

Abstract
Paragangliomas are neuroendocrine tumors of the autonomic nervous system that are variably clinically functional and have a potential for metastasis. Up to 40% occur in the setting of a hereditary syndrome, most commonly due to germline mutations in succinate dehydrogenase (SDHx) genes. Immunohistochemically, paragangliomas are characteristically GATA3-positive and cytokeratin-negative, with loss of SDHB expression in most hereditary cases. In contrast, the rare paragangliomas arising in the cauda equina (CEP) or filum terminale region have been shown to be hormonally silent, clinically indolent, and have variable keratin expression, suggesting these tumors may represent a separate pathologic entity. We retrospectively evaluated seventeen CEPs from eleven male and six female patients with a median age of 38 years (range 21–82), none with a family history of neuroendocrine neoplasia. Six of the seventeen tumors demonstrated prominent gangliocytic or ganglioneuromatous differentiation. By immunohistochemistry, none of the CEPs showed GATA3 positivity or loss of SDHB staining; all seventeen CEPs were cytokeratin positive. Genome-wide DNA methylation profiling was performed on twelve of the tumors and compared with publicly available genome-wide DNA methylation data. Clustering analysis showed that CEPs form a distinct epigenetic group, separate from paragangliomas of extraspinal sites, pheochromocytomas, and other neuroendocrine neoplasms. Copy number analysis revealed diploid genomes in the vast majority of CEPs, whereas extraspinal paragangliomas were mostly aneuploid with recurrent trisomy 1q and monosomies of 1p, 3, and 11, none of which were present in the cohort of CEP. Together, these findings indicate that CEPs likely represent a distinct entity. Future genomic studies are needed to further elucidate the molecular pathogenesis of these tumors.
Keywords: cauda equina paraganglioma, filum terminale, neuroendocrine tumor, molecular neuropathology, succinate dehydrogenase, DNA methylation profiling
INTRODUCTION
Pheochromocytomas and paragangliomas are rare neural crest-derived neuroendocrine tumors arising from the adrenal medulla or paraganglia, respectively. Paragangliomas of the sympathetic paraganglia are most commonly located in abdomen, pelvis, or thorax, whereas paragangliomas of parasympathetic ganglia are most commonly located in the head and neck (e.g. carotid body tumor) [3, 19]. Paragangliomas, particularly of the sympathetic type, are often hormonally active and secrete catecholamines, similar to pheochromocytomas. By histology, paragangliomas often demonstrate a nested to organoid “zellballen” growth pattern of chief cells with surrounding sustentacular cells. The characteristic immunohistochemical profile of paragangliomas and pheochromocytomas is that they are cytokeratin negative, have synaptophysin, chromogranin, GATA3, and tyrosine hydroxylase positive chief cells, and have S100 positive sustentacular cells and, to a lesser extent, chief cells [3, 9]. Of clinical importance, a subset of paragangliomas demonstrate aggressive clinical behavior and all paragangliomas are considered to be at risk for metastasis [12, 19]. Approximately 40% of paragangliomas are associated with germline mutations causing hereditary pheochromocytoma-paraganglioma syndromes, most commonly with mutations in succinate dehydrogenase (SDH) subunit genes [5, 11, 18]. As the majority of hereditary paragangliomas demonstrate loss of SDHB immunoreactivity, recommendations for immunohistochemically screening all paragangliomas have been published [3, 14, 25, 26, 29].
Paragangliomas very rarely occur in the spinal cord, characteristically in the lumbar, cauda equina, or filum terminale regions. Prior descriptions of these “cauda equina paragangliomas” (CEPs) have shown striking histologic similarity to paragangliomas of other sites, including the characteristic zellballen pattern; given their rarity at this site and the often perivascular arrangement of chief cells, some have been mistaken for ependymoma [6, 10, 22, 30]. However, in contrast to paragangliomas at other sites, all CEPs reported to date have been sporadically occurring, hormonally silent, and very rarely recur or metastasize [1, 27, 28], with none reported to have metastasized outside of the central nervous system. By immunohistochemistry, multiple small series have found CEPs to be positive for cytokeratins [9, 20, 24], including a recent study by Mamilla et al. examining three CEPs and showing them to be cytokeratin positive, GATA3 negative, and tyrosine hydroxylase negative [20]. Importantly, CEPs have not been associated with hereditary paraganglioma syndromes; a single case of CEP arising in a patient with SDHD mutation has been reported, but whether this tumor arose from biallelic loss of SDHD is unclear [21]. Taken together, these findings suggest that CEPs may be a separate entity from paragangliomas of other sites and from pheochromocytomas.
The aim of this study was to determine the extent to which the immunohistochemical, DNA methylation, and chromosomal copy number profiles of CEP differ from extra-spinal paraganglioma and pheochromocytoma. Toward this goal, we retrospectively examined seventeen CEPs by immunohistochemistry for cytokeratin, GATA3, and SDHB. We evaluated twelve of these tumors by genome-wide DNA methylation profiling, which was compared against publicly available epigenetic data from 19 additional CEPs alongside extra-spinal paragangliomas, pheochromocytomas, other neuroendocrine neoplasms, and other common spinal cord neoplasms including various ependymoma subtypes. Finally, using the same data sets, we compared the chromosomal copy number of CEPs and extra-spinal paragangliomas.
MATERIALS AND METHODS
Tumor samples
Seventeen cauda equina paragangliomas (CEPs), defined as intradural spinal paragangliomas of the lumbar, cauda equina, filum terminale, or sacral regions, were retrieved from the surgical (N=8) and consult (N=9) neuropathology files at UCSF with local IRB approval. All tumor specimens were fixed in 10% neutral-buffered formalin and embedded in paraffin.
