

Abstract
Cervical cancer (CC) is a common gynecological malignancy for which prognostic and therapeutic biomarkers are urgently needed. The signature based on immune-related lncRNAs (IRLs) of CC has never been reported. This study is aimed at establishing an IRL signature for patients with CC. A cohort of 326 CC and 21 normal tissue samples with corresponding clinical information was included in this study. Twenty-eight IRLs were collected according to the Pearson correlation analysis between the immune score and lncRNA expression (p < 0.01). Four IRLs (BZRAP1-AS1, EMX2OS, ZNF667-AS1, and CTC-429P9.1) with the most significant prognostic values (p < 0.05) were identified which demonstrated an ability to stratify patients into the low-risk and high-risk groups by developing a risk score model. It was observed that patients in the low-risk group showed longer overall survival (OS) than those in the high-risk group in the training set, valid set, and total set. The area under the curve (AUC) of the receiver operating characteristic curve (ROC curve) for the four-IRL signature in predicting the one-, two-, and three-year survival rates was larger than 0.65. In addition, the low-risk and high-risk groups displayed different immune statuses in GSEA. These IRLs were also significantly correlated with immune cell infiltration. Our results showed that the IRL signature had a prognostic value for CC. Meanwhile, the specific mechanisms of the four IRLs in the development of CC were ascertained preliminarily.
1. Introduction
Cervical cancer (CC) is a malignant gynecologic tumor threatening the health of women. The morbidity and mortality for CC rank fourth worldwide among women [1]. Infection with high-risk human papillomavirus (HPV), especially HPV16 and HPV18, is the main etiologic risk factor for CC and plays an important role in diagnostic tests [2]. Surgery is the main treatment method for CC in early stages while advanced-stage CC can be treated with radiotherapy, chemotherapy, or concurrent chemoradiation, thereby improving the survival rate of CC patients [3]. However, a considerable number of CC patients have poor prognosis due to metastasis or recurrence within two years after treatment [4]. Hence, effective prevention to reduce morbidity and individual treatments to improve the prognosis of CC are important for obstetricians and gynecologists.
The immune system can recognize tumor antigens expressed on the surface of tumor cells. It generates an immune response via the activation of effector cells and triggers the release of a series of effector molecules to attack and eliminate tumor cells and to inhibit tumor growth [5]. The immune imbalance in the tumor microenvironment plays an important role in the occurrence and development of cancer. With the development of cellular molecular biology and immunology, immunotherapy has become a new treatment approach for cervical cancer [6]. Tumor immunotherapy acts mainly by increasing the immunogenicity of tumor cells and the effect of cell damage sensitivity and stimulates and enhances the antitumor immune response, with the aid of biological agents; doping effects of immune cells and molecules into the body, together, the body's immune system can not only kill cancer cells remaining small but also can prevent tumor metastasis and recurrence [7].
Long noncoding RNAs (lncRNAs), defined arbitrarily as transcripts lacking protein-coding potential, are RNAs with more than 200 nucleotides [8]. lncRNAs are not only associated with the invasion, migration, and proliferation of CC but also are involved in autophagy and epithelial-mesenchymal transition (EMT) [9]. Emerging studies have demonstrated that lncRNAs also modulate the immune response to tumors. For instance, the downregulated lncRNA AGER has been demonstrated to be closely related to T cell status in lung cancer [10]. lncRNA GM16343 is regulated by interleukin 36β to strengthen the antitumor immune response of CD8+ T cells [11]. lncRNA LINK-A has been demonstrated to downregulate antigen presentation and intrinsic suppression in triple-negative breast cancer [12]. lncRNAs can also affect the development of cancer by regulating NK cells. lncRNA GAS5 can inhibit tumor growth, and the overexpression of GAS5 can regulate miR-544/RUNX3 to enhance the killing effect of NK cells in liver cancer [13]. lncRNAs can also modulate tumor immunity by regulating Treg cells. The expression of miR-448 can be increased by interfering with lncRNA SNHG1, which downregulates the expression of indoleamine 2,3-dioxygenase (IDO), inhibits the differentiation of Treg cells, and reduces the immune escape of breast cancer cells [14]. EGFR is an important member of the tyrosine kinase receptor family. By binding to EGFR specifically, LNC-EGFR promotes Treg cell differentiation and promotes the immune escape of liver cancer cells [15]. lncRNAs can affect the tumor microenvironment and thus play an important role in immunotherapy. Nonetheless, the effect research on immune-related lncRNAs in CC is rarely reported.
The purpose of our research was to identify an IRL signature, which might serve as prognostic and therapeutic biomarkers in CC. We developed a prognostic signature and mechanisms of IRLs using single-sample gene set enrichment analysis (ssGSEA), survival analysis, a Cox regression risk model, gene set enrichment analysis (GSEA), ceRNA network, and other analysis methods.
2. Materials and Methods
2.1. Datasets and Preprocessing
The RNA sequencing profiles associated with CC were obtained from The Cancer Genome Atlas (TCGA) [16] (https://toil.xenahubs.net), which consisted of 306 CC and 13 normal tissue samples. The low-expression genes were filtered, and the genes whose expression level was greater than 0 in more than a third of the samples were retained. Additionally, the RNAs were identified as mRNAs or lncRNAs based on their annotation information in the GENCODE database [17] (https://www.gencodegenes.org/). GPL570 (HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 Array platform was used to obtain the microarray dataset GSE6791 from the Gene Expression Omnibus (GEO) repository (Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/) [18], of which twenty CC and eight normal tissue samples were included. All 326 CC samples and 21 normal tissue samples were contained with corresponding clinical information. For the RNA sequencing profiles obtained from TCGA TARGET GTEx, empirical Bayes and linear regression along with Benjamini and Hochberg multiple comparison methods from the limma package [19] (version 3.10.3, http://www.bioconductor.org/packages/2.9/bioc/html/limma.html) were performed to gain adjusted p value and |logFC| (adj.p.value < 0.05, ∣logFC | >0.585). For GSE6791, GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) was used with the SeqMap [20] tool to map probes to mRNA and lncRNA sequences. The immune-related genes (IRGs) were downloaded from the InnateDB database (http://www.innatedb.com) [21]. Foc`using on screening genes that were up- and downregulated consistently, we used Venn analysis to select the intersection genes of the aforementioned datasets. Single-sample gene set enrichment analysis (ssGSEA) [22] was used to identify the immune scores (IS) of each sample. Pearson's correlation coefficient between lncRNAs and IS was calculated for each corresponding samples to identify immune-related lncRNAs (IRLs) (p < 0.01).
2.2. Signature Development of IRLs
Univariate Cox regression analysis with hazard ratio (HR) was gained from overall survival (OS) and OS time from the candidate IRLs. HR > 1 indicated that expression was higher and the risk and the survival rate were lower. lncRNAs that are upregulated in the tumor should, in principle, have an HR greater than 1. The Kaplan–Meier analysis was generated by survminer (version 0.4.3) in the R package based on the expression value, survival time, and survival status to determine the optimal cut point. The log-rank test was performed based on survival (version 2.42-6) in the R package to sort the IRLs with a significant prognostic value (p < 0.05); then, the survival curves were drawn. Multivariate Cox regression analysis was performed based on the expression value, OS, and OS time of IRLs in each sample. Subsequently, the individual prognostic risk model for corresponding samples was established. Exprgene indicated the expression of corresponding IRL for each sample. All the samples were divided into the low-risk and high-risk groups according to the median of risk scores of the following study. All the samples in TCGA were regarded as a total set. Training and valid sets were constructed by dividing the total set equally into two parts to validate the risk score formula. The Kaplan–Meier survival curves of all three sets were drawn to determine the prognostic difference between the risk groups. The one-year, two-year, and three-year survival receiver operating characteristic (ROC) curves predicted by the risk model were drawn.
2.3. Clinical and Pathological Characteristics of the Risk Score Model
Clinical and pathological characteristics, including age, pathologic M, pathologic N, pathologic T, clinical stage, neoplasm histologic grade, neoplasm cancer status, primary therapy outcome success, radiation therapy, tobacco smoking history, along with the risk score, the immune score, and the expression of IRLs in each sample were included in the analysis for illustration of a heat map. We observed the differences in clinical pathological characteristics in risk groups or the risk score of clinical pathological characteristics using Student's t-test. A scatter plot was drawn using GraphPad Prism 5 [23] based on significant clinicopathological characteristics.
2.4. Nomogram Model Construction and Visualization
Univariate Cox regression analyses were performed, respectively, based on the risk groups and clinicopathological factors including age, cancer type (adenocarcinoma and squamous cell carcinoma), FIGO stage, TNM stages, histologic grade, radiation therapy, and smoking history. Multivariate Cox regression analysis was conducted on the factors with p < 0.05. Using the IRL signature along with the factors with p < 0.05, a nomogram was constructed to visualize the results of multivariate Cox regression analysis more clearly.
2.5. Gene Set Enrichment Analysis
To elucidate the biological differences between risk groups, a gene set enrichment analysis (GSEA) [24] of “c5.bp.v7.1.symbols.gmt” background was carried out using GSEA (version 4.0.3) software. A nominal p value < 0.05 (NOM p val < 0.05) was considered significant. We focused on selecting immune-related terms for display.
2.6. Evaluation of Infiltrating Immune Cells
To further observe the differences in the abundance of infiltrating immune cells of the risk groups, we used the CIBERSORT algorithm, a deconvolution method [25], coupled with LM22 that distinguished 22 immune cell subpopulations from CIBERSORT (a web server), and a heat map for all the samples was drawn using ggplot2 (version 3.2.1). Student's t-test was applied to find significant immune cell subpopulations in risk groups to chart violin plot by boxplot (version 0.3.2) in the R package.
2.7. Construction of an IRL-Associated ceRNA Network
The ceRNA network was constructed to explore the association among IRLs, miRNAs, and mRNAs based on the ceRNA hypothesis [26]. First, the Pearson correlation coefficient analysis between the IRLs and immune-related DEGs was performed to obtain lncRNA-mRNA pairs (r > 0 and adj.p.value < 0.01). The target miRNAs of IRLs were predicted using DIANA-LncBase v2 [27] (http://carolina.imis.athenainnovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex-experimental). The mRNAs targeted by miRNAs were predicted by integrating miRWalk2.0 [28] (version:3.4.0, http://chianti.ucsd.edu/cytoscape-3.4.0/), miRanda, RNA22, and TargetScan databases. Only miRNA-mRNA pairs recognized by all four databases were considered candidate targets. Subsequently, the ceRNA network based on the same miRNA of the lncRNA-miRNA and miRNA-mRNA pairs was established and visualized using Cytoscape (version:3.4.0, http://chianti.ucsd.edu/cytoscape-3.4.0/) [29]. Sorting the mRNA with a significant prognostic value (p < 0.05) in the ceRNA network, the Kaplan–Meier analysis was generated using survminer (version 0.4.3) to determine the optimal cut point, and the log-rank test was performed based on survival (version 2.42-6) in the R package.
3. Results
3.1. Differential Analysis of Genes
In the aggregate, 1995 differentially expressed lncRNAs, with corresponding clinical information, were extracted from the TCGA and GSE6791 databases mentioned above. Of these, 567 lncRNAs were highly expressed and 1428 lncRNAs were expressed at low levels in the CC samples (Table 1). A total of 64 lncRNAs were obtained from the intersection of the two databases (Figure 1(a)), among which 10 lncRNAs were consistently upregulated and 54 lncRNAs were consistently downregulated in the two databases. A total of 1040 IRGs were identified from the InnateDB. The ssGSEA analysis was based on 201 IRGs obtained from the intersection of the TCGA and InnateDB databases (Figure 1(b)) to compute IS. Finally, a cohort of 28 IRLs was obtained based on the Pearson correlation coefficient analysis between the 64 lncRNAs and IS of 201 IRGs (p < 0.01).
Table 1.
Differential mRNAs and lncRNAs.
| mRNA | lncRNA | |
|---|---|---|
| TCGA | ||
| Up | 3305 | 472 |
| Down | 4163 | 733 |
| Total | 7468 | 1205 |
| GSE6791 | ||
| Up | 4680 | 95 |
| Down | 3133 | 695 |
| Total | 7813 | 790 |
Figure 1.
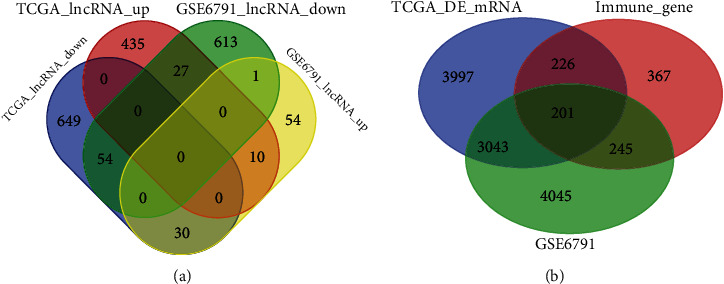
Venn analysis. (a) Venn diagram of differential lncRNAs. (b) Venn diagram of differential mRNA and immune-related genes.
3.2. Univariate Cox Regression and Kaplan–Meier Survival Analysis
Univariate Cox regression and Kaplan–Meier survival analysis were performed for the cohort of 28 IRLs. Four IRLs (BZRAP1-AS1, EMX2OS, ZNF667-AS1, and CTC-429P9.1) with low expression were found to be in accordance with our expectation and were then included in the signature development. The results are shown in Table 2. The Kaplan–Meier survival analysis revealed that four IRLs were related to OS in CC significantly (p < 0.05) (Figure 2). The four IRLs were defined as protective factors due to their HRvalue < 1, which showed that the high expression of the four IRLs was associated with lower OS.
Table 2.
K-M analysis, univariate Cox regression analysis, and Pearson correlation analysis of 4-IRLs.
| lncRNA | K-M analysis | Univariate Cox regression analysis | Pearson correlation analysis | Differential expression analysis | ||||
|---|---|---|---|---|---|---|---|---|
| p value | HR | Lower.95 | Upper.95 | p value | r | p.adj.value | Up_down | |
| BZRAP1-AS1 | 0.0002 | 0.82 | 0.72 | 0.93 | 0.00 | 0.32 | 2.84E-08 | Down_lnc |
| EMX2OS | 0.0010 | 0.91 | 0.82 | 1.00 | 0.05 | -0.25 | 3.01E-05 | Down_lnc |
| ZNF667-AS1 | 0.0019 | 0.87 | 0.77 | 0.99 | 0.03 | -0.17 | 0.007 | Down_lnc |
| CTC-429P9.1 | 0.0159 | 0.93 | 0.82 | 1.06 | 0.27 | -0.24 | 5.15E-05 | Down_lnc |
Figure 2.
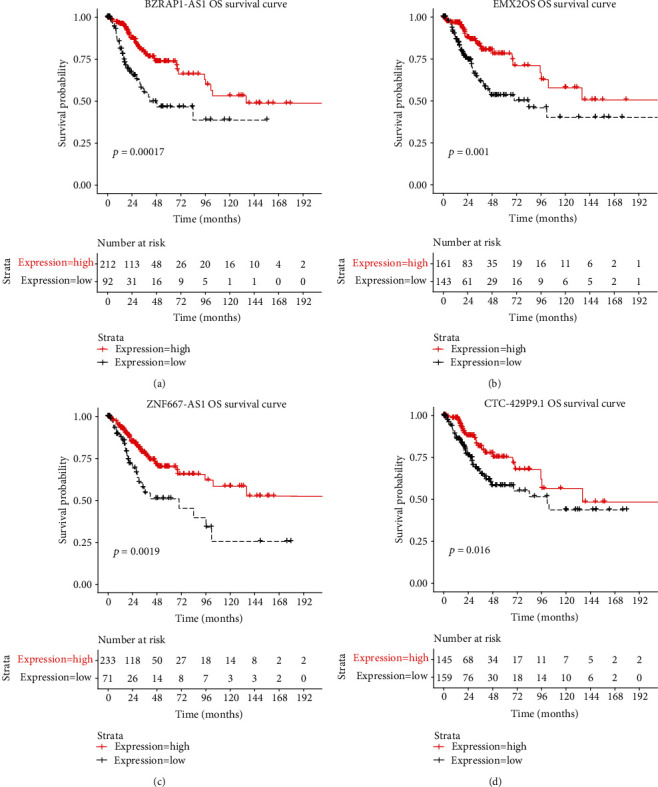
Kaplan–Meier curve of 4 IRLs.
3.3. Construction of the Prognostic Risk Model and Validation Using Four IRLs
A total of 304 TCGA samples were regarded as the total set so that there were both 152 samples in the training set and the valid set as described in the methods. The regression coefficient β was first generated from the training set (βZNF667−AS1 = −0.152565, βEMX2OS = −0.019887, βBZRAP1−AS1 = −0.17831, and βCTC−429P9.1 = 0.0096755; Table 3) to establish the risk score (RS) formula. Hence, the prognostic risk model for corresponding samples was established using the following formula:
| (1) |
Table 3.
β of each of the 4 IRLs.
| Ensemble_ID | lncRNA | β |
|---|---|---|
| ENSG00000166770.10 | ZNF667-AS1 | -0.152565 |
| ENSG00000229847.8 | EMX2OS | -0.019887 |
| ENSG00000265148.5 | BZRAP1-AS1 | -0.17831 |
| ENSG00000269427.1 | CTC-429P9.1 | 0.0096755 |
Exprgene indicated the expression value of the corresponding IRL for each sample. An RS higher than the median was identified as a high-risk group while an RS lower than the medium was identified as a low-risk group. Using this approach, a risk model signature based on four IRLs was constructed, which was further validated in the total set and valid set using the same β to confirm the prediction potential of the 4-IRL signature. The Kaplan–Meier survival curves based on the log-rank test revealed that OS in the low-risk group was markedly longer than that in the other group in all three sets (pTraining−set = 0.0068, pValid−set = 0.02, and pTotal−set = 0.0015; Figures 3(a)–3(c)). The one-year, two-year, and three-year survival ROC curves predicted by the risk model indicated that the AUCs were larger than 0.65 (0.695, 0.66, and 0.676; Figure 3(d)), thus predicting that the risk score model could efficiently forecast over 65% of OS for CC patients. Therefore, the risk model signature based on four IRLs was accurate in predicting OS of CC patients.
Figure 3.
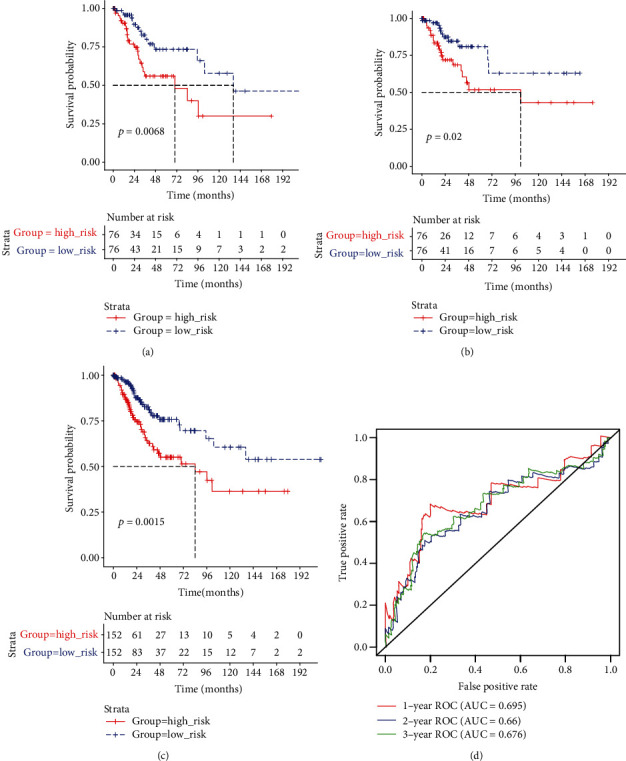
The 4-IRL prognostic risk model and validating. (a) Kaplan–Meier survival curves of OS among CC patients from different groups stratified by the signature in the training set, the valid set, and the total set. (b) Time-dependent receiver operating characteristic (ROC) curve for predicting overall survival (OS) of the risk model.
3.4. Clinicopathological Characteristics of the Risk Score Model
A heat map was constructed by combining the expression values of four IRLs and their clinicopathological characteristics (Figure 4(a)). The higher the RS, the lower the IS. RS and IS values showed a significant negative correlation based on the scatterplot of the correlation coefficient (r = −0.14, p = 0.01631; Figure 4(b)). The scatterplot for the distribution of IS in risk groups showed that the IS of the high-risk group was significantly lower than that of the low-risk group (Figure 4(c)). Furthermore, the high expression of the four IRLs could be seen with low RS, which suggested that the upregulation of the four IRLs were associated with better prognosis. In contrast, the low expression of the four IRLs had the opposite consequence. An RS contrast of the neoplasm histologic grade showed that the RS in stages IIB-III-IV was significantly greater than that in stages I-II-IIA (Figure 4(c)). This part of the result suggests that a high-risk score has an adverse effect on prognosis, which may be caused by a decrease in immune score.
Figure 4.
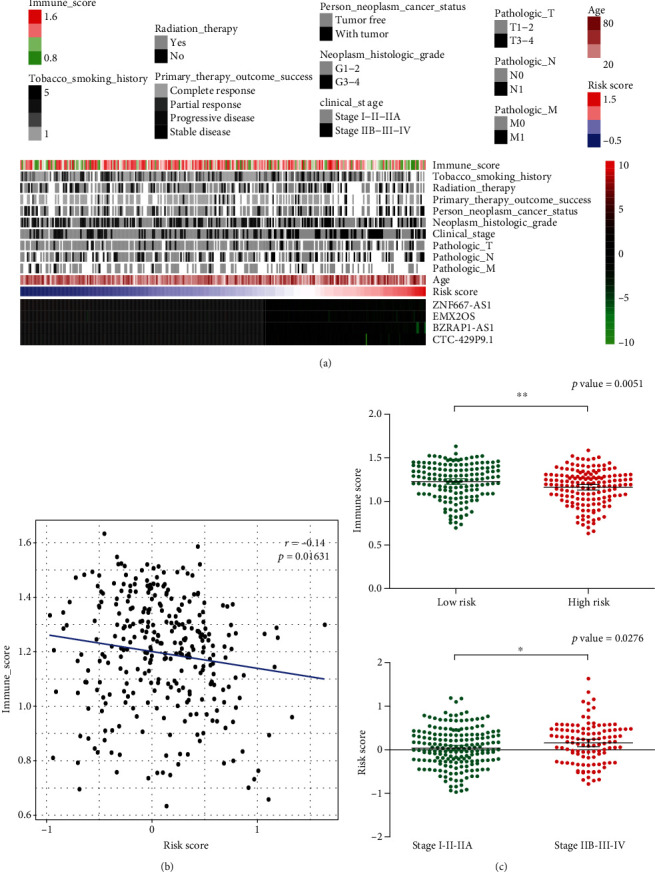
Clinical pathological characteristics of the risk score model. (a) Distribution of the immune score, clinicopathologic features, 4-IRL expression, and risk score. (b) Correlations between the risk score and immune score. (c) Immune score in different risk groups and risk score in tumor different stages.
3.5. Nomogram Model Construction and Visualization
The univariate and multivariate Cox regression analyses of clinicopathological characteristics and the four-IRL signature for the total TCGA dataset demonstrated that the four-IRL signature was an independent risk factor for CC patients (p < 0.05, Table 4). In univariate Cox analysis, FIGO stage and TNM stages were risk factors for CC patients (p < 0.05), whereas their prognostic values were not validated in the multivariate Cox analysis. Nonetheless, radiation therapy and a history of tobacco smoking were not correlated with prognosis independently. To better predict prognosis at one-, three-, and five-year OS of CC patients, we constructed a nomogram of variables such as the four-IRL signature, age, and FIGO stage (Figure 5).
Table 4.
Univariate and multivariate Cox regression analyses of clinical parameters.
| Variables | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| Coeff | HR (95% CI) | p value | Coeff | HR (95% CI) | p value | |
| Risk score | 0.854 | 2.349 (1.317-4.191) | 0.004 | 0.819 | 2.268 (1.101-4.674) | 0.026 |
| Age | 0.014 | 1.014 (0.996-1.032) | 0.117 | -0.003 | 0.997 (0.973-1.022) | 0.812 |
| Cancer type | 0.040 | 1.041 (0.531-2.041) | 0.907 | 0.049 | 1.050 (0.452-2.438) | 0.910 |
| FIGO stage | 0.362 | 1.436 (1.141-1.806) | 0.002 | 0.159 | 1.173 (0.834-1.648) | 0.359 |
| Tumor grade | 0.216 | 1.241 (0.969-1.589) | 0.087 | 0.093 | 1.098 (0.82-1.47) | 0.530 |
| M stage | 0.419 | 1.520 (1.130-2.044) | 0.006 | 0.237 | 1.268 (0.89-1.806) | 0.188 |
| N stage | 0.674 | 1.962 (1.406-2.738) | <0.001 | 0.362 | 1.436 (0.881-2.341) | 0.146 |
| T stage | 0.398 | 1.488 (1.222-1.813) | <0.001 | 0.171 | 1.186 (0.872-1.613) | 0.277 |
| Radiotherapy | 0.289 | 1.335 (0.735-2.426) | 0.343 | -0.256 | 0.774 (0.368-1.629) | 0.500 |
| Smoke | 0.130 | 1.139 (0.696-1.862) | 0.605 | -0.031 | 0.970 (0.484-1.943) | 0.930 |
Figure 5.
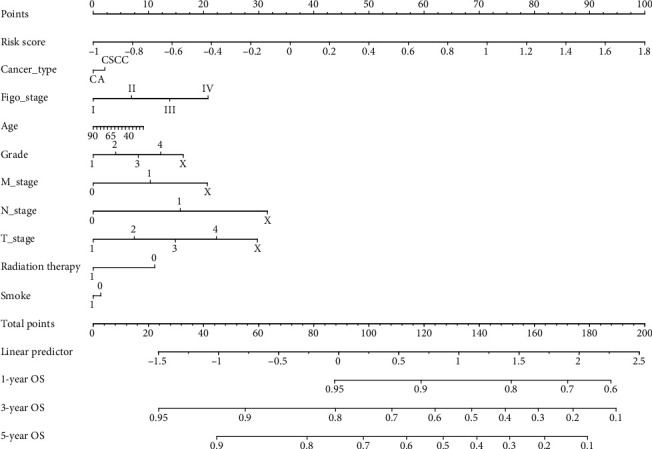
A nomogram based on the signature and clinical information.
3.6. Gene Set Enrichment Analysis
GSEA analysis of the two risk groups was carried out to predict enrichment status disparities of molecular mechanism functions. The enrichment analysis showed that 118 biological functions were markedly enriched in the low-risk group, whereas only one biological function (GO_ATP_SYNTHESIS_COUPLED_ELECTRON_TRANSPORT) was enriched in the high-risk group. In the enrichment status enriched in the low-risk group, we sought out couple immune-related responses as shown in Figure 6.
Figure 6.
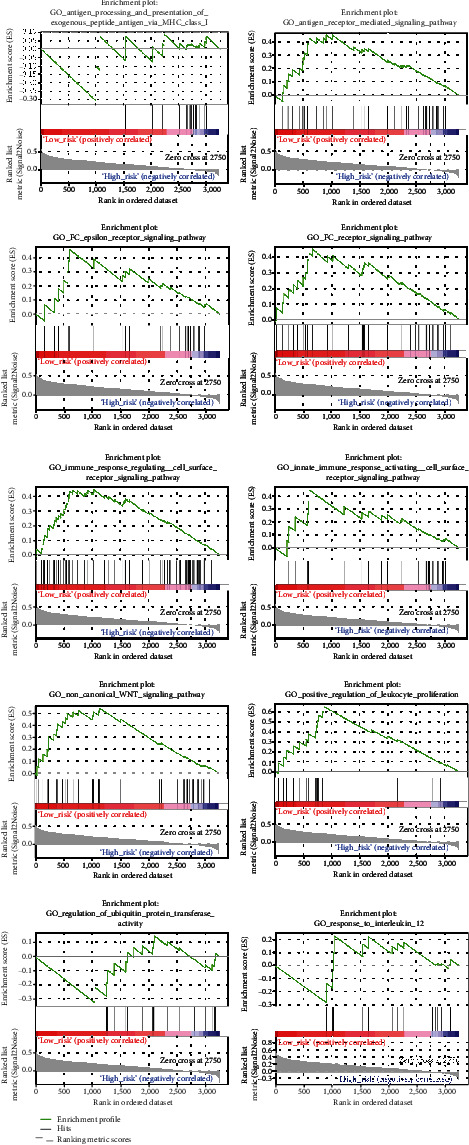
Immunologic characteristics regulated via the GSEA.
3.7. Evaluation of Infiltrating Immune Cells
Previous studies have shown that infiltrating immune cells are closely related to the prognosis and treatment of malignant tumors [29]. From the GSEA analysis in our study, we discovered that the four-IRL signature was associated with many immune characteristics. Hence, the abundance of twenty-two infiltrating immune cells (Figure 7(a)) were estimated which showed that the abundance of nine infiltrating immune cells (B cell naïve, B cell memory, T cell CD4 memory resting, NK cells resting, macrophages M1, dendritic cells activated, mast cells resting, mast cells activated, and neutrophils) was significantly different (p < 0.05) between the risk groups (Figure 7(b)). The abundance of four infiltrating immune cells (B cell naïve, T cell CD4 memory resting, macrophages M1, and mast cells resting) in the low-risk group were significantly higher than that in the high-risk group as shown by Student's t-test (p < 0.05).
Figure 7.
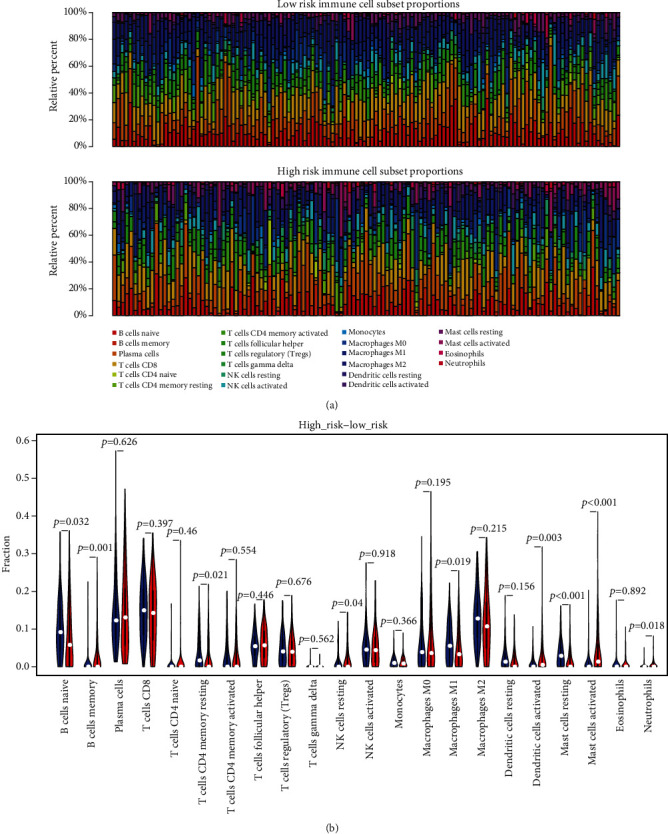
Evaluation of tumor-infiltrating immune cells. (a) The landscape of immune infiltration in risk groups. (b) The difference of 22 tumor-infiltrating immune cells among risk groups.
3.8. Construction of an IRL-Associated ceRNA Network
Four-IRLs, forty-five hub mRNAs, and thirty-eight miRNAs were involved in the ceRNA network (Figure 8). A total of 46 lncRNA-miRNA pairs, 232 miRNA-mRNA pairs, and 55 lncRNA-mRNA pairs were identified. The downregulation of 12 mRNAs in the ceRNA network was significantly related to OS in CC. Among them, the downregulation of seven mRNAs (CXCL12, FREM1, IGF1, IRF4, NFATC2, NTN1, and STAT6) had an adverse effect on prognosis, which was in line with what was expected (Figure 9).
Figure 8.
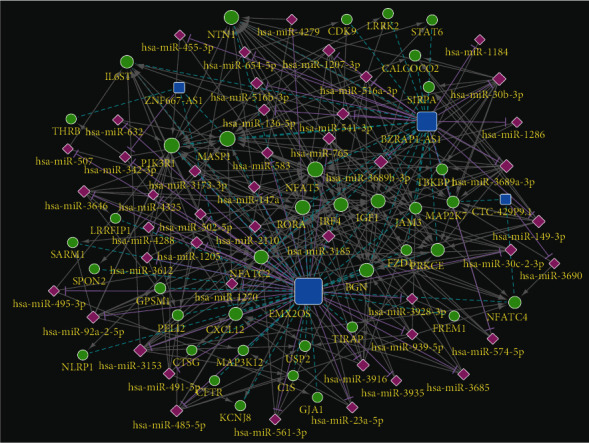
ceRNA network of 4-IRLs.
Figure 9.
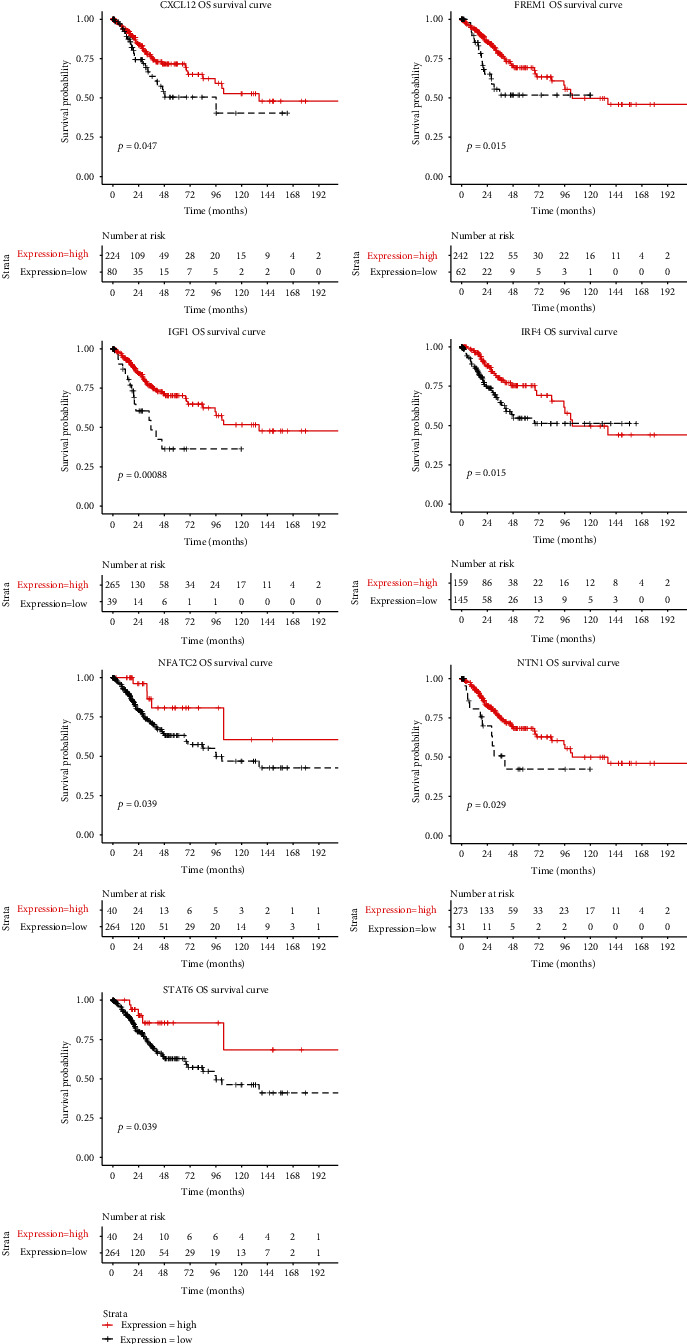
Kaplan–Meier curve of 7 mRNAs.
4. Discussion
In this study, a cohort of 326 CC and 21 normal tissue samples from two datasets (TCGA, GSE6791) were included to identify the differential lncRNAs for patients with CC. A total of 1040 IRGs were collected from the InnateDB. Four IRLs (BZRAP1-AS1, EMX2OS, ZNF667-AS1, and CTC-429P9.1) were identified after ssGSEA analysis, Pearson correlation coefficient analysis, univariate Cox regression analysis, and Kaplan–Meier survival analysis between the lncRNAs and IS. The prognostic risk model based on the four IRLs could divide CC patients into two risk groups according to the median RS, which was validated by dividing the total samples equally. The univariate and multivariate cox regression analyses of clinicopathological characteristics showed that age, AJCC stage, and the four-IRL signature were all independent prognostic factors. A nomogram was constructed based on age, AJCC stage, and four-IRL signature to predict OS for CC patients more clearly. We also found that RS and IS showed significant negative correlation, which indicated that high RS had an adverse effect on prognosis due to a decrease in IS.
In our study, the four IRLs (BZRAP1-AS1, EMX2OS, ZNF667-AS1, and CTC-429P9.1) were identified to be protective against CC; only ZNF667-AS1 had been previously reported in CC. In the existing literature, ZNF667-AS1 with low expression has been identified to be negatively correlated with the OS, tumor size, and FIGO stage in CC [30]. Additionally, ZNF667-AS1 can competitively bind to miR-93-3p, which targets PEG3, to regulate the progression of CC [31]. Recent research has shown that inhibiting PEG3 would promote the immune escape of cancer cells [32]. BZRAP1-AS1 was found to be a novel biomarker associated with prostate cancer (PC), being downregulated in PC samples [33]. It was shown, however, to be highly expressed in hepatocellular carcinoma and inhibited the transcription of THBS1 by recruiting DNMT3b to its promoter region [34]. Previous research has revealed that the downregulation of EMX2OS in classical papillary thyroid cancer might independently predict shorter recurrence-free survival [35], while the overexpression of EMX2OS in ovarian cancer and EMX2OS/miR-654/AKT3 axis may target PD-L1 (programmed cell death protein 1) to suppress the initiation and progression of cancer [36]. Accumulating evidences have suggested that Thrombospondin-1 (THBS1) may affect tumor immunity [37]. PD1-PDL1 (PD1 ligand) has already been shown to be an important immune checkpoint pathway, which can be used by cancer cells to evade immune attacks [38]. Thus, BZRAP1-AS1 and EMX2OS may play a dual role in cancer and directly or indirectly regulate tumor immunology. By contrast, no reports concerning CTC-429P9.1 in cancer have been published, and therefore, the role of CTC-429P9.1 remains unclear.
In recent years, a small number of researches have reported that lncRNAs can directly or indirectly affect the tumor microenvironment of cervical cancer. LOC105374902 induced by TNF-α, a multiple functional cytokine which can regulate inflammation and immunity of cancer, was found to promote the malignant behavior of cervical cancer cells by acting as a sponge of miR-1285-3p [39]. lncRNA SNHG14 was shown to be associated with the activation of the JAK-STAT pathway in cervical tumor cells [40]. STAT3-binding sequence in the enhancer region of lncRNA MALAT1 was demonstrated to be crucial for the IL-6- or STAT3-induced MALAT1 promoter activation in cervical cancer HeLa cells [41]. HOTAIR was identified to promote the overactivation of the Wnt/β-catenin signaling pathway by the downregulation of PCDH10, SOX17, AJAP1, and MAGI2 and also TET [42]. lncRNA HIPK1-AS has been proved to regulate the inflammatory process of cervical cancer [43]. Generally, there are still relatively few immune-related lncRNAs in CC.
Our GSEA analysis showed that certain immune-related enrichment statuses were dramatically enriched in the low-risk group. Noncanonical WNT signaling has been demonstrated to be closely associated with cancer stem cell survival, bulk tumor expansion, and invasion/metastasis, which have the potential for tumor immunology [44]. Ubiquitination has been shown to be crucial for tumor immunity [45].
The ceRNA network was established to explore specific mechanisms in the development of CC based on the competing endogenous RNA theory. Combining the mRNAs of the most outstanding prognosis with the connectivity of miRNAs, the following ceRNA relationships have been established, which should be used as a follow-up verification: EMX2OS/hsa-miR-3153/CXCL12, EMX2OS/hsa-miR-3928-3p/FREM1, BZRAP1-AS1/hsa-miR-30b-3p/IGF1, EMX2OS/hsa-miR-92a-2-5p/IRF4, BZRAP1-AS1/hsa-miR-30b-3p/NFATC2, BZRAP1-AS1/hsa-miR-30b-3p/NTN1, and BZRAP1-AS1/hsa-miR-541-3p/STAT6. CXCL12 binds primarily to CXCR4 which can cause a mass of signal routing including the immunity [46]. FREM1 has been authenticated as an immune-related gene which may be a potential target for immunotherapy in breast cancer and clear cell renal cell carcinoma [47, 48]. The insulin-like growth factor 1 (IGF1) pathway has been proven to contribute to the suppression of immune tumor microenvironment (TME) in gynecologic cancers [49]. IRF4+ Tregs have been confirmed to be correlated with poor prognosis in patients with multiple cancers [50]. NFATC2 regulates IL-21 expression in human CD4+ CD45RO+ T lymphocytes [51]. STAT6 is a factor converge on intracellular determinants of cell functions and drives the recruitment and polarization of tumor-associated macrophages (TAMs) [52]. Thus, our IRLs need to be further investigated.
Compared to a certain number of studies carried out to explore IRL signatures in several human malignancies [53–59], this was the first study based on a comprehensive analysis that focused on IRL signature in CC. However, our study has some limitations. Additional datasets are needed to validate our study. The specific molecular mechanisms in which these genes may be involved should be verified through in vitro and in vivo experiments.
5. Conclusions
We established a four-IRL-based signature with a prognostic value for CC, which could stratify CC patients into the low- and high-risk groups. Meanwhile, the specific mechanisms of the four IRLs in the development of CC were preliminarily ascertained. This may provide the basis for tumor prevention and immunotherapy in the future.
Acknowledgments
The author, Jianfeng Zheng, gratefully acknowledges Miss Lynn for providing assistance with English writing.
Abbreviations
- CC:
Cervical cancer
- IRL:
Immune-related lncRNA
- IS:
Immune score
- ssGSEA:
Single-sample gene set enrichment analysis
- OS:
Overall survival
- AUC:
Area under the curve
- ROC:
Receiver operating characteristic
- HPV:
Human papillomavirus
- EMT:
Epithelial-mesenchymal transition
- GSEA:
Gene set enrichment analysis
- TCGA:
The Cancer Genome Atlas
- GEO:
Gene expression omnibus
- HR:
Hazard ratio.
Data Availability
The RNA sequencing profiles are able to be gained from The Cancer Genome Atlas (TCGA) (https://toil.xenahubs.net) and Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). The immune-related genes can be downloaded from the InnateDB database (http://www.innatedb.com).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
JZ, BC, NZ, and XZ contributed to data analysis, interpretation, and drafting. JZ and JT contributed to study design, study supervision, and final approval of the manuscript. All authors read and approved the final manuscript.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Hu Z., Ma D. The precision prevention and therapy of HPV-related cervical cancer: new concepts and clinical implications. Cancer Medicine. 2018;7(10):5217–5236. doi: 10.1002/cam4.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vu M., Yu J., Awolude O. A., Chuang L. Cervical cancer worldwide. Current Problems in Cancer. 2018;42(5):457–465. doi: 10.1016/j.currproblcancer.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Liontos M., Kyriazoglou A., Dimitriadis I., Dimopoulos M. A., Bamias A. Systemic therapy in cervical cancer: 30 years in review. Critical Reviews in Oncology/Hematology. 2019;137:9–17. doi: 10.1016/j.critrevonc.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Barabas A. Z., Cole C. D., Lafreniere R. Antibody-initiated beneficial and harmful immune responses. Immunologic Research. 2018;66(6):783–789. doi: 10.1007/s12026-018-9037-0. [DOI] [PubMed] [Google Scholar]
- 6.Ventriglia J., Paciolla I., Pisano C., et al. Immunotherapy in ovarian, endometrial and cervical cancer: state of the art and future perspectives. Cancer Treatment Reviews. 2017;59:109–116. doi: 10.1016/j.ctrv.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 7.O'Donnell J. S., Teng M. W. L., Smyth M. J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nature Reviews Clinical Oncology. 2019;16(3):151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 8.Kopp F., Mendell J. T. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172(3):393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tornesello M. L., Faraonio R., Buonaguro L., et al. The role of microRNAs, long non-coding RNAs, and circular RNAs in cervical cancer. Frontiers in Oncology. 2020;10:p. 150. doi: 10.3389/fonc.2020.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan Z., Liu L., Nie W., et al. Long non-coding RNA AGER-1 functionally upregulates the innate immunity gene AGER and approximates its anti-tumor effect in lung cancer. Molecular Carcinogenesis. 2018;57(3):305–318. doi: 10.1002/mc.22756. [DOI] [PubMed] [Google Scholar]
- 11.Mao D., Hu C., Zhang J., et al. Long noncoding RNA GM16343 promotes IL-36β to regulate tumor microenvironment by CD8+T cells. Technology in Cancer Research & Treatment. 2019;18, article 1533033819883633 doi: 10.1177/1533033819883633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu Q., Ye Y., Chan L. C., et al. Oncogenic lncRNA downregulates cancer cell antigen presentation and intrinsic tumor suppression. Nature Immunology. 2019;20(7):835–851. doi: 10.1038/s41590-019-0400-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang P., Xiang L., Chen W., et al. LncRNA GAS5 enhanced the killing effect of NK cell on liver cancer through regulating miR-544/RUNX3. Innate Immunity. 2019;25(2):99–109. doi: 10.1177/1753425919827632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei X., Wang X., Li H. LncRNA SNHG1 regulates the differentiation of Treg cells and affects the immune escape of breast cancer via regulating miR-448/IDO. International Journal of Biological Macromolecules. 2018;118, Part A:24–30. doi: 10.1016/j.ijbiomac.2018.06.033. [DOI] [PubMed] [Google Scholar]
- 15.Jiang R., Tang J., Chen Y., et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nature Communications. 2017;8(1, article 15129) doi: 10.1038/ncomms15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldman M. J., Craft B., Hastie M., et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nature Biotechnology. 2020;38(6):675–678. doi: 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrow J., Frankish A., Gonzalez J. M., et al. GENCODE: the reference human genome annotation for the ENCODE project. Genome Research. 2012;22(9):1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrett T., Troup D. B., Wilhite S. E., et al. NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucleic Acids Research. 2007;35(D1):D760–D765. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ritchie M. E., Phipson B., Wu D., et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015;43(7, article e47) doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang H., Wong W. H. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24(20):2395–2396. doi: 10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breuer K., Foroushani A. K., Laird M. R., et al. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Research. 2013;41(D1):D1228–D1233. doi: 10.1093/nar/gks1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshihara K., Shahmoradgoli M., Martínez E., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nature Communications. 2013;4(1, article 2612) doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berkman S. J., Roscoe E. M., Bourret J. C. Comparing self-directed methods for training staff to create graphs using Graphpad Prism. Journal of Applied Behavior Analysis. 2019;52(1):188–204. doi: 10.1002/jaba.522. [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A., Tamayo P., Mootha V. K., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen B., Khodadoust M. S., Liu C. L., Newman A. M., Alizadeh A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods in Molecular Biology. 2018;1711:243–259. doi: 10.1007/978-1-4939-7493-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tay Y., Rinn J., Pandolfi P. P. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paraskevopoulou M. D., Vlachos I. S., Karagkouni D., et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Research. 2016;44(D1):D231–D238. doi: 10.1093/nar/gkv1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dweep H., Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nature Methods. 2015;12(8):p. 697. doi: 10.1038/nmeth.3485. [DOI] [PubMed] [Google Scholar]
- 29.Jochems C., Schlom J. Tumor-infiltrating immune cells and prognosis: the potential link between conventional cancer therapy and immunity. Experimental Biology and Medicine. 2011;236(5):567–579. doi: 10.1258/ebm.2011.011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao L. P., Li R. H., Han D. M., et al. Independent prognostic factor of low-expressed LncRNA ZNF667-AS1 for cervical cancer and inhibitory function on the proliferation of cervical cancer. European Review for Medical and Pharmacological Sciences. 2017;21(23):5353–5360. doi: 10.26355/eurrev_201712_13920. [DOI] [PubMed] [Google Scholar]
- 31.Li Y. J., Yang Z., Wang Y. Y., Wang Y. Long noncoding RNA ZNF667-AS1 reduces tumor invasion and metastasis in cervical cancer by counteracting microRNA-93-3p-dependent PEG3 downregulation. Molecular Oncology. 2019;13(11):2375–2392. doi: 10.1002/1878-0261.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang F., Wang T., Du P., Fan H., Dong X., Guo H. M2 bone marrow-derived macrophage-derived exosomes shuffle microRNA-21 to accelerate immune escape of glioma by modulating PEG3. Cancer Cell International. 2020;20(1):p. 93. doi: 10.1186/s12935-020-1163-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan J., Jin X., Wang K. Integrated bioinformatics analysis of potential biomarkers for prostate cancer. Pathology Oncology Research. 2019;25(2):455–460. doi: 10.1007/s12253-017-0346-8. [DOI] [PubMed] [Google Scholar]
- 34.Wang W., Chen G., Wang B., et al. Long non-coding RNA BZRAP1-AS1 silencing suppresses tumor angiogenesis in hepatocellular carcinoma by mediating THBS1 methylation. Journal of Translational Medicine. 2019;17(1):p. 421. doi: 10.1186/s12967-019-02145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu Y., Feng C., Liu T., Zhang B., Yang L. The downregulation of lncRNA EMX2OS might independently predict shorter recurrence-free survival of classical papillary thyroid cancer. PLoS One. 2018;13(12, article e0209338) doi: 10.1371/journal.pone.0209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan M., Fang M., Wang C., Wang H., Li M. LncRNA EMX2OS induces proliferation, invasion and sphere formation of ovarian cancer cells via regulating the miR-654-3p/AKT3/PD-L1 axis. Cancer Management and Research. 2020;12:2141–2154. doi: 10.2147/CMAR.S229013. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Huang T., Sun L., Yuan X., Qiu H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget. 2017;8(48):84546–84558. doi: 10.18632/oncotarget.19165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X., Shao C., Shi Y., Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. Journal of Hematology & Oncology. 2018;11(1):p. 31. doi: 10.1186/s13045-018-0578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng Y., Ma J., Fan H., et al. TNF-α-induced lncRNA LOC105374902 promotes the malignant behavior of cervical cancer cells by acting as a sponge of miR-1285-3p. Biochemical and Biophysical Research Communications. 2019;513(1):56–63. doi: 10.1016/j.bbrc.2019.03.079. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y. Y., Li M., Xu Y. D., Shang J. LncRNA SNHG14 promotes the development of cervical cancer and predicts poor prognosis. European Review for Medical and Pharmacological Sciences. 2019;23(9):3664–3671. doi: 10.26355/eurrev_201905_17790. [DOI] [PubMed] [Google Scholar]
- 41.Hao Y., Yan Z., Zhang A., et al. IL-6/STAT3 mediates the HPV18 E6/E7 stimulated upregulation of MALAT1 gene in cervical cancer HeLa cells. Virus Research. 2020;281, article 197907 doi: 10.1016/j.virusres.2020.197907. [DOI] [PubMed] [Google Scholar]
- 42.Salmerón-Bárcenas E. G., Illades-Aguiar B., Del Moral-Hernández O., Ortega-Soto A., Hernández-Sotelo D. HOTAIR knockdown decreased the activity Wnt/β-catenin signaling pathway and increased the mRNA levels of its negative regulators in Hela cells. Cellular Physiology and Biochemistry. 2019;53(6):948–960. doi: 10.33594/000000188. [DOI] [PubMed] [Google Scholar]
- 43.Zhou B., Yu Y., Yu L., Que B., Qiu R. Sipi soup inhibits cancer-associated fibroblast activation and the inflammatory process by downregulating long non-coding RNA HIPK1-AS. Molecular Medicine Reports. 2018;18(2):1361–1368. doi: 10.3892/mmr.2018.9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (review) International Journal of Oncology. 2017;51(5):1357–1369. doi: 10.3892/ijo.2017.4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mansour M. A. Ubiquitination: friend and foe in cancer. The International Journal of Biochemistry & Cell Biology. 2018;101:80–93. doi: 10.1016/j.biocel.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 46.Zhou W., Guo S., Liu M., Burow M. E., Wang G. Targeting CXCL12/CXCR4 Axis in tumor immunotherapy. Current Medicinal Chemistry. 2019;26(17):3026–3041. doi: 10.2174/0929867324666170830111531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z., Li J., He T., Ding J. Bioinformatics identified 17 immune genes as prognostic biomarkers for breast cancer: application study based on artificial intelligence algorithms. Frontiers in Oncology. 2020;10:p. 330. doi: 10.3389/fonc.2020.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo J., Xie Y., Zheng Y., et al. Comprehensive insights on pivotal prognostic signature involved in clear cell renal cell carcinoma microenvironment using the ESTIMATE algorithm. Cancer Medicine. 2020;9(12):4310–4323. doi: 10.1002/cam4.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yahya M. A., Sharon S. M., Hantisteanu S., Hallak M., Bruchim I. The role of the insulin-like growth factor 1 pathway in immune tumor microenvironment and its clinical ramifications in gynecologic malignancies. Frontiers in Endocrinology. 2018;9:p. 297. doi: 10.3389/fendo.2018.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvisi G., Brummelman J., Puccio S., et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. The Journal of Clinical Investigation. 2020;130(6):3137–3150. doi: 10.1172/JCI130426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.el-Said H., Fayyad-Kazan M., Aoun R., et al. miR302c, Sp1, and NFATc2 regulate interleukin-21 expression in human CD4+CD45RO+ T lymphocytes. Journal of Cellular Physiology. 2019;234(5):5998–6011. doi: 10.1002/jcp.27151. [DOI] [PubMed] [Google Scholar]
- 52.Szebeni G. J., Vizler C., Kitajka K., Puskas L. G. Inflammation and cancer: extra- and intracellular determinants of tumor-associated macrophages as tumor promoters. Mediators of Inflammation. 2017;2017:13. doi: 10.1155/2017/9294018.9294018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y., Jiang T., Zhou W., et al. Pan-cancer characterization of immune-related lncRNAs identifies potential oncogenic biomarkers. Nature Communications. 2020;11(1, article 1000) doi: 10.1038/s41467-020-14802-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen Y., Peng X., Shen C. Identification and validation of immune-related lncRNA prognostic signature for breast cancer. Genomics. 2020;112(3):2640–2646. doi: 10.1016/j.ygeno.2020.02.015. [DOI] [PubMed] [Google Scholar]
- 55.Wang W., Zhao Z., Yang F., et al. An immune-related lncRNA signature for patients with anaplastic gliomas. Journal of Neuro-Oncology. 2018;136(2):263–271. doi: 10.1007/s11060-017-2667-6. [DOI] [PubMed] [Google Scholar]
- 56.Wei C., Liang Q., Li X., et al. Bioinformatics profiling utilized a nine immune-related long noncoding RNA signature as a prognostic target for pancreatic cancer. Journal of Cellular Biochemistry. 2019;120(9):14916–14927. doi: 10.1002/jcb.28754. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y., Zhang L., Xu Y., Wu X., Zhou Y., Mo J. Immune-related long noncoding RNA signature for predicting survival and immune checkpoint blockade in hepatocellular carcinoma. Journal of Cellular Physiology. 2020;235(12):9304–9316. doi: 10.1002/jcp.29730. [DOI] [PubMed] [Google Scholar]
- 58.Zhong Z., Hong M., Chen X., et al. Transcriptome analysis reveals the link between lncRNA-mRNA co-expression network and tumor immune microenvironment and overall survival in head and neck squamous cell carcinoma. BMC Medical Genomics. 2020;13(1):p. 57. doi: 10.1186/s12920-020-0707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou M., Zhang Z., Zhao H., Bao S., Cheng L., Sun J. An immune-related six-lncRNA signature to improve prognosis prediction of glioblastoma multiforme. Molecular Neurobiology. 2017;55(5):3684–3697. doi: 10.1007/s12035-017-0572-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The RNA sequencing profiles are able to be gained from The Cancer Genome Atlas (TCGA) (https://toil.xenahubs.net) and Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). The immune-related genes can be downloaded from the InnateDB database (http://www.innatedb.com).