GROMOS96 54a7 force field (Schmid et al., 2011) was used for MD simulation studies of two system 50 ns each. The PRODRG2 Server (Schuttelkopf & Van Aalten, 2004) was used to generate the topology of A2 and A4. Each system was placed in the centre of cubic box having distance of 1.0 nm between protein and edge of the simulation box and solvated with explicit water molecules, with the SPC solvation model (Mark & Nilsson, 2001). Before proceeding towards energy minimization, all the systems were neutralized by adding Na+/Cl− ions accordingly. Steepest descent method was used for energy minimization of each system. MD simulations with NVT (isochoric-isothermal) and NPT (isobaric-isothermal) ensembles (N = constant particle number, V = Volume, P = Pressure, T = Temperature) were performed for 1 ns to equilibrate the protein-ligand system for constant volume, pressure (1 atm) and temperature (300 K). To calculate electrostatic interaction, Particle Mesh Ewald (PME) algorithm (Darden et al., 1993; Essmann et al., 1995) was used with grid spacing of 1.6 Å and a cutoff of 10 Å and LINCS method (Hess et al., 1997) was used to restrain the bond length. Finally, 50 ns of production MD was performed for each system and trajectories were saved at every 2 ps.
4.3.1. Binding energy calculation for A2 and A4
G_mmpbsa (Kumari et al., 2014) was developed using two widely used open source software i.e. GROMACS and APBS and it has similar user interface like other GROMACS tools. The tool calculates components of binding energy using MM-PBSA method except the entropic term and energetic contribution of each residue to the binding using energy decomposition scheme. Binding free energy estimation of all the docked complexes was done by MM/PBSA method through g_mmpbsa module. For each simulated system, to calculate average binding energy by using bootstrap analysis, a total of 200 snapshots were taken from the last 10 ns of the trajectory on the intervals of 50 ps. The module calculates electrostatic interactions, van der Waals interactions, polar solvation energy and non-polar solvation energy.
4.4. ADME calculation
ADME parameters of the designed ligands were calculated using the SwissADME (http://www.swissadme.ch) (Daina et al., 2017) accessed on- 5th May 2020. All the compounds were drawn in Chem Draw software and smiles were taken as input, before being used in SwissADME. Lipinski’s rule of five (Lipinski et al., 1997).
Supplementary Material
Acknowledgements
AK acknowledges Bioinformatics laboratory, Biotechnology Division of CSIR-CIMAP, Lucknow. AD, AJ and AS acknowledge to GITAM University, Visakhapatnam, India for the support. AS also acknowledges Kalyanashis Jana for fruitful discussions.
Funding Statement
AD, AJ and AS also thank the Department of Science and Technology, India under DST-FIST Program 2014 with Grant No. SR/FST/ETI-373/2016. The Hebrew University of Jerusalem, Israel for Corona Fund to GB.
Author contributions
The manuscript was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript.
Disclosure statement
There are no conflicts to declare.
References
- Afifi, T. H., Okasha, R. M., Ahmed, H. E. A., Ilaš, J., Saleh, T., & Abd-El-Aziz, A. S. (2017). Structure-activity relationships and molecular docking studies of chromene and chromene based azo chromophores: A novel series of potent antimicrobial and anticancer agents. EXCLI Journal, 16, 868–902. 10.17179/excli2017-356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal, M., Singh, V., Sharma, S. K., Sharma, P., Ansari, M. Y., Jadav, S. S., Yasmin, S., Sreenivasulu, R., Hassan, M. Z., Saini, V., & Ahsan, M. J. (2016). Design and synthesis of new 2,5-disubstituted-1,3,4-oxadiazole analogues as anticancer agents. Medicinal Chemistry Research, 25(10), 2289–2303. [Google Scholar]
- Anand, P. S., Sethukumar, A., Kumar, C. U., Krishnasamy, K., Senthan, S., Manikandan, G., & Prakasam, B. A. (2019). Synthesis, spectroscopic investigation, computational, stereochemical and biological studies of 1, 3-dimethyl-2, 6-diphenylpiperidin-4-one N (4′)-cyclohexylsemicarbazone: Crystal structure and Hirshfeld surface analysis. Chemical Data Collections, 21, 100216. [Google Scholar]
