

Abstract
The development of advanced gene and cell therapies for the treatment of genetic diseases requires reliable animal and cellular models to test their efficacy. Moreover, the availability of the target human primary cells of these therapies is reduced in many diseases. The development of endonucleases that can cut into specific sites of the cell genome, as well as the repair of the generated break by non-homologous end-joining, results in a variety of outcomes, insertions, deletions, and inversions that can induce the disruption of any specific gene. Among the many methods that have been developed for gene editing, CRISPR-Cas9 technology has become one of the most widely used endonuclease tools due to its easy design and its low cost. It has also been reported that the use of two guides, instead of just the one required, reduces the outcomes of non-homologous end joining mainly to the precise genomic sequences between the cutting sites of the guides used. We have explored this strategy to generate useful cellular and animal models. Different distances between the two guides have been tested (from 8 to 500 bp apart), and using the optimal range of 30–60 bp we have obtained a human primary cellular model of a genetic disease, pyruvate kinase deficiency, where the availability of the target cells is limited. We have also generated an in vivo model of glycolate oxidase (GO) deficiency, which is an enzyme involved in the glyoxylate metabolism following the same strategy. We demonstrate that the use of two-guide CRISPR-Cas9-induced non-homologous end joining is a feasible and useful tool for disease modeling, and it is most relevant to those diseases in which it is difficult to get the cells that will be genetically manipulated.
Graphical Abstract
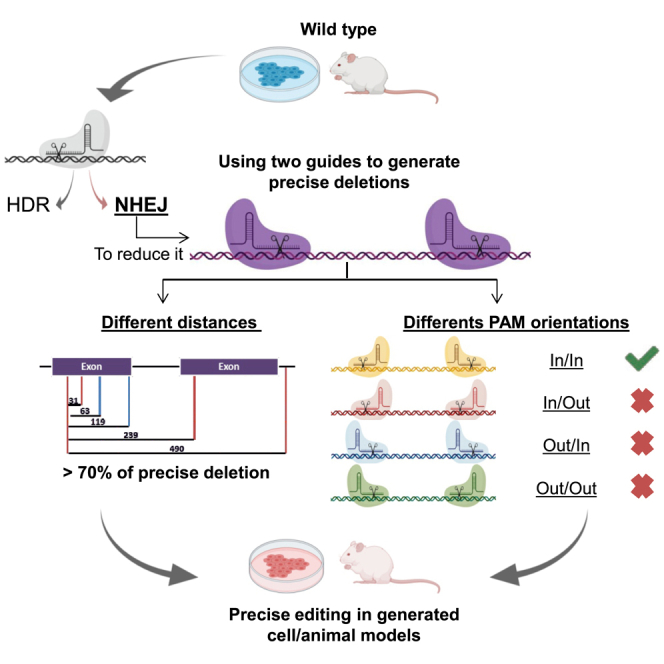
A CRISPR-Cas9 system generates precise deletions (up to 90% efficiency) when two guides are used simultaneously by means of non-homologous end joining. The PAM-in/PAM-in Cas9 orientation is the best combination to generate precise deletions. This gene-editing procedure facilitates the generation of homogeneously genetically defined cell and animal models.
Introduction
Basic biology and the study of the function of the genome have been based on the availability of function-deficient models, either for cells or organisms, and the association of these losses of function with gene mutations. The availability of these models has allowed the research of human genetic diseases and even the development of gene therapy strategies for their treatment.1 The availability of endonucleases that can be directed to interact with great precision in the cell genome has allowed the generation of knockout (KO) models of any desired gene or position.2, 3, 4
Above all, CRISPR technology has become one of the most widespread gene-editing tools in recent years thanks to its easy design and its low cost. The action of these endonucleases produces cleavage in both DNA strands in a precise manner according to the guide RNA (gRNA) position.5, 6, 7 The DNA cell machinery repairs these breaks either by non-homologous end joining (NHEJ), an error-prone process, or by homology-directed repair, which precisely corrects the damage. Both mechanisms have been extensively used to either eliminate the expression of a specific gene or to introduce new genetic material in a precise position of the cell genome. Repair by the NHEJ machinery results in a high variety of insertions or deletions (indels) and sporadically inversions. This capacity to alter the original sequence has made these nucleases an excellent tool for the generation of knockout models from bacteria to the human genome.8, 9, 10 Moreover, the deletion of specific regions that results in a recovery of function, either by eliminating a premature stop codon11 or by deleting specific gene regulators or silencers,5,12,13 has been suggested as an alternative therapy for specific diseases.11,14,15 In fact, a gene therapy clinical trial for the re-expression of fetal globin in adult β-thalassemia patients by means of knocking out the BCL11A protein regulator is already underway.16
NHEJ repairs double-strand breaks (DSBs) in a non-predictive way, introducing indels that can be extremely variable. Different reports have shown that the generation of two DSBs can facilitate NHEJ repair action.12,13,17 Moreover, it has been proposed as a potential therapeutic option by eliminating mutated exons and recovering an almost normal although functional protein.14,15 Guo et al.18 studied the efficacy of this NHEJ-precise deletion (NHEJ-PD) and how this process acts when guides are separated by 23–148 bp, with precise deletion of the DNA material between the two guide cuts being the most common event. Thus, the use of two guides that could delete a defined DNA fragment and alter the open reading frame of the affected locus in an efficient and pseudo-controlled way could be used to generate knockout models for the study of the biology of the cell or for the generation of cellular or animal models of rare diseases.
Herein, we report the use of NHEJ-PD to generate cellular and animal models of genetic diseases. In addition, we study the optimal distances and the impact of the CRISPR-Cas9 machinery orientation in the NHEJ-PD.
