

Abstract
Patients with gout are at a higher risk of cardiovascular disease, which is associated with hyperlipidemia. Management of gout in Taiwan is poor, and the association between urate-lowering therapy (ULT) among gout patients and hyperlipidemia is unclear. We conducted a retrospective cohort study using data from the Longitudinal Health Insurance Database (LHID) of Taiwan on new-onset gout patients and a comparison cohort without gout. A Cox proportional hazards model was used to analyze differences in the risk of hyperlipidemia between patients with and without gout after considering related comorbidities. We also examined the ULT medications on the hepatic expression of lipogenesis-related genes. After adjusting for potential confounders, the case group (44,413 patients) was found to have a higher risk of hyperlipidemia than the control cohort (177,652 patients) [adjusted hazards ratio (aHR) = 2.55]. Gout patients without antigout treatment had significantly higher risk of hyperlipidemia than the control cohort (aHR = 3.10). Among gout patients receiving ULT, except those receiving probenecid (aHR = 0.80), all had significantly lower risk of hyperlipidemia than gout patients without ULT (all aHR < 0.90). Using real-time polymerase chain reaction, we found that most of the antigout drugs decreased the expression of hepatic genes related to lipogenesis in differentiated HepaRG cells. These data indicate that these antigout drugs reduce hyperlipidemia in gout patients, partly via the reduction in expression of lipogenesis-related genes, leading to improved blood lipid profiles. We provide evidence of the strong association between gout and hyperlipidemia and highlight the need for appropriate treatment guidelines.
1. Introduction
Gout is a common form of severe, inflammatory arthritis in adults, especially in males [1]. It is characterized by the crystallization of uric acid (UA), a product of purine metabolism, as a result of chronic elevation of serum UA levels above the saturation point for urate crystal formation. Urate crystal deposits accumulate within small and large joints and may lead to irreversible dysfunction of peripheral joints, subcutaneous tophi, urate nephropathy, and urolithiasis [1, 2]. The disease burden is large and is likely to continue to increase in developed countries, affecting 0.53–6.1% of the population due to changes in lifestyle and diet [3].
Gout-related comorbidities and their treatment may have an effect on the development of gout and on the choice of therapeutic agents for gout. Drugs used to treat acute attacks of gout-related pain and prevent future pain attacks include nonsteroidal anti-inflammatory drugs (NSAIDs), consisting of over-the-counter options for an acute attack; colchicine for an acute attack or prescribed at a low daily dose to prevent future attacks; and corticosteroids to control gout inflammation and pain for patients who cannot take NSAIDs or colchicine [4]. However, urate-lowering therapy (ULT) may be recommended for patients who frequently experience gout attacks and to reduce gout-related complications. It includes xanthine oxidase inhibitors (XOIs) such as allopurinol and febuxostat, to block and lower UA production, and uricosurics such as benzbromarone, sulfinpyrazone, and probenecid, to remove UA from the kidneys [4].
A meta-analysis revealed that gout was associated with an increased risk of myocardial infarction (MI) [relative risk (RR) 1.45; 95% confidence interval (CI), 1.19–1.75] in both men and women [5]. In addition, a gradual increase in risk of MI was observed for younger age at gout onset (age 20–44 years, RR 2.82; 95% CI, 1.38–5.79) [5]. Gout was associated with an excess risk of cardiovascular disease- (CVD-) related mortality [hazard ratio (HR) 1.29, 95% CI 1.14–1.44] and coronary heart disease- (CHD-) related mortality (HR 1.42, 95% CI 1.22–1.63), which remained statistically significant after adjusting for traditional cardiovascular risk factors in a meta-analysis report [6]. Epidemiological studies originating from big data in healthcare support the association between gout and CVD. The prevalence of gout in the general population of Taiwan is 1 in 16 people; although the incidence has decreased recently, the prevalence remains unchanged [7]. Furthermore, asymptomatic hyperuricemia in elderly Chinese subjects without comorbidities was also linked to a significant risk of stroke events [8].
Several clinical reports have shown that hypercholesterolemia and hypertriglyceridemia are associated with an increased risk of gout [9, 10]. Hyperuricemia is an independent risk factor associated with the prevalence and parameters of metabolic syndrome [8, 11]. Hyperlipidemia [low levels of high-density lipoprotein cholesterol (HDL-C), high levels of triglycerides (TGs), total cholesterol (TC), and low-density lipoprotein cholesterol (LDL-C)] is one of the main characteristics of metabolic syndrome. This could be associated with higher incidence of stroke and CVD [12] as observed in an animal model [13].
A high LDL-C level is a strong risk factor for CVD, and hence, determining its risk factors is clinically relevant as it is an important therapeutic target for preventing CVD [14, 15]. Approximately 25–60% of patients with gout have higher levels of serum TC and TGs [16]; thus, gout control may be beneficial for blood lipid control. However, despite the high prevalence of gout, well-understood pathogenesis, and well-established ULT, gout is still ignored by doctors in primary care [2], who often treat gout as an acute disease, rather than as a chronic disease with major adverse consequences. Management of gout in Taiwan is poor, with only 1/5 affected patients being treated with ULT [7].
Liver X receptors (LXRs) are members of a nuclear receptor superfamily that function as ligand-activated transcription factors. There are two isoforms of LXRs, LXRα (encoded by NR1H3) and LXRβ (encoded by NR1H2) [17]. LXRβ is expressed ubiquitously, whereas LXRα is highly and specifically expressed in tissues with high levels of lipid metabolism that are responsible for de novo synthesis and excretion of cholesterol, such as the liver, adipose tissue, intestine, macrophages, and lungs [17]. Activation of LXRα results in the development of steatosis, which is mediated by the hepatic lipogenic pathway, primarily through sterol regulatory element binding protein 1 (SREBP-1c) [18]. In addition, hepatic expression of LXRα, SREBP-1c, and their target genes, such as acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), and stearoyl-CoA desaturase (SCD), has been found to be significantly upregulated in liver biopsies from nonalcoholic fatty liver disease (NAFLD) patients [19]. Ultrasonographic monitoring revealed hepatic steatosis in approximately 50% of hyperlipidemia patients, with hypertriglyceridemia being frequently associated with this condition [20]. Patients with NAFLD had significantly higher levels of LDL-C, TG, and TC, but lower levels of HDL-C; indeed, the increase in TG levels was positively related to the severity of NAFLD [20]. Thus, the most important role of LXRα is the maintenance of lipid homeostasis as it regulates the balance of lipid-metabolism genes.
As the relationship between gout and hyperlipidemia is not fully elucidated in Asian populations and the Nutrition and Health Survey in Taiwan reported prevalence of gout in 8.2% of men and 2.3% of women in Taiwan from 2005 to 2008 [21], it is important for physicians to understand the relationship between gout and hyperlipidemia. The objective of our study was to investigate the relationship between gout and hyperlipidemia by assessing the risk of development of hyperlipidemia in gout patients using the data obtained from the Taiwan National Health Insurance Research Database (NHIRD), which contains comprehensive information on diagnoses, prescriptions, and hospitalization of the entire population in Taiwan. Using this database, we conducted a population-based cohort study with a large sample drawn from Taiwanese adults and characterized the in vitro effects of ULT medications on the hepatic expression of lipogenesis-related genes that may play important roles in blood lipid homeostasis.
2. Materials and Methods
2.1. Data Source
The NHIRD was established in 1995 by the National Health Insurance Administration, Ministry of Health and Welfare, and consists of comprehensive information covering more than 99% insured persons who reside in Taiwan; it is also known as the Longitudinal Health Insurance Database (LHID). Between March 1995 and the end of 2013, the LHID included registration files and claim data on demographics, dates of clinical visits, and details of disease diagnoses and medical procedures for approximately 28.75 million (living and deceased) beneficiaries registered in the database. Upon request, the National Health Research Institute (the data holder of NHIRD) randomly selected a sample comprising one million individuals with a diagnosis of gout from NHIRD for use in this cohort study. This sample contained all original claim data of outpatient departments, emergency departments, and hospitalization.