- Badavath, V. N., Baysal, I., Ucar, G., Sinha, B. N., & Jayaprakash, V. (2016. a). Monoamine oxidase inhibitory activity of novel pyrazoline analogues: Curcumin based design and synthesis. ACS Medicinal Chemistry Letters, 7(1), 56–61. 10.1021/acsmedchemlett.5b00326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badavath, V. N., Baysal, I., Uçar, G., Mondal, S. K., Sinha, B. N., & Jayaprakash, V. (2016. b). Monoamine oxidase inhibitory activity of ferulic acid amides: Curcumin-based design and synthesis. Archiv Der Pharmazie, 349(1), 9–19. 10.1002/ardp.201500317 [DOI] [PubMed] [Google Scholar]
- Badavath, V. N., Ucar, G., Sinha, B. N., Mondal, S. K., & Jayaprakash, V. (2016. c). Monoamine oxidase inhibitory activity of novel pyrazoline analogues: Curcumin based design and synthesis-II. ChemistrySelect, 1(18), 5879–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badavath, V. N., Baysal, I., Ucar, G., Sinha, B. N., & Jayaprakash, V. (2015). Monoamine oxidase inhibitory activity of novel pyrazoline analogues: Curcumin based design and synthesis. ACS Medicinal Chemistry Letters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badavath, V. N., Nath, C., Ganta, N. M., Ucar, G., Sinha, B. N., & Jayaprakash, V. (2017). Design, synthesis and MAO inhibitory activity of 2-(arylmethylidene)-2,3-dihydro-1-benzofuran-3-one derivatives. Chinese Chemical Letters, 28(7), 1528–1532. 10.1016/j.cclet.2017.02.009 [DOI] [Google Scholar]
- Bauer, D. J., & Sadler, P. W. (1960). The structure-activity relationships of the antiviral chemotherapeutic activity of isatin beta-thiosemicarbazone. British Journal of Pharmacology and Chemotherapy, 15(1), 101–110. 10.1111/j.1476-5381.1960.tb01216.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. Journal of Chemical Physics, 98(7), 5648–5652. [Google Scholar]
- Beedie, S. L., Peer, C. J., Pisle, S., Gardner, E. R., Mahony, C., Barnett, S., Ambrozak, A., Gütschow, M., Chau, C. H., Vargesson, N., & Figg, W. D. (2015). Anticancer properties of a novel class of tetrafluorinated thalidomide analogues. Molecular Cancer Therapeutics, 14(10), 2228–2237. 10.1158/1535-7163.MCT-15-0320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi, I., Kivity, S., Langevitz, P., & Shoenfeld, Y. (2012). Hydroxychloroquine: From malaria to autoimmunity. Clinical Reviews in Allergy & Immunology, 42(2), 145–153. 10.1007/s12016-010-8243-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt, P., Kumar, M., & Jha, A. (2018). Design, synthesis and anticancer evaluation of oxa/thiadiazolylhydrazones of barbituric and thiobarbituric acid: A collective in vitro and in silico approach. ChemistrySelect, 3(25), 7060–7065. [Google Scholar]
- Bhatt, P., Sen, A., & Jha, A. (2020). Design and ultrasound assisted synthesis of novel 1, 3, 4‐oxadiazole drugs for anti‐cancer activity. ChemistrySelect, 5(11), 3347–3354. 10.1002/slct.201904412 [DOI] [Google Scholar]
- Bhattacharjee, M. K. (2016). Chemistry of antibiotics and related drugs (Vol. 219). Springer. [Google Scholar]
- Blair, J. A., Rauh, D., Kung, C., Yun, C.-H., Fan, Q.-W., Rode, H., Zhang, C., Eck, M. J., Weiss, W. A., & Shokat, K. M. (2007). Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nature Chemical Biology, 3(4), 229–238. 10.1038/nchembio866 [DOI] [PubMed] [Google Scholar]
- Burger, A. (1979). Burger’s medicinal chemistry. Wiley. [Google Scholar]
