

Abstract
Gastroesophageal cancers are leading causes of cancer death. Our attempts at adopting molecularly based treatment approaches have been slow and ineffective even though we begin to identify specific targetable gene mutations and pathways. It is clear that we should no longer treat all gastroesophageal cancers as a homogeneous disease, which is what we do when we use non-specific chemotherapy. However, we currently cannot monitor successful gene/pathway targeting, nor understand how/when tumors develop resistance, nor predict which patients will derive maximal benefit. To improve outcomes, we must precisely detail the heterogeneity of these tumors to then individualize cancer therapy as well as develop novel avenues to study and predict treatment effects in individual patients. To this end, patient-derived organoids, in which tumor cells from individual patients are grown in a Petri dish, are a new versatile system that allows for timely expandability, detailed molecular characterization, and genetic manipulation with the promise of enabling predictive assessment of treatment response. In this review, we will explore the development and basic techniques for organoid generation, and discuss the current and potential future applications of this exciting technology to study the basic science of carcinogenesis and to predict/guide cancer patient care in the clinics.
Keywords: targeted therapy, precision oncology, cancer model, tumor heterogeneity, cancer evolution, personalized medicine
Introduction
Cancers of the upper gastrointestinal tract represent a substantial fraction of cancer incidence and mortality world-wide (Bray et al., 2018) and in the USA (Siegel et al., 2019). Symptoms are often insidious (Wanebo et al., 1993; Rustgi and El-Serag, 2014) resulting in many patients presenting with advanced disease, especially in the USA where endoscopic screening is not routine (Leung et al., 2008; Zhang et al., 2018). In addition, there has been an alarming increase in both stomach and esophageal cancers in certain US demographics over the past several decades (Pohl and Welch, 2005; Anderson et al., 2010). Despite the substantial clinical impact of upper gastrointestinal cancers, the foundation of treatment remains one-size-fits-all cytotoxic chemotherapy, with our best regimens still only affording patients a median overall survival of just under 12 months (Wagner et al., 2010; Ter Veer et al., 2016). In other solid tumors, targeted therapies (e.g. based on specific altered molecular pathways) have been a welcome addition to traditional chemotherapy, increasing not only survival metrics but quality of life as well (Cunningham et al., 2004; Piccart-Gebhart et al., 2005; Maemondo et al., 2010; Chapman et al., 2011; Shaw et al., 2013; Schwaederle et al., 2015). Unfortunately, the wave of new targeted therapeutic agents has not resulted in a survival benefit in advanced gastroesophageal cancers (Samson and Lockhart, 2017)—a surprising fact, given the high prevalence of potentially targetable molecular alterations these tumors harbor (Cancer Genome Atlas Research Network, 2014; Cancer Genome Atlas Research Network et al., 2017).
One reason for the lag between our molecular understanding and the clinical availability of improved treatments is that large clinical trials may mask responses of individual patient subgroups. One molecular target that has yielded positive clinical benefit is the HER2/ERBB2 receptor tyrosine kinase pathway, resulting in trastuzumab becoming the only targeting agent approved in the frontline setting for advanced gastroesophageal cancers (Bang et al., 2010). This study used a biomarker-driven strategy (high HER2 protein expression) to enrich for a subset of advanced gastroesophageal cancer patients that would derive the maximal clinical benefit. It seems unlikely that gastroesophageal adenocarcinomas are somehow uniquely resistant to biomarker-driven targeted therapy, rather it is likely our lack of understanding that is causing the frustration in the field. Thus, novel experimental approaches are needed to improve our ability to truly pair cohorts of patients harboring specific molecular pathway signatures with targeted therapy.
Specifically, to increase our therapeutic arsenal for gastroesophageal cancer, we need to invoke precision oncology, an emerging philosophy that is resulting in a paradigm shift in oncology treatment strategy (Woodcock and LaVange, 2017). However, our current methods to individualize cancer treatments are still largely based on genomic mutational data obtained from tissue sampling that is limited in quantity, obtained at discreet time points during the disease, and often not reflective of overall tumor heterogeneity (Conley and Doroshow, 2014). In this review, we will present a new tool to apply for precision oncology: the patient-derived organoid (PDO). We will discuss the development of this technology from a historical perspective and current applications. Specifically, we will highlight the novel ways in which PDOs might be purposed to address many of the most difficult and poignant questions in solid tumor oncology. Finally, we will posit how PDOs may translate from bench to the bedside to truly enable oncologists to tailor treatment for individual cancer patients.
The development of mouse and human gastrointestinal organoids
Before expanding on the implications and applications of patient-derived tumor organoids, let us define the term ‘organoid’. Lancaster and Knoblich (2014) termed an organoid as: (i) composed of specific cell types, (ii) able to reproduce some function of the organ, and (iii) organized in a similar fashion as the organ. In other words, an organoid is a ‘collection of organ-specific cell types that develops from stem cells or organ progenitors and self-organizes through cell sorting and spatially restricted lineage commitment in a manner similar to in vivo’. Organoids can be generated either directly by growing cells from specific organs or by starting with pluripotent stem cells (from embryonic stem cell lines or induced via reprogramming of adult cells) and then differentiating them step-by-step toward specific mature organ phenotypes (Clevers, 2016; McCauley and Wells, 2017; Schutgens and Clevers, 2020). As we are concerned largely with using PDOs to understand cancer, we will focus on organoids derived directly from normal and tumor tissues.
The ability to culture and propagate adult stem cells originated from seminal studies in the mouse intestines from the Clevers lab including the discovery that multipotent adult intestinal stem cells express a Wnt signaling receptor: Leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5) (Barker et al., 2007). Identification of this marker facilitated the design of in vitro culture conditions that mimic Wnt signaling to propagate these multipotent intestinal stem cells (Sato et al., 2009). A basic understanding of gastrointestinal development and the unique signaling pathways needed to generate each anatomic segment has enabled culturing of organoids from various tissues (McGrath and Wells, 2015). Basic tenets of gastrointestinal organoid culturing include: (i) a source of Wnt signaling using Wnt3a and R-spondin, which is an extracellular matrix-associated protein that works with LGR5 to promote Wnt signaling; (ii) need for additional growth factors (e.g. EGF); (iii) inhibition of the BMP pathway using Noggin; (iv) inhibition of anoikis using ROCK inhibitor; and (v) physical growth support (most commonly the extracellular matrix mimetic, Matrigel) (Figure 1). With minor variations on these themes, organoids from multiple murine tissues have been generated (Clevers, 2016). Specifically, in the mouse stomach, LGR5-positive antral cells have been used to generate antral/pyloric organoids with the additional requirement of FGF10 and gastrin (Barker et al., 2010). Organoids have also been generated from the stomach body (corpus) (Stange et al., 2013). Murine esophageal organoids have been grown through the conventional Wnt3a, R-spondin, Noggin, and growth factor media culturing technique (DeWard et al., 2014) and by an alternative method using keratinocyte serum-free media (KSFM) supplemented with EGF and calcium without the addition of Wnt3a, R-spondin, or Noggin (Kasagi et al., 2018).
Figure 1.