Denominator data were based on the Registry of Beneficiaries, a part of NHIRD with records of the demographics, insurance status, residence, and socioeconomic data of all beneficiaries. However, ethnicity data were not available. The data were anonymized and deidentified prior to analysis; therefore, the need for patient consent was waived. Diagnostic codes were defined based on the International Classification of Diseases, 9th Revision, Clinical Modification (ICD-9-CM) codes. The Research Ethics Committee of China Medical University Hospital in Taiwan approved the study (CMUH-104-REC2-115-R3).
2.2. Study Population
The date of diagnosis of gout was taken as the index date, and a total of 44,413 patients, who were diagnosed with gout (ICD-9-CM: 274) more than once in outpatient visits or at least once in inpatient visits and who were without hyperlipidemia (ICD-9-CM: 272) before the index date, between January 1, 2000, and December 31, 2013, were identified as the case cohort. Each case was randomly matched with 4 patients without gout during the same study period, and a total of 177,652 patients were recognized as the comparison cohort. Matching variables included sex, age, and index date, and we adopted two study cohorts by frequency matching on age with every 5-year span; hence, the index date of each sample in the comparison cohort was identical to the index date of the corresponding case. The end date of the follow-up period was the date on which hyperlipidemia occurred in patients; patients died or withdrew from NHIRD, or December 31, 2013, whichever was earlier.
The covariates of comorbidity comprised hypertension (ICD-9-CM: 401 to 405), stroke (ICD-9-CM: 430 to 438), diabetes mellitus (ICD-9-CM: 250), chronic obstructive pulmonary disease (COPD) (ICD-9-CM: 491, 492, and 496), coronary artery disease (CAD) (ICD-9-CM: 410 to 414), alcohol-related illness (ICD-9-CM: 291, 303, 305, 571.0, 571.1, 571.2, 571.3, 790.3, and V11.3), and asthma (ICD-9-CM: 493). Patients diagnosed with any of these comorbidities more than once in outpatient visits or at least once in inpatient visits before the index date were included in the study. Antigout treatments were determined by their Anatomical Therapeutic Chemical classification codes. The medications included in the analysis that available in Taiwan were allopurinol, febuxostat, benzbromarone, sulfinpyrazone, probenecid, and colchicine.
2.3. Chemical and Cell Culture
All chemicals were purchased from Sigma-Aldrich (St. Louis, Missouri, USA) and were of the highest-purity grade available. Chemicals were dissolved in dimethyl sulfoxide (DMSO) at appropriate concentrations before use. Human hepatoma HepaRG™ cells were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, USA). Frozen cells were thawed and maintained in Williams' E medium (Sigma-Aldrich, St. Louis, Missouri, USA) supplemented with 10% FetalClone™ II serum (Hyclone™, GE Healthcare, Chicago, Illinois, USA), 1 × L-glutamine, 5 μg/mL human insulin, and 50 μM hydrocortisone hemisuccinate without antibiotics for 2 weeks. Next, the medium was replaced with the aforementioned medium plus 2% DMSO for two more weeks to induce differentiated hepatocyte-like properties. Cells were cultured in a humidified atmosphere of 5% CO2 at 37°C. Cell viability was assessed using p-nitrophenylphosphate (PNPP) in an acid phosphatase (ACP) assay, as previously reported [22].
2.4. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR) Analysis
To evaluate the effects of ULT (allopurinol, febuxostat, benzbromarone, sulfinpyrazone, probenecid, and colchicine) on hepatic lipogenesis-related gene expression, mRNA levels were measured. Total RNA was extracted from differentiated HepaRG under various treatment conditions using a Direct-zol™ RNA MiniPrep kit (ZYMO Research, Irvine, CA, USA) by following the manufacturer's protocol. The quantity and purity of RNA were confirmed by calculating the ratio of the absorbance at 260 nm to absorbance at 280 nm. Total RNA (1 μg) was subjected to synthesis of first-strand cDNA using a MultiScribe™ reverse transcriptase kit (Thermo Fisher Scientific, Waltham, MA, USA). Expression of SREBP-1c, SCD, FAS, fatty acid elongase (FAE), adenosine 5′-triphosphate (ATP) citrate lyase (ACLY), ACC, LXRα, and β-actin was analyzed by qRT-PCR using Luminaris Color HiGreen qPCR master mix (Thermo Fisher Scientific, Waltham, MA, USA) in a StepOnePlus™ Real-Time PCR System by using standard procedures. Each pair of specific primers used for real-time PCR analysis is listed in Table 1. The amount of target cDNA in each sample was calculated by determining a fractional PCR threshold cycle number (Ct value). The relative mRNA levels were normalized to those of β-actin, and the target cDNA expression was calculated using 2−(Ct target gene–Ct β-actin). Data are presented as fold change compared to the control group.
Table 1.
Sequences of PCR primers.
| Gene | Species | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|---|
| SREBP-1c | Human | CGC TCC TCC ATC AAT GAC AA | TGC AGA AAG CGA ATG TAG TCG AT |
| SCD | Human | CCG ACG TGG CTT TTT CTT CT | GCG TAC TCC CCT TCT CTT TGA C |
| FAS | Human | ACA TCA TCG CTG GTG GTC TG | GGA GCG AGA AGT CAA CAC GA |
| FAE | Human | TTC CGA GTC TCC CGG AAG T | ACA GCC CAT CAG CAT CTG AGT |
| ACLY | Human | GTG TGG ACG TGG GTG ATG TG | TTG ATG TCC TCA GGA TTC AGT TTC |
| ACC | Human | CTC TTG ACC CTG GCT GTG TAC TAG | TGA GTG CCG TGC TCT GGA T |
| LXRα | Human | CGA TC GAG GTG ATG CTT CTG | GGC AAA GTC TTC CCG GTT AT |
| β-Actin | Human | CCT GGC ACC CAG CAC AAT | GCC GAT CCA CAC GGA GTA CT |
2.5. Statistical Analysis
For intercohort comparisons, Student's t-tests and chi-square tests were used for continuous variables and categorical variables, respectively. Cumulative incidence rates and curves of hyperlipidemia were estimated and plotted by the Kaplan-Meier method, and log-rank tests were used to compare differences in time-to-event distributions between case and comparison cohorts. Cox proportional hazards models were developed to calculate HRs with 95% CIs of hyperlipidemia in patients, with or without gout. Adjusted HRs were calculated with the following covariates: age, sex, and comorbidities of hypertension, stroke, diabetes, COPD, CAD, alcohol-related illness, and asthma, to evaluate the risk of hyperlipidemia between case and comparison cohorts.
Subanalyses stratified by sex, age group, comorbidity, and antigout group were also performed to assess the association of gout with the subsequent risk of hyperlipidemia. Two-sided significance levels of all tests were set at 0.05. All data were analyzed using SAS 9.4 software (SAS Institute Inc., Cary, NC, USA), and cumulative incidence curves were plotted using R software (R version 3.5.3, GNU package).
For in vitro studies, data obtained from separate measurements were reported as the mean ± standarderror (SE). The P value for each experimental comparison was determined using analysis of variance, followed by the Least Significant Difference test for multiple comparisons. All P values were determined relative to the vehicle control group, as indicated in the figures. All statistical analyses were performed using SPSS for Windows, version 20.0 (IBM SPSS, Armonk, NY, USA). A P value less than 0.05 was considered statistically significant.
3. Results
3.1. Baseline Characteristics: Demographic and Association Findings
In this study, approximately 22.5% of patients were female, and the remaining were male. Among patients with or without gout, 57.6%, 24.1%, and 18.3% of patients were in the ≤49-, 50–64-, and ≥65-year-old age groups, respectively. The average age in the case cohort was 47.9 ± 16.9 years, and the average age in the comparison cohort was and 47.3 ± 17.1 years. The percentages of patients with hypertension, stroke, diabetes, COPD, CAD, alcohol-related illness, and asthma in the case cohort were significantly different from those in the comparison cohort (all P < 0.001). Approximately 14.3%, 0.21%, 43.4%, 2.87%, 0.51%, and 50.6% of patients received allopurinol, febuxostat, benzbromarone, sulfinpyrazone, probenecid, and colchicine, respectively (Table 2).
Table 2.
Demographic characteristics, comorbidities, and medication in patients with and without gout.
| Variable | Gout | P value | |
|---|---|---|---|
| No | Yes | ||
| N = 177652 | N = 44413 | ||
| Sex | N (%) | N (%) | 0.99 |
| Female | 39952 (22.5) | 9988 (22.5) | |
| Male | 137700 (77.5) | 34425 (77.5) | |
| Age, mean (SD)# | 47.3 (17.1) | 47.9 (16.9) | <0.001 |
| Stratified age | 0.99 | ||
| ≤49 | 102312 (57.6) | 25578 (57.6) | |
| 50-64 | 42884 (24.1) | 10721 (24.1) | |
| 65+ | 32456 (18.3) | 8114 (18.3) | |
| Comorbidity | |||
| Hypertension | 19455 (11.0) | 6396 (14.4) | <0.001 |
| Stroke | 6654 (3.75) | 2063 (4.65) | <0.001 |
| Diabetes | 4786 (2.69) | 1674 (3.77) | <0.001 |
| COPD | 8137 (4.58) | 2507 (5.64) | <0.001 |
| CAD | 7870 (4.43) | 2839 (6.39) | <0.001 |
| Alcohol-related illness | 7922 (4.46) | 2728 (6.14) | <0.001 |
| Asthma | 5049 (2.84) | 1723 (3.88) | <0.001 |
| Medication | |||
| Allopurinol | 6346 (14.3) | ||
| Febuxostat | 93 (0.21) | ||
| Benzbromarone | 19286 (43.4) | ||
| Sulfinpyrazone | 1275 (2.87) | ||
| Probenecid | 226 (0.51) | ||
| Colchicine | 22482 (50.6) | ||
Chi-square test. #Student's t-test.
The Kaplan-Meier curves are shown in Figure 1. The cumulative incidence of hyperlipidemia was lower in patients without gout than in patients with gout, and the log-rank test for the comparison of cumulative incidence curves revealed that P < 0.001. The average follow-up periods of the comparison and case cohorts were 7.76 ± 3.93 years and 6.60 ± 4.11 years, respectively.
Figure 1.
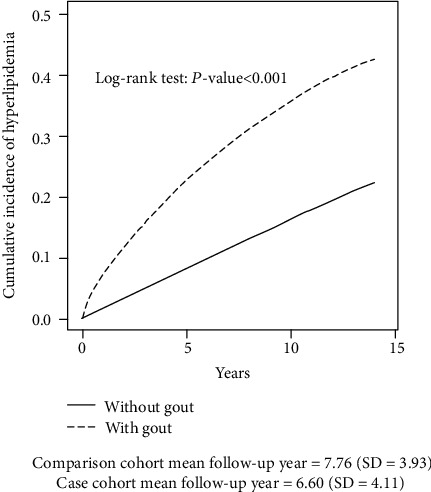
Comparison of cumulative incidence of hyperlipidemia between patients, with and without gout, using the Kaplan-Meier method. Comparison cohort mean follow-up year = 7.76 (SD = 3.93). Case cohort mean follow-up year = 6.60 (SD = 4.11).
Table 3 displays the incidence rates and HRs of hyperlipidemia in the case and comparison cohorts with different demographic characteristics. After adjustment, patients with gout were found to have significantly higher risk of developing hyperlipidemia than patients without gout (aHR = 2.55, 95% CI = 2.50–2.61, P < 0.001) regardless of sex (aHR = 2.15, 95% CI = 2.06–2.24, P < 0.001 in females; aHR = 2.70, 95% CI = 2.64–2.77, P < 0.001 in males), age (aHR = 3.08, 95% CI = 2.99–3.18, P < 0.001 in the ≤49 years' age group; aHR = 2.18, 95% CI = 2.10–2.26, P < 0.001 in the 50–64 years' age group; and aHR = 1.88, 95% CI = 1.79–1.98, P < 0.001 in the ≥65 years' age group), or comorbidity (aHR = 3.37, 95% CI = 3.28–3.46, P < 0.001 in the noncomorbidity group; aHR = 1.54, 95% CI = 1.49–1.60, P < 0.001 in the comorbidity group).
Table 3.
Comparison of incidence and hazard ratio of hyperlipidemia stratified by sex, age, and comorbidity between patients with and without gout.
| Variable | Gout | Crude HR (95% CI) | Adjusted HR† (95% CI) | |||||
|---|---|---|---|---|---|---|---|---|
| No | Yes | |||||||
| Event | PY | Rate# | Event | PY | Rate# | |||
| All | 24485 | 1378799 | 17.8 | 13639 | 293172 | 46.5 | 2.61 (2.55, 2.66)∗∗∗ | 2.55 (2.50, 2.61)∗∗∗ |
| Sex | ||||||||
| Female | 7080 | 282065 | 25.1 | 3217 | 59166 | 54.4 | 2.14 (2.06, 2.23)∗∗∗ | 2.15 (2.06, 2.24)∗∗∗ |
| Male | 17405 | 1096734 | 15.9 | 10422 | 234006 | 44.5 | 2.80 (2.73, 2.87)∗∗∗ | 2.70 (2.64, 2.77)∗∗∗ |
| Stratified age | ||||||||
| ≤49 | 10087 | 850974 | 11.9 | 7382 | 191709 | 40.6 | 3.44 (3.34, 3.55)∗∗∗ | 3.08 (2.99, 3.18)∗∗∗ |
| 50-64 | 9290 | 312176 | 29.8 | 4160 | 63233 | 65.8 | 2.19 (2.11, 2.27)∗∗∗ | 2.18 (2.10, 2.26)∗∗∗ |
| 65+ | 5108 | 215649 | 23.7 | 2097 | 48229 | 43.5 | 1.82 (1.73, 1.91)∗∗∗ | 1.88 (1.79, 1.98)∗∗∗ |
| Comorbidity‡ | ||||||||
| No | 13645 | 1003452 | 13.6 | 10840 | 375347 | 28.9 | 3.40 (3.31, 3.49)∗∗∗ | 3.37 (3.28, 3.46)∗∗∗ |
| Yes | 8639 | 181008 | 47.7 | 5000 | 112164 | 44.6 | 1.57 (1.52, 1.62)∗∗∗ | 1.54 (1.49, 1.60)∗∗∗ |
PY: person-years; Rate#: incidence rate, per 1,000 person-years; Crude HR: crude hazard ratio; Adjusted HR†: multivariable analysis including age, sex, and comorbidities of hypertension, stroke, diabetes, COPD, CAD, alcohol-related illness, and asthma; Comorbidity‡: patients with any one of the comorbidities of hypertension, stroke, diabetes, COPD, CAD, alcohol-related illness, and asthma were classified as the comorbidity group. ∗∗∗P < 0.001.