Results
The Use of Guide Pairs Allows Precise Deletion of DNA Sequences between the Two Induced DSBs
As a first attempt we studied whether the use of two guides simultaneously could reduce the heterogeneity of the NHEJ activity to inactivate a certain gene. We chose the PKLR gene, which is responsible for the expression of liver and the erythroid pyruvate kinase enzymes (L-type pyruvate kinase [LPK] and R-type pyruvate kinase [RPK], respectively). Mutations in this gene are responsible for the rare disease pyruvate kinase deficiency (PKD). Aiming to mimic mutations occurring in PKD patients, we looked for exons where mutation clusters were present. From these, we selected exon 9 of the PKLR gene, the exon with the second-highest number of mutations reported (33 different along the exon). This gave us in silico gRNAs with the better efficacy and off-target scores. Two gRNAs were then designed to cut into two hotspots of exon 9 where several mutations where grouped (Figure 1A; Figure S1A).
Figure 1.

Design and Use of CRISPR gRNAs in HEK293T Cell Line
(A) Exon 9 of the human PKLR gene is represented. Vertical lines and arrows are reported mutations, corresponding to one or more than 1 nt affected, respectively. Horizontal dashed lines represent the two gRNAs used. Vertical dashed lines represent predicted cut sites of gRNAs selected. (B) Sorting of green fluorescence based on the ZsGreen present in the electroporated plasmids. A 530/40- versus 580/30-nm dot plot was used to distinguish ZsGreen fluorescence from autofluorescence. The figure represents the ZsGreen fluorescence of the cell electroporated with the plasmid DNA containing only the Cas9 and the ZsGreen cDNAs (C), or containing the Cas9, the ZsGreen cDNAs and the sequences of the guide I (I) or the guide II (II). (C) Surveyor assay of the DNA obtained from the cells electroporated with the plasmids containing no guide, guide I, guide II, or both guides together (I+II). The gene editing generates a characteristic band pattern for guide I (160 bp + 135 bp), for guide II (191 bp + 104 bp), and a mixed pattern when both are used at the same time (160 bp + 104 bp). (D) Scheme of the band sizes that appear in (C) showing the 31-bp deletion resulting after the simultaneous electroporation of guides I and II.
Human HEK293T cells were electroporated with all-in-one DNA plasmids containing sequences for one gRNA, spCas9 nuclease, and green fluorescent protein (ZsGreen). Plasmids were electroporated individually or together. Seventy-two hours after the electroporation cells were sorted based on ZsGreen expression (Figure 1B) and expanded up to confluence in six-well plates. A surveyor assay was conducted in order to determine the activity of the designed guides and their combination of the two. When guides were electroporated individually, the expected pattern of surveyor DNA fragments was obtained with high cutting efficiencies (40% for guide I and 59% for guide II; Figures 1C and 1D). When the guides were electroporated together, surveyor DNA fragments obtained were the combination of the cut of the two guides (30% of efficacy) with the loss of the DNA fragment between the cut sites of the two guides (31-bp fragment). No evidence of fragments resulting from the cut of only one guide was found (Figures 1C and 1D). These results confirmed previous results, thus demonstrating that the deletion of the DNA sequences between two guides, when used simultaneously, utilizing the CRISPR-Cas9 system, was highly frequent and the most common event of the NHEJ repair mechanism. The presence of a non-edited wild-type (WT) band after ZsGreen sorting (Figure 1C, 294-bp band) corroborated the necessity of selecting the ZsGreen-high population to maximize the chances of selecting only edited cells.
In order to define the joint between the two generated DSBs, we amplified the region of the joint and cloned it in PCR plasmids for analysis by Sanger sequencing. All colonies analyzed that contained non-WT sequences presented the precise joint between the two DNA sides (NHEJ-PD; Figure S1B).
Distance between the Two Guides (30–500 bp) Does Not Affect the Generation of Precise Deletions
To test whether the process was cell-dependent, we performed a similar study using K562 (chronic myelogenous leukemia cell line). Furthermore, in order to determine whether the generation of the NHEJ-PD, being the most common event, was dependent on the distance between the guides used, five additional pairs were designed separated by 30–500 bp. These pairs used the same upstream guide. gRNA pairs were designed to delete regions at exon 9, intron 9, and exon 10 of the PKLR gene (Figure 2A).
Figure 2.

DNA and Protein Analysis of K562 Cells Electroporated with Five Guide Pairs
(A) Left: scheme of the five pairs tested. In dark purple: PKLR gene, with exons represented as boxes and introns as horizontal lines. Light purple represents mutations reported in exon 9. Vertical lines indicate the position of the guides used; the different colors represent the PAM position with respect to the deletion; red lines indicate PAM “in” (inside the deletion sequence), and blue lines indicate PAM “out” (outside the deletion sequence). Numbers above black horizontal lines are the size of the deletion generated in each combination. Right: list of the guides used, as well as size of the deletion generated and its PAM disposition. (B) PCR amplicons of the region of all combinations. Top: pair number. Red indicates the size of the precise deletion. C, control sample, i.e., K562 cells electroporated with Cas9-P2A-ZsGreen without guides and sorted. Pairs 1–4 were evaluated performing a 320-bp PCR, while pair 5 was checked by a 700-bp PCR. (C) Next-generation sequencing (NGS) results from variants of the pairs tested, represented in more than 1%. Dark purple indicates precise deletion; light blue indicates variants with modifications around expected deletion. Data are presented as mean ± SD. A Tukey’s multiple comparison test was used to evaluate differences between groups (n = 2 each group). No statistical differences were found. (D) Western blot of RPK pairs tested. WT, non-electroporated K562 cells; C, control sample, i.e., K562 cells electroporated with Cas9-P2A-ZsGreen without guides and sorted. Black numbers indicate different pairs tested; red idicates sizes of RPK protein, complete (62 kDa) or after physiological N-terminal excision (57 kDa). GAPDH and γ-tubulin were used as loading controls.