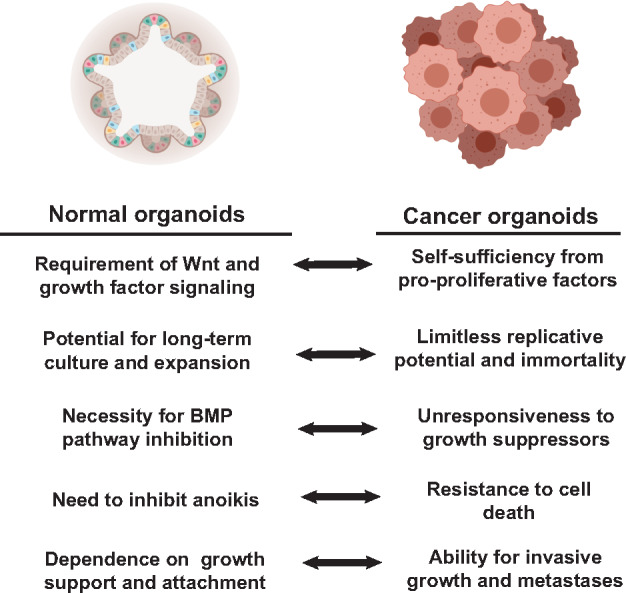
Comparison between growth parameters and culture requirements of normal vs. cancer organoid cultures. Normal organoid culture conditions are designed to allow indefinite expansion, mimicking certain aspects of carcinogenesis. Cancers by definition are prone to indefinite expansion in the absence of external queues, so when cancers are cultured in organoid conditions, they may variably not require certain culture media components. All figures are created with BioRender.com.
Methods for deriving human intestinal organoids necessitated adaptations of the murine protocol with additional factors required including nicotinamide, inhibition of ALK5/4/7 (a.k.a. TGFBR1, ACVR1B, ACVR1C) through the small-molecule inhibitor A83-01, and inhibition of p38 MAPK signaling using the small-molecular inhibitor SB202190 (Sato et al., 2011). For human stomach organoids, p38 inhibition with SB202190 was not needed, and nicotinamide paradoxically allowed initial organoid formation but limited the longevity of the cultured organoids (Bartfeld et al., 2015). Successful culturing of human esophageal organoids was shown using the KSFM media-based method (Kasagi et al., 2018). It is important to note that these cultured human organoids remain faithful histologically and genetically over long-term culture conditions through multiple passages spanning several months (Sato et al., 2011; Bartfeld et al., 2015). So far, whatever somatic mutations or polymorphisms present seem to be correlated with the original age of the source tissue (Blokzijl et al., 2016), not subsequent passaging. Currently, organoids have been generated from a vast array of both murine and human tissues (Clevers, 2016; Li and Izpisua Belmonte, 2019).
We will now focus the remainder of our review on a discussion of the current and future oncology applications of organoid culturing techniques. There is a central seeming contradiction in our use of cancer organoids that can be seen from the perspective of the so-called ‘Hallmarks of Cancer’ (Hanahan and Weinberg, 2011). Tumors are defined as different from non-tumor tissue by virtue of their ‘sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis’. Thus, the requirements for growing normal organoids are what tumors have often evolved not to need (Figure 1). In other words, we are growing normal tissue organoids in culture conditions designed to mimic indefinite growth like tumors, but cancers inherently possess those features. We will discuss the implications of this tumor‒non-tumor relationship within the perspective of studies that have addressed the unique nature of cancer organoids.
Culturing human cancer organoids and establishment of cancer biobanks
The feasibility of culturing human cancer specimens originated from pioneering work in the intestines (Sato et al., 2011). ‘Tumoroids’ were developed by culturing murine intestinal adenomas using the Lgr5-GFP-ires-CreER/APCfl/fl mouse model (Barker et al., 2009). This culturing method was then used as a foundation to generate organoids derived from human colon cancer resection/biopsy specimens (Sato et al., 2011). Interestingly, human cancer organoids had fewer and different growth medium requirements compared to normal human colon organoids, including independence from Wnt3A, R-spondin, Noggin, nicotinamide, and gastrin with varied dependence on EGF, A83-01, or SB202190, consistent with tumors evolving factor independence (Figure 1). The Clevers Lab subsequently established 22 colorectal cancer organoids (grown in media lacking Wnt3A) and 19 matched ‘normal’ tissue controls from 20 patients (van de Wetering et al., 2015). They demonstrated that histologically, the cancer organoids maintained aspects of the original tumor organization including cystic vs. solid architecture. In addition, genomic sequencing studies demonstrated that these cancer organoids shared a high degree of similarity compared to biopsied primary tumor samples (0.88 median frequency of concordance). The 22 colorectal cancer organoids encompassed the common mutations found in colorectal cancers and also the major molecular subtypes (Cancer Genome Atlas Network, 2012). Thus, tumor organoids were established as a novel model system to study cancer biology that mimicked the histology, genomic variations, and gene expression patterns of the primary tumors from which they were derived.
While a high level of molecular concordance was demonstrated, there were key discordances between cancer organoids and primary tumor samples. For example, the somatic mutational differences between each organoid and primary tumor sample range from 0% to 38% with some of these different mutations involving cancer-related genes including APC, SMAD4, and POLE (van de Wetering et al., 2015). It is unclear whether these differences are due to unique clonal populations that are enriched in the primary tumor and not present in the organoid population, or vice versa. These differences might also represent additional stromal or normal tissue contamination present in the primary tumor samples. Another important observation from van de Wetering et al. (2015) was that for many of the paired organoids derived from cancer and adjacent normal tissue, the normal tissue organoids paradoxically outgrew the cancer organoids. To address this discrepancy, the colorectal cancer organoids were maintained in culture media lacking Wnt3a in an attempt to preferentially select for cancer growth conditions given the high prevalence of APC pathway mutations (i.e. alterations that allow the tumor cells to grow independent of exogenous Wnt) seen in colorectal cancers (Powell et al., 1992; Cancer Genome Atlas Network, 2012). These observations highlight that cancer organoids have key molecular differences compared to primary tumor samples, and the important issue of adjacent tissue contamination.
Multiple groups have established additional biobanks of colorectal cancer organoids. Some have conformed to the culture methodology from van de Wetering et al. (2015) using media lacking Wnt3a (Weeber et al., 2015; Fujii et al., 2016; Schutte et al., 2017; Ooft et al., 2019; Yao et al., 2019), while others have established biobanks of gastrointestinal cancers using media containing Wnt3a (Vlachogiannis et al., 2018). Biobanks of colorectal cancer organoids have also been established with media initially containing Wnt3a along with R-spondin and Noggin, with those factors all removed upon passaging (Ganesh et al., 2019). Clearly, media conditions are critical for our interpretation of results from cancer organoid studies, yet the implications of all the varying methodologies are still unclear and deserve further analyses. We have summarized key, common media components for both normal and cancer gastrointestinal organoids generated from mice and humans (Figure 2).
Figure 2.

Major components of culture media for normal and cancer GI organoids derived from humans and mice. The culture media composition is essential for the establishment and maintenance of normal and cancer organoids. We detail here the important differences in culturing techniques used to culture normal and cancer GI organoids from humans and mice including different approaches to generate organoids from the same starting tissues.