Table 4 shows the comparison among gout patients with and without antigout treatment and the comparison between nongout patients and gout patients without antigout treatment. Gout patients without antigout treatment showed significantly higher risk of hyperlipidemia than nongout patients (aHR = 3.10, 95% CI = 3.00–3.20, P < 0.001). Among gout patients, those with antigout treatment, except those receiving probenecid (aHR = 0.80, 95% CI = 0.64-1.00), had significantly lower risk of hyperlipidemia than gout patients without antigout treatment (aHR = 0.04, 95% CI = 0.01–0.17, P < 0.001 in patients receiving febuxostat; aHR = 0.57, 95% CI = 0.51–0.64, P < 0.001 in patients receiving sulfinpyrazone; aHR = 0.57, 95% CI = 0.54–0.61, P < 0.001 in patients receiving allopurinol; aHR = 0.89, 95% CI = 0.86–0.93, P < 0.001 in patients receiving benzbromarone; and aHR = 0.72, 95% CI = 0.68–0.76, P < 0.001 in patients receiving colchicine). In addition, among gout patients, those with different numbers of antigout combinations had significantly lower risk of hyperlipidemia than gout patients without antigout treatment (aHR = 0.86, 95% CI = 0.82–0.89, P < 0.001 in patients receiving one antigout treatment; aHR = 0.71, 95% CI = 0.68–0.75, P < 0.001 in patients receiving two combinations; aHR = 0.51, 95% CI = 0.47–0.55, P < 0.001 in patients receiving three combinations; and aHR = 0.35, 95% CI = 0.25–0.47, P < 0.001 in patients receiving four combinations of antigout treatments).
Table 4.
Incidence, crude, and adjusted hazard ratio of hyperlipidemia compared among gout patients with or without antigout treatment and compared between gout patients without antigout treatment and nongout patients.
| Variables | N | Event | PY | Rate# | Crude HR (95% CI) | Adjusted HR† (95% CI) | Adjusted HR† (95% CI) |
|---|---|---|---|---|---|---|---|
| Nongout | 177652 | 24485 | 1378799 | 17.8 | 1 | 1 | |
| Gout without the selected antigout treatment | 12617 | 4279 | 73332 | 58.4 | 3.26 (3.15, 3.36)∗∗∗ | 3.10 (3.00, 3.20)∗∗∗ | 1 |
| Gout with antigout treatment | |||||||
| Febuxostat | 93 | 2 | 860 | 2.33 | 0.13 (0.03, 0.53)∗∗ | 0.12 (0.03, 0.47)∗∗ | 0.04 (0.01, 0.17)∗∗∗ |
| Probenecid | 226 | 77 | 1810 | 42.6 | 2.40 (1.92, 3.00)∗∗∗ | 2.42 (1.93, 3.02)∗∗∗ | 0.80 (0.64, 1.00) |
| Sulfinpyrazone | 1244 | 301 | 9579 | 31.4 | 1.77 (1.58, 1.98)∗∗∗ | 1.74 (1.55, 1.95)∗∗∗ | 0.57 (0.51, 0.64)∗∗∗ |
| Allopurinol | 5885 | 1459 | 46659 | 31.3 | 1.76 (1.67, 1.86)∗∗∗ | 1.70 (1.62, 1.80)∗∗∗ | 0.57 (0.54, 0.61)∗∗∗ |
| Benzbromarone | 15352 | 5193 | 103671 | 50.1 | 2.81 (2.73, 2.89)∗∗∗ | 2.78 (2.69, 2.86)∗∗∗ | 0.89 (0.86, 0.93)∗∗∗ |
| Colchicine | 8996 | 2328 | 57261 | 40.7 | 2.27 (2.18, 2.37)∗∗∗ | 2.29 (2.19, 2.39)∗∗∗ | 0.72 (0.68, 0.76)∗∗∗ |
| Number of antigout treatments | |||||||
| 1 | 17125 | 5314 | 107897 | 49.3 | 2.76 (2.67, 2.84)∗∗∗ | 2.67 (2.59, 2.75)∗∗∗ | 0.86 (0.82, 0.89)∗∗∗ |
| 2 | 11670 | 3359 | 85575 | 39.3 | 2.21 (2.13, 2.29)∗∗∗ | 2.22 (2.14, 2.30)∗∗∗ | 0.71 (0.68, 0.75)∗∗∗ |
| 3 | 2764 | 646 | 24083 | 26.8 | 1.52 (1.40, 1.64)∗∗∗ | 1.53 (1.41, 1.65)∗∗∗ | 0.51 (0.47, 0.55)∗∗∗ |
| 4 | 234 | 41 | 2252 | 18.2 | 1.03 (0.76, 1.40) | 1.02 (0.75, 1.38) | 0.35 (0.25, 0.47)∗∗∗ |
| 5 | 3 | 0 | 34 | 0.00 | — | — | — |
PY: person-years; Rate#: incidence rate, per 1,000 person-years; Crude HR: crude hazard ratio; Adjusted HR†: multivariable analysis including age, sex, and comorbidities of hypertension, stroke, diabetes, COPD, CAD, alcohol-related illness and asthma. ∗∗P < 0.01; ∗∗∗P < 0.001.
3.2. Effects of Antigout Drugs on Lipogenesis-Related Gene Expressions
In this study, we also evaluated the effects of allopurinol, febuxostat, benzbromarone, sulfinpyrazone, probenecid, and colchicine on lipogenesis-related gene expression. The concentrations we used were based on the maximum serum drug concentrations [23–27]. As hepatic toxicity of these drugs is well-recognized [28], we examined their toxicity towards human hepatoma HepaRG cells in a cell viability assay. In this assay, the concentrations of these antigout drugs were 14.7 and 22.1 μM (for allopurinol); 5.66 and 13.09 μM (for febuxostat); 4.24 and 7.73 μM (for benzbromarone); 32.24 and 56.04 μM (for sulfinpyrazone); 243.9 and 520.6 μM (for probenecid); and 7.51 × 10−4 and 0.075 μM (for colchicine). HepaRG cells were treated with these antigout drugs for 24 h, followed by the ACP assay to assess their cytotoxicity. The ACP assay showed no significant difference in viability of control cells and cells treated with the antigout drugs (Figure 2).
Figure 2.

Viability of HepaRG cells following exposure to antigout drugs. HepaRG cells were exposed to allopurinol (14.7 and 22.1 μM), febuxostat (5.66 and 13.09 μM), benzbromarone (4.24 and 7.73 μM), sulfinpyrazone (32.24 and 56.04 μM), probenecid (243.9 and 520.6 μM), and colchicine (7.51 × 10−4 and 0.075 μM) for 24 h. Cell viability was monitored by measuring cellular acid phosphatase activity using p-nitrophenylphosphate as a substrate. Data shown are the mean ± standarderror (SE) (n = 3).
Expression of hepatic lipogenesis-related genes, SREBP-1c, SCD, FAS, FAE, ACLY, ACC, and LXRα, was measured in differentiated HepaRG cells by using real-time PCR. Differentiated HepaRG cells were treated with the antigout drugs for 24 h, total RNA was harvested, and expression of the indicated genes was analyzed. SREBP-1c is a hepatic transcription factor that regulates lipid homeostasis through several target lipogenic genes. T0901317 (T090), a synthetic LXRα agonist, significantly induces the expression of the target genes [29], SREBP-1c, SCD, FAS, FAE, ACLY, and ACC, and hence, was used as a positive control. However, mRNA levels for SREBP-1c, SCD, FAS, FAE, ACLY, ACC, and LXRα in the cells treated with most of these antigout drugs were significantly lower than those in the untreated cells, especially at the higher concentrations (Figures 3(a)–3(g)). However, ACC expression was not affected by the higher concentration of probenecid, while expression of LXRα, the master regulator of lipid homeostasis in hepatocytes, was induced by the higher concentration of probenecid. These data indicate that antigout treatment may partly reduce hepatic lipogenesis-related gene expression in gout patients, resulting in improved blood lipid profiles.