DNA plasmid constructs containing gRNAs and Cas9 were electroporated into K562 cells. Twenty-four hours post-transduction, cells were sorted based on ZsGreen expression and expanded as described above. Editing efficacy was tested by PCR amplification of the affected region. Guide pairs showed a good capacity to delete gDNA, decreasing the size of the PCR product at the expected sizes. Indeed, WT size products were not found (Figure 2B).
To clearly elucidate the outcomes in the gene editing with the different guide pairs, PCR amplicons of the joint sequences were analyzed by next-generation sequencing (NGS). Guide pair number 1, whose precise deletion was predicted on 31 bp, presented only two events. The most represented event is the mentioned precise deletion, with an 84.60% frequency. The other recorded event was also a deletion of 32 bp that was the same 31-bp deletion without an additional A at 5′ of the deleted sequence (precise deletion+1). Similar results were obtained with the other guide pair combinations (guide pair 2, 63-bp deletion, 69.70%; guide pair 3, 119-bp deletion, 74.05%; guide pair 4, 239-bp deletion, 69.18%; guide pair 5, 490-bp deletion, 95.84%) (Figure 2C). In all other combinations, the remaining events, while different from the precise deletion, were merely slight modifications surrounding it (Figure S2A). When considering precise deletion and precise deletion+1 species together, the efficiency was always higher than 75% of the sequences analyzed in all of the cases (Figure S2C).
An additional guide pair with cutting sites separated by 8 bp was also tested. Due to the proximity of the two cutting sites, the gRNAs were overlapping. In this case, indels detected were around one or the other cut site. No precise deletions or deletions containing the fragment between the cut sites were observed (Figure S2B).
Finally, to corroborate the capacity of these deletions to knockout the gene, a western blot assay of the edited populations was performed. By in silico studies, guide pair 1 should generate premature stop codons, guide pairs 2 and 3 would generate aberrant proteins deleting part of exon 9, and guide pairs 4 and 5 would generate a protein without exon 9. We performed western blots using a polyclonal antibody (Ab) against RPK to be able to detect any smaller aberrant RPK protein smaller than the original one. No RPK protein was detected in any of the edited samples, indicating that any combination of guides was capable of completely eliminating the production of the protein (Figure 2D).
PAM Orientation of the Guides Affects the Efficacy of Precise Deletions
One of the potential explanations for why a short distance between the two guides is less efficient in generating precise deletions could be the difficulties accessing the two cutting sites at the same time due to allosteric or space impediments. To study these potential difficulties, we designed and cloned gRNAs spaced approximately 30 bp apart that facilitated the Cas9-DNA interactions in all four possible orientations, protospacer-adjacent motif (PAM)-in/PAM-in (or-1), PAM-in/PAM-out (or-2), PAM-out/PAM-in (or-3), and PAM-out/PAM-out (or-4) (Figure 3A). Human K562 cells were electroporated with pairs of all-in-one DNA plasmids containing sequences for one gRNA, a spCas9 nuclease, and ZsGreen. Twenty-four hours after electroporation cells were sorted based on ZsGreen expression and expanded as above. Total DNA was extracted from the four different combinations. Three different experiments were conducted with each combination. NGS and bioinformatics analysis were performed to define the incidence of NHEJ-PD and additional indels in the genomic region. The combination or-1 (PAM-in/PAM-in) showed the best NHEJ-PD efficiency, and the or-4 combination (PAM-out/PAM-out) was the worst one, including a high proportion of non-edited sequences. Combinations or-2 (PAM-in/PAM-out) and or-3 (PAM-out/PAM-in) were also efficient, but the percentages of indels different from the precise deletion were higher (Figure 3B; Figure S3A). The or-4 combination had overlapping guides, and we supposed that sequence competition between Cas9 proteins could be taking place, thus impairing the cutting efficiency. Then, we generated new guides that avoided gRNA overlapping. This new combination did not generate precise deletions (Figure 3). Interestingly, if we analyze the cases where only one guide cut, the guide-oriented PAM-in was always more efficient (data not shown). As observed above, when we consider the NHEJ-PD plus the NHEJ-PD+1 together, the percentages of edited sequences were close to 100% in the or-1 combination (PAM-in/PAM-in) (Figure S3B). Altogether, these experiments show that steric difficulties could be taking place that limit the efficacy of the cutting activity of the guides, suggesting that orientation of the PAM and positioning of the Cas9 nuclease on the DNA would be important and should be considered when the generation of short DNA deletions by NHEJ is intended.
Figure 3.

DNA Analysis of K562 Cells Electroporated with Four Guide Pairs Separated by 28–34 bp Providing All of the PAM-In and PAM-Out Combinations
(A) Scheme of the four different combinations tested. Light colors indicate guides; dark colors indicate PAM sequences. The orientations of the PAMs are represented on the left, being “in” when the PAM is inside of the precise deletion and “out” when it is outside. (B) Next-generation sequencing results from variants of the different combinations, represented in more than 1%. Dark purple indicates precise deletion; light purple indicates variants with modifications around expected deletion; grey indicates WT. Data are presented as mean ± SEM. A Tukey’s multiple comparison test was used to evaluate differences between groups.