Cancer organoids have been established now from a variety of solid tumor tissues (Drost and Clevers, 2018). We will focus here on those derived from gastroesophageal cancers. Bartfeld et al. (2015) showed the ability to culture gastric cancer organoids using media similar to that for normal human stomach organoids, consisting of Wnt3a, R-spondin-1, Noggin, EGF, FGF10, gastrin, and A83-01. Gao et al. (2018) showed the feasibility of generating patient-derived gastric cancer organoids from endoscopic biopsy and surgical resection samples. These studies were expanded upon by Yan et al. (2018) by establishing a biobank of 63 organoids from 34 patients with gastric cancer comprising normal, dysplastic, primary tumor, and lymph node metastatic tissues using the same media conditions as Bartfeld et al. (2015). Histologic, genomic, and transcriptomic analyses were performed showing concordance between the gastric cancer organoids and frozen tumor samples. The cancer organoids in this study maintained genomic stability over long-term culture in terms of both somatic mutations and copy number variants. Yan et al. (2018) also noted contamination of cancer organoids with non-tumor epithelial and stromal cells. Unlike colorectal cancer organoids, gastric cancer organoids could not be easily selected with media lacking Wnt3a. Thus, the authors employed several enrichment techniques including changing digestion times, preferential biopsy sampling, manual selection and replating, and use of nutlin-3a, a small-molecular inhibitor of the p53 inhibitor MDM2, to select organoids with TP53 mutations. Additional gastric cancer biobanks have been generated (Nanki et al., 2018; Seidlitz et al., 2019) using a similar approach as Yan et al. (2018). Other groups are using variations including media lacking A83-01 (Steele et al., 2019) or an alternative technique utilizing conditioned media from a supportive cell line (murine L cells) genetically modified to secrete Wnt, R-spondin, and Noggin (L-WRN cells; Miyoshi and Stappenbeck, 2013) to develop gastric cancer biobanks (Corso et al., 2019).
Tumor organoids from esophageal adenocarcinoma have also been developed. Li et al. (2018) established 10 esophageal adenocarcinoma organoids from surgical resection specimens showing histologic, genomic, and transcriptomic concordance with primary tumors using culture conditions similar to those established to generate normal human intestinal organoids. Although we focus mostly on adenocarcinoma, we note briefly that studies have also established organoids from both esophageal and oropharyngeal squamous cell cancers using a culturing technique established for the normal murine esophagus (Kijima et al., 2019).
Thus, these key studies have shown the feasibility of growing organoids that match their source tumors with establishment of cancer biobanks that encompass the patient-to-patient heterogeneity of these diseases. Standardization of the protocols including methods to avoid adjacent normal epithelial contamination and strategies to select for cancer organoid growth will be important future considerations (Drost and Clevers, 2018). Several strategies have already been discussed to enrich cancer organoids, but the theme is the ability of cultured cancer cells to gain independence from the principles of normal organoid growth (Figure 1). Specifically, there is the unique ability of cancer organoids to grow without ‘niche factors’. Fujii et al. (2016) generated colorectal cancer organoids using an optimization protocol to specifically test dependence on these niche factors (Wnt3a, R-spondin 1, Noggin, EGF, SB202190, and A83-01) and hypoxia conditions. They found high variability among colorectal cancer organoids generated from different patients in terms of factor dependence. Not only were factors found to be dispensable for some tumors, but factors including A83-01 and normoxia conditions were found to be detrimental to the growth of some cancer organoids. In addition, organoids derived from higher stage colorectal cancers exhibited greater factor independence. The basis of factor independence was, not surprisingly, found to depend on oncogenic activation of the respective signaling pathways in question. Similarly, a gastric cancer organoid biobank demonstrated genotypic factors contributing to EGF, FGF10, Wnt3a, and R-spondin independence (Nanki et al., 2018). In addition, emerging work using biomimetic hydrogel growth scaffolds afford new potential to study and optimize specific cancer growth conditions (Li and Kumacheva, 2018). Insights from these studies provide opportunities to select and enrich cancer organoids based on stage and aggressiveness, and from non-cancerous organoids (derived from adjacent normal tissue or areas of metaplasia/dysplasia). Specifically, Nanki et al. (2018) used nutlin-3 to enrich for TP53-mutated tumoroids, ROCK inhibitor-free culture media to enrich for RHO pathway-altered tumoroids, TGF-β without A83-01 to enrich for TGF-β-insensitive ones, and EGF and FGF10-free culture media to enrich for growth factor pathway-constitutively activated gastric cancer organoids.
Again, the emerging challenge in the tumor organoid field is how to standardize culture conditions across and within laboratories, while allowing the natural diversity of tumors in their in vivo state to be recapitulated in vitro. This will be a difficult task given the complexity of patient-to-patient (interpatient) organoid variation in tumor mutations and growth factor dependence, and also varying heterogeneity of each same patient-derived (intrapatient) tumor organoid, just as tumors in patients can vary in primary vs. various metastatic sites and even within each tumor focus (Figure 3). Thus, tumor organoids from a single patient will likely encompass subclones with wholly unique culturing conditions and selective advantages. The strength of the organoid system is the ability to recapitulate the cellular heterogeneity of the original tissue as seen with normal intestines; but, as seen in organoid culturing of normal gastric organoids, maintaining this representative heterogeneity through subsequent passaging is often difficult (Schumacher et al., 2015). How do we determine standard culture techniques that would be inclusive enough to allow establishment and maintenance of organoids derived from different patients obtained from heterogeneous sampled tumor sites, whereas be exclusive enough to preferentially enrich for growth and select against normal tissue organoid growth? The answers may be a standard approach with multiple different culturing media conditions that are matched to cancer tumoroids based on information obtained in the clinic, such as cancer stage, metastatic site, and mutational profile. These issues will be critical for cancer organoid growth to move from the experimental, research-lab-based realm into a clinical, diagnostic technique used in medical labs where quality assurance and compliance issues are paramount.
Figure 3.

Organoids can help study cancer heterogeneity and evolution. Cancer heterogeneity exists patient to patient (‘interpatient’) and within the same patient between different metastatic sites, as a function of time during treatment course, and even within regions of the same tumor site (‘intrapatient’). Such heterogeneity can be modeled by generating organoids from single cells that can each be grown as subclones that model a tumor cell population within the patient. On the other hand, growing organoids in bulk from tumors may reflect the overall behavior and response of a tumor as a whole and preserve key subclone‒subclone interactions and overall tumor clonal architecture. Circulating tumor cells often travel as single cells that can be grown in bulk conditions or grown as subclones without the need for cell separation. In either case, organoids can provide valuable and clinically applicable information regarding therapy response at the clonal level, though this tendency of tumor cells (and thus organoids derived from them) to be heterogeneous must be kept in mind when interpreting results. For example, in the hypothetical plot, the yellow subclone is responsive to therapy, whereas the orange subclone continues to grow.
Tumor organoids to study carcinogenesis
Murine models still represent arguably the most important system to study the mechanisms of carcinogenesis. When well-defined genetic alterations drive cancer formation (e.g. if most tumors in an organ depend on the same key driver mutation), genetically engineered mouse models can often recapitulate the development of cancers in vivo; key examples include pancreatic (mutations in KRAS; Hingorani et al., 2005) and colorectal (mutations in the Wnt pathway; Su et al., 1992) malignancies. However, for gastroesophageal cancers, the mutational landscape is, in most cases, complex with distinct and often disparate molecular pathways that lead to cancer formation (Cancer Genome Atlas Research Network, 2014; Cancer Genome Atlas Research Network et al., 2017). This has made the development of representative mouse models difficult, and only recently have advanced gastric and esophageal mouse models of tumorigenesis been generated (Moon et al., 2019; Seidlitz et al., 2019). While these murine models represent important new tools to understand gastroesophageal cancers, they have limitations. Central is the fact that it is not clear whether these murine models of gastroesophageal cancers progress to cancer in a manner that incorporates the precancerous lesions seen in humans, i.e. atrophic gastritis with spasmolytic polypeptide expressing metaplasia (SPEM) and intestinal metaplasia (Correa, 1992; Jin and Mills, 2018, 2019). They also do not appear ultimately to develop the aggressive metastatic cancer phenotype characteristic of most late-stage human tumors (Hayakawa et al., 2013).