Figure 3.

Expression of hepatic lipid metabolism-related genes following treatment with T0901317 and antigout drugs. Differentiated HepaRG cells were treated for 24 h with T0901317 (10 μM), allopurinol (14.7 and 22.1 μM), febuxostat (5.66 and 13.09 μM), benzbromarone (4.24 and 7.73 μM), sulfinpyrazone (32.24 and 56.04 μM), probenecid (243.9 and 520.6 μM), and colchicine (7.51 × 10−4 and 0.075 μM). Following treatment, RNA was extracted, and the expression levels of (a) SREBP-1c, (b) SCD, (c) FAS, (d) FAE, (e) ACLY, (f) ACC, and (g) LXRα were analyzed by quantitative reverse transcription-polymerase chain reaction. Values were normalized to the expression of β-actin, with the β-actin levels of dimethyl sulfoxide- (DMSO-) treated cells set at 1. Results are expressed as the means ± standarderror (SE) (n = 3). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 compared with cells treated with DMSO. SREBP-1c: sterol regulatory element binding protein 1; SCD: stearoyl-CoA desaturase-1; FAS: fatty acid synthase; FAE: fatty acid elongase; ACLY: adenosine 5′-triphosphate (ATP) citrate lyase; ACC: acetyl-CoA carboxylase; LXRα: liver X receptor alpha.
4. Discussion
To the best of our knowledge, there are few studies that have investigated the development of hyperlipidemia in gout patients and the medications used for gout therapy in Asian adults. Our study demonstrates the strong association between gout and the risk of hyperlipidemia by comprehensively adjusting for confounders. In this study, examining the correlation between gout and the risk of hyperlipidemia under antigout drug treatment, we observed the following: (1) we used a nationwide, population-based cohort study to comprehensively survey an Asian population and observed a strong association between gout and the incidence of hyperlipidemia; (2) overall incidence risk of hyperlipidemia in gout patients was 2.55-fold compared to that in nongout populations; (3) without ULT, the incidence of hyperlipidemia was higher in patients with gout than in patients without gout (aHR = 3.10, 95% CI = 3.00–3.20, P < 0.001); (4) with ULT, the risk for hyperlipidemia in gout patients was significantly lower when compared to gout patients without ULT (all aHR < 1); (5) using the hepatocyte-derived HepaRG cell line, we found that these antigout drugs significantly reduced the expression of lipogenic-related genes, LXRα, SREBP-1c, SCD, FAS, FAE, ACLY, and ACC, thus contributing to improved blood lipid profiles; (6) we investigated a single cohort consisting of a large number of participants for a long duration of the study period, and the outcome was adjusted for most confounding factors; (7) and physicians should be made aware of the benefits of antigout treatment for blood lipid levels and cardiovascular prognosis, so as to reduce the risk of CVD.
In a population-based study conducted in USA, researchers estimated that the annual incidence of gout was 0.45 (1977 to 1978), 0.62 (1995 to 1996), and 1.6 and 0.2 (men and women, respectively, 1948 to 1980) per 1000 person-years [30, 31]. In the UK, the overall gout incidence was estimated to be 1.4 per 1,000 person-years in 1981 [32], 1.31 in 1999 [33], and 1.77 in 2012 [34]. In contrast, the incidence of gout has rarely been estimated in populations of other ethnicities. Researchers from Taiwan had used the NHIRD to identify the prevalence and incidence of gout for each calendar year from 2005 to 2010 [7]. Of 23,371,362 beneficiaries in 2010, there were 1,458,569 prevalent and 56,595 incident cases of gout, giving a prevalence of 6.24% (95% CI = 6.23-6.25) and an incidence of 2.74 (95% CI = 2.72-2.76) per 1,000 person-years [7]. Among prevalent gout cases in 2010, only 22.93% (95% CI = 22.87-23.00) were prescribed ULT, suggesting that poor management of gout is a common problem in Taiwan.
Sustained reduction in serum UA levels is critical for the long-term management of gout. The recommended target serum UA level is <360 μmol/L (or< 6.5 mg/dL), resulting in dissolution of monosodium urate crystals, suppression of acute gout pain, and resolution of gouty tophi [35]. Reduction in serum UA levels can be achieved by decreasing UA production with the use of XOIs, allopurinol and febuxostat; increasing excretion of UA with the use of uricosurics, benzbromarone, sulfinpyrazone, and probenecid; or metabolism of UA to the more water-soluble allantoin with the use of recombinant uricases, pegloticase and rasburicase (these two drugs were not available in Taiwan).
Uricosuric agents are more commonly prescribed than XOIs in Taiwan. In Taiwan, 1 in 16 people have gout, but only 1 in 5 affected people are treated with ULT [7]. Among patients who received ULT treatment, 60.08% received uricosuric agents alone, 28.54% received XOIs, and 11.38% received both. Our results found that 28.4% of gout patients did not receive any ULT, and 52.9%, 18.8%, and 28.3% received uricosuric agents, XOIs, and colchicine alone. As the database did not include nonprescribed drugs (such as NSAIDs), we focused on the aforementioned prescription drugs in our study.
Data from previous studies show that allopurinol, benzbromarone, and febuxostat can decrease blood levels of TC and TGs [36]. Thus, as statins and fenofibrate are expensive medications, these antigout drugs could facilitate the management of both gout and hyperlipidemia in gout patients. Therefore, we examined the effects of these drugs on the expression of hepatic lipogenesis-related genes.
A few studies have been conducted to investigate the effects of these antigout drugs on lipid metabolism regulation. New Zealand rabbits fed with a high fat diet showed significantly decreased serum TC and TG levels after administration of febuxostat for 4 weeks through antioxidant and anti-inflammatory mechanisms [37]. Lanaspa et al. [38] reported that high intracellular UA levels induce the mitochondrial translocation of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunit, NADPH oxidase 4 (Nox4), thus leading to increased oxidative stress and mitochondrial dysfunction and subsequent citrate release into the cytosol for de novo lipogenesis and TG synthesis [38]. In their study, they proposed that fructose induces UA production and TG synthesis; UA either alone or as a by-product generated by the initial phosphorylation of fructose, from 0 to 750 μmol/liter (4–12 mg/dL), can also induce lipid synthesis independent of fructose. They challenged hepatocytes with fructose in the presence of allopurinol to block UA production; allopurinol effectively blocked the production of UA upon exposure of the cells to fructose. Although allopurinol may have off-target effects (due to its antioxidant properties), the prevention of fructose-induced lipogenesis appears to be dependent on its ability to reduce UA levels because the subsequent addition of UA restored the metabolic effects of fructose. Notably, TG accumulation was also inhibited by allopurinol, indicating that UA may play a key role in the lipogenic ability of fructose through the stimulation of NADPH oxidase. Consistent with this finding, UA also stimulates oxidative stress in the mitochondria and further decreases the mitochondrial membrane potential. Fructose-induced oxidative stress was blocked by allopurinol. High levels of UA (750μmol/liter ≈ 12mg/dL) also induced significant changes in mitochondrial morphology as determined via electron microscopy. Compared with the controls, the UA-treated HepG2 cells exhibited mitochondria that were shorter and smaller, suggesting mitochondrial fragmentation [38]. The authors also found that lowering the intracellular UA levels could prevent fat accumulation in fructose-exposed HepG2 cells and thus might also prevent fatty liver in an animal model of hyperuricemia and metabolic syndrome. Allopurinol effectively reduced the intrahepatic UA levels, TG content, and hepatic cholesterol content. These findings corresponded with the significantly decreased activity of NOX4 in the mitochondria of mice receiving allopurinol [38]. The authors also determined whether hyperuricemia could predict NAFLD independent of obesity by examining a population undergoing hemodialysis, as hyperuricemia is common even when obesity is absent; they found that serum UA levels were associated with the increasing prevalence of hepatic steatosis independent of obesity [38]. Clinically, the UA in patients decreases upon challenge with antigout treatment; moreover, we found that antigout drugs reduce lipogenesis-related gene expression as well as the upper control elements, SREBP-1c and LXRα, suggesting that these drugs can affect earlier gene expression. These dual effects of antigout treatments, direct decreases in lipogenesis gene expression and UA production and, thus reduced lipogenesis might be beneficial for gout patients.
Furthermore, XO-mediated conversion of hypoxanthine/xanthine to UA is associated with the generation of hydrogen peroxide [39], and UA forms reactive intermediates, including alkylating species, by reacting with peroxynitrite [40]. Other potential mechanisms have been proposed to explain how UA can modulate LDL-C levels, which are ameliorated by the XOI, allopurinol, by decreasing lipid peroxidation rates [41]. Nevertheless, the exact mechanism by which UA increases LDL-C and TG levels is still unclear.
The most frequently used urate-lowering drug, allopurinol, is effective, easy to administer (once-daily dosing), inexpensive, and generally well-tolerated. However, it usually causes severe cutaneous adverse reactions, such as hypersensitivity syndrome and Stevens-Johnson syndrome, and is associated with human leukocyte antigen HLA-B∗5801, especially in Han Chinese (20% of Han Chinese population are HLA-B∗5801 carriers) [42]. As suggested by the Taiwan Department of Health, we applied the molecular diagnosis of the patients' genotype before allopurinol administration so as to reduce the side effects of this drug. Such recommendations have been made about the patients' genotype before drug administration. Moreover, allopurinol has been associated with a significant reduction in CVD-related mortality (RR = 0.74, 95% CI = 0.61–0.90) [43] and is also beneficial in ischemic heart disease patients [44].
Another XOI, febuxostat, was approved by the European Medicines Agency in 2008 and the United States Food and Drug Administration in 2009 for the treatment of gout [35]. It has shown a superior urate-lowering effect compared to allopurinol, but information on its effects on blood cholesterol or TGs in gout patients is limited [45].
Benzbromarone, sulfinpyrazone, and probenecid lower serum UA levels by increasing renal urate excretion. Benzbromarone is not available in many countries because of concerns about its hepatotoxicity, and it is not recommended for patients with liver and renal dysfunction [46]. This risk could be reduced by increasing the dose gradually and monitoring liver function. Our results reveal that viability of HepaRG cells was not significantly affected by the concentration of benzbromarone that is equivalent to its Cmax concentration in humans. However, benzbromarone reduced the expression of LXRα and its target lipogenic-related genes. In contrast, probenecid induced the expression of LXRα and did not affect gene expression of ACC even at a higher concentration. Thus, benzbromarone may contribute to the lower risk of hyperlipidemia in gout patients in our study.
Colchicine is another commonly used drug for acute gout treatment. In the Acute Gout Flare Receiving Colchicine Evaluation study, although low- (1.8 mg total) and high- (4.8 mg total) dose colchicine regimens were effective in pain reduction, low-dose colchicine had fewer adverse effects [47]. Gout patients often have related medical conditions that may promote the development of gout and make treatment more challenging. The treatment options for acute gout and prevention of gout need to be adjusted based on patients' comorbidities and their coprescribed drugs.
Hyperuricemia is frequently reported in patients with CVD and is associated with CVD-related mortality, suggesting that it may have a direct vascular effect [48, 49]. A previous report on hypercholesterolemia and hypertriglyceridemia patients treated with febuxostat, allopurinol, and benzbromarone revealed that febuxostat significantly decreased serum TC and TG levels, whereas allopurinol and benzbromarone only modestly decreased TG levels, but not TC levels, especially after 3–5 weeks of treatment [36]. No serious adverse events were observed. Our study included a long follow-up period of up to 6.60 and 7.76 years for the case and control groups, respectively, which may have increased the likelihood of observing hyperlipidemia treatment with antigout drugs significantly reduced the risk of hyperlipidemia.
This study has some strengths that are noteworthy. As this study was based on a large and representative population cohort extracted from the Taiwan NHI system, it eliminates bias from selection, nonresponse, or poor recall. The LHID has been shown to have good levels of accuracy and completeness in recording prescriptions and clinical diagnoses. In addition, we adjusted for many potentially confounding factors that are associated with hyperlipidemia and selected 4 times the number of nongout patients to form the control group. Both the case and control groups were studied for a long observation period. This study shows that gout patients had a higher risk of hyperlipidemia with a narrower and statistically significant CI.
This is the first study to examine an Asian population by using a population-based, nationwide cohort study to evaluate the effects of antigout therapy on hyperlipidemia in gout patients. However, our study has several limitations. First, patients' information concerning lifestyle, smoking status, alcohol consumption, environmental exposure, body mass index, and family history of hyperlipidemia was unavailable in the LHIRD. The aforementioned factors may contribute as confounding factors to the development of hyperlipidemia. LHIRD claims data are used mainly for administrative billing purposes. Therefore, the additional information was anonymous, and it was not possible to directly contact the patients for additional information. Second, no data on diets were available in this study, and dietary intake may contribute to the patients' lipid profiles. Third, exact laboratory data in the LHIRD were limited; therefore, we did not know the serum UA status in gout patients after ULT treatment. Fourth, the LHIRD lacks complete information regarding patients' liver disease by Child-Pugh classification, which might highly correlate with the development of hyperlipidemia. Therefore, our study reduced the confounding effects of the medications by adjusting for comorbidities. In addition, the severity of gout and disease duration may also affect blood lipid levels. This is an epidemiological study that can only provide evidence to show the association between gout and hyperlipidemia and cannot show a causal relationship. It is necessary to obtain more information from other databases or questionnaires to conduct a prospective study or randomized controlled trial to investigate such a relationship between gout, ULT, and hyperlipidemia. Therefore, the essential part of the optimization of care of patients with gout is likely to be physician education to improve knowledge and promote interest in gout. However, our results provide evidence that recommendations and guidelines for clinical management of gout patients can effectively mitigate long-term consequences of hyperlipidemia such as the possibility of developing CVD. Although we cannot identify the levels of each type of lipid or the levels of serum UA, we identified gout patients without ULT who may have had high levels of UA, which could be correlated with high serum LDL-C, TG, and TC levels and with low levels of HDL-C. Peng et al. identified HDL-C as a protective factor for CVD risk, which is inversely correlated to serum UA levels, but positively correlated with levels of LDL-C and TGs [8]. Thus, their findings suggest that a greater atherogenic ability is linked to a predisposition to CVD. Our study indicates that gout might intensify many pathophysiological mechanisms associated with the development of hyperlipidemia and consequently CVD. Thus, treatment of gout with ULT might be also beneficial for blood lipid levels through the inhibition of lipogenesis-related gene expression.
5. Conclusion
This population-based cohort study demonstrates a substantial association between gout, ULT, and the risk of hyperlipidemia. We confirm that management of gout in Taiwan remains poor, with 28.4% of gout patients not being prescribed ULT, which could contribute to elevated serum UA levels and increased gout flares with major adverse consequences such as hyperlipidemia. Due to the strong association between gout and hyperlipidemia, appropriate treatment guidelines such as changes in lifestyle, diet, and measurement of serum parameters should be developed to improve serum UA levels and understand the therapeutic mechanism of action of these drugs. We provide evidence of the benefits of these common antigout drugs for hyperlipidemia in gout patients through the reduction in the expression of lipogenesis-related genes.