NHEJ-PD Generates a Cellular Model of PKD-like CD34 Primary Cells
Next, we wanted to test whether the NHEJ-PD would also occur in primary cells and whether this mechanism could be used to generate cellular models of genetic diseases in primary cells. To this end, we applied the guide pair 1 to knockout the PKLR gene in human cord blood CD34+ (CB-CD34+) cells. CD34+ cells are responsible for the generation of all hematopoietic lineages, including the erythroid lineage where RPK expression is required for the proper production of mature erythroid cells. Reduction or lack of RPK function leads to PKD. Healthy CB-CD34+ cells were electroporated. 5%–10% efficiency was obtained based on ZsGreen high fluorescence expression. Positive cells were then sorted (94%–98% purity after sorting) and differentiated toward erythroid lineage. Afterward, the modified region was analyzed by deep sequencing. Similar to the results obtained in immortalized cell lines, NHEJ-PD was consistently achieved in edited CB-CD34+ cells, which is easily traceable by following the decrease of the size of the PCR product (Figure 4A). Furthermore, the proportion of precise deletions was followed by the analysis of the cleavage produced by BstXI, whose target sequence is generated after precise deletion and is not present in the WT or in other deletions such as the 32-bp one (Figure 4B). Interestingly, the most frequent event recorded in these primary cells was the same as that found in the cell line studies (31-bp deletion, 82% ± 2%; 32-bp deletion, 16% ± 4%). In addition, in one of the samples two other variants were found, representing around 1.5% of the events. Both were deletions, one of 40 bp and another of 55 bp, which were also centered in the precise deletion region (Figure 4C). Thus, the use of these two selected guides demonstrated the capacity for consistently limiting the editing events to just these two that, afterward, generate stop codons (Figure 4D).
Figure 4.

Primary CD34+ Cells Modified by Pair 1 gRNAs and Differentiated toward the Erythroid Lineage
(A) Exon 9 PCR amplicons from different experiments of hematopoietic progenitor cells differentiated in vitro toward erythroid lineage and edited with guides I + II (pair 1). C, control cells electroporated with Cas9- P2A-ZsGreen without guides. I + II and C are the ZsGreen+ fraction selected by sorting. WT, non-electroporated cells. Pair 1 shows edited cells from different experiments. (B) Top: sequences resulting from the two main deletions produced after the gene edit. Blue indicates the region covered by guide I; purple indicates the region covered by guide II; a sequence in gray indicates the BstXI recognition site; and light red indicates a differential nucleotide, not present in the 32-bp deletion, that is required for BstXI recognition. Bottom: lane 1, molecular weight reference; lanes 2–4, hematopoietic progenitor cells differentiated in vitro toward erythroid lineage and edited with guides I + II; lanes 5–7, control cells electroporated with Cas9-P2A-ZsGreen without guides. ZsGreen, + or – fractions separated by sorting; + or – BstXI, treated or not with the enzyme. Red triangles indicate DNA cut after enzyme digestion. (C) Next-generation sequencing results from two edited DNA samples; colors represent each of the deletions found named Del 31, Del 32, Del 40, and Del 55. Data are presented as mean ± SEM. A Tukey’s multiple comparison test was used to evaluate differences between groups. (D) Amino acid sequence resulting after 31- and 32-bp deletions. Red amino acids are the alerted ones with respect to the WT. Both deletions generate premature stop codons represented by dots on a red background.
In order to check whether the edited cells reproduced the in vitro PKD phenotype, CD34-PKLR−/− cells were cultured in specific media to allow erythroid differentiation (Figure S4). After a 2-week differentiation protocol, cells were analyzed in terms of cell morphology, immunophenotype, and pyruvate kinase activity. Non-edited CB-CD34+ cells and bone marrow CD34 cells obtained from a PKD patient were also differentiated following the same protocol. No differences were observed in the in vitro differentiation of the different samples, demonstrating that PKD progenitors differentiate normally in vitro under the conditions used (Figure 5A). Similarly, analysis of cytospins performed at the end of the culture corroborated the same degree of differentiation in all of the groups, independent of its deficiency in PKD. The most representative cell type was the orthochromatic erythroblast population (Figure 5B).
Figure 5.

Immunophenotype and Activity of Hematopoietic Cells Edited and Differentiated toward Erythroid Lineage
(A) Left: percentage of cells considered erythroid based on their CD36/CD45 staining (which includes cells in transition from CD45+/CD36+ to CD45−/CD45−; values are normalized with respect to control samples). Right: erythroid maturation followed by CD71/CD235a staining. Immature, CD71+/CD235a+ (orange); mature, CD71−/CD235a+ (red); transition from immature to mature (purple). n = 6. PKD, cells differentiated from patient bone marrow sample; pair 1, cells electroporated with guides I + II; C, control cells electroporated with Cas9-P2A-ZsGreen without guides; + or −, ZsGreen positive or negative fraction separated by sorting; WT, non-electroporated/sorted cells. (B) Cytospin after 14 days of erythroid differentiation: 1, polychromatophilic erythroblast; 2, orthochromatic erythroblast; 3, reticulocyte. (C) PK/HK activity of edited cells after erythroid differentiation. Green circles indicate ZsGreen+ fraction cells electroporated with the two guides from different experiments; purple square indicates PKD cells differentiated from patient bone marrow sample; red triangles indicate control cells. Measures are normalized with respect to PK/HK of control samples, which were considered equal to 1 (∗∗∗∗p < 0.0001; pair 1 edited cells, n = 6; PKD patient cells, n = 1; control cells, n = 9).
Finally, edited, control, and PKD patient cells were tested for pyruvate kinase activity. PKD activity was tested together with the hexokinase (HK) activity to normalize the increased PK activity of the most immature erythroblasts. The PK/HK ratio of the edited cells showed a decreased enzymatic activity comparable with the values obtained in the PKD patient sample differentiated in parallel, which was approximately 0.6 times the values of the healthy control cells (Figure 5C). Overall, NHEJ-PD after Cas9 DNA breaks by means of the use of guide pair combination generates an in vitro cellular model that resembles the characteristics of patient-deficient cells, which would be very valuable for further testing of alternative therapeutic strategies.