Given the limitations of mouse models of tumorigenesis in the stomach and esophagus, there is excitement about the potential for using human organoids for this purpose. For example, it should be possible to grow organoids from normal, metaplastic/precancerous, dysplastic, and neoplastic tissues from various sites in the same patient. The genomic and transcriptomic natures of each of these could be determined, and key variations could be altered by genetic manipulations to determine which genes are most critical for each phenotype (Driehuis and Clevers, 2017). In this section, we detail some of the efforts to use organoids in this way to study cancer development and tumor evolution.
Several groups have detailed the differences between tumor and precancerous tissues (areas of metaplasia or dysplasia in the stomach) or lesions (adenomas in the intestine) through organoid generation (Fujii et al., 2016; Nanki et al., 2018; Yan et al., 2018). They show that organoids derived from these precancerous lesions have relatively few mutations compared to cancer organoids and harbor transcriptomic differences unique from cancer and normal organoids. In addition, the niche factor requirements for these precancerous lesions were shown to be less than normal tissue-derived organoids but more than cancer tissue-derived organoids (Fujii et al., 2016). Thus, a thorough characterization of these precursor organoids may provide a snapshot of the molecular steps of carcinogenesis.
Other work has more directly explored the origins of cancer using organoids through genetic manipulation. Matano et al. (2015) introduced the most commonly mutated genes (APC, SMAD4, TP53, KRAS, and PIK3CA) seen in colorectal cancers into normal human intestinal organoids using CRISPR–Cas9 technology. The authors showed that genetically altering normal organoids caused them to acquire niche factor independence and gain oncogenic potential. These acquired cancer characteristics were also reflected in the ability of the transformed organoids to form tumors upon xenografting into immunodeficient mice. CRISPR–Cas9 genetic editing has also been used to model serrated adenoma progression to colorectal cancers through BRAF oncogene activation (Fessler et al., 2016). This technique also enables sequential introduction of mutations seen during oncogenesis. Seino et al. (2018) used CRISPR–Cas9 to introduce KRAS, CDKN2A, TP53, and SMAD4 mutations in normal pancreatic organoids. As the normal pancreatic organoids accumulate these sequential mutations, they displayed more aggressive histologic features, niche factor independence, and ability to engraft upon xenotransplantation. Additional groups have also used genetic manipulation of organoids to recapitulate mutational signatures of cancer. Drost et al. (2017) used CRISPR–Cas9 technology to delete MLH1 and NTHL1 in normal human colon organoids to mimic mismatch repair deficiency and base excision repair deficiency, respectively. These altered colon organoids displayed mutational accumulation and profiles that matched those seen in mismatch repair-deficient or germline NTHL1-mutated human cancers. In gastric cancer, Nanki et al. (2018) have been able to investigate complex genotype‒phenotype relationships including the role of CDH1- and RHOA-inactivating mutations in diffuse gastric cancer using genetic manipulation of normal gastric organoids. They showed that organoids with CDH1 mutations displayed a degree of ROCK inhibitor independence (reflection of anoikis independence); however, the double knockout organoids (with both CDH1- and RHOA-inactivating mutations) had enhancement of this phenotype, suggesting that RHOA inactivation is a necessary step in CDH1 mutation-driven gastric carcinogenesis. The use of genetic manipulation has been applied to not only normal tissue-derived organoids, but also organoids derived from precancerous lesions. Using organoids derived from human Barrett’s esophagus, the precursor lesion to esophageal adenocarcinoma (Spechler and Souza, 2014), Liu et al. (2018) introduced APC mutations via CRISPR–Cas9 to mimic Wnt pathway activation, a common molecular pathway seen in esophageal carcinogenesis. They showed that the mutated organoids had demonstration of cancer-like histology and growth behavior. The application of genomic editing to normal and precancerous tissue-derived organoids provides a powerful tool to recapitulate known aspects of carcinogenesis and elucidate novel oncogenes and tumor suppressors.
The formation of cancer is an important aspect of cancer biology; however, the issues of cancer evolution and tumor heterogeneity are equally important, especially when viewed through the lens of the patient during their disease course (Figure 3). Such clonal changes that occur within tumors from various sites within a single patient are difficult to study, but cancer organoids have provided novel insights and have enable modeling of this complex process (Figure 3). Roerink et al. (2018) generated colorectal cancer organoids from single-dissociated cancer cells derived from multiple tumor regions. These clonal organoids were then shown to have unique genomic, transcriptomic, and epigenetic differences, and, using this information, a map of the overall tumor molecular architecture was constructed. The potential clinical impact of recognizing these subclonal populations was manifested in how each subclone responded differently to common colorectal cancer treatments including chemotherapeutics and small-molecule targeted inhibitors. A similar method of generating monoclonal cancer organoids from different regions of colorectal cancers was used to track clonal chromosomal stability and overall genomic changes (Bolhaqueiro et al., 2019). The authors uncovered intratumor differences in genomic stability and modeled this genomic heterogeneity in vitro using PDOs. An alternative method to track intratumor clonal changes would be to initiate organoids from a larger bulk sampling of the primary tumor rather than from single cell isolation. Such bulk organoids would be expected to grow as a heterogeneous mix of tumor subclonal populations that could be tracked over time with various degrees of clonal stability and fluctuation (Li et al., 2018).
In addition to exploring changes in the primary tumor populations over time, investigators have begun to analyze the clonal evolution from primary tumor to various metastatic sites. The feasibility of generating cancer organoids from metastases that retain the unique molecular features of that site has been shown (Weeber et al., 2015). Yan et al. (2018) compared the molecular profiles and clonal architecture of cancer organoids generated from lymph node biopsies to organoids generated from the primary tumor. The authors found examples of shared clonality between the metastatic site and the primary tumor, as well as examples of clonal heterogeneity distinct from the primary tumor. The versatility of organoid generation enables derivation of cancer organoids from either single cell or a larger heterogeneous mix of cancer cell populations, and the diversity of cancer organoids faithfully reflects the diversity of the starting material. Such approaches should eventually help us track heterogeneity across and within tumor foci throughout the disease and treatment course for individual patients.
In the field of solid tumor oncology, there has been great recent interest in identifying and characterizing circulating tumor cells. Because such cells are easily accessible via blood draws and because they can emerge from diverse tumor foci, they have emerged as an appealing platform to explore heterogeneity of tumor burden in patients (Keller and Pantel, 2019; Pantel and Alix-Panabieres, 2019). Specifically, blood-borne tumor cells are potentially shed from different portions of the primary tumor as well as any metastatic site, and thus they could potentially be used as a ‘dip stick’ to assess subclonal cancer diversity. A variety of different molecular analyses can be performed on these cells including culturing them in vitro. For example, circulating tumor cells from breast (Yu et al., 2014) and colon cancers (Cayrefourcq et al., 2015; Soler et al., 2018) have been cultured under non-adherent hypoxic conditions using growth factor (EGF and FGF)-supplemented culture media. However, successful culture of these cells has been limited to patients with >300 cancer cells per 7.5 ml of blood (Cayrefourcq et al., 2015). Application of cancer organoid culturing technique and methodology may allow for more robust culture viability of these circulating tumor cells. As a proof of concept, using established organoid culturing conditions including growth on Matrigel support and modified culture media (containing R-spondin, Noggin, EGF, FGF, A83-01, and SB202190) has allowed in vitro expansion and characterization of prostate cancer circulating tumor cells at much lower cell densities (Gao et al., 2014). Further studies are needed to establish the scope and limitations of working with organoids from these circulating tumor cells. If technical issues can be further overcome, circulating tumor cells represent a source of starting material that might have several advantages vs. culturing from biopsy or resection specimens.
In summary, organoid culture technique allows generation of monoclonal homogeneous tumoroids as well as mixed heterogeneous populations from any number of metastatic or primary cancer sites. Genetically manipulating tumoroids will allow further detailed insights into the origins and drivers of cancer development. In addition, organoids are also amenable to cryopreservation and expansion providing a means to augment initially small, limited samples to generate a potentially limitless biobank for detailed molecular analyses. These features of PDOs make them not only novel models to study cancer biology, but also an emerging translation tool for cancer treatment development.
Tumor organoids as a translation tool in oncology
The landscape of cancer treatment has shifted dramatically over the past decade, as detailed molecular characterization has revealed both shared and unique gene programs and pathways harbored by tumors (Hoadley et al., 2018). These detailed molecular characterizations have in turn translated to new efficacious, safe, and well-tolerated molecular targeted therapies for many malignancies including lung cancer, melanoma, breast cancer, and colorectal cancer (Cunningham et al., 2004; Piccart-Gebhart et al., 2005; Maemondo et al., 2010; Chapman et al., 2011; Shaw et al., 2013). As our basic cancer biology understanding increases, new innovative treatments are emerging that will need to be validated in the clinics. This monumental task of testing each new regimen for safety and efficacy in patients has resulted in ‘too many clinical trials’ (Kolata, 2017). The combinatorial regimens that need to be tested are simply outpacing our availability of patients to test them resulting in potentially delayed approval of new lifesaving treatments. In fact, adaptations have been made by regulatory agencies to adopt new surrogate end points such as progression-free survival rather than overall survival for oncology clinical trial design in an attempt to accommodate this challenge and accelerate this process (Jena et al., 2017).
While it is important to continue to develop new oncology treatments, an equally important task will be to identify the subset of patients that will derive the maximal benefit from each available treatment regimen and to tailor cancer treatment to the individual. Many times in oncology clinical trials, a brute force tactic of enrolling more patients is adopted in an attempt to demonstrate small statistically significant efficacy improvements. This strategy is obviously costly yet still often fails, as reflected in the unsuccessful clinical trials of many molecularly targeted therapeutic agents in gastroesophageal cancer (Samson and Lockhart, 2017). When an efficacy biomarker is available to enrich for patients who will more likely respond, then these trials are more likely to result in a meaningful positive result as seen in the ToGA study with HER2 expression positivity used as a surrogate inclusion criteria for trastuzumab response (Bang et al., 2010). However, there are few predictive biomarkers available to guide most cancer treatments (La Thangue and Kerr, 2011).
Simply put, there are too many treatments available and oftentimes no way to know which one will be the best for any individual patient. PDOs are a novel model for translational oncology. They are easy to generate, fast-growing, and expandable, making them an ideal tool for not only oncology drug screening, but also development of predictive personalized cancer therapy. In this section, we will discuss current studies using organoids to assess oncology treatments and posit about the future applications of organoids to guide individual cancer treatment.
The initial exploration of organoids as a tool for guiding cancer treatment was to use them for large-scale drug screens. These initial studies were unbiased screens in terms of examining a variety of approved and exploratory chemotherapeutics and molecularly targeted agents. In addition, these early studies lacked correlative clinical data of actual patient responses for comparison. van de Wetering et al. (2015) used their biobank of 22 colorectal cancer organoids derived from 20 patients (discussed above) to screen 85 compounds including drugs in clinical use such as 5-FU and additional investigatory compounds such as nutlin-3a, an MDM2 inhibitor. By virtue of having available detailed molecular profiling or each tumor organoid, the authors were able to make correlations between organoid responses (IC50 values) with genomic alterations including TP53 loss-of-function mutations associated with resistance to the MDM2 inhibitor nutlin-3a and KRAS mutations associated with resistance to anti-EGFR inhibitors. Similar drug sensitivity screens have been performed for upper gastrointestinal cancers using esophageal adenocarcinoma organoids to assess both compound sensitivities and correlation with mutational data (Li et al., 2018) and gastric adenocarcinomas to assess response to chemotherapeutics (Seidlitz et al., 2019).
Integration of patient data would be the next important step to validate organoids as a suitable preclinical model, i.e. comparing in vitro organoid sensitivities to cancer treatment with in vivo responses of the patient from which the organoids were derived to the same treatment. Vlachogiannis et al. (2018) generated colorectal and gastroesophageal cancer organoids from patients undergoing treatment in phase 1 or 2 clinical trials. The authors were able to make 21 direct treatment comparisons between patient responses and PDO responses. They noted that organoids generated from patients that had progressed on chemotherapy (paclitaxel or 5-FU/cisplatin) were refractory to these same treatments reflected in higher GI50 values. The authors showed patient and PDO response correlation with not only standard chemotherapeutics, but also molecularly targeted agents used in colorectal cancers including cetuximab, an anti-EGFR drug that is only effective for KRAS wild-type colorectal cancers. Interestingly, in this study, one patient without alteration in the RAS pathway (KRAS wild-type by clinical molecular pathology biomarker testing), who was treated with cetuximab per standard of care, had cancer organoids that showed unexpected extreme resistance to EGFR inhibition. This in vitro organoid response actually mirrored and was predictive of the patient’s unexpected refractoriness to EGFR inhibition. Thus, use of cancer organoids may enable improved predictive ability beyond the current genomic mutational analyses-driven biomarkers. Similar strong correlations were shown between patients and PDOs for several other approved colorectal cancer treatments including regorafenib (a multi-tyrosine kinase inhibitor) and TAS-102 (an oral chemotherapeutic). As a whole, PDOs displayed remarkable similarity to actual patient responses to standard chemotherapeutics and molecularly targeted agents; they displayed 100% sensitivity, 93% specificity, 88% positive predictive value, and 100% negative predictive value. Similar predictive correlation results have been shown in other cancers. Yan et al. (2018) assessed patient and organoid response data in response to standard chemotherapy for gastric cancers. They found that organoids derived from two patients responsive to cisplatin and 5-FU chemotherapy showed similar in vitro efficacy. However, organoids derived from the third patient unresponsive to chemotherapy displayed similar treatment refractoriness. Overall, these studies demonstrate the tremendous potential of PDOs as a tool for treatment prediction in oncology.
Clinically, immune checkpoint blockade has become a major therapeutic option for a wide variety of cancers including gastroesophageal cancers (Postow et al., 2015). In the USA, only one immunotherapy agent, pembrolizumab, a humanized monoclonal antibody that blocks the programmed cell death 1 (PD-1) receptor on lymphocytes, is approved as a third-line option for metastatic gastroesophageal adenocarcinomas (Shitara et al., 2018; Kojima et al., 2019). There is a clear clinical interest in improving these responses and understanding mechanisms of immune therapy resistance/response in gastroesophageal cancers. PDOs are currently being developed to address these challenging questions. The organoid models we have been describing have been developed to culture epithelial cells and, by extension, the tumor cells of solid tumors, which are largely epithelial in nature. However, interactions between the tumor cells and immune and stromal components can also be modeled in organoid co-culture systems. For example, Ootani et al. (2009) created a system using collagen gel matrix, minced murine tissues (neonatal and adult), and basic media without the addition of exogenous factors (e.g. Wnt3a, R-spondin 1, Noggin, growth factors, or small-molecule inhibitors) to culture long-term murine intestinal and gastric organoids with stromal components (Katano et al., 2013). The explanted tissues were embedded in the collagen matrix and cultured above the media fluid level in a 3D assembly to allow establishment of an air‒liquid interface (Li et al., 2016). This 3D air‒liquid interface organoid culturing technique has been used to study cancer, first using organoids derived from genetically engineered mouse models of intestine, pancreas, and stomach cancers (Li et al., 2014). Subsequently, a biobank of air‒liquid interface-grown human cancer organoids has been generated from a variety of human cancers including colorectal, stomach, and esophageal (Neal et al., 2018). The authors showed that this method of organoid culturing was able to preserve primary tumor histology and genomic alterations, and most importantly, that these organoid cultures contained tumor microenvironment components including cancer-associated fibroblasts and tumor-infiltrating lymphocytes. Neal et al. (2018) were then able to use these air‒liquid interface cancer organoid cultures to model immunotherapy response using the PD-1 blocking antibody, nivolumab, by showing lymphocyte activation and tumor cell death. An alternative approach has been developed in which peripheral blood lymphocytes and cancer organoids derived from the same patients are co-cultured (Dijkstra et al., 2018; Holokai et al., 2019). The rapid development of these various organoid co-culturing techniques has provided a novel platform to study immunotherapy resistance pathways (Jenkins et al., 2018) and develop novel immuno-oncology therapies (Gonzalez-Exposito et al., 2019; Schnalzger et al., 2019).
The use of patient-derived cancer organoids to forecast patient response will benefit patients with advanced non-curative metastatic cancers in which standard of care lines of treatments is often limited and is quickly exhausted. However, predictive PDO treatment responses may also be clinically applicable and guide treatment decisions for patients with localized and curative disease. A key example of an arena in which organoid-based therapeutics might be helpful is demonstrated by the current state-of-the-art approach to locally advanced rectal cancers. These cancers have been shown to respond so well to total neoadjuvant therapy in which radiation and chemotherapy are given prior to planned surgery that the necessity for subsequent resection has been questioned (Cercek et al., 2018; Smith et al., 2019). PDOs could be developed as a tool to dictate the type of neoadjuvant theory applied. Toward that aim, Ganesh et al. (2019) developed a biobank of human rectal cancer organoids from 65 patients and xenografted these organoids into mice to model chemotherapy response. Using organoids derived from both chemosensitive and chemoresistant patients, the authors were able to recapitulate chemotherapy sensitivity. Yao et al. (2019) similarly derived rectal cancer organoids from 80 patients undergoing neoadjuvant chemoradiation, directly comparing organoid in vitro responses to radiation, 5-FU, and irinotecan with actual patient clinical responses. Incredibly, the authors found that these PDOs demonstrated strong correlation with patient clinical outcomes with 78% sensitivity, 92% specificity, and 84% accuracy. These key studies establish the potential use of organoids as a clinical decision tool for a variety of cancers. For example, in esophageal cancer, a similar trend toward ‘definitive’ chemoradiation presents another potential opportunity for organoids to detect a non-surgical approach to tumor ablation (van Hagen et al., 2012; Conroy et al., 2014).
There is clear promise that PDOs can serve as an avatar to identify and predict patient responses. The ability to implement PDOs into current clinical oncology treatment algorithms will be an important consideration moving forward (Pauli et al., 2017). Using organoids to identify patients that will be treatment-refractory will allow for the timely selection of and rational planning for alternative treatment strategies with potential applicability for all cancer patients (Figure 4). Patients might benefit not only from receiving individualized efficacious treatments but also from the avoidance of toxicities from ineffective treatments.
Figure 4.

Proposed integration of PDOs into clinical oncology. Organoids hold promise to impact clinical oncology decision-making in both localized and advanced disease. Curative treatment of localized disease often involves multi-modality treatment including systemic treatment after (‘adjuvant’) or before (‘neoadjuvant’) surgical resection. Generating patient organoids in the adjuvant setting from the surgical resection specimen or in the neoadjuvant setting from initial diagnostic biopsy samples will allow screening of systemic treatments to predict regimens with maximal curative efficacy and to avoid ineffective treatments. For patients with advanced disease, organoids can be generated from multiple sites (primary tumor and accessible metastatic sites). These patients can then begin standard of care first-line treatment. During this time, PDOs can be used to screen established and novel therapies including chemotherapeutics, targeted agents, and immunotherapy, to individualize the choice of treatment prior to disease progression. This iterative approach can be adopted for each subsequent line of therapy.
Perspective
Here, we have reviewed how organoids are emerging from a basic science tool to a promising tool for precision oncology. Our focus has been applications for gastroesophageal cancers, using studies in colorectal cancers as a guidepost. The use of PDOs has already and will continue to improve our fundamental knowledge of cancer; however, many key questions remain. How do we integrate organoid models with previous models such as cancer cell lines and patient-derived xenograft models? Are the other cancer biology models still needed, or will our understanding of organoids (with inclusion of stromal, matrix, and immune elements) grow to more faithfully reproduce all components of a tumor, thereby rendering the older models obsolete (Drost and Clevers, 2018; Tuveson and Clevers, 2019)? Not only do organoids have the potential to greatly improve translational and clinical oncology, but they can be used to study basic mechanisms of carcinogenesis from precancerous lesions as well. Given that cancer organoids are a means to model a complex system, how do we maintain the utility of organoids as an accessible model while preserving the complexity and heterogeneity of cancer, the totality of which defines treatment outcome and patient survival? For example, will it be possible to standardize media or culture conditions, or will these have to be modified for each tumor? Will culture media depend on the stage or the tumor, or the site from which the organoids are generated? Finally, given the clinical potential of cancer organoids, how should they be integrated into the clinics in terms of standard of care treatment algorithms, clinical trial design, and drug development?
Conclusions
The lack of novel treatments for many solid tumors including gastroesophageal cancers is directly reflected by our inability to accurately model tumor behavior. PDOs provide the means to now grow cancers quickly from limited starting material while reflecting overall tumor heterogeneity and allowing molecular analyses and interventions. These are exciting times for oncology in terms of the rapid development of novel cancer treatments. Organoids have the potential to further fuel these advancements to provide safe, efficacious, and potentially curative individual patient-based cancer treatments.
Funding
Support is provided by the NIDDK R01s (DK094989, DK105129, and DK110406) P30 (DK052574), Alvin J. Siteman Cancer Center/Barnes Jewish Hospital Foundation Cancer Frontier Fund, NIH NCI (P30 CA091842 and U54 CA163060), The Barnard Trust, and DeNardo Education & Research Foundation grants to J.C.M.
Conflict of interest: none declared.
