

Supplemental Digital Content is available in the text.
Keywords: acute coronary syndrome, coronary artery disease, myocardial infarction, percutaneous coronary intervention, troponin
Abstract
Objective:
Arterial thrombosis leading to ischemic injury worsens the prognosis of many patients with cardiovascular disease. PZ-128 is a first-in-class pepducin that reversibly inhibits PAR1 (protease-activated receptor 1) on platelets and other vascular cells by targeting the intracellular surface of the receptor. The TRIP-PCI (Thrombin Receptor Inhibitory Pepducin in Percutaneous Coronary Intervention) trial was conducted to assess the safety and efficacy of PZ-128 in patients undergoing cardiac catheterization with intent to perform percutaneous coronary intervention.
Approach and Results:
In this randomized, double-blind, placebo-controlled, phase 2 trial, 100 patients were randomly assigned (2:1) to receive PZ-128 (0.3 or 0.5 mg/kg), or placebo in a 2-hour infusion initiated just before the start of cardiac catheterization, on top of standard oral antiplatelet therapy. Rates of the primary end point of bleeding were not different between the combined PZ-128 doses (1.6%, 1/62) and placebo group (0%, 0/35). The secondary end points of major adverse coronary events at 30 and 90 days did not significantly differ but were numerically lower in the PZ-128 groups (0% and 2% in the PZ-128 groups, 6% and 6% with placebo, p=0.13, p=0.29, respectively). In the subgroup of patients with elevated baseline cardiac troponin I, the exploratory end point of 30-day major adverse coronary events + myocardial injury showed 83% events in the placebo group versus 31% events in the combined PZ-128 drug groups, an adjusted relative risk of 0.14 (95% CI, 0.02–0.75); P=0.02.
Conclusions:
In this first-in-patient experience, PZ-128 added to standard antiplatelet therapy appeared to be safe, well tolerated, and potentially reduced periprocedural myonecrosis, thus providing the basis for further clinical trials.
Registration:
URL: https://www.clinicaltrials.gov. Unique identifier: NCT02561000.
Highlights.
A ceiling of net clinical benefit associated with aspirin and P2Y12 receptor blockers indicates a need for targeting alternative pathways to prevent life-threatening ischemic complications in patients with severe coronary artery disease.
Thrombin, the most potent mediator of both hemostasis and thrombosis, targets PAR1 (protease-activated receptor 1), but there are no approved PAR1 inhibitors for treatment of acute coronary syndromes or patients undergoing percutaneous coronary intervention.
Thrombin Receptor Inhibitory Pepducin in Percutaneous Coronary Intervention is the first randomized, placebo-controlled trial in humans to test the safety and efficacy of a pepducin compound which targets the inside surface of a G-protein–coupled receptor, namely PAR1.
PZ-128 was well tolerated in coronary artery disease and acute coronary syndromes patients undergoing percutaneous coronary intervention and may confer a potential benefit in protecting against myocardial injury along with suppressing platelet thrombus formation in higher-risk patients.
Consistent with its mechanism of interacting with the PAR1 receptor on the inside surface of platelets, the pepducin effects were prolonged with >50% inhibition at 24 hours but were reversible and could be overcome with higher amounts of PAR1 agonist at all time points.
Cardiovascular disease remains the major underlying cause of death in the United States and Europe with 41% to 44% of these deaths due to ischemic coronary heart disease.1,2 Despite highly prevalent use of antithrombotics and lipid-lowering agents, recurrent atherothrombosis remains an area of major unmet medical need with a 5-year mortality rate of 36% to 47% after a first myocardial infarction (MI) in patients ≥45 years old.1 Patients with suspected acute coronary syndrome (ACS) admitted to hospital emergency departments with myocardial injury (myonecrosis) identified by cardiac troponin assay will experience a 17% to 19% rate of MI or death from cardiovascular causes within 1 year.3
Antiplatelet therapy plays a central role in preventing thrombosis and recurrent MI in ACS and high-risk coronary artery disease (CAD), and nonculprit lesion MI.4–6 Platelets regulate normal hemostasis after injury and play a dominant role in arterial thrombus formation. Acute plaque rupture exposes subendothelial collagen which promotes platelet activation and formation of thrombi at the site of vascular damage. Compared with clopidogrel, more potent ADP (adenosine diphosphate) receptor inhibitors, such as prasugrel, ticagrelor, and cangrelor, reduce the risk of ischemic events by 20% in patients with ACS.6,7 However, intensified blockade of P2Y12 with these agents is associated with greater bleeding and other side effects, such as dyspnea.8 Many patients taking these drugs still sustain thrombotic events and would benefit from therapeutics that suppress both platelet activation and myonecrosis.
Parenteral PAR1 receptor inhibition is emerging as a new therapeutic strategy to regulate thrombin-induced platelet activation during percutaneous coronary intervention (PCI) and ACS.9 Currently, there are no approved PAR1 inhibitors for treatment of ACS or patients undergoing PCI. The oral PAR1 inhibitor vorapaxar10–12 was associated with a reduction in risk of MI, stroke, cardiovascular death, and revascularization in stable patients with a previous MI or peripheral arterial disease and was approved by the Food and Drug Administration for these 2 indications. The limitations of vorapaxar include a very long pharmacodynamic half-life of ≥4 weeks and oral administration leading to a slow onset of pharmacodynamic effects during the PCI procedure and an elevated risk of bleeding.13 There have been no reports of fast-acting parenteral PAR1 blockade in humans. The ability to rapidly and reversibly inhibit PAR1 signaling by a parenteral strategy would be an ideal in the high-risk patient undergoing PCI.
PZ-128 is a first-in-class pepducin inhibitor of PAR1 intended for use in patients with CAD/ACS undergoing coronary interventions.14,15 Pepducins are lipidated peptides which target the cytoplasmic surface of their cognate receptor.16,17 Pepducin technology comprises a short peptide derived from a GPCR (G-protein–coupled receptor) intracellular loop, tethered to a hydrophobic moiety. This structure allows the PZ-128 to anchor in the cell membrane lipid bilayer, rapidly flip across to the inner leaflet in a reversible manner, and target the GPCR/G-protein interface to modulate coupling of receptor with G protein (Figure 1).18 PZ-128 has shown significant efficacy in a number of preclinical models including suppressing acute arterial thrombosis in guinea pigs and nonhuman primates and reducing atherosclerotic plaque burden in mice.14,19 Here, we report the results of the TRIP-PCI clinical study (Thrombin Receptor Inhibitory Pepducin in PCI), which evaluated 2 dose levels of PZ-128 (0.3 and 0.5 mg/kg) compared with a placebo in adult patients with CAD/ACS referred for cardiac catheterization and PCI. The objectives were to assess the safety, tolerability, and efficacy of intravenous PZ-128 pepducin in this phase 2 clinical trial.
Figure 1.
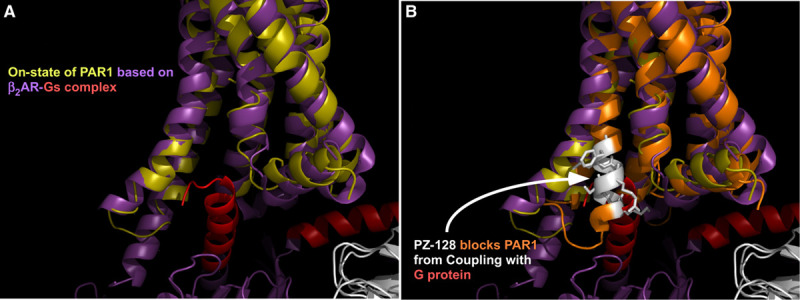
PZ-128. A, The activated structure of PAR1 (protease-activated receptor 1; on-state: chartreuse) bound to intracellular G protein was modeled on the x-ray structure of the β2-adrenergic receptor (purple) bound to Gs (red) and the x-ray structure of PAR1 in the (B) off-state (orange).33 PZ-128 (white), is a N-palmitoylated peptide (palmitate-KKSRALF) derived from the juxtamembrane region of the third intracellular (i3) loop-transmembrane 6 segments of PAR1.14 PZ-128 rapidly flips across the lipid bilayer to the intracellular surface where it specifically inhibits signaling between PAR1 and associated G proteins.
Materials and Methods
Study Design
The data that support the findings of this study are available from the corresponding author upon reasonable request.