NHEJ-PD Generates an In Vivo Knockout Animal Model
In order to study whether the use of two closed guides to minimize the variability of NHEJ could also work in vivo, we addressed the knockout of the mouse Hao1 gene, which encodes for the glycolate oxidase (GO), in the liver of Agxt−/− mice. For this purpose mice were injected with two AAV8 vectors expressing two different gRNAs and the Staphylococcus aureus Cas9 protein under the control of a liver-specific promoter restricting Cas9 expression to the hepatocytes and previously tested individually.19 Both gRNAs targeted opposite strands of exon 2 of the mouse Hao1 gene, were in a PAM-out orientation, and DSBs were 64 bp apart (Figure 6A). An overall efficiency of 55% editing was obtained in the livers of the treated animals, similar to the editing efficiency to each vector when administered individually (Figure 6B, left). NGS analysis of the livers of animals treated with both vectors revealed that inside the edited cells, most of the sequences presented the deletion of the 64 bp harbored between the two DSBs (Figure 6B, right). Moreover, these genetic modifications led to a decrease in Hao1 mRNA levels (Figure 6C) and to the complete elimination of GO protein analyzed by western blot (Figure 6D).
Figure 6.

In Vivo Generation of NHEJ-PD Generates a Hao1 Gene Knockout in Mice
(A) Scheme of the two guides tested in Hao1 gene. Light colors indicate guides (Hao1-g1 and Hao1-g2); dark colors indicate PAM sequences. Within the pair scheme there are three differentiated parts. Top: WT sequence and the guides used. Middle: WT and precise deletion sequence with its frequency. Bottom: indels near precise deletion and its frequency. Next-generation sequencing was performed for two different groups of mice, i.e., a group treated with a saline solution (n = 4) and a group treated intravenously (i.v.) with AAV8s expressing the two guides and the SaCas9 each at a dose of 5 × 1012 vg/kg (n = 5). Stars represent the WT sequence, and scripts are deletions respective to the WT sequence. (B) Next-generation sequencing results from variants of the different combinations. Genotype of the different groups. Gray indicates WT mice; green indicates edited mice. (C) Percentage of the different edition variant. Dark purple shows precise deletion; light purple shows variants with modifications around the expected deletion. (C) Quantification of Hao1 mRNA expression levels analyzed by qRT-PCR in animals treated with saline or the combination of Hao1-g1 and Hao1-g2. (D) Western blot analysis of GO protein levels in representative animals treated with saline and the combination of Hao1-g1 and Hao1-g2. GAPDH was used as a loading control as previously described19. Data are presented as mean ± SEM and Tukey's Multiple Comparison statistical test was used to evaluate differences between groups.
Discussion
In this study, we have shown that the use of two gRNAs to drive Cas9 cutting in adult cells facilitates the precise deletion of the sequences between them and reduces, or even eliminates, the heterogeneity of error-prone NHEJ, which allows the knockdown or knockout of the desired gene and the generation of cellular or animal models with an almost homogeneous genotype. Previous works have indicated the preference for NHEJ repair by the precise deletion between the two DSBs generated when two guides are used simultaneously.18 Herein, we demonstrate that this characteristic facilitates the generation of cellular and animal models with a more homogeneous and controlled genotype modification.
We have observed that the NHEJ-PD process generated by the two cuts occurs in different cell types, in primary and in immortalized cell lines, in different species, and using different Cas9 proteins (either Streptococcus pyogenes [spCas9] or Staphylococcus aureus Cas9 [saCas9]). Thus, the process seems to be mainly dependent on the NHEJ mechanism.
The combination of NHEJ-PD and the design of gRNAs to drive the activity of the Cas9 protein to very specific genome sites allows the prediction and generation of genotypes that could very precisely resemble mutations already described in humans. In this study, we selected a specific region in exon 9 that generated mutations in the same location as a cluster of genetic mutations found in PKD patients. This procedure is 10-fold more efficient than the introduction of specific mutations by means of homology directed repair and has allowed us to apply it to primary human hematopoietic progenitors. This procedure allows us to then generate primary cells carrying almost identical human mutations in normal cells from healthy donors for those samples that are difficult or not ethically justified, and these could be used as cellular models for testing newly available therapeutic strategies. Also, NHEJ-PD permits the selection of specific sites whose alterations could generate stop codons nearby, thus facilitating the generation of complete knockout cellular models in a more efficient way. Moreover, we have demonstrated that the same strategy applied to animal models is also very efficient.
Previous studies have suggested that a minimum distance between the two cutting sites is required for optimal NHEJ-PD. We have observed a similar requirement with an almost total avoidance of precise deletion when cutting site guides were designed 8 bp apart. In addition, we also demonstrate that the orientation of the guides, and consequently the orientation of the Cas9 enzyme, is also important for optimal NHEJ-PD. Higher efficiency was obtained when both guides had the PAM sequence located in the sequence to be deleted, what we called PAM-in. The worst cutting efficiency was observed in the opposite orientation, PAM-out/PAM-out. Taking into account that the last 3 bp were common for both guides, steric interferences could be responsible for this worse efficacy. However, when we designed gRNAs similarly located but without overlapping sequences, no precise deletions were obtained either. Moreover, in those cases where PAM-in and PAM-out guides were used simultaneously, the PAM-in guide was more efficient than the PAM-out guide. Thus, factors such as steric interactions but also orientation toward the sequence to be deleted would be important for the efficiency of the CRISPR-Cas9 system to generate NHEJ-PD.
The use of guide pairs and the generation of NHEJ-PD have been also proposed as a therapeutic tool to precisely eliminate specific sequences that would allow the restoration of an almost completely functional protein. Mutations in the dystrophin gene have been precisely eliminated by removing the sequence between exons 45 and 55 with the use of two guides that restore the function and the Duchenne muscular dystrophy phenotype.15 The same strategy has been followed in epidermolysis bullosa, a recessive disorder caused by frameshift mutations in the COL7A1 gene. The excision of the mutated sequences at exon 80 restores collagen function and recovers a normal phenotype in keratinocytes.14 Our results clearly support this strategy and point out the importance of strategy design in terms of CAs9 positioning.
A potential drawback of the use of two guides is that the amount of off-target mutations is multiplied by two. The optimization of online tools for the design of gRNAs and the use of better systems to characterize them will help to improve their usefulness.