- Bzówka, M., Mitusińska, K., Raczyńska, A., Samol, A., Tuszyński, J. A., & Góra, A. (2020). Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. International Journal of Molecular Sciences, 21(9), 3099. 10.3390/ijms21093099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L.-R., Wang, Y.-C., Lin, Y. W., Chou, S.-Y., Chen, S.-F., Liu, L. T., Wu, Y.-T., Kuo, C.-J., Chen, T. S.-S., & Juang, S.-H. (2005). Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorganic & Medicinal Chemistry Letters, 15(12), 3058–3062. 10.1016/j.bmcl.2005.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, N., Zhou, M., Dong, X., Qu, J., Gong, F., Han, Y., Qiu, Y., Wang, J., Liu, Y., Wei, Y., Xia, J., Yu, T., Zhang, X., & Zhang, L. (2020. a). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. The Lancet, 395(10223), 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. W., Yiu, C.-P., & Wong, K.-Y. (2020. b). Prediction of the 2019-nCoV 3C-like protease (3CLpro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res, 9, 129. 10.12688/f1000research.22457.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z.-M., Fu, J.-F., Shu, Q., Chen, Y.-H., Hua, C.-Z., Li, F.-B., Lin, R., Tang, L.-F., Wang, T.-L., Wang, W., Wang, Y.-S., Xu, W.-Z., Yang, Z.-H., Ye, S., Yuan, T.-M., Zhang, C.-M., & Zhang, Y.-Y. (2020. c). Diagnosis and treatment recommendations for pediatric respiratory infection caused by the 2019 novel coronavirus. World Journal of Pediatrics: WJP, 16(3), 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortegiani, A., Ingoglia, G., Ippolito, M., Giarratano, A., & Einav, S. (2020). A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. Journal of Critical Care, 57, 279–283. 10.1016/j.jcrc.2020.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, W., Zhang, B., Jiang, X.-M., Su, H., Li, J., Zhao, Y., Xie, X., Jin, Z., Peng, J., Liu, F., Li, C., Li, Y., Bai, F., Wang, H., Cheng, X., Cen, X., Hu, S., Yang, X., Wang, J., … Liu, H. (2020). Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science (New York, N.Y.), 368(6497), 1331–1335. 10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717. 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden, T., York, D., & Pedersen, L. (1993). Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98(12), 10089–10092. 10.1063/1.464397 [DOI] [Google Scholar]
- De Clercq, E. (2006). Potential antivirals and antiviral strategies against SARS coronavirus infections. Expert Review of Anti-Infective Therapy, 4(2), 291–302. 10.1586/14787210.4.2.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, L., Song, B., Wu, J., Wu, Z., Zhu, Y., Chen, X., & Hu, D. (2016). Synthesis and antiviral activity of novel thioether derivatives containing 1, 3, 4-oxadiazole/thiadiazole and emodin moieties. Phosphorus, Sulfur, and Silicon and the Related Elements, 191(6), 904–907. 10.1080/10426507.2015.1114944 [DOI] [Google Scholar]
- Einarsson, K. (1994). Effect of ursodeoxycholic acid on hepatic cholesterol metabolism. Scandinavian Journal of Gastroenterology, 29(sup 204), 19–23. 10.3109/00365529409103620 [DOI] [PubMed] [Google Scholar]
- Enogieru, A. B., Haylett, W., Hiss, D. C., Bardien, S., & Ekpo, O. E. (2018). Rutin as a potent antioxidant: Implications for neurodegenerative disorders. Oxidative Medicine and Cellular Longevity, 2018, 6241017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., & Pedersen, L. G. (1995). A smooth particle mesh Ewald method. The Journal of Chemical Physics, 103(19), 8577–8593. 10.1063/1.470117 [DOI] [Google Scholar]
- Fan, H.-H., Wang, L.-Q., Liu, W.-L., An, X.-P., Liu, Z.-D., He, X.-Q., Song, L.-H., & Tong, Y.-G. (2020). Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chinese Medical Journal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, A., Sellner, M., Neranjan, S., Smieško, M., & Lill, M. A. (2020). Potential inhibitors for novel coronavirus protease identified by virtual screening of 606 million compounds. International Journal of Molecular Sciences, 21(10), 3626. 10.3390/ijms21103626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischmann, T. O., Hruza, A., Duca, J. S., Ramanathan, L., Mayhood, T., Windsor, W. T., Le, H. V., Guzi, T. J., Dwyer, M. P., Paruch, K., Doll, R. J., Lees, E., Parry, D., Seghezzi, W., & Madison, V. (2008). Structure-guided discovery of cyclin-dependent kinase inhibitors. Biopolymers, 89(5), 372–379. 10.1002/bip.20868 [DOI] [PubMed] [Google Scholar]
- Forli, W., Halliday, S., Belew, R., & Olson, A. J. (2012). AutoDock (Version 4.2). Citeseer.
