

Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent liver disease world-wide, affecting 20–25% of the adult population. In 25% of patients, NAFLD progresses to non-alcoholic steatohepatitis (NASH), which increases the risk for the development of cirrhosis, liver failure and hepatocellular carcinoma. In patients with NASH, liver fibrosis is the main determinant of mortality. Here, we review how interactions between different liver cells culminate in fibrosis development in NASH, focusing on triggers and consequences of hepatocyte-macrophage-hepatic stellate cell (HSC) crosstalk. We will discuss pathways through which stressed and dead hepatocytes instigate the profibrogenic crosstalk with HSC and macrophages including the reactivation of developmental pathways such as TAZ, Notch and hedgehog; how clearance of dead cells in NASH via efferocytosis may affect inflammation and fibrogenesis; and insights into HSC and macrophage heterogeneity revealed by single cell RNA-sequencing. Finally, we will summarize options to therapeutically interrupt this profibrogenic hepatocyte-macrophage-HSC network in NASH.
BACKGROUND
With nearly 40% of the world’s population being overweight or obese, non-alcoholic fatty liver disease (NAFLD) is becoming a rapidly growing health problem, affecting ≈25% of the world’s adult population1, i.e. ~1.5 billion people. Chronic liver disease (CLD) due to non-alcoholic steatohepatitis (NASH), an advanced form of NAFLD, is expected to become the leading cause of liver transplantation in the US2,3, whereas CLD caused by HBV and HCV are declining due to improved treatments and HBV vaccination programs. Despite the extent of the problem, there are currently no approved therapies for NAFLD and NASH4,5. With several studies showing fibrosis as main determinant of mortality in NASH3,6,7, fibrosis has become a major focus in the NASH arena. Moreover, regulatory agencies such as FDA and EMA are mandating trials in advanced NASH so that the effect of interventions on measurable outcomes, including fibrosis, can be determined8,9. While correcting the underlying metabolic alterations is likely the best treatment for NAFLD, hepatocytes are in part replaced by fibrotic scar tissue in advanced NASH and are severely altered, rendering therapeutic targeting of underlying metabolic abnormalities less efficient. Therefore, increasing emphasis has been put on therapies that improve NASH fibrosis - which may be achieved by improving metabolic abnormalities, liver injury or inflammation, and/or by direct antifibrogenics5. Additional targets lie outside of the liver, such as the microbiome/gut-liver axis to decrease energy extraction or improve inflammation5,10–12; the CNS to limit food intake5; adipose and muscle to improve metabolism and inter-organ crosstalk13,14; and kidney to lose calories, e.g. via SGLT2 inhibition15.
Fibrosis is the result of excessive production of extracellular matrix (ECM) that is not adequately balanced by degradation, thus resulting in net accumulation. In the liver, hepatic stellate cells (HSC) constitute the main source of ECM-producing fibroblasts in models of toxic and biliary liver disease and NAFLD16–19. Portal fibroblasts represent only a minor source of liver fibroblasts in most studies18,19 and they are not known to play a role in NASH. ECM represents a complex network of ECM proteins that include 20 genetically distinct types fibrillary and non-fibrillar collagen; noncollagenous glycoproteins such as elastin, laminin, and fibronectin; glycosaminoglycans such as hyaluronan; proteoglycans such as aggrecan, fibromodulin, decorin, biglycan, glypicans, and syndecans 20. In addition to increased amounts, the composition of the ECM also changes in fibrosis20,21, with increases in embryonic or wound-healing associated ECM and increased crosslinks, which render ECM more resistant to degradation, contributing to the slow and often incomplete reversibility of advanced fibrosis. Besides acting as a three-dimensional scaffold that provides structure and determines boundaries, ECM provides important cues to surrounding cells. Notably, ECM controls cell survival, proliferation and differentiation and possibly metabolic pathways, acting through via specific cell surface receptors such as integrins or via the modulation of mechanosensitive signaling pathways22,23. Moreover, matricellular proteins such as thrombospondins, connective tissue growth factor, osteopontin and ECM-associated growth factors such as HGF may also affect hepatocytes and non-parenchymal cells. While alterations of the liver “matrisome” have been described in alcoholic liver disease 21, its composition in NASH remains to be determined. Hence, altered ECM composition in the fibrotic or cirrhotic NASH liver can impact liver function, regeneration and carcinogenesis24,25. The majority of clinical studies focus on complications associated with fibrosis, such as the replacement of liver parenchyma (contributing to liver failure) as well as increased stiffening and vascular resistance (contributing to portal hypertension). However, the underlying pathobiology is complex and not fully understood, and may also encompass protective functions of HSC and ECM/matrisome. It is conceivable that the role of HSC and ECM in NASH changes from restorative in early stages to disease-promoting in later stages. While fibrosis and cirrhosis are viewed as the common end-stage of different forms of CLD, it is not known whether there are disease-specific characteristics in regards to HSC activation, ECM and matrisome relevant for human NASH.
Hepatic stellate cell activation in NASH
HSC represent the dominant hepatic fibrogenic cell population contributing about 80–95% of collagen-producing myofibroblasts in different mouse models of fibrosis including NASH18. HSC are responsible for most of the architectural changes that characterize the fibrotic or cirrhotic NASH liver, in particular the deposition of the type I collagen-rich ECM, which contributes to typical complications such as portal hypertension and loss of functional liver mass. With HSC activation and fibrogenesis representing a unifying element in the response to hepatic injury between different liver diseases, it is conceivable that HSC activation is a conserved process, retaining high similarities between different liver diseases. This concept has been suggested for BDL- and CCl4-induced liver injury26 and recently for NASH-driven fibrosis27. At the same time, recent single cell RNA-sequencing (scRNA-seq) studies have revealed heterogeneity within the fibrogenic cell population, even though this needs further confirmation in larger cohorts. In patients, scRNA-seq has showed multiple profibrogenic cell populations including HSC, mesothelia/portal fibroblasts, vascular smooth muscle cells and scar-associated mesenchymal cells28. scRNA-seq in mice has shown fibrogenic cells to largely consist of HSC and only a small proportion of a portal fibroblast-like cell population in multiple models including NASH fibrosis29 (and unpublished results, RFS). Moreover, there is cellular heterogeneity in regards to activation and proliferation in both mice and patients28,29 as well as heterogeneity linked to anatomical localization in specific zones30 or interaction with other cell types such as scar-associated macrophages in specific locations28. While some of this heterogeneity may reflect the transition between different states (resting vs proliferating vs activated), it is likely that there is also functional heterogeneity. Hence both a fibrogenic core program, operating in virtually all HSC, as well as disease-, location-, context- or patient-specific functions may exist in parallel.
Transforming growth factor-β (TGFβ) is the most potent fibrogenic cytokine and a key driver of HSC activation and liver fibrosis31. TGFβ is released in its latent form by several hepatic cell populations31, and is locally activated by HSC expressing integrin aV32. Profibrogenic effects of TGFβ are mediated by SMAD-dependent pathways, by MAPK17 and possibly by YAP-dependent pathways33. Similar to toxic and biliary liver fibrosis, pharmacologic inhibition of TGFβ reduces NASH-induced fibrosis, albeit only partially, and combined inhibition of TGFβ and IL-13 signaling attenuates the fibrotic machinery more efficiently than TGFβ alone34. Platelet-derived growth factor (PDGF) represents a second fibrosis-promoting cytokine31. HSC express high levels of PDGF receptors, whose activation potently stimulates HSC proliferation and migration31. However, the specific role of PDGF signaling in NASH remains to be established. Emerging data show a key role of YAP in HSC activation as demonstrated by reduced fibrosis in mice treated with YAP inhibitor verteporfin and reduced HSC culture activation treated with YAP siRNA or verteporfin35,36. As YAP is also strongly upregulated in hepatocytes and cholangiocytes, it is not clear to what degree the reduction of fibrosis in verteporfin-treated mice is due to inhibition of YAP in HSC versus other cell types. Moreover, the contribution of HSC YAP/TAZ to NASH fibrosis as well as triggers of YAP/TAZ activation in HSC remain elusive. Other activators of HSC in NASH include Indian hedgehog (IHH, induced by TAZ in hepatocytes), sonic hedgehog (SHH, expressed by ballooning hepatocytes) and osteopontin (induced by hepatocyte Notch and TAZ)37–39. A plethora of additional mediators including CTGF, Il-17 and angiotensin II also contributes directly to HSC activation17,40,41 but their role in NASH remains to be determined. In summary, it appears that cellular events in hepatocytes and possibly also macrophages differ substantially between NASH and other liver diseases, whereas the activation of HSC may follow a more conserved pattern with similarities between NASH and other liver diseases and more subtle disease- and/or patient-specific variations. However, further studies in mice and patients are needed to substantiate this concept, in particular larger-scale scRNA-seq studies that compare various cell populations in patients with different liver diseases.
Cellular networks driving HSC activation and fibrosis as well as fibrosis resolution in NASH.
Cell death and inflammation are key drivers of fibrosis in NASH and other forms of CLD42–44. Both are triggered, sensed and responded to by cellular networks consisting of distinct resident and non-resident cells (Fig.1). Accordingly, cellular networks - rather than a single cell type – regulate fibrosis development in NASH. While our review will focus on the hepatocyte-macrophage-HSC network as most important driver of fibrosis in NASH, several other cell populations affect HSC activation and fibrosis in NASH42 (Fig.1). The contribution of immune cells is best exemplified by the decrease of CCl4- and NASH-induced liver fibrosis in RAG2−/−42 and RAG1−/− mice45, which lack mature B, T and NKT cells. As such B cells46, NKT cells45,47,48, platelets49,50 and type 2 innate lymphoid cells (ILC2)51 promote liver fibrosis, whereas NK cells may restrict fibrosis via killing of HSC52–54. In NASH, NKT cell promote fibrosis by increasing steatosis and hepatocellular damage45,48. Platelets promote NASH fibrosis by increasing lipid accumulation and immune cell infiltration49. Moreover, LSEC normally suppress HSC activation and lose this suppressive state in fibrosis55,56. While LSEC acquire lose their fenestrae at early stages of NAFLD57,58, their functional role in NASH fibrosis remains to be determined.
Fig.1. The cellular network regulating HSC activation and fibrosis in NASH.
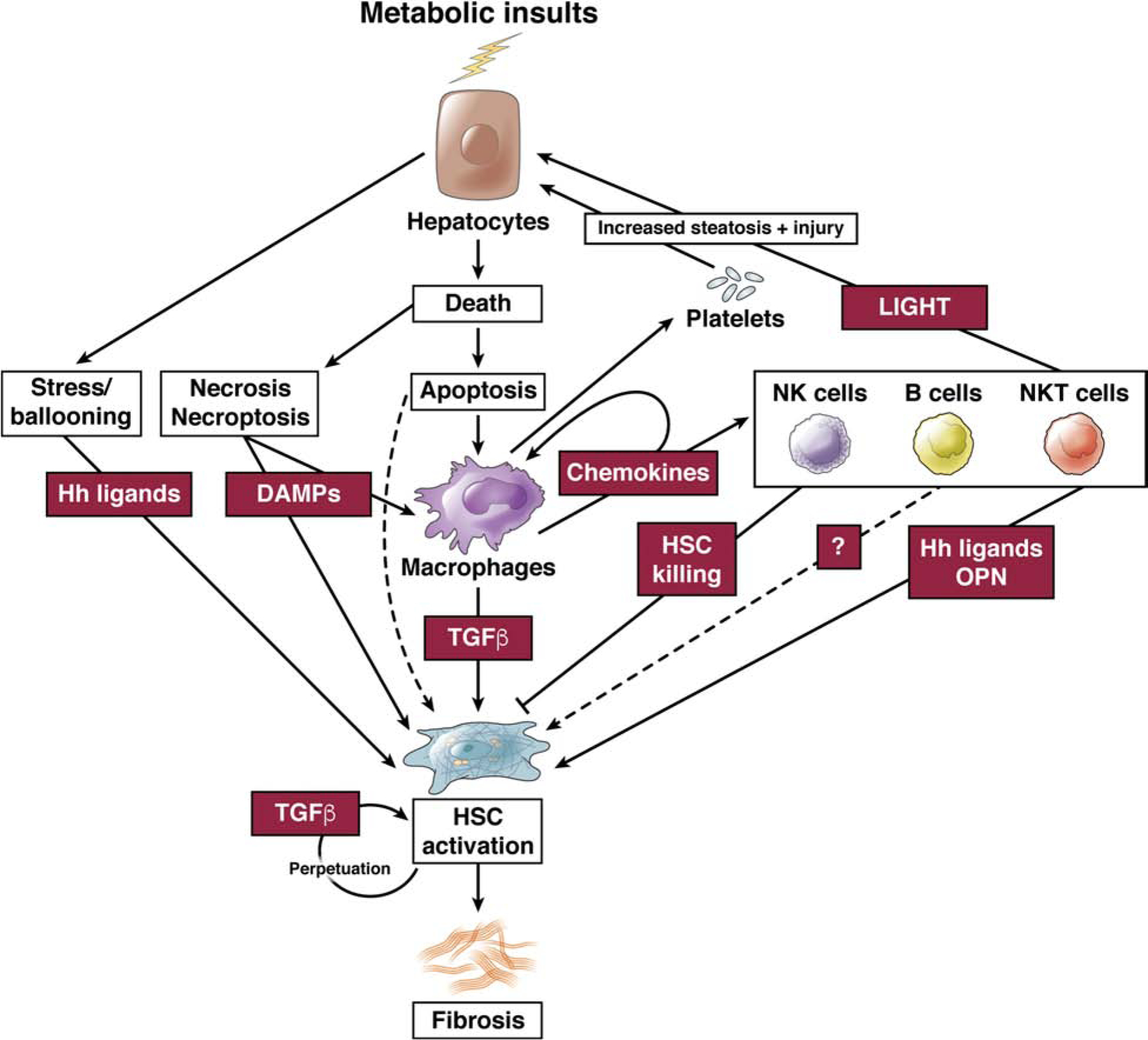
Metabolic insults promote hepatocyte steatosis and injury, activating a multi-cellular network consisting of macrophages, NKT cells, NK cells, B cells and NK cells that control HSC activation and the development of fibrosis.
Similar to other liver diseases (i.e. viral hepatitis, biliary obstruction or autoimmune hepatitis), successful treatment of the underlying disease results in fibrosis regression, as demonstrated in patients undergoing bariatric surgery59–62. Unlike patients with viral hepatitis, where successful treatment of the underlying disease can lead to the reversal of cirrhosis, such data are currently not available for NASH. Similar to the multi-cellular network involved in fibrogenesis, fibrosis resolution involves multiple cell types, in particular pro-resolution and ECM-degrading macrophages, promoting HSC apoptosis63 and HSC deactivation64,65 as well as the degradation of ECM66. These underlying mechanisms and cell-cell interactions in fibrosis regression have been reviewed in detail elsewhere66,67 and there is currently no insight whether fibrosis regression, HSC apoptosis and deactivation in NASH differs from other diseases. The fact that alterations of hepatocytes (via improving hepatocyte health), macrophages (via a switch from fibrotic to fibrosis-resolving macrophage) and HSC (via HSC apoptosis and deactivation) contribute to fibrosis resolution suggest that the hepatocyte-macrophage-HSC network not only stands in the center of fibrogenesis but represents the core network in fibrosis resolution. While there are additional signals feeding into this cellular network such as above-discussed liver cell types, the gut-liver axis - via microbial signals and metabolism, FXR activation and FGF1968 - as well as the adipose tissue - via adipokines and possibly via newly identified adipocyte-released lipid-filled vesicles (AdExos) that affect macrophage differentiation69 - we will focus on cell-cell crosstalk within the liver as well as means of therapeutically targeting it in this review.
How stressed, “undead” and dead hepatocytes trigger HSC activation and fibrosis in NASH
Hepatocytes contribute to HSC activation via multiple mechanisms. Most notably, hepatocyte stress and death promote inflammation, resulting in recruitment of macrophages and their secretion of profibrogenic mediators such as TGFβ, thus putting the hepatocyte-macrophage-HSC network at the center of the fibrogenic response in NASH (Fig.1). However, there is also evidence for HSC activation occurring through direct interactions of stressed or dead hepatocytes with HSC (Fig.1). This may be through the release of profibrogenic DAMPs43,44,70 or other profibrogenic mediators such as Hh ligands and osteopontin37–39, or via apoptotic bodies71,72, which may directly act on HSC. It is possible that (i) these are relevant in settings where there is little macrophage-derived TGFβ pathway, (ii) they occur in parallel to TGFβ-mediated HSC activation, resulting in stronger fibrogenesis, or (iii) hepatocyte-derived signals amplify TGFβ activation or TGFβ signaling in HSC.
Hepatocyte death as trigger for cell-cell networks that promote fibrosis.
Hepatocellular stress and death and the subsequent induction of fibrosis is common to all types of CLD including NASH. Similar to chronic HBV and HCV infection73–78, elevated levels of serum ALT, a surrogate marker for hepatocyte death, predict the presence of liver fibrosis and risk for fibrosis progression in NASH79,80. Vice versa, normalization of ALT levels after lifestyle intervention is significantly associated with fibrosis improvement in NASH81. As one third of patients with NASH fibrosis have normal ALT, it appears that although hepatocyte death is associated with NASH and fibrosis development, current methods to detect cell death are not sufficiently sensitive and/or that additional events may drive disease progression. Hepatocyte death comes in many flavors including apoptosis, necroptosis, necrosis, pyroptosis and ferroptosis. While it was initially suggested that apoptosis is the most prominent form of cell death in NASH82 and knockout of caspase 3 protected from several - but not all - aspects of MCD-induced NASH83, recent studies have also found evidence for necroptosis in murine and human NASH, and its contribution to NASH84. Therefore, it is likely that multiple forms of hepatocyte death act as key triggers in NASH44, but that they may differentially affect cell-cell communication leading to inflammation and fibrogenesis43,44. Dead hepatocytes can be removed by efferocytosis by professional phagocytes such as macrophages, triggering TGFβ release; or they can spill their contents in a less controlled fashion as in the case of necrosis, necroptosis and other non-apoptotic forms of cell death, thereby triggering a wide range of signals, in particular inflammation, in resident and non-resident cell types of the liver43,44. Cell-cell communication triggered by hepatocyte apoptosis and subsequent efferocytosis remain uncharacterized in NASH fibrosis. One essentially unanswered question is whether efferocytosis contributes to protective and antifibrotic cell-cell communication by removing dead hepatocytes and reducing inflammatory signaling induced via DAMPs from hepatocytes that underwent apoptosis, necroptosis and/or secondary necrosis; or whether it is part of a fibrosis-promoting response via activation of a TGFβ-secreting macrophage-HSC network (discussed in detail below). Finally, reports demonstrating activation of HSC after their engulfment of apoptotic bodies in vitro71 and in vivo72 suggests direct links between hepatocyte death and HSC activation. As efferocytosis can be carried out by epithelial cells, it is also conceivable that hepatocytes have a role in efferocytosis of dead hepatocytes in NASH85.
Stressed, injured or “undead” hepatocytes as trigger for fibrosis.
Hepatocyte ballooning is one of the most characteristic features of NASH. Ballooning is associated with higher risk for NASH and fibrosis development86,87 and decreased long-term survival6. While ballooned hepatocyte may be on the path to cell death and/or more sensitive to apoptosis, they are generally considered to be injured but living hepatocytes88,89 and have hence been termed as “undead” hepatocytes 90. Ballooned hepatocytes secrete sonic hedgehog (SHH), which promotes HSC activation37, but it is likely that they secrete additional profibrogenic ligands. While above-described associations suggest a key contribution to NASH fibrosis, is it conceivable that ballooned “undead” hepatocytes are a surrogate for hepatocyte stress, implying that hepatocyte stress drives fibrosis development in NASH without the need for hepatocytes to reach a state of ballooning or death. As such, ER stress is induced in steatotic livers from high-fat diet-fed mice91, and ER stress is common feature of fatty liver and NASH in patients92. Hepatocyte ER stress contributes to steatosis, hepatoycte death, inflammation and fibrosis93, suggesting that long-term and maladaptive ER stress triggers key features of NASH. Moreover, decreased expression of mitofusin 2, observed in NASH patients and mice on high-fat diet, may contribute to NASH by increasing steatosis and fibrosis in an ER stress-dependent manner via reduced transfer of phosphatidylserine from ER to mitochondria94. Hepatocyte ER stress also induces caspase 2, which contributes to NASH development95.
Hepatocyte signaling pathways that promote HSC activation and fibrosis in NASH.
TAZ.
TAZ, a paralogue of YAP and key component of the HIPPO-YAP/TAZ-TEAD signaling cascade, is strongly upregulated in hepatocytes in mouse models and patients with NASH38. Interestingly, TAZ upregulation appears to be confined to NASH as there was no increase in TAZ protein expression in CCl4-induced liver injury38. Moreover, TAZ was not upregulated in simple steatosis, suggesting that TAZ could be involved in the transition from simple steatosis to NASH38. Indeed, the functional contribution of hepatocyte TAZ to NASH development was proven by hepatocyte-specific deletion and silencing, with therapeutic efficacy even in advanced stages38,96. TAZ may affect fibrogenesis in NASH through multiple mechanisms (Fig.2): TAZ silencing reduces the expression of its target Indian Hedgehog (IHH), which exerted profibrogenic effects on HSC in vitro; conversely, NASH-induced fibrosis in vivo was suppressed by IHH silencing38. Further, IHH, like TAZ, is increased in human NASH but not simple steatosis38,97. Another study reported reduced fibrosis in a NASH-driven HCC model after IHH deletion in hepatocytes98. In addition to reducing IHH, TAZ silencing also decreased hepatocyte death and inflammation38. Owing to its effects on hepatocyte death, inflammation and HSC activation, hepatocyte TAZ appears to be a key hub in disease-promoting cell-cell communication in NASH and NASH fibrosis. TAZ upregulation occurs predominantly through transcription-independent pathways38, but the pathways upregulating TAZ in NASH remain unknown. As TAZ is not upregulated in NASH in simple steatosis or models of toxic liver injury suggest that TAZ increases are not explained by triglycerides or hepatocyte injury but linked to yet undefined factors associated with NASH. Moreover, functional interactions of TAZ with its paralogue YAP as well as other fibrosis-promoting pathways including Notch need to be further explored in order to determine whether there is a general reactivation of developmental pathways in NASH. In this context, a recent study showed a key role for hepatocyte YAP and its target Cyr61 in CCl4-induced liver fibrosis99.
Fig.2. The role of hepatocyte TAZ and Notch in NASH fibrosis.
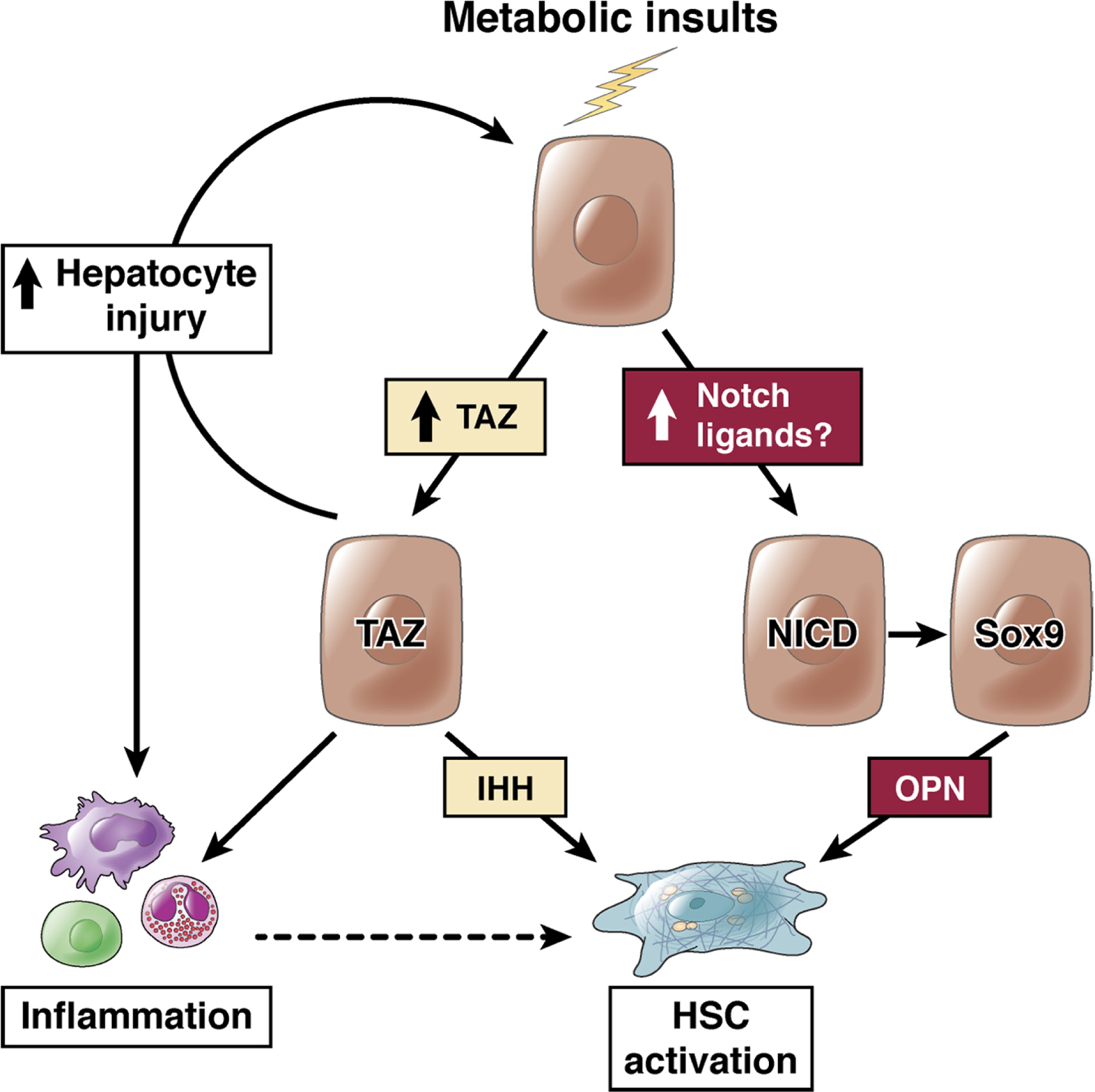
Metabolic insults lead to the activation of TAZ and Notch in hepatocytes. Increased hepatocytes expression of TAZ in NASH (but not simple steatosis) directly leads to HSC activation via the release of IHH, and additionally promotes hepatocyte injury and inflammation, which may indirectly promote HSC activation. Notch activity, driven by cell-surface ligands on a neighboring cell, leads to Sox9-dependent increase in osteopontin secretion, to activate HSC.
Notch.
Notch is a developmental pathway with roles in cell fate decisions100, contributing to the differentiation of hepatocyte progenitors towards cholangiocytes101. Whereas Notch activity is nearly absent in hepatocytes in the healthy adult liver and mildly elevated in simple steatosis 102, it is substantially increased in murine and human NASH39. Hepatocyte-specific Notch loss-of-function mouse models showed attenuated NASH-associated liver fibrosis without affecting cell death and inflammation39, thus distinguishing the effects of Notch on NASH-induced fibrosis from TAZ, which affects all these parameters38 (Fig.2). Analysis of the secretome of Notch-activated hepatocytes revealed an increase of osteopontin, which was responsible for the majority of the profibrogenic effects of Notch activation (Fig.2), both in vitro and in vivo39. Mechanisms of increased Notch activity are not as yet clear, but as ligand availability is normally limiting, the positive association of liver JAG1 expression (but not other Notch ligands)103 with propensity to NASH/fibrosis is intriguing. Also noteworthy is the finding that Notch activation increases FoxO1 activation at gluconeogenic promoters, leading to glucose intolerance104, which may partially explain the well-appreciated association between type 2 diabetes and accelerated NASH pathology.
Hh.
The hedgehog (Hh) pathway exerts fundamental morphogenic and mitogenic roles in tissue development, homeostasis, and repair105. Hh exerts important roles in hepatic injury responses and fibrogenesis106. As described above, human NASH liver, but not steatotic liver, expresses IHH, and causation data in vitro and in mice show a pro-fibrotic role of IHH in NASH31. In NASH patients, ballooned hepatocytes express Hh ligand SHH and are surrounded by Gli2-positive, i.e. Hh-activated myofibroblasts. ER stress, a common feature of NASH, results in increased expressed of SHH37. These findings suggest that stressed and ballooned hepatocytes generate Hh ligands which act as paracrine pro-fibrogenic factors for Hh-responsive stromal cells. Accordingly, hepatic SHH expression correlates with ballooning, Mallory-Denk bodies, fibrosis, ductular reaction, lymphocytic infiltration and serum AST107,108. However, patients with holoprosencephaly, a disorder that is often caused by inactivating mutations of SHH signaling, display increased liver pathology, in particular steatosis109. Likewise, Gli2 heterozygosity increases liver steatosis109. Together, these findings suggest a dual role of SHH in NAFLD, to suppress hepatic triglyceride accumulation while promoting the development of fibrosis. Whether SHH and IHH exert similar or distinct roles in NASH, remains to be further evaluated. Moreover, it is not clear whether all effects of Hh ligands SHH and IHH are directly on HSC or whether they may also promote HSC activation and fibrosis indirectly.
Crosstalk between hepatocyte TAZ, Notch, Hh and other profibrogenic pathways.
The YAP/TAZ and Notch pathway can interact, either by transcriptional coregulation of common target genes or via the YAP/TAZ-mediated transcriptional upregulation of Notch ligands110. For instance, YAP upregulates Notch2 to promote transdifferentiation of hepatocytes towards the cholangiocyte fate111. However, interactions between Notch and TAZ, two key drivers of NASH development, remain unexplored in NASH. Notch promotes fibrogenesis without altering liver injury whereas TAZ affects both processes, suggesting distinct mechanisms of action. Further studies are needed to understand whether YAP/TAZ upregulate Notch in NASH and thereby promote fibrosis and whether Hh signaling intersects with YAP/TAZ and/or Notch in NASH. Moreover, further studies are needed to determine whether the activation of these different development pathways is triggered by conserved upstream regulators. Similarly, TAZ, Notch and Hh pathways are thought to promote HSC activation and fibrosis independently of TGFβ, with TAZ and Notch signaling via IHH and osteopontin, respectively. But whether TGFβ signaling is amplified by TAZ-IHH- and Notch-OPN-mediated signals, and whether dual inhibition represents a rationale therapeutic strategy requires further study.
DAMPs.
Although it has been suggested that DAMPs from dying or stressed hepatocytes may promote liver fibrosis43,44,70, there is currently no convincing evidence to pinpoint a key role of classical DAMPs. As such, recent studies clearly show no effect of HMGB1 deletion on liver fibrosis112,113. However, the concept that hepatocyte-released DAMPs act on specific receptors on macrophages or HSC to promote fibrosis directly or indirect. Indeed, we have identified a profibrogenic HSC-enriched DAMP receptor (RFS, unpublished data).
How metabolic alterations trigger NASH-promoting pathways and cellular crosstalk
It is widely accepted that hepatic insulin resistance contributes to the development of NAFLD and that this is at least in part explained by selective insulin resistance in hepatocytes, which lose their ability to suppress glucose production in response to insulin while retaining the capacity to drive lipogenesis and increasing de novo lipogenesis114. This may be explained by Notch-driven hepatic insulin resistance104 and de novo lipogenesis115, but identification of molecular regulators of this selective insulin resistance paradox has proven elusive. Recent data support a role for the Akt Ser473 phosphatase, PHLPP2, as a molecular uncoupler of insulin-mediated repression of gluconeogenesis and activation of lipogenesis. Akt is a critical signaling node in determining insulin action– within minutes 116, Akt phosphorylates FoxO1 to repress glucose production; later, Akt activates mTORC1 signaling by phosphorylation of TSC2117, leading to increased Srebp1c activity at lipogenic gene promoters. PHLPP2 levels are decreased in obese liver118; adenoviral rescue reduces lipogenesis, whereas hepatocyte-specific PHLPP2 deletion is sufficient to cause fatty liver even in mice fed a normal chow diet119. Remarkably, PHLPP2 gain- and loss-of-function mice show unchanged glucose homeostasis. These data suggest that insulin signaling is not paradoxically selective in obesity, but rather that downstream elements determine relative effects on glucose production and lipogenesis.
But while this mechanism explains hepatocyte steatosis, it remains elusive which metabolic alterations trigger NASH promoting pathways and why the majority of people with steatosis do not develop NASH. Lipid droplets are often considered an inert storage site that puts lipids out of harm’s way, but a strong overload of this protective system could trigger the activation of NASH-promoting pathways such as TAZ, Notch or Hh. Thus, while steatosis is the key defining feature of NAFLD and NASH, it remains uncertain whether it is merely a required element (“first hit”) that sensitizes the liver to injury by mediators such as endotoxin or TNF (“second hit”) and progression to NASH. Alternatively, lipids may directly contribute to disease progression. Even though the majority of studies have not found a relationship between the degree of steatosis and NASH development3,6, the loss of steatotic hepatocytes in more advanced stages due to ECM accumulation and “burnt-out” disease confound these analyses. Indeed, recent studies found a positive correlation between the degree of hepatic steatosis, fibrosis development120 and liver disease mortality121. These findings are in line with epidemiologic data showing an influence of single nucleotide polymorphisms in PNPLA3 or TMSF6, two genes with roles in lipid metabolism, on fibrosis development122,123.
Hepatic diacylglycerol, triacylglycerol, saturated free fatty acids and free cholesterol increase in NAFLD and NASH124. It is now believed that triglycerides contribute to steatosis but not to injury and fibrosis125,126, whereas “bad” fats such as saturated fatty acid 127,128 or free cholesterol129 trigger lipotoxicity. Consistent with the concept that triglyceride-containing lipid droplets serve as a protective buffer, mice lacking diacylglycerol acyltransferase 2 with inability to convert FFAs into inert intracellular triacylglycerol show increased injury and fibrosis125. Mechanisms by which harmful lipids trigger NASH-promoting signaling pathways in hepatocytes - such as TAZ, Notch and Hh - and the subsequent activation of the NASH-promoting multicellular network described above remain elusive (Fig.2). It is conceivable that each NASH-promoting signaling pathway is activated by specific lipids; or that multiple harmful lipids, possibly in conjunction with additional hits, converge, triggering the activation of multiple NASH-promoting signaling pathways in parallel. Accordingly, several key hepatocyte proteins involved in lipid metabolism, including ACC-1/2, FXR/FGF19/FXR4, SCD-1, are promising therapeutic targets that are investigated in clinical trials5, thus highlighting the key role of hepatocyte lipid metabolism as initiator of the fibrosis-promoting hepatocyte-macrophage-HSC crosstalk in NASH.
In addition to affecting hepatocytes, metabolic changes may affect inflammatory cells and may – in concert with their effects on hepatocytes - contribute to NASH development. Although “metabolic activation” of NKT and T cells in NASH has been suggested 45, no evidence exists that this constitutes a direct metabolic activation independent of hepatocyte steatosis and injury. However, there is evidence supporting a role for lipid-mediated activation of macrophages and HSC. Saturated fatty acids as well as peroxidized lipids induce a proinflammatory macrophage phenotype130. In NASH, free cholesterol accumulates in macrophages, some of which is esterified to cholesteryl fatty acid esters, which causes them to resemble foam cell macrophages in atherosclerotic lesions131. Moreover, cholesterol crystals within lipid droplets of dead hepatocytes induce the formation of crown-like structures, resulting in macrophage activation with high levels of NLRP3 and caspase 1132. Oxidized LDL is sequestered in Kupffer cells lysosomes when injected into hyperlipidemic mice, resulting induced inflammatory gene expression133. In HSC, free cholesterol increases after feeding mice high-cholesterol diets, contributing to HSC activation and fibrosis via increased expression TLR4 or amplification of TLR4 signaling134–136. In summary, it appears that cholesterol not only contributes to NASH- and fibrosis-promoting changes in hepatocytes but also in other cell types such as HSC and macrophages. Although the relative contribution in these different cell types remains to be determined, it is likely that hepatocytes are the main target of NASH-promoting lipids.
Macrophages in HSC activation, fibrogenesis and fibrosis resolution in NASH
While the majority of macrophages in the healthy state are liver-resident yolk sac-derived Kupffer cells137, there is a marked infiltration of bone marrow-derived macrophages in the setting of liver injury, including NASH130. scRNA-seq studies have revealed macrophage heterogeneity showing the presence of Kupffer cells, tissue monocytes, and CD9+ TREM2+ NASH-associated macrophages (NAM)/scar-associated macrophages (SAM) in mice and patients28,138, the latter being expanded in cirrhotic livers and accumulating within fibrotic septa. Liver macrophages contribute to toxic liver fibrosis as demonstrated by pharmacologic and genetic ablation as well as by knockout of mediators of macrophage recruitment139–142. Similarly, macrophage depletion or inhibition of macrophage recruitment via CCR2/CCR5 inhibition suppresses fibrogenesis murine NASH143,144. TGFβ represents the most potent mediator of HSC activation and fibrosis and is enriched in macrophages31. Moreover, SAM express multiple other fibrosis-promoting genes including IL1B, SPP1, PDGFB and TNFSF1228. Conversely, HSC/fibroblasts, which are in close proximity to SAM, express cognate receptors that recognize IL-1B, SPP1, PDGFB and TNFSF12, rendering them highly responsive to these SAM-secreted profibrogenic factors and making the SMA-HSC/fibroblast axis an essential contributor to fibrogenesis28. As such, macrophage-derived IL-1β may promote the survival of HSC, thus increasing the pool of ECM-producing myofibroblasts142; SPP1, encoding osteopontin directly promotes HSC activation145; TNFSF12 and PDGFB promote HSC proliferation28. It is likely that the SAM-HSC axis acts in concert with other cell types and that for example TGFβ may also derived from cells such as activated HSC31 and contributes to the “perpetuation” of HSC activation. In NASH, macrophages exert additional functions that may modulate fibrogenesis such as the modulation of hepatic insulin sensitivity, and hepatic inflammation146 (discussed below).
Liver macrophages not only contribute to HSC activation and fibrosis but also to ECM degradation during the regression stage. CD11b+F4/80+Ly6Chi (mice) or CD14+/CD16+/CCR2+ (human) macrophage populations147 are fibrogenic, contrasting the murine CD11b+ F4/80+ Ly-6Clo population of restorative macrophages (the human restorative macrophage phenotype is not well-defined) that contributes to ECM degradation148. Accordingly, infusion of bone marrow-derived macrophages into mice increased hepatic recruitment of endogenous macrophages and neutrophils, thereby upregulating levels of MMP9, MMP13 and IL-10 in the scar, resulting in improved murine liver fibrosis149. Similar restorative functions can be seen in patients, as evidence by a recent trial in which infusion of macrophages was not only safe but also let to a decrease in MELD score in some participants150. Even though it appears that profibrogenic macrophages can be converted to pro-resolution macrophages in the liver148,151, the signals that regulate the switch to the resolving phenotype are currently not understood.
Macrophages linking hepatocyte damage to HSC activation in NASH via efferocytosis.
Even though saturated fatty acids, cholesterol, and oxidized lipids may directly trigger macrophage activation in NASH, it appears that NASH-induced hepatocyte damage and cell death are the dominant drivers of progression and fibrosis43,44, triggering macrophage recruitment, activation and subsequent macrophage-mediated HSC activation. One of the main purported functions of macrophages is the clearance of dead cells152. While apoptotic cell death is considered non-reactive, there is a wide body of literature linking NASH to hepatocyte apoptosis, and hepatocyte apoptosis to fibrosis44. Interestingly, the non-inflammatory nature of apoptosis is promoted by active suppression of inflammation and activation of inflammation resolution and possibly fibrogenesis-promoting pathways following the engulfment of apoptotic cells via efferocytosis. Apoptotic cells trigger the recruitment of macrophages through the release of soluble “find me” signals such as ATP (recognized by P2Y2 receptor) , CX3CL1 (recognized by CX3CR1) and sphingosine-1-phosphate (recognized by multiple sphingosine-1-phosphate receptors)152,153. Specific “find me” signals released by apoptotic hepatocytes in NASH are currently not known. The subsequent engulfment of apoptotic cells by “eat me” signals is largely triggered by the presence of phosphatidylserine on the outer leaflet of apoptotic cell membranes, which either directly binds to receptors on macrophages such as TIM-4 and αvβ5 integrin or engage with proteins such as GAS6 and PROS1, which in turn engage and activate receptors of the TAM family receptors on macrophages, such as MerTK152,153. In addition to the removal of apoptotic cells – which per se is anti-inflammatory - efferocytosis actively suppresses inflammation via the secretion of anti-inflammatory cytokines such as TGFβ152. Through this TGFβ pathway, efferocytosis may not only blunt inflammation but also promote HSC activation and fibrogenesis. This could be interpreted as attempt to resolve inflammation while repairing tissue injury in the setting of apoptotic liver injury (Fig.3). There is currently no literature that has experimentally tested the role of efferocytosis and efferocytosis-induced TGFβ in the liver even though one study demonstrated the presence of hepatic macrophages containing annexin V, suggesting macrophage phagocytosis of apoptotic fat-laden hepatocytes154. However, it is also conceivable that efferocytosis is a protective response in NAFLD which removes dead hepatocytes, thereby preventing the release of DAMPs and subsequent DAMP-mediated inflammation and fibrosis. If this were the case, failure to efficiently efferocytose and/or a switch from apoptotic cell death to more inflammatory cell death modes such as necroptosis, necrosis or ferroptosis might contribute to disease progression and fibrosis in NASH. It is also possible that efferocytosis of apoptotic hepatocytes – providing profibrogenic TGFβ – and non-apoptotic cell death – resulting in disease-promoting inflammation - exist in parallel and cooperatively promote the development of liver fibrosis. If efferocytosis turns out to be pro-fibrotic, it may be necessary to target multiple receptors to achieve significant therapeutic effects in NASH as each phase of efferocytosis employs multiple and often redundant receptors. It will also be important to understand whether other pathways in hepatocytes, such as the activation of TAZ and Notch but also specific metabolic alterations or ballooning, cooperate with or modulate efferocytosis or efferocytosis-mediated induction of macrophage-derived TGFβ.
Fig.3. Efferocytosis of dead hepatocytes as promoter of HSC activation and fibrosis.
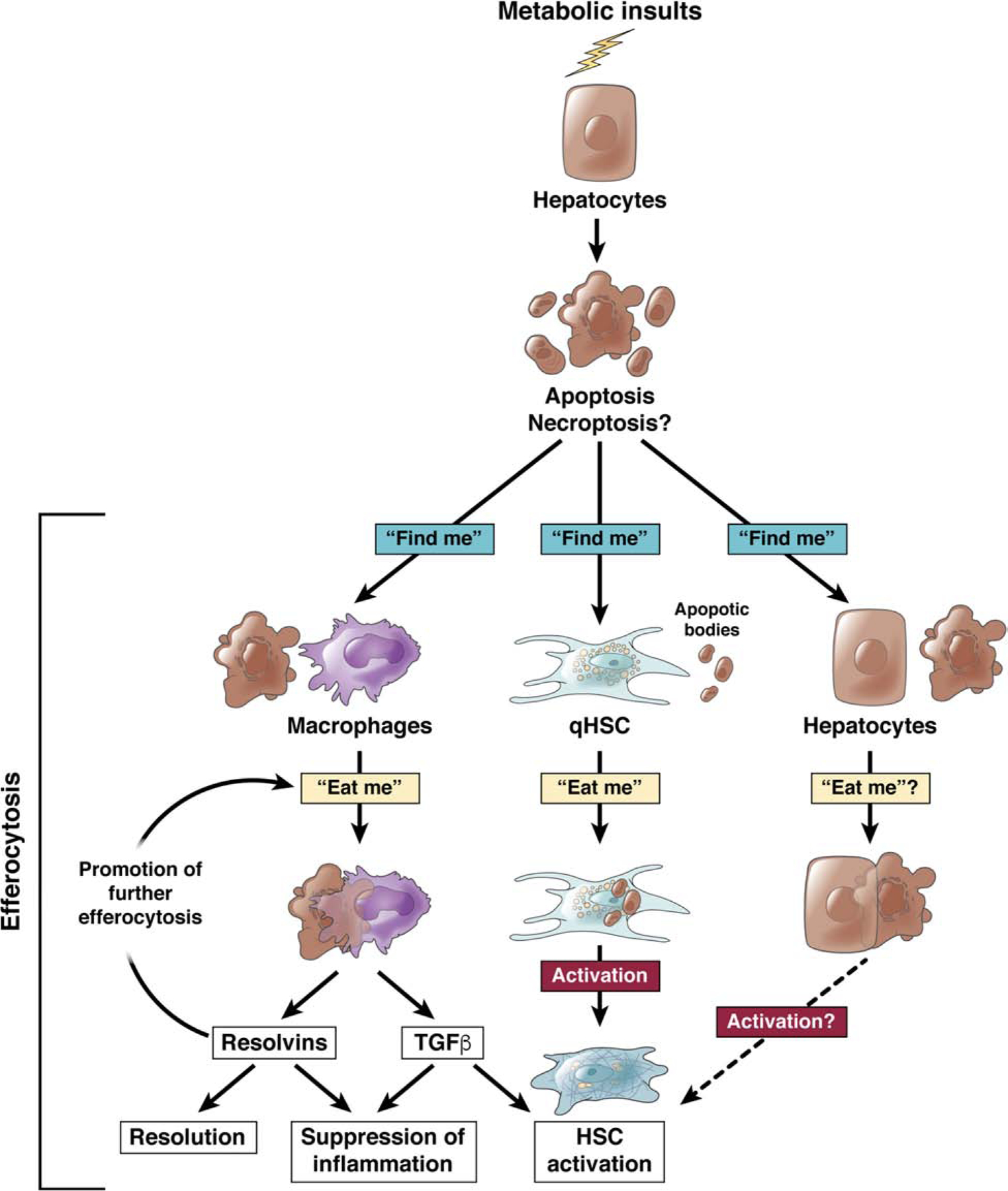
Apoptotic hepatocytes may be detected and engulfed by macrophages in response to “find me” and “eat me” signal in a process termed efferocytosis. Efferocytosis is most commonly exerted by professional phagocytes such as macrophages, where it leads to the release of resolvins – suppressing inflammation and promoting resolution - and TGFβ - suppressing inflammation and promoting HSC activation. Efferocytosis has also been suggested to occur in HSC and promote their activation. It is conceivable that efferocytosis could occur in hepatocytes, directly or indirectly affecting HSC activation.
Macrophages as instigators of inflammation and metabolic alterations in NASH
Besides directly affecting HSC and fibrosis, macrophages also regulate hepatic inflammation and metabolism, which may indirectly contribute to fibrogenesis. Hepatic macrophages have an important role in regulating inflammation in NASH. Liver macrophages express high levels of TLR4, making them highly responsive to gut-derived LPS, which is increased in NAFLD and NASH11,155, and contributing to high production of TNF and IL-1β and promotion of inflammation by macrophages in NAFLD130. Macrophage YAP contributes to hepatic inflammation but not steatosis or fibrosis in a HFD model of NASH156. The lacking effects of macrophage YAP on fibrosis in this study were attributed to low fibrosis induction by this model156. In addition, macrophage TNF and IL-1β may enhance fibrosis by directly promoting HSC activation and/or survival142,157. Moreover, macrophage-derived inflammatory mediators may also influence hepatocyte steatosis157,158. As such, macrophage-derived IL-1β downregulates PPARα, thereby leading to reduced fatty acid oxidation and increased triglyceride accumulation in hepatocytes158. Accordingly, macrophage depletion decreased steatosis and hepatic insulin resistance158,159. In conclusion, the regulation of hepatic inflammation, steatosis, hepatocyte death, and fibrosis by macrophage-derived cytokines are likely intimately linked and act in concert to drive NASH progression.
HSC effects on other cell types regulating liver injury, inflammation, regeneration, metabolism and liver function in NASH
While HSC are well-characterized as executors of fibrogenesis, it is not known how HSC affect other aspects of NASH. It is possible that the initial role of HSC in NAFLD is restorative via signals to other liver cells, in particular hepatocytes. As such, HSC are a main source of hepatocyte growth factor (HGF). Although the role HSC-derived HGF has not yet been studied, its receptor Met exerts an essential role in hepatocyte regeneration and survival in NASH and other diseases160–162. HSC-derived collagen promotes hepatocyte survival via Erk163 and possibly via integrin- and mechanosensitive signaling pathways. Lastly, it is not known whether HSC and HSC-derived ECM affect hepatocyte function and metabolism in NASH. Consistent with their close proximity to endothelial and hepatocytes, HSC could promote or inhibit hepatic steatosis by affecting inhibiting lipid shuttling into or out of hepatocytes.
Targeting cell-cell crosstalk in NASH
Given the key role of cell-cell crosstalk in NASH and the difficulty to non-selectively inhibit key fibrogenic pathways such as TGFβ due to severe side effects, targeting key intercellular pathways that trigger or maintain these disease-promoting interactions in may be a promising treatment strategy for NASH fibrosis (Fig.4). Hepatocytes represent a key cell driver initating NASH progression and can nowadays be targeted in a cell-specific manner by GalNAc-coupled siRNA, which has recently been approved by the FDA for TTR amyloidosis164. This approach can be applied to many hepatocyte pathways that trigger NASH fibrosis. Hepatocyte-specific silencing of TAZ may be useful as prevention as well as treatment as shown by GalNAc-siRNA silencing of TAZ in murine NASH fibrosis in mice96 and would interrupt several arms of the multi-cellular network that promotes NASH fibrosis, including hepatocyte death, inflammation and HSC activation (Fig.4). Targeting hepatocyte Notch may also be promising for NASH fibrosis but would require delivery of hepatocyte-targeted Notch inhibitors (Fig.4). In contrast, select targeting of pathways that mediate hepatocyte-HSC crosstalk such as SHH, IHH or osteopontin is likely to only achieve partial inhibition of NASH fibrosis due to the moderate profibrogenic potency of each pathway. Targeting key pathways that contribute to hepatocyte metabolic stress or death via inhibition of ACC, ASK-1, SCD-1 or via activation of FXR, FGF19, PPARα/δ or PPARα/γ signaling may indirectly inhibit the NASH fibrosis-promoting crosstalk between hepatocytes, macrophages and HSC (Fig.4; reviewed in5). Macrophages and efferocytosis pathways seem attractive targets as source and potential driver, respectively of TGFβ release in NASH fibrosis. However, we first need to learn more about the role of efferocytosis in NASH as it might also exert protective effects. Therefore, inhibition of macrophage recruitment by blockade of chemokine receptors such as CCR2 and CCR5 (Fig.4) seems a more feasible strategy to block cell-cell communication at the level of macrophages. CCR2/5 antagonist Cenicriviroc improved macrophage recruitment, steatosis, NAS score and fibrosis in mouse models of NASH144,165 and increased the percentage of patients who had improvement in fibrosis by ≥1 stage166. Currently, there are no approaches to specifically target macrophage-HSC crosstalk in NASH. However, given that liver macrophage-selective silencing via nanoparticles is feasible167, one could also envision macrophage-specific silencing of TGFβ as therapy for NASH fibrosis (Fig.4), although this strategy may increase liver inflammation. Infusion of macrophages is another possibility that may activate cell-cell networks leading to increased MMP and IL-10 expression and subsequent decreases in fibrosis and is currently tested in patients with cirrhosis of various etiologies150.Targeting of HSC is likely useful for directly inhibiting fibrogenesis in this cell type, rather than inhibiting cell-cell crosstalk in NASH, and is currently tested using a vitamin A-coupled lipid nanoparticle containing siRNA against HSP47, which leads to collagen misfolding and HSC death (NCT02227459). Finally, anti-platelet therapy using ticragelor or aspirin+clopidogrel may be beneficial in NASH, interrupting the crosstalk with hepatocytes and immune cells, thereby decreasing steatosis, inflammation and injury49. In summary, direct targeting of cell-cell communication or indirect targeting – via upstream triggers within hepatocytes that initiate this NASH-promoting cell-cell crosstalk - appear to be promising strategies for treating NASH fibrosis. However, further efforts are needed to establish the safest and most potent approaches.
Fig.4. Therapeutic inhibition of NASH by targeting intercellular networks.
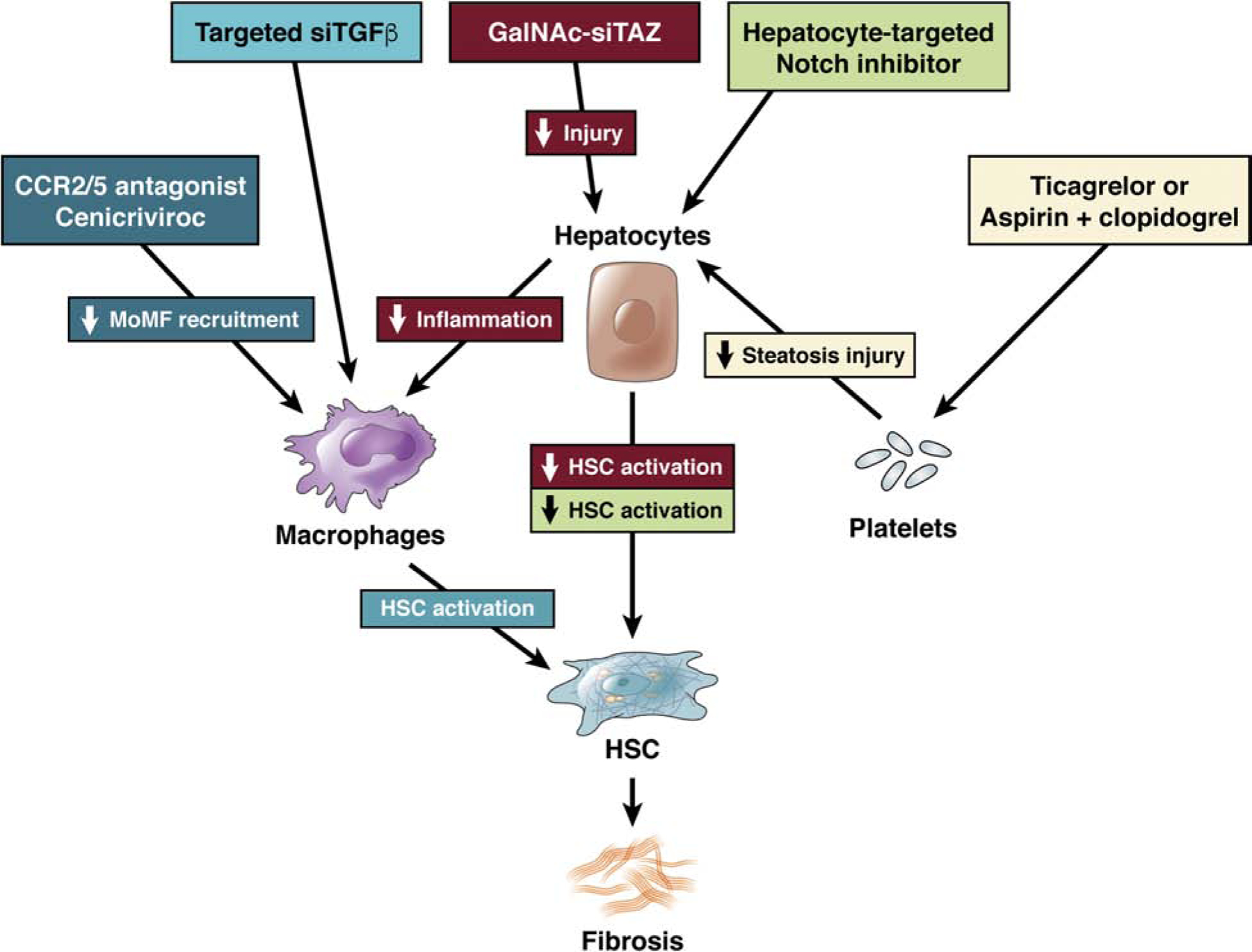
Targeting metabolic pathways, either in hepatocytes (ACC, SCD1, FXR/FGF19) or upstream (PPARα/δ or PPARα/γ) will improve hepatocyte metabolism and health and thereby indirectly reduce the fibrosis-promoting crosstalk with macrophages and HSC. Pathways that more directly initiate fibrosis-promoting crosstalk may be targeted at different levels.. Hepatocyte-specific TAZ silencing via GalNAc-coupled siRNA may lead to a reduction of IHH-mediated HSC activation as well as reduced inflammation and hepatocyte injury in NASH. Hepatocyte-targeted Notch inhibitors may decrease HSC activation in NASH fibrosis. Targeting the recruitment of monocyte-derived macrophages (MoMF) via CCR2/5 antagonist Cenicriviroc or the release of TGFβ from macrophages may reduce HSC activation in NASH. Targeting of the platelet-hepatocyte crosstalk via anti-platelet therapies such as Ticagrelor or aspirin+clopidogrel may reduce hepatocyte steatosis and injury, and a subsequent secondary reduction of HSC activation in NASH.
Acknowledgments
Grant support: 5 R01DK116620 (to IT and RFS), 5 R01CA200597 (to RFS); R01 DK103818 and R01 DK119767 (to UBP)
Abbreviations:
- NAFLD
non-alcoholic fatty liver disease (NAFLD)
- NASH
non-alcoholic steatohepatitis
- HSC
hepatic stellate cell
- ECM
extracellular matrix
- TGFβ
transforming growth factor-β
- IHH
Indian hedgehog
- SHH
sonic hedgehog
- Hh
hedgehog
- LSEC
liver sinusoidal endothelial cells
- BDL
bile duct ligation
Biographies



Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures: Nothing to disclose.
Writing Assistance: None
REFERENCES
- 1.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11–20. [DOI] [PubMed] [Google Scholar]
- 2.Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, Ahmed A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547–55. [DOI] [PubMed] [Google Scholar]
- 3.Diehl AM, Day C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N Engl J Med 2017;377:2063–2072. [DOI] [PubMed] [Google Scholar]
- 4.Schuppan D, Pinzani M. Anti-fibrotic therapy: lost in translation? J Hepatol 2012;56 Suppl 1:S66–74. [DOI] [PubMed] [Google Scholar]
- 5.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015;149:389–97 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, Eslam M, Gonzalez-Fabian L, Alvarez-Quinones Sanz M, Conde-Martin AF, De Boer B, McLeod D, Hung Chan AW, Chalasani N, George J, Adams LA, Romero-Gomez M. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018;155:443–457 e17. [DOI] [PubMed] [Google Scholar]
- 8.Rinella ME, Tacke F, Sanyal AJ, Anstee QM, participants of the AEW. Report on the AASLD/EASL joint workshop on clinical trial endpoints in NAFLD. J Hepatol 2019. [DOI] [PubMed]
- 9.Administration USDoHaHSFaD, (CDER) CfDEaR. Nonalcoholic Steatohepatitis with Compensated Cirrhosis: Developing Drugs for Treatment Guidance for Industry. 2019.
- 10.Canfora EE, Meex RCR, Venema K, Blaak EE. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol 2019;15:261–273. [DOI] [PubMed] [Google Scholar]
- 11.Kolodziejczyk AA, Zheng D, Shibolet O, Elinav E. The role of the microbiome in NAFLD and NASH. EMBO Mol Med 2019;11. [DOI] [PMC free article] [PubMed]
- 12.Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol 2016;13:412–25. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi H, Kotani K, Tanaka K, Egucih Y, Anzai K. Therapeutic Approaches to Nonalcoholic Fatty Liver Disease: Exercise Intervention and Related Mechanisms. Front Endocrinol (Lausanne) 2018;9:588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boutari C, Perakakis N, Mantzoros CS. Association of Adipokines with Development and Progression of Nonalcoholic Fatty Liver Disease. Endocrinol Metab (Seoul) 2018;33:33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheen AJ. Beneficial effects of SGLT2 inhibitors on fatty liver in type 2 diabetes: A common comorbidity associated with severe complications. Diabetes Metab 2019;45:213–223. [DOI] [PubMed] [Google Scholar]
- 16.Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A 1985;82:8681–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, Schwabe RF. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wells RG, Schwabe RF. Origin and function of myofibroblasts in the liver. Semin Liver Dis 2015;35:97–106. [DOI] [PubMed] [Google Scholar]
- 20.Friedman SL. Extracellular matrix In: Dufour J-FCP-A, ed. Signaling Pathways in Liver Diseases. Chichester, UK:: Wile, 2015:85–96. [Google Scholar]
- 21.Massey VL, Dolin CE, Poole LG, Hudson SV, Siow DL, Brock GN, Merchant ML, Wilkey DW, Arteel GE. The hepatic “matrisome” responds dynamically to injury: Characterization of transitional changes to the extracellular matrix in mice. Hepatology 2017;65:969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogel V Unraveling the Mechanobiology of Extracellular Matrix. Annu Rev Physiol 2018;80:353–387. [DOI] [PubMed] [Google Scholar]
- 23.Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009;326:1216–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filliol A, Schwabe RF. Contributions of Fibroblasts, Extracellular Matrix, Stiffness, and Mechanosensing to Hepatocarcinogenesis. Semin Liver Dis 2019. [DOI] [PubMed]
- 25.Affo S, Yu LX, Schwabe RF. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol 2017;12:153–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, Schwabe RF. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 2007;132:1937–46. [DOI] [PubMed] [Google Scholar]
- 27.Marcher AB, Bendixen SM, Terkelsen MK, Hohmann SS, Hansen MH, Larsen BD, Mandrup S, Dimke H, Detlefsen S, Ravnskjaer K. Transcriptional regulation of Hepatic Stellate Cell activation in NASH. Sci Rep 2019;9:2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, Taylor RS, Efremova M, Vento-Tormo R, Carragher NO, Kendall TJ, Fallowfield JA, Harrison EM, Mole DJ, Wigmore SJ, Newsome PN, Weston CJ, Iredale JP, Tacke F, Pollard JW, Ponting CP, Marioni JC, Teichmann SA, Henderson NC. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019. [DOI] [PMC free article] [PubMed]
- 29.Krenkel O, Hundertmark J, Ritz TP, Weiskirchen R, Tacke F. Single Cell RNA Sequencing Identifies Subsets of Hepatic Stellate Cells and Myofibroblasts in Liver Fibrosis. Cells 2019;8. [DOI] [PMC free article] [PubMed]
- 30.Dobie R, J. W-K, Henderson BEP, Smith M Jr, K.P., Portman J, Wallenborg K, Picelli S, Zagorska A, Budas GR, Harrison EM, Mole DJ, Wigmore S, Ramachandran P, Ponting CP, Teichmann SA, Marioni JC, Henderson NC. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Reports 2019;In Press. [DOI] [PMC free article] [PubMed]
- 31.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- 32.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, Maher JJ, Iredale JP, Lacy-Hulbert A, Adams RH, Sheppard D. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 2013;19:1617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu HX, Yao Y, Bu FT, Chen Y, Wu YT, Yang Y, Chen X, Zhu Y, Wang Q, Pan XY, Meng XM, Huang C, Li J. Blockade of YAP alleviates hepatic fibrosis through accelerating apoptosis and reversion of activated hepatic stellate cells. Mol Immunol 2019;107:29–40. [DOI] [PubMed] [Google Scholar]
- 34.Hart KM, Fabre T, Sciurba JC, Gieseck RL 3rd, Borthwick LA, Vannella KM, Acciani TH, de Queiroz Prado R, Thompson RW, White S, Soucy G, Bilodeau M, Ramalingam TR, Arron JR, Shoukry NH, Wynn TA. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-beta. Sci Transl Med 2017;9. [DOI] [PubMed]
- 35.Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, Hoorens A, Reynaert H, Halder G, van Grunsven LA. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 2015;63:679–88. [DOI] [PubMed] [Google Scholar]
- 36.Martin K, Pritchett J, Llewellyn J, Mullan AF, Athwal VS, Dobie R, Harvey E, Zeef L, Farrow S, Streuli C, Henderson NC, Friedman SL, Hanley NA, Piper Hanley K. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat Commun 2016;7:12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rangwala F, Guy CD, Lu J, Suzuki A, Burchette JL, Abdelmalek MF, Chen W, Diehl AM. Increased production of sonic hedgehog by ballooned hepatocytes. J Pathol 2011;224:401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, Tabas I. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab 2016;24:848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, Schwabe RF, Tabas I, Valenti L, Lavine JE, Pajvani UB. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed]
- 40.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, Brenner DA. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest 2003;112:1383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, Osterreicher CH, Stickel F, Ley K, Brenner DA, Kisseleva T. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012;143:765–776 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 2015;61:1066–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765–783 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, Lorentzen A, Einer C, Schulz S, Clavel T, Protzer U, Thiele C, Zischka H, Moch H, Tschop M, Tumanov AV, Haller D, Unger K, Karin M, Kopf M, Knolle P, Weber A, Heikenwalder M. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014;26:549–64. [DOI] [PubMed] [Google Scholar]
- 46.Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, Casola S, Shlomchik MJ, Koteliansky V, Hochman PS, Ibraghimov A. Attenuated liver fibrosis in the absence of B cells. J Clin Invest 2005;115:3072–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, Xie G, Philips G, Chan IS, Karaca GF, Pereira Tde A, Chen Y, Mi Z, Kuo PC, Choi SS, Guy CD, Abdelmalek MF, Diehl AM. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012;61:1323–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maricic I, Marrero I, Eguchi A, Nakamura R, Johnson CD, Dasgupta S, Hernandez CD, Nguyen PS, Swafford AD, Knight R, Feldstein AE, Loomba R, Kumar V. Differential Activation of Hepatic Invariant NKT Cell Subsets Plays a Key Role in Progression of Nonalcoholic Steatohepatitis. J Immunol 2018;201:3017–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, Leone V, Peiseler M, Surewaard BGJ, Rath D, Ali A, Wolf MJ, Drescher H, Healy ME, Dauch D, Kroy D, Krenkel O, Kohlhepp M, Engleitner T, Olkus A, Sijmonsma T, Volz J, Deppermann C, Stegner D, Helbling P, Nombela-Arrieta C, Rafiei A, Hinterleitner M, Rall M, Baku F, Borst O, Wilson CL, Leslie J, O’Connor T, Weston CJ, Adams DH, Sheriff L, Teijeiro A, Prinz M, Bogeska R, Anstee N, Bongers MN, Notohamiprodjo M, Geisler T, Withers DJ, Ware J, Mann DA, Augustin HG, Vegiopoulos A, Milsom MD, Rose AJ, Lalor PF, Llovet JM, Pinyol R, Tacke F, Rad R, Matter M, Djouder N, Kubes P, Knolle PA, Unger K, Zender L, Nieswandt B, Gawaz M, Weber A, Heikenwalder M. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 2019;25:641–655. [DOI] [PubMed] [Google Scholar]
- 50.Yoshida S, Ikenaga N, Liu SB, Peng ZW, Chung J, Sverdlov DY, Miyamoto M, Kim YO, Ogawa S, Arch RH, Schuppan D, Popov Y. Extrahepatic platelet-derived growth factor-beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology 2014;147:1378–92. [DOI] [PubMed] [Google Scholar]
- 51.Eisenach JC, Nugent M, Miller FA Jr., Mucha P Jr. Echocardiographic evaluation of patients with blunt chest injury: correlation with perioperative hypotension. Anesthesiology 1986;64:364–6. [DOI] [PubMed] [Google Scholar]
- 52.Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006;130:435–52. [DOI] [PubMed] [Google Scholar]
- 53.Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, Horani A, Nassar M, Friedman SL, Safadi R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol 2006;45:60–71. [DOI] [PubMed] [Google Scholar]
- 54.Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta 2013;1832:1061–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008;48:920–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, Gaarde WA, Deleve LD. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012;142:918–927 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, Tamaki K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest 2015;95:1130–44. [DOI] [PubMed] [Google Scholar]
- 58.Cogger VC, Mohamad M, Solon-Biet SM, Senior AM, Warren A, O’Reilly JN, Tung BT, Svistounov D, McMahon AC, Fraser R, Raubenheimer D, Holmes AJ, Simpson SJ, Le Couteur DG. Dietary macronutrients and the aging liver sinusoidal endothelial cell. Am J Physiol Heart Circ Physiol 2016;310:H1064–70. [DOI] [PubMed] [Google Scholar]
- 59.Fakhry TK, Mhaskar R, Schwitalla T, Muradova E, Gonzalvo JP, Murr MM. Bariatric surgery improves nonalcoholic fatty liver disease: a contemporary systematic review and meta-analysis. Surg Obes Relat Dis 2019;15:502–511. [DOI] [PubMed] [Google Scholar]
- 60.Klein S, Mittendorfer B, Eagon JC, Patterson B, Grant L, Feirt N, Seki E, Brenner D, Korenblat K, McCrea J. Gastric bypass surgery improves metabolic and hepatic abnormalities associated with nonalcoholic fatty liver disease. Gastroenterology 2006;130:1564–72. [DOI] [PubMed] [Google Scholar]
- 61.Liu X, Lazenby AJ, Clements RH, Jhala N, Abrams GA. Resolution of nonalcoholic steatohepatits after gastric bypass surgery. Obes Surg 2007;17:486–92. [DOI] [PubMed] [Google Scholar]
- 62.de Almeida SR, Rocha PR, Sanches MD, Leite VH, da Silva RA, Diniz MT, Diniz Mde F, Rocha AL. Roux-en-Y gastric bypass improves the nonalcoholic steatohepatitis (NASH) of morbid obesity. Obes Surg 2006;16:270–8. [DOI] [PubMed] [Google Scholar]
- 63.Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C, Arthur MJ. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 1998;102:538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, Pradere JP, Friedman RA, Schwabe RF. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012;143:1073–83 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kisseleva T, Brenner DA. Inactivation of myofibroblasts during regression of liver fibrosis. Cell Cycle 2013;12:381–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campana L, Iredale JP. Regression of Liver Fibrosis. Semin Liver Dis 2017;37:1–10. [DOI] [PubMed] [Google Scholar]
- 67.Zoubek ME, Trautwein C, Strnad P. Reversal of liver fibrosis: From fiction to reality. Best Pract Res Clin Gastroenterol 2017;31:129–141. [DOI] [PubMed] [Google Scholar]
- 68.Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, Knight R. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 2018;15:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Flaherty SE 3rd, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 2019;363:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arriazu E, Ge X, Leung TM, Magdaleno F, Lopategi A, Lu Y, Kitamura N, Urtasun R, Theise N, Antoine DJ, Nieto N. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 2017;66:1123–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest 2003;83:655–63. [DOI] [PubMed] [Google Scholar]
- 72.Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, Torok NJ. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006;43:435–43. [DOI] [PubMed] [Google Scholar]
- 73.Tai DI, Lin SM, Sheen IS, Chu CM, Lin DY, Liaw YF. Long-term outcome of hepatitis B e antigen-negative hepatitis B surface antigen carriers in relation to changes of alanine aminotransferase levels over time. Hepatology 2009;49:1859–67. [DOI] [PubMed] [Google Scholar]
- 74.Fattovich G, Olivari N, Pasino M, D’Onofrio M, Martone E, Donato F. Long-term outcome of chronic hepatitis B in Caucasian patients: mortality after 25 years. Gut 2008;57:84–90. [DOI] [PubMed] [Google Scholar]
- 75.Ghany MG, Kleiner DE, Alter H, Doo E, Khokar F, Promrat K, Herion D, Park Y, Liang TJ, Hoofnagle JH. Progression of fibrosis in chronic hepatitis C. Gastroenterology 2003;124:97–104. [DOI] [PubMed] [Google Scholar]
- 76.Hui CK, Belaye T, Montegrande K, Wright TL. A comparison in the progression of liver fibrosis in chronic hepatitis C between persistently normal and elevated transaminase. J Hepatol 2003;38:511–7. [DOI] [PubMed] [Google Scholar]
- 77.Wiese M, Berr F, Lafrenz M, Porst H, Oesen U. Low frequency of cirrhosis in a hepatitis C (genotype 1b) single-source outbreak in germany: a 20-year multicenter study. Hepatology 2000;32:91–6. [DOI] [PubMed] [Google Scholar]
- 78.Wiese M, Fischer J, Lobermann M, Gobel U, Grungreiff K, Guthoff W, Kullig U, Richter F, Schiefke I, Tenckhoff H, Zipprich A, Berg T, Muller T, East German HCVSG. Evaluation of liver disease progression in the German hepatitis C virus (1b)-contaminated anti-D cohort at 35 years after infection. Hepatology 2014;59:49–57. [DOI] [PubMed] [Google Scholar]
- 79.Amarapurka DN, Amarapurkar AD, Patel ND, Agal S, Baigal R, Gupte P, Pramanik S. Nonalcoholic steatohepatitis (NASH) with diabetes: predictors of liver fibrosis. Ann Hepatol 2006;5:30–3. [PubMed] [Google Scholar]
- 80.Argo CK, Northup PG, Al-Osaimi AM, Caldwell SH. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J Hepatol 2009;51:371–9. [DOI] [PubMed] [Google Scholar]
- 81.Canestrari F, Cucchiarini L, Giacchi R, Di Quirico M, Biagiarelli B. [Erythrocyte ATPase activity as a function of the age of the subjects and the cell]. Boll Soc Ital Biol Sper 1988;64:801–7. [PubMed] [Google Scholar]
- 82.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003;125:437–43. [DOI] [PubMed] [Google Scholar]
- 83.Thapaliya S, Wree A, Povero D, Inzaugarat ME, Berk M, Dixon L, Papouchado BG, Feldstein AE. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Dig Dis Sci 2014;59:1197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP, Canbay A, Reeves HL, Luedde M, Tacke F, Trautwein C, Heikenwalder M, Luedde T. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med 2014;6:1062–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davies SP, Reynolds GM, Stamataki Z. Clearance of Apoptotic Cells by Tissue Epithelia: A Putative Role for Hepatocytes in Liver Efferocytosis. Front Immunol 2018;9:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Caldwell S, Ikura Y, Dias D, Isomoto K, Yabu A, Moskaluk C, Pramoonjago P, Simmons W, Scruggs H, Rosenbaum N, Wilkinson T, Toms P, Argo CK, Al-Osaimi AM, Redick JA. Hepatocellular ballooning in NASH. J Hepatol 2010;53:719–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kakisaka K, Suzuki Y, Fujiwara Y, Abe T, Yonezawa M, Kuroda H, Ishida K, Sugai T, Takikawa Y. Evaluation of ballooned hepatocytes as a risk factor for future progression of fibrosis in patients with non-alcoholic fatty liver disease. J Gastroenterol 2018;53:1285–1291. [DOI] [PubMed] [Google Scholar]
- 88.Machado MV, Cortez-Pinto H. Cell death and nonalcoholic steatohepatitis: where is ballooning relevant? Expert Rev Gastroenterol Hepatol 2011;5:213–22. [DOI] [PubMed] [Google Scholar]
- 89.Lackner C Hepatocellular ballooning in nonalcoholic steatohepatitis: the pathologist’s perspective. Expert Rev Gastroenterol Hepatol 2011;5:223–31. [DOI] [PubMed] [Google Scholar]
- 90.Hirsova P, Gores GJ. Ballooned hepatocytes, undead cells, sonic hedgehog, and vitamin E: therapeutic implications for nonalcoholic steatohepatitis. Hepatology 2015;61:15–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011;473:528–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008;134:568–76. [DOI] [PubMed] [Google Scholar]
- 93.Maiers JL, Malhi H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin Liver Dis 2019;39:235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hernandez-Alvarez MI, Sebastian D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, Plana N, Veiga SR, Hernandez V, Vasconcelos N, Peddinti G, Adrover A, Jove M, Pamplona R, Gordaliza-Alaguero I, Calvo E, Cabre N, Castro R, Kuzmanic A, Boutant M, Sala D, Hyotylainen T, Oresic M, Fort J, Errasti-Murugarren E, Rodrigues CMP, Orozco M, Joven J, Canto C, Palacin M, Fernandez-Veledo S, Vendrell J, Zorzano A. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019;177:881–895 e17. [DOI] [PubMed] [Google Scholar]
- 95.Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, Kaufman RJ, Saltiel AR, Karin M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018;175:133–145 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang X, Sommerfeld MR, Jahn-Hofmann K, Cai B, Filliol A, Remotti HE, Schwabe RF, Kannt A, Tabas I. A Therapeutic Silencing RNA Targeting Hepatocyte TAZ Prevents and Reverses Fibrosis in Nonalcoholic Steatohepatitis in Mice. Hepatol Commun 2019:In press. [DOI] [PMC free article] [PubMed]
- 97.Khajehahmadi Z, Mohagheghi S, Nikeghbalian S, Geramizadeh B, Khodadadi I, Karimi J, Ghaffari ME, Tavilani H. Downregulation of hedgehog ligands in human simple steatosis may protect against nonalcoholic steatohepatitis: Is TAZ a crucial regulator? IUBMB Life 2019;71:1382–1390. [DOI] [PubMed] [Google Scholar]
- 98.Chong YC, Lim TE, Fu Y, Shin EM, Tergaonkar V, Han W. Indian Hedgehog links obesity to development of hepatocellular carcinoma. Oncogene 2019;38:2206–2222. [DOI] [PubMed] [Google Scholar]
- 99.Mooring M, Fowl BH, Lum SZC, Liu Y, Yao K, Softic S, Kirchner R, Bernstein A, Singhi AD, Jay DG, Kahn CR, Camargo FD, Yimlamai D. Hepatocyte Stress Increases Expression of YAP and TAZ in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology 2019. [DOI] [PMC free article] [PubMed]
- 100.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science 1999;284:770–6. [DOI] [PubMed] [Google Scholar]
- 101.Tanimizu N, Miyajima A. Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J Cell Sci 2004;117:3165–74. [DOI] [PubMed] [Google Scholar]
- 102.Valenti L, Mendoza RM, Rametta R, Maggioni M, Kitajewski C, Shawber CJ, Pajvani UB. Hepatic notch signaling correlates with insulin resistance and nonalcoholic fatty liver disease. Diabetes 2013;62:4052–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, Murphy SK, Ashley-Koch AE, Choi SS, Michelotti GA, Hampton DD, Chen Y, Tillmann HL, Hauser MA, Abdelmalek MF, Diehl AM. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014;59:471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pajvani UB, Shawber CJ, Samuel VT, Birkenfeld AL, Shulman GI, Kitajewski J, Accili D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat Med 2011;17:961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pak E, Segal RA. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev Cell 2016;38:333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Machado MV, Diehl AM. Hedgehog signalling in liver pathophysiology. J Hepatol 2018;68:550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guy CD, Suzuki A, Abdelmalek MF, Burchette JL, Diehl AM, Nash CRN. Treatment response in the PIVENS trial is associated with decreased Hedgehog pathway activity. Hepatology 2015;61:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Estep M, Mehta R, Bratthauer G, Alaparthi L, Monge F, Ali S, Abdelatif D, Younoszai Z, Stepanova M, Goodman ZD, Younossi ZM. Hepatic sonic hedgehog protein expression measured by computer assisted morphometry significantly correlates with features of non-alcoholic steatohepatitis. BMC Gastroenterol 2019;19:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guillen-Sacoto MJ, Martinez AF, Abe Y, Kruszka P, Weiss K, Everson JL, Bataller R, Kleiner DE, Ward JM, Sulik KK, Lipinski RJ, Solomon BD, Muenke M. Human germline hedgehog pathway mutations predispose to fatty liver. J Hepatol 2017;67:809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Totaro A, Castellan M, Di Biagio D, Piccolo S. Crosstalk between YAP/TAZ and Notch Signaling. Trends Cell Biol 2018;28:560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD. Hippo pathway activity influences liver cell fate. Cell 2014;157:1324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hernandez C, Huebener P, Pradere JP, Antoine DJ, Friedman RA, Schwabe RF. HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J Clin Invest 2018;128:2436–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khambu B, Huda N, Chen X, Antoine DJ, Li Y, Dai G, Kohler UA, Zong WX, Waguri S, Werner S, Oury TD, Dong Z, Yin XM. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J Clin Invest 2018;128:2419–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Santoleri D, Titchenell PM. Resolving the Paradox of Hepatic Insulin Resistance. Cell Mol Gastroenterol Hepatol 2019;7:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pajvani UB, Qiang L, Kangsamaksin T, Kitajewski J, Ginsberg HN, Accili D. Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat Med 2013;19:1054–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem 2005;280:20589–95. [DOI] [PubMed] [Google Scholar]