It is interesting to note the discordance of Indel percentage and mRNA and protein level reduction observed in the in vivo experiments, which are also reflected in the in vitro ones. In the case of in vivo treatments, these discrepancies have previously been reported19, 20, 21 and they are attributed to different factors. First, only hepatocytes were targeted, but the analysis of indels was performed in all liver cells, including non-parenchymal cells, and the analysis in hepatocytes has been reported to increase the detected indel frequency.22 Additionally, chromatin status-related differences in editing efficiency have been found,23 implying that transcriptionally active sites would be more accessible, and therefore more efficiently edited. In terms of the effect of the animal model on editing efficiency, we have not explored editing with our system in WT C57BL/6 mice. Although the target sequence is conserved in the Agxt−/− model, the possibility exists of different transcriptional activity as a compensatory mechanism of the metabolic deficit of AGXT, which has not been reported but could affect indel frequencies.
In summary, we demonstrate in the present study that the use of guide pairs in combination with the Cas9 nuclease generates NHEJ-PD in adult cells that facilitates the generation of cellular and animal models of specific mutations found in human diseases. Moreover, these combinations could be used as a therapeutic tool for the treatment of genetic diseases where the excision of a defined sequence restores the function of the modified protein.
Materials and Methods
Cell Lines
The HEK293T cell line (human embryonic kidney cell line competent to replicate vectors carrying the SV40 T antigen; ATCC, CRL-3219) was cultured in Iscove’s modified Dulbecco’s medium (IMDM; Gibco), HyClone fetal bovine serum (10%; GE Healthcare), and penicillin/streptomycin (1%; Gibco). Cells were maintained at 5 × 105 cells/mL concentration.
The K562 cell line (chronic myelogenous leukemia; ATCC, CCL-243) was cultured in IMDM (Gibco), HyClone (10%; GE Healthcare), and penicillin/streptomycin (1%; Gibco). Cells were maintained at 1 × 105 to 1 × 106 cells/mL.
Incubation conditions were the same for all cell lines used: 37°C, 5% CO2, and 95% relative humidity.
Animals
Agxt1−/− mice (B6.129SvAgxttm1Ull) were bred and maintained in a pathogen-free facility with free access to standard chow and water. Agxt1−/− mice were genotyped as described.24 A dose of 5 × 1012 vector genomes (vg)/kg of each AAV8 vector carrying Hao1-specific CRISPR-Cas9 system previously described 19 (final dose of 1 × 1013 vg/kg) was administered to 12- to 14-week-old Agxt1−/− male animals by intravenous injection. Animals were sacrificed and livers harvested 21 days after the administration of the vectors. All experimental procedures were approved by the Ethics Committee of the University of Navarra and the Institute of Public Health of Navarra according to European Council guidelines. All experiments using animal models complied with all relevant ethical regulations.
Design and Generation of CRISPR-Cas9 Tools
For the experiments performed in vitro in cell lines, two different plasmids were used to clone specific guides. The plasmids used contained gRNA and RNA scaffold expression driven by the U6 promoter and also the Cas9-ZsGreen sequences linked by a P2A (porcine teschovirus-1’s motif that produces co-translational “cleavage” of both proteins) under the cytomegalovirus (CMV) promoter. The different guides were designed based on Zhang Lab’s program (MIT) against the PKLR gene and chosen by score and its location in the gene.
The vectors and gRNAs used for the generation of a knockout model in vivo were previously designed and individually characterized.22 Briefly, single guide RNAs (sgRNAs) targeting exonic regions of the Hao1 gene were designed and selected using Benchling software (https://www.benchling.com), which consisted of 21-nt sequences upstream of the 5′-NNGRRT-3′ PAM sequence of SaCas9. The pX602-AAV-TBG::NLS-SaCas9-NLS-HA-OLLAS-bGHpA;U6::BsaIsgRNA plasmid that contains the saCas9 expressed under the tTBG promoter, the sgRNA under the U6 promoter, and inverted terminal repeat (ITR) sequences for AAV vector production was a gift from Feng Zhang (Addgene plasmid #61593).19 These plasmids were packaged in AAV8 particles following the standard protocol in the field.25
Human Hematopoietic Progenitor Medium
Human hematopoietic progenitors were cultured in StemSpan serum-free expansion medium (SFEM) I (STEMCELL Technologies) supplemented with human stem cell factor (hSCF, 100 ng/mL; EuroBioSciences), recombinant human thrombopoeitin (hTPO, 100 ng/mL; EuroBioSciences), human FMS-like tyrosine kinase 3 ligand (hFlt3-ligand, 100 ng/mL; EuroBioSciences), GlutaMAX (1%; Gibco), and penicillin/streptomycin (1%; Gibco). Cells were maintained at 1 × 106 cells/mL concentration. Incubation conditions were the following: 37°C, 5% CO2, and 95% relative humidity.