Immunohistochemical stains
The immunohistochemical stained sections evaluated in this study included a combination of those performed by the submitting institution prior to consultation and those performed at UCSF Medical Center using commercial antibodies and standard clinical protocols. GATA3 and SDHB immunohistochemical stains were performed at UCSF Medical Center on all thirteen cases. Immunohistochemical stains performed at UCSF Medical Center used the following parameters: Chromogranin A (clone LK2H10, Cell Marque, 1:100, 15-minute incubation, 10-minute ER1 pretreatment); SDHB (clone 21A11AE7, Abcam, 1:200 dilution, 15-minute incubation, 20-minute ER2 pretreatment); Keratin cocktail (clones AE1/AE3 and CAM5.2, DAKO/Becton, 1:100 dilution, 15-minute incubation, 20-minute ER1 pretreatment); Ki-67 (Clone MIB1, DAKO, 1:50 dilution, 30-minute incubation, 20-minute ER2 pretreatment); S100 (polyclonal, DAKO, 1:2000, 30-minute incubation, no pretreatment); and GATA3 (L50–823, Ventana, undiluted, 60-minute incubation, 64-minute 95 degrees Celsius in CC1 reagent). Immunostaining was performed on a Leica Bond-III automated stainer for all antibodies except GATA3, which was performed on Ventana Ultra. Diaminobenzidine was used as the chromogen, followed by hematoxylin counterstain.
DNA Methylation Profiling and Copy Number Analysis
Tumor tissue was selectively punched from formalin-fixed, paraffin-embedded blocks from 12 of the cauda equina paragangliomas using 2.0 mm disposable biopsy punches (Integra Miltex Instruments, cat# 33–31-P/25). Genomic DNA was then extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen). 250 ng of this genomic DNA was bisulfite converted using the EZ DNA Methylation kit following the manufacturer’s recommended protocol (Zymo Research). Bisulfite converted DNA was then amplified, fragmented, and hybridized to Infinium EPIC 850k Human DNA Methylation BeadChips following the manufacturer’s recommended protocol (Illumina).
In addition to the DNA methylation profiles generated from our cohort of 12 cauda equina paragangliomas, publicly available DNA methylation files produced from Illumina Infinium 450k beadchips in .idat format were obtained from The Cancer Genome Atlas (TCGA) data portal and Gene Expression Omnibus (GEO) spanning 16 tumor entities including cauda equina paragangliomas, extra-spinal paragangliomas, pheochromocytomas, other neuroendocrine tumor types, and other common spinal cord tumor types [7, 8, 15, 23]. Raw methylation data files were then processed in R statistical environment (v3.6.0) using the minfi package (v1.30.0) [2]. The detection p-value for each sample was computed. All samples had detection p values less than 0.05. Additional quality control was performed by calculating the median log (base2) intensities for methylated and unmethylated signals for each array. All samples had unmethylated and methylated median intensity values above 10 that were used for analysis. Beta density plots for all samples before and after normalization were also examined for quality control. Functional normalization with NOOB background correction [13] and dye-bias normalization was performed. Probe filtering was performed after normalization. Specifically, probes located on sex chromosomes, containing nucleotide polymorphisms (dbSNP132 Common) within five base pairs of and including the targeted CpG site, or mapping to multiple sites on hg19 (allowing for one mismatch), as well as cross reactive probes were removed from analysis.
The analysis included 31 cauda equina paragangliomas, 30 extra-spinal paragangliomas, 148 pheochromocytomas, 81 adrenocortical carcinomas, 14 Ewing sarcomas, 8 malignant melanotic schwannian tumors, 90 meningiomas, 28 myxopapillary ependymomas, 104 neuroblastomas, 32 pancreatic neuroendocrine tumors, 38 pilocytic astrocytomas of the midline epigenetic subgroup, 95 pituitary adenomas, 23 schwannomas, 11 small cell lung carcinomas, 27 spinal ependymomas, and 9 spinal subependymomas (tSNE sample manifest in Supplementary Table 1). Row-wise standard deviation was calculated for each of the 349,948 filtered probes across all 769 samples, and the 30,477 most differentially methylated probes with standard deviation >0.21 were selected for clustering analysis. R implementation of t-distributed stochastic neighbor embedding (tSNE) analysis was used for dimensionality reduction in our sample set to cluster samples with similar CpG methylation patterns. tSNE analysis was performed with the following analysis parameters using Rtsne package (version 0.15): perplexity = 15; max_iter = 8,000; theta = 0 [17]. The tSNE plot was visualized with ggplot2 (v 3.2.0) [https://ggplot2.tidyverse.org/]. The conumee Bioconductor package version 1.18.0 was used for chromosomal copy number analysis from the methylation array data, with manual curation of chromosomal gains, losses, and focal amplifications and deletions for each tumor sample [16].
RESULTS
Patient cohort and clinical features
The cohort of seventeen patients included eleven males and six females with a median age of 41 years (range 21 to 82) (Table 1). Tumor sizes ranged in maximum dimension from 1.3 cm to 9.0 cm. For the 11 patients with available clinical follow-up (range 3 to 159 months), two experienced local recurrence: patient #10 at 36 months and patient #15 at 348 months. No patients developed metastatic disease or died due to disease during the period of clinical follow-up. While one patient (#13) had both a personal and family history of papillary thyroid carcinoma, none of the patients in this cohort had a family history of neuroendocrine neoplasia. Two patients (#8 and #13) underwent clinical genetic testing at commercial laboratories, including assessment of SDHA, SDHAF2, SDHB, SDHC, SDHD, RET, VHL, FH, and NF1 genes among others, and were both reported to be negative for pathogenic variants.
Table 1.
Patient cohort and clinical information of seventeen cauda equina paragangliomas (CEPs), including personal and family history of neoplasia, maximum size of tumor on imaging, whether gross total resection (GTR) was achieved, length of clinical follow-up, evidence of recurrence, and clinical status at last follow-up. All seventeen cases were assessed by immunohistochemistry (IHC) and twelve cases were studied by DNA methylation profiling as indicated.