The TRIP-PCI trial was a prospective, multicenter randomized, parallel-group, double-blind, placebo-controlled trial to evaluate the safety and efficacy of a single, intravenous infusion of PZ-128 at 2 dose levels (0.3 or 0.5 mg/kg) in adults with CAD/ACS undergoing nonemergent cardiac catheterization ± PCI. The TRIP-PCI trial was conducted at 3 clinical sites in the United States.
Institutional review board approval was obtained before study initiation at each clinical site. The study was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonization E6 guidelines for Good Clinical Practice, and applicable US Food and Drug Administration regulations. All of the study patients provided written informed consent, including US Health Insurance Portability and Accountability Act (1996) authorization, before participation in the study.
Participant Eligibility
Eligible patients included men and women (18 years or older) with stable CAD (stable angina), no angina with abnormal stress test/imaging (other), unstable angina (negative or unknown cardiac biomarker status), or non–ST-segment–elevation myocardial infarction (NSTEMI; positive cardiac biomarker) according to the American College of Cardiology Foundation/American Heart Association20 who were scheduled to undergo nonemergent coronary angiography with the intention to perform PCI if clinically indicated. Patients with any history of stroke or transient ischemic attack were excluded. A comprehensive list of inclusion and exclusion criteria is provided in the Data Supplement.
Randomization and Masking
Eligible patients were randomized within a 14-day screening interval before cardiac catheterization, in order of enrollment via a remote, web-based software platform. Patients were randomly assigned to one of 4 parallel groups (2:2:1:1) to receive either PZ-128 0.3 mg/kg, PZ-128 0.5 mg/kg, placebo 0.3 mg/kg, or placebo 0.5 mg/kg as a single infusion of Investigational Product, based on a computer-generated randomization scheme provided by an independent statistician. Randomization was stratified according to 2 variables: clinical site and urgency of the cardiac catheterization procedure (elective versus urgent).
Participating patients, clinical site investigators and personnel, and the sponsor were blinded to treatment assignments, with the exception of designated clinical site investigational pharmacists who received the patients’ study drug assignments through a restricted role login within a web-response system and prepared the parenteral solutions of PZ-128 or matching placebo. The pepducin was reconstituted from lyophilized powder (48 mg/vial) as micellular nanoparticles with sterile water for injection. Weight-adjusted dosing volumes were diluted into a 250 mL sterile bag of 5% dextrose injection USP resulting in colorless liquids which could not be visually distinguished from placebo.
Study Protocol
On the day of the procedure, all patients received a dose of aspirin orally (81–325 mg) before the initiation of the study drug, which was to commence between 0 and 60 minutes before the start of coronary angiography. Regardless of whether a PCI was performed, the entire 2-hour study drug infusion was to be given without modification to the dose or rate of administration. Allowances were made for a temporary interruption of the infusion up to 1 hour or discontinuation of the study drug if clinically indicated. Background treatment was provided at the discretion of the local clinical investigator according to the current standard-of-care guidelines. If patients underwent a PCI (defined as guidewire crossing the lesion), investigators administered unfractionated or low-molecular-weight heparin anticoagulant (direct thrombin inhibitors were not permitted) followed by a loading dose of an oral P2Y12 inhibitor per investigator preference (clopidogrel, prasugrel, or ticagrelor). Food and Drug Administration-approved bare metal, drug-eluting, or biodegradable coronary stents were implanted. A GP (glycoprotein)IIb/IIIa inhibitor was allowed only as a rescue or bailout therapy to manage thrombotic complications arising during PCI. Patients for whom PCI was not indicated or performed that day based upon the coronary angiography findings were managed medically or recommended for coronary artery bypass grafting (CABG). Following cardiac catheterization ± PCI, investigators employed maintenance antiplatelet therapy with aspirin and oral P2Y12 inhibitor. cTnI (Cardiac troponin I) biomarker blood samples were collected at predose, 2 hours, 4 to 6 hours, discharge (same day) or 24 hours, and 14 days after the start of study drug and measured (Abbott Architect assay; 99th URL=0.028 ng/mL, % coefficient of variation=14.0, limit of detection=0.01 ng/mL) at a central, certified clinical chemistry lab. Patients were seen for an outpatient study visit at 14 days and were contacted via telephone at 30 and 90 days after administration of the study drug for clinical end points (efficacy and safety) and adverse outcomes.
Study End Points
The primary end point was the incidence of TIMI (Thrombolysis In Myocardial Infarction) major or minor bleeding not related to CABG up to 30 days after study drug treatment. Secondary safety end points included TIMI minimal bleeding, clinically significant bleeding, and TIMI bleeding related to CABG up to 30 days. Other safety outcome measures included spontaneously reported adverse events and clinical and laboratory assessments, including vital signs, ECGs, physical examination, blood chemistry, hematology, and coagulation monitoring.
A key secondary efficacy end point was the occurrence of 5-point (5P)-major adverse coronary events (MACE; cardiovascular death, nonfatal MI, nonfatal stroke, recurrent ischemia requiring hospitalization, or urgent coronary revascularization) up to 30 and 90 days. Other secondary efficacy end points included individual components of the composite 5P-MACE end point and stent thrombosis up to 30 and 90 days, ex vivo platelet aggregation and pharmacokinetics.
An independent clinical events committee, blinded to treatment assignment, adjudicated all suspected bleeding events and efficacy end point events identified by investigators or triggered by monitoring procedures according to predefined and standardized criteria utilizing patient medical records/source documentation (see Data Supplement for further details). In particular, a diagnosis of nonfatal or PCI-related (within 48 hours) MI was made by applying the Third Universal Definition of MI21 to documented clinical evidence (symptoms, ECG changes, angiography, or imaging) and cTnI concentrations (local or central lab value determined using a conventional nonhigh sensitivity assay). In addition to all of the planned, predetermined end points listed above, an exploratory post hoc ischemic end point included 30-day MACE plus the occurrence of myocardial injury within 48 hours defined by increasing levels of cTnI (>99th percentile URL threshold(s)21) in absence of ischemic, angiographic or imaging findings as defined by the Third Universal Definition of MI. An independent data and safety monitoring board oversaw the individual and collective safety of the patients in the study by reviewing unadjudicated bleeding events, efficacy end point events, and other adverse events through 90 days.
Pharmacokinetics and Pharmacodynamics
Two clinical sites possessing the capability to perform light transmittance aggregometry participated in the pharmacokinetics/pharmacodynamics substudy to assess platelet function and measure plasma drug concentrations in randomized patients. Blood samples were collected at predose, 0.5 hours, 1 hour, 2 hours, 4 to 6 hours, 24 hours or discharge (same day), and 14 days after initiation of study drug. Research laboratory technicians, blinded to treatment allocation, measured ex vivo inhibition of platelet aggregation induced by multiple agonists in recalcified platelet-rich plasma derived from whole blood samples anticoagulated with sodium citrate (0.4% final) and EDTA (ethylenediaminetetraacetic acid; 0.25 mmol/L final) as described.15 Whole blood samples collected in EDTA for plasma separation were quantified for PZ-128 by an ultra-high performance liquid chromatography-tandem mass spectrometric method.15
Statistical Analysis
TRIP-PCI was designed as a proof-of-concept phase 2 trial to estimate the bleeding risk of the PZ-128 pepducin on top of standard aspirin and P2Y12 inhibitor therapy in patients undergoing cardiac catheterization ± PCI. Power calculations were used to evaluate the size of noninferiority margins that could be detected using the assumption that the incidence of major plus minor bleeding among placebo-treated subjects would range from 2% to 6% (Data Supplement). Safety data were summarized and analyzed as treated, which included randomized patients who received any amount of study drug (n=97). Efficacy data (MACE and stent thrombosis) were summarized by intention-to-treat, which included randomized patients with at least one outcome assessment (n=98). The post hoc efficacy evaluation of MACE plus myocardial injury using cTnI was analyzed by a modified intention-to-treat analysis, which included randomized patients who underwent cardiac catheterization ± PCI and received any amount of study drug and had sufficient cTnI biomarker data available to assess cTnI event status from baseline up to 48 hours (n=95). The Kaplan-Meier analysis of MACE plus myocardial injury at 90 days was performed using a modified intent-to-treat population, which included randomized patients with at least one follow-up end point in the high-risk CAD/ACS subgroup (n=85). Pharmacokinetics (n=20) and pharmacodynamics (platelet aggregation; n=56) data were summarized and analyzed as treated among the pharmacokinetics/pharmacodynamics substudy population. Baseline characteristics were summarized and analyzed by intention-to-treat which included all randomized patients irrespective of initiation of treatment (n=100).