- Gangireddy, M. R., Mantipally, M., Gundla, R., Badavath, V. N., Paidikondala, K., & Yamala, A. (2019). Design and synthesis of piperazine‐linked imidazo [1, 2‐a] pyridine derivatives as potent anticancer agents. ChemistrySelect, 4(46), 13622–13629. [Google Scholar]
- Gautret, P., Lagier, J.-C., Parola, P., Hoang, V. T., Meddeb, L., Mailhe, M., Doudier, B., Courjon, J., Giordanengo, V., Vieira, V. E., Tissot Dupont, H., Honoré, S., Colson, P., Chabrière, E., La Scola, B., Rolain, J.-M., Brouqui, P., & Raoult, D. (2020). Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. International Journal of Antimicrobial Agents, 56(1), 105949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerova, M. S., Stateva, S. R., Radonova, E. M., Kalenderska, R. B., Rusew, R. I., Nikolova, R. P., Chanev, C. D., Shivachev, B. L., Apostolova, M. D., & Petrov, O. I. (2016). Combretastatin A-4 analogues with benzoxazolone scaffold: Synthesis, structure and biological activity. European Journal of Medicinal Chemistry, 120, 121–133. 10.1016/j.ejmech.2016.05.012 [DOI] [PubMed] [Google Scholar]
- Ghosh, A. K., Osswald, H. L., & Prato, G. (2016). Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. Journal of Medicinal Chemistry, 59(11), 5172–5208. 10.1021/acs.jmedchem.5b01697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya, A. E., Baker, S. C., Baric, R., Groot, R. J., de Drosten, C., Gulyaeva, A. A., Haagmans, B. L., Lauber, C., Leontovich, A. M., Neuman, B. W., & Penzar, D. (2020). Severe acute respiratory syndrome-related coronavirus: The species and its viruses–a statement of the Coronavirus Study Group.
- Hall Jr, D. C., & Ji, H.-F. (2020). A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Medicine and Infectious Disease, 35, 101646. 10.1016/j.tmaid.2020.101646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, B., Bekker, H., Berendsen, H. J. C., & Fraaije, J. G. E. M. (1997). LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18(12), 1463–1472. [DOI] [Google Scholar]
- Hess, B., Kutzner, C., Van Der Spoel, D., & Lindahl, E. (2008). GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation, 4(3), 435–447. 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- Huey, R., Morris, G. M., Olson, A. J., & Goodsell, D. S. (2007). A semiempirical free energy force field with charge-based desolvation. Journal of Computational Chemistry, 28(6), 1145–1152. 10.1002/jcc.20634 [DOI] [PubMed] [Google Scholar]
- Huynh, T., Wang, H., & Luan, B. (2020). In silico exploration of the molecular mechanism of clinically oriented drugs for possibly inhibiting SARS-CoV-2's main protease. The Journal of Physical Chemistry Letters, 11(11), 4413–4420. 10.1021/acs.jpclett.0c00994 [DOI] [PubMed] [Google Scholar]
- Ivanova, Y., Momekov, G., Petrov, O., Karaivanova, M., & Kalcheva, V. (2007). Cytotoxic Mannich bases of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones. European Journal of Medicinal Chemistry, 42(11–12), 1382–1387. 10.1016/j.ejmech.2007.02.019 [DOI] [PubMed] [Google Scholar]
- Jha, A., & Ramarao, T. A. (2018). Expeditious microwave-assisted synthesis and bio-evaluation of novel bis (trifluoromethyl) phenyl-triazole-pyridine hybrid analogues by the click chemistry approach. Research on Chemical Intermediates, 44(1), 585–599. 10.1007/s11164-017-3121-2 [DOI] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., & Peng, C. (2020. a). Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. [DOI] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020. b). Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature, 582(7811), 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Keogh-Brown, M. R., & Smith, R. D. (2008). The economic impact of SARS: How does the reality match the predictions? Health Policy (Amsterdam, Netherlands), 88(1), 110–120. 10.1016/j.healthpol.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharb, R., Shahar Yar, M., & Chander Sharma, P. (2011). Recent advances and future perspectives of triazole analogs as promising antiviral agents. Mini Reviews in Medicinal Chemistry, 11(1), 84–96. 10.2174/138955711793564051 [DOI] [PubMed] [Google Scholar]
- Kok, S. H. L., Gambari, R., Chui, C. H., Yuen, M. C. W., Lin, E., Wong, R. S. M., Lau, F. Y., Cheng, G. Y. M., Lam, W. S., Chan, S. H., Lam, K. H., Cheng, C. H., Lai, P. B. S., Yu, M. W. Y., Cheung, F., Tang, J. C. O., & Chan, A. S. C. (2008). Synthesis and anti-cancer activity of benzothiazole containing phthalimide on human carcinoma cell lines. Bioorganic & Medicinal Chemistry, 16(7), 3626–3631. 10.1016/j.bmc.2008.02.005 [DOI] [PubMed] [Google Scholar]
- Kumari, R., Kumar, R., Consortium, O. S. D. D., & Lynn, A. (2014). g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling, 54(7), 1951–1962. 10.1021/ci500020m [DOI] [PubMed] [Google Scholar]
- Lakshmithendral, K., Saravanan, K., Elancheran, R., Archana, K., Manikandan, N., Arjun, H. A., Ramanathan, M., Lokanath, N. K., & Kabilan, S. (2019). Design, synthesis and biological evaluation of 2-(phenoxymethyl)-5-phenyl-1,3,4-oxadiazole derivatives as anti-breast cancer agents. European Journal of Medicinal Chemistry, 168, 1–10. 10.1016/j.ejmech.2019.02.033 [DOI] [PubMed] [Google Scholar]
- Lane, M. E., Yu, B., Rice, A., Lipson, K. E., Liang, C., Sun, L., Tang, C., McMahon, G., Pestell, R. G., & Wadler, S. (2001). A Novel cdk2-selective inhibitor, SU9516, induces apoptosis in colon carcinoma cells. Cancer Research, 61(16), 6170–6177. [PubMed] [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B, 37(2), 785–789. 10.1103/PhysRevB.37.785 [DOI] [PubMed] [Google Scholar]
- Li, Y., Zhang, J., Wang, N., Li, H., Shi, Y., Guo, G., Liu, K., Zeng, H., & Zou, Q. (2020). Therapeutic drugs targeting 2019-nCoV main protease by high-throughput screening. BioRxiv. [Google Scholar]
- Li, Z., Zhan, P., & Liu, X. (2011). 1,3,4-oxadiazole: A privileged structure in antiviral agents. Mini Reviews in Medicinal Chemistry, 11(13), 1130–1142. 10.2174/138955711797655407 [DOI] [PubMed] [Google Scholar]
- Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23(1–3), 3–25. 10.1016/S0169-409X(96)00423-1 [DOI] [PubMed] [Google Scholar]
- Liu, K., Tang, M., Liu, Q., Han, X., Jin, H., Zhu, H., Li, Y., He, L., Ji, H., & Zhou, B. (2020). Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discovery, 6(1), 1–4. 10.1038/s41421-019-0132-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., & Wang, X.-J. (2020). Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. Journal of Genetics and Genomics = Yi Chuan Xue Bao, 47(2), 119–121. 10.1016/j.jgg.2020.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallikarjuna, N., Vasudharani, D., & Ambrish, S. (2020). Screening of chloroquine, hydroxychloroquine and its derivatives for their binding affinity to multiple SARS-CoV-2 protein drug targets. Journal of Biomolecular Structure and Dynamics, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark, P., & Nilsson, L. (2001). Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. The Journal of Physical Chemistry A, 105(43), 9954–9960. 10.1021/jp003020w [DOI] [Google Scholar]
- Martin, F., & Zipse, H. (2005). Charge distribution in the water molecule-a comparison of methods. Journal of Computational Chemistry, 26(1), 97–105. 10.1002/jcc.20157 [DOI] [PubMed] [Google Scholar]
- Mendel, D. B., Laird, A. D., Xin, X., Louie, S. G., Christensen, J. G., Li, G., Schreck, R. E., Abrams, T. J., Ngai, T. J., Lee, L. B., Murray, L. J., Carver, J., Chan, E., Moss, K. G., Haznedar, J. O., Sukbuntherng, J., Blake, R. A., Sun, L., Tang, C., … Cherrington, J. M. (2003). In vivo antitumor activity of SU11248 antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 9(1), 327–337. [PubMed] [Google Scholar]
- Mittal, L., Kumari, A., Srivastava, M., Singh, M., & Asthana, S. (2020). Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. Journal of Biomolecular Structure and Dynamics, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munusamy, S., Badavath, V. N., Maji, S., Sekar, M., & Shabbir, M. (2019). Novel halogenated pyrido [2, 3-a] carbazole with enhanced aromaticity as potent anticancer and antioxidant: Rationale design and microwave assisted synthesis. New Journal of Chemistry, 43(44), 17231–17240. [Google Scholar]