| Patient | Age (yrs) | Sex | Location | Size (max cm) | GTR | Recurrence | Follow-up (mos) |
|---|---|---|---|---|---|---|---|
| 1 | 47 | Female | L3 | 4 cm | Yes | None | 89 |
| 2 | 30 | Male | L1 | N/A | Yes | None | 159 |
| 3 | 41 | Male | L2-L3 | 3.0 | Yes | None | 98 |
| 4 | 48 | Female | L2-L3 | N/A | N/A | Unknown | N/A |
| 5 | 66 | Male | L4 | 1.3 | Yes | None | 64 |
| 6 | 82 | Female | L3-L4 | 0.9 | Yes | None | 69 |
| 7 | 38 | Male | L3 | 1.7 | Yes | None | 27 |
| 8 | 38 | Male | L3 | 1.6 | Yes | None | 12 |
| 9 | 62 | Male | L3-L4 | 1.0 | Yes | None | 35 |
| 10 | 34 | Male | L5-S2 | 9.0 | Yes | Local recurrence at 36 mos | 36 |
| 11 | 21 | Male | L3-L4 | 2.0 | Yes | None | 40 |
| 12 | 35 | Female | L3 | 2.6 | Yes | None | 13 |
| 13 | 32 | Female | L5 | 2.1 | Yes | None | 3 |
| 14 | 74 | Male | L5 | 3.0 | Yes | None | 6 |
| 15* | 37 | Male | L4-L5 | N/A | Yes | Local recurrence at 348 mos | 348 |
| 16 | 35 | Female | L4 | N/A | N/A | Unknown | N/A |
| 17 | 63 | Male | L4-L5 | 4.8 | Yes | Unknown | None |
| Patient | Personal history of neoplasia | Family history of neoplasia | Clinical status at last follow-up | IHC studies | Methylation profiling |
|---|---|---|---|---|---|
| 1 | None | None | Died of unrelated disease | Yes | Yes |
| 2 | None | None | Alive | Yes | No |
| 3 | None | None | Alive | Yes | Yes |
| 4 | Unknown | Unknown | Unknown | Yes | Yes |
| 5 | None | None | Alive | Yes | Yes |
| 6 | Basal cell carcinoma | None | Alive | Yes | No |
| 7 | None | None | Alive | Yes | Yes |
| 8 | None | None | Alive | Yes | Yes |
| 9 | None | None | Alive | Yes | Yes |
| 10 | None | None | Alive | Yes | Yes |
| 11 | None | None | Alive | Yes | Yes |
| 12 | None | None | Alive | Yes | Yes |
| 13 | Papillary thyroid carcinoma | Papillary thyroid carcinoma (Mother) | Alive | Yes | Yes |
| 14 | None | None | Alive | Yes | Yes |
| 15* | None | None | Alive | Yes | No |
| 16 | Unknown | Unknown | Unknown | Yes | No |
| 17 | None | None | Alive | Yes | No |
For patient 15, the recurrent tumor was analyzed for this study, as the initial tumor was unavailable for review.
Histologic and immunohistochemical features
On histologic evaluation, the most common appearance of CEPs was a proliferation of round to epithelioid chief cells growing in nests and sometimes with perivascular nucleus-free zones, mimicking perivascular pseudorosettes (Fig. 1). Nuclei were typically round and regular with ‘salt and pepper’ chromatin and moderate amphophilic to eosinophilic cytoplasm. Sustentacular cells were difficult to identify on H&E stains, but S100 often highlighted scattered small cells with thin processes partially surrounding the chief cell nests. Six of the seventeen (35%) CEPs additionally showed prominent gangliocytic differentiation (Table 2, Fig. 1), with three of these six additionally showing Schwann cell (ganglioneuromatous) differentiation.
Fig. 1.
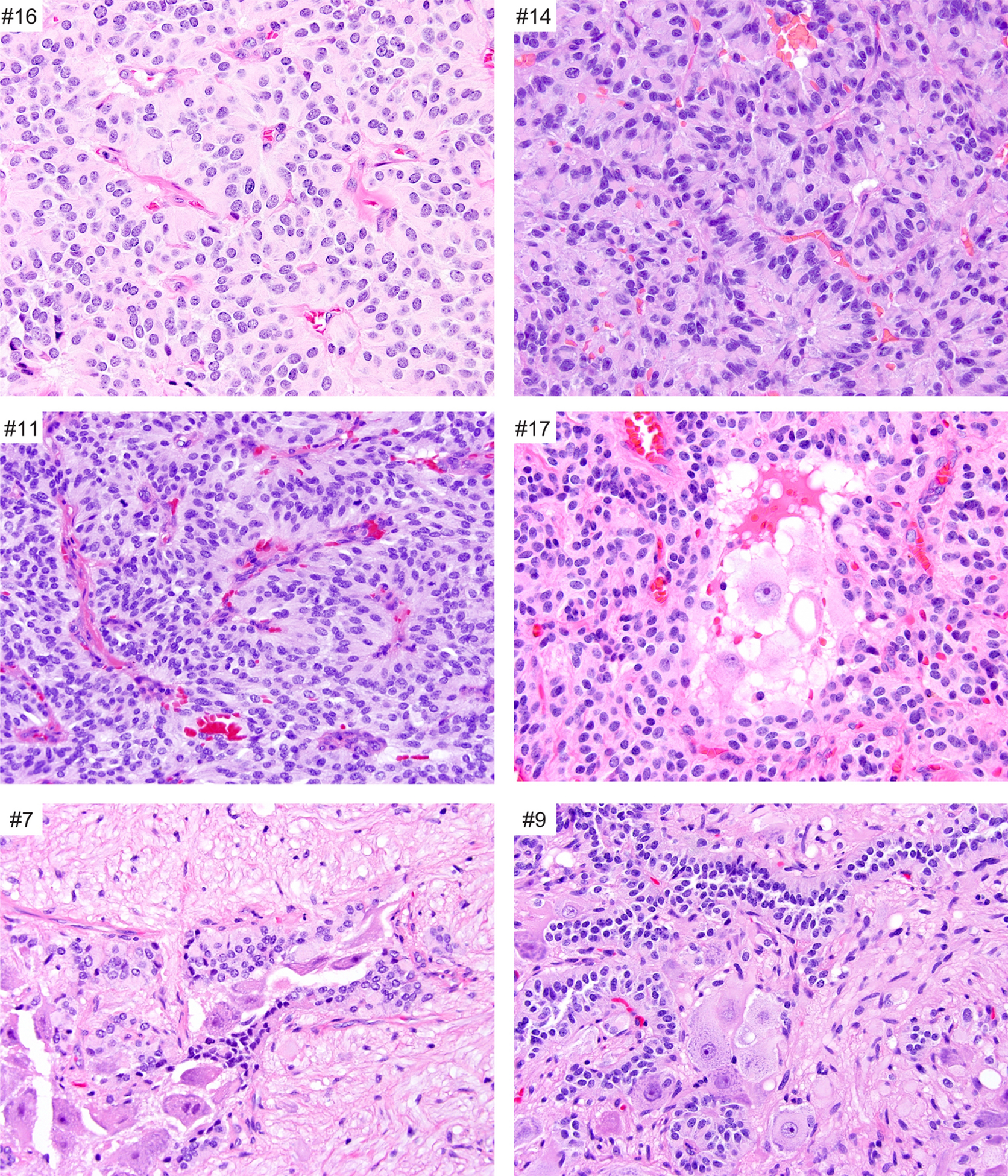
Histologic features of CEPs. Most CEPs are composed of a dominant population of chief cells that have round to elongate, relatively uniform nuclei and moderate eosinophilic cytoplasm. The chief cells are arranged in organoid or Zellballen pattern. A subset of CEPs demonstrate a gangliocytic (#17) or a ganglioneuromatous component (#7 and #9).