Student t test or Mann-Whitney U test was used to compare continuous variables between groups, and the Fisher exact test was used to compare the categorical variables between 2 groups. Differences between the changes of parameters were analyzed using 2-way repeated-measures ANOVA mixed-effects models with Bonferroni post hoc test correction. Logistic regression adjusted for age and total coronary atherosclerotic burden (total lesions of ≥50% stenosis) was used to compare the relative risks in the post hoc efficacy analysis where a nominal P≤0.05 was considered statistically significant. The Kaplan-Meier cumulative event probability over time was used for the MACE plus myocardial injury events plot, and the Gehan-Breslow-Wilcoxon test was used to produce the hazard ratio and associated P value. For platelet aggregation, repeated measures, 2-way ANOVA mixed-effects models, with dose of PZ-128 and time (global P value) and between group comparisons with placebo (P value versus 0.0 mg/kg) were conducted. Statistical analyses were performed using SAS version 9.3 (SAS Institute Inc, Cary, NC).
Results
Study Design and Dose Selection in the TRIP-PCI Trial
The trial was a parallel-group, multicenter, randomized, double-blind, placebo-controlled phase 2 study. The study used one recruitment stage to simultaneously evaluate safety at 2 dose levels of PZ-128 (0.3 and 0.5 mg/kg; Figure 2) which were expected to produce ample pharmacodynamic target engagement on PAR1 based on the previous phase 1 experience using ex vivo platelet aggregation assays.15
Figure 2.
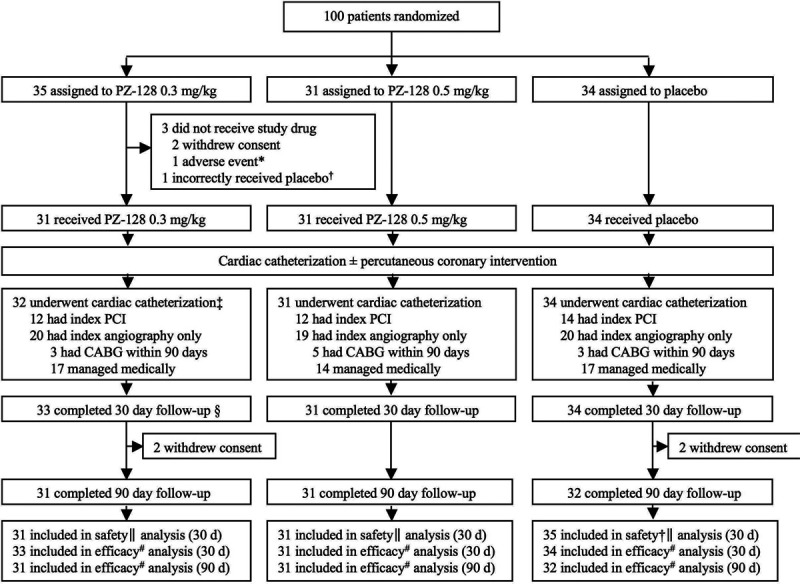
Trial profile. CABG indicates coronary artery bypass grafting; and PCI, percutaneous coronary intervention. *Transient ischemic attack (exclusion criteria) before start of study drug caused cardiac catheterization procedure to be canceled, and patient was followed for 30 d as part of the intent-to-treat population (efficacy). †Patient included in safety analysis for placebo group (n=35). ‡These 32 patients include the one patient† that incorrectly received placebo. §Of the 35 patients assigned to 0.3 mg/kg PZ-128, 2 patients withdrew consent, leaving 33 patients who completed 30-d follow-up. ∥Thrombolysis in myocardial infarction major plus minor bleeding as treated. #Major adverse cardiac events and stent thrombosis by intention-to-treat.
Patient Characteristics in the TRIP-PCI Trial
The TRIP-PCI trial enrolled 100 patients presenting for nonemergent cardiac catheterization and meeting criteria for eligibility as defined by the protocol (Data Supplement) across 3 clinical sites in the United States. The first patient was randomized and dosed on May 27, 2016, and the final patient was dosed on February 19, 2018, with last follow-up contact on May 25, 2018. Patients were assigned to receive PZ-128 0.3 mg/kg (n=35), PZ-128 0.5 mg/kg (n=31), and placebo (n=34; Figure 2). Of the 100 patients randomized, 97 (97%) received the entire 2-hour infusion of PZ-128 (0.3 or 0.5 mg/kg) or placebo and underwent the index cardiac catheterization procedure (Figure 2). Two patients (2%) withdrew consent to participate in the trial immediately after randomization before dosing due to reconsideration. One patient (1%) experienced an adverse event shortly after randomization, was not dosed with the Investigational Product (PZ-128 or placebo), and did not undergo cardiac catheterization but was followed in the trial for 30 days for intent-to-treat. Of the 97 patients who received the Investigational Product infusion and underwent the cardiac catheterization, 38 (39%) received a PCI that day. Out of the 59 patients who did not undergo PCI, 11 (11%) patients went on to have surgical intervention via CABG within the 90-day duration of the trial. Forty-eight (49%) received medical management out of which 3 subsequently ended up getting a PCI between 2-90 days. There was no unblinding done in any patient for CABG or for any reason in the entire study.
The baseline demographics and clinical characteristics of the study population are shown in Table 1. Although there were no significant baseline differences between the combined active (0.3 mg/kg + 0.5 mg/kg) and placebo groups, body mass index was generally higher in the placebo group, while age, smoking history, dyslipidemia, and cTnI tended to be higher in the active drug pool (Table 1). The proportions of patients with an initial diagnosis at presentation of stable angina (39%), unstable angina (27%), NSTEMI (19%), and other/no angina (eg, positive functional tests; 15%) were similar across the groups. Of note, the higher dose level of 0.5 mg/kg comprised a higher-risk group with 26% NSTEMI compared with placebo (18% NSTEMI). Although not significant due to the small numbers, there were 3-times more diabetics in the 0.3 mg/kg PZ-128 group compared with the 0.5 mg/kg PZ-128 group but almost twice as many NSTEMI in the 0.5 mg/kg PZ-128 group compared with the 0.3 mg/kg PZ-128 group. The average TIMI risk score among the 46 patients with ACS was 3.4±1.3 with 70% having a moderately high-risk score of 3 to 4. The index diagnostic angiography was successfully completed in all dosed patients with 88% performed via radial arterial access (Table 2). Overall, the number of lesions with ≥50% stenosis averaged 2.9 (range 0–12) per patient with the PZ-128 group having a higher average (3.1 versus 2.5 placebo) as well as proportion of individuals with ≥5 lesions (31% versus 17% placebo). The duration of the entire cardiac catheterization ± PCI was similar across dose groups with a median time of 32 minutes. The procedure typically ended 1 hour before the blinded Investigational Product infusion was completed. More than half of the patients had dual antiplatelet therapy (52/97, 54%) during the index hospitalization and were routinely discharged on antiplatelet therapy (95%) with 39% aspirin only and 56% dual antiplatelet therapy (favoring clopidogrel 31% versus ticagrelor/prasugrel 25%).
Table 1.