- Nath, C., Badavath, V. N., Thakur, A., Ucar, G., Acevedo, O., Mohd Siddique, M. U., & Jayaprakash, V. (2018). Curcumin-based pyrazoline analogues as selective inhibitors of human monoamine oxidase A. MedChemComm, 9(7), 1164–1171. 10.1039/C8MD00196K [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak, B. V., Ciftci-Yabanoglu, S., Bhakat, S., Timiri, A. K., Sinha, B. N., Ucar, G., Soliman, M. E. S., & Jayaprakash, V. (2015). Monoamine oxidase inhibitory activity of 2-aryl-4H-chromen-4-ones. Bioorganic Chemistry, 58, 72–80. 10.1016/j.bioorg.2014.11.008 [DOI] [PubMed] [Google Scholar]
- Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., … Raghavachari, K. (2016). Gaussian 16, Revision A. 03. Gaussian, Inc. [Google Scholar]
- Pandeya, S. N., Sriram, D., Nath, G., & De Clercq, E. (2000). Synthesis, antibacterial, antifungal and anti-HIV activities of norfloxacin Mannich bases. European Journal of Medicinal Chemistry, 35(2), 249–255. 10.1016/s0223-5234(00)00125-2 [DOI] [PubMed] [Google Scholar]
- Pauwels, R., Balzarini, J., Baba, M., Snoeck, R., Schols, D., Herdewijn, P., Desmyter, J., & De Clercq, E. (1988). Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. Journal of Virological Methods, 20(4), 309–321. [DOI] [PubMed] [Google Scholar]
- Pessoa, C., Ferreira, P. M. P., Lotufo, L. V. C., de Moraes, M. O., Cavalcanti, S. M. T., Coêlho, L. C. D., Hernandes, M. Z., Leite, A. C. L., De Simone, C. A., Costa, V. M. A., & Souza, V. M. O. (2010). Discovery of phthalimides as immunomodulatory and antitumor drug prototypes. ChemMedChem, 5(4), 523–528. 10.1002/cmdc.200900525 [DOI] [PubMed] [Google Scholar]
- Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y., & Jung, S.-H. (2016). An overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. Journal of Medicinal Chemistry, 59(14), 6595–6628. 10.1021/acs.jmedchem.5b01461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp, F. D. (1969). Synthesis of potential antineoplastic agents. XX. Compounds related to the 3-o-nitrophenylhydrazone of isatin. Journal of Medicinal Chemistry, 12(1), 182–184. 10.1021/jm00301a054 [DOI] [PubMed] [Google Scholar]
- Rasul, A., Millimouno, F. M., Ali Eltayb, W., Ali, M., Li, J., & Li, X. (2013). Pinocembrin: A novel natural compound with versatile pharmacological and biological activities. BioMed Research International, 2013, 379850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raugi, D. N., Smith, R. A., Gottlieb, G. S., & Group, U. (2016). Four amino acid changes in HIV-2 protease confer class-wide sensitivity to protease inhibitors. Journal of Virology, 90(2), 1062–1069. 10.1128/JVI.01772-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, I. (1997). Atomic charges derived from different methods: A comparative study applied to SO2 heterocycles. International Journal of Quantum Chemistry, 62(5), 477–487. [Google Scholar]
- Scavone, C., Brusco, S., Bertini, M., Sportiello, L., Rafaniello, C., Zoccoli, A., Berrino, L., Racagni, G., Rossi, F., & Capuano, A. (2020). Current pharmacological treatments for COVID‐19: What’s next? British Journal of Pharmacology, 177(21), 4813–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelman, W. R., Liu, G., Wilding, G., Morris, T., Phung, D., & Dreicer, R. (2011). A phase I study of zibotentan (ZD4054) in patients with metastatic, castrate-resistant prostate cancer. Investigational New Drugs, 29(1), 118–125. 10.1007/s10637-009-9318-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, N., Eichenberger, A. P., Choutko, A., Riniker, S., Winger, M., Mark, A. E., & van Gunsteren, W. F. (2011). Definition and testing of the GROMOS force-field versions 54A7 and 54B7. European Biophysics Journal: EBJ, 40(7), 843–856. 10.1007/s00249-011-0700-9 [DOI] [PubMed] [Google Scholar]
- Schuttelkopf, A. W., & Van Aalten, D. M. F. (2004). PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica Section D: Biological Crystallography, 60(8), 1355–1363. [DOI] [PubMed] [Google Scholar]