Acknowledgments
This study was supported by the Ministry of Science and Technology, Taiwan, R.O.C. (MOST107-2320-B-039-042-MY3); China Medical University Hospital, Taichung, Taiwan (DMR-107-109); China Medical University, Taichung, Taiwan (CMU109-MF-92); Show Chwan Memorial Hospital, Changhua, Taiwan (SRD-109026); and Taichung Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation (TTCRD109-25) and partially supported by Taiwan Ministry of Health and Welfare Clinical Trial Center (MOHW109-TDU-B-212-114004), MOST Clinical Trial Consortium for Stroke (MOST108-2321-B-039-003), and Tseng-Lien Lin Foundation, Taichung, Taiwan.
Data Availability
The data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
Y.J.F., T.Y.W., and Y.P.L. are responsible for the conceptualization. T.Y.W., C.L.L., C.Y.S., J.R.L., and Y.P.L. are assigned to the methodology. C.L.L. and Y.P.L. acquired the software. Y.J.F. and T.Y.W. performed the validation. C.L.L., C.Y.S., and J.R.L. performed the formal analysis. T.Y.W. and Y.P.L. performed the investigation. C.L.L. and Y.P.L. acquired the resources. C.L.L. and Y.P.L. performed the data curation. All authors did the writing—original draft preparation, review and editing, and visualization. Y.J.F., T.Y.W., and Y.P.L. are responsible for the supervision. Y.P.L. is assigned to the project administration. Y.J.F., T.Y.W., and Y.P.L. are responsible for the funding acquisition. Yi-Jen Fang and Tien-Yuan Wu contributed equally to this work.
References
- 1.Choi H. K., Atkinson K., Karlson E. W., Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in Men. Archives of Internal Medicine. 2005;165(7):742–748. doi: 10.1001/archinte.165.7.742. [DOI] [PubMed] [Google Scholar]
- 2.Doherty M., Jansen T. L., Nuki G., et al. Gout: why is this curable disease so seldom cured? Annals of the Rheumatic Diseases. 2012;71(11):1765–1770. doi: 10.1136/annrheumdis-2012-201687. [DOI] [PubMed] [Google Scholar]
- 3.Richette P., Bardin T. Gout. Lancet. 2010;375(9711):318–328. doi: 10.1016/S0140-6736(09)60883-7. [DOI] [PubMed] [Google Scholar]
- 4.Stamp L. K., Chapman P. T. Gout and its comorbidities: implications for therapy. Rheumatology. 2012;52(1):34–44. doi: 10.1093/rheumatology/kes211. [DOI] [PubMed] [Google Scholar]
- 5.Liu S. C., Xia L., Zhang J., et al. Gout and risk of myocardial infarction: a systematic review and meta-analysis of cohort studies. PLoS One. 2015;10(7, article e0134088) doi: 10.1371/journal.pone.0134088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clarson L. E., Chandratre P., Hider S. L., et al. Increased cardiovascular mortality associated with gout: a systematic review and meta-analysis. European Journal of Preventive Cardiology. 2014;22(3):335–343. doi: 10.1177/2047487313514895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuo C. F., Grainge M. J., See L. C., et al. Epidemiology and management of gout in Taiwan: a nationwide population study. Arthritis Research & Therapy. 2015;17(1):p. 13. doi: 10.1186/s13075-015-0522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng T. C., Wang C. C., Kao T. W., et al. Relationship between hyperuricemia and lipid profiles in US adults. BioMed Research International. 2015;2015:7. doi: 10.1155/2015/127596.127596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen S. Y., Chen C. L., Shen M. L., Kamatani N. Trends in the manifestations of gout in Taiwan. Rheumatology. 2003;42(12):1529–1533. doi: 10.1093/rheumatology/keg422. [DOI] [PubMed] [Google Scholar]
- 10.Jung J. H., Song G. G., Ji J. D., et al. Metabolic syndrome: prevalence and risk factors in Korean gout patients. The Korean Journal of Internal Medicine. 2018;33(4):815–822. doi: 10.3904/kjim.2016.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cibičková L., Langová K., Vaverková H., Kubíčková V., Karásek D. Correlation of uric acid levels and parameters of metabolic syndrome. Physiological Research. 2017;66(3):481–487. doi: 10.33549/physiolres.933410. [DOI] [PubMed] [Google Scholar]
- 12.Abeles A. M., Pillinger M. H. Gout and cardiovascular disease: crystallized confusion. Current Opinion in Rheumatology. 2019;31(2):118–124. doi: 10.1097/BOR.0000000000000585. [DOI] [PubMed] [Google Scholar]
- 13.Sinan Deveci O., Kabakci G., Okutucu S., et al. The association between serum uric acid level and coronary artery disease. International Journal of Clinical Practice. 2010;64(7):900–907. doi: 10.1111/j.1742-1241.2009.02263.x. [DOI] [PubMed] [Google Scholar]
- 14.Stone N. J., Robinson J. G., Lichtenstein A. H., et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 supplement 2):S1–S45. doi: 10.1161/01.cir.0000437738.63853.7a. [DOI] [PubMed] [Google Scholar]
- 15.Lloyd-Jones D. M., Morris P. B., Ballantyne C. M., et al. 2017 Focused update of the 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. Journal of the American College of Cardiology. 2017;70(14):1785–1822. doi: 10.1016/j.jacc.2017.07.745. [DOI] [PubMed] [Google Scholar]
- 16.Matsubara K., Matsuzawa Y., Jiao S., Takama T., Kubo M., Tarui S. Relationship between hypertriglyceridemia and uric acid production in primary gout. Metabolism. 1989;38(7):698–701. doi: 10.1016/0026-0495(89)90110-8. [DOI] [PubMed] [Google Scholar]
- 17.Willy P. J., Umesono K., Ong E. S., Evans R. M., Heyman R. A., Mangelsdorf D. J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes and Development. 1995;9(9):1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- 18.Schultz J. R., Tu H., Luk A., et al. Role of LXRs in control of lipogenesis. Genes and Development. 2000;14(22):2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higuchi N., Kato M., Shundo Y., et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatology Research. 2008;38(11):1122–1129. doi: 10.1111/j.1872-034X.2008.00382.x. [DOI] [PubMed] [Google Scholar]
- 20.Changchien C. S., Wang J. H., Tsai T. L., Hung C. H. Correlation between fatty liver and lipidemia in Taiwanese. Journal of Medical Ultrasound. 2003;11(2):60–65. doi: 10.1016/S0929-6441(09)60043-6. [DOI] [Google Scholar]
- 21.Chuang S. Y., Lee S. C., Hsieh Y. T., Pan W. H. Trends in hyperuricemia and gout prevalence: Nutrition and Health Survey in Taiwan from 1993-1996 to 2005-2008. Asia Pacific Journal of Clinical Nutrition. 2011;20(2):301–308. [PubMed] [Google Scholar]
- 22.Li Y. W., Wang C. H., Chen C. J., et al. Effects of antiepileptic drugs on lipogenic gene regulation and hyperlipidemia risk in Taiwan: a nationwide population-based cohort study and supporting in vitro studies. Archives of Toxicology. 2018;92(9):2829–2844. doi: 10.1007/s00204-018-2263-3. [DOI] [PubMed] [Google Scholar]
- 23.Wells B. G., Dipiro J. T., Schwinghammer T. L., Dipiro C. V. Pharmacotherapy Handbook. 9th. The McGraw-Hill Companies, Inc.; 2012. [Google Scholar]