Baseline Patient Characteristics and Periprocedural Medications
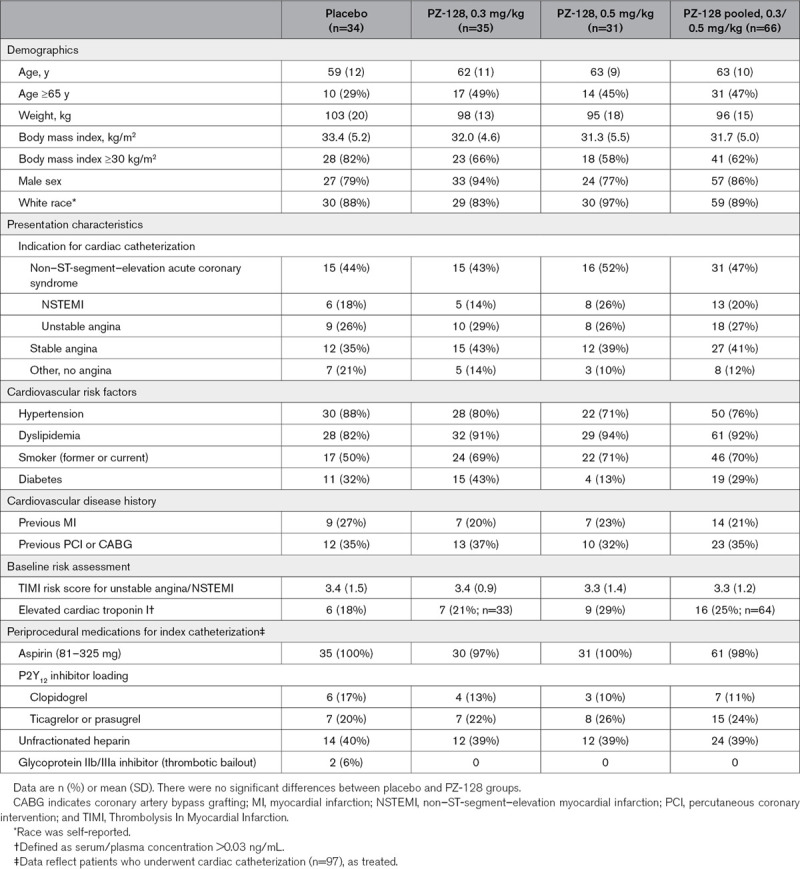
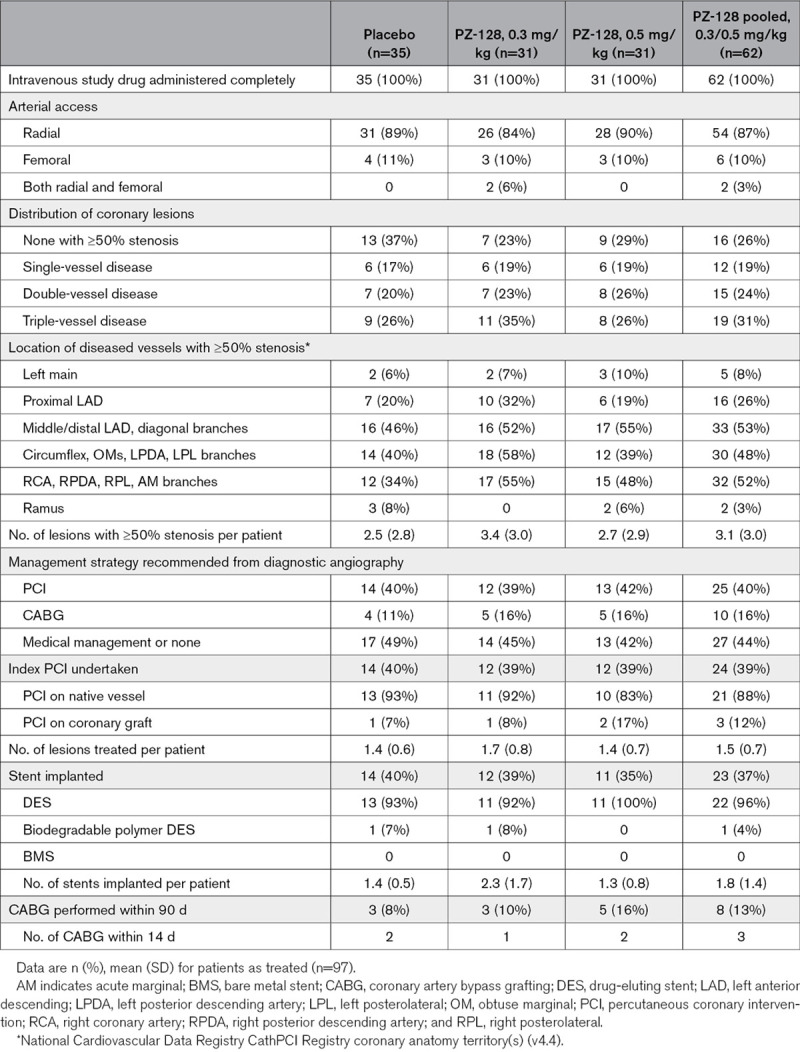
Table 2.
Angiographic and Procedural Characteristics in Patients Undergoing Cardiac Catheterization
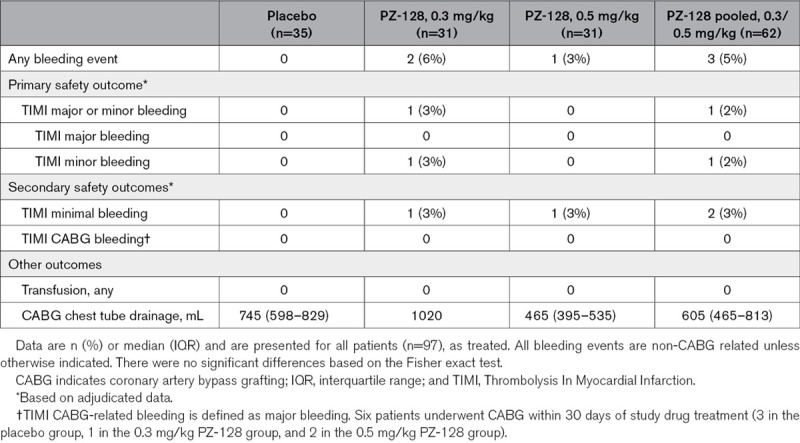
Primary and Secondary Safety Outcomes
The primary end point of the study, namely the incidence of TIMI major plus minor bleeding was 1.0% (1/97) among all patients: 1.6% (1/62) in the PZ-128-treated patients and 0% (0/35) in placebo-treated patients and was not significantly different (Table 3). There were no fatal or major bleeding events or transfusions. There was one minor bleed at the arterial access site in the PZ-128 0.3 mg/kg dose group and no minor bleeds in the high dose (0.5 mg/kg) PZ-128 arm. The incidence of TIMI minimal bleeding (secondary safety end point) was 2.1% (2/97)—one occurred in the 0.3 mg/kg dose group and the other at 0.5 mg/kg, with no bleeding observed in the placebo cohort. CABG was performed on 11 patients ranging from 2 to 51 days, and in 5 patients within 14 days of receiving study drug (Table 2). There were no CABG-related TIMI major bleeding events (0/11) or transfusions required in any patient (0/97). Median 24-hour chest tube drainage in CABG patients was similar between active PZ-128 (425 mL) and placebo groups (420 mL), as was the total drainage (605 and 745 mL, respectively, Table 3).
Table 3.
Bleeding Outcomes From Treatment Onset to 30 Days
Secondary and Exploratory Efficacy Outcomes
The overall incidence of the predetermined secondary end point of MACE—cardiovascular death, MI (nonfatal), stroke (nonfatal), recurrent ischemia requiring hospitalization, or urgent coronary revascularization—at 30 days according to dose group were 5.9% placebo (2/34) and 0% (0/64) PZ-128 group (P=0.13), and at 90 days they were 6.1% (2/33) placebo and 1.6% (1/62) PZ-128 (P=0.29; Table I in the Data Supplement). There was no stent thrombosis (0/37) during the trial.
As shown in the exploratory end point post hoc analysis of Figure 3, PZ-128 was associated with numerically less MACE plus myonecrosis events (Table I in the Data Supplement) among high-risk CAD (diabetes, smoking, prior MI) and NSTE-ACS patients (unstable angina + NSTEMI), patients with positive baseline cardiac biomarkers (NSTEMI/elevated baseline cTnI) and older patients (age ≥75 years). In the high-risk CAD plus ACS patient group (n=83), there were 8/27=30% events in the placebo arm versus 14/56 events=25% in the combined 0.3+0.5 mg/kg PZ-128 dose group (adjusted relative risk [RR]=0.89 [95% CI, 0.44–1.9]; P=0.77). Event rates occurring in the individual high-risk groups of patients with diabetes, smoking, or prior MI ranged from 22% to 29% in the placebo group versus 7% to 17% in the combined drug group, an RR=0.29 to 0.32 (P=0.29–0.32). The ACS subjects (n=42) had a placebo event rate of 43% and PZ-128 event rate of 32% (RR=0.89 [95% CI, 0.39–2.0]; P=0.77). The NSTEMI patients (n=15) had a placebo event rate of 80% versus 40% in the PZ-128 group (RR=0.23 [95% CI, 0.04–1.2]; P=0.08).