Erythroid Differentiation Protocol
Human hematopoietic progenitors were differentiated toward erythroid lineage using four different media. The first medium, used on days 1–7, was based on StemSpan SFEM I (STEMCELL Technologies) supplemented with hSCF (50 ng/mL; EuroBioSciences), hFlt3-ligand (16.7 ng/mL; EuroBioSciences), bone morphogenetic protein 4 (BMP-4, 6.7 ng/mL; PeproTech), human interleukin-3 (hIL-3, 6.7 ng/mL; EuroBioSciences), human interleukin-11 (hIL-11, 6.7 ng/mL; EuroBioSciences), and human erythropoietin (hEPO, 1.3 U/mL; Amgen). Cells were then transferred to the second medium in which they were cultured on days 7–14. The second medium was based on IMDM glutamine supplemented (IMDM GlutaMAX supplemented; Gibco) enriched with BSA (1%; Sigma), insulin (0.01 mg/mL; Sigma), human transferrin (0.2 mg/mL; Sigma), β-mercaptoethanol (91 μM; Gibco), penicillin/streptomycin (1%; Gibco), lipid mixture 1 (1×; Sigma), ethanolamine (0.004%, Sigma), hSCF (5 ng/mL; EuroBioSciences), hIL-3 (6.7 ng/mL; EuroBioSciences), hIL-11 (6.7 ng/mL; EuroBioSciences), hEPO (1.3 U/mL; Amgen), insulin-like growth factor-1 (IGF-1, 20 ng/mL; PeproTech), and hydrocortisone (1 μM; Sigma). After day 14 and for 2 days, cells were transferred to the third medium that was also based on IMDM GlutaMAX medium (Gibco) supplemented with BSA (1%; Sigma), insulin (0.01 mg/mL; Sigma), human transferrin (0.2 mg/mL; Sigma), β-mercaptoethanol (91 μM; Gibco), penicillin/streptomycin (1%; Gibco), lipid mixture 1 (1×; Sigma), ethanolamine (0.004%; Sigma), and hEPO (10 U/mL; Amgen). Final enucleation was carried out for 5 days on the fourth medium that was also based on IMDM GlutaMAX medium (Gibco) supplemented with BSA (1%; Sigma), insulin (0.01 mg/mL; Sigma), human transferrin (0.2 mg/mL; Sigma), β-mercaptoethanol (91 μM; GIBCO), penicillin/streptomycin (1%; Gibco), lipid mixture 1 (1×; Sigma), and ethanolamine (0.004%; Sigma).
All media were filtered through 0.22-μm filters before use. Sampling and analysis were done within this third week. During erythroid differentiation, cells were maintained between 4 × 105 and 4 × 106 cell/mL concentration. Incubation conditions were also 37°C, 5% CO2, and 95% relative humidity.
Erythroid Differentiation Immunophenotype
The immunophenotype of hematopoietic progenitors differentiated toward the erythroid lineage was analyzed by flow cytometry using the following Ab combination: CD36-phycoerythrin (PE), clone CB38 (NL07), BD Pharmingen, 1 μL of Ab/100 μL; CD45-allophycocyanin (APC)-Cy7, clone HI30, BioLegend, 1 μL of Ab/100 μL; CD71-PE-Cy5, clone M-A712, BD Pharmingen, 1 μL of Ab/100 μL; CD235a-PE-Cy7, clone HI264, BioLegend, 1 μL of Ab/100μL). Cells were resuspended in a final volume of 100 μL, labeled for 30 min at 4°C, and washed with PBA (PBS with 0.2% sodium azide [Merck] and 1% BSA [Sigma]). DAPI (final concentration of 1 μg/mL) was added and events were recorded with an LSRFortessa (BD Biosciences). Data were analyzed by FlowJo software (BD Biosciences).
Electroporation of the K562 Cell Line
DNA delivery was carried out on a 20-μL volume format using an Amaxa 4D-Nucleofector system and cell line Nucleofector solution SF (Lonza). The total amount of DNA electroporated was 3 μg (1.5 μg for each plasmid in the case of using two plasmids at the same time). 2 × 105 cells were electroporated following the manufacturer’s protocol under the FF-120 program.
Electroporation of Primary Cells
Hematopoietic progenitor cells were electroporated with CRISPR-Cas9 plasmids, as previously described, using Amaxa Nucleofector I (Lonza) and following the manufacturer’s protocols. Cells were electroporated in cuvettes (electroporation volume of 100 μL) using a human CD34+ cells kit (Lonza) and the U08 electroporation program. The total amount of plasmid used was 11.26 μg; hence, when two plasmids were electroporated at the same time, 5.63 μg of each was added to the mix. After electroporation, cells were cultured on a 24-well plate (Falcon) at 2 mL of final volume. In the case of the experiments with selection based on ZsGreen reporter expression, sorting was performed 24 h after electroporation. Aggregates were eliminated before sorting using a 40-μm cell strainer (Falcon).
Deep Sequencing Analysis
NGS for a single cell suspension was carried out by STABVIDA and GeneWiz Companies. PCR products were treated and purified according to manufacturer's protocols. Samples were used for a library construction and sequenced with v3 chemistry in the lllumina MiSeq platform. Only variants represented in more than 1% were considered.
For deep sequencing of liver cells, total genomic DNA was extracted from the livers using a NucleoSpin tissue DNA extraction kit (Macherey-Nagel) according to the manufacturer’s instructions. A first PCR round was performed using primers that amplified 390 bp of the mouse Hao1 gene (containing the targeted region in the middle of the amplicon) carrying the universal adaptor M13 sequence (underlined) in their 5′ end (forward, 5′-GTTGTAAAACGACGGCCAGTAGACCAATGTTTGTCAGAGG-3′ and reverse, 5′-CACAGGAAACAGCTATGACCTAAAAGCATCCTAGGAAGGG-3′). Platinum Taq polymerase (Invitrogen) was used. Subsequently, a second PCR was performed using primers targeting the universal M13 sequence and adding unique MID (multiplex identifier) barcodes to each sample. Sample pools were NGS sequenced on the MiSeq platform (Illumina, San Diego, CA, USA) following the manufacturer’s protocol.
Author Contributions
S.L.-M. and I.O.-P. designed and performed experiments and wrote and reviewed the manuscript; N.Z. and L.T. performed experiments and reviewed the manuscript; A.G.-T. performed experiments and prepared reagents; O.A. and R.S.-D. performed flow cytometry and cell sorting procedures; R.T. and J.C.R. provided reagents; E.O. and J.M. helped with erythroid differentiation experiments; J.A.B. and G.G.-A. suggested ideas and reviewed the manuscript; and J.-C.S. obtained financial support, designed experiments, and wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors would like to thank Miguel A. Martin for the careful maintenance of NSG mice, and Aurora de la Cal, María del Carmen Sánchez, Soledad Moreno, Nadia Abu-sabha, Montserrat Aldea, and Sergio Losada for dedicated administrative help. This work was supported by grants from the Ministerio de Economía, Comercio y Competitividad y Fondo Europeo de Desarrollo Regional (FEDER) (SAF2017-84248-P), the Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (Red TERCEL; RD16/0011/0011), and the Comunidad de Madrid (AvanCell, B2017/BMD-3692). The authors also thank Fundación Botín for promoting translational research at the Hematopoietic Innovative Therapies Division of the CIEMAT. CIBERER is an initiative of the Instituto de Salud Carlos III and the Fondo Europeo de Desarrollo Regional (FEDER).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.10.007.
