

Summary:
Acute kidney injury is a major contributor of chronic kidney disease development. The pathogenesis of acute kidney injury and chronic kidney disease shows significant similarities. Both conditions are associated with a defect in cellular metabolism, such as fatty acid oxidation and mitochondrial oxidative phosphorylation in kidney tubule cells and a marked increase in infiltrating immune cells. Here, we discuss how inflammatory cytokines and macrophages contribute to epithelial injury and metabolic defects. In addition, we discuss the role of mitochondrial damage and cytosolic leakage of the mitochondrial DNA activating the innate immune pathway such as cyclic guanosine monophosphate–adenosine monophosphate synthase/stimulator of interferon genes. The interplay between inflammation and metabolism appears to be critical for kidney disease development.
Keywords: cGAS-STING, kidney injury, inflammation, innate immune system, metabolic dysregulation
More than 30 million people in the United States suffer from chronic kidney disease (CKD). Of these, diabetic kidney disease (DKD) and hypertensive kidney disease account for close to 75% of cases. CKD is the 10th leading cause of death worldwide. Acute kidney injury (AKI) is differentiated from CKD based on its temporal course (because it occurs during a matter of days rather than months to years). Over the past 2 decades little progress has been made in reducing the burden of AKI and CKD. The lack of progress likely stems from our poor understanding of the mechanisms of kidney dysfunction.1,2
Although studies on the genetics of CKD and AKI have not yielded common genes, unbiased microarray-based gene expression analysis has indicated dysregulation of a few common pathways. These include the activation of immune cells and lower expression of genes governing cellular metabolism.3–5 Here, we briefly review observations indicating the activation of inflammatory pathways and the role of dysregulated metabolism in kidney disease. Finally, we discuss some of the recent findings that indicate an intimate connection between these two key pathways in kidney disease development.
DYSREGULATED METABOLISM IN CKD
The renal glomerulus is an approximately 60-kDa selective filter. This broad permeability requires long tubule segments to reclaim more than 99% of filtered small molecules, which is a highly energy-demanding process. To meet this high energy demand, the proximal and distal tubule epithelial cells almost exclusively rely on fatty acid oxidation (FAO) and are equipped with a large number of mitochondria to generate adenosine 5′-triphosphate (ATP) via oxidative phosphorylation.6–9 Gene expression studies performed on human kidney samples with varying degrees of kidney dysfunction and fibrosis have indicated a strong linear correlation between kidney function and expression of genes involved in FAO and mitochondrial oxidative phosphorylation. Similarly, animal model studies have shown a strong correlation between gene and protein expression of FAO and their transcriptional regulators such as peroxisome proliferator-activated receptor α (PPARA) and peroxisome proliferator-activated receptor γ coactivator 1-α (PPARGC1A) and kidney fibrosis.4 The causal role of FAO in AKI, CKD, and kidney fibrosis has been investigated extensively using genetic and pharmacologic approaches in rodents. For example, transgenic expression of PPARGC1A, one of the key transcriptional regulators of mitochondrial biogenesis and FAO, protects mice from ischemia-induced AKI, folic acid–induced AKI and CKD, and unilateral ureteral obstruction–induced kidney fibrosis.6,8 Genetic overexpression of PPARA in proximal tubule cells protects mice from ischemia or cisplatin-induced AKI or unilateral ureteral obstruction–induced kidney fibrosis.10,11 Pharmacologic activators of PPARA such as fenofibrate also effectively ameliorate kidney damage in AKI and CKD models. Furthermore, fenofibrate has been shown to reduce albuminuria and the rate of kidney function decline in patients with DKD, indicating the key role of these pathways in patients.12 Manipulation of upstream and downstream molecular pathways such as liver kinase B1 (also known as serine/threonine kinase 11), 5′ adenosine monophosphate-activated protein kinase, or carnitine palmitoyltransferase 1, which improve FAO, and the energetic capacity of proximal tubules, also have shown effectiveness in rodent kidney disease models.13,14 These studies strongly and consistently concluded that changes in tubule epithelial metabolism directly contribute to CKD and fibrosis development.13 Because tubule cells cannot readily switch to glycolysis, reduced FAO causes lower cellular ATP levels. The cellular energy deficit leads to a functional compromise, cellular dedifferentiation, and kidney disease development (Fig. 1). Consistent with the notion that FAO plays a key role in ATP generation and kidney tubule health, genetic deletion of estrogen-related receptor γ, a key transcription factor that regulates metabolism in kidney tubules, causes severe structural defect without leading to end-stage kidney failure.15 Surprising to these findings is that genetic deletion of PPARA, PPARG, farnesoid X receptor, liver X receptor, estrogen-related receptor alpha, or PPARGC1A does not lead to an observable renal phenotype in the absence of injury.8,16,17 These animals are more susceptible to kidney injury and develop more severe fibrosis after injury, supporting the key role of epithelial metabolism in kidney disease development. Nevertheless, it is still hard to reconcile the lack of phenotype in these knockout animals with the key role of these transcriptional regulators in kidney disease development and the strong protection observed in animals with transgenic expression of the same transcription factors. These observations clearly indicate the need for further studies to elucidate the relationship between kidney tubule cell metabolism and CKD, such as defining key compensatory pathways or underappreciated metabolic reserve in kidney tubules.
Figure 1.
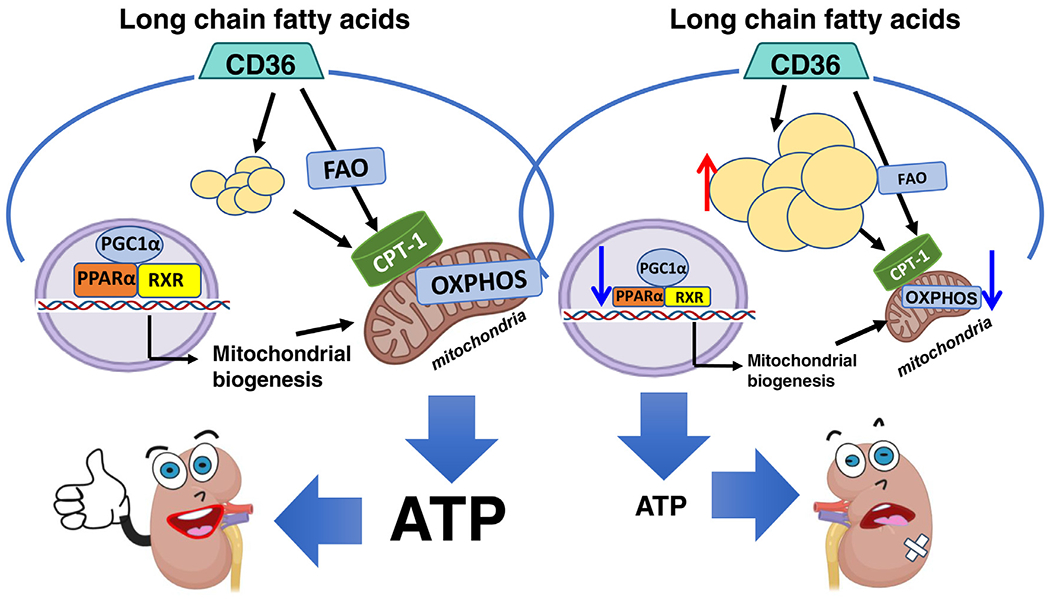
FAO and metabolism play a key role in tubule and kidney health. Proximal tubule epithelial cells have a high-energy need to transport filtered solutes. Kidney epithelial cells rely on FAO and mitochondrial oxidative phosphorylation to meet this high ATP demand. AKI and CKD are characterized by a defect in fatty acid oxidation and low ATP levels. This defect in metabolism contributes to epithelial dedifferentiation and renal failure. Abbreviations: CPT-1, carnitine palmitoyltransferase 1; OXPHOS, oxidative phosphorylation; PGC1α, proliferator-activated receptor γ co-activator 1α (i.e., PPARGC1A); RXR, retinoid X receptor.
INNATE IMMUNE SYSTEM ACTIVATION
Gene expression profiling of human kidney samples identified a strong and reproducible activation of immune and inflammatory pathways, such as the innate immune cells. The innate immune system is the dominant immune response found in primitive multicellular organisms. Natural killer cells, mast cells, eosinophils, basophils, and the phagocytic cells including macrophages, neutrophils, and dendritic cells are part of the innate immune system that identifies and eliminates pathogens. Innate immune cells act as a physical barrier, and release cytokines to recruit other lymphoid immune cells such as B and T cells to the site of infection to evoke adaptive immune response via antigen presentation. They also activate the complement cascade, which promotes clearance of antibody complexes and dead cells.
Janeway’s18,19 “infectious non-self” model proposed that antigen-presenting cells discriminate between self and nonself through germline-encoded pattern recognition receptors (PRRs) that recognized pathogen-associated molecular patterns on bacteria. The Danger Model, proposed by Matzinger,20 introduced the notion that the immune system recognizes and reacts to danger and damage rather than just to self and nonself. Thus, in the noninfectious setting, damage-associated molecular patterns (DAMPs) are released by various states of cellular injury, ischemia, or stress, and are recognized by PRRs, leading to a noninfectious inflammatory response.21 In the kidney, sterile inflammatory responses can be initiated when DAMPs are recognized by PRRs on various cells including renal tubular epithelial cells. In the context of AKI and CKD, DAMPs theoretically could arise from metabolic dysregulation, cellular stress, and any form of cellular injury.
MACROPHAGES
Macrophages are involved in the detection and phagocytosis of pathogens and cellular debris. They are found in essentially all tissues and play a diverse role in development, homeostasis, repair, and inflammation. The metabolic dysregulation and cellular injury present in chronic inflammatory states such as insulin resistance and diabetes provide signals to activate proinflammatory macrophages and perpetuate renal injury.
In the 1980s, Vlassara et al22,23 showed that hyperglycemia-associated advanced glycosylation end products are taken up and degraded by macrophages, which then release cytokines such as tumor necrosis factor α, interleukin 1β (IL1β), and IL6.22 Adoptive transfer experiments in rats have shown that transferred macrophages play a direct role in promoting mesangial cell proliferation and proteinuria.24 Irradiation of macrophages in streptozotocin-induced diabetic rats showed a decrease in mesangial area and type IV collagen messenger RNA.25 Both mouse and human studies have indicated that macrophage accumulation correlated positively with kidney disease and disease progression.26
In renal injury, macrophage infiltration is driven primarily by signals from DAMPs27 and chemokines such as monocyte chemoattractant protein 1 (MCP-1, also known as chemokine [C-C motif] ligand 2 [CCL2]).28 Human studies have shown that an increase in urinary CCL2 expression correlates with macrophage infiltration in the kidney.29 Blockade of CCL2 reduces macrophage infiltration and ameliorates kidney disease in rodent models and patients. CCL2 blockade has been achieved via use of Spiegelmers, which are mirror image L-ribose oligonucleotides that can block the transcription of target molecules. The Spiegelmer emapticap pegol (NOX-E36), which blocks CCL2, was shown to decrease albuminuria in diabetic patients in a phase IIa trial.30 However, these studies were not followed up by phase III clinical trials. Monocytes, macrophages, and podocytes also express the cognate receptor for CCL2 (CCR2).31 In mouse models, genetic deletion or pharmacologic blockade of CCR2 has been shown to decrease renal fibrosis in a variety of models.32 In a phase II, randomized, double-blind, clinical trial, the CCR2 antagonist CCX140-B was shown to reduce albuminuria when administered on top of standard of care.33 Another chemokine, CCL5, also known as Regulated upon Activation, Normal T cell Expressed, and Secreted also was implicated in the pathogenesis of renal disease.34 Polymorphisms in its receptor, CCR5 were shown to associate with better outcomes in type 2 diabetic subjects.35,36 A double antagonist of CCR2 and CCR5 also has been developed by Pfizer (Cambridge, MA) and tested in patients with DKD. The CCR2/CCR5 double-antagonist PF-04634817 had a modest effect on albuminuria in a phase II, randomized, double-blind, placebo-controlled trial that was halted early.37 Overall, animal models and phase II studies have indicated an important role for macrophages and CCL2 chemokines in kidney disease, it seems, however, the phase II human studies were not taken to the next level to conclusively show the role of these immune pathways in patients with kidney disease.
Macrophages often are characterized as classically activated, proinflammatory M1 macrophages and alternatively activated, anti-inflammatory M2 macrophages. The designation of M1 macrophages originates back to the discovery because they are activated by proinflammatory T-helper cell 1 cytokines such as interferon-γ and microbial signals such as lipopolysaccharide. M2 macrophages were found to be activated by T-helper cell 2 cytokines, IL4/IL13.38 In kidney injury models and DKD, macrophages have been shown to polarize toward the proinflammatory M1 phenotype (Fig. 2).39 The plastic nature of macrophages allows them to move back and forth between these proinflammatory and anti-inflammatory phenotypes depending on the external signals.
Figure 2.

Macrophages polarize toward a proinflammatory phenotype in kidney disease. Th1 cytokines and microbial signals such as lipopolysaccharide (LPS) promote macrophage polarization toward the proinflammatory M1 phenotype in kidney injury. M1 macrophages release proinflammatory cytokines leading to further kidney injury. Macrophages can be polarized toward the M2 phenotype; however, macrophages are plastic in nature and may move back and forth between proinflammatory and anti-inflammatory phenotype depending on external signals. Abbreviations: ACE-i, angiotensin-converting enzyme inhibitor; IFN-γ, interferon γ; TGF-β, transforming growth factor β; Th1, T-helper cell 1; TNF-a, tumor necrosis factor a; TZD, thiazolidinedione.
In addition to macrophage blockade, macrophage polarization also can be targeted pharmacologically. For instance, thiazolidinediones activates PPARG to promote M2 cell fate of macrophages.40 In in vitro studies, statins and angiotensin-converting enzyme inhibitors such as enalapril also promote M2 macrophage polarization.41,42 Understanding drivers of macrophage polarity is an active area of research and new methods such as single-cell sequencing will help to answer this important question.
THE COMPLEMENT SYSTEM
The complement system is a feature of the innate immune system that consists of plasma proteins and its activation opsonizes pathogens through phagocytosis, ultimately, leading to inflammatory responses to fight infection. Complement activation occurs via three pathways: the lectin pathway, the classic pathway, and the alternative pathway, all of which eventually lead to generation of the membrane attack complex (MAC). The kidney can synthesize most of the components of the complement pathway.43
Although the complement system activation mostly occurs at the proteolytic level, surprisingly we also could detect an increase in transcript levels of complement pathway components in diseased human kidney samples. Complement transcript levels strongly correlated with the degree of complement deposition observed in diabetic glomeruli observed in approximately half of all analyzed DKD cases.3 It is unclear whether the complement system activation is linked causally to disease development or progression.44 Complement C3 glomerular deposition can be observed in both type 1 and type 2 diabetic mouse models.45 Furthermore, plasma and urine complement components are increased in patients with biopsy-proven diabetic nephropathy.45 Urinary complement components were shown to be increased in patients with diabetic nephropathy at levels comparable with patients with autoimmune glomerulonephritis and correlated positively with degree of proteinuria.46 It is believed that abnormal glycation of CD59 explains the increase in MAC activation in diabetes. Nonglycated CD59 has an inhibitory role on the MAC.47,48 Increase in glycation of CD59 in the setting of diabetes appears to lose its regulatory function, which leads to MAC-induced growth factor release from endothelial cells with downstream proinflammatory effects.47,48
Complement-directed therapeutics have been clinically approved for rare complement-associated kidney diseases such as hemolytic uremic syndrome. Eculizumab is a monoclonal antibody directed against C5, which ultimately prevents generation of C5a and the MAC. Use of complement-targeted therapies in DKD has gained significant popularity and hopefully will be tested in clinical trials. Animal model studies support the role of complements in CKD, DKD, and kidney fibrosis. Previously, it was shown that inhibition of complement activation in a rat model by preventing complement C5 activation decreased mesangial expansion and albuminuria. A more recent study showed that C3a-receptor blockade reduced renal fibrosis, progression of renal disease, and albuminuria.49
Activation of the adaptive immune system through a process known as antigen presentation takes more time and creates a long-lasting immune response against a specific pathogen. The key players, such as B and T lymphocytes, are remarkably diverse, highly specific, and able to recognize self from nonself. We are at early stages of characterizing the role of lymphocytes in kidney disease development. The massive diversity of these cells and significant differences between murine and human lymphocytes is a critical limitation to define the role of these cells in patient samples.
INFLAMMATION AND METABOLIC DYSREGULATION
There is a critical interplay between metabolism and inflammation, which probably is best characterized in fat tissues. Obesity is characterized as a state of low-level inflammation. This inflammatory milieu has been proposed to be an important contributor of metabolic dysregulation in the context of diabetes. Seminal reports brought to light a new understanding of the role of adipose inflammation, particularly macrophage-mediated inflammation, in obesity.50 These articles showed that macrophages infiltrate adipose tissue at the onset of weight gain and directly contribute to and perpetuate the inflammatory state of fat, eventually leading to critical changes in metabolism such as systemic insulin resistance and obesity in mouse models and human beings.51 Macrophages play diverse roles in metabolic reprogramming of fat cells, from lipid buffering to insulin resistance.52,53 Although the role of adipose tissue macrophages in metabolic dysregulation of fat tissue is fairly well established, the increase in macrophage number and inflammatory cytokines could play a similar role in altering epithelial metabolism of kidney epithelial cells as well.
Recently, a different relationship between inflammation and metabolism has been described. FAO and oxidative phosphorylation genes were decreased notably in kidneys of patients with DKD and hypertensive CKD.3,4 Mitochondrially encoded genes showed a stronger correlation with kidney fibrosis than nuclear-encoded mitochondrial genes.54 The inverse relationship between metabolic gene expression and fibrosis was recapitulated in mouse kidney disease models. To model the metabolic dysregulation observed in animal models and patients with CKD, the mitochondrial transcription factor (Tfam) was deleted from kidney tubule cells during late stages of kidney development. Because Tfam is the key transcriptional regulator in the mitochondria, it was expected that the loss of mitochondria would lead to compromised bioenergetics in the tubules, causing epithelial dedifferentiation and fibrosis. Surprisingly, animals appeared normal at birth and died at approximately 12 weeks of age. Cellular ATP levels were decreased by 6 weeks of age and mitochondria appeared dysmorphic. Despite the massive metabolic dysregulation, significant kidney fibrosis development coincided with an increase in cytokine expression and the influx of innate immune cells such as macrophages.54 These results indicated the key role of inflammation in fibrosis development and the direct role of mitochondrial alterations inducing the inflammatory environment and fibrosis (Fig. 3). In vitro studies indicated that deletion of Tfam in culture tubule cells lead to the leakage of mitochondrial DNA into the cytoplasm. Because the mitochondrial DNA is an ancient bacterial (circular) DNA, it is recognized as foreign DNA by the cellular defense pathways in the cytosol of epithelial cells. One such defense pathway is cyclic guanosine monophosphate–adenosine monophosphate synthase, which recognizes cytosolic double-stranded DNA in cytosol. Activated cyclic guanosine monophosphate–adenosine monophosphate synthase converts ATP and guanosine 5′-triphosphate into cyclic guanosine monophosphate–5′ adenosine monophosphage, which functions as a secondary messenger that binds and activates stimulator of interferon genes (STING), an adapter protein on the endoplasmic reticulum membrane.55 STING then triggers a signaling cascade, which leads to production of a battery of immune and inflammatory mediators, including type I and type II interferons.56 In the context of kidney disease, STING resulted in an early but short activation of interferon response but later showed sustained nuclear factor-κB activation, leading to the release of proinflammatory cytokines. This strong proinflammatory immune response resulted in an influx of macrophages and other immune cells leading to kidney fibrosis and dysfunction development. To show the role of STING activation, double-knockout mice were generated by deleting STING in Tfam knockout mice.54 These double-knockout mice had improved survival and significant improvement in their kidney function.
Figure 3.

Mitochondrial defects contribute to immune system activation and renal failure. Mitochondrial defects, such as loss of mitochondrial transcription factor (Tfam), leads to cytosolic release of mitochondrial DNA (mtDNA). Cytosolic DNA is recognized by the cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS)/stimulator of interferon genes (STING) pathway, leading to the activation of nuclear factor-κB (NF-κb), resulting in cytokine release, cell death, influx of immune cells, and, ultimately, renal failure. Abbreviations: OXPHOS, oxidative phosphoryation; TFAM, mitochondrial transcription factor.
Several new small molecular inhibitors have been developed targeting the STING pathway. Administration of pharmacologic inhibitors of STING was associated with renal and survival benefit in the Tfam knockout mice. Finally, to demonstrate a more general role for STING in CKD and fibrosis development, the authors also have shown lesser fibrosis in STING knockout mice in the folic acid injection–induced kidney fibrosis model. Pharmacologic inhibition of STING also showed a partial benefit in the folic acid–induced kidney fibrosis model. Overall these studies provide a strong rationale for targeting STING in the context of kidney disease.54 Although excellent pharmacologic inhibitors have been developed for STING, based on its critical role in innate immunity, such as type I interferon production upon intracellular pathogen infection (ie, by viruses, mycobacteria, and intracellular parasites), the safety of such molecules would need to be characterized in patients. Overall, these are interesting observations indicating the critical role of metabolic dysregulation as a key driver of inflammation and downstream kidney disease development and provide new avenues for therapeutic targeting.
CONCLUSIONS
In conclusion, AKI is a key contributor of CKD. Multiple molecular pathways are shared between AKI and CKD. Expression profiling studies of human kidney samples and animal models highlighted the activation of inflammation and immune cells and the decrease in expression of metabolic genes. Kidney tubule cells have high metabolic needs and keenly rely on fatty acid oxidation for energy generation. Reducing inflammation was associated with significant renal benefit in animal studies and in early clinical trials. Recent reports also have indicated that the mitochondrial and metabolic defect observed in tubule epithelial cells contributes to the leakage of mitochondrial DNA into the cytoplasm, which is recognized as foreign DNA by the cytosolic DNA-sensing pathway and contributed to the release of inflammatory and profibrotic cytokines and, ultimately, kidney disease development. There is a strong interplay between metabolism and inflammation, and it seems that targeting one of them also will have a significant effect on the other, resulting in disease development.
Acknowledgments
Financial support: Supported by research grant 5T32DK007006-45 from the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (G.Z.Q.). Work in Katalin Susztak’s laboratory is supported by the Juvenile Diabetes Research organization and by the National Institutes of Health grants R01 DK076077, R01 DK087635, and DP3 DK108220.
Footnotes
Conflict of interest statement: Work performed in the Susztak laboratory is supported by Gilead, Regeneron, GSK, Boehringer Ingelheim, and Bayer.
REFERENCES
- 1.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124:2333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breyer MD, Susztak K. The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov. 2016;15:568–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60:2354–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beckerman P, Qiu C, Park J, Ledo N, Ko YA, Park AD, et al. Human kidney tubule-specific gene expression based dissection of chronic kidney disease traits. EBioMedicine. 2017;24:267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ledo N, Ko YA, Park AS, Kang HM, Han SY, Choi P, et al. Functional genomic annotation of genetic risk loci highlights inflammation and epithelial biology networks in CKD. J Am Soc Nephrol. 2015;26:692–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran M, Parikh SM. Mitochondrial biogenesis in the acutely injured kidney. Nephron Clin Pract. 2014;127:42–5. [DOI] [PubMed] [Google Scholar]
- 8.Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121:4003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531:528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lakhia R, Yheskel M, Flaten A, Quittner-Strom EB, Holland WL, Patel V. PPARalpha agonist fenofibrate enhances fatty acid beta-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol. 2018;314:F122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou X, Shen YH, Li C, Wang F, Zhang C, Bu P, et al. PPARalpha agonist fenofibrate protects the kidney from hypertensive injury in spontaneously hypertensive rats via inhibition of oxidative stress and MAPK activity. Biochem Biophys Res Commun. 2010;394:653–9. [DOI] [PubMed] [Google Scholar]
- 12.Frazier R, Mehta R, Cai X, Lee J, Napoli S, Craven T, et al. Associations of fenofibrate therapy with incidence and progression of CKD in patients with type 2 diabetes. Kidney Int Rep. 2019;4:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han SH, Wu MY, Nam BY, Park JT, Yoo TH, Kang SW, et al. PGC-1alpha protects from notch-induced kidney fibrosis development. J Am Soc Nephrol. 2017;28:3312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han SH, Malaga-Dieguez L, Chinga F, Kang HM, Tao J, Reidy K, et al. Deletion of Lkb1 in renal tubular epithelial cells leads to CKD by altering metabolism. J Am Soc Nephrol. 2016;27:439–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Lupino K, Wilkins BJ, Qiu C, Liu J, Omura Y, et al. Genomic integration of ERRgamma-HNF1beta regulates renal bioenergetics and prevents chronic kidney disease. Proc Natl Acad Sci USA. 2018;115:E4910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guan Y, Breyer MD. Peroxisome proliferator-activated receptors (PPARs): novel therapeutic targets in renal disease. Kidney Int. 2001;60:14–30. [DOI] [PubMed] [Google Scholar]
- 17.Guan Y, Zhang Y, Schneider A, Davis L, Breyer RM, Breyer MD. Peroxisome proliferator-activated receptor-gamma activity is associated with renal microvasculature. Am J Physiol Renal Physiol. 2001;281:F1036–46. [DOI] [PubMed] [Google Scholar]
- 18.Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. [DOI] [PubMed] [Google Scholar]
- 19.Janeway CA Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13:11–6. [DOI] [PubMed] [Google Scholar]
- 20.Matzinger P The danger model: a renewed sense of self. Science. 2002;296:301–5. [DOI] [PubMed] [Google Scholar]
- 21.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–78. [DOI] [PubMed] [Google Scholar]
- 22.Vlassara H, Brownlee M, Cerami A. Macrophage receptor-mediated processing and regulation of advanced glycosylation end-product (AGE)-modified proteins: role in diabetes and aging. Prog Clin Biol Res. 1989;304:205–18. [PubMed] [Google Scholar]
- 23.Vlassara H, Li YM, Imani F, Wojciechowicz D, Yang Z, Liu FT, et al. Identification of galectin-3 as a high-affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol Med. 1995;1:634–46. [PMC free article] [PubMed] [Google Scholar]
- 24.Ma FY, Ikezumi Y, Nikolic-Paterson DJ. Macrophage signaling pathways: a novel target in renal disease. Semin Nephrol. 2010;30:334–44. [DOI] [PubMed] [Google Scholar]
- 25.Sassy-Prigent C, Heudes D, Mandet C, Belair MF, Michel O, Perdereau B, et al. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes. 2000;49:466–75. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen D, Ping F, Mu W, Hill P, Atkins RC, Chadban SJ. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton). 2006;11:226–31. [DOI] [PubMed] [Google Scholar]
- 27.Rosin DL, Okusa MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol. 2011;22:416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cassini MF, Kakade VR, Kurtz E, Sulkowski P, Glazer P, Torres R, et al. Mcp1 promotes macrophage-dependent cyst expansion in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2018;29:2471–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wada T, Furuichi K, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, et al. A new anti-inflammatory compound, FR167653, ameliorates crescentic glomerulonephritis in Wistar-Kyoto rats. J Am Soc Nephrol. 2000;11:1534–41. [DOI] [PubMed] [Google Scholar]
- 30.Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant. 2017;32:307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tarabra E, Giunti S, Barutta F, Salvidio G, Burt D, Deferrari G, et al. Effect of the monocyte chemoattractant protein-1/CC chemokine receptor 2 system on nephrin expression in streptozotocin-treated mice and human cultured podocytes. Diabetes. 2009;58:2109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitagawa K, Wada T, Furuichi K, Hashimoto H, Ishiwata Y, Asano M, et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol. 2004;165:237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol. 2015;3: 687–96. [DOI] [PubMed] [Google Scholar]
- 34.Anders HJ, Belemezova E, Eis V, Segerer S, Vielhauer V, Perez de Lema G, et al. Late onset of treatment with a chemokine receptor CCR1 antagonist prevents progression of lupus nephritis in MRL-Fas(lpr) mice. J Am Soc Nephrol. 2004;15:1504–13. [DOI] [PubMed] [Google Scholar]
- 35.Muntinghe FL, Gross S, Bakker SJ, Landman GW, van der Harst P, Bilo HJ, et al. CCR5Delta32 genotype is associated with outcome in type 2 diabetes mellitus. Diabetes Res Clin Pract. 2009;86:140–5. [DOI] [PubMed] [Google Scholar]
- 36.Muntinghe FL, Verduijn M, Zuurman MW, Grootendorst DC, Carrero JJ, Qureshi AR, et al. CCR5 deletion protects against inflammation-associated mortality in dialysis patients. J Am Soc Nephrol. 2009;20:1641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gale JD, Gilbert S, Blumenthal S, Elliott T, Pergola PE, Goteti K, et al. Effect of PF-04634817, an oral CCR2/5 chemokine receptor antagonist, on albuminuria in adults with overt diabetic nephropathy. Kidney Int Rep. 2018;3:1316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gordon S Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. [DOI] [PubMed] [Google Scholar]
- 39.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22:317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stienstra R, Duval C, Keshtkar S, van der Laak J, Kersten S, Muller M. Peroxisome proliferator-activated receptor gamma activation promotes infiltration of alternatively activated macrophages into adipose tissue. J Biol Chem. 2008;283:22620–7. [DOI] [PubMed] [Google Scholar]
- 41.Fujita E, Shimizu A, Masuda Y, Kuwahara N, Arai T, Nagasaka S, et al. Statin attenuates experimental anti-glomerular basement membrane glomerulonephritis together with the augmentation of alternatively activated macrophages. Am J Pathol. 2010;177: 1143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cucak H, Nielsen Fink L, Hojgaard Pedersen M, Rosendahl A. Enalapril treatment increases T cell number and promotes polarization towards M1-like macrophages locally in diabetic nephropathy. Int Immunopharmacol. 2015;25:30–42. [DOI] [PubMed] [Google Scholar]
- 43.Chen SF, Chen M. Complement activation in progression of chronic kidney disease. Adv Exp Med Biol. 2019;1165:423–41. [DOI] [PubMed] [Google Scholar]
- 44.Niewczas MA, Pavkov ME, Skupien J, Smiles A, Md Dom ZI, Wilson JM, et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat Med. 2019;25:805–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly KJ, Liu Y, Zhang J, Dominguez JH. Renal C3 complement component: feed forward to diabetic kidney disease. Am J Nephrol. 2015;41:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelletier K, Bonnefoy A, Chapdelaine H, Pichette V, Lejars M, Madore F, et al. Clinical value of complement activation biomarkers in overt diabetic nephropathy. Kidney Int Rep. 2019; 4:797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qian YM, Qin X, Miwa T, Sun X, Halperin JA, Song WC. Identification and functional characterization of a new gene encoding the mouse terminal complement inhibitor CD59. J Immunol. 2000;165:2528–34. [DOI] [PubMed] [Google Scholar]
- 48.Qin X, Goldfine A, Krumrei N, Grubissich L, Acosta J, Chorev M, et al. Glycation inactivation of the complement regulatory protein CD59: a possible role in the pathogenesis of the vascular complications of human diabetes. Diabetes. 2004;53:2653–61. [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Wang K, Liang X, Li Y, Zhang Y, Zhang C, et al. Complement C3 produced by macrophages promotes renal fibrosis via IL-17A secretion. Front Immunol. 2018;9:2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeyda M, Stulnig TM. Adipose tissue macrophages. Immunol Lett. 2007;112:61–7. [DOI] [PubMed] [Google Scholar]
- 51.Bouloumie A, Casteilla L, Lafontan M. Adipose tissue lymphocytes and macrophages in obesity and insulin resistance: makers or markers, and which comes first? Arterioscler Thromb Vasc Biol. 2008;28:1211–3. [DOI] [PubMed] [Google Scholar]
- 52.Furuhashi M, Fucho R, Gorgun CZ, Tuncman G, Cao H, Hotamisligil GS. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest. 2008;118:2640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. 2008;14:1225–30. [DOI] [PubMed] [Google Scholar]
- 54.Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 2019;30: 784–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bai J, Liu F. The cGAS-cGAMP-STING pathway: a molecular link between immunity and metabolism. Diabetes. 2019;68:1099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657–74. [DOI] [PubMed] [Google Scholar]