Figure 3.
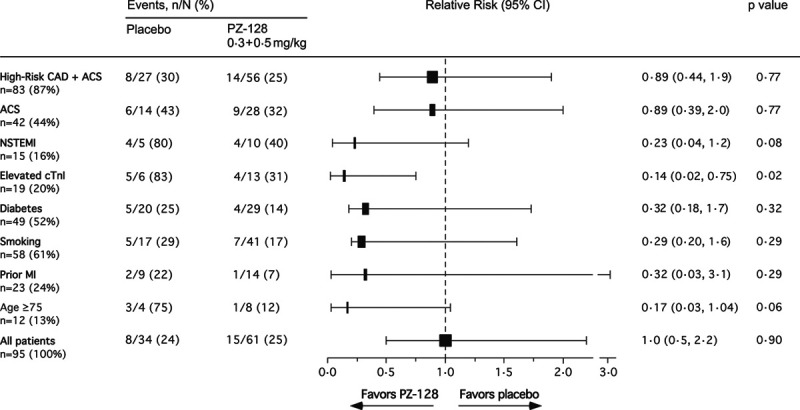
Stratified analysis of major adverse cardiac events (MACE) plus myocardial injury at 30 d across subgroups. A 5P-MACE is defined as a composite of death from cardiovascular causes, nonfatal myocardial infarction (MI), nonfatal stroke, recurrent ischemia with hospitalization, or urgent coronary revascularization. Myocardial injury is defined as an elevation of cTnI (cardiac troponin I) of >5×99% URL (>0.14 ng/mL) in patients with a normal baseline cTnI (<0.028 ng/mL) or >20% increase above an elevated baseline cTnI according to the Third Universal Definition of Myocardial Infarction. Analysis was performed in a modified intent-to-treat population which comprised randomized patients who underwent cardiac catheterization ± percutaneous coronary intervention (PCI) and received any amount of investigational product and had adequate cTnI research biomarker data available for evaluation (n=95). Solid box width is proportional to number of patients in subgroup. Adjusted relative risk (RR) with 95% CI and P values adjusted for age and total coronary atherosclerotic burden (29-segment CASS [Coronary Artery Surgery Study]) using logistic regression (LR). Prior MI did not regress by LR, and χ2 analysis was used instead. P value for age ≥75 was adjusted only for CASS score in the LR model. Diabetes includes prediabetes. Smoking includes former and current. Elevated cTnI patients were those patients with a diagnosis of NSTEMI (non–ST-segment–elevation myocardial infarction) determined by the clinical site before randomization and index catheterization plus additional patients whose elevated cTnI levels were identified from the central research lab baseline sample in the absence of local clinical biomarker assessment. ACS indicates acute coronary syndromes; and CAD, coronary artery disease.
Analysis of the subgroup of patients with elevated baseline cTnI values (n=19) comprised mainly of patients with NSTEMI, showed that the placebo group had 83% (5/6) events versus 31% (4/13) events in the combined PZ-128 group, an RR=0.14 (95% CI, 0.02–0.75; P=0.02; Figure 3). In patients ≥75 years old, there was an RR=0.17 (95% CI, 0.03–1.04; P=0.06) for the PZ-128 cohort (1/8 events) versus placebo (3/4 events).
Figure 4 shows the Kaplan-Meier estimates of the time-to-event for MACE/myonecrosis for the placebo versus combined PZ-128 treatment groups in the high-risk CAD/ACS patients (n=85) over 90 days. There was a nonsignificant decrease in hazard ratio=0.81 (95% CI, 0.32–2.1), P=0.61 of a MACE/myonecrosis event in the PZ-128-treated cohorts versus placebo over 90 days. The majority of the events occurred in the first 24 hours due to the overall dominance of myocardial injury/necrosis based on elevations of cTnI detected during the periprocedural period shortly after cardiac catheterization ± PCI. While most of the benefit of PZ-128 occurred in the first 30 days, the difference from placebo was maintained at 90 days.
Figure 4.
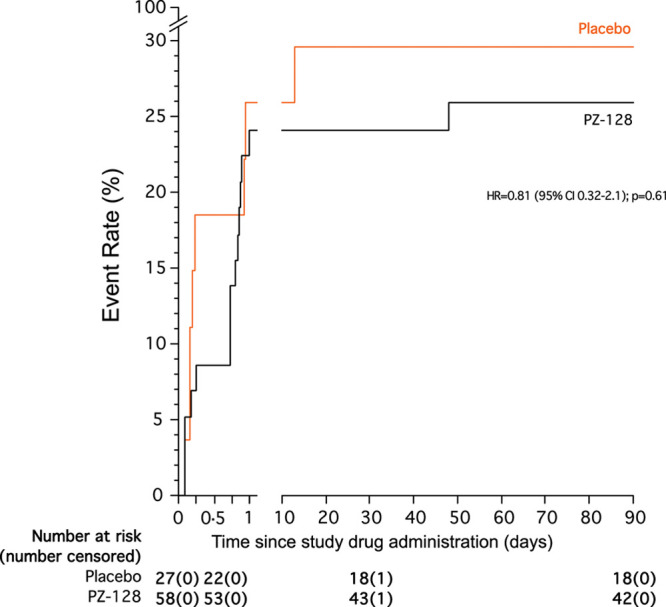
Kaplan-Meier plot for major adverse cardiac events (MACE) plus myocardial injury over 90 d. Kaplan-Meier curves show the cumulative incidence of 5P-MACE plus myocardial injury through 90 d of follow-up after treatment with study drug. The efficacy analysis was conducted in coronary artery disease (CAD) patients with high-risk features or acute coronary syndrome (ACS) who underwent cardiac catheterization ± percutaneous coronary intervention (PCI) and received the study drug as an adjunct to standard antiplatelet and anticoagulant therapies (n=85). The P value was calculated by the Gehan-Breslow-Wilcoxon test.
Platelet Aggregation and Pharmacokinetics of PZ-128
PZ-128 at a dose of 0.5 mg/kg provided significant (P<0.01) inhibition of final platelet aggregation to PAR1 agonist, 5 μmol/L SFLLRN, at 2 hours (end of infusion) and at 24 hours/discharge, and to 20 µmol/L SFLLRN at 24 hours/discharge (P<0.001) compared with placebo (Figure 5A, Table II in the Data Supplement). Onset of inhibition of PAR1 showed 34% mean inhibition of platelet aggregation to 5 μmol/L SFLLRN by the end of the 2 hours infusion and peak inhibition of 61% by 24 hours/discharge. The inhibitory effects of PZ-128 were dose-dependent with lower mean inhibition of platelet aggregation to 5 μmol/L SFLLRN at 1 hour through 14-day time points in the 0.3 mg/kg group as compared to the 0.5 mg/kg group. Inhibition of PAR1 by PZ-128 was reversible with 50% recovery of aggregation at 14 days (32% inhibition of platelet aggregation) relative to 24 hours (61% inhibition of platelet aggregation) in the 0.5 mg/kg dose group (Figure 5A). At the high PAR1 agonist concentration of 20 µmol/L SFLLRN, there was 61% recovery of aggregation by 14 days (14%) relative to 24 hours (36%) with 0.5 mg/kg PZ-128 (Table II in the Data Supplement). PZ-128 was specific for PAR1 with no significant effects on aggregation to any other platelet agonists to the ADP, PAR4 and collagen receptors as compared to placebo (Figure 5B and 5C, Table II in the Data Supplement). The mean PZ-128 plasma concentrations in each treatment group are shown in Figure 5D, and a summary of the pharmacokinetic parameters of PZ-128 is presented in Table III in the Data Supplement. Peak drug levels were proportional to dose and occurred at the end of the 2 hours infusion in both PZ-128 dose levels with a terminal t1/2 of elimination of 1.20 to 1.31 hours. PZ-128 was undetectable in plasma at the 24-hour and 14-day time points.
Figure 5.