- Selvam, P., Murgesh, N., Chandramohan, M., De Clercq, E., Keyaerts, E., Vijgen, L., Maes, P., Neyts, J., & Ranst, M. V. (2008). In vitro antiviral activity of some novel isatin derivatives against HCV and SARS-CoV viruses. Indian Journal of Pharmaceutical Sciences, 70(1), 91–94. 10.4103/0250-474X.40339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafim, R. A. M., de Oliveira, T. F., Loureiro, A. P. M., Krogh, R., Andricopulo, A. D., Dias, L. C., & Ferreira, E. I. (2017). Molecular modeling and structure–activity relationships studies of bioisoster hybrids of N-acylhydrazone and furoxan groups on cruzain. Medicinal Chemistry Research, 26(4), 760–769. 10.1007/s00044-016-1776-7 [DOI] [Google Scholar]
- Sheahan, T. P., Sims, A. C., Zhou, S., Graham, R. L., Pruijssers, A. J., Agostini, M. L., Leist, S. R., Schäfer, A., Dinnon, K. H., Stevens, L. J., Chappell, J. D., Lu, X., Hughes, T. M., George, A. S., Hill, C. S., Montgomery, S. A., Brown, A. J., Bluemling, G. R., Natchus, M. G., … Baric, R. S. (2020). An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Science Translational Medicine, 12(541), eabb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki, K., & Daikoku, T. (2020). Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacology & Therapeutics, 107512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekanth, K., & Jha, A. (2020). Microwave assisted synthesis and antimicrobial activity of novel pyrrolidine derivatives. Russian Journal of General Chemistry, 90(1), 129–134. 10.1134/S107036322001020X [DOI] [Google Scholar]
- Tamokou, J-d-D., Tsemeugne, J., Fondjo, E. S., Sarkar, P., Kuiate, J.-R., Djintchui, A. N., Sondengam, B. L., & Bag, P. K. (2016). Antibacterial and cytotoxic activities and SAR of some azo compounds containing thiophene backbone. Pharmacologia, 7(4), 182–192. [Google Scholar]
- Teitz, Y., Ronen, D., Vansover, A., Stematsky, T., & Riggs, J. L. (1994). Inhibition of human immunodeficiency virus by N-methylisatin-beta 4':4'-diethylthiosemicarbazone and N-allylisatin-beta-4':4'-diallythiosemicarbazone. Antiviral Research, 24(4), 305–314. 10.1016/0166-3542(94)90077-9 [DOI] [PubMed] [Google Scholar]
- Tsuzuki, S., Uchimaru, T., Tanabe, K., & Yliniemela, A. (1996). Comparison of atomic charge distributions obtained from different procedures: Basis set and electron correlation effects. Journal of Molecular Structure: Theochem, 365(2–3), 81–88. 10.1016/0166-1280(96)04500-9 [DOI] [Google Scholar]
- Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., & Berendsen, H. J. C. (2005). GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 26(16), 1701–1718. 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- Varma, R. S., & Nobles, W. L. (1967). Synthesis and antiviral and antibacterial activity of certain N-dialkylaminomethylisatin beta-thiosemicarbazones. Journal of Medicinal Chemistry, 10(5), 972–974. 10.1021/jm00317a061 [DOI] [PubMed] [Google Scholar]
- Venkatarao, V., Kumar, L., Jha, A., & Sridhar, G. (2019). Synthesis and biological evaluation of chalcone fused quinoline derivatives as anticancer agents. Chemical Data Collections, 22, 100236. 10.1016/j.cdc.2019.100236 [DOI] [Google Scholar]
- Vishnu Nayak, B., Ciftci-Yabanoglu, S., Singh Jadav, S., Jagrat, M., Sinha, B. N., Ucar, G., & Jayaprakash, V. (2013). Monoamine oxidase inhibitory activity of 3,5-biaryl-4,5-dihydro-1H-pyrazole-1-carboxylate derivatives. European Journal of Medicinal Chemistry, 69, 762–767. 10.1016/j.ejmech.2013.09.010 [DOI] [PubMed] [Google Scholar]