Supplemental Information
References
- 1.Keeler A.M., ElMallah M.K., Flotte T.R. Gene therapy 2017: progress and future directions. Clin. Transl. Sci. 2017;10:242–248. doi: 10.1111/cts.12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakuma T., Woltjen K. Nuclease-mediated genome editing: at the front-line of functional genomics technology. Dev. Growth Differ. 2014;56:2–13. doi: 10.1111/dgd.12111. [DOI] [PubMed] [Google Scholar]
- 3.Silva G., Poirot L., Galetto R., Smith J., Montoya G., Duchateau P., Pâques F. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr. Gene Ther. 2011;11:11–27. doi: 10.2174/156652311794520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng D., Yan C., Pan X., Mahfouz M., Wang J., Zhu J.K., Shi Y., Yan N. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science. 2012;335:720–723. doi: 10.1126/science.1215670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mojica F.J.M., Montoliu L. On the origin of CRISPR-Cas technology: from prokaryotes to mammals. Trends Microbiol. 2016;24:811–820. doi: 10.1016/j.tim.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Shibata M., Nishimasu H., Kodera N., Hirano S., Ando T., Uchihashi T., Nureki O. Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat. Commun. 2017;8:1430. doi: 10.1038/s41467-017-01466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wassef M., Luscan A., Battistella A., Le Corre S., Li H., Wallace M.R., Vidaud M., Margueron R. Versatile and precise gene-targeting strategies for functional studies in mammalian cell lines. Methods. 2017;121–122:45–54. doi: 10.1016/j.ymeth.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Islam W. CRISPR-Cas9; an efficient tool for precise plant genome editing. Mol. Cell. Probes. 2018;39:47–52. doi: 10.1016/j.mcp.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Gilles A.F., Schinko J.B., Averof M. Efficient CRISPR-mediated gene targeting and transgene replacement in the beetle Tribolium castaneum. Development. 2015;142:2832–2839. doi: 10.1242/dev.125054. [DOI] [PubMed] [Google Scholar]
- 11.Mettananda S., Fisher C.A., Hay D., Badat M., Quek L., Clark K., Hublitz P., Downes D., Kerry J., Gosden M. Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia. Nat. Commun. 2017;8:424. doi: 10.1038/s41467-017-00479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canver M.C., Bauer D.E., Dass A., Yien Y.Y., Chung J., Masuda T., Maeda T., Paw B.H., Orkin S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014;289:21312–21324. doi: 10.1074/jbc.M114.564625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geisinger J.M., Turan S., Hernandez S., Spector L.P., Calos M.P. In vivo blunt-end cloning through CRISPR/Cas9-facilitated non-homologous end-joining. Nucleic Acids Res. 2016;44:e76. doi: 10.1093/nar/gkv1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonafont J., Mencía Á., García M., Torres R., Rodríguez S., Carretero M., Chacón-Solano E., Modamio-Høybjør S., Marinas L., León C. Clinically relevant correction of recessive dystrophic epidermolysis bullosa by dual sgRNA CRISPR/Cas9-mediated gene editing. Mol. Ther. 2019;27:986–998. doi: 10.1016/j.ymthe.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ousterout D.G., Kabadi A.M., Thakore P.I., Majoros W.H., Reddy T.E., Gersbach C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sangamo Therapeutics . 2018. A study to assess the safety, tolerability, and efficacy of ST-400 for treatment of transfusion-dependent beta-thalassemia (TDT)https://clinicaltrials.gov/ct2/show/NCT03432364?term=BCL11A&cond=B-Thalassemia&draw=2&rank=1 [Google Scholar]
- 17.Zheng Q., Cai X., Tan M.H., Schaffert S., Arnold C.P., Gong X., Chen C.Z., Huang S. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques. 2014;57:115–124. doi: 10.2144/000114196. [DOI] [PubMed] [Google Scholar]
- 18.Guo T., Feng Y.L., Xiao J.J., Liu Q., Sun X.N., Xiang J.F., Kong N., Liu S.C., Chen G.Q., Wang Y. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 2018;19:170. doi: 10.1186/s13059-018-1518-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finn J.D., Smith A.R., Patel M.C., Shaw L., Youniss M.R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 21.Jarrett K.E., Lee C.M., Yeh Y.H., Hsu R.H., Gupta R., Zhang M., Rodriguez P.J., Lee C.S., Gillard B.K., Bissig K.D. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci. Rep. 2017;7:44624. doi: 10.1038/srep44624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zabaleta N., Barberia M., Martin-Higueras C., Zapata-Linares N., Betancor I., Rodriguez S., Martinez-Turrillas R., Torella L., Vales A., Olagüe C. CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Nat. Commun. 2018;9:5454. doi: 10.1038/s41467-018-07827-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X., Rinsma M., Janssen J.M., Liu J., Maggio I., Gonçalves M.A.F.V. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016;44:6482–6492. doi: 10.1093/nar/gkw524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salido E.C., Li X.M., Lu Y., Wang X., Santana A., Roy-Chowdhury N., Torres A., Shapiro L.J., Roy-Chowdhury J. Alanine-glyoxylate aminotransferase-deficient mice, a model for primary hyperoxaluria that responds to adenoviral gