

Abstract
The arrival of mass cytometry (MC) and, more recently, spectral flow cytometry (SFC) has revolutionized the study of cellular, functional and phenotypic diversity, significantly increasing the number of characteristics measurable at the single-cell level. As a consequence, new computational techniques such as dimensionality reduction and/or clustering algorithms are necessary to analyze, clean, visualize and interpret these high-dimensional data sets. In this small comparison study, we investigated splenocytes from the same sample by either MC or SFC and compared both high-dimensional data sets using expert gating, t-distributed stochastic neighbor embedding (t-SNE), uniform manifold approximation and projection (UMAP) analysis and FlowSOM. When we downsampled each data set to their equivalent cell numbers and parameters, our analysis yielded highly comparable results. Differences between the data sets only became apparent when the maximum number of parameters in each data set were assessed, due to differences in the number of recorded events or the maximum number of assessed parameters. Overall, our small comparison study suggests that mass cytometry and spectral flow cytometry both yield comparable results when analyzed manually or by high-dimensional clustering or dimensionality reduction algorithms such as t-SNE, UMAP or FlowSOM. However, large scale studies combined with an in-depth technical analysis will be needed to assess differences between these technologies in more detail.
Keywords: Spectral flow cytometry, mass cytometry, high dimensional data analysis, t-SNE, UMAP, FlowSOM
Introduction
Flow cytometry has been an important technology for cellular analysis. In the 1980s 3 markers could be analyzed at a time (1) and this number increased over the following 30 years. However, the number of laser lines, the breadth of fluorescent dye emission profiles, their subsequent spillover and limited hardware, restricted most experiments to no more than 14 markers. In 2009, the development of mass cytometry (MC, also called cytometry by time-of-flight or CyTOF) which uses metal-conjugated antibodies, led to an unprecedented increase in the number of analytes that could be assessed (2). Over the last few years, advances in flow cytometry instrumentation and fluorophore availability also increased dramatically, allowing for the design of panels with up to 30 fluorophores (3,4). Among those advances has been the development of spectral flow cytometry (SFC), which is particularly useful for distinguishing fluorophores with a high degree of spectral overlap (5–8). In contrast to conventional flow cytometry, SFC assesses the full visible-light spectrum of each fluorophore and uses unmixing technology with non-square compensation matrices to differentiate individual markers (6).
The lack of autofluorescence, minimal overlap between tags that would typically require compensation, and the large range of metal tags available for MC, have made MC an important discovery tool for immunologists. However, high costs, sample loss, low acquisition speed and sample destruction upon analysis, are practical limitations that also need to be considered. In turn, flow cytometry panels that include 12 or more fluorescent probes suffer from increased spectral spillover. Additionally, autofluorescence signatures from cells or tissues can further reduce signal resolution. SFC has overcome some of these limitations by detecting the emission spectrum of each fluorophore over a wide range of wavelengths, creating a spectral fingerprint for each dye. Thus, SFC is able to discriminate fluorophores with very similar emission peaks and provides multi-parametric analysis with smaller compensation requirements [9, 10]. Previous comparisons between SFC and conventional flow cytometry have concluded that SFC provides distinct advantages in the analysis of solid tissues, such as the heart and intestines, due to its improved signal-to-noise ratio and autofluorescence correction tools (9).
To visualize and interpret high dimensional data sets generated by conventional flow cytometry, MC or single-cell RNA sequencing, dimensionality reduction and/or cell clustering algorithms are commonly used [11, 12]. These include t-distributed stochastic linear embedding (t-SNE) (11), uniform manifold approximation and projection (UMAP) (12) and FlowSOM (13).
The t-SNE algorithm reduces data dimensionality by incorporating all relevant parameters for each cell, while maintaining the overall data structure. Thus, cells with more similar properties are plotted closer together, which creates a two-dimensional map that provides a visual representation of the single-cell data, similar to a biaxial plot, with the position of cells reflecting their proximity in high-dimensional space (14). As a limitation, t-SNE often fails to preserve the global geometry of the data, meaning that the relative position of clusters on the t-SNE plot is arbitrary (15). Data representation using the UMAP algorithm yields comparable data to t-SNE in terms of visualization quality, but in addition, tends to preserve the global geometry of the data more accurately and has superior run time performance (12), although some of the differences might be due to different initialization parameters (16).
In contrast, the FlowSOM algorithm creates a self-organizing map (SOM) using an unsupervised technique for clustering and dimensionality reduction to identify unique cellular subsets and visualize relationships. Using a two-level clustering and star charts, this algorithm helps to obtain a clear overview of how markers are expressed on all cells, and clusters subpopulations (13).