References
- Anderson W.F., Camargo M.C., Fraumeni J.F. Jr, et al. (2010). Age-specific trends in incidence of noncardia gastric cancer in US adults. JAMA 303, 1723–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang Y.J., Van Cutsem E., Feyereislova A., et al. (2010). Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697. [DOI] [PubMed] [Google Scholar]
- Barker N., Huch M., Kujala P., et al. (2010). Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6, 25–36. [DOI] [PubMed] [Google Scholar]
- Barker N., Ridgway R.A., van Es J.H., et al. (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611. [DOI] [PubMed] [Google Scholar]
- Barker N., van Es J.H., Kuipers J., et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Bartfeld S., Bayram T., van de Wetering M., et al. (2015). In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148, 126–136.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F., de Ligt J., Jager M., et al. (2016). Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhaqueiro A.C.F., Ponsioen B., Bakker B., et al. (2019). Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 51, 824–834. [DOI] [PubMed] [Google Scholar]
- Bray F., Ferlay J., Soerjomataram I., et al. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Analysis Working Group: Asan University, BC Cancer Agency, et al. (2017). Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrefourcq L., Mazard T., Joosse S., et al. (2015). Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res. 75, 892–901. [DOI] [PubMed] [Google Scholar]
- Cercek A., Roxburgh C.S.D., Strombom P., et al. (2018). Adoption of total neoadjuvant therapy for locally advanced rectal cancer. JAMA Oncol. 4, e180071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman P.B., Hauschild A., Robert C., et al. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. (2016). Modeling development and disease with organoids. Cell 165, 1586–1597. [DOI] [PubMed] [Google Scholar]
- Conley B.A., Doroshow J.H. (2014). Molecular analysis for therapy choice: NCI MATCH. Semin. Oncol. 41, 297–299. [DOI] [PubMed] [Google Scholar]
- Conroy T., Galais M.P., Raoul J.L., et al. (2014). Definitive chemoradiotherapy with FOLFOX versus fluorouracil and cisplatin in patients with oesophageal cancer (PRODIGE5/ACCORD17): final results of a randomised, phase 2/3 trial. Lancet Oncol. 15, 305–314. [DOI] [PubMed] [Google Scholar]
- Correa P. (1992). Human gastric carcinogenesis: a multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 52, 6735–6740. [PubMed] [Google Scholar]
- Corso S., Isella C., Bellomo S.E., et al. (2019). A comprehensive PDX gastric cancer collection captures cancer cell-intrinsic transcriptional MSI traits. Cancer Res. 79, 5884–5896. [DOI] [PubMed] [Google Scholar]
- Cunningham D., Humblet Y., Siena S., et al. (2004). Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 351, 337–345. [DOI] [PubMed] [Google Scholar]
- DeWard A.D., Cramer J., Lagasse E. (2014). Cellular heterogeneity in the mouse esophagus implicates the presence of a nonquiescent epithelial stem cell population. Cell Rep. 9, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra K.K., Cattaneo C.M., Weeber F., et al. (2018). Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E., Clevers H. (2017). CRISPR/Cas 9 genome editing and its applications in organoids. Am. J. Physiol. Gastrointest. Liver Physiol. 312, G257–G265. [DOI] [PubMed] [Google Scholar]
- Drost J., Clevers H. (2018). Organoids in cancer research. Nat. Rev. Cancer 18, 407–418. [DOI] [PubMed] [Google Scholar]
- Drost J., van Boxtel R., Blokzijl F., et al. (2017). Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 358, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler E., Drost J., van Hooff S.R., et al. (2016). TGFβ signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol. Med. 8, 745–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M., Shimokawa M., Date S., et al. (2016). A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 18, 827–838. [DOI] [PubMed] [Google Scholar]
- Ganesh K., Wu C., O'Rourke K.P., et al. (2019). A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 25, 1607–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D., Vela I., Sboner A., et al. (2014). Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Lin M., Rao M., et al. (2018). Development of patient-derived gastric cancer organoids from endoscopic biopsies and surgical tissues. Ann. Surg. Oncol. 25, 2767–2775. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Exposito R., Semiannikova M., Griffiths B., et al. (2019). CEA expression heterogeneity and plasticity confer resistance to the CEA-targeting bispecific immunotherapy antibody cibisatamab (CEA-TCB) in patient-derived colorectal cancer organoids. J. Immunother. Cancer 7, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y., Fox J.G., Gonda T., et al. (2013). Mouse models of gastric cancer. Cancers 5, 92–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani S.R., Wang L., Multani A.S., et al. (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Hoadley K.A., Yau C., Hinoue T., et al. (2018). Cell-of-origin patterns dominate the molecular classification of 10000 tumors from 33 types of cancer. Cell 173, 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holokai L., Chakrabarti J., Broda T., et al. (2019). Increased programmed death-ligand 1 is an early epithelial cell response to Helicobacter pylori infection. PLoS Pathog. 15, e1007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena A.B., Zhang J., Lakdawalla D.N. (2017). The trade-off between speed and safety in drug approvals. JAMA Oncol. 3, 1465–1466. [DOI] [PubMed] [Google Scholar]
- Jenkins R.W., Aref A.R., Lizotte P.H., et al. (2018). Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R.U., Mills J.C. (2018). Are gastric and esophageal metaplasia relatives? The case for Barrett's stemming from SPEM. Dig. Dis. Sci. 63, 2028–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R.U., Mills J.C. (2019). The cyclical hit model: how paligenosis might establish the mutational landscape in Barrett's esophagus and esophageal adenocarcinoma. Curr. Opin. Gastroenterol. 35, 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasagi Y., Chandramouleeswaran P.M., Whelan K.A., et al. (2018). The esophageal organoid system reveals functional interplay between notch and cytokines in reactive epithelial changes. Cell. Mol. Gastroenterol. Hepatol. 5, 333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katano T., Ootani A., Mizoshita T., et al. (2013). Establishment of a long-term three-dimensional primary culture of mouse glandular stomach epithelial cells within the stem cell niche. Biochem. Biophys. Res. Commun. 432, 558–563. [DOI] [PubMed] [Google Scholar]
- Keller L., Pantel K. (2019). Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 19, 553–567. [DOI] [PubMed] [Google Scholar]
- Kijima T., Nakagawa H., Shimonosono M., et al. (2019). Three-dimensional organoids reveal therapy resistance of esophageal and oropharyngeal squamous cell carcinoma cells. Cell. Mol. Gastroenterol. Hepatol. 7, 73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima T., Muro K., Francois E., et al. (2019). Pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer: Phase III KEYNOTE-181 study. J. Clin. Oncol. 37, 2. [DOI] [PubMed] [Google Scholar]
- Kolata G. (2017, August 12). A cancer conundrum: too many drug trials, too few patients. The New York Times, p. A1. Retrieved from http://www.nytimes.com.
