

Abstract
The highly pathogenic avian H5N1 influenza viruses have been recognized as a potential pandemic threat to humans, and to the poultry industry since 1997. H5 viruses consist of a high mutation rate, so universal vaccine designing is very challenging. Here, we describe a vaccinomics approach to design a novel multi-epitope influenza vaccine, based on the highly conserved regions of surface glycoprotein, Hemagglutinin (HA). Initially, the HA protein sequences from Bangladeshi origin were retrieved and aligned by ClustalW. The sequences of 100% conserved regions extracted and analyzed to select the highest potential T-cell and B-cell epitope. The HTL and CTL analyses using IEDB tools showed that DVWTYNAELLVLMEN possesses the highest affinity with MHC class I and II alleles, and it has the highest population coverage. The docking simulation study suggests that this epitope has the potential to interact with both MHC class I and MHC class II. The B-cell epitope prediction provides a potential peptide, GAIAGFIEGGWQGM. We further retrieved HA sequences of 3950 avian and 250 human H5 isolates from several populations of the world, where H5 was an epidemic. Surprisingly, these epitopes are more than 98% conserved in those regions which indicate their potentiality as a conserved vaccine. We have proposed a multi-epitope vaccine using these sequences and assess its stability and potentiality to induce B-cell immunity. In vivo study is necessary to corroborate this epitope as a vaccine, however, setting forth groundwork for wet-lab studies essential to mitigate pandemic threats and provide cross-protection of both avian and humans against H5 influenza viruses.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-02544-3) contains supplementary material, which is available to authorized users.
Keywords: Peptide vaccine, H5N1, Pandemic, Hemagglutinin, Epitope, Immunoinformatics
Introduction
Influenza virus A is a virus of the Orthomyxoviridae family (Presti et al. 2009). Influenza virus causes an acute respiratory disease and continually circulates and changes in several animal hosts, including wild birds, poultry, pigs, horses, and humans. Their genotypes are based on the nature of their surface glycoproteins, Hemagglutinin (HA), and Nuraminidase (NA) (Lamb 2001). There are 18 different HAs and 11 NAs which are serologically distinguishable, in which antibodies to one virus genotype do not react with another.
Influenza H5 viruses are avian viruses, hosted by birds, but may infect several species of mammals. Among nine known subtypes of H5 viruses (H5N1, H5N2, H5N3, H5N4, H5N5, H5N6, H5N7, H5N8, and H5N9), LPAI (Low Pathogenic Avian Influenza) are the most identified worldwide in wild birds and poultry, but occasionally HPAI (Highly Pathogenic Avian Influenza) viruses are also detected. The first reported case of human illness with a highly pathogenic H5N1 virus infection occurred in May 1997 in Hong Kong (Yuen et al. 1998). They spread widely throughout the countries of Asia, the Middle East, Africa, and Europe, and evolved rapidly into 10 distinct clades (0–9). H5N1 viruses are recognized as threats to humans, wild birds, and poultry in Bangladesh since 2007. H5N1 clade 2.2.2 circulated from 2007 to 2011, while clade 2.3.2.1a circulated from 2011 to the present (Marinova-Petkova et al. 2015). The virulence of H5 viruses in poultry and humans has raised concerns about their potential to cause an influenza pandemic. In a yearly outbreak, influenza spreads around the world, resulting in about 3–5 million human cases of severe illness and about 250,000 to 500,000 deaths (WHO 2017). A total of 858 human cases and 453 deaths worldwide from avian influenza A virus subtype, H5N1 were reported by WHO from 2003–2017. From them, the same subtype of H5N1 infection caused eight human cases and one death in Bangladesh (WHO 2019). H5 viruses are endemic in Bangladesh and have one of the highest numbers of reported outbreaks in poultry (OIE 2013).
Current therapeutic options for the treatment of influenza virus infections are limited (Fry et al. 2015). Vaccination is the most effective way to prevent infection and severe outcomes of influenza H5 viruses, but they possess a high mutation rate. Current vaccine formulations predominantly rely on eliciting neutralizing antibodies that target the highly variable head domain (HA1 subunit) of the surface-expressed viral glycoprotein hemagglutinin (Vergara-Alert et al. 2012). They are generally exclusively effective against infectious influenza viral populations that match the vaccine strain (Wiersma et al. 2015; Sridhar et al. 2015). Therefore, a particular influenza vaccine usually confers protection for no more than a few years. These vaccine types are strain-specific, and their efficacy relies heavily on the inclusion of antigens (viruses or their proteins) or transfection of the cell with plasmid DNA. As a result of antigenic drift or shift, frequent changes occurred in influenza viruses, limit protection due to the low correlation between the vaccine antigens, and the current circulating wild-type influenza virus. Commercially available strain-specific vaccines show a relatively reduced clinical efficacy due to poor matching between the vaccine and circulating strains (Manzoli et al. 2007; Demicheli et al. 2018). Therefore, the development of effective vaccines against pandemic H5 viruses is challenging. These cumulative limitations are the driving force for the development of novel vaccines based on the viral peptide, which omits the use of HPAI H5 viruses.
Most recently, one approach that focuses on the identification of specific epitopes expressed by infectious pathogens has significantly advanced the development of peptide-based vaccines. Improved understanding of the molecular basis of antigen recognition and Major Histocompatibility Complex (MHC)-binding motifs has resulted in the development of rationally designed vaccines (Petrovsky and Brusic 2002). In various clinical studies, peptide-based vaccines demonstrated their effectiveness against various infectious diseases, including malaria (Kashala et al. 2002), hepatitis B (Engler et al. 2001), HIV (Gahery et al. 2006), multiple sclerosis (Bourdette et al. 2005), and tuberculosis (Robinson and Amara 2005). A CTL response to epitope-based vaccines is MHC molecule dependent. A vaccine intended for a broad human and avian population should include T-cell epitope(s) that will induce responses in the vast majority of the avian and human population. This can be achieved by selecting several T-cell epitopes that are specific to the prevalent MHC genotypes in that population (Keogh et al. 2001).
The surface glycoprotein of H5 influenza viruses, HA is an attractive target for vaccine development as neutralizing antibodies produced against HA during protection in humans (Gerhard 2001; Padilla-Quirarte et al. 2019). The mature HA has a globular head domain known as HA1 responsible for receptor binding, whereas the stem domain predominantly comprises the HA2 subunit. To create a universal vaccine, which would obviate a yearly vaccine, the highly conserved stalk domain of HA, HA2 subunit has been shown to be a potential region (Wright et al. 2007; Bridges et al. 2000). A novel approach, multi-epitope-based vaccine represents inducing specific cellular immunity and highly potent neutralizing antibodies (Vakili et al. 2019). A previous study showed that a multi-epitope vaccine protects against H5 influenza viruses (Hassan et al. 2017).
Herein, we shall describe a vaccinomics approach to design a novel multi-epitope influenza vaccine, based on a conserved region of the HA protein. The major aim of this study was to design a peptide vaccine specific for H5 viruses in Bangladesh. We have also assessed the potentiality of the Bangladesh specific epitope as a vaccine in the other regions of the world where they are endemic.
Methods
The methodology of the entire study has been described in a flow diagram (Fig. 1).
Fig. 1.

Flow diagram of the methodology
Sequence retrieval and multiple sequence alignment
The hemagglutinin protein sequences of different strains of influenza H5 viruses were retrieved from the Influenza Research Database (IRD) (Squires et al. 2012). Initially, HA protein sequences isolated from Bangladeshi human, avian (duck, mallard, chicken, quail, and migratory bird) and environment representative to influenza H5N1, H5N2, H5N6 viruses were analyzed to search conserved regions within HA proteins. All the HA protein sequences aligned by multiple sequence alignment program ClustalW using the BioEdit 7.2 program with a number of bootstraps value 1000 (Hall et al. 2011).
T-cell epitope prediction and affinity with MHC
CTL-epitope prediction is very important as rational vaccine design requires selective and specific targets that can deflate the volume of the experimental work. The epitope prediction from the respective conserved sequences and their affinity score with MHC class I and class II allele was calculated following the previously used approaches (Oany et al. 2017; Hossain et al. 2018). Concisely, the potential cytotoxic T-lymphocyte (CTL) epitopes predicted by the NetCTL v1.2 server (Larsen et al. 2007) utilizing the conserved sequences of the protein. The most antigenic T-cell epitope was selected depending on the combined score of the algorithms for MHC class I binding, the transporter of antigenic peptide (TAP) transport efficiency, and proteasomal C-terminal cleavage prediction. We have selected the top epitope for each HLA supertypes. The threshold level was set at 0.75 to find the epitopes for 12 MHC class I supertypes.
T-cell epitope prediction tools from Immune Epitope Database and Analysis Resource (IEDB-AR) were also used to predict the affinity with MHC class I (Buus et al. 2003) and MHC class II (Wang et al. 2008, 2010a). The stabilized matrix method (SMM) was utilized to measure the half-maximal inhibitory concentration (IC50) of the preselected 9.0-mer epitope binding to MHC class I. We selected the IEDB recommended method while analyzing affinity with the specific HLA-DP, HLA-DQ, and HLA-DR loci of MHC class II. For MHC class II binding analysis, Fifteen-mer epitopes were designed depending on the preselected 9-mer epitope considering the most conserved region in the influenza strains. The epitopes consisting of IC50 < 250 nM for the MHC class I and IC50 < 100 nM for MHC class II alleles, respectively, were selected for further analysis. The MHC class I binding was crosschecked by the software EPISOPT. We also utilized PREDIVAC, the MHC class II binding prediction tool to evaluate their affinity with HLA_DRB_1.
Population coverage analysis
Population coverage was evaluated by the IEDB population coverage calculation tool for each epitope (Bui et al. 2006). We selected area_country_ethnicity for the query, and the combined score for MHC classes I and II was used to determine the population coverage of the whole world population as well as an individual country.
Homology modeling and structural frustration analysis
A homology model of the hemagglutinin protein built by MODELLER v9 (Sali et al. 1995) and the simulated model evaluated by the PROCHECK server (Laskowski et al. 1996). The disordered region in the protein sequences measured by DISOPRED v3 (Ward et al. 2004). The protein frustratometer server (Jenik et al. 2012) exploited to detect the stability and energy differences in the 3D structure of the protein. The prediction of the transmembrane region was performed by TMHMM and TMpred.
Molecular docking analysis and HLA allele interaction
CABSDOCK WEB SERVER [http://biocomp.chem.uw.edu.pl/CABSdock] employed to perform molecular docking studies using the best possible epitope obtained from the above analyses. We have picked the HLA-B*15:01 molecules as a candidate for MHC class I, and the HLA-DRB01*01:01 as a candidate for MHC class II to dock with the selected epitope. The docking of HLA-DRB1*01:01 (PDB structure 1FYT) and HLA-B*15:01 (PDB structure- 1XR8) with naturally bound peptides was used as a control docking model for MHC class I and MHC class II, respectively. We selected the crystal structure, 1XR8 that included the HLA-B*1501 in Complex with Peptides from Human UbcH6 and Epstein–Barr virus EBNA-3 from the Protein Data Bank (PDB) database. On the other hand, as a candidate for MHC class II, the crystal structure 1FYT was selected that comprised a complex of a human alpha/beta-t-cell receptor, influenza HA antigen peptide, and HLA-DR1 (Berman et al. 2000). Then, the structure of MHC class I and MHC class II molecule was extracted using PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC) for the final docking purpose. The 9-mer epitope and the 15-mer epitope with their MHC class I and II molecule, respectively, submitted to CABS-dock with 50 simulation cycles. The docked peptides in the binding groove of HLAs with the lowest RMSD value were selected. The CABS-dock server stipulates a docking simulation of the binding site that allows full flexibility of the peptide and occasionally allows minor fluctuations of the receptor backbone. It also validates the model stability by calculating the RMSD value and cluster density.
B-cell epitope prediction
The prediction of linear B-cell epitopes from the conserved regions was performed using the ABCPred server (https://webs.iiitd.edu.in/raghava/ abcpred/ABC_submission.html).
Conservancy analyses
This study covers influenza viruses isolated from different countries which are repeatedly reported H5 virus outbreaks in avian, and some cases in humans, like, China, Viet Nam, Cambodia, Bangladesh, Thailand, India, Indonesia, Egypt, USA, etc. IEDB conservancy analysis tool was employed to measure the conservancy of all the anticipated epitopes in this study.
Designing of multi-epitope vaccine and analyses of its properties
We selected the best epitope from the T-cell and B-cell epitopes depending on the affinity with MHC class I and II alleles, population coverage, and conservancy. The epitopes are linked by the AAY linker and a Cysteine residue set at the N-terminal of the epitope. The cysteine residue will be required to conjugate the peptide with the carrier protein via a disulfide linkage. The ProtParam server was utilized to evaluate various physiochemical parameters of the multi-epitope vaccine, e.g., molecular weight, theoretical isoelectric point (pI), in vitro, and in vivo half-life, instability and aliphatic index, and grand average of hydropathicity (GRAVY).
Modeling, refinement, and validation of Vaccine-structure
The secondary structural properties of the multi-epitope vaccine construct assessed by the SOPMA server. The 3D model of the vaccine was constructed using the homology modeling tool—iTASSER (Yang et al. 2015). The model is refined using the Galaxy Refine server (http://galaxy.seoklab.org/). This server performs the repacking and molecular dynamics simulation to relax the structure, a CASP10-based refinement technique. The tertiary structures of the vaccines were validated using ProSA-web (Wiederstein and Sippl 2007). The server evaluated the overall quality of the model in the form of a z score. If the z scores of the predicted model are outside the range of the characteristic for native proteins, it indicates the erroneous structure. Furthermore, the Ramachandran plot analysis of the predicted model was performed using the PROCHECK server (https://servicesn.mbi.ucla.edu/PROCHECK/) to determine its overall quality.
Allergenicity investigation and b-cell epitope prediction
The allergenicity of the proposed vaccine was assessed by the Allertop server [https://www.ddg-pharmfac.net/AllerTOP/], where the support vector machine (SVM) algorithm-based analyses were utilized (Liao and Noble 2003). We have also checked the allergenicity of the peptides by the online server-based tool AlgPred (https://webs.iiitd.edu.in/raghava/algpred/submission.html) utilizing the prediction approach available in the server; (1) Mapping of IgE epitopes and PID, (2) MEME/MAST motif, and (3) Blast search on allergen representative peptides (ARPs). The selected T-cell epitope (15-mer) was checked for suitability as the B-cell epitope by IEDB-AR using several sequence-based web tools (Chou and Fasman 1978; Kolaskar and Tongaonkar 1990; Emini et al. 1985; Parker et al. 1986; Karplus 1985). We have also checked the discontinuous B-cell epitope prediction by Ellipro in IEDB server (http://tools.iedb.org/ellipro/).
Results
Sequence retrieval and identification of conserved regions
As our main concern was to design an epitope vaccine specific for the Bangladeshi H5 strains, we retrieved 220 HA sequences from IRD that were Bangladesh specific. All the sequences aligned by ClustalW and the conserved regions were identified. We have identified nine regions in the Haemagglutinin protein that are 100% conserved in the Bangladeshi isolates (Fig. 2). These conserved sequences were mainly analyzed further to identify the most potential epitope that might act as a vaccine against H5.
Fig. 2.

The features of the Hemagglutinin protein of the H5N1 influenza virus. The conserved regions of HA proteins with their sequences and positions have been shown. There are 9 regions including a 24 amino acid, and a 30 amino acid region (blue colored sequences) found to be 100% conserved in the HA-2 domain of Hemagglutinin sequences in Bangladeshi H5 isolates (N = 220) circulating from 2007 to 2018. The B-cell epitope (GAIAGFIEGGWQGM) and the T-cell epitope (DVWTYNAELLVLMEN) selected are finally shown in a bold font. The signal peptide (green) is located at the N-terminal and the transmembrane region (red) is located at the C-terminal of the protein
T-cell epitope identification
The T-cell epitopes were identified by the NetCTLv1.2 server, where the epitope prediction was restricted to 12 MHC class I supertypes. The top 11 epitopes (Table 1) for each HLA supertype were picked out based on the combined score, listed for further analysis. There was no epitope found for the B7 supertype with a combined score above the threshold value of 0.75. The most conserved epitopes are shown in an alignment (Fig. 3).
Table 1.
The predicted T-cell epitopes of Hemagglutinin protein by the NetCTL server based on the combined score
| Supertypes | Start position | Peptide sequence | Predictions score (combined) |
|---|---|---|---|
| A1 | 359 | WQGMVDGWY | 0.8988 |
| A2 | 428 | KMEDGFLDV | 1.4408 |
| A3 | 263 | FIAPEYAYK | 1.2590 |
| A24 | 438 | TYNAELLVL | 1.2966 |
| A26 | 431 | DGFLDVWTY | 1.5072 |
| B8 | 425 | LNKKMEDGF | 1.0565 |
| B27 | 361 | GMVDGWYGY | 0.8978 |
| B39 | 439 | YNAELLVLM | 0.7586 |
| B44 | 429 | MEDGFLDVW | 1.9448 |
| B58 | 351 | IAGFIEGGW | 1.4175 |
| B62 | 346 | GLFGAIAGF | 1.4017 |
Here, epitopes for the 11 different HLA supertypes have been represented. The epitopes for a single HLA supertype (B7) having a combined score lower than the threshold value 0.075 excluded. The top epitope for each HLA supertype has been represented
Fig. 3.

Multiple Sequence Alignment shows the location of the different 9-mer MHC class I epitopes within the hemagglutinin (HA) proteins of the influenza H5 virus. The selected potential vaccine candidate DVWTYNAELLVLMEN (red box) and GAIAGFIEGGWQGM (blue box) has been shown by a transparent box
MHC restriction and cluster analysis
Both the MHC class I and MHC class II-restricted alleles were predicted by IEDB analysis resource based on the IC50 value. All the predicted epitopes in Table 1 were evaluated for the analyses of MHC interaction. The MHC class I epitopes are summarized in Table 2 and Table S1. Furthermore, the interacted alleles were reevaluated by cluster analysis using MHCcluster v2.0 and shown in Figure S1A, as a heat map, and in Figure S1B, as a dynamic tree. These analyses create a cluster among the HLA molecules to show the major functional relationship between the HLA alleles for the selected peptide (Thomsen et al. 2013; Oany et al. 2017). The MHC class 1 interaction has been crosschecked by EPISOPT software, and the result is shown in Table S3.
Table 2.
The potential CD8 + T-cell epitopes and CD4 + T-cell epitopes with their number of interacting MHC class I and II alleles
| MHC class I Epitope |
No. of alleles | MHC class II Epitope (Core peptide in bold font) |
No. of allelesa |
|---|---|---|---|
| WQGMVDGWY | 04 | IEGGWQGMVDGWYGY | 0 |
| KMEDGFLDV | 05 | LNKKMEDGFLDVWTY | 0 |
| FIAPEYAYK | 06 | NGNFIAPEYAYKIVK | 41 |
| TYNAELLVL | 06 | VWTYNAELLVLMENE | 0 |
| DGFLDVWTY | 05 | KKMEDGFLDVWTYNA | 0 |
| LNKKMEDGF | 02 | NLNKKMEDGFLDVWT | 0 |
| GMVDGWYGY | 07 | IEGGWQGMVDGWYGY | 0 |
| YNAELLVLM | 03 | DVWTYNAELLVLMEN | 246 |
| MEDGFLDVW | 02 | NKKMEDGFLDVWTYN | 0 |
| IAGFIEGGW | 05 | FGAIAGFIEGGWQGM | 0 |
| GLFGAIAGF | 06 | RGLFGAIAGFIEGGW | 0 |
The IC50 < 250 nM and IC50 < 100 nM used for MHC class I and II alleles, respectively
aThe epitopes that have IC50 value < 100 nM are mentioned
The MHC class II epitopes are summarized in Table 2 and Table S2. Two 15-mer peptide candidates were chosen considering the threshold IC50 values along with the high number of MHC class II alleles. The peptides NGNFIAPEYAYKIVK, and DVWTYNAELLVLMEN were predicted to have high affinity with the MHC class II allele that can interact with 41, and 246 MHC class II alleles, respectively. The interaction with MHC class II has been validated by a software PREDIVAC, which predicts based on the specificity-determining residue (SDR) concept. The predivac scores of the two core peptides FIAPEYAYK and YNAELLVLM showed a considerable affinity with HLA_DRB_1 (Table S2). Contemplating the MHC class I allele as well as MHC class II allele-based analyses, we suggested that DVWTYNAELLVLMEN peptide has the best score; however, NGNFIAPEYAYKIVK has also vaccine potential from all the peptides selected initially.
Population coverage analysis
The prediction of both MHC class I- and MHC class II-based coverage of the selected epitopes was performed by IEDB analysis resource for the world population. The epitope DVWTYNAELLVLMEN consists of the highest population coverage of 99.73% for the whole world population and 100.0% for the South Asian population; on the other hand, NGNFIAPEYAYKIVK has 94.64% population coverage for the whole world and 98.85% for the South Asian population (Table 3). We have also assessed the population coverage for both the peptides for different regions of the world (Table 3). The result showed that the former epitope has more than 90% population coverage in Central Africa, East Africa, North Africa, West Africa, Central America, North America, South America, Europe, East Asia, Northeast Asia, South Asia, Southwest Asia, and Oceania.
Table 3.
Population coverage results for the peptide DVWTYNAELLVLMEN and NGNFIAPEYAYKIVK
| Population/area | Combined coverage of DVWTYNAELLVLMEN | Combined coverage of NGNFIAPEYAYKIVK |
|---|---|---|
| Central Africa | 100.0% | 90.3% |
| Central America | 99.96% | 22.49% |
| East Africa | 100.0% | 84.31% |
| East Asia | 92.54% | 65.42% |
| Europe | 100.0% | 99.87% |
| North Africa | 99.17% | 38.03% |
| North America | 100.0% | 99.95% |
| Northeast Asia | 100.0% | 93.59% |
| Oceania | 100.0% | 91.01% |
| South Africa | 11.65% | 45.75% |
| South America | 99.95% | 95.51% |
| South Asia | 100.0% | 98.85% |
| Southeast Asia | 69.34% | 65.03% |
| Southwest Asia | 99.72% | 49.6% |
| West Africa | 100.0% | 92.84% |
| West Indies | 5.69% | 27.65% |
| World | 99.73% | 94.64% |
Modeling of hemagglutinin, model validation, and structural frustration analysis
MODELLER modeled the three-dimensional structure of the hemagglutinin protein through the best multiple template-based modeling approaches (Fig S2A). The model was validated by the PROCHECK server represented as Ramachandran plot and is illustrated in Figure S3. Here, the amino acid residues were observed within the favored region. Moreover, the model was also evaluated for the frustration analysis and is illustrated in Figure S4. The disorder of the protein sequences was measured by the DISOPRED server to retrieve disorder among the targeted sequences (Fig S5). Both analyses showed that the potential peptide was placed in a stable part of the protein. Moreover, the proposed epitope was shown to be on the surface of HA protein (Fig S2B), and it is not located in the transmembrane region rather than existed in the extracellular region (Fig S2A and Fig S6).
Molecular docking analysis
The core epitope, YNAELLVLM, and its 15-mer extension, DVWTYNAELLVLMEN were docked with selected MHC class I and MHC class II alleles. CABSDOCK WEB SERVER generated a different layout of the docked peptide, and the best one was picked for the final calculation. The docking interface was visualized with the PyMOL Molecular Graphics System. There are several polar and non-polar interactions identified in the docking simulation analyses. The polar contacts were extracted by PyMOL and visualized in the figures. The Ala 3 and Val 7 residues of 9-mer peptide (YNAELLVLM) interacted with Arg 97 and Tyr 84 residues of HLA-B*15:01, respectively. There is also a network of polar contacts created by Asn 70 of HLA-B*15:01 with the Asn 2 and Tyr-1 residues of the 9-mer peptide (Fig. 4). The docking of HLA-B*15:01 with the peptide have Average RMSD = 1.13, Maximum RMSD = 3.36, and Cluster density = 88.87. We also performed control docking of a natural peptide, LEKARGSTY of Epstein–Barr virus EBNA-3 with the same HLA, and found the average RMSD of 1.51 and maximum RMSD of 4.32 that was quite similar to our designed peptide (Fig S9 and Fig S10).
Fig. 4.

Docking analysis of the proposed epitope YNAELLVLM (blue) and HLA-B*15:01 allele. a, b Showing the oriented view of the interaction ensuring the perfect binding. c Showing the cartoon view. d Representing the interaction between the amino acid residues of HLA (red) and the peptide (blue). The polar interactions are marked by green dashes
On the other hand, the Tyr 5 of 15-mer epitope (DVWTYNAELLVLMEN) interacted with the Asn 69 and Val 65 residues, whereas Met 13 interacted with Gln 9 residues of HLA-DRB01*01:01 through polar interaction. Moreover, Asn 15 amino acid residue from the epitope has polar contact with both Ser-53 and Glu-55 of the HLA-DRB molecule (Fig. 5). The docking of HLA-DRB01*01:01 with the peptide have average RMSD = 2.40, Maximum RMSD = 11.93, and Cluster density = 44.99. We also performed control docking of a natural peptide, PKYVKQNTLKLAT of HA protein of Influenza, and found that the Average RMSD = 3.43, and Maximum RMSD = 10.74 that was also similar to our designed 15-mer peptide (Fig S11 and Fig S12). The comparative view of RMSD and cluster density of test docking and control docking is presented in Table S7.
Fig. 5.

Docking analysis of the proposed epitope DVWTYNAELLVLMEN (red) and HLA-DRB*01:01 allele. a, b Oriented view of the interaction ensuring the perfect binding. c Cartoon view. d Interaction between the amino acid residues of HLA (Cyan) and the peptide (red). The polar interactions are marked by pink dashes
B-cell epitope prediction
We have utilized ABCpred to identify 10–14 amino acid long potential B-cell epitopes and found 13 epitopes. We selected the top two B-cell epitopes (H5B1, and H5B2), considering the scores found from the server. The epitope SSMPFHNIHP (H5B1) has a score of 0.76, and GAIAGFIEGGWQGM (H5B2) has a score of 0.79 for inducing B-cell response.
Conservancy analysis
The conservancy of all the proposed epitopes for Bangladeshi isolates is measured by the IEDB conservancy analysis tool and represented in Table S4. As the 9-mer T-cell epitopes and B-cell epitopes were selected from conserved regions of the hemagglutinin proteins, they were 100% conserved. During the selection of 15-mer epitopes, we added the most conserved amino acids from the flanking region of the 9-mer epitope. Among the 15-mer epitopes, DVWTYNAELLVLMEN and VWTYNAELLVLMENE were shown to have 100% conservancy, whereas the epitope NGNFIAPEYAYKIVK has 86.36% conservancy among H5 virus isolated in Bangladesh (Table S4). The location of all the 9-mer epitopes is illustrated in a multiple sequence alignment of hemagglutinin proteins, as shown in Fig. 3. Here, we showed only the desired sequences for appropriate annotation. Furthermore, we retrieved 4200 Hemagglutinin sequences (3950 isolated from humans and 250 isolated from non-human populations), where, H5 viruses were endemic. The results showed that the T-cell epitope DVWTYNAELLVLMEN (H5T1) is 98.93% conserved, and NGNFIAPEYAYKIVK (H5T2) is 78.69% conserved in all the countries included (Table S5). The B-cell epitope H5B1 is 94.12%, and H5B2 is 98.98% conserved (Table S6).
Designing of multi-epitope vaccine and analyses of its properties
The top T-cell epitope H5T1 and B-cell epitope H5B2 are adjoined by AAY linker. An N-terminal cysteine was added to find the benefit of conjugating the vaccine with a carrier protein (Fig. 6a). We performed a sequence-based allergenicity assessment by the AllerTop v. 2.0 web server and AlgPred server that concluded that the modified peptide was non-allergenic in nature. The secondary structural information of the vaccine was analyzed by SOPMA (Fig. 6b). A three-dimensional model of the vaccine has been generated by iTASSER and refined by the Galaxy refine server (Fig. 6c). ProSA-web was adopted to analyze the model quality (Fig. 6d). The results revealed a z score of − 0.14. The overall quality of the finalized model of the multi-epitope vaccine constructs was checked by Ramachandran plot analysis. The results revealed 80.8%, 15.4%, 3.8%, and 0.0% in the favored, additionally allowed, generously allowed, and disallowed regions, respectively (Fig S13). The resulting multi-epitope vaccine has been analyzed for various physicochemical properties. The chimeric vaccine was 33 amino acids long with a molecular weight of 3.59 kDa and a pI of 3.50. The estimated half-life was 1.2 h for mammalian reticulocytes, in vitro, > 20 h for yeast, in vivo, and > 10 h for Escherichia coli, in vivo. The instability index was computed to be 34.28, indicating that the peptide is stable in nature. The aliphatic index was 91.82, and the Grand average of hydropathicity (GRAVY) was 0.464.
Fig. 6.

a Schematic view of the final vaccine construct. The T-cell and B-cell epitopes were included in the vaccine construct, and they were linked via AAY linker (L). A cysteine residue was added at the N-terminal of the vaccine required for conjugation with a carrier molecule. The construct is 33 amino acid long, and that is non-allergic in nature predicted by the AllerTop v.2 and AlgPred server. b Secondary structure properties of the vaccine model by SOPMA. c 3D model of the final vaccine constructs, i. the preliminary form generated by iTASSER, ii. Refined by Galaxy refine server, and iii. Merged. d Validation of the structure using PROSA with a Z score of − 0.14
We used the sequence-based approaches for the suitability of the multi-epitope vaccine for B-cell induction. The vaccine was analyzed by Chou & Fasman Beta-Turn Prediction that showed the antigenic property of the epitope with a maximum of 1.137 (Fig. 7a). The Kolaskar and Tongaonkar antigenicity scale was utilized for assessing the antigenic property of the epitope with a maximum of 1.125 (Fig. 7b). Another important benchmark for being a potential B-cell epitope is peptide surface accessibility. Therefore, we exploited the prediction method, Emini surface accessibility of the predicted peptide with a maximum propensity score of 3.104 (Fig. 7c). The Parker hydrophilicity prediction is also utilized with a maximum score of 2.829 that strengthens our prediction about the epitope for eliciting B-cell response and is illustrated in Fig. 7d. The Karplus & Schulz Flexibility Prediction was also utilized that predicted a maximum score of 1.027 (Fig. 7e). We have also checked the discontinuous B-cell epitope prediction by ElliPro server and found that three discontinuous B-cell epitopes exist in the chimeric vaccine (Table S8).
Fig. 7.
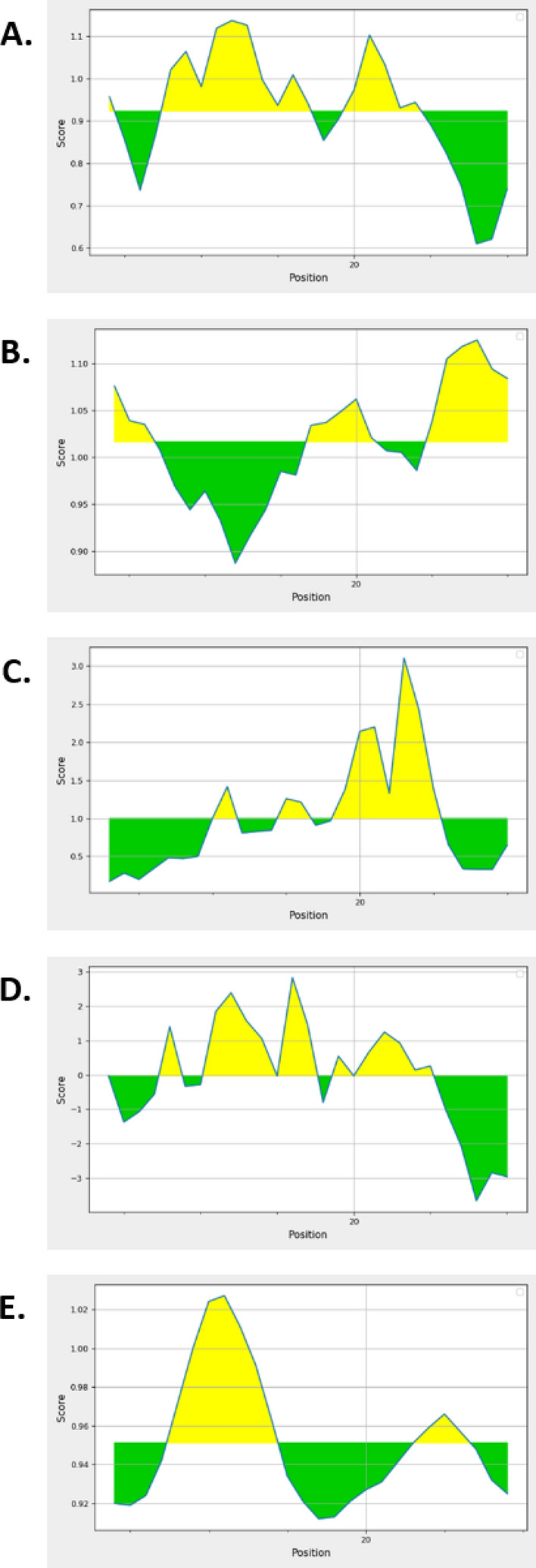
Prediction of B-cell epitope for the multi-epitope vaccine. The column showed the analyses of the respective multi-epitope peptides with a Chou and Fasman Beta-Turn Prediction, b Kolaskar and Tongaonkar antigenicity prediction, c Emini surface accessibility prediction, d Parker hydrophilicity prediction, and e Karplus and Schulz Flexibility Prediction. Notes: the x-axis and y-axis of the plot reflect the sequence location and antigenic propensity, respectively. The areas over the threshold line are antigenic, shown by yellow color
Discussion
Influenza H5 viruses cause respiratory infections that range from asymptomatic to lethal in humans. Perhaps, the influenza virus possesses the greatest risk of threats to human and avian populations among the current infectious disease globally, and virologists warned of the possibility of a new and devastating influenza pandemic (Fedson 2018). Around 60 countries have reported active outbreaks of H5 viruses in domestic poultry, wild birds, and humans over the 20 years since its first appearance in China in 1996 (Olsen et al. 2006; Durand et al. 2015; OIE 2019).
Influenza H5 viruses are avian origin and infect avian species. Human infection of the H5 virus is relatively low that makes the population vulnerable to the spread of H5 viruses because of not being primed as of lack of previous exposure, which causes immunization more challenging. The amino acid changes that have been identified in surface hemagglutinin protein (HA), switch of receptor binding preference from avian receptor SAα2, 3 Gal to human receptor SAα2, 6Gal that make them the pandemic potential of H5 viruses (CDC 2012). The Global Influenza Programme (GIP) recommends the development of an influenza vaccine for each distinct clade of H5 viruses (Baz et al. 2013). Research is required to delineate the extent of cross-protection as well as cross-reactive immunity conferred by vaccines based on heterologous clades of H5 viruses. The purpose of the present study was to utilize vaccinomics approaches to design novel epitope-based vaccine candidates based on the conserved region of the HA subunit of the surface glycoprotein from different types of H5 viruses with different clades. This vaccine could provide cross-protection against all H5 viruses within all clades specifically for Bangladesh, and then assess its potentiality for the world.
Many strategies have been taken for developing a vaccine against H5 viruses including, (a) inactivated vaccine (Treanor et al. 2006), (b) live attenuated vaccines (Rudenko et al. 2008), (c) recombinant protein-based vaccines (Treanor et al. 2001), (d) virus-like particle vaccines (Kang et al. 2009), (e) DNA vaccine (Kim and Jacob 2009), (f) vector-based vaccine (Hoelscher et al. 2006), (g) protein subunit vaccine (Belshe et al. 2011), and (h) in silico-based vaccines (Shahsavandi et al. 2015; Tambunan et al. 2016).
Initially, we started work with the HA sequences of Bangladeshi origin, as these viruses are very diverse in nature, and the predicted vaccine should work at least in the Bangladeshi population. We retrieved 220 HA protein sequences of H5 viruses including, H5N1, H5N2, and H5N6, available in the database and analyzed for conserved regions. As expected, most of the regions of Bangladeshi HA proteins of H5 viruses were found to be diverse, except a 24aa, a 30aa, and seven small islets of 11–15aa regions. We next utilized the immunoinformatic tools to find the most immunogenic peptide that could elicit an immune response in most of the regions of the world, including South Asia. From the conserved regions, the core epitopes FIAPEYAYK and YNAELLVLM (in 15.0-mer form, NGNFIAPEYAYKIVK, and DVWTYNAELLVLMEN, respectively) found to be the most potent and highly interacting human leukocyte antigen (HLA) candidates for both MHC class I and MHC class II molecules, interacted with 6 and 3 MHC class I along with 41 and 246 MHC class II alleles, respectively. Accumulating both allele-based analyses, we demonstrated DVWTYNAELLVLMEN peptide to have the best score; however, NGNFIAPEYAYKIVK has also the vaccine potential from all the peptides selected initially.
As the 9.0-mer epitopes selected from conserved regions of the hemagglutinin proteins, all epitopes were 100% conserved. We have tried to design 15.0-mer epitopes with different forms based on the two most potential epitopes. Among the 15.0-mer epitopes, DVWTYNAELLVLMEN, and VWTYNAELLVLMENE (core peptide YNAELLVLM), were shown to have 100% conservancy, whereas the epitope NGNFIAPEYAYKIVK had 86.36% conservancy (Table S4).
The 3D model of conserved domains was built through MODELLER and validated by the PROCHECK server through the Ramachandran plot. The model displayed the perfect position of the epitope on the surface of the protein structure (Fig S2). The result indicates their surface accessibility and increased earlier interaction with the immune system. Moreover, the analysis from the DISOPRED and frustration index; there are no disorders and energy frustration in the epitope domain of the sequences and predicted model which has strengthened our prediction (Fig S4 and Fig S5).
To design a universal vaccine that would protect against H5 viruses worldwide, the most important thing is to analyze population coverage of the suggested epitopes. The vaccine candidates must have wider population coverage to get the acceptability. In our analysis, our proposed epitope DVWTYNAELLVLMEN has the highest population coverage of 99.73–100% for the whole world population and South Asian population, respectively. On the other hand, NGNFIAPEYAYKIVK has 94.64% population coverage for the whole world, and 98.85% for the South Asian population (Table 3). In terms of different regions of the world, DVWTYNAELLVLMEN has shown wider population coverage in Northeast Asia, South Asia, Southwest Asia, Central Africa, East Africa, West Africa, North Africa, Central America, North America, South America, and Oceania (Table 3). These outputs showed that the predicted epitopes would have broader coverage in vitro. However, it shows population coverage of only 11.65% in South Africa, 5.69% in the West Indies, and 69.34% in Southeast Asia indicating its inefficiency in these regions.
As the peptide DVWTYNAELLVLMEN shows 100% conservancy in Bangladesh and 100% population coverage in South Asia, we predict that it would be a good vaccine for this region. Afterward, we assessed its potentiality as a vaccine for other regions of the world. In addition to Bangladeshi isolates, we retrieved 4200 HA sequences of H5 isolates, and search for the conservancy of this peptide that appears to an interesting output. According to conservancy analysis, this 15.0-mer epitope is highly conserved among H5 viruses isolated from avian, human, and other species circulated in Bangladesh, China, Japan, USA, India, Thailand, Egypt, Viet Nam, Cambodia, etc. where H5 viruses are epidemic (Table S5 and Table S6). Study data show that the proposed epitope would be a potential candidate for human and non-human populations for treating all of the H5 viruses.
Therefore, we propose this epitope as a vaccine in most of the regions of the world, except, South Africa, West Indies, and Southeast Asia. Among the encoded eleven influenza A proteins, nine are usually identified in the virion (Palese and Shaw 2007). The lipid envelope of the virus particle is embedded with two major surface glycoproteins, HA and NA, whereas M1 is beneath the membrane and M2 exists at a minor level on the membrane (Skehel and Schild 1971; Zebedee and Lamb 1988). Therefore, HA proteins of H5 viruses are common interest to be targeted as a vaccine that could be recognized in the whole set of H5 viruses. The present analyses thus demonstrate the importance of the peptide DVWTYNAELLVLMEN, as this is more conserved and with high population coverage among the H5 viruses. Molecular docking revealed the strong interaction between predicted both 9.0-mer and 15.0-mer epitopes and MHC molecules with the perfect orientation. Moreover, our proposed epitope has shown interaction with the MHC molecules, which enhance the prediction of MHC binding.
We have also predicted B-cell epitopes by ABCpred (Table 4). These peptides were 100% conserved in Bangladesh, as we selected these from the identified sequences described in Fig. 2. The two top-scoring epitopes further checked for conservancy in the clades of endemic regions of the world. It shows that H5B1 and H5B2 have 94.12% and 98.98% conservancy, respectively. Therefore, these peptides can not only be used in Bangladesh but also have a large utility in the world endemic regions. Then, we added the top T-cell and B-cell epitopes together by the appropriate linker and added cysteine at the N-terminal of the peptide, as it will be required during conjugation with a carrier protein. The resulting multi-epitope vaccine is 33 aa peptide, mostly stable in nature. We found that the fused peptide is non-allergenic in nature, analyzed by several strategies that exist in AllerTop and AlgPred server. The three-dimensional model shows two β-strand regions, and the model was verified by Procheck and Prosa (Fig. 6). It has the potential to induce B-cell response evaluated by multiple well-recognized approaches from IEDB (Fig. 7 and Table S8).
Table 4.
The B-cell epitope prediction by ABCpred with their respective scores
| Peptide prediction by ABCpred | Score |
|---|---|
| NGNFIAPEYA | 0.64 |
| SSMPFHNIHP | 0.76 |
| IGECPKYVKS | 0.66 |
| GLFGAIAGFI | 0.62 |
| IAGFIEGGWQ | 0.60 |
| GAIAGFIEGGWQGM | 0.79 |
| IEGGWQGMVDGWYG | 0.52 |
| LDVWTYNAEL | 0.67 |
| NLNKKMEDGF | 0.60 |
| DGFLDVWTYNAELL | 0.72 |
| VWTYNAELLVLMEN | 0.62 |
| GLSLWMCSNG | 0.66 |
| AGLSLWMCSNGSLQ | 0.63 |
Bold values indicate the highest scores from the enlisted peptides
The major strength of our study is the strong conservancy analyses that dictate the appropriate use of this vaccine in Bangladesh. Furthermore, the conservancy among the world endemic regions also suggests its utility in those regions. Selecting the small peptides from a conserved region is less prone to be mutated early, which is useful for the high mutation prone virus, like, influenza. Several epitope-based vaccine designs were already performed on Hemagglutinin proteins of H1, H2, and H5 (Babon et al. 2012; Vergara-Alert et al. 2012; Wang et al. 2010b); however, they have not targeted exclusively H5 viruses, and they chose big sequences sufficient to compromise the conservancy among the same set of HA of H5 viruses. A lot of peptides are also reported in IEDB; most of them are away from the 100% conserved region revealed in this study and/or big peptides. A portion of our peptides, especially GLFGAIAGF, found to be immunogenic in these studies suggested that this epitope-based conserved vaccine can give protection against circulating viruses (Qiu et al. 2017; Prabhu et al. 2009). The T-cell and B-cell inducing properties of this vaccine can efficiently help both human and avian species to induce protection in their immune system. Therefore, our suggested epitope has the potential to elicit an immune response in vitro.
Conclusion
The study suggests that the proposed multi-epitope vaccine is highly conserved among HA proteins of H5 viruses, and it might mitigate pandemic threats and provide cross-protection of both avian and humans against different types of H5 viruses within different clades. This strategy could be utilized to find a conserved vaccine from the viruses that are highly diverse in nature. The vaccine can not only be used in Bangladesh but also in the endemic regions of the world. This type of immunoinformatic approach reduces a huge work for wet-lab experiments. Further study is required to validate the immunogenicity of this peptide.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary file1 (DOCX 25 KB) Fig S1: Cluster analysis of the HLA alleles for MHC class 1 molecule through (A) heat map and (B) dynamic tree representation. Epitopes are clustered on the basis of interaction with HLA and shown as red color indicating strong interaction with appropriate annotation. The yellow zone indicates a weaker interaction. Here, all the available alleles are shown only. In (B), HLA molecules are clustered on the basis of their preference for interaction with appropriate peptides. Fig S2: The three-dimensional model of a Hemagglutinin protein of H5 virus. A. Different regions are shown by colors, HA subunit 1 (cyan), HA subunit 2 (green), Transmembrane region (magenta), and Peptide DVWTYNAELLVLMEN (Red). B. The superficial localities of the epitope, DVWTYNAELLVLMEN (Red), indicate their surface location of the peptide. Fig S3: Ramachandran plot of the predicted three-dimensional model of Haemagglutinin protein, which shows that most of the residues are in the allowed region of the plot, proving the validity of the model. Fig S4: The configurational frustration index of the anticipated model of the HA protein. (A) This analysis determines the stability and energy differences of the 3D structure of the protein. Different colors correspond with their frustration index. The highly frustrated regions marked with red color, the minimally frustrated regions marked with green color, and the dark gray color regions are not frustrated. (B) The location of the selected epitope is indicated by magenta color. The epitope DVWTYNAELLVLMEN is external to the frustrated regions and ensuring stability. Fig S5: Disorder prediction of the amino acid sequences of Hemagglutinin of Influenza virus A. Here, the potential epitopes localized outside of the disordered regions (260-274 and 435-449) and securing their potentiality for being an effective vaccine candidate. Notes: Amino acids in the input sequence are considered disordered when the dotted line is above the gray dashed line, that is, when the confidence score is 0.5. Fig S6: The transmembrane region prediction of selected Hemagglutinin protein by (A) TMHMM and (B) TMpred. Fig S7: Contact maps between the 9-mer peptide (YNAELLVLM) with HLA-B*15:01. Here, the distances between two amino acids closer than 4.5A are shown. The polar interactions between amino acids of the epitope and HLA are indicated by the red font. Fig S8: Contact maps between the 15-mer peptide (DVWTYNAELLVLMEN) with HLA-DRB*01:01. Here, the distances between two amino acids closer than 4.5 A are shown. The polar interactions between amino acids of the epitope and HLA are indicated by the red font. Fig S9: Docking analysis of the natural epitope LEKARGSTY (light green), and HLA-B*15:01 allele. This docking analysis has been performed as a control experiment for interaction with MHC class I. (A) and (B) Representing the oriented view of the interaction and assuring the perfect binding. (C) Representing the cartoon view. (D) Embodying the interacted residues (red) with the peptide (light green). The polar interactions are marked by blue dashes. Fig S10: Contact maps between the natural epitope LEKARGSTY, and HLA-B*15:01 allele. Here, the distances between two amino acids closer than 4.5 A are shown. The polar interactions between amino acids of the epitope and HLA are indicated by the red font. Fig S11: Docking analysis of the natural epitope PKYVKQNTLKLAT (pink), and HLA- DRB01*01:01 allele. This docking analyses have been performed as control experiment for interaction with MHC class I. (A) and (B) Representing the oriented view of the interaction and assuring the perfect binding. (C) Representing the cartoon view. (D) Embodying the interacted residues (cyan) with the peptide (pink). The polar interactions are marked by red dashes. Fig S12: Contact maps between the natural epitope PKYVKQNTLKLAT, and HLA- DRB01*01:01 allele. Here, the distances between two amino acids closer than 4.5 A are shown. The polar interactions between amino acids of the epitope and HLA are indicated by the red font. Fig S13: Ramachandran plot analysis of the model using Procheck server showing 80.8%, 15.4%, 3.8%, and 0.0% in the favored, additional allowed, generously allowed, and disallowed region, respectively.
Acknowledgements
The author reports no acknowledgments of this work.
Author contributions
MSBI and MM carried out the immunoinformatic analyses, and drafted the manuscript. MEH helped in the design of the study and performed critical revision. KMKK conceived, designed, and guided the study; analyzed the data, and drafted and critically revised the manuscript; All authors read and approved the final manuscript.
Funding
No funding was obtained for this study.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
As samples from human or animals had not been used in this study, ethical clearance is not applicable.
Footnotes
Md. Shaid Bin Islam and Mojnu Miah contributed equally to this manuscript.
Contributor Information
Md. Shaid Bin Islam, Email: shaidbin@gmail.com.
Mojnu Miah, Email: mojnumiah.bge@gmail.com.
Mohammad Enayet Hossain, Email: enayet.hossain@icddrb.org.
K. M. Kaderi Kibria, Email: km_kibria@yahoo.com.
References
- Babon JA, Cruz J, Ennis FA, Yin L, Terajima M. A human CD4+ T cell epitope in the influenza hemagglutinin is cross-reactive to influenza A virus subtypes and to influenza B virus. J Virol. 2012;86(17):9233–9243. doi: 10.1128/JVI.06325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baz M, Luke CJ, Cheng X, Jin H, Subbarao K. H5N1 vaccines in humans. Virus Res. 2013;178(1):78–98. doi: 10.1016/j.virusres.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshe RB, Frey SE, Graham I, Mulligan MJ, Edupuganti S, Jackson LA, Wald A, Poland G, Jacobson R, Keyserling HL, Spearman P, Hill H, Wolff M. Safety and immunogenicity of influenza A H5 subunit vaccines: effect of vaccine schedule and antigenic variant. J Infect Dis. 2011;203(5):666–673. doi: 10.1093/infdis/jiq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdette D, Edmonds E, Smith C, Bowen J, Guttmann CR, Nagy Z, Simon J, Whitham R, Lovera J, Yadav V. A highly immunogenic trivalent T cell receptor peptide vaccine for multiple sclerosis. Mult Scler J. 2005;11(5):552–561. doi: 10.1191/1352458505ms1225oa. [DOI] [PubMed] [Google Scholar]
- Bridges CB, Thompson WW, Meltzer MI, Reeve GR, Talamonti WJ, Cox NJ, Lilac HA, Hall H, Klimov A, Fukuda K. Effectiveness and cost-benefit of influenza vaccination of healthy working adults: a randomized controlled trial. JAMA. 2000;284(13):1655–1663. doi: 10.1001/jama.284.13.1655. [DOI] [PubMed] [Google Scholar]
- Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformat. 2006;7:153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buus S, Lauemoller SL, Worning P, Kesmir C, Frimurer T, Corbet S, Fomsgaard A, Hilden J, Holm A, Brunak S. Sensitive quantitative predictions of peptide-MHC binding by a 'Query by Committee' artificial neural network approach. Tissue Antigens. 2003;62(5):378–384. doi: 10.1034/j.1399-0039.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- CDC (2012) H5N1 genetic changes inventory: a tool for influenza surveillance and preparedness. Centers for Disease Control and Prevention, Atlanta, GA [cited 2012 June 26]. Available from https://www.cdc.gov/flu/pdf/avianflu/h5n1-inventory.pdf
- Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- Demicheli V, Jefferson T, Ferroni E, Rivetti A, Di Pietrantonj C. Vaccines for preventing influenza in healthy adults. Cochrane Database Syst Rev. 2018;2(2):CD001269. doi: 10.1002/14651858.CD001269.pub6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand LO, Glew P, Gross D, Kasper M, Trock S, Kim IK, Bresee JS, Donis R, Uyeki TM, Widdowson M-A. Timing of influenza A (H5N1) in poultry and humans and seasonal influenza activity worldwide, 2004–2013. Emerg Infect Dis. 2015;21(2):202. doi: 10.3201/eid2102.140877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55(3):836–839. doi: 10.1128/JVI.55.3.836-839.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler OB, Dai WJ, Sette A, Hunziker IP, Reichen J, Pichler WJ, Cerny A. Peptide vaccines against hepatitis B virus: from animal model to human studies. Mol Immunol. 2001;38(6):457–465. doi: 10.1016/S0161-5890(01)00081-5. [DOI] [PubMed] [Google Scholar]
- Fedson DS. Influenza, evolution, and the next pandemic. Evol Med Public Health. 2018;2018(1):260–269. doi: 10.1093/emph/eoy027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry AM, Zhong W, Gubareva LV. Advancing treatment options for influenza: challenges with the human influenza challenge. J Infect Dis. 2015;211(7):1033–1035. doi: 10.1093/infdis/jiu543. [DOI] [PubMed] [Google Scholar]
- Gahery H, Daniel N, Charmeteau B, Ourth L, Jackson A, Andrieu M, Choppin J, Salmon D, Pialoux G, Guillet J-G. New CD4+ and CD8+ T cell responses induced in chronically HIV type-1-infected patients after immunizations with an HIV type 1 lipopeptide vaccine. AIDS Res Hum Retroviruses. 2006;22(7):684–694. doi: 10.1089/aid.2006.22.684. [DOI] [PubMed] [Google Scholar]
- Gerhard W. The role of the antibody response in influenza virus infection. Curr Top Microbiol Immunol. 2001;260:171–190. doi: 10.1007/978-3-662-05783-4_9. [DOI] [PubMed] [Google Scholar]
- Hall T, Biosciences I, Carlsbad C. BioEdit: an important software for molecular biology. GERF Bull Biosci. 2011;2(1):60–61. [Google Scholar]
- Hassan AO, Amen O, Sayedahmed EE, Vemula SV, Amoah S, York I, Gangappa S, Sambhara S, Mittal SK. Adenovirus vector-based multi-epitope vaccine provides partial protection against H5, H7, and H9 avian influenza viruses. PLoS ONE. 2017;12(10):e0186244. doi: 10.1371/journal.pone.0186244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelscher MA, Garg S, Bangari DS, Belser JA, Lu X, Stephenson I, Bright RA, Katz JM, Mittal SK, Sambhara S. Development of adenoviral-vector-based pandemic influenza vaccine against antigenically distinct human H5N1 strains in mice. Lancet. 2006;367(9509):475–481. doi: 10.1016/S0140-6736(06)68076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MU, Omar TM, Oany AR, Kibria KMK, Shibly AZ, Moniruzzaman M, Ali SR, Islam MM. Design of peptide-based epitope vaccine and further binding site scrutiny led to groundswell in drug discovery against Lassa virus. 3 Biotech. 2018;8(2):81. doi: 10.1007/s13205-018-1106-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenik M, Parra RG, Radusky LG, Turjanski A, Wolynes PG, Ferreiro DU. Protein frustratometer: a tool to localize energetic frustration in protein molecules. Nucleic Acids Res. 2012;40:W348–351. doi: 10.1093/nar/gks447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SM, Yoo DG, Lipatov AS, Song JM, Davis CT, Quan FS, Chen LM, Donis RO, Compans RW. Induction of long-term protective immune responses by influenza H5N1 virus-like particles. PLoS ONE. 2009;4(3):e4667. doi: 10.1371/journal.pone.0004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PASG. Prediction of chain flexibility in proteins. Naturwissenschaften. 1985;72:212–213. doi: 10.1007/BF01195768. [DOI] [Google Scholar]
- Kashala O, Amador R, Valero MV, Moreno A, Barbosa A, Nickel B, Daubenberger CA, Guzman F, Pluschke G, Patarroyo ME. Safety, tolerability and immunogenicity of new formulations of the Plasmodium falciparum malaria peptide vaccine SPf66 combined with the immunological adjuvant QS-21. Vaccine. 2002;20(17–18):2263–2277. doi: 10.1016/S0264-410X(02)00115-9. [DOI] [PubMed] [Google Scholar]
- Keogh E, Fikes J, Southwood S, Celis E, Chesnut R, Sette A. Identification of new epitopes from four different tumor-associated antigens: recognition of naturally processed epitopes correlates with HLA-A∗ 0201-binding affinity. J Immunol. 2001;167(2):787–796. doi: 10.4049/jimmunol.167.2.787. [DOI] [PubMed] [Google Scholar]
- Kim JH, Jacob J. DNA vaccines against influenza viruses. Curr Top Microbiol Immunol. 2009;333:197–210. doi: 10.1007/978-3-540-92165-3_10. [DOI] [PubMed] [Google Scholar]
- Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1–2):172–174. doi: 10.1016/0014-5793(90)80535-Q. [DOI] [PubMed] [Google Scholar]
- Lamb RA (2001) Orthomyxoviridae: the viruses and their replication. Fields Virol 1353–1395. https://ci.nii.ac.jp/naid/10013153254/en/
- Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformat. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8(4):477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Liao L, Noble WS. Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J Comput Biol. 2003;10(6):857–868. doi: 10.1089/106652703322756113. [DOI] [PubMed] [Google Scholar]
- Manzoli L, Schioppa F, Boccia A, Villari P. The efficacy of influenza vaccine for healthy children: a meta-analysis evaluating potential sources of variation in efficacy estimates including study quality. Pediatr Infect Dis J. 2007;26(2):97–106. doi: 10.1097/01.inf.0000253053.01151.bd. [DOI] [PubMed] [Google Scholar]
- Marinova-Petkova A, Shanmuganatham K, Feeroz MM, Jones-Engel L, Hasan MK, Akhtar S, Turner J, Walker D, Seiler P, Franks J. The continuing evolution of H5N1 and H9N2 influenza viruses in Bangladesh between 2013 and 2014. Avian Dis. 2015;60(1s):108–117. doi: 10.1637/11136-050815-Reg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oany AR, Pervin T, Mia M, Hossain M, Shahnaij M, Mahmud S, Kibria KMK. Vaccinomics approach for designing potential peptide vaccine by targeting Shigella spp. serine protease autotransporter subfamily protein SigA. J Immunol Res. 2017 doi: 10.1155/2017/6412353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OIE (2019) Update on avian influenza in animals (types H5 and H7). Available from https://www.oie.int/animal-health-in-the-world/update-on-avianinfluenza/2019
- Olsen B, Munster VJ, Wallensten A, Waldenström J, Osterhaus AD, Fouchier RA. Global patterns of influenza A virus in wild birds. Science. 2006;312(5772):384–388. doi: 10.1126/science.1122438. [DOI] [PubMed] [Google Scholar]
- Padilla-Quirarte HO, Lopez-Guerrero DV, Gutierrez-Xicotencatl L, Esquivel-Guadarrama F. Protective Antibodies Against Influenza Proteins. Front Immunol. 2019;10:1677. doi: 10.3389/fimmu.2019.01677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P, Shaw ML. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Field virology. 5. Philadelphia: Lippincott Williams and Wilkins; 2007. pp. 1647–1689. [Google Scholar]
- Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25(19):5425–5432. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- Petrovsky N, Brusic V. Computational immunology: the coming of age. Immunol Cell Biol. 2002;80(3):248–254. doi: 10.1046/j.1440-1711.2002.01093.x. [DOI] [PubMed] [Google Scholar]
- Prabhu N, Prabakaran M, Ho HT, Velumani S, Qiang J, Goutama M, Kwang J. Monoclonal antibodies against the fusion peptide of hemagglutinin protect mice from lethal influenza A virus H5N1 infection. J Virol. 2009;83(6):2553–2562. doi: 10.1128/JVI.02165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presti RM, Zhao G, Beatty WL, Mihindukulasuriya KA, Da Rosa APT, Popov VL, Tesh RB, Virgin HW, Wang D. Quaranfil, Johnston Atoll, and Lake Chad viruses are novel members of the family Orthomyxoviridae. J Virol. 2009;83(22):11599–11606. doi: 10.1128/JVI.00677-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Duvvuri VR, Gubbay JB, Webby RJ, Kayali G, Bahl J. Lineage-specific epitope profiles for HPAI H5 pre-pandemic vaccine selection and evaluation. Influenza Other Respir Viruses. 2017;11(5):445–456. doi: 10.1111/irv.12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson HL, Amara RR. T cell vaccines for microbial infections. Nat Med. 2005;11(4s):S25. doi: 10.1038/nm1212. [DOI] [PubMed] [Google Scholar]
- Rudenko L, Desheva J, Korovkin S, Mironov A, Rekstin A, Grigorieva E, Donina S, Gambaryan A, Katlinsky A. Safety and immunogenicity of live attenuated influenza reassortant H5 vaccine (phase I-II clinical trials) Influenza Other Respir Viruses. 2008;2(6):203–209. doi: 10.1111/j.1750-2659.2008.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23(3):318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- Shahsavandi S, Ebrahimi MM, Sadeghi K, Mahravani H. Design of a heterosubtypic epitope-based peptide vaccine fused with hemokinin-1 against influenza viruses. Virol Sin. 2015;30(3):200–207. doi: 10.1007/s12250-014-3504-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Schild GC. The polypeptide composition of influenza A viruses. Virology. 1971;44(2):396–408. doi: 10.1016/0042-6822(71)90270-4. [DOI] [PubMed] [Google Scholar]
- Squires RB, Noronha J, Hunt V, García-Sastre A, Macken C, Baumgarth N, Suarez D, Pickett BE, Zhang Y, Larsen CN. Influenza research database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respir Viruses. 2012;6(6):404–416. doi: 10.1111/j.1750-2659.2011.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar S, Brokstad KA, Cox RJ. Influenza vaccination strategies: comparing inactivated and live attenuated influenza vaccines. Vaccines. 2015;3(2):373–389. doi: 10.3390/vaccines3020373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambunan US, Sipahutar FR, Parikesit AA, Kerami D. Vaccine Design for H5N1 Based on B- and T-cell Epitope Predictions. Bioinform Biol Insights. 2016;10:27–35. doi: 10.4137/BBI.S38378bbi-10-2016-027[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Lundegaard C, Buus S, Lund O, Nielsen M. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics. 2013;65(9):655–665. doi: 10.1007/s00251-013-0714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M. Safety and immunogenicity of an inactivated subvirion influenza A (H5N1) vaccine. N Engl J Med. 2006;354(13):1343–1351. doi: 10.1056/NEJMoa055778. [DOI] [PubMed] [Google Scholar]
- Treanor JJ, Wilkinson BE, Masseoud F, Hu-Primmer J, Battaglia R, O'Brien D, Wolff M, Rabinovich G, Blackwelder W, Katz JM. Safety and immunogenicity of a recombinant hemagglutinin vaccine for H5 influenza in humans. Vaccine. 2001;19(13–14):1732–1737. doi: 10.1016/s0264-410x(00)00395-9. [DOI] [PubMed] [Google Scholar]
- Vakili B, Nezafat N, Zare B, Erfani N, Akbari M, Ghasemi Y, Rahbar MR, Hatam GR. A new multi-epitope peptide vaccine induces immune responses and protection against Leishmania infantum in BALB/c mice. Med Microbiol Immunol. 2019;209(1):69–79. doi: 10.1007/s00430-019-00640-7. [DOI] [PubMed] [Google Scholar]
- Vergara-Alert J, Argilaguet JM, Busquets N, Ballester M, Martin-Valls GE, Rivas R, Lopez-Soria S, Solanes D, Majo N, Segales J, Veljkovic V, Rodriguez F, Darji A. Conserved synthetic peptides from the hemagglutinin of influenza viruses induce broad humoral and T-cell responses in a pig model. PLoS ONE. 2012;7(7):e40524. doi: 10.1371/journal.pone.0040524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4(4):e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B. Peptide binding predictions for HLA DR. DP and DQ molecules BMC Bioinformatics. 2010;11:568. doi: 10.1186/1471-2105-11-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TT, Tan GS, Hai R, Pica N, Ngai L, Ekiert DC, Wilson IA, Garcia-Sastre A, Moran TM, Palese P. Vaccination with a synthetic peptide from the influenza virus hemagglutinin provides protection against distinct viral subtypes. Proc Natl Acad Sci U S A. 2010;107(44):18979–18984. doi: 10.1073/pnas.1013387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20(13):2138–2139. doi: 10.1093/bioinformatics/bth195bth195[pii]. [DOI] [PubMed] [Google Scholar]
- WHO (2017) WHO "Influenza (Seasonal) Fact sheet N°211". Retrieved on 10 June 2017. (accessed)
- WHO (2019) Cumulative number of confirmed human cases for avian influenza A(H5N1) reported to WHO, 2003–2019 [cited 2019 November 25]. Available from https://www.who.int/influenza/human_animal_interface/2019_11_25_tableH5N1.pdf?ua=1
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersma L, Rimmelzwaan GF, De Vries RD. Developing universal influenza vaccines: hitting the nail, not just on the head. Vaccines. 2015;3(2):239–262. doi: 10.3390/vaccines3020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Organization for Animal Health (OIE) (2013) Outbreaks of highly pathogenic avian influenza (subtype H5N1) in poultry notified to the OIE from the end of 2003 to 11 April 2013 [cited 2013 May 26]. http://www.oie.int/fileadmin/Home/eng/Animal_Health_in_the_World/docs/pdf/graph_avian_influenza/graphs_HPAI_26_05_2013.pdf
- Wright PF, Neumann G, Kawaoka Y. Orthomyxoviruses. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields virology. 5. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 161–740. [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen K, Chan P, Peiris M, Tsang D, Que T, Shortridge K, Cheung P, To W, Ho E, Sung R. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. The Lancet. 1998;351(9101):467–471. doi: 10.1016/S0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- Zebedee SL, Lamb RA. Influenza A virus M2 protein: monoclonal antibody restriction of virus growth and detection of M2 in virions. J Virol. 1988;62(8):2762–2772. doi: 10.1128/JVI.62.8.2762-2772.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1 (DOCX 25 KB) Fig S1: Cluster analysis of the HLA alleles for MHC class 1 molecule through (A) heat map and (B) dynamic tree representation. Epitopes are clustered on the basis of interaction with HLA and shown as red color indicating strong interaction with appropriate annotation. The yellow zone indicates a weaker interaction. Here, all the available alleles are shown only. In (B), HLA molecules are clustered on the basis of their preference for interaction with appropriate peptides. Fig S2: The three-dimensional model of a Hemagglutinin protein of H5 virus. A. Different regions are shown by colors, HA subunit 1 (cyan), HA subunit 2 (green), Transmembrane region (magenta), and Peptide DVWTYNAELLVLMEN (Red). B. The superficial localities of the epitope, DVWTYNAELLVLMEN (Red), indicate their surface location of the peptide. Fig S3: Ramachandran plot of the predicted three-dimensional model of Haemagglutinin protein, which shows that most of the residues are in the allowed region of the plot, proving the validity of the model. Fig S4: The configurational frustration index of the anticipated model of the HA protein. (A) This analysis determines the stability and energy differences of the 3D structure of the protein. Different colors correspond with their frustration index. The highly frustrated regions marked with red color, the minimally frustrated regions marked with green color, and the dark gray color regions are not frustrated. (B) The location of the selected epitope is indicated by magenta color. The epitope DVWTYNAELLVLMEN is external to the frustrated regions and ensuring stability. Fig S5: Disorder prediction of the amino acid sequences of Hemagglutinin of Influenza virus A. Here, the potential epitopes localized outside of the disordered regions (260-274 and 435-449) and securing their potentiality for being an effective vaccine candidate. Notes: Amino acids in the input sequence are considered disordered when the dotted line is above the gray dashed line, that is, when the confidence score is 0.5. Fig S6: The transmembrane region prediction of selected Hemagglutinin protein by (A) TMHMM and (B) TMpred. Fig S7: Contact maps between the 9-mer peptide (YNAELLVLM) with HLA-B*15:01. Here, the distances between two amino acids closer than 4.5A are shown. The polar interactions between amino acids of the epitope and HLA are indicated by the red font. Fig S8: Contact maps between the 15-mer peptide (DVWTYNAELLVLMEN) with HLA-DRB*01:01. Here, the distances between two amino acids closer than 4.5 A are shown. The polar interactions between amino acids of the epitope and HLA are indicated by the red font. Fig S9: Docking analysis of the natural epitope LEKARGSTY (light green), and HLA-B*15:01 allele. This docking analysis has been performed as a control experiment for interaction with MHC class I. (A) and (B) Representing the oriented view of the interaction and assuring the perfect binding. (C) Representing the cartoon view. (D) Embodying the interacted residues (red) with the peptide (light green). The polar interactions are marked by blue dashes. Fig S10: Contact maps between the natural epitope LEKARGSTY, and HLA-B*15:01 allele. Here, the distances between two amino acids closer than 4.5 A are shown. The polar interactions between amino acids of the epitope and HLA are indicated by the red font. Fig S11: Docking analysis of the natural epitope PKYVKQNTLKLAT (pink), and HLA- DRB01*01:01 allele. This docking analyses have been performed as control experiment for interaction with MHC class I. (A) and (B) Representing the oriented view of the interaction and assuring the perfect binding. (C) Representing the cartoon view. (D) Embodying the interacted residues (cyan) with the peptide (pink). The polar interactions are marked by red dashes. Fig S12: Contact maps between the natural epitope PKYVKQNTLKLAT, and HLA- DRB01*01:01 allele. Here, the distances between two amino acids closer than 4.5 A are shown. The polar interactions between amino acids of the epitope and HLA are indicated by the red font. Fig S13: Ramachandran plot analysis of the model using Procheck server showing 80.8%, 15.4%, 3.8%, and 0.0% in the favored, additional allowed, generously allowed, and disallowed region, respectively.