

Abstract
Glioblastoma (WHO grade IV glioma) is the most common malignant primary brain tumor in adults. Survival has remained largely static for decades, despite significant efforts to develop new effective therapies. Immunotherapy, and especially immune checkpoint inhibitors and programmed cell death (PD)-1/PD-L1 inhibitors have transformed the landscape of cancer treatment and improved patient survival in a number of different cancer types. With the exception of few select cases (e.g., patients with Lynch syndrome) the neuro-oncology community is still awaiting evidence that PD-1 blockade can lead to meaningful clinical benefit in glioblastoma. This lack of progress in the field is likely to be due to multiple reasons, including inherent challenges in brain tumor drug development, the blood-brain barrier, the unique immune environment in the brain, the impact of corticosteroids, as well as inter- and intra-tumoral heterogeneity. Here we critically review the clinical literature, address the unique aspects of glioma immunobiology and potential immunobiological barriers to progress, and contextualize new approaches to increase the efficacy of PD-1/PD-L1 inhibitors in glioblastoma that may identify gaps and testable relevant hypotheses for future basic and clinical research and to provide a novel perspective to further stimulate pre-clinical and clinical research to ultimately help patients with glioma, including glioblastoma, which is arguably one of the greatest areas of unmet need in cancer. Moving forward, we need to build on our existing knowledge by conducting further fundamental glioma immunobiology research in parallel with innovative and methodologically sound clinical trials.
Keywords: glioma, glioblastoma, PD-1, PD-L1, immunotherapy
Introduction
Glioblastoma (GBM; grade IV glioma) is the most common and aggressive type of malignant primary brain tumor. The standard of care for glioblastoma is maximal safe surgical resection followed by radiotherapy plus temozolomide and adjuvant temozolomide, achieves a median overall survival of 15 months (1–3). Despite recent advances in multi-modality therapy for glioblastoma, incorporating surgery, radiotherapy, systemic therapy, tumor treating fields, and supportive care, median survival remains static at about 15 months, and long-term survival is rare (1). Two of three patients have an unmethylated O6-methylguanine-DNA methyltransferase (MGMT) promotor that renders them less likely to benefit from DNA alkylating agents such as temozolomide or nitrosoureas, and their median survival is even shorter, at about 12 months (4).
Cancer escapes immunosurveillance via several mechanisms, including activation of immune checkpoint pathways that suppress anti-tumor immune responses. Immune checkpoint inhibitors reinstitute the immune response by disrupting co-inhibitory signaling pathways aiming to eliminate tumor cells. Immune checkpoint blockade has transformed the landscape in several cancers, including cancers with brain metastases (5). The main immune checkpoints that have been successfully targeted with monoclonal antibodies are programmed death-1 (PD-1), PD-ligand 1 (PD-L1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4). Immunotherapy, especially with PD-1/PD-L1 inhibitors, has transformed the management of many cancers, and consequently there has been considerable investigation and research into immune-based therapeutic approaches for glioma, and specifically glioblastoma. However, to date these therapies have not demonstrated a major benefit for these patients.
Here, we critically appraise the preclinical and clinical literature to systematically understand the lack of success in glioblastoma clinical trials, especially studies including immune checkpoint inhibitors. The aim is to identify gaps and testable relevant hypotheses for future basic and clinical research and to provide a novel perspective on how to make progress in this area of unmet need.
Clinical Trials Targeting PD-1/PD-L1 in Glioblastoma
Several phase I/II trials in recurrent and newly diagnosed glioblastoma are evaluating anti-PD-1 agents such as nivolumab, pembrolizumab, cemiplimab or anti-PD-L1 antibodies including atezolizumab, avelumab, durvalumab, alone or in combination with other therapies.
Combinations with standard treatments are often developed to test specific synergy hypotheses, such as simultaneous PD-1-inhibition and anti-angiogenic therapy (bevacizumab) and/or radiotherapy. Examples of this approach include a phase II trial evaluating the anti-PD-L1 antibody durvalumab in five different cohorts of patients (6), including combinations with radiotherapy for newly diagnosed glioblastoma or with bevacizumab for recurrent glioblastoma. Numerous studies of immune checkpoint inhibitors, in newly diagnosed and recurrent glioblastoma are ongoing (Table 1) and some mature results of phase III studies (Table 2) with nivolumab have been reported.
Table 1;
Some ongoing clinical Trials including PD-1/ PD-L1 inhibitors in gliomas and glioblastoma
| NCT Number | Study Title | Phase | Size (n) |
|---|---|---|---|
| NCT03426891 | Pembrolizumab and Vorinostat Combined With Temozolomide for Newly Diagnosed Glioblastoma | 1 | 32 |
| NCT03491683 | INO-5401 and INO-9012 Delivered by Electroporation (EP) in Combination With Cemiplimab (REGN2810) in Newly-Diagnosed Glioblastoma | 1/2 | 52 |
| NCT03532295 | Epacadostat in Combination With Radiation Therapy and Avelumab in Patients With Recurrent Gliomas | 1/2 | 55 |
| NCT03665545 | Pembrolizumab in Association With the IMA950/Poly-ICLC for Relapsing Glioblastoma | 1/2 | 24 |
| NCT02794883 | Tremelimumab and Durvalumab in Combination or Alone in Treating Patients With Recurrent Malignant Glioma | 2 | 36 |
| NCT03493932 | Cytokine Microdialysis for Real-Time Immune Monitoring in Glioblastoma Patients Undergoing Checkpoint Blockade | 1 | 15 |
| NCT03661723 | Pembrolizumab and Reirradiation in Bevacizumab Naive and Bevacizumab Resistant Recurrent Glioblastoma | 2 | 60 |
| NCT03018288 | Radiation Therapy Plus Temozolomide and Pembrolizumab With and Without HSPPC-96 in Newly Diagnosed Glioblastoma | 2 | 108 |
| NCT03557359 | Nivolumab for Recurrent or Progressive IDH Mutant Gliomas | 2 | 37 |
| NCT03743662 | Nivolumab With Radiation Therapy and Bevacizumab for Recurrent MGMT Methylated Glioblastoma | 2 | 94 |
| NCT03174197 | Atezolizumab in Combination With Temozolomide and Radiation Therapy in Treating Patients With Newly Diagnosed Glioblastoma | 1|/2 | 60 |
| NCT03636477 | A Study of Ad-RTS-hIL-12 With Veledimex in Combination With Nivolumab in Subjects With Glioblastoma; a Substudy to ATI001–102 | 1 | 18 |
| NCT03233152 | Intra-tumoral Ipilimumab Plus Intravenous Nivolumab Following the Resection of Recurrent Glioblastoma | 1 | 6 |
| NCT03722342 | TTAC-0001 and Pembrolizumab Combination Trial in Recurrent Glioblastoma | 1 | 20 |
| NCT03047473 | Avelumab in Patients With Newly Diagnosed Glioblastoma | 2 | 30 |
| NCT03707457 | Biomarker-Driven Therapy Using Immune Activators With Nivolumab in Patients With First Recurrence of Glioblastoma | 1 | 30 |
| NCT03718767 | Nivolumab in People With IDH-Mutant Gliomas With and Without Hypermutator Phenotype | 2 | 95 |
| NCT03341806 | Avelumab With Laser Interstitial Therapy for Recurrent Glioblastoma | 1 | 30 |
| NCT04047706 | Radiation, nivolumab, and BMS986205 (an indoleamine 2,3 dioxygenase 1 inhibitor) | 1 | 30 |
| NCT03367715 | Nivolumab, Ipilimumab, and Short-course Radiotherapy in Adults With Newly Diagnosed, MGMT Unmethylated Glioblastoma | 2 | 24 |
| NCT03673787 | A Trial of Ipatasertib in Combination With Atezolizumab | 1/2 | 51 |
| NCT02829931 | Hypofractionated Stereotactic Irradiation With Nivolumab, Ipilimumab and Bevacizumab in Patients With Recurrent High Grade Gliomas | 1 | 26 |
| NCT03291314 | Combination of Avelumab and Axitinib for the Treatment of Patients With Recurrent Glioblastoma | 2 | 52 |
| NCT03430791 | Trial of Combination TTF(Optune), Nivolumab Plus/Minus Ipilimumab for Bevacizumab-naive, Recurrent Glioblastoma | 2 | 60 |
Table 2;
Some completed clinical trials including PD-1/ PD-L1 inhibitors in gliomas and glioblastoma
| NEWLY DIAGNOSED GLIOBLASTOMA TRIALS | |||
|---|---|---|---|
| AGENTS | PHASE | TRIAL | COMMENTS |
| Nivolumab Compared to TMZ in patients with unmethylated MGMT promotor | 3 | CheckMate-498 NCT02617589 |
550 patients randomized This study confirmed the safety of PD-1 blockade plus radiotherapy for newly diagnosed GBM patients, but did not demonstrate either an overall survival or progression-free survival advantage3 |
| TMZ and RT with Nivolumab or Placebo, in patients with methylated MGMT promotor | 3 | CheckMate-548 NCT02667587 | 693 patients randomized The study did not demonstrate an improvement in progression-free survival, but its overall survival findings are pending further maturation of study follow-up data3 |
| RECURRENT GLIOBLASTOMA TRIALS | |||
| AGENTS | PHASE | TRIAL | COMMENTS |
| Nivolumab versus bevacizumab | III | Chechmate-143 NCT02017717 |
|
| Single-agent atezolizumab | I | NCT04047706 | 16 patients enrolled had received prior chemotherapy, and 50% prior bevacizumab. Ten patients (63%) experienced a treatment-related event. None was Grade 4–5 events30 |
| bifunctional molecules targeting PD-L1 and TGF-β (bintrafusp alfa) 41 | IB | NCT02517398 | 35 patients enrolled Two patients had a PR, and 9 had SD The most common TRAEs were gingival bleeding (17.1%), asthenia (14.3%), pruritus, and rash (each 11.4%). |
Abbreviations: MGMT: O6-methylguanine-DNA methyltransferase, PR: Partial Response, RT: Radiation Therapy, SD: Stable Disease, TMZ: Temozolomide, TRAE: Treatment Related Adverse Events,
In addition to the effect of perioperative corticosteroid administration, surgery itself may dampen the immune response, with suppression of cellular immunity as one of the host responses to surgical stress (7). Trials have also been conducted with neoadjuvant (pre-operative) administration of PD-1 inhibitors to induce a more pronounced immune response compared to the usual post-resection approach (Table 3).
Table 3;
Examples of Completed Neoadjuvant Clinical Trials including PD-1/ PD-L1 inhibitors in glioblastoma
| NEADJUVANT GLIOBLASTOMA TRIALS | |||
|---|---|---|---|
| AGENTS | PHASE | TRIAL | COMMENTS |
| A Pre-surgical dose of nivolumab followed by post-surgical nivolumab | II | NCT02550249 | A 30 patient single-arm trial. An increase in expression of chemokine transcripts, higher immune cell infiltration, and augmented T-cell receptor clonal diversity among tumor-infiltrating T lymphocytes, was reported in patients receiving nivolumab compared to those who did not. (92) |
| Pre- and post-operative pembrolizumab versus post-operative therapy alone | II | NCT02852655 | 35 patient enrolled Better survival in the presurgical-surgical PD-1(93), together with an upregulation of T-cell and interferon-γ-related gene expression, but downregulation of cell cycle-related gene expression within the tumor. This was not seen in the control group. Peripheral markers of immune engagement were observed more often in the neoadjuvant group than in patients treated only in the post-operative setting. |
| pre- and post-operative pembrolizumab | II | NCT02337686 | 15 patients enrolled Indirect signs of immune engagement was observed. The study concluded that anti-PD-1 monotherapy is insufficient for an immunomodulatory response in the majority of GBM patients, likely due to paucity of T-cells within the tumor microenvironment and a CD68+ macrophage preponderance (94) |
In summary, the phase II and III studies in GBM with nivolumab have not demonstrated a meaningful benefit. No other phase III studies with PD-1/PD-L1 inhibitors are planned or have been conducted.
Why are PD-1/PD-L1 Inhibitors Ineffective in Glioblastoma?
Challenges with pre-clinical and clinical trial design paradigms
Evaluating immunotherapy relies on pre-clinical models to identify targets, conduct mechanistic studies, and to optimize delivery and pharmacokinetic aspects. These models have helped develop the CTLA-4 and PD-L1/PD-1 inhibitors (8,9). GBM immune biology may not be well reflected in the models that are frequently utilized. A commonly used animal model is the GL261 glioma, which is moderately immunogenic and expresses clonotypic, homogeneous, and robust levels of PD-L1, which is not the case in human gliomas (10). Some pre-clinical models are developed by implanting cancer cell lines either in the flank or in the brain of mice. Tumors that are successfully implanted rarely represent the GBM immune complexity seen in humans. Common pre-clinical models are relatively easy to evaluate as subcutaneous implants. Mice can be engineered to approximate, but not completely represent, human GBM. Knocking out suppressor genes or induction of somatic mutations can result in de novo tumors, but they do not fully represent the accumulation of genetic aberrations that develop in human GBM. The immune system is dependent on the tumor microenvironment (TME), which is challenging to reproduce in animal models.
Humanized mice models using both tumor and immune systems that are patient-specific are an improvement to study immunotherapies but they are limited by cost, low engraftment rates and slow tumor growth. It is also unclear whether humanized murine models consistently recapitulate the tumor microenvironment of human tumors. Zhai et al, demonstrate the use of humanized mice partially reconstituted with a human immune system and intracranially-engrafted with human glioblastoma. The authors demonstrate that the tumor-infiltrating T cells increase immunosuppression in glioblastoma. This preclinical study suggests that it is not enough to enhance T cell infiltration, but if successful, one must then deal with the consequence of the T cell-dependent increase of glioblastoma-induced immune suppression.
Another study treated immunocompetent orthotopic mouse models of glioblastoma with an indoleamine 2,3 dioxygenase 1 (IDO1) enzyme inhibitor, combined with a PD1 inhibitor and radiotherapy(11). The study demonstrated a durable survival benefit from this novel three-agent treatment. Of note, efficacy required IDO1 enzyme inhibition in non-glioblastoma cells, rather than tumor cells. An ongoing clinical trial [NCT04047706] aims to replicate these findingsAnother factor making interpretation of preclinical findings difficult is the challenge of surgical resection of murine tumors derived from syngeneic glioma cells. This deficiency limits extrapolation of preclinical studies for the subset of human patients who are able to undergo surgical tumor debulking either before or after initiation of immune checkpoint blockade.
The scarcity of representative pre-clinical models has led to the initiation of clinical trials evaluating immunotherapies that have shown promise in other cancers without definitive pre-clinical rationale in GBM. Clinical trials are often initiated with uncontrolled, open label (unblinded), non-randomized, small sample size trials that have inherent selection and other biases. This in turn will have a high type I error rate, with ineffective therapies showing false positive results that may surreptitiously build the case to start larger studies. Large studies minimize risk of bias and false positive results will be less likely to be reproduced. Unfortunately, the true effect size or lack of activity only becomes apparent after hundreds of patients have participated and millions of research dollars have been invested over the course of several years. There are other barriers to translating pre-clinical ideas to human trials. For example, immune priming, activation, and memory responses may require sequencing of therapies that are challenging to evaluate using the traditional clinical trial paradigms. Another challenge in glioblastoma is the inability to collect biopsies at different time-points as it is often done in other solid tumors (at baseline before the immunotherapy, after a few weeks of treatment, and often at recurrence).
Challenges with immunotherapy combination studies
Trials need rational combinations backed by plausible pre-clinically tested hypotheses. Integration of pre-clinical and clinical studies to evaluate promising therapies in pre-clinical models with a “bench to bedside and back to bench” approach may detect early signals or declare ineffective combinations in a more rational manner. Combinations, which may include a therapy that has single-agent activity, are challenging to assess in phase I trials due to factors such as patient heterogeneity, selection bias, and lack of randomization. Newer methods of drug development such as randomization into phase I trials may need to be explored (12,13).
Unlike chemotherapy, effective combination immunotherapies may include agents that are unlikely to independently induce a radiologic response. These combinations may not require individual agent activity. Some of the therapies in the combination may have an effect only by sensitizing to a second agent (e.g., the CD40 agonistic antibodies).
Immunosuppression in Glioblastoma
Patients with GBM are known to have significant deficits in cell-mediated immunity within the lymphocyte population, and specifically the CD4 cells.
Tumor-infiltrating lymphocytes
Tumors can be defined depending on immune infiltration and are classified as immune desert, immune-excluded, and hot tumors, depending on the presence of tumor-infiltrating lymphocytes (TILs) in tumor specimens (14). A tumor that is characterized as an immune desert can arise from immune ignorance, tolerance, or absence of T-cell priming or activation (14). In some studies, the numbers of TILs suggest a positive correlation with better prognosis (15–17). While the presence of lymphocytes in a tumor sample may represent immune engagement, these lymphocytes might be exhausted in GBM and thus may not yield a favorable immune effect. Subsets of TILs may differ, as shown in a study where the level of CD8+ T-cells and tumor grade were inversely correlated, while the level of CD4+ T-cells and tumor grade were positively correlated(18) (19). TILs or CD8+ T-cell infiltrates have been proposed as a surrogate for the presence of antigen, immune activation, and trafficking to the TME. They are positively correlated with response to therapy in other tumors, particularly melanoma (20) and lung cancer (21). Their exact role in gliomas requires further investigation.
T-regulatory cells (Tregs) are a highly diverse and plastic subset of CD4 T-cells and are important in immune tolerance. The thymus-derived natural Tregs (nTregs), characterized by high constitutive expression of FoxP3, cause contact-dependent cytokine-independent immunosuppression through CTLA-4 and PD-L1 pathways (22).
T-cell exhaustion
A study of T-cell exhaustion in GBM in TILs and peripheral blood lymphocytes isolated from 21 patients evaluated multiple immune checkpoint markers, including PD-1, CTLA-4, and TIM-3, and reported upregulation of multiple immune checkpoints. The T-cell transcriptional profile approximated virus-induced exhaustion (23). Another study that reported reduced absolute CD4 counts in 20 patients with GBM also observed that the fraction containing CD4+CD25+FoxP3+CD45RO+ T-cells was increased compared to healthy controls (24). T-cells from these patients demonstrated anergy or secreted Th2 polarizing cytokines on stimulation. Removal of the Treg population reversed this cytokine signature. Treg removal eliminated T-cell proliferative defects and restored TH2 cytokine shifts, where T-cells from patients with glioblastoma could function in vitro at levels equivalent to those of healthy controls.
Another group used glioma samples to demonstrate that the nTreg population is predominant in glioma with high FoxP3 expression (25). Thymectomy appeared to help reduce Treg levels in the GL261 mouse cell line injected intracranially (26). An analogous approach to restore T-cell function is currently being explored in a trial of lymphodepletion in combination with PD-1 inhibition (NCT02664363) (27).
One study found no correlation with levels of Tregs and prognosis, suggesting that gliomas may also mediate immunosuppression though other mechanisms (28). Some cancers employ loss or downregulation of class I MHC to evade immune destruction (29). PD-L1/PD-1 inhibitor activity enables an endogenous immune response that relies on previously primed CD8+ T-cells that specifically recognize cancer cells. These immune cells can either be re-invigorated or expanded by PD-L1/PD-1 inhibition, leading to recognition and targeting of cancer(30) (31).
Immune desert tumors
Immune desert cancers like GBM, hormone receptor positive breast cancer, and prostate cancer respond poorly to immune checkpoint inhibitors (32–34) for a number of potential reasons, including lack of immune infiltration and impaired antigen presentation (low MHC class I) and may have high tumor cell proliferation.
Distinct mechanisms associated with the immune desert tumors include tumor-intrinsic WNT/β-catenin signaling that may reduce the chemokine gradient required for recruiting CD103+ dendritic cells (including CXCL1, CXCL2, and CCL4) (35). This impacts early T-cell priming and infiltration into the TME. Rapidly proliferating immune desert tumors alter the metabolic conditions in the TME to create a hostile environment for the function and proliferation of T-cells (36). T-cells depend on similar glycolytic pathways as tumor cells for survival (37). Lactate production, presence of an acidic TME, and lipogenesis may alter the immunometabolism of T-cells, affecting TCR engagement, T effector activation, differentiation, and proliferation, ultimately resulting in reduction or absence of TILs(30) (36).
Immune ignorance is also associated with the presence of a stem cell-like phenotype in tumors (“stemness”) (38). Stemness correlates with higher intratumoral heterogeneity, possibly by protecting antigenic clones from elimination by the immune system. Stemness is associated with cell intrinsic suppression of endogenous retroviruses, type I interferon signaling, and increased expression of multiple immunosuppressive pathways, and may provide a mechanistic link between antigenicity, intratumoral heterogeneity, and immune suppression across cancers.
The TGF-β pathway
The transforming growth factor-β (TGF-β) pathway is known to be a driver of glioma tumorigenicity and immunosuppression. TGF-β secretion is also one of the characteristics of immune desert tumors, in addition to lack of TILs and the presence of myeloid-derived suppressor cells and VEGF-associated tumor angiogenesis (39,40). The first description of TGF-β isoform 2 was as a glioblastoma-derived T-cell suppressor factor, measured in GBM cell lines and patient serum (41). TGFβ was noted to suppress T-cell proliferation through IL-2-dependent and independent pathways. The TGF-β receptor is highly expressed on glioma cells and RNA silencing of TGF-β reduces glioma proliferation, migration, and invasiveness. A clinical trial (n=150) that treated GBM patients with lomustine with or without galunisertib, a TGF-β receptor inhibitor, failed to demonstrate improved overall survival relative to placebo (42). New approaches to targeting TGF-β together with PD-L1 are ongoing (43).
The immunosuppressive effect of corticosteroids
GBM patients typically receive systemic corticosteroid therapy immediately before and after a craniotomy. Steroids modulate the immune system, causing lymphopenia, including reducing the absolute number of T-cells (44). Corticosteroids are also routinely used to control vasogenic edema in gliomas, as well as for the treatment of a variety of autoimmune diseases, immune-related adverse events triggered by checkpoint inhibition, or cytokine release syndrome. It is very common for GBM patient to have received, or to continue to receive, relatively high doses of corticosteroids, which may diminish T-cell activity, including that associated with immune checkpoint inhibitors.
There are data to support the hypothesis that systemic corticosteroids used to treat immune-related adverse events may not compromise the therapeutic benefit of immune checkpoint inhibitor therapy (45). However, it is challenging to ascertain whether benefit was achieved before or after steroids were given. Individuals who develop immune-related adverse events may have more engaged immune systems (46). This creates an inherent bias, especially in retrospective analyses. Assessment of the impact of steroids on immunotherapy will also need to take the timing of steroid administration into account. While corticosteroids may not completely remove the anti-cancer immune response, they can potentially attenuate it. This might be acceptable in an immune dominant tumor environment (such as melanoma), but this effect may be more significant in immune desert tumors such as GBM.
Other unclear aspects include the effects of the type, timing, and overall dose of corticosteroids on response to immunotherapies. These questions are important but difficult to answer in clinical trials.
Macrophages, Microglia and Myeloid-Derived Suppressor Cells (MDSCs)
Microglia and bone marrow derived macrophages in the brain are mononuclear phagocytes that are increasingly recognized to be important in diseases of the brain. Peripheral immune cells can infiltrate the CNS in certain conditions, but microglial precursors migrate to and reside in the brain parenchyma. Brain resident macrophages and microglia are essential part of the innate immune system against various insults. Parenchymal microglial cells and recruited monocyte-derived macrophages from the periphery exhibit disease-specific phenotypic characteristics and are subpopulations based on their molecular signatures. An abundance of myeloid over lymphoid lineage cells during CNS pathology, including gliomas, is a feature of brain immunity as compared to extra-CNS immunity(47).
Macrophages are traditionally divided into M1 and M2 subtypes: M1-type macrophages have anti-tumor features and secrete pro-inflammatory factors such as IL-6, IL-8 and TNF-α, whereas M2-type macrophages have tumor-promoting features and secrete anti-inflammatory factors such as IL-10 and TGF-β, promoting tumor cell growth and migration(48). This classification might be an oversimplification and there likely exists a continuum of myeloid cell phenotype including a non-polarized subset(49).
MDSCs play a major role on tumor microenvironment immunosuppression in blood and tumors from glioblastoma patients. MDSC subsets may differentially drive immune suppression in a gender-specific manner with monocytic (mMDSC) versus granulocytic (gMDSC) subsets having different contributions(50). In a mouse GBM study, mMDSCs were enriched in the male tumors, while gMDSCs were elevated in blood of females(50). Only female mice had extended survival with depletion of gMDSCs(50). mMDSCs could be targeted with anti-proliferative agents in males while blockade in females inhibited the gMDSC function. In this study, human patient data showed that proliferating mMDSCs were predominant in male tumors. A high gMDSC/IL-1b gene signature correlated with worse prognosis of the female patients.
PD-L1 is expressed not only by glioblastoma cells but also can be detected in extracellular vesicles (EVs). GBM EV-mediate immunosuppression by inducing immunosuppressive monocytes rather than inhibition of T-cells(51). PD-L1 expression is important for the induction of specific immunosuppressive monocyte populations but immunosuppression through EVs is complex and likely extends beyond that mediated by PD-L1(51).
A recent study reported that resistance to immune checkpoint blockade that is driven by macrophages is established by CD4 T cell suppression and Treg expansion in the tumor microenvironment via the PD-L1/PD-1/CD80 axis(52). This study also reported heterogeneity of response in syngeneic tumors and suggest that targeting PD-L1-expressing tumor-associated macrophages might overcome resistance to immune checkpoint blockade.
There are ongoing efforts to exploit these observations therapeutically, including combinations of macrophage/monocytes modulation with immune checkpoint blockade.
Immune Biomarkers
PD-L1 immunohistochemistry
Tumors with high expression of PD-L1 or those with increased tumor-infiltrating immune cells might be considered inflamed and theoretically may respond to PD-L1 and PD-1 inhibitors. While this true to a degree in melanoma, lung, and some other cancers, it has not been demonstrated in gliomas. Tissue testing of PD-L1 expression in general, but also in glioma, has been challenging, with various antibodies and techniques employing variable cut points for positivity (53). In one study, about 61% of gliomas had at least 1% of tumor cells expressing PD-L1 (10), with 38% of gliomas exhibiting at least 5% PD-L1 expression. PD-1, PD-L1, and TIL appear to be positively correlated with tumor grade in all gliomas, and levels of expression are higher in GBM (54). In other cancers, responses can be identified in cohorts that do not express PD-1 or PD-L1 (55). It is also uncertain whether expression of PD-1 and/or PD-L1 on infiltrating immune and myeloid cells should be considered (56).
The capacity to present antigens, such as the expression of MHC class I on the surface of tumor cells and an intact HLA, is also essential, but difficult to assess for clinical purposes. Expression of the gene encoding PD-L1 increases post transcriptionally in human glioma after loss of phosphatase and tensin homolog (PTEN) and activation of the phosphatidylinositol-3-OH kinase (PI(3)K) pathway. In one study specimens from glioblastoma patients had levels of PD-L1 protein that correlated with PTEN loss, and tumor-specific T cells lysed human glioma targets expressing wild-type PTEN more effectively than those expressing mutant PTEN. This may suggest a mechanism linking loss of the tumor suppressor PTEN with immunoresistance, mediated in part by PD-L1(57).
Tumor Mutational Burden (TMB)
In melanoma, there is a positive correlation between the median frequency of somatic mutations in tumors and overall response to anti-PD-1 therapies (58). A higher mutational load in tumors may result in more tumor antigens, increasing immunogenicity (58). Higher mutational load in tumors has also been associated with increased survival and long-term benefit from immunotherapy (59). Further, RNA sequencing of lung, ovarian, breast, colorectal, brain, and renal cancers has shown that tumor immunogenic mutation load was associated with survival (60). Mutational load is thought to be a surrogate marker of neoantigen expression. Recognition of neoantigens is a major factor in the activity of immunotherapies (61); and therefore neoantigen load may be a useful biomarker to facilitate the development of therapeutic approaches that selectively enhance T-cell reactivity and may translate to a higher benefit from checkpoint blockade. An increase in mutations as we age is well documented, not only in GBM, but in cancer in general (62,63). This trend can only be seen in exome-wide analysis, not in the commercial next generation sequencing panels that identify genes that are frequently mutated as part of the cancer process (64). To test this hypothesis, a phase II trial is currently randomizing 102 elderly GBM patients (≥65 years) in a 2:1 fashion to short course radiotherapy followed by temozolomide with or without nivolumab (65).
However, techniques and cut points for defining mutational load are not harmonized, so other more easily measured indices have been proposed, such as determining mutations in the exonuclease domain of polymerase E that lead to hypermutation and neoantigen load(18) (66). Individuals with germline Lynch syndrome mismatch repair (MMR) defects have long been recognized to be at increased risk of central nervous system (CNS) tumors (67,68).
Nonetheless, tumors with very high TMB, of which tumors with microsatellite instability (MSI) represent the highest fraction of the TMB continuum, respond favorably to PD blockade and this has led to the approval of pembrolizumab in any MSI-high tumors, regardless of the organ of origin (69). Indeed, patients with biallelic dMMR in glioblastoma have responded to nivolumab (70).
A small percentage of newly diagnosed tumors and a larger proportion of tumors during and after standard therapy with radiation and alkylating agents may have DNA repair defects of various types, including a “hypermutator” phenotype that has also been described in GBM specimens with MSH6 mutations (71). Another subset of GBMs with a potential hypermutator phenotype are lower-grade gliomas that recur after treatment with alkylator therapy (72). Theoretically, this may make these tumors responsive to immune checkpoint blockade (73).
Touat et al(74) analyzed molecular determinants of mutational burden and signatures in 10,294 gliomas. Two main pathways to hypermutation were described, one associated with constitutional defects in DNA polymerase and mismatch repair (MMR) genes, and a more common post-treatment pathway, associated with acquired resistance driven by MMR defects in chemotherapy-sensitive gliomas that relapse following temozolomide.
Experimentally, mutational signature of post-therapy hypermutated gliomas was reintroduced by temozolomide-induced damage in cells with MMR deficiency. MMR-deficient gliomas that lacked prominent T cell infiltrates had extensive intratumoral heterogeneity, poor patient survival, and a low rate of response to PD-1 blockade. Bulk analyses did not detect MSI in MMR-deficient gliomas, but single-cell whole-genome sequencing analysis of post-treatment hypermutated glioma cells identified microsatellite mutations. This study shows that chemotherapy may introduce hypermutated populations without promoting a response to PD-1 inhibitors. Touat et al defined high TMB as ≥17.0 mut. per Mb. However, the cutoff for TMB is not well defined and response to PD-1 inhibitors in inflamed tumors may differ from that in non-inflamed tumors (75). Intratumoral heterogeneity may have a greater impact on immune response and may predict immunotherapy outcomes better than mutational burden (38). All of these complexities make the value of TMB as a biomarker questionable, with respect to highly heterogeneous tumors such as GBM, even in those with MMR defects.
There have been no prospective studies in gliomas, including spontaneously arising gliomas without a predisposing germline alteration, which have addressed the associations of mutational load and dMMR. Retrospective series have shown disappointing results of patients with high TMB but without germline Lynch syndrome or MSI-high status treated with immune checkpoint blockers (76).
Other Biomarkers
It is possible that PD-1/PD-L1/TMB and others are bystanders and thus not the ideal biomarkers in a disease like GBM. Other indicators of immune engagement, such as inflammation markers, might be more valuable (Figure 1). One example may be assessment of the role of inflammasomes, which are important for CD8+ T-cell activation in response to PD-1 blockade. This can induce a PD-L1-NLRP3 inflammasome signaling cascade that ultimately may lead to the recruitment of granulocytic myeloid-derived suppressor cells into tumor tissues (77). There may be other (yet unknown) and underexplored mechanisms dampening a potential anti-tumor immune response in GBM.
Figure 1.
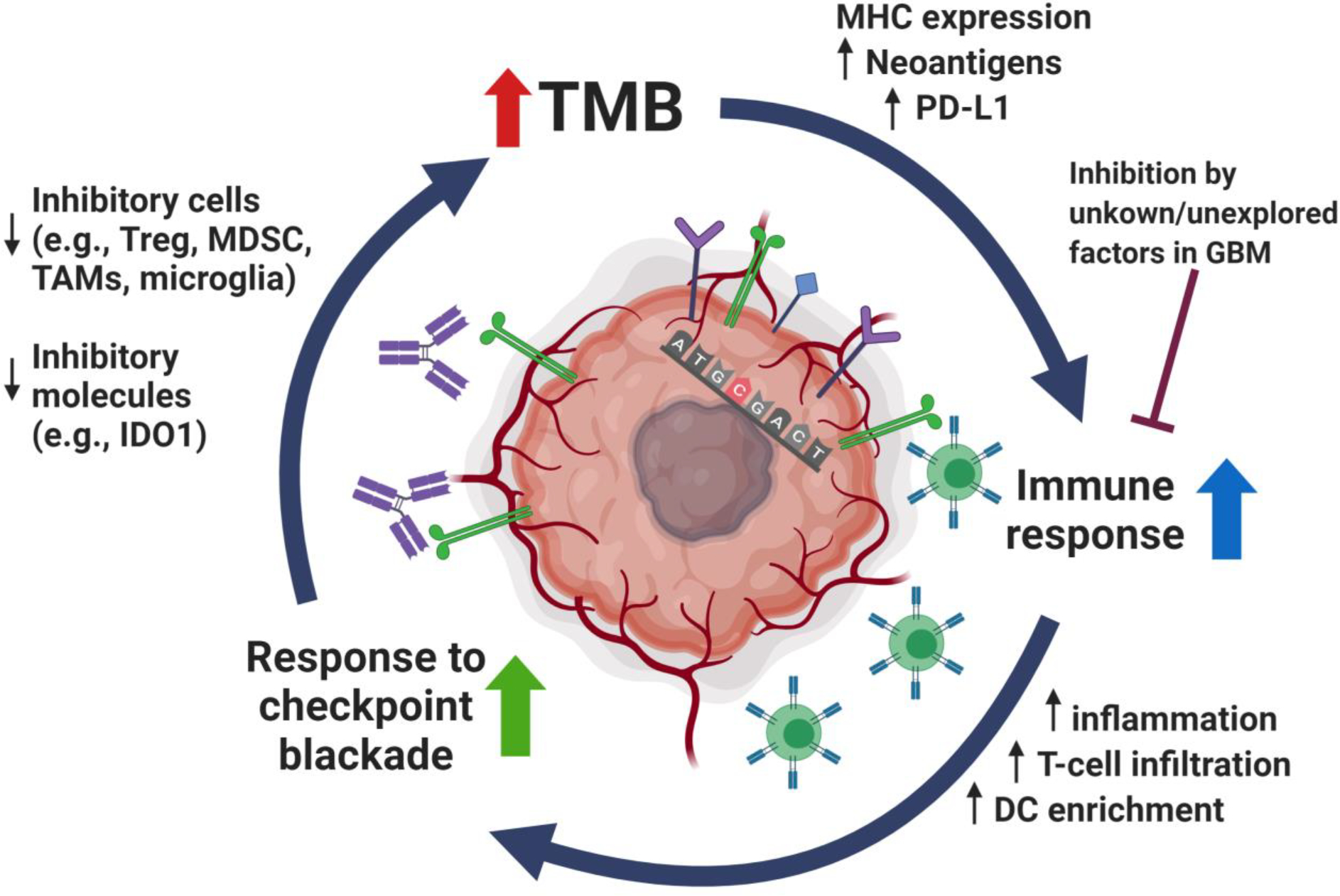
The immune response to glioblastoma and cancer in general, is a dynamic and complex process involving multiple interactions as outlined. The cancer immunity cycle encompasses stimulatory and inhibitory components with an immune regulatory feedback mechanisms. There are unique aspects specific to the brain (e.g., microglia, brain specific immune trafficking… etc.), that pose additional challenges to the development of effective immunotherapy strategies for treatment of glioblastoma. Adapted from an image created with BioRender.com. Abbreviations; GBM: Glioblastoma; TMB: Tumor Mutational Burden; MHC: Major Histocompatibility Complex; PD-L1: Programmed Death Ligand 1; GBM: Glioblastoma; DC: Dendritic Cells; IDO1: Indoleamine 2,3-Dioxygenase 1; TAMs: Tumor Associated Macrophages; MDSC: Myeloid-Derived Suppressor Cells; Tregs: Regulatory T cells.
The Blood-Brain Barrier (BBB)
The blood-brain barrier (BBB) prevents the passive movement of cells and molecules that could induce inflammation or damage to the CNS. Endothelial cells, and the intact tight junctions between them, interact to maintain the integrity of the BBB, but do respond to environmental changes. Brain pathologies may permit the passage of circulating monocytes and lymphocytes from the periphery (57,58). Passage of T-cells across the BBB involves adhesion molecules and chemokines that might vary for different immune cell populations; e.g., CD8+ T-cells seem to be dependent on α4 integrin (78).
PD-1 and PD-L1 antibodies are relatively large molecules and may not achieve high intratumoral concentrations, which could diminish their therapeutic benefit. However, they may not need to cross the BBB if they have a potent systemic immune effect. They may achieve some of their goals by acting on T-cells that are circulating or localized within lymphatic structures.
Various invasive and noninvasive approaches have been used for drug and gene delivery into the brain parenchyma with or without altering BBB homeostasis (79,80). However, the macroscopically normal peritumoral zone is recognized as the primary site of GBM recurrence (81). Immune surveillance, antigen presentation, and the role of myeloid cells in the CNS are complex and requires lymphatic drainage to lymph nodes (82). Unlike extracranial organs, the brain contains a sparse lymphatic network. Cerebrospinal fluid is drained through the meningeal lymphatics into the deep cervical lymph nodes where brain-derived antigens and peripheral T-cells interact (83). The vascular endothelial growth factor-C (VEGF-C; a growth factor for lymphatic vessels), can influence immune interactions via the BBB (84). GL261 mice treated to increase VEGF-C expression were able to reject intracranial GBM tumor implants (84). These mice also reject a re-challenge with flank tumor implants, demonstrating the generation of memory responses. The survival benefit in mice with only intracranial tumors treated with VEGF-C/anti-PD-1 combination therapy was similar to that in mice with both intracranial and flank tumors treated with checkpoint inhibitor therapy alone. Ligation of the deep cervical lymph nodes abrogated the VEGF-C benefit in mice with intracranial tumors, but not mice with both intracranial and flank implants. T-cell priming through expression of VEGF-C in the cerebrospinal fluid, or through a flank tumor, enables checkpoint inhibition in the CNS. However, in these experiments, a tumor that was confined to the CNS at steady state (e.g., GBM), did not benefit from an immune checkpoint inhibitor alone (84).
In contrast to GBM, combined anti-PD-1 and anti-CTLA-4 have extracranial and intracranial activity in melanoma and lung cancer(85). Mice with both a flank and an intracranial tumor respond better to combination immunotherapy than those with just intracranial melanoma (86). This may mean that immune checkpoint inhibitors are effective intracranially if the relevant immune signatures against the tumor are present to induce an extracranial immune response.
PD-1 Blockade in Glioblastoma: Ongoing Approaches
While the results of single-agent PD-1 blockade remain unsatisfactory to date, a number of new strategies are being considered, including the combination of PD-1 blockade with other immune checkpoint inhibitors, as well as targeted agents, radiation therapy, and chemotherapies.
Combination therapy with two or more therapeutic agents is a cornerstone of cancer therapy. There are barriers to the conduct of combination studies. These include toxicity, challenges with combination trial design, and the interpretation of endpoints. Endpoints, including the objective response rate, progression-free survival, and overall survival, have not evolved to optimally assess responses to immunotherapy. Trials are often underpowered and may require prolonged follow-up, often lasting multiple years (87). Response assessment does not take into account the potential for immune checkpoint inhibitors to lead to either a delayed response (e.g., continued tumor growth followed by tumor shrinkage) or “pseudoprogression” (e.g., apparent tumor growth driven by tumor inflammation followed by tumor shrinkage)(30). It is yet to be seen if these issues will be addressed by the prospective use and validation of immune-related response criteria such as the Immunotherapy Response Assessment in Neuro-Oncology (iRANO)(88). Validation requires large datasets and iterative modifications and ultimately may be optimized for the immunotherapy setting.
A number of immunotherapies other than immune checkpoint blockers are currently under investigation alone, or in combination with PD-1 blockade. These include viral therapies, cancer vaccines, adoptive T-cells, and targeting ubiquitous viral antigens, such as those from cytomegalovirus (CMV), as well as therapies that aim to induce broad-based immune responses. These approaches may include targeting components of the innate immune system through oncolytic viruses, Toll-like receptors, the inflammasome, and other means to generate pathogen-associated molecular patterns and/or exploit self-molecules known as damage-associated molecular patterns to achieve immunogenic cell death (89,90).
Conclusions
While there is an urgent unmet need for glioma and GBM clinical research to find therapeutic breakthroughs, investment will need to continue in fundamental basic science in order to increase our understanding of the immunobiology of gliomas. More pre-clinical novel animal and humanized models are needed to better evaluate immune checkpoint inhibitors and combination immunotherapies (Table 4).
Table 4;
Summary of the issues, barriers, recommendations and future directions
| The Major Biological Barriers; Why PD-1 inhibitors were successful in many cancers but not in GBM? |
|
| Challenges in Identifying Reliable Immune Biomarkers in GBM |
|
| Recommendations and Future Directions |
|
Phase I and phase II trials of PD-1/PD-L1 inhibitors, either alone or in combination, have reported safety and also some promising efficacy outcomes, but larger studies with adequate statistical power are needed.
Biomarkers for targeted therapies are usually based on a genetic aberration in the target itself (e.g., BRAF mutations, ALK translocations). These are reported clinically as either positive or negative assay results. This is different from immune biomarkers, which may need to be expressed as a matrix of PD-L1 expression, TMB, and tumor-derived interferon-γ gene signatures. While some have suggested developing an immunogram (91) that includes all components of the immune system, identification of specific pathway drivers, such as TGFβ, myeloid biology, WNT/β-catenin, or altered tumor metabolism that requires complex approaches, including metabolic, biochemical, cellular and nucleic acid-based assays.
Capitalizing on the success of PD-1 blockade in other malignancies, new approaches will need to build on new insights in glioma immunobiology to develop the next generation of clinical trials to help improve the outcome of people suffering from this devastating disease in a meaningful way.
Acknowledgement:
The authors would like to than Sherilyn Goldstone for her editorial assistance and Kelly Hotchkiss for creating figure 1 with Biorender.com.
Conflicts of interests:
MK reports consultant or advisory roles for AbbVie, Ipsen, Pfizer Roche, and Jackson Laboratory for Genomic Medicine; research funding from AbbVie, Bristol-Myers Squibb (BMS), and Specialized Therapeutics.
MW has received research grants from AbbVie, Adastra, BMS, Dracen, Merck, Sharp & Dohme (MSD), Merck (EMD), Novocure, Piqur, and Roche; and honoraria for lectures or advisory board participation or consulting from AbbVie, Basilea, BMS, Celgene, MSD, EMD, Novocure, Orbus, Roche, and Tocagen.
DAR has received financial compensation for advisory board participation from AbbVie, Advantagene, Agenus, Amgen, Bayer, BMS, Celldex, DelMar, EMD Serono, Genentech/Roche, Inovio, Merck, Merck KGaA, Monteris, Novocure, Oncorus, Oxigene, Regeneron, Stemline, and Taiho Oncology, Inc. He has also received institutional research support from Acerta Phamaceuticals, Agenus, Celldex, EMD Serono, Incyte, Inovio, Midatech, Omniox, and Tragara.
JHS is a paid consultant for BMS and holds ownership interest (including patents) in Istari Oncology and Annias Therapeutics.
Funding sources: The authors are supported by the following National Institutes of Health (NIH) grants to Duke University: P50-CA190991 (SPORE; JHS, MK), P01-CA225622 (JHS, MK), U01-NS090284 (JHS), R01-NS099463 (JHS), R01-CA175517 (JHS), and R01-CA235612 (JHS, MK).
References:
- 1.Weller M, Van Den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. The lancet oncology 2017;18(6):e315–e29. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England journal of medicine 2005;352(10):987–96. [DOI] [PubMed] [Google Scholar]
- 3.Wen PY, Weller M, Lee EQ, Alexander BA, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro-Oncology 2020. doi 10.1093/neuonc/noaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, De Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. New England Journal of Medicine 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 5.Fecci PE, Sampson JH. The current state of immunotherapy for gliomas: an eye toward the future: JNSPG 75th Anniversary Invited Review Article. Journal of neurosurgery 2019;131(3):657–66. [DOI] [PubMed] [Google Scholar]
- 6.Reardon DA, Kaley TJ, Dietrich J, Lim M, Dunn GP, Gan HK, et al. Phase 2 study to evaluate the clinical efficacy and safety of MEDI4736 (durvalumab) in patients with glioblastoma (GBM). ASCO Meeting Abstracts 2016;34(15_suppl):TPS2080. [Google Scholar]
- 7.Ogawa K, Hirai M, Katsube T, Murayama M, Hamaguchi K, Shimakawa T, et al. Suppression of cellular immunity by surgical stress. Surgery 2000;127(3):329–36. [DOI] [PubMed] [Google Scholar]
- 8.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271(5256):1734–6. [DOI] [PubMed] [Google Scholar]
- 9.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of experimental medicine 2000;192(7):1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nduom EK, Wei J, Yaghi NK, Huang N, Kong L-Y, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol 2016;18(2):195–205 doi 10.1093/neuonc/nov172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ladomersky E, Zhai L, Lenzen A, Lauing KL, Qian J, Scholtens DM, et al. IDO1 inhibition synergizes with radiation and PD-1 blockade to durably increase survival against advanced glioblastoma. Clinical cancer research 2018;24(11):2559–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meropol NJ, Somer RA, Gutheil J, Pelley RJ, Modiano MR, Rowinsky EK, et al. Randomized phase I trial of recombinant human keratinocyte growth factor plus chemotherapy: potential role as mucosal protectant. Journal of Clinical Oncology 2003;21(8):1452–8. [DOI] [PubMed] [Google Scholar]
- 13.Levy J, Licini L, Haelterman E, Moris P, Lestrate P, Damaso S, et al. Safety and immunogenicity of an investigational 4-component Staphylococcus aureus vaccine with or without AS03B adjuvant: results of a randomized phase I trial. Human vaccines & immunotherapeutics 2015;11(3):620–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature 2017;541(7637):321–30. [DOI] [PubMed] [Google Scholar]
- 15.Brooks WH, Markesbery WR, Gupta GD, Roszman TL. Relationship of lymphocyte invasion and survival of brain tumor patients. Annals of neurology 1978;4(3):219–24 doi 10.1002/ana.410040305. [DOI] [PubMed] [Google Scholar]
- 16.Boker DK, Kalff R, Gullotta F, Weekes-Seifert S, Mohrer U. Mononuclear infiltrates in human intracranial tumors as a prognostic factor. Influence of preoperative steroid treatment. I. Glioblastoma. Clinical neuropathology 1984;3(4):143–7. [PubMed] [Google Scholar]
- 17.Palma L, Di Lorenzo N, Guidetti B. Lymphocytic infiltrates in primary glioblastomas and recidivous gliomas. Incidence, fate, and relevance to prognosis in 228 operated cases. J Neurosurg 1978;49(6):854–61 doi 10.3171/jns.1978.49.6.0854. [DOI] [PubMed] [Google Scholar]
- 18.Tan AC, Heimberger AB, Khasraw M. Immune checkpoint inhibitors in gliomas. Current oncology reports 2017;19(4):23. [DOI] [PubMed] [Google Scholar]
- 19.Han S, Zhang C, Li Q, Dong J, Liu Y, Huang Y, et al. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. British journal of cancer 2014;110(10):2560–8 doi 10.1038/bjc.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. Journal of translational medicine 2011;9:204 doi 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruffini E, Asioli S, Filosso PL, Lyberis P, Bruna MC, Macri L, et al. Clinical significance of tumor-infiltrating lymphocytes in lung neoplasms. The Annals of thoracic surgery 2009;87(2):365–71; discussion 71–2 doi 10.1016/j.athoracsur.2008.10.067. [DOI] [PubMed] [Google Scholar]
- 22.Perng P, Lim M. Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Frontiers in oncology 2015;5:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clinical Cancer Research 2018;24(17):4175–86 doi 10.1158/1078-0432.ccr-17-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer research 2006;66(6):3294–302. [DOI] [PubMed] [Google Scholar]
- 25.Yue Q, Zhang X, Ye H-x, Wang Y, Du Z-g, Yao Y, et al. The prognostic value of Foxp3+ tumor-infiltrating lymphocytes in patients with glioblastoma. Journal of neuro-oncology 2014;116(2):251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wainwright DA, Sengupta S, Han Y, Lesniak MS. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro-oncology 2011;13(12):1308–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suryadevara CM, Desai R, Abel ML, Riccione KA, Batich KA, Shen SH, et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology 2018;7(6):e1434464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clinical cancer research: an official journal of the American Association for Cancer Research 2008;14(16):5166–72 doi 10.1158/1078-0432.ccr-08-0320. [DOI] [PubMed] [Google Scholar]
- 29.Aptsiauri N, Ruiz-Cabello F, Garrido F. The transition from HLA-I positive to HLA-I negative primary tumors: the road to escape from T-cell responses. Current opinion in immunology 2018;51:123–32. [DOI] [PubMed] [Google Scholar]
- 30.Hegde PS, Chen DS. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020;52(1):17–35. [DOI] [PubMed] [Google Scholar]
- 31.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukas RV, Rodon J, Becker K, Wong ET, Shih K, Touat M, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. Journal of Neuro-Oncology 2018;140(2):317–28 doi 10.1007/s11060-018-2955-9. [DOI] [PubMed] [Google Scholar]
- 33.Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau H-T, Forero-Torres A, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Research and Treatment 2018;167(3):671–86 doi 10.1007/s10549-017-4537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansen A, Massard C, Ott P, Haas N, Lopez J, Ejadi S, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Annals of Oncology 2018;29(8):1807–13. [DOI] [PubMed] [Google Scholar]
- 35.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523(7559):231–5 doi 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 36.Sugiura A, Rathmell JC. Metabolic barriers to T cell function in tumors. The Journal of Immunology 2018;200(2):400–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011;35(6):871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miranda A, Hamilton PT, Zhang AW, Pattnaik S, Becht E, Mezheyeuski A, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proceedings of the National Academy of Sciences 2019;116(18):9020–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaur S, Chang T, Singh SP, Lim L, Mannan P, Garfield SH, et al. CD47 Signaling Regulates the Immunosuppressive Activity of VEGF in T Cells. The Journal of Immunology 2014;193(8):3914–24 doi 10.4049/jimmunol.1303116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nature Reviews Clinical Oncology 2018;15(5):325–40 doi 10.1038/nrclinonc.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuppner MC, Hamou MF, Bodmer S, Fontana A, De Tribolet N. The glioblastoma‐derived T‐cell suppressor factor/transforming growth factor beta2 inhibits the generation of lymphokineactivated killer (LAK) cells. International journal of cancer 1988;42(4):562–7. [DOI] [PubMed] [Google Scholar]
- 42.Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O, et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-oncology 2016;18(8):1146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khasraw M, Weller M, Estelles DL, Kolibaba K, Lee C, Gedye C, et al. ATIM-16. PHASE 1 STUDY RESULTS OF M7824 (MSB0011359C), A BIFUNCTIONAL FUSION PROTEIN TARGETING TGF-AND PD-L1, AMONG PATIENTS WITH RECURRENT GLIOBLASTOMA (rGBM). Neuro-Oncology 2018;20(suppl_6):vi4–vi. [Google Scholar]
- 44.Olnes MJ, Kotliarov Y, Biancotto A, Cheung F, Chen J, Shi R, et al. Effects of Systemically Administered Hydrocortisone on the Human Immunome. Scientific Reports 2016;6(1):23002 doi 10.1038/srep23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freeman-Keller M, Kim Y, Cronin H, Richards A, Gibney G, Weber JS. Nivolumab in Resected and Unresectable Metastatic Melanoma: Characteristics of Immune-Related Adverse Events and Association with Outcomes. Clinical Cancer Research 2016;22(4):886–94 doi 10.1158/1078-0432.ccr-15-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013;39(1):1–10. [DOI] [PubMed] [Google Scholar]
- 47.Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, et al. Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro-Oncology 2019;22(2):180–94 doi 10.1093/neuonc/noz212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nature Reviews Immunology 2018;18(4):225–42 doi 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- 49.Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016;1(2) doi 10.1172/jci.insight.85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bayik D, Zhou Y, Park C, Hong C, Vail D, Silver DJ, et al. Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discovery 2020:CD-19–1355 doi 10.1158/2159-8290.cd-19-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Himes BT, Peterson TE, de Mooij T, Garcia LMC, Jung M-Y, Uhm S, et al. The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro-Oncology 2020. doi 10.1093/neuonc/noaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aslan K, Turco V, Blobner J, Sonner JK, Liuzzi AR, Núñez NG, et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nature communications 2020;11(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol 2015;17(8):1064–75 doi 10.1093/neuonc/nou307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garber ST, Hashimoto Y, Weathers SP, Xiu J, Gatalica Z, Verhaak RG, et al. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol 2016. doi 10.1093/neuonc/now132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16(5):275–87 doi 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clinical cancer research: an official journal of the American Association for Cancer Research 2013;19(12):3165–75 doi 10.1158/1078-0432.ccr-12-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature medicine 2007;13(1):84–8. [DOI] [PubMed] [Google Scholar]
- 58.Champiat S, Ferté C, Lebel-Binay S, Eggermont A, Soria JC. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. Oncoimmunology 2014;3(1):e27817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. New England Journal of Medicine 2014;371(23):2189–99 doi doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome research 2014;24(5):743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schumacher Ton N, Kesmir C, van Buuren Marit M. Biomarkers in Cancer Immunotherapy. Cancer Cell 2015;27(1):12–4. [DOI] [PubMed] [Google Scholar]
- 62.Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proceedings of the National Academy of Sciences 2013;110(6):1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome research 2015;25(3):316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khasraw M, McDonald K, Yip S, Verhaak R, Heimberger A, Hall M, et al. RBTT-07. NUTMEG: A RANDOMISED PHASE II STUDY OF NIVOLUMAB AND TEMOZOLOMIDE (TMZ) VS TMZ ALONE IN ELDERLY PATIENTS WITH NEWLY DIAGNOSED GLIOBLASTOMA (GBM): TRIAL IN PROGRESS. Neuro-Oncology 2018;20(suppl_6):vi235–vi. [Google Scholar]
- 66.Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, et al. Association of polymerase e–mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA oncology 2015;1(9):1319–23. [DOI] [PubMed] [Google Scholar]
- 67.Network NCC. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Genetic/familial high-risk assessment: colorectal, Version 2.2015. 2016. [DOI] [PubMed]
- 68.Leung SY, Chan TL, Chung LP, Chan ASY, Fan YW, Hung KN, et al. Microsatellite Instability and Mutation of DNA Mismatch Repair Genes in Gliomas. The American Journal of Pathology 1998;153(4):1181–8 doi 10.1016/S0002-9440(10)65662-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prasad V, Kaestner V, Mailankody S. Cancer drugs approved based on biomarkers and not tumor type—FDA approval of pembrolizumab for mismatch repair-deficient solid cancers. JAMA oncology 2018;4(2):157–8. [DOI] [PubMed] [Google Scholar]
- 70.Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2016. doi 10.1200/jco.2016.66.6552. [DOI] [PubMed] [Google Scholar]
- 71.Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, et al. Loss of the Mismatch Repair Protein MSH6 in Human Glioblastomas Is Associated with Tumor Progression during Temozolomide Treatment. Clinical Cancer Research 2007;13(7):2038–45 doi 10.1158/1078-0432.ccr-06-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014;343(6167):189–93 doi 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee V, Murphy A, Le DT, Diaz LA Jr. Mismatch Repair Deficiency and Response to Immune Checkpoint Blockade. The oncologist 2016. doi 10.1634/theoncologist.2016-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020. doi 10.1038/s41586-020-2209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nature Genetics 2019;51(2):202–6 doi 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ahmad H, Fadul CE, Schiff D, Purow B. Checkpoint inhibitor failure in hypermutated and mismatch repair-mutated recurrent high-grade gliomas. Neuro-Oncology Practice 2019;6(6):424–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Theivanthiran B, Evans KS, DeVito NC, Plebanek MP, Sturdivant M, Wachsmuth LP, et al. A tumor-intrinsic PD-L1-NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. The Journal of Clinical Investigation 2020. doi 10.1172/JCI133055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mastorakos P, McGavern D. The anatomy and immunology of vasculature in the central nervous system. Science Immunology 2019;4(37):eaav0492 doi 10.1126/sciimmunol.aav0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pouliopoulos AN, Wu S-Y, Burgess MT, Karakatsani ME, Kamimura HA, Konofagou EE. A Clinical System for Non-invasive Blood–Brain Barrier Opening Using a Neuronavigation-Guided Single-Element Focused Ultrasound Transducer. Ultrasound in medicine & biology 2020;46(1):73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vogelbaum MA, Aghi MK. Convection-enhanced delivery for the treatment of glioblastoma. Neuro-oncology 2015;17(suppl_2):ii3–ii8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lemée J-M, Clavreul A, Menei P. Intratumoral heterogeneity in glioblastoma: don’t forget the peritumoral brain zone. Neuro-Oncology 2015;17(10):1322–32 doi 10.1093/neuonc/nov119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nature reviews Immunology 2012;12(9):623–35 doi 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 83.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015;523(7560):337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Song E, Mao T, Dong H, Boisserand LSB, Antila S, Bosenberg M, et al. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. New England Journal of Medicine 2018;379(8):722–30 doi 10.1056/NEJMoa1805453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taggart D, Andreou T, Scott KJ, Williams J, Rippaus N, Brownlie RJ, et al. Anti–PD-1/anti–CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8+ T cell trafficking. Proceedings of the National Academy of Sciences 2018;115(7):E1540–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gauci M-L, Lanoy E, Champiat S, Caramella C, Ammari S, Aspeslagh S, et al. Long-Term Survival in Patients Responding to Anti–PD-1/PD-L1 Therapy and Disease Outcome upon Treatment Discontinuation. Clinical Cancer Research 2019;25(3):946–56 doi 10.1158/1078-0432.ccr-18-0793. [DOI] [PubMed] [Google Scholar]
- 88.Okada H, Weller M, Huang R, Finocchiaro G, Gilbert MR, Wick W, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. The Lancet Oncology 2015;16(15):e534–e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nature Reviews Cancer 2020;20(1):12–25 doi 10.1038/s41568-019-0224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature 2019;574(7776):45–56 doi 10.1038/s41586-019-1593-5. [DOI] [PubMed] [Google Scholar]
- 91.Blank CU, Haanen JB, Ribas A, Schumacher TN. The “cancer immunogram”. Science 2016;352(6286):658–60. [DOI] [PubMed] [Google Scholar]
- 92.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nature medicine 2019;25(3):470–6. [DOI] [PubMed] [Google Scholar]
- 93.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. 2019;25(3):477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de Groot J, Penas-Prado M, Alfaro-Munoz KD, Hunter K, Pei B, O’brien B, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro-Oncology 2019. doi 10.1093/neuonc/noz185. [DOI] [PMC free article] [PubMed] [Google Scholar]