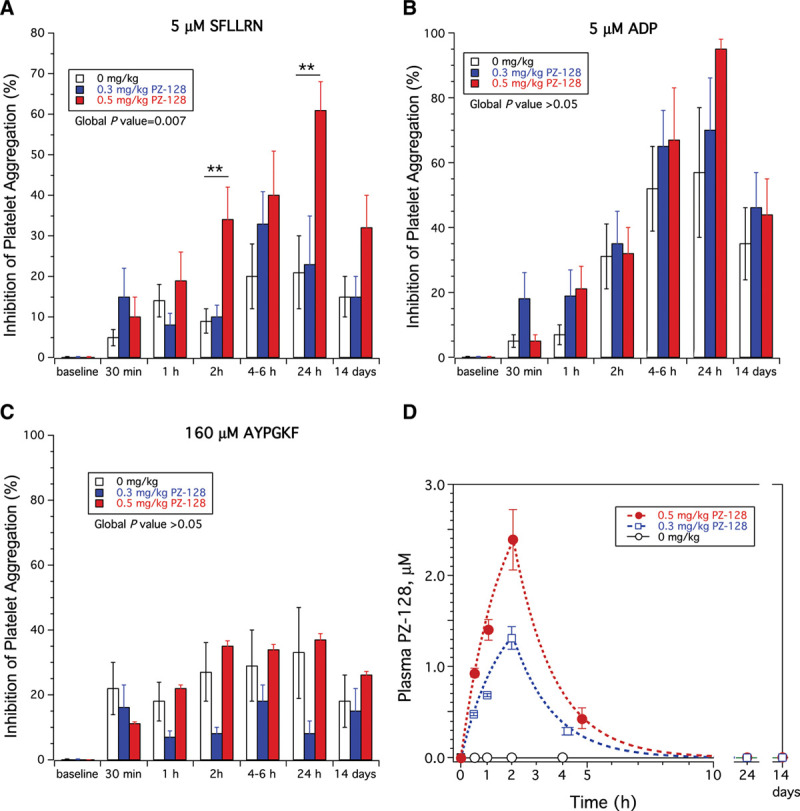
Inhibition of platelet aggregation and pharmacokinetics in TRIP-PCI (Thrombin Receptor Inhibitory Pepducin in Percutaneous Coronary Intervention) patients. Ex vivo aggregation was conducted in platelet-rich plasma by light transmission aggregometry and final aggregation at 7 min normalized to baseline (t=0) for each patient. Inhibition of platelet aggregation (mean±SD) for the placebo (n=20), 0.3 mg/kg PZ-129 (n=19), and 0.5 mg/kg (n=17) cohorts was determined for (A) 5 µmol/L PAR1 (protease-activated receptor 1) agonist peptide, SFLLRN, (B) 5 µmol/L ADP (adenosine diphosphate), (C) 160 µmol/L PAR4 agonist peptide, AYPGKF. The global P value was determined by repeated-measures 2-way ANOVA, **P<0.01 for 0.5 mg/kg PZ-128 vs placebo. D, Plasma levels of PZ-128 in the 3 dose cohorts were determined by mass spectrometry and curves fit to a single-compartment model by nonlinear regression.
Additional Safety and Tolerability Outcomes Measured by Adverse Events
PZ-128 was well tolerated at both dose levels with no observed allergic drug reactions. Most frequently reported treatment-emergent adverse events occurring in the combined PZ-128 groups through 30 days were oral paresthesia, hypotension, and pruritic rash. There were no significant differences between the PZ-128 and placebo groups with respect to routine clinical laboratory assessments, vital signs, or ECG parameters.
Discussion
PZ-128 is a first-in-class, cell-penetrating, membrane-tethered pepducin that targets the intracellular surface of PAR1 to prevent coupling with G proteins. In TRIP-PCI, we showed that PZ-128 was well tolerated and did not cause a significant elevation in bleeding in CAD/ACS patients undergoing cardiac catheterization ± PCI even when administered on top of dual antiplatelet drugs (aspirin and P2Y12 inhibitor) and heparin. Likewise, there was no elevated bleeding observed in the patients that underwent CABG instead of PCI after receiving PZ-128. Although this phase 2 study was not powered to specifically detect clinical efficacy, a potential reduction in MACE plus myocardial injury events at 30 days was noted in the PZ-128 cohort relative to placebo in patients who had elevated baseline cTnI levels (ie, NSTEMI), in a post hoc analysis.
Contrary to thrombin inhibitors (bivalirudin, hirudin, argatroban, and dabigatran), PAR1 antagonists do not interfere with the procoagulant activity of thrombin cleavage of fibrinogen.22 Because thrombin-dependent fibrin generation is largely unaffected by inhibition of PAR1, a thrombin-receptor antagonist should be safer a priori than a thrombin inhibitor22,23 in treating arterial thrombosis. PZ-128 did not prevent initial platelet adhesion to exposed collagen surfaces but prevented large occlusive thrombi from forming.24 PZ-128 significantly suppressed platelet aggregation to PAR1 agonist, SFLLRN, at 2- to 24-hour time points in TRIP-PCI but did not inhibit aggregation to PAR4, ADP, or collagen agonists. These findings suggest that PZ-128 can permit the formation of initial platelet monolayer necessary for control of bleeding but block pathological thrombus propagation that occurs at the site of plaque rupture or iatrogenic endothelial denudation. Furthermore, synergistic protective effects against carotid artery thrombosis using dual therapy of PZ-128 plus clopidogrel was shown previously in guinea pig.14 Similar to these findings, the PAR1 inhibitor SCH 602539 was shown to synergistically inhibit thrombosis in a Folts model in monkeys in combination with the P2Y12 inhibitor cangrelor.25 These synergistic effects between PAR1 and P2Y12 were predicted by platelet functional studies showing that PAR1 preferentially contributes to shape change, calcium flux and early GPIIb-IIIa activation, whereas P2Y12 stabilizes the high affinity state of GPIIb-IIIa leading to irreversible aggregation.4,26 Therefore, inhibiting both pathways should have a strong synergistic protective effect in PCI patients. In this regard, there was an extra 15% to 38% inhibition of ADP-dependent aggregation at the 4 to 6 hours and 24-hour time points in the 0.5 mg/kg PZ-128 versus placebo groups yielding nearly complete inhibition (95%) of mean final ADP aggregation at the 24-hour point relative to placebo (60%) This indicates synergism of ADP platelet inhibition with dual antiplatelet therapy (PZ-128/P2Y12 inhibitor) on top of aspirin in the periprocedural time window. This extra synergism on ADP inhibition was not observed in recent studies with the Food and Drug Administration-approved dose (2.08 mg) of vorapaxar at even up to 30 days of repeat administration.13
Despite the critical importance of PAR1 in mediating platelet-thrombosis and myocardial ischemia, there are no approved PAR1 inhibitors for the treatment of NSTE-ACS (unstable angina, NSTEMI), STEMI, or patients undergoing PCI. The orally active vorapaxar was approved in the United States as an adjunctive treatment for the secondary prevention of atherothrombotic events in patients with prior MI or peripheral arterial disease and no history of stroke. The TRACER study (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome) investigated the ability of vorapaxar to reduce 5P-MACE on top of standard antiplatelet therapy in patients with high-risk NSTE-ACS.12 In TRACER, vorapaxar significantly reduced the risk of the secondary end point of cardiovascular death, MI, or stroke. However, there was a significant increase in moderate and severe bleeding with vorapaxar compared with placebo, including intracerebral hemorrhage.12 The effects of vorapaxar in preventing the composite of cardiovascular death, MI, stroke, or urgent coronary revascularization in stable patients with established atherothrombotic disease was assessed in the Thrombin Receptor Antagonist TRA 2°P-TIMI 50 study.11 In TRA 2°P-TIMI 50, the original primary end point of cardiovascular death, MI, stroke, or recurrent ischemia leading to revascularization was significantly reduced in the vorapaxar group versus placebo as was the reordered primary end point of 3-year cardiovascular death, MI, or stoke. Again, there was an increased risk of moderate or severe bleeding and increased rate of intracerebral hemorrhage in the vorapaxar group.11 These data suggested that while vorapaxar is effective at reducing ischemic outcomes in a broad population of patients with acute and chronic atherothrombosis in combination with other antiplatelet therapy (eg, aspirin and P2Y12), this comes with the cost of an increased risk of clinically significant bleeding, due in large part to the extremely long ≥4 weeks pharmacodynamic half-life of vorapaxar. The ability to rapidly and reversibly inhibit PAR1 signaling by an intravenous strategy such as PZ-128 would be an ideal in the high-risk patient undergoing cardiac intervention and may translate into a superior reduction in adverse thrombotic event occurrence. For the highest risk groups, namely patients with NSTEMI and STEMI, who may derive the largest clinical benefit from a fast-acting reversible PAR1 agent, the routine use of the strongest antiplatelet drugs, namely intravenous GPIIb/IIIa inhibitors,27 is no longer recommended in combination with heparin, due to a less clear benefit in the modern era of more effective ADP receptor (P2Y12) antagonists, safer interventional techniques (ie, radial approach), and the associated significant increase in bleeding risk.5
In support of the notion that PAR1 may play an additional role in mediating cardiac ischemic injury, post hoc efficacy analysis of 30-day MACE plus myocardial injury events in the subgroup of patients with elevated baseline cTnI values showed a potential treatment effect for PZ-128 with RR=0.14 (95% CI, 0.02–0.75, P=0.02) compared with placebo. This positive effect could be due to a type 1 error due to the low number of patients, however, all of the other high-risk subgroup analyses (high-risk CAD+ACS; ACS; NSTEMI; diabetes; smoking; prior MI; age ≥75) also showed a relative risk reduction of a MACE+myocardial injury event which favors PZ-128 over placebo (with P values ranging from 0.06 to 0.89). Indeed, PAR1 has been shown to directly mediate myocardial ischemia/reperfusion injury28,29 and cardiac remodeling and hypertrophy30 in rodent models of MI which lack PAR1 on their platelets. PAR1-deficiency reduced dilation of the left ventricle and improved left ventricular function after ischemia/reperfusion injury similar to PZ-128 but had no effect on scar formation at 2 weeks.30 It is possible that infarct differences at 2 hours may not be the same at 2 weeks or longer in rodents and humans, to be examined in future studies with PZ-128. Complete genetic deficiency of PAR1 may have effects on cardiovascular function in mice including 50% embryonic lethality at E12.5, and potential compensation on hemodynamic effects in surviving animals, which could account for differences in wound healing processes that occur over chronic time periods.31 Strande et al28 demonstrated that treatment with SCH79797 before or during ischemia significantly reduced infarct size associated with ischemia/reperfusion injury in rat. The PAR1 inhibitor also attenuated left ventricular dilation and improved left ventricular systolic function of reperfused rat myocardium at 28 days with a decrease in scar size.29 Overall, these findings indicate both platelet-dependent and independent effects of PAR1 inhibitors that could potentially translate into reduced cardiac events.
This was a proof-of-concept study in 100 patients undergoing cardiac catheterization ± PCI that was primarily intended to show safety and evaluate bleeding risk of intravenous PZ-128 pepducin on top of standard-of-care antiplatelet therapies. A limitation was the bleeding rate was extremely low (0%–1.6%), in part, due to the high use of standard-of-care radial access (88%). Therefore, the study was not sufficiently powered for formal testing of noninferiority for the primary end point at 30 days (would require >600 patients) or to detect clinically meaningful differences in TIMI bleeding events between treatment groups. Given the small numbers of NSTEMI subjects in TRIP-PCI, a larger study that includes a high proportion of NSTEMI subjects will be needed to validate whether PZ-128 can confer a potential dual benefit in protecting against cardiac ischemia/acute myocardial injury along with suppressing thrombus formation in patients presenting with more significant coronary disease. It is noteworthy that patients with acute myocardial injury according the Third or Fourth Universal Definition of MI have a comparable rate of cardiovascular death or MI at 1 year as patients with type 1 MI (14%–17%) due to occlusive ischemic events.32
In conclusion, the favorable initial safety profile, and potentially protective effects against myocardial ischemia/necrosis associated with PZ-128 indicates that the TRIP-PCI intervention provides the basis for additional clinical trials. Establishing rapid and reversible receptor blockade with a parenterally administered PAR1 blocker may translate into a superior reduction in adverse post-PCI thrombotic event occurrence with acceptable bleeding. Pepducins, as exemplified by PZ-128, therefore represent a new approach to modulating signal transduction from the inside surface of the cell, potentially expanding the scope of GPCR therapeutics to treat a much wider range of severe and life-threatening illnesses, such as acute arterial thrombosis.
Acknowledgments
We thank the patients, study coordinators, and investigators who participated in TRIP-PCI (Thrombin Receptor Inhibitory Pepducin in Percutaneous Coronary Intervention), and Nga Nguyen, Rachel Bell, and Udaya Tantry for performing platelet aggregometry. We are especially grateful to David Simon for performing statistical analyses.
Sources of Funding
The TRIP-PCI trial (Thrombin Receptor Inhibitory Pepducin in Percutaneous Coronary Intervention) was funded through the National Heart, Lung, and Blood Institute by P50HL110789 and R01HL136485 (Dr Kuliopulos), and the National Heart, Lung, and Blood Institute (NHLBI) SMARTT program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
None.
Supplemental Material
Online Expanded Methods
Online Tables I–III
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- ACS
- acute coronary syndromes
- CABG
- coronary artery bypass grafting
- CAD
- coronary artery disease
- cTnI
- cardiac troponin I
- GP
- glycoprotein
- GPCR
- G-protein-coupled receptor
- MI
- myocardial infarction
- NSTEMI
- non-ST-segment-elevation myocardial infarction
- PAR1
- protease-activated receptor 1
- PCI
- percutaneous coronary intervention
- TIMI
- Thrombolysis In Myocardial Infarction
- TRIP-PCI
- Thrombin Receptor Inhibitory Pepducin in Percutaneous Coronary Intervention
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.315168.
For Sources of Funding and Disclosures, see page 3002.
Contributor Information
Paul A. Gurbel, Email: Pgurbel@lifebridgehealth.org.
Jeffrey J. Rade, Email: jeffrey.rade@umassmed.edu.
Carey D. Kimmelstiel, Email: ckimmelstiel@tuftsmedicalcenter.org.
Susan E. Turner, Email: turnersusanelizabeth@gmail.com.
Kevin P. Bliden, Email: kbliden@lifebridgehealth.org.
Elizabeth K. Fletcher, Email: e.fletcher01@yahoo.com.au.
Daniel H. Cox, Email: dancox284@gmail.com.
Lidija Covic, Email: lcovic@tuftsmedicalcenter.org.
References
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, et al. ; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659 [DOI] [PubMed] [Google Scholar]
- 2.Timmis A, Townsend N, Gale CP, Torbica A, Lettino M, Petersen SE, Mossialos EA, Maggioni AP, Kazakiewicz D, May HT, et al. ; European Society of Cardiology. European Society of Cardiology: cardiovascular disease statistics 2019. Eur Heart J. 2020;41:12–85. doi: 10.1093/eurheartj/ehz859 [DOI] [PubMed] [Google Scholar]
- 3.Shah ASV, Anand A, Strachan FE, Ferry AV, Lee KK, Chapman AR, Sandeman D, Stables CL, Adamson PD, Andrews JPM, et al. ; High-STEACS Investigators. High-sensitivity troponin in the evaluation of patients with suspected acute coronary syndrome: a stepped-wedge, cluster-randomised controlled trial. Lancet. 2018;392:919–928. doi: 10.1016/S0140-6736(18)31923-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurbel PA, Kuliopulos A, Tantry US. G-protein-coupled receptors signaling pathways in new antiplatelet drug development. Arterioscler Thromb Vasc Biol. 2015;35:500–512. doi: 10.1161/ATVBAHA.114.303412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reed GW, Rossi JE, Cannon CP. Acute myocardial infarction. Lancet. 2017;389:197–210. doi: 10.1016/S0140-6736(16)30677-8 [DOI] [PubMed] [Google Scholar]
- 6.Scirica BM, Bergmark BA, Morrow DA, Antman EM, Bonaca MP, Murphy SA, Sabatine MS, Braunwald E, Wiviott SD. Nonculprit lesion myocardial infarction following percutaneous coronary intervention in patients with acute coronary syndrome. J Am Coll Cardiol. 2020;75:1095–1106. doi: 10.1016/j.jacc.2019.12.067 [DOI] [PubMed] [Google Scholar]
- 7.Bhatt DL, Stone GW, Mahaffey KW, Gibson CM, Steg PG, Hamm CW, Price MJ, Leonardi S, Gallup D, Bramucci E, et al. ; CHAMPION PHOENIX Investigators. Effect of platelet inhibition with cangrelor during PCI on ischemic events. N Engl J Med. 2013;368:1303–1313. doi: 10.1056/NEJMoa1300815 [DOI] [PubMed] [Google Scholar]
- 8.Storey RF, Bliden KP, Patil SB, Karunakaran A, Ecob R, Butler K, Teng R, Wei C, Tantry US, Gurbel PA; ONSET/OFFSET Investigators. Incidence of dyspnea and assessment of cardiac and pulmonary function in patients with stable coronary artery disease receiving ticagrelor, clopidogrel, or placebo in the ONSET/OFFSET study. J Am Coll Cardiol. 2010;56:185–193. doi: 10.1016/j.jacc.2010.01.062 [DOI] [PubMed] [Google Scholar]
- 9.Mackman N, Bergmeier W, Stouffer GA, Weitz JI. Therapeutic strategies for thrombosis: new targets and approaches. Nat Rev Drug Discov. 2020;19:333–352. doi: 10.1038/s41573-020-0061-0 [DOI] [PubMed] [Google Scholar]
- 10.Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J, Niederman A, Ziada KM, Berman G, Strony J, Joseph D, et al. ; TRA-PCI Investigators. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet. 2009;373:919–928. doi: 10.1016/S0140-6736(09)60230-0 [DOI] [PubMed] [Google Scholar]
- 11.Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KA, Lipka LJ, Liu X, Nicolau JC, et al. ; TRA 2P–TIMI 50 Steering Committee and Investigators. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404–1413. doi: 10.1056/NEJMoa1200933 [DOI] [PubMed] [Google Scholar]
- 12.Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, et al. ; TRACER Investigators. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20–33. doi: 10.1056/NEJMoa1109719 [DOI] [PubMed] [Google Scholar]
- 13.Bliden K, Chaudhary R, Kuliopulos A, Tran H, Taheri H, Tehrani B, Rosenblatt A, Navarese E, Tantry US, Gurbel PA. Effects of vorapaxar on clot characteristics, coagulation, inflammation, and platelet and endothelial function in patients treated with mono- and dual-antiplatelet therapy. J Thromb Haemost. 2020;18:23–35. doi: 10.1111/jth.14616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang P, Gruber A, Kasuda S, Kimmelstiel C, O’Callaghan K, Cox DH, Bohm A, Baleja JD, Covic L, Kuliopulos A. Suppression of arterial thrombosis without affecting hemostatic parameters with a cell-penetrating PAR1 pepducin. Circulation. 2012;126:83–91. doi: 10.1161/CIRCULATIONAHA.112.091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gurbel PA, Bliden KP, Turner SE, Tantry US, Gesheff MG, Barr TP, Covic L, Kuliopulos A. Cell-penetrating pepducin therapy targeting PAR1 in subjects with coronary artery disease. Arterioscler Thromb Vasc Biol. 2016;36:189–197. doi: 10.1161/ATVBAHA.115.306777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc Natl Acad Sci USA. 2002;99:643–648. doi: 10.1073/pnas.022460899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. 2002;8:1161–1165. doi: 10.1038/nm760 [DOI] [PubMed] [Google Scholar]
- 18.O’Callaghan K, Kuliopulos A, Covic L. Turning receptors on and off with intracellular pepducins: new insights into G-protein-coupled receptor drug development. J Biol Chem. 2012;287:12787–12796. doi: 10.1074/jbc.R112.355461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rana R, Huang T, Koukos G, Fletcher EK, Turner SE, Shearer A, Gurbel PA, Rade JJ, Kimmelstiel CD, Bliden KP, et al. Noncanonical matrix metalloprotease 1-protease-activated receptor 1 signaling drives progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2018;38:1368–1380. doi: 10.1161/ATVBAHA.118.310967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannon CP, Brindis RG, Chaitman BR, Cohen DJ, Cross JT, Jr, Drozda JP, Jr, Fesmire FM, Fintel DJ, Fonarow GC, Fox KA, et al. ; American College of Cardiology Foundation/American Heart Association Task Force on Clinical Data Standards; American College of Emergency Physicians; Emergency Nurses Association; National Association of Emergency Medical Technicians; National Association of EMS Physicians; Preventive Cardiovascular Nurses Association; Society for Cardiovascular Angiography and Interventions; Society of Cardiovascular Patient Care; Society of Thoracic Surgeons. 2013 ACCF/AHA key data elements and definitions for measuring the clinical management and outcomes of patients with acute coronary syndromes and coronary artery disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Acute Coronary Syndromes and Coronary Artery Disease Clinical Data Standards). Circulation. 2013;127:1052–1089. doi: 10.1161/CIR.0b013e3182831a11 [DOI] [PubMed] [Google Scholar]
- 21.Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, Katus HA, Lindahl B, Morrow DA, Clemmensen PM, et al. ; Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. Circulation. 2012;126:2020–2035. doi: 10.1161/CIR.0b013e31826e105822923432 [Google Scholar]
- 22.Kimmelstiel C, Zhang P, Kapur NK, Weintraub A, Krishnamurthy B, Castaneda V, Covic L, Kuliopulos A. Bivalirudin is a dual inhibitor of thrombin and collagen-dependent platelet activation in patients undergoing percutaneous coronary intervention. Circ Cardiovasc Interv. 2011;4:171–179. doi: 10.1161/CIRCINTERVENTIONS.110.959098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petzold T, Thienel M, Konrad I, Schubert I, Regenauer R, Hoppe B, Lorenz M, Eckart A, Chandraratne S, Lennerz C, et al. Oral thrombin inhibitor aggravates platelet adhesion and aggregation during arterial thrombosis. Sci Transl Med. 2016;8:367ra168 doi: 10.1126/scitranslmed.aad6712 [DOI] [PubMed] [Google Scholar]
- 24.Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O’Callaghan K, Covic L, Kuliopulos A. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chintala M, Strony J, Yang B, Kurowski S, Li Q. SCH 602539, a protease-activated receptor-1 antagonist, inhibits thrombosis alone and in combination with cangrelor in a Folts model of arterial thrombosis in cynomolgus monkeys. Arterioscler Thromb Vasc Biol. 2010;30:2143–2149. doi: 10.1161/ATVBAHA.110.203414 [DOI] [PubMed] [Google Scholar]
- 26.Covic L, Singh C, Smith H, Kuliopulos A. Role of the PAR4 thrombin receptor in stabilizing platelet-platelet aggregates as revealed by a patient with Hermansky-Pudlak syndrome. Thromb Haemost. 2002;87:722–727. [PubMed] [Google Scholar]
- 27.Kimmelstiel C, Badar J, Covic L, Waxman S, Weintraub A, Jacques S, Kuliopulos A. Pharmacodynamics and pharmacokinetics of the platelet GPIIb/IIIa inhibitor tirofiban in patients undergoing percutaneous coronary intervention: implications for adjustment of tirofiban and clopidogrel dosage. Thromb Res. 2005;116:55–66. doi: 10.1016/j.thromres.2004.11.011 [DOI] [PubMed] [Google Scholar]
- 28.Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts. Basic Res Cardiol. 2007;102:350–358. doi: 10.1007/s00395-007-0653-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J, Strande JL. Protease-activated receptor 1 inhibition by SCH79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J Cardiovasc Pharmacol Ther. 2013;18:460–475. doi: 10.1177/1074248413485434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, Andrade-Gordon P, Kotzsch M, Spring D, Luther T, et al. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation. 2007;116:2298–2306. doi: 10.1161/CIRCULATIONAHA.107.692764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114:1070–1077. doi: 10.1161/CIRCULATIONAHA.105.574830 [DOI] [PubMed] [Google Scholar]
- 32.Chapman AR, Adamson PD, Shah ASV, Anand A, Strachan FE, Ferry AV, Ken Lee K, Berry C, Findlay I, Cruikshank A, et al. ; High-STEACS Investigators. High-sensitivity cardiac troponin and the universal definition of myocardial infarction. Circulation. 2020;141:161–171. doi: 10.1161/CIRCULATIONAHA.119.042960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Leger AJ, Baleja JD, Rana R, Corlin T, Nguyen N, Koukos G, Bohm A, Covic L, Kuliopulos A. Allosteric activation of a G protein-coupled receptor with cell-penetrating receptor mimetics. J Biol Chem. 2015;290:15785–15798. doi: 10.1074/jbc.M115.636316 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.