It is also important to establish a quality control pipeline to process, normalize and transform the raw data, as only high quality data sets can be reliably analyzed by high-dimensional data analysis algorithms (3,17). This is to prevent artefacts, such as the effects of spreading errors (18,19), that can mistakenly be classified as distinct cell populations by high-dimensional analysis algorithms (20).
While several studies have highlighted the potential of mass cytometry for describing cellular subsets in great detail (21–26), only a few of these studies have directly compared mass cytometry with conventional fluorescence cytometry data (27–31). In these studies, comparable frequencies of cell subpopulations were identified when comparing traditional flow cytometry and MC technologies (30,31) and also, when analyzing proteins of the cell cycle (28). Furthermore, quantitative flow cytometry and MC provided reasonably consistent antibody binding results for two lyophilized cell preparations (29).
As the use of SFC is becoming more frequent (9,32–34), yet few comparisons between SFC and other high-dimensional data sets exist, we designed a small comparison study to assess high-dimensional MC and SFC data by expert gating and common analysis tools like t-SNE, UMAP and FlowSOM.
Materials and methods
Cell isolation and sample processing
Murine C57BL/6 spleens (5 samples) were harvested at steady state from 2 different experiments (2 samples from the first experiment and 3 samples from a second experiment) and digested 20 minutes at 37 °C with 100μg/ml Liberase TL and 100μg/ml DNAse I (both from Roche). Red blood cells were lyzed (Qiagen) and samples were gently resuspended in media composed of 90% fetal bovine serum (FBS) (Invitrogen) containing 10% dimethyl sulfoxide (DMSO) (Sigma-Aldrich) and transferred to sterile cryogenic vials. For each sample, cells were divided in two distinct cryotubes to be able to analyze half of the same sample with each technology. Cell freezing containers were used to limit rate of freezing to a 1–3 °C per minute temperature drop. After 24 h incubation at −70 °C, the cryovials were transferred to −140°C liquid nitrogen for long term storage. Half of the cryotubes were sent to Sydney Cytometry Facility to be analyzed using a Helios CyTOF instrument, and the other half were used at the Malaghan Institute of Medical Research to be analyzed using a 3-laser Aurora spectral flow cytometer. Cells were rapidly thawed in a 37 °C water bath, and diluted in pre-warmed medium (IMDM supplemented with 10% FBS) (Invitrogen). Residual DMSO was removed by centrifugation at 400 g for 5 minutes and pelleted cells were resuspended in media with 10% FBS and allowed to recover for 30 minutes at 37 °C and 5% CO2 prior to staining (35).
Spectral Flow Cytometry
Cells were incubated with the viability dye NIR Zombie (BioLegend) for 15 minutes at room temperature. Cells were then washed once in FACs buffer (FCS 1%, NaN3 0.01% and EDTA 2mM in PBS) and incubated in Fc block for 5 minutes at room temperature. Samples were stained with surface antibodies (Table 1A) in FACs buffer with Brilliant Buffer Plus (BD Biosciences) for 20 minutes at 4 °C. Ki67, FoxP3 and TNFα were stained intracellularly following the fixation and permeabilization of cells with the FoxP3/transcription factor staining buffer kit (eBioscience) according to the manufacturer’s instructions. Cells were resuspended in FACs buffer, filtered through a 70 μm filter (ThermoFisher), and analyzed on a 3-laser Aurora spectral flow cytometer (Cytek Biosciences) using SpectroFlo software version 2.1 at the Malaghan Institute of Medical Research, Wellington, New Zealand.
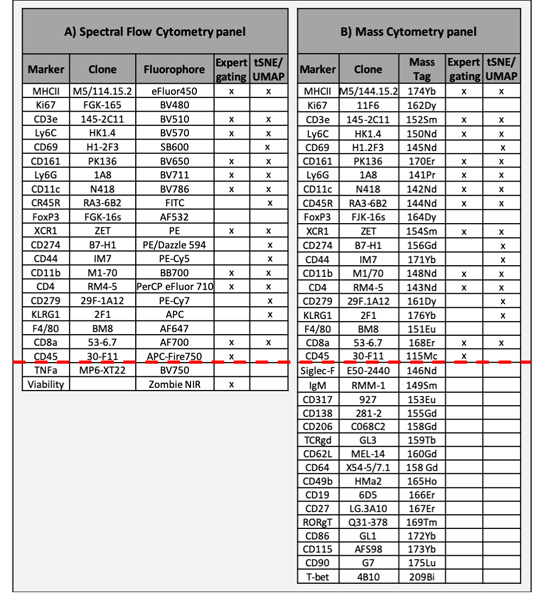
Table 1. Antibody panels used for SFC and MC.