- 24.Uchida S., Shimada K., Misaka S., et al. Benzbromarone pharmacokinetics and pharmacodynamics in différent cytochrome P450 2C9 genotypes. Drug Metabolism and Pharmacokinetics. 2010;25(6):605–610. doi: 10.2133/dmpk.DMPK-10-NT-040. [DOI] [PubMed] [Google Scholar]
- 25.Dieterle W., Faigle J. W., Mory H., Richter W. J., Theobald W. Biotransformation and pharmacokinetics of sulfinpyrazone (Anturan®) in man. European Journal of Clinical Pharmacology. 1975;9(2-3):135–145. doi: 10.1007/BF00614010. [DOI] [PubMed] [Google Scholar]
- 26.Selen A., Amidon G. L., Wellingx P. G. Pharmacokinetics of probenecid following oral doses to human volunteers. Journal of Pharmaceutical Sciences. 1982;71(11):1238–1242. doi: 10.1002/jps.2600711114. [DOI] [PubMed] [Google Scholar]
- 27.Rochdi M., Sabouraud A., Baud F. J., Bismuth C., Scherrmann J. M. Toxicokinetics of colchicine in humans: analysis of tissue, plasma and urine data in ten cases. Human & Experimental Toxicology. 1992;11(6):510–516. doi: 10.1177/096032719201100612. [DOI] [PubMed] [Google Scholar]
- 28.Strilchuk L., Fogacci F., Cicero A. F. Safety and tolerability of available urate-lowering drugs: a critical review. Expert Opinion on Drug Safety. 2019;18(4):261–271. doi: 10.1080/14740338.2019.1594771. [DOI] [PubMed] [Google Scholar]
- 29.Ferré P., Foufelle F. SREBP-1c transcription factor and lipid homeostasis: clinical perspective. Hormone Research. 2007;68(2):72–82. doi: 10.1159/000100426. [DOI] [PubMed] [Google Scholar]
- 30.Abbott R. D., Brand F. N., Kannel W. B., Castelli W. P. Gout and coronary heart disease: the Framingham Study. Journal of Clinical Epidemiology. 1988;41(3):237–242. doi: 10.1016/0895-4356(88)90127-8. [DOI] [PubMed] [Google Scholar]
- 31.Arromdee E., Michet C. J., Crowson C. S., O'Fallon W. M., Gabriel S. E. Epidemiology of gout: is the incidence rising? The Journal of Rheumatology. 2002;29(11):2403–2406. [PubMed] [Google Scholar]
- 32.Stewart O. J., Silman A. J. Review of UK data on the rheumatic DISEASES-4 Gout. Rheumatology. 1990;29(6):485–488. doi: 10.1093/rheumatology/29.6.485. [DOI] [PubMed] [Google Scholar]
- 33.Mikuls T. R., Farrar J. T., Bilker W. B., Fernandes S., Schumacher H. R., Saag K. G. Gout epidemiology: results from the UK General Practice Research Database, 1990-1999. Annals of the Rheumatic Diseases. 2005;64(2):267–272. doi: 10.1136/ard.2004.024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo C. F., Grainge M. J., Mallen C., Zhang W., Doherty M. Rising burden of gout in the UK but continuing suboptimal management: a nationwide population study. Annals of the Rheumatic Diseases. 2015;74(4):661–667. doi: 10.1136/annrheumdis-2013-204463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson P. C., Stamp L. K. The management of gout: much has changed. Australian Family Physician. 2016;45(5):299–302. [PubMed] [Google Scholar]
- 36.Wu J., Zhang Y. P., Qu Y., Jie L. G., Deng J. X., Yu Q. H. Efficacy of uric acid-lowering therapy on hypercholesterolemia and hypertriglyceridemia in gouty patients. International Journal of Rheumatic Diseases. 2019;22(8):1445–1451. doi: 10.1111/1756-185X.13652. [DOI] [PubMed] [Google Scholar]
- 37.Heikal M. M., Shaaban A. A., Elkashef W. F., Ibrahim T. M. Effect of febuxostat on biochemical parameters of hyperlipidemia induced by a high-fat diet in rabbits. Canadian Journal of Physiology and Pharmacology. 2019;97(7):611–622. doi: 10.1139/cjpp-2018-0731. [DOI] [PubMed] [Google Scholar]
- 38.Lanaspa M. A., Sanchez-Lozada L. G., Choi Y. J., et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. The Journal of Biological Chemistry. 2012;287(48):40732–40744. doi: 10.1074/jbc.M112.399899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelley E. E., Khoo N. K., Hundley N. J., Malik U. Z., Freeman B. A., Tarpey M. M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radical Biology & Medicine. 2010;48(4):493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gersch C., Palii S. P., Imaram W., et al. Reactions of peroxynitrite with uric acid: formation of reactive intermediates, alkylated products and triuret, and in vivo production of triuret under conditions of oxidative stress. Nucleosides, Nucleotides & Nucleic Acids. 2009;28(2):118–149. doi: 10.1080/15257770902736400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minami M., Ishiyama A., Takagi M., Omata M., Atarashi K. Effects of allopurinol, a xanthine oxidase inhibitor, on renal injury in hypercholesterolemia-induced hypertensive rats. Blood Pressure. 2009;14(2):120–125. doi: 10.1080/08037050510008878. [DOI] [PubMed] [Google Scholar]
- 42.Chiu M. L., Hu M., Ng M. H., et al. Association between HLA-B∗58:01 allele and severe cutaneous adverse reactions with allopurinol in Han Chinese in Hong Kong. British Journal of Dermatology. 2012;167(1):44–49. doi: 10.1111/j.1365-2133.2012.10894.x. [DOI] [PubMed] [Google Scholar]
- 43.Thanassoulis G., Brophy J. M., Richard H., Pilote L. Gout, allopurinol use, and heart failure outcomes. Archives of Internal Medicine. 2010;170(15):1358–1364. doi: 10.1001/archinternmed.2010.198. [DOI] [PubMed] [Google Scholar]
- 44.Rentoukas E., Tsarouhas K., Tsitsimpikou C., Lazaros G., Deftereos S., Vavetsi S. The prognostic impact of allopurinol in patients with acute myocardial infarction undergoing primary percutaneous coronary intervention. International Journal of Cardiology. 2010;145(2):257–258. doi: 10.1016/j.ijcard.2009.08.037. [DOI] [PubMed] [Google Scholar]
- 45.Saviola G., Benucci M., Abdi-Ali L., Sacco S., Cera I., Rossini M. Can febuxostat reduce triglyceride and cholesterol serum levels in gouty patients? Journal of Clinical Rheumatology. 2014;20(7):394–395. doi: 10.1097/RHU.0000000000000168. [DOI] [PubMed] [Google Scholar]
- 46.Azevedo V. F., Kos I. A., Vargas-Santos A. B., da Rocha Castelar Pinheiro G., dos Santos Paiva E. Benzbromarone in the treatment of gout. Advances in Rheumatology. 2019;59(1, article 37) doi: 10.1186/s42358-019-0080-x. [DOI] [PubMed] [Google Scholar]
- 47.Terkeltaub R. A., Furst D. E., Bennett K., Kook K. A., Crockett R. S., Davis M. W. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis & Rheumatology. 2010;62(4):1060–1068. doi: 10.1002/art.27327. [DOI] [PubMed] [Google Scholar]
- 48.Zhang S., Wang Y., Cheng J., et al. Hyperuricemia and cardiovascular disease. Current Pharmaceutical Design. 2019;25(6):700–709. doi: 10.2174/1381612825666190408122557. [DOI] [PubMed] [Google Scholar]
- 49.Wu K.-M., Hsu Y.-M., Ying M.-C., et al. High-density lipoprotein ameliorates palmitic acid-induced lipotoxicity and oxidative dysfunction in H9c2 cardiomyoblast cells via ROS suppression. Nutrition & Metabolism. 2019;16(1):p. 36. doi: 10.1186/s12986-019-0356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included within the article.