Table 2.
Histologic and immunohistochemical findings in seventeen CEPs, including presence of gangliocytic or ganglioneuromatous differentiation.
| Patient | Gangliocytic | Ganglioneuromatous | Chromogranin | SDHB | GATA3 | Keratin cocktail | *S100 | Ki-67 |
|---|---|---|---|---|---|---|---|---|
| 1 | Yes | No | Positive | Retained | Negative | Focal | Positive | 1% |
| 2 | No | No | Positive | Retained | Negative | Diffusely positive | Positive | 2% |
| 3 | No | No | Positive | Retained | Negative | Focal/patchy | Focal | 2% |
| 4 | Yes | No | Positive | Retained | Negative | Diffusely positive | Positive | 3% |
| 5 | No | No | Positive | Retained | Negative | Diffusely positive | Positive | 3% |
| 6 | No | No | Positive | Retained | Negative | Diffusely positive (Paranuclear) | Positive | 1% |
| 7 | Yes | Yes | Positive | Retained | Negative | Focal | Positive | 1% |
| 8 | No | No | Positive | Retained | Negative | Diffusely positive | Positive | 4% |
| 9 | Yes | Yes | Positive | Retained | Negative | Diffusely positive | Positive | 1% |
| 10 | No | No | Positive | Retained | Negative | Focal | Positive | 5% |
| 11 | No | No | Positive | Retained | Negative | Focal | Focal | 9% |
| 12 | Yes | No | Positive | Retained | Negative | Diffusely positive | Positive | 8% |
| 13 | No | No | Positive | Retained | Negative | Focal | Focal | 8% |
| 14 | No | No | Positive | Retained | Negative | Diffusely positive (Paranuclear) | Positive | 6% |
| 15 | No | No | Positive | Retained | Negative | Diffusely positive | Positive | 2% |
| 16 | No | No | Positive | Retained | Negative | Diffuse positive (paranuclear) | Negative | 8% |
| 17 | Yes | No | Positive | Retained | Negative | Diffusely positive (paranuclear) | Positive | 6% |
S100 positivity in the CEPs refers to staining seen in sustentacular cells.
On immunohistochemical evaluation, all CEPs (17/17) were chromogranin-positive (Fig. 2, Table 2, Table 3). Nearly all paragangliomas (16/17) showed positivity for S100, often more intensely positive in sustentacular cells and variably positive in chief cells. However, in contrast to paragangliomas described for other sites, 17/17 CEPs were completely negative for GATA3 (Fig. 2). All seventeen (17/17) cases showed retained SDHB expression. All CEPs (17/17) showed positivity for keratin with variable staining patterns, including focal staining or rare positive cells (6/17) and diffuse, strong staining (11/17), four of which had prominent paranuclear staining. Ki-67 labeling indices ranged from 1% to 9% (median 3%).
Fig. 2.
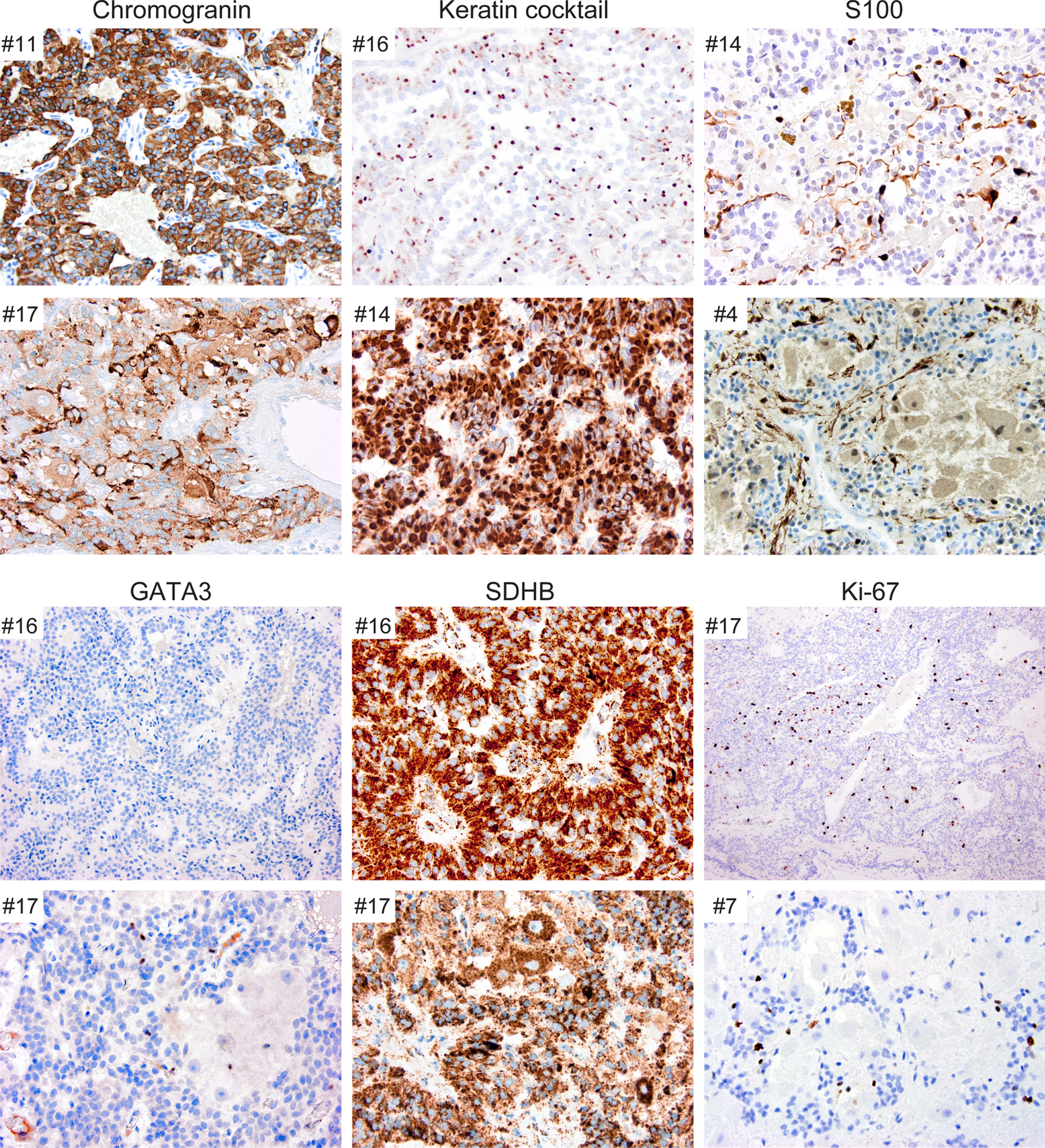
Immunohistochemical features of CEPs. All CEPs were chromogranin-positive, keratin-positive to varying degrees, and SDHB-retained. All cases showed S100 positivity with strong staining in sustentacular cells and weaker staining in the chief cells. GATA3 staining was negative in all cases. Ki-67 labeling varied from less than 1% to 9%.
Table 3.
Summary of histologic and immunohistochemical findings in seventeen CEPs.
| Gangliocytic differentiation | 6/17 (35%) |
| Ganglioneuromatous differentiation | 3/17 (17%) |
| Chromogranin expression | 17/17 (100%) |
| SDHB loss of expression | 0/17 (0%) |
| GATA3 expression | 0/17 (0%) |
| Keratin cocktail positivity | 17/17 (100%) |
| S100 positivity in sustentacular cells | 16/17 (94%) |
DNA methylation clustering analysis
We performed genome-wide DNA methylation profiling of twelve CEPs from our cohort as indicated in Table 1. We additionally obtained publicly available genome-wide DNA methylation profiles of an independent cohort of 19 CEPs, as well as 30 extra-spinal paragangliomas and 148 pheochromocytomas, and also other neuroendocrine tumor types (e.g. pituitary adenoma, small cell lung carcinoma, pancreatic neuroendocrine tumor), and other common spinal cord tumors (e.g. pilocytic astrocytoma, schwannoma, meningioma, conventional ependymoma, myxopapillary ependymoma, and subependymoma) (sample manifest including data source for the 769 tumors is listed in Supplementary Table 1) [7, 8, 11, 15, 23]. This publicly available cohort of nineteen CEPs evaluated for DNA methylation clustering included 10 male and 9 female patients with a median age of 42 years (range 18 to 73) (Supplementary Table 2). The 31 total CEPs from both cohorts all clustered together as an epigenetic group that was distinct from extra-spinal paragangliomas, pheochromocytomas, and all other tumor types included in the analyses (Fig. 3).
Fig. 3.
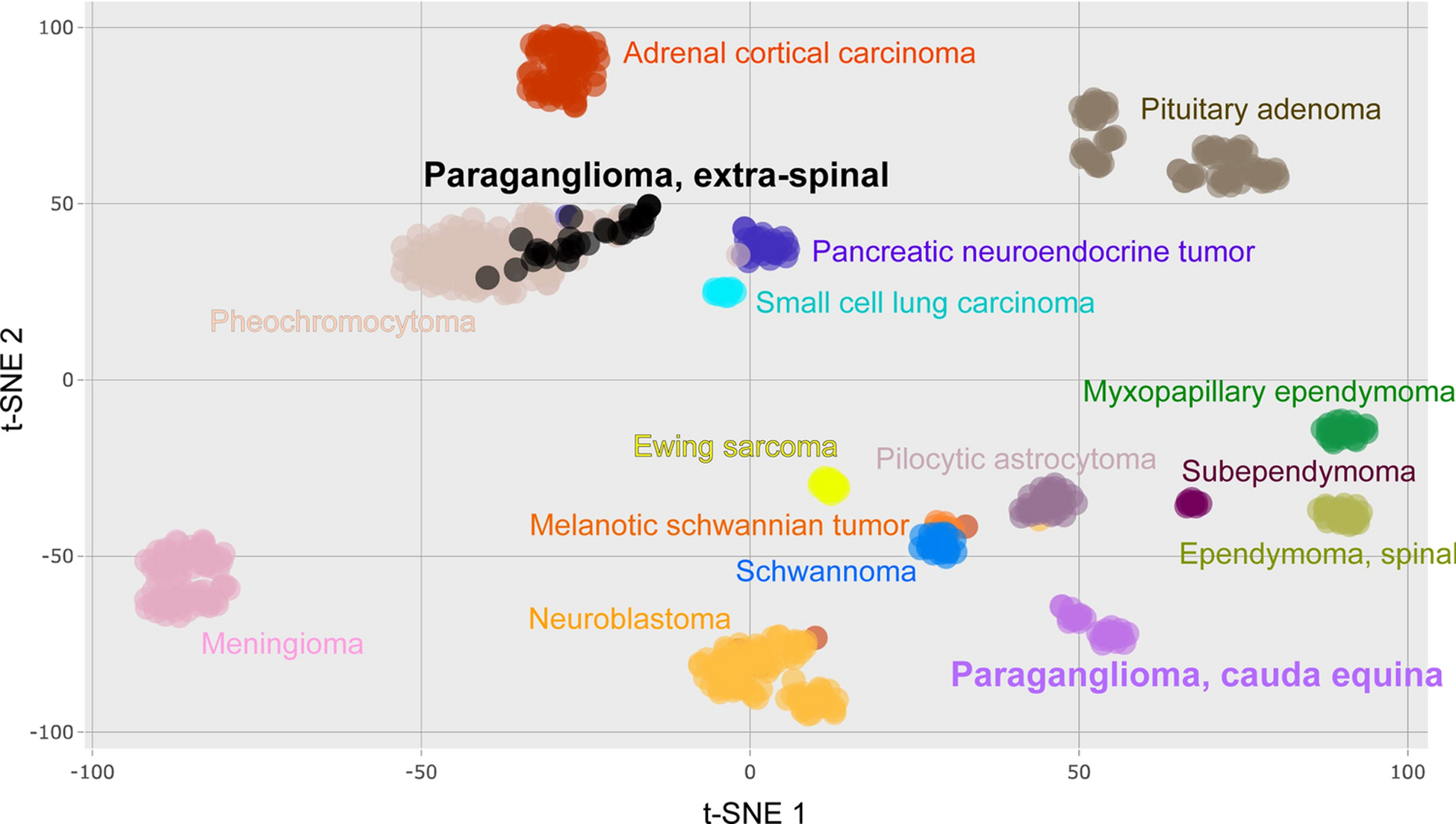
DNA methylation cluster analysis of CEPs and other tumors. tSNE plot of genome-wide DNA methylation profiles showing that CEP forms a distinct epigenetic cluster compared to extra-spinal paraganglioma, pheochromocytoma, other neuroendocrine neoplasms, nerve sheath tumors, and ependymomas, among other tumor types.
Genomic copy number analysis
The chromosomal copy number status of the 31 CEPs was assessed using the DNA methylation array data (Table 4, Fig. 4). 28 of the tumors (90%) demonstrated a diploid genome without chromosomal gains, losses, or focal amplifications or deletions. One tumor demonstrated trisomy of chromosome 4 as the solitary aberration, while another tumor demonstrated loss of proximal 21q as the solitary aberration. The remaining tumor demonstrated multiple aberrations. The copy number status of 30 extra-spinal paragangliomas was also assessed, which revealed chromosomal imbalances in 28/30 tumors (93%) and multiple recurrent aberrations (Supplementary Table 3). These included trisomy of chromosome 1q in 6/30 tumors (20%), monosomy of chromosome 1p in 19/30 tumors (63%), monosomy of chromosome 3 in 16/30 tumors (53%), and monosomy of chromosome 11 in 15/30 tumors (50%).
Table 4.
Chromosomal copy number changes for the 31 paraganglioma of the cauda equine
| Sample identifier | Chromosomal gains | Chromosomal losses | Focal amplifications | Focal deep deletions |
|---|---|---|---|---|
| UCSF CEP #1, GSM4729525 | none | none | none | none |
| UCSF CEP #3, GSM4729524 | none | proximal 21 q | none | none |
| UCSF CEP #4, GSM4729515 | none | none | none | none |
| UCSF CEP #5, GSM4729523 | none | none | none | none |
| UCSF CEP #7, GSM4729522 | none | none | none | none |
| UCSF CEP #8, GSM4729521 | none | none | none | none |
| UCSF CEP #9, GSM4729520 | none | none | none | none |
| UCSF CEP #10, GSM4729519 | none | none | none | none |
| UCSF CEP #11, GSM4729518 | none | none | none | none |
| UCSF CEP #12, GSM4729517 | none | none | none | none |
| UCSF CEP #13, GSM4729516 | none | none | none | none |
| UCSF CEP #14, GSM4729514 | distal 7q, distal 9q, 15q | 3, 10q, distal 11q, distal 18q | none | none |
| DKFZ GSM2403733 | none | none | none | none |
| DKFZ GSM2403739 | none | none | none | none |
| DKFZ GSM2403989 | none | none | none | none |
| DKFZ GSM2403990 | none | none | none | none |
| DKFZ GSM2403991 | none | none | none | none |
| DKFZ GSM2403992 | none | none | none | none |
| DKFZ GSM2403993 | none | none | none | none |
| DKFZ GSM2403994 | none | none | none | none |
| DKFZ GSM2403995 | none | none | none | none |
| DKFZ GSM2404003 | 4 | none | none | none |
| DKFZ GSM2404005 | none | none | none | none |
| DKFZ GSM2404006 | none | none | none | none |
| DKFZ GSM2404007 | none | none | none | none |
| DKFZ GSM2404008 | none | none | none | none |
| DKFZ GSM2404110 | none | none | none | none |
| DKFZ GSM2404141 | none | none | none | none |
| DKFZ GSM2404272 | none | none | none | none |
| DKFZ GSM2404273 | none | none | none | none |
| DKFZ GSM2404284 | none | none | none | none |
Fig. 4.
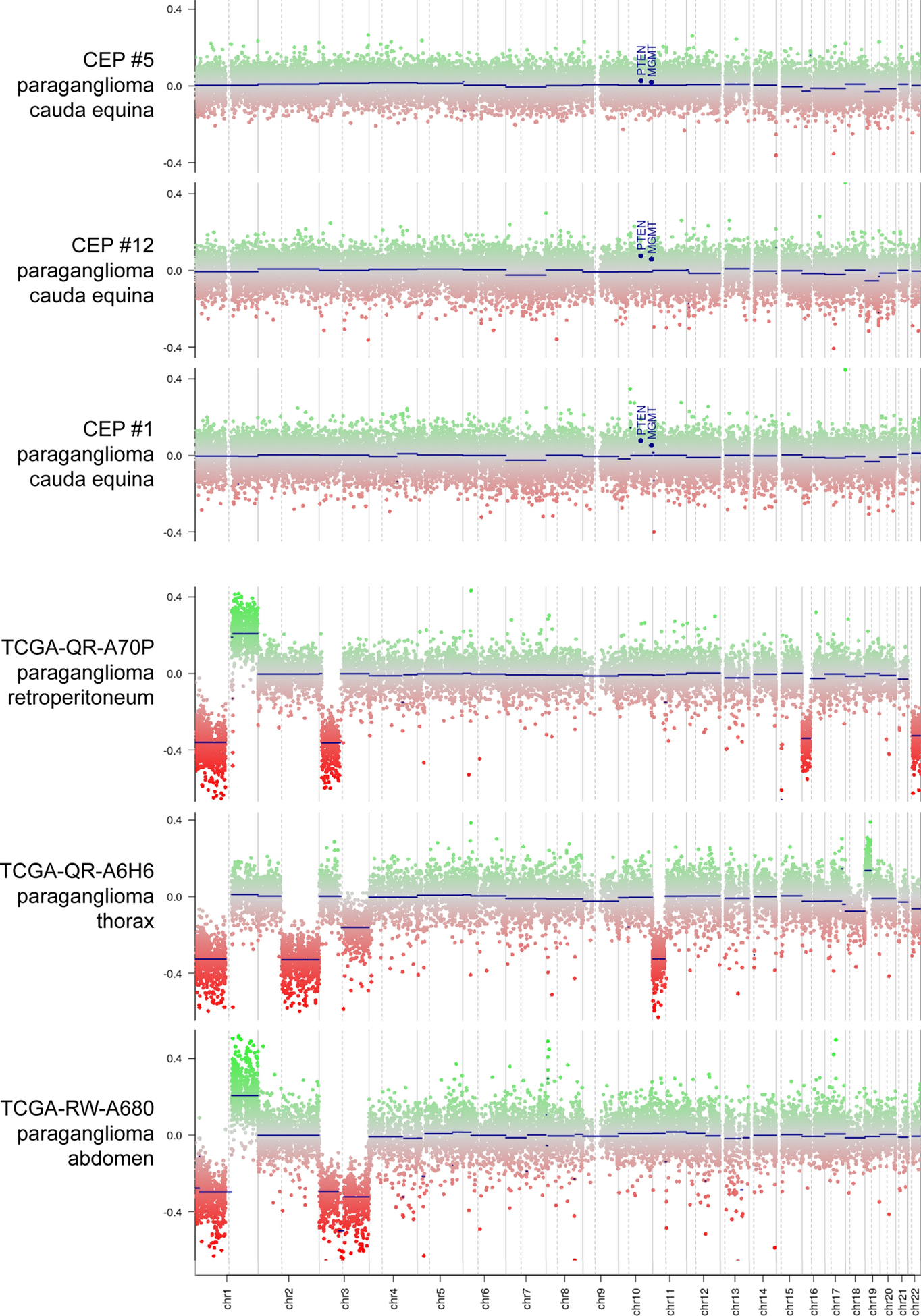
Chromosomal copy number analysis of CEP and extra-spinal paraganglioma. Shown are three representative genome-wide copy number plots from CEPs demonstrating a flat diploid genome, and three representative genome-wide copy number plots from extra-spinal paragangliomas demonstrating multiple chromosomal imbalances including recurrent gain of 1q and losses of 1p, 3, and 11.
DISCUSSION
The findings in this study suggest that CEPs may be biologically distinct from other paragangliomas and pheochromocytomas. Histologically, six of the seventeen (35%) examined CEPs demonstrated a gangliocytic component and occasionally even a Schwann cell component, whereas these features are almost never reported at other sites with the exception of the duodenum [4, 20]. Immunohistochemically, negative GATA3 staining and positive cytokeratin staining distinguishes CEP from paragangliomas at other sites. Similarly, none of the CEPs showed loss of SDHB expression, indicating that these are likely not associated with germline or somatic mutations inactivating any of SDH subunits. This also fits well with prior observations that hereditary paragangliomas do not arise in this location [21].
Additionally, our results show that the CEPs are epigenetically distinct from extra-spinal paragangliomas, pheochromocytomas, other neuroendocrine tumors, and glial or ependymal neoplasms of the spinal cord. This is in contrast to extra-spinal paragangliomas which all demonstrated epigenetic profiles overlapping with those of pheochromocytoma, suggesting a similar ontology of these two neoplasms. Furthermore, the vast majority of CEP demonstrate diploid genomes, whereas most extra-spinal paragangliomas are aneuploid tumors harboring recurrent cytogenetic aberrations including trisomy 1q and monosomies of 1p, 3, and 11.
Due to limited data, it is not entirely clear whether CEPs should be considered a subtype of paraganglioma, an epithelial neuroendocrine tumor, or a completely distinct biologic entity. Principally, the cell of origin for this tumor within the cauda equina or filum terminale is unknown. Native paraganglia are not known to exist within the spinal cord and, beyond its morphologic similarities, the extent to which these tumors are truly derived from paraganglia or neural-crest remains unknown. Nevertheless, CEPs appear immunohistochemically distinct from and show no cytogenetic or epigenetic overlap with extra-spinal paraganglioma and pheochromocytoma. The observation that gangliocytic paragangliomas of the duodenum are also keratin positive and GATA3 negative [20] suggests these could be related entities as epithelial neuroendocrine tumors. However, DNA methylation profiles for duodenal cases are not currently available for comparison. Nonetheless, CEPs did not show methylation clustering overlap with other epithelial neuroendocrine tumors, such as pancreatic neuroendocrine tumor and pituitary adenoma. We considered the possibility that CEPs originate from the rare ependymal rests in this region, although methylation clustering showed no overlap with any ependymal neoplasms including spinal subependymoma, conventional spinal ependymoma, and myxopapillary ependymoma. Similarly, although CEPs more commonly exhibit gangliocytic and Schwann cell differentiation, they showed no overlap with schwannomas or neuroblastomas on DNA methylation profiling in our analysis. Given these unknowns, we are unable to propose an alternative nomenclature for CEP, lumbar paraganglioma, or paraganglioma of the filum terminale at this time. Nonetheless, our data adds to the growing literature that these tumors are highly distinct from paragangliomas elsewhere. Additional genetic studies are needed to determine if there is a common driver mutation or gene fusion in CEPs.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by the Department of Pathology, University of California San Francisco clinical research endowment fund. This study was also supported in part by the NIH Director’s Early Independence Award from the Office of the Director, National Institutes of Health (DP5 OD021403) to D.A.S.; a Developmental Research Program Award from the UCSF Brain Tumor SPORE from the National Cancer Institute, National Institutes of Health (P50 CA097257) to D.A.S.; and the UCSF Glioblastoma Precision Medicine Program from the Sandler Foundation.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
CONFLICT OF INTEREST
None of the authors have any conflicts of interest to disclose. All procedures performed in studies involving human participants were in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
DATA AVAILABILITY STATEMENT
DNA methylation array data files for the 12 cauda equina paragangliomas generated as part of this study are available from the Gene Expression Omnibus (GEO) repository under accession number GSE156358 (https://www.ncbi.nlm.nih.gov/geo/).
REFERENCES
- 1.Akbik O, Floruta C, Chohan M, SantaCruz K, Carlson A (2016) A Unique Case of an Aggressive Gangliocytic Paraganglioma of the Filum Terminale. Case Rep Surg 2016. 10.1155/2016/1232594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. 30:1363–1369. 10.1093/bioinformatics/btu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asa S, Ezzat S, Mete O (2018) The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. J Clin Med 7:280 10.3390/jcm7090280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanco Sampascual S, Bereciartua E, Fernández Ramos JR, Martínez Cadilla J, Testillano Tarrero M, Moretó Canela M (1995) Duodenal gangliocytic paraganglioma. Rev Esp Enferm Dig 87:813–815. 10.14309/crj.0000000000000272 [DOI] [PubMed] [Google Scholar]
- 5.Burnichon N, Abermil N, Buffet A, Favier J, Gimenez-Roqueplo A-P (2012) The genetics of paragangliomas. Eur Ann Otorhinolaryngol Head Neck Dis 129:315–318. 10.1016/j.anorl.2012.04.007 [DOI] [PubMed] [Google Scholar]
- 6.Calenbergh F, Plets C, Ardon H, Sciot R (2011) Paraganglioma of the cauda equina region: A report of three cases. Surg Neurol Int 2:96 10.4103/2152-7806.82989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. 10.1038/nature26000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan CS, Laddha SV., Lewis PW, Koletsky MS, Robzyk K, Da Silva E, et al. (2018) ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat Commun 9 10.1038/s41467-018-06498-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dermawan JK, Mukhopadhyay S, Shah AA (2019) Frequency and extent of cytokeratin expression in paraganglioma: an immunohistochemical study of 60 cases from 5 anatomic sites and review of the literature. Hum Pathol 93:16–22. 10.1016/j.humpath.2019.08.013 [DOI] [PubMed] [Google Scholar]
- 10.Fiorini F, Lavrador JP, Vergani F, Bhangoo R, Gullan R, Reisz Z, et al. (2020) Primary Lumbar Paraganglioma: Clinical, Radiologic, Surgical, and Histopathologic Characteristics from a Case Series of 13 Patients. World Neurosurg. 10.1016/j.wneu.2020.05.144 [DOI] [PubMed] [Google Scholar]
- 11.Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. (2017) Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 31:181–193. 10.1016/j.ccell.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fliedner SMJ, Lehnert H, Pacak K (2010) Metastatic paraganglioma. Semin Oncol 37:627–637. 10.1053/j.seminoncol.2010.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. (2014) Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol 15 10.1186/s13059-014-0503-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY, et al. (2010) Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol 41:805–14. 10.1016/j.humpath.2009.12.005 [DOI] [PubMed] [Google Scholar]
- 15.Henrich KO, Bender S, Saadati M, Dreidax D, Gartlgruber M, Shao C, et al. (2016) Integrative genome-scale analysis identifies epigenetic mechanisms of transcriptional deregulation in unfavorable neuroblastomas. Cancer Res 76:5523–5537. 10.1158/0008-5472.CAN15-2507 [DOI] [PubMed] [Google Scholar]
- 16.Hovestadt V, Zapatka M conumee: Enhanced copy-number variation analysis using Illumina DNA methylation arrays. R package version 1.9.0 http://bioconductor.org/packages/conumee/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krijthe J (2015) Rtsne: T-Distributed Stochastic Neighbor Embedding using a Barnes-Hut implementation. https://github.com/jkrijthe/Rtsne [Google Scholar]
- 18.Lefebvre M, Foulkes WD (2014) Pheochromocytoma and paraganglioma syndromes: Genetics and management update. Curr Oncol 21 10.3747/co.21.1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lloyd R, Osamura R, Kloppel G, Rosai J (2017) WHO Classification of Tumours of Endocrine Organs, 4th ed International Agency for Research on Cancer, Lyon [Google Scholar]
- 20.Mamilla D, Manukyan I, Fetsch PA, Pacak K, Miettinen M (2020) Immunohistochemical distinction of paragangliomas from epithelial neuroendocrine tumors—gangliocytic duodenal and cauda equina paragangliomas align with epithelial neuroendocrine tumors. Hum Pathol 103:72–82. 10.1016/j.humpath.2020.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masuoka J, Brandner S, Paulus W, Soffer D, Vital A, Chimelli L, et al. (2001) Germline SDHD mutation in paraganglioma of the spinal cord. Oncogene 20:5084–5086. 10.1038/sj.onc.1204579 [DOI] [PubMed] [Google Scholar]
- 22.Mishra T, Goel NA, Goel AH (2014) Primary paraganglioma of the spine: A clinicopathological study of eight cases. J Craniovertebr Junction Spine 5:20–24. 10.4103/0974-8237.135211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, VanAller GS, et al. (2015) A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 28:57–69. 10.1016/j.ccell.2015.06.002 [DOI] [PubMed] [Google Scholar]
- 24.Orrell J, Hales S (1992) Paragangliomas of the cauda equina have a distinctive cytokeratin immunophenotype. Histopathology 21:479–481. 10.1111/j.1365-2559.1992.tb00436.x [DOI] [PubMed] [Google Scholar]
- 25.Pai R, Manipadam MT, Singh P, Ebenazer A, Samuel P, Rajaratnam S (2014) Usefulness of Succinate dehydrogenase B (SDHB) immunohistochemistry in guiding mutational screening among patients with pheochromocytoma-paraganglioma syndromes. APMIS 122:1130–1135. 10.1111/apm.12269 [DOI] [PubMed] [Google Scholar]
- 26.Papathomas TG, Oudijk L, Persu A, Gill AJ, Van Nederveen F, Tischler AS, et al. (2015) SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod Pathol 28:807–821. 10.1038/modpathol.2015.41 [DOI] [PubMed] [Google Scholar]
- 27.Roche PH, Figarella-Branger D, Regis J, Peragut JC (1996) Cauda equina paraganglioma with subsequent intracranial and intraspinal metastases. Acta Neurochir (Wien) 138:475–479. 10.1007/BF01420312 [DOI] [PubMed] [Google Scholar]
- 28.Strommer K, Brandner S, Sarioglu A, Sure U, Yonekawa Y (1995) Symptomatic cerebellar metastasis and late local recurrence of a cauda equina paraganglioma. In: J. Neurosurg [DOI] [PubMed] [Google Scholar]
- 29.Udager AM, Magers MJ, Goerke DM, Vinco ML, Siddiqui J, Cao X, et al. (2018) The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma-pheochromocytoma syndromes. Hum Pathol 71:47–54. 10.1016/j.humpath.2017.10.013 [DOI] [PubMed] [Google Scholar]
- 30.Yang SY, Jin YJ, Park SH, Jahng TA, Kim HJ, Chung CK (2005) Paragangliomas in the cauda equina region: Clinicopathoradiologic findings in four cases. J Neurooncol 72:49–55. 10.1007/s11060-004-2159-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.