- 117.Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, Manning BD. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 2011;14:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim K, Qiang L, Hayden MS, Sparling DP, Purcell NH, Pajvani UB. mTORC1-independent Raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat Commun 2016;7:10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim K, Ryu D, Dongiovanni P, Ozcan L, Nayak S, Ueberheide B, Valenti L, Auwerx J, Pajvani UB. Degradation of PHLPP2 by KCTD17, via a Glucagon-Dependent Pathway, Promotes Hepatic Steatosis. Gastroenterology 2017;153:1568–1580 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ajmera V, Park CC, Caussy C, Singh S, Hernandez C, Bettencourt R, Hooker J, Sy E, Behling C, Xu R, Middleton MS, Valasek MA, Faulkner C, Rizo E, Richards L, Sirlin CB, Loomba R. Magnetic Resonance Imaging Proton Density Fat Fraction Associates With Progression of Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2018;155:307–310 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Unalp-Arida A, Ruhl CE. Noninvasive fatty liver markers predict liver disease mortality in the U.S. population. Hepatology 2016;63:1170–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, Vogt TF, Hobbs HH, Cohen JC. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007;46:1081–90. [DOI] [PubMed] [Google Scholar]
- 125.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li YX, Diehl AM. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007;45:1366–74. [DOI] [PubMed] [Google Scholar]
- 126.McClain CJ, Barve S, Deaciuc I. Good fat/bad fat. Hepatology 2007;45:1343–6. [DOI] [PubMed] [Google Scholar]
- 127.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774–88. [DOI] [PubMed] [Google Scholar]
- 128.Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem 2009;284:5637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab 2006;4:185–98. [DOI] [PubMed] [Google Scholar]
- 130.Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, Schuppan D, Gronbaek H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 2019;16:145–159. [DOI] [PubMed] [Google Scholar]
- 131.Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J Lipid Res 2013;54:1326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, Lee SP, Savard C. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res 2017;58:1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bieghs V, Walenbergh SM, Hendrikx T, van Gorp PJ, Verheyen F, Olde Damink SW, Masclee AA, Koek GH, Hofker MH, Binder CJ, Shiri-Sverdlov R. Trapping of oxidized LDL in lysosomes of Kupffer cells is a trigger for hepatic inflammation. Liver Int 2013;33:1056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Teratani T, Tomita K, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, Tominaga S, Hiroi S, Irie R, Okada Y, Kurihara C, Ebinuma H, Saito H, Hokari R, Sugiyama K, Kanai T, Miura S, Hibi T. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012;142:152–164 e10. [DOI] [PubMed] [Google Scholar]
- 135.Furuhashi H, Tomita K, Teratani T, Shimizu M, Nishikawa M, Higashiyama M, Takajo T, Shirakabe K, Maruta K, Okada Y, Kurihara C, Watanabe C, Komoto S, Aosasa S, Nagao S, Yamamoto J, Miura S, Hokari R. Vitamin A-coupled liposome system targeting free cholesterol accumulation in hepatic stellate cells offers a beneficial therapeutic strategy for liver fibrosis. Hepatol Res 2018;48:397–407. [DOI] [PubMed] [Google Scholar]
- 136.Teratani T, Tomita K, Furuhashi H, Sugihara N, Higashiyama M, Nishikawa M, Irie R, Takajo T, Wada A, Horiuchi K, Inaba K, Hanawa Y, Shibuya N, Okada Y, Kurihara C, Nishii S, Mizoguchi A, Hozumi H, Watanabe C, Komoto S, Nagao S, Yamamoto J, Miura S, Hokari R, Kanai T. Lipoprotein Lipase Up-regulation in Hepatic Stellate Cells Exacerbates Liver Fibrosis in Nonalcoholic Steatohepatitis in Mice. Hepatol Commun 2019;3:1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015;518:547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, Ji Y, Li C, Guo L, Zhou L, Chen Z, Leon-Mimila P, Chung MT, Kurabayashi K, Opp J, Campos-Perez F, Villamil-Ramirez H, Canizales-Quinteros S, Lyons R, Lumeng CN, Zhou B, Qi L, Huertas-Vazquez A, Lusis AJ, Xu XZS, Li S, Yu Y, Li JZ, Lin JD. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell 2019;75:644–660 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rivera CA, Bradford BU, Hunt KJ, Adachi Y, Schrum LW, Koop DR, Burchardt ER, Rippe RA, Thurman RG. Attenuation of CCl(4)-induced hepatic fibrosis by GdCl(3) treatment or dietary glycine. Am J Physiol Gastrointest Liver Physiol 2001;281:G200–7. [DOI] [PubMed] [Google Scholar]
- 140.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 2005;115:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 2007;13:1324–32. [DOI] [PubMed] [Google Scholar]
- 142.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R, Dragomir AC, Aloman C, Schwabe RF. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013;58:1461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 2012;302:G1310–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, Liepelt A, Lefebvre E, Luedde T, Hellerbrand C, Weiskirchen R, Longerich T, Costa IG, Anstee QM, Trautwein C, Tacke F. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018;67:1270–1283. [DOI] [PubMed] [Google Scholar]
- 145.Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, Mi Z, Pereira TA, Zdanowicz M, Malladi P, Chen Y, Moylan C, Jung Y, Bhattacharya SD, Teaberry V, Omenetti A, Abdelmalek MF, Guy CD, Adams DH, Kuo PC, Michelotti GA, Whitington PF, Diehl AM. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011;53:106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Reports 2019:30–43. [DOI] [PMC free article] [PubMed]
- 147.Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, Merad M, Luedde T, Trautwein C, Tacke F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009;50:261–74. [DOI] [PubMed] [Google Scholar]
- 148.Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, Williams MJ, Dunbar DR, Manning JR, van Rooijen N, Fallowfield JA, Forbes SJ, Iredale JP. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A 2012;109:E3186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, Ramachandran P, Van Deemter M, Hume DA, Iredale JP, Forbes SJ. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology 2011;53:2003–15. [DOI] [PubMed] [Google Scholar]
- 150.Moroni F, Dwyer BJ, Graham C, Pass C, Bailey L, Ritchie L, Mitchell D, Glover A, Laurie A, Doig S, Hargreaves E, Fraser AR, Turner ML, Campbell JDM, McGowan NWA, Barry J, Moore JK, Hayes PC, Leeming DJ, Nielsen MJ, Musa K, Fallowfield JA, Forbes SJ. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat Med 2019. [DOI] [PubMed]
- 151.Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, Ransohoff RM, Charo IF, Jenne CN, Kubes P. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med 2015;212:447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Morioka S, Maueroder C, Ravichandran KS. Living on the Edge: Efferocytosis at the Interface of Homeostasis and Pathology. Immunity 2019;50:1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Akalu YT, Rothlin CV, Ghosh S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol Rev 2017;276:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jindal A, Bruzzi S, Sutti S, Locatelli I, Bozzola C, Paternostro C, Parola M, Albano E. Fat-laden macrophages modulate lobular inflammation in nonalcoholic steatohepatitis (NASH). Exp Mol Pathol 2015;99:155–62. [DOI] [PubMed] [Google Scholar]
- 155.Yu LX, Schwabe RF. The gut microbiome and liver cancer: mechanisms and clinical translation. Nat Rev Gastroenterol Hepatol 2017;14:527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Song K, Kwon H, Han C, Chen W, Zhang J, Ma W, Dash S, Gandhi CR, Wu T. YAP in Kupffer cells enhances the production of pro-inflammatory cytokines and promotes the development of non-alcoholic steatohepatitis. Hepatology 2019. [DOI] [PMC free article] [PubMed]
- 157.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010;139:323–34 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, Staels B, Kersten S, Muller M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010;51:511–22. [DOI] [PubMed] [Google Scholar]
- 159.Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, Scott DK, O’Doherty RM. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010;59:347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Giebeler A, Boekschoten MV, Klein C, Borowiak M, Birchmeier C, Gassler N, Wasmuth HE, Muller M, Trautwein C, Streetz KL. c-Met confers protection against chronic liver tissue damage and fibrosis progression after bile duct ligation in mice. Gastroenterology 2009;137:297–308, 308 e1–4. [DOI] [PubMed] [Google Scholar]
- 161.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A 2004;101:4477–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kroy DC, Schumacher F, Ramadori P, Hatting M, Bergheim I, Gassler N, Boekschoten MV, Muller M, Streetz KL, Trautwein C. Hepatocyte specific deletion of c-Met leads to the development of severe non-alcoholic steatohepatitis in mice. J Hepatol 2014;61:883–90. [DOI] [PubMed] [Google Scholar]
- 163.Bourbonnais E, Raymond VA, Ethier C, Nguyen BN, El-Leil MS, Meloche S, Bilodeau M. Liver fibrosis protects mice from acute hepatocellular injury. Gastroenterology 2012;142:130–139 e4. [DOI] [PubMed] [Google Scholar]
- 164.Ledford H Gene-silencing technology gets first drug approval after 20-year wait. Nature 2018;560:291–292. [DOI] [PubMed] [Google Scholar]
- 165.Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, Chou HL, Hashiguchi T, Plato C, Poulin D, Richards T, Yoneyama H, Jenkins H, Wolfgang G, Friedman SL. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS One 2016;11:e0158156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, Francque S, Farrell G, Kowdley KV, Craxi A, Simon K, Fischer L, Melchor-Khan L, Vest J, Wiens BL, Vig P, Seyedkazemi S, Goodman Z, Wong VW, Loomba R, Tacke F, Sanyal A, Lefebvre E. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018;67:1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Foerster F, Bamberger D, Schupp J, Weilbacher M, Kaps L, Strobl S, Radi L, Diken M, Strand D, Tuettenberg A, Wich PR, Schuppan D. Dextran-based therapeutic nanoparticles for hepatic drug delivery. Nanomedicine (Lond) 2016;11:2663–2677. [DOI] [PubMed] [Google Scholar]