- La Thangue N.B., Kerr D.J. (2011). Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat. Rev. Clin. Oncol. 8, 587–596. [DOI] [PubMed] [Google Scholar]
- Lancaster M.A., Knoblich J.A. (2014). Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125. [DOI] [PubMed] [Google Scholar]
- Leung W.K., Wu M.S., Kakugawa Y., et al. (2008). Screening for gastric cancer in Asia: current evidence and practice. Lancet Oncol. 9, 279–287. [DOI] [PubMed] [Google Scholar]
- Li M., Izpisua Belmonte J.C. (2019). Organoids—preclinical models of human disease. N. Engl. J. Med. 380, 569–579. [DOI] [PubMed] [Google Scholar]
- Li X., Francies H.E., Secrier M., et al. (2018). Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 9, 2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Ootani A., Kuo C. (2016). An air–liquid interface culture system for 3D organoid culture of diverse primary gastrointestinal tissues. Methods Mol. Biol. 1422, 33–40. [DOI] [PubMed] [Google Scholar]
- Li X.N., Nadauld L., Ootani A., et al. (2014). Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 20, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Kumacheva E. (2018). Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 4, eaas8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Cheng Y., Abraham J.M., et al. (2018). Modeling Wnt signaling by CRISPR–Cas9 genome editing recapitulates neoplasia in human Barrett epithelial organoids. Cancer Lett. 436, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maemondo M., Inoue A., Kobayashi K., et al. (2010). Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388. [DOI] [PubMed] [Google Scholar]
- Matano M., Date S., Shimokawa M., et al. (2015). Modeling colorectal cancer using CRISPR–Cas9-mediated engineering of human intestinal organoids. Nat. Med. 21, 256–262. [DOI] [PubMed] [Google Scholar]
- McCauley H.A., Wells J.M. (2017). Pluripotent stem cell-derived organoids: using principles of developmental biology to grow human tissues in a dish. Development 144, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath P.S., Wells J.M. (2015). SnapShot: GI tract development. Cell 161, 176–176.e1. [DOI] [PubMed] [Google Scholar]
- Miyoshi H., Stappenbeck T.S. (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 8, 2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon H., Zhu J., Donahue L.R., et al. (2019). Krt5+/Krt15+ foregut basal progenitors give rise to cyclooxygenase-2-dependent tumours in response to gastric acid stress. Nat. Commun. 10, 2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanki K., Toshimitsu K., Takano A., et al. (2018). Divergent routes toward Wnt and R-spondin niche independency during human gastric carcinogenesis. Cell 174, 856–869.e17. [DOI] [PubMed] [Google Scholar]
- Neal J.T., Li X., Zhu J., et al. (2018). Organoid modeling of the tumor immune microenvironment. Cell 175, 1972–1988.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooft S.N., Weeber F., Dijkstra K.K., et al. (2019). Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 11, eaay2574. [DOI] [PubMed] [Google Scholar]
- Ootani A., Li X., Sangiorgi E., et al. (2009). Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 15, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantel K., Alix-Panabieres C. (2019). Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nat. Rev. Clin. Oncol. 16, 409–424. [DOI] [PubMed] [Google Scholar]
- Pauli C., Hopkins B.D., Prandi D., et al. (2017). Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 7, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccart-Gebhart M.J., Procter M., Leyland-Jones B., et al. (2005). Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 353, 1659–1672. [DOI] [PubMed] [Google Scholar]
- Pohl H., Welch H.G. (2005). The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J. Natl Cancer Inst. 97, 142–146. [DOI] [PubMed] [Google Scholar]
- Postow M.A., Callahan M.K., Wolchok J.D. (2015). Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 33, 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell S.M., Zilz N., Beazer-Barclay Y., et al. (1992). APC mutations occur early during colorectal tumorigenesis. Nature 359, 235–237. [DOI] [PubMed] [Google Scholar]
- Roerink S.F., Sasaki N., Lee-Six H., et al. (2018). Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556, 457–462. [DOI] [PubMed] [Google Scholar]
- Rustgi A.K., El-Serag H.B. (2014). Esophageal carcinoma. N. Engl. J. Med. 371, 2499–2509. [DOI] [PubMed] [Google Scholar]
- Samson P., Lockhart A.C. (2017). Biologic therapy in esophageal and gastric malignancies: current therapies and future directions. J. Gastrointest. Oncol. 8, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Stange D.E., Ferrante M., et al. (2011). Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141, 1762–1772. [DOI] [PubMed] [Google Scholar]
- Sato T., Vries R.G., Snippert H.J., et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. [DOI] [PubMed] [Google Scholar]
- Schnalzger T.E., de Groot M.H., Zhang C., et al. (2019). 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 38, e100928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M.A., Aihara E., Feng R., et al. (2015). The use of murine-derived fundic organoids in studies of gastric physiology. J. Physiol. 593, 1809–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutgens F., Clevers H. (2020). Human organoids: tools for understanding biology and treating diseases. Annu. Rev. Pathol. 15, 211–234. [DOI] [PubMed]
- Schutte M., Risch T., Abdavi-Azar N., et al. (2017). Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 8, 14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaederle M., Zhao M., Lee J.J., et al. (2015). Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J. Clin. Oncol. 33, 3817–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidlitz T., Merker S.R., Rothe A., et al. (2019). Human gastric cancer modelling using organoids. Gut 68, 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino T., Kawasaki S., Shimokawa M., et al. (2018). Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 22, 454–467.e6. [DOI] [PubMed] [Google Scholar]
- Shaw A.T., Kim D.W., Nakagawa K., et al. (2013). Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 368, 2385–2394. [DOI] [PubMed] [Google Scholar]
- Shitara K., Ozguroglu M., Bang Y.J., et al. (2018). Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 392, 123–133. [DOI] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. (2019). Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34. [DOI] [PubMed] [Google Scholar]
- Smith J.J., Strombom P., Chow O.S., et al. (2019). Assessment of a watch-and-wait strategy for rectal cancer in patients with a complete response after neoadjuvant therapy. JAMA Oncol. 5, e185896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler A., Cayrefourcq L., Mazard T., et al. (2018). Autologous cell lines from circulating colon cancer cells captured from sequential liquid biopsies as model to study therapy-driven tumor changes. Sci. Rep. 8, 15931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spechler S.J., Souza R.F. (2014). Barrett's esophagus. N. Engl. J. Med. 371, 836–845. [DOI] [PubMed] [Google Scholar]
- Stange D.E., Koo B.K., Huch M., et al. (2013). Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell 155, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele N.G., Chakrabarti J., Wang J., et al. (2019). An organoid-based preclinical model of human gastric cancer. Cell. Mol. Gastroenterol. Hepatol. 7, 161–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L.K., Kinzler K.W., Vogelstein B., et al. (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668–670. [DOI] [PubMed] [Google Scholar]
- Ter Veer E., Haj Mohammad N., van Valkenhoef G., et al. (2016). The efficacy and safety of first-line chemotherapy in advanced esophagogastric cancer: a network meta-analysis. J. Natl Cancer Inst. 108, djw166. [DOI] [PubMed] [Google Scholar]
- Tuveson D., Clevers H. (2019). Cancer modeling meets human organoid technology. Science 364, 952–955. [DOI] [PubMed] [Google Scholar]
- van de Wetering M., Francies H.E., Francis J.M., et al. (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hagen P., Hulshof M.C., van Lanschot J.J., et al. (2012). Preoperative chemoradiotherapy for esophageal or junctional cancer. N. Engl. J. Med. 366, 2074–2084. [DOI] [PubMed] [Google Scholar]
- Vlachogiannis G., Hedayat S., Vatsiou A., et al. (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A.D., Unverzagt S., Grothe W., et al. (2010). Chemotherapy for advanced gastric cancer. Cochrane Database Syst. Rev. 8, CD004064. [DOI] [PubMed] [Google Scholar]
- Wanebo H.J., Kennedy B.J., Chmiel J., et al. (1993). Cancer of the stomach. A patient care study by the American College of Surgeons. Ann. Surg. 218, 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber F., van de Wetering M., Hoogstraat M., et al. (2015). Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl Acad. Sci. USA 112, 13308–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock J., LaVange L.M. (2017). Master protocols to study multiple therapies, multiple diseases, or both. N. Engl. J. Med. 377, 62–70. [DOI] [PubMed] [Google Scholar]
- Yan H.H.N., Siu H.C., Law S., et al. (2018). A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 23, 882–897.e11. [DOI] [PubMed] [Google Scholar]
- Yao Y., Xu X., Yang L., et al. (2019). Patient-derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell Stem Cell 26, 17–26.e6. [DOI] [PubMed] [Google Scholar]
- Yu M., Bardia A., Aceto N., et al. (2014). Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Li M., Chen S., et al. (2018). Endoscopic screening in Asian countries is associated with reduced gastric cancer mortality: a meta-analysis and systematic review. Gastroenterology 155, 347–354.e9. [DOI] [PubMed] [Google Scholar]