Marker, Clone, Fluorophore, MC Tag and methods of post-acquisition data analysis are indicated. The red line discriminates between markers common to both data sets or unique to a single data set.
 |
Mass Cytometry
Thawed splenocytes were first incubated with cisplatin for viability assessment (25 mmol/L, Enzo Life Sciences), then washed in FACs buffer and incubated with Fc block for 30 minutes at 4 °C. Staining occurred in 50 μL FACs buffer for 30 minutes at 4 °C using the antibodies listed in Table 1B. Cells were intracellularly stained using eBioscience FoxP3/transcription factor staining buffer kit according to the manufacturer’s instructions, then washed with FACs buffer and fixed overnight in 4% paraformaldehyde containing Cell-ID Intercalator-Ir (0.125 mM 191/193Ir; Fluidigm), to identify nucleated cells. The next day, and after multiple washes with FACS buffer and Milli-Q water, cells were filtered through a 35 μm nylon mesh cell-strainer cap (BD Biosciences) and then analyzed on a Helios CyTOF 1.0 mass cytometer (Fluidigm Sciences) using CyTOF software version version 6.7 at the Sydney Cytometry Facility, Sydney, Australia.
Data Processing, High-Dimensional and Statistical Analysis
All SFC and MC FCS files were uploaded and evaluated using Cytobank software (36). Data were manually transformed to arcsinh scales with varying cofactors for visualization purposes: MC cofactors ranged from 15 to 50, while SFC cofactors ranged from 150 to 10,000.
To run high-dimensional algorithms, data pre-processing included the application of an arcsinh transformation with a standard cofactor of 5 (MC data) (23) or 6000 (SFC). Data sets were downsampled to have the same number of events (19.858 events/fcs file) using FlowJo version 10.6.1. t-SNE maps and UMAP maps were created using the 16 common antibodies and antibody clones listed in Table 1. t-SNE analysis was performed in Cytobank with a perplexity of 50 (typical values are between 5 and 50 (14)) and 7,500 iterations and were manually optimized to improve t-SNE map appearance. t-SNE scales were normalized to −800 to 800 for each marker of the SFC and MC data sets. UMAP analysis was performed in FlowJo version 10.6.1 with default settings of nearest neighbors of 15 and minimum distance of 0.5 using the Euclidean distance function (12). UMAP scales for each marker were as follows [SFC: B220 (−742 to 74790), XCR1 (−1833 to 44838), CD44 (925.6 to 4×106), CD279 (−1084 to 41613), KLRG1 (−2278 to 11380), CD8 (−5253 to 52293), CD3 (−3626 to 88681), Ly6G (−2144 to 60818), CD11c (−2163 to 41943), NK1.1 (−2317 to 38061), Ly6c (−1204 to 2×106), MHCII (−1667 to 4×106), CD4 (−2261 to 2×106), CD11b (−6701 to 5 ×106), CD274 (−909 to 44060), CD69 (−1870 to 8072), Ki67 (−2764 to 4278), FoxP3 (−1661 to 2525), F4/80 (−3740 to 32465); MC: CD274 (0 to 87.56), KLRG1 (0 to 32.9), CD11b (0 to 578.4), Ly6G (0 to 292.3), CD11c (0 to 125.6), CD4 (0 to 185.2), B220 (90 to 98.02), CD69 (0 to 11.69), CD8 (0 to 131.3), NK1.1 (0 to 108.4), MHCII (0 to 4695), CD279 (0 to 38.71), Ly6c (0 to 634.6), CD44 (0 to 613.5), XCR1 (0 to 48.93), CD3 (0 to 214.3), F4/80 (0 to 38.37), Ki67 (0 to 693.5) and FoxP3 (0 to 21.72)]. For FlowSOM analysis, Cytobank and the R packages FlowSOM (37) and flowCore (38) were used. Numbers of cells per cluster (Range) and f-scores, which summarize the Precision and Recall value compared to the previous clustering iteration were implemented in FlowSOM and were determined for squared matrix clusters ranging from 4–625 FlowSOM clusters (for details see https://github.com/SameOldSamOld/CyTOFAuroraComparison). GraphPad Prism software version 8.3.1 was used to compare population percentages. The concordance correlation coefficient was calculated using the epiR package (https://CRAN.R-project.org/package=epiR).
Ethics statement
All animal experiments carried out at the Malaghan Institute of Medical Research adhered to the regulations of the Ministry of Primary Industries New Zealand (license 24432) and were approved by the Animal Welfare Ethical Review Board of the Victoria University of Wellington.
Source Data
CyTOF and SFC datasets used for manual and high-dimensional analysis can be downloaded from FlowRepository (https://flowrepository.org/id/FR-FCM-Z2GZ) and scripts to reproduce the analyses are available on GitHub (https://github.com/SameOldSamOld/CyTOFAuroraComparison).
Results and discussion
The use of SFC is becoming more frequent (9,33,34,39), yet few comparisons between SFC and other high-dimensional data sets exist. In order to generate a side-by-side comparison between SFC and MC data, we collected a small data set from two independent experiments (n=3+2) using murine C57BL/6 splenocytes at steady state. Spleens were processed and single cell suspensions were independently stained with either a 37-parameter panel for MC, or a 22-parameter panel for SFC (Table 1) and 50,000 events per sample were acquired for MC at the Sydney Cytometry Facility, Australia or 1,000,000 events were recorded for SFC at the Malaghan Institute of Medical Research, Wellington, New Zealand.
We compared both data sets by expert gating and found that both data sets allowed for the identification of major immune cell populations such as B cells, CD4 and CD8 T cells, neutrophils, NK cells, DCs and monocytes (Figure 1A). Importantly, the frequency of all gated populations was highly comparable between our SFC and MC data sets, despite their significant difference in recorded events, and were closely correlated (Concordance correlation coefficient (CCC) = 0.99, and CCC=0.91 for cellular frequencies below <10%) (Figure 1B and C). We furthermore plotted the mean of the observed cell proportions based on the 5 samples per data set and compared them between the SFC and MC data using a two-tail difference between means t-test (figure 1D). We observed no significant difference between any of the cell types, suggesting that the difference in initial sampling did not impact the identification of the assessed cells.
Figure 1. Expert gating strategy for SFC and MC data and their correlation.

(A) Representative biaxial plots showing the gating strategy for SFC and MC data sets and population frequencies. Intact single cells were pre-gated on Forward and Side Scatter Area and Height properties for SFC, Event Length and Intercalator Uptake for MC. Single cells were then assessed for viability and expression of CD45. (B) Gated cell populations for all samples (n=3+2 from two independent experiments) were compared between both data sets and the concordance correlation coefficient was calculated to measure the similarity between both data sets. (C) Values plotted for cell populations with a frequency below 10% were plotted separately, to allow for better visualization. (D) Mean observed cell proportions for SFC and MC samples (n=3+2 from two independent experiments) were plotted and the 95% confidence intervals are shown. A calculation of the two-tail difference between means t-test showed no significant difference between any of the cell populations from the SFC and MS data sets (smallest p-value was 0.223 for DC1s).
To further compare our SFC and MC data sets we used high-dimensional data analysis algorithms, such as t-SNE, UMAP and FlowSOM and compared the common markers present in both data sets side-by-side.
To improve the runtime of the algorithms and normalize the data, samples were first downsampled to the same number of events and concatenated within each group. We initially used t-SNE analysis to visualize each data set. Due to the random generation of the plot, both data sets showed a different island distribution, but displayed major similarities regarding t-SNE density plots and expert gating overlays for both SFC and MC data sets (Figure 2A and 2C). Despite the aleatory distribution of the different clusters, all major cell populations were detected and were present at very similar proportions (Figure 2B and 2D and Suppl. Figure 1A and 1B).
Figure 2. Comparison of marker expression by t-SNE and UMAP from two comparable MC and SFC data sets.

For both data sets, CD45+/Single/Live cells were used for analysis, downsampled to equal cell numbers and the individual samples concatenated. (A) t-SNE analysis was performed with 7,500 iterations with perplexity of 50 and displayed in 2D plots using the resultant t-SNE 1 and t-SNE 2 dimensions according to the per cell expression of 16 proteins. Expression levels of CD3, CD4, Ly6G, NK1.1, CD11c, Ly6c and B220 and the population frequency (±SD) of positively stained populations are shown. t-SNE scales were normalized to −800 to 800 for each marker and visualized using a rainbow heat scale (arcsinh). (B) Expert gating overlay of CD4 T cells, CD8 T cells, neutrophils, natural killer (NK) cells, dendritic cells (DCs), inflammatory monocytes and B cells on t-SNE dimensions 1 and 2. (C) UMAP analysis was performed with default FlowJO settings of nearest neighbours of 15 and minimum distance of 0.5 using the Euclidean distance function and displayed in 2D plots using the resultant UMAP 1 and UMAP 2 dimensions according to the per cell expression of 16 proteins. Expression levels of CD3, CD4, Ly6G, NK1.1, CD11c, Ly6c and B220 and the population frequency (±SD) of positively stained populations are shown. UMAP scales were manually normalized for each marker (see Materials and methods for details) and visualized using a rainbow heat scale (arcsinh). (D) Expert gating overlay of CD4 T cells, CD8 T cells, neutrophils, natural killer (NK) cells, dendritic cells (DCs), inflammatory monocytes and B cells on UMAP dimensions 1 and 2.
We also analyzed our data sets using the more recently developed UMAP analysis (12). Similar to our t-SNE analysis, all cell populations could be visualized and were present in comparable frequencies compared to our t-SNE and expert gating analysis. However, SFC and MC plots again showed different island distributions, due to random plot generation.
Data integration efforts in the single-cell RNA sequencing field have enabled the overlay of different data types, allowing for a combined analysis. However, we and others have to date been unable to replicate this approach using cytometry data, due to the very different data structure between SFC and MC data sets, especially in regards to low signal near zero, and the low number of overlapping features and similar principle components between different data sets.
When we interrogated the positioning of cell populations within the UMAP or t-SNE space we noticed that cell clusters were projected at different distances to each other. For example, CD8 DCs cluster farther away from T cells in the UMAP projection, whereas CD4 T cells and CD8 T cells, were closer together. When we compared the relationship of these populations by expert gating we found that the distances projected within the UMAP space were more representative of the biological differences between these cell types, than compared with t-SNE. Our observations were therefore in line with previous reports that suggested that the UMAP algorithm tends to preserve the global geometry of the data better and projects biologically similar populations closer together (14,40). However, this difference might be attributed to different choices of initialization, as recently suggested (16).
Another commonly used visualization tool is FlowSOM, which creates a self-organizing map using an unsupervised technique for clustering and dimensionality reduction to identify unique cellular subsets and visualize relationships (13). However, an input requirement for the FlowSOM algorithm is the number of clusters the data is grouped into. We therefore incremented the number of clusters and utilised the F measure algorithm implemented in FlowSOM, which compares the f-score between two clustering results, as well as the range of cells per cluster, to evaluate how data is distributed within the different number of clusters (Figure 3A and B). We observed an f-score above 0.6 when low numbers of clusters (4-81) were used (Figure 3A). However, cells were distributed within a wide range, fluctuating from several 100 to several 1000 cells per clusters, with little biological relevance due to underclustering (Figure 3B). We noticed a plateau in the f-score when our data was clustered in 100–144 clusters, which resulted in a range of 200–1000 cells per cluster. From 169 clusters onwards we detected a noticeable drop in the f-score and started to observe small clusters with less than 50 cells per cluster. Above 300 clusters, overclustering problems became more and more apparent, which included cyclic FlowSOM star charts and very small clusters which only included 2 cells per cluster.
Figure 3. Iterations of FlowSOM clustering of the SFC and MC data sets.

For both data sets, CD45+/Single/Live cells were used for analysis and down-sampled to equal cell numbers. (A) f-scores, which summarize the Precision and Recall value compared to the previous clustering iteration were calculated for squared matrix clusters ranging from 4–625 FlowSOM clusters. (B) The Range including the median, maximum and minimum number of cells per clustering iteration (4–625 FlowSOM clusters) are shown. (C) FlowSOM analysis was performed with a default setting of 100 clusters and overlayed with CD4 T cells, CD8 T cells, neutrophils, natural killer (NK) cells, dendritic cells (DCs), inflammatory monocytes and B cells from expert gating analysis.
We therefore selected the default setting of 100 FlowSOM clusters, as previously reported (13), for our analysis, which displayed an f-score of 0.54 for the SFC and 0.58 for our MC data, with a range of 337–893 and 104–1308 cells per cluster, respectively (Figure 3A and B). Our FlowSOM analysis revealed that both data sets were organized into a similar number and size of clusters for each cell population, similar to our previous analysis with t-SNE and UMAP (Figure 3C). We also observed that the position of the branchpoints differed between both data sets, but manual analysis suggested that this was likely caused by the aleatory distribution of the clustering method, which is a common feature of many high-dimensional clustering algorithms.
Our study has demonstrated that SFC and MC data yield highly comparable results on a small data set. This is a favourable technical result as it suggests that both technologies can be used to interrogate complex immune cell phenotypes. We anticipate that similar results can also be expected using larger data sets, as long as the technical limitations of each technology are correctly taken into account. It would be especially interesting to compare stimulated versus unstimulated samples or analyse tissues after infection, where a greater degree of change is expected. These studies are needed to ensure that the concordance seen in our experiments comparing data acquired via SFC or MC is maintained during a state of flux. Once the panels for SFC or MC have been optimized and the same antibody clones are used, similar results should be expected when performing high-dimensional data analysis on either technology platform, as suggested by our study. If a decision has to be made to use one platform over the other, several practical factors need to be taken into consideration such as: tissue/sample and technology availability, desired marker number, cell loss during staining and speed of acquisition.
In summary, our study indicates that both SFC and MC technologies allow for the acquisition of high-dimensional data sets, which can be represented and analysed in similar ways, resulting in comparable t-SNE, UMAP and FlowSOM plots. However, due to differences in the data structure of these two technologies, an overlay of both data sets has so far not been possible, which makes minor differences between both data sets hard to detect. Importantly, the number of recorded events significantly affected the ability to detect smaller cell populations and their analysis (Supplementary Figure 2), and large-scale studies will be required to assess subtle differences between SFC and MC technologies in more detail.
Supplementary Material
Supplementary Figure 1. Comparison of marker expression by t-SNE and UMAP from two comparable MC and SFC data sets. For both data sets, CD45+/Single/Live cells were used for analysis and downsampled to equal cell numbers. (A) t-SNE analysis was performed with 7,500 iterations with perplexity of 50 and displayed in 2D plots using the resultant t-SNE 1 and t-SNE 2 dimensions according to the per cell expression of 16 proteins. Expression levels of XCR1, CD44, KLRG1, CD8, CD279, MHCII, CD11b, CD274, CD69 and F4/80 are shown for t-SNE (A) and UMAP (B) with scales normalized as indicated in the materials and methods section.
Supplementary Figure 2. Analysis of specific populations using SFC or MC data. Separate t-SNE plots with 7,500 iterations and a perplexity of 50 were calculated for total CD4 T cells and dendritic cells (DCs) and displayed in 2D using the resultant t-SNE 1 and t-SNE 2 dimensions. The expression level of cell-type specific markers for subsetting was further highlighted for each cell type.
Acknowledgements
This work was enabled by the Hugh Green Cytometry Centre and we wish to thank the Hugh Green Foundation and the Maurice Wilkins Center for funding this study. The authors are also very grateful to Thomas Ashhurst and Caryn Van Vreden from the Sydney Cytometry Facility for CyTOF support and Paul Wallace for feedback on the manuscript.
Footnotes
Conflict of Interest
Jonathan M. Irish was a co-founder and board member of Cytobank Inc. and received research support from Incyte Corp, Janssen, and Pharmacyclics. The authors declare that there are no other conflicts of interest.
References
- 1.Steinkamp JA, Stewart CC, Crissman HA. Three-color fluorescence measurements on single cells excited at three laser wavelengths. Cytometry 2005;2:226–231. Available at: http://doi.wiley.com/10.1002/cyto.990020405. Accessed May 1, 2019. [DOI] [PubMed] [Google Scholar]
- 2.Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends Immunol. 2012;33:323–32. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22476049. Accessed December 10, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brummelman J, Haftmann C, Núñez NG, Alvisi G, Mazza EMC, Becher B, Lugli E. Development, application and computational analysis of high-dimensional fluorescent antibody panels for single-cell flow cytometry. Nat. Protoc. 2019;14:1946–1969. Available at: http://www.nature.com/articles/s41596-019-0166-2. Accessed July 8, 2019. [DOI] [PubMed] [Google Scholar]
- 4.Nettey L, Giles AJ, Chattopadhyay PK. OMIP-050: A 28-color/30-parameter Fluorescence Flow Cytometry Panel to Enumerate and Characterize Cells Expressing a Wide Array of Immune Checkpoint Molecules. Cytom. Part A 2018;93:1094–1096. Available at: http://www.ncbi.nlm.nih.gov/pubmed/30347136. Accessed May 2, 2019. [DOI] [PubMed] [Google Scholar]
- 5.Robinson JP, Patsekin V, Gregori G, Rajwa B, Jones J. Multispectral Flow Cytometry: Next Generation Tools for Automated Classification. Microsc. Microanal. 2005;11:10–11. [Google Scholar]
- 6.Novo D, Grégori G, Rajwa B. Generalized unmixing model for multispectral flow cytometry utilizing nonsquare compensation matrices. Cytom. Part A 2013;83A:508–520. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23526804. Accessed August 21, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nolan JP, Condello D. Spectral flow cytometry. Curr. Protoc. Cytom. 2013;Chapter 1:Unit1.27. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23292705. Accessed May 1, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Futamura K, Sekino M, Hata A, Ikebuchi R, Nakanishi Y, Egawa G, Kabashima K, Watanabe T, Furuki M, Tomura M. Novel full-spectral flow cytometry with multiple spectrally-adjacent fluorescent proteins and fluorochromes and visualization of in vivo cellular movement. Cytometry. A 2015;87:830–42. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26217952. Accessed July 29, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmutz S, Valente M, Cumano A, Novault S. Spectral Cytometry Has Unique Properties Allowing Multicolor Analysis of Cell Suspensions Isolated from Solid Tissues Hoffman RM, editor. PLoS One 2016;11:e0159961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber LM, Robinson MD. Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytom. Part A 2016;89:1084–1096. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27992111. Accessed July 26, 2019. [DOI] [PubMed] [Google Scholar]
- 11.Amir ED, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013;31:545–52. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23685480. Accessed January 23, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McInnes L, Healy J, Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. 2018. Available at: http://arxiv.org/abs/1802.03426.
- 13.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 2015;87:636–645. [DOI] [PubMed] [Google Scholar]
- 14.Van Der Maaten L, Hinton G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. Available at: http://www.jmlr.org/papers/volume9/vandermaaten08a/vandermaaten08a.pdf. Accessed May 1, 2019. [Google Scholar]
- 15.Kobak D, Berens P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. Available at: 10.1038/s41467-019-13056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobak D, Linderman GC. UMAP does not preserve global structure any better than t-SNE when using the same initialization. bioRxiv 2019:2019.12.19.877522. [Google Scholar]
- 17.Ashhurst TM, Smith AL, Jonathan N, King C. High-Dimensional Fluorescence Cytometry. 2017:1–38. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen R, Perfetto S, Mahnke YD, Chattopadhyay P, Roederer M. Quantifying spillover spreading for comparing instrument performance and aiding in multicolor panel design. Cytom. Part A 2013;83 A:306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perfetto SP, Roederer M. Increased immunofluorescence sensitivity using 532 nm laser excitation. Cytom. Part A 2007;71A:73–79. Available at: http://doi.wiley.com/10.1002/cyto.a.20358. Accessed December 12, 2019. [DOI] [PubMed] [Google Scholar]
- 20.Mazza EMC, Brummelman J, Alvisi G, Roberto A, De Paoli F, Zanon V, Colombo F, Roederer M, Lugli E. Background fluorescence and spreading error are major contributors of variability in high-dimensional flow cytometry data visualization by t-distributed stochastic neighboring embedding. Cytom. Part A 2018;93:785–792. Available at: http://www.ncbi.nlm.nih.gov/pubmed/30107099. Accessed August 21, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KWW, Low D, Ruedl C, Riccardi-Castagnoli P, Poidinger M, Greter M, Ginhoux F, Newell EW. High-dimensional analysis of the murine myeloid cell system. Nat. Immunol. 2014;15:1181–1189. [DOI] [PubMed] [Google Scholar]
- 22.Mingueneau M, Krishnaswamy S, Spitzer MH, Bendall SC, Stone EL, Hedrick SM, Pe’er D, Mathis D, Nolan GP, Benoist C. Single-cell mass cytometry of TCR signaling: Amplification of small initial differences results in low ERK activation in NOD mice. Proc. Natl. Acad. Sci. U. S. A. 2014;111:16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bendall SC, Simonds EF, Qiu P, Amir ED, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011;332:687–96. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21551058. Accessed March 5, 2019.21551058 [Google Scholar]
- 24.Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, Dekker CL, Mackey S, Maecker H, Swan GE, Davis MM, Norman PJ, Guethlein LA, Desai M, Parham P, Blish CA. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci. Transl. Med. 2013;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by Time-of-Flight Shows Combinatorial Cytokine Expression and Virus-Specific Cell Niches within a Continuum of CD8+ T Cell Phenotypes. Immunity 2012;36:142–152. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22265676. Accessed March 5, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stern L, McGuire H, Avdic S, Rizzetto S, Fazekas de St Groth B, Luciani F, Slobedman B, Blyth E. Mass Cytometry for the assessment of immune reconstitution after hematopoietic stem cell transplantation. Front. Immunol. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polikowsky HG, Wogsland CE, Diggins KE, Huse K, Irish JM. Cutting Edge: Redox Signaling Hypersensitivity Distinguishes Human Germinal Center B Cells. J. Immunol. 2015;195:1364–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Behbehani GK, Bendall SC, Clutter MR, Fantl WJ, Nolan GP. Single-cell mass cytometry adapted to measurements of the cell cycle. Cytom. Part A 2012;81 A:552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Abbasi F, Ornatsky O, Cole KD, Misakian M, Gaigalas AK, He HJ, Marti GE, Tanner S, Stebbings R. Human CD4 + lymphocytes for antigen quantification: Characterization using conventional flow cytometry and mass cytometry. Cytom. Part A 2012;81 A:567–575. [DOI] [PubMed] [Google Scholar]
- 30.Nicholas KJ, Greenplate AR, Flaherty DK, Matlock BK, Juan JS, Smith RM, Irish JM, Kalams SA. Multiparameter analysis of stimulated human peripheral blood mononuclear cells: A comparison of mass and fluorescence cytometry. Cytom. Part A 2016;89:271–280. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26599989. Accessed August 14, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gadalla R, Noamani B, MacLeod BL, Dickson RJ, Guo M, Xu W, Lukhele S, Elsaesser HJ, Razak ARA, Hirano N, McGaha TL, Wang B, Butler M, Guidos CJ, Ohashi PS, Siu LL, Brooks DG. Validation of CyTOF against flow cytometry for immunological studies and monitoring of human cancer clinical trials. Front. Oncol. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferrer-Font L, Mehta P, Harmos P, Schmidt AJ, Chappell S, Price KM, Hermans IF, Ronchese F, le Gros G, Mayer JU. High-dimensional analysis of intestinal immune cells during helminth infection. Elife 2020;9 Available at: https://elifesciences.org/articles/51678. Accessed February 21, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hemmers S, Schizas M, Azizi E, Dikiy S, Zhong Y, Feng Y, Rudensky AY. IL-2 production by self-reactive CD4 thymocytes scales regulatory T cell generation in the thymus. 2019;216:2466–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lippitsch A, Chukovetskyi Y, Baal N, Bein G, Hackstein H. Unique high and homogenous surface expression of the transferrin receptor CD71 on murine plasmacytoid dendritic cells in different tissues. Cell. Immunol. 2017;316:41–52. [DOI] [PubMed] [Google Scholar]
- 35.Kadić E, Moniz RJ, Huo Y, Chi A, Kariv I. Effect of cryopreservation on delineation of immune cell subpopulations in tumor specimens as determinated by multiparametric single cell mass cytometry analysis. BMC Immunol. 2017;18 Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5288879/pdf/12865_2017_Article_192.pdf. Accessed October 20, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Gassen S, Callebaut B, Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. 2019. [DOI] [PubMed] [Google Scholar]
- 38.Ellis B, Haaland P, Hahne F, Le Meur N, Gopalakrishnan N, Spidlen J, Jiang M, Finak G, Granjeaud S. flowCore: Basic structures for flow cytometry data. R package version 1.50.0. 2019. Available at: https://git.bioconductor.org/packages/flowCore. Accessed February 21, 2020. [Google Scholar]
- 39.Ferrer-Font L, Mehta P, Harmos P, Schmidt A, Price KM, Hermans IF, Ronchese F, Gros G Le, Mayer JU. Single-cell analysis of intestinal immune cells during helminth infection. bioRxiv 2019;19:746479 Available at: https://www.biorxiv.org/content/10.1101/746479v1. Accessed December 16, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, Newell EW. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019;37:38–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Comparison of marker expression by t-SNE and UMAP from two comparable MC and SFC data sets. For both data sets, CD45+/Single/Live cells were used for analysis and downsampled to equal cell numbers. (A) t-SNE analysis was performed with 7,500 iterations with perplexity of 50 and displayed in 2D plots using the resultant t-SNE 1 and t-SNE 2 dimensions according to the per cell expression of 16 proteins. Expression levels of XCR1, CD44, KLRG1, CD8, CD279, MHCII, CD11b, CD274, CD69 and F4/80 are shown for t-SNE (A) and UMAP (B) with scales normalized as indicated in the materials and methods section.
Supplementary Figure 2. Analysis of specific populations using SFC or MC data. Separate t-SNE plots with 7,500 iterations and a perplexity of 50 were calculated for total CD4 T cells and dendritic cells (DCs) and displayed in 2D using the resultant t-SNE 1 and t-SNE 2 dimensions. The expression level of cell-type specific markers for subsetting was further highlighted for each cell type.