- Wang, F., Chen, C., Tan, W., Yang, K., & Yang, H. (2016). Structure of main protease from human coronavirus NL63: Insights for wide spectrum anti-coronavirus drug design. Scientific Reports, 6, 22677. 10.1038/srep22677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Fang, X., Ge, L., Cao, F., Zhao, L., Wang, Z., & Xiao, W. (2018). Antitumor, antioxidant and anti-inflammatory activities of kaempferol and its corresponding glycosides and the enzymatic preparation of kaempferol. PLoS One, 13(5), e0197563. 10.1371/journal.pone.0197563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M., Cao, R., Zhang, L., Yang, X., Liu, J., Xu, M., Shi, Z., Hu, Z., Zhong, W., & Xiao, G. (2020). Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research, 30(3), 269–271. 10.1038/s41422-020-0282-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., Wang, Y., Wang, Q., Xu, Y., Li, M., Li, X., Zheng, M., Chen, L., & Li, H. (2020. a). Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica. B, 10(5), 766–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., Hu, Y., Tao, Z.-W., Tian, J.-H., Pei, Y.-Y., Yuan, M.-L., Zhang, Y.-L., Dai, F.-H., Liu, Y., Wang, Q.-M., Zheng, J.-J., Xu, L., Holmes, E. C., & Zhang, Y.-Z. (2020. b). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z., Zhao, S.-J., & Liu, Y. (2019). 1, 2, 3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. European Journal of Medicinal Chemistry, 183, 111700. [DOI] [PubMed] [Google Scholar]
- Xu, J., Shi, P.-Y., Li, H., & Zhou, J. (2020. a). Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infectious Diseases, 6(5), 909–915. 10.1021/acsinfecdis.0c00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z., Peng, C., Shi, Y., Zhu, Z., Mu, K., Wang, X., & Zhu, W. (2020. b). Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. BioRxiv. [Google Scholar]
- Xue, X., Yu, H., Yang, H., Xue, F., Wu, Z., Shen, W., Li, J., Zhou, Z., Ding, Y., Zhao, Q., Zhang, X. C., Liao, M., Bartlam, M., & Rao, Z. (2008). Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. Journal of Virology, 82(5), 2515–2527. 10.1128/JVI.02114-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadagiri, B., Gurrala, S., Bantu, R., Nagarapu, L., Polepalli, S., Srujana, G., & Jain, N. (2015). Synthesis and evaluation of benzosuberone embedded with 1,3,4-oxadiazole, 1,3,4-thiadiazole and 1,2,4-triazole moieties as new potential anti proliferative agents. Bioorganic & Medicinal Chemistry Letters, 25(10), 2220–2224. 10.1016/j.bmcl.2015.03.032 [DOI] [PubMed] [Google Scholar]
- Yang, S., Chen, S.-J., Hsu, M.-F., Wu, J.-D., Tseng, C.-T K., Liu, Y.-F., Chen, H.-C., Kuo, C.-W., Wu, C.-S., Chang, L.-W., Chen, W.-C., Liao, S.-Y., Chang, T.-Y., Hung, H.-H., Shr, H.-L., Liu, C.-Y., Huang, Y.-A., Chang, L.-Y., Hsu, J.-C., … Hsu, M.-C. (2006). Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. Journal of Medicinal Chemistry, 49(16), 4971–4980. 10.1021/jm0603926 [DOI] [PubMed] [Google Scholar]
- Zhang, H., Saravanan, K. M., Yang, Y., Hossain, M. T., Li, J., Ren, X., Pan, Y., & Wei, Y. (2020. a). Deep learning based drug screening for novel coronavirus 2019-nCov. Interdisciplinary Sciences, Computational Life Sciences, 12, 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., Lin, D., Kusov, Y., Nian, Y., Ma, Q., Wang, J., von Brunn, A., Leyssen, P., Lanko, K., Neyts, J., de Wilde, A., Snijder, E. J., Liu, H., & Hilgenfeld, R. (2020. b). α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. Journal of Medicinal Chemistry, 63(9), 4562–4578. 10.1021/acs.jmedchem.9b01828 [DOI] [PubMed] [Google Scholar]
- Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., Becker, S., Rox, K., & Hilgenfeld, R. (2020. c). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, 368(6489), 409–412. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.-M., Qiu, M., Sun, J., Zhang, Y.-B., Yang, Y.-S., Wang, X.-L., Tang, J.-F., & Zhu, H.-L. (2011). Synthesis, biological evaluation, and molecular docking studies of 1,3,4-oxadiazole derivatives possessing 1,4-benzodioxan moiety as potential anticancer agents. Bioorganic & Medicinal Chemistry, 19(21), 6518–6524. [DOI] [PubMed] [Google Scholar]
- Zhou, L., Liu, Y., Zhang, W., Wei, P., Huang, C., Pei, J., Yuan, Y., & Lai, L. (2006). Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. Journal of Medicinal Chemistry, 49(12), 3440–3443. 10.1021/jm0602357 [DOI] [PubMed] [Google Scholar]
- Zhou, P., Yang, X.-L., Wang, X.-G., Hu, B., Zhang, L., Zhang, W., Si, H.-R., Zhu, Y., Li, B., Huang, C.-L., Chen, H.-D., Chen, J., Luo, Y., Guo, H., Jiang, R.-D., Liu, M.-Q., Chen, Y., Shen, X.-R., Wang, X., … Shi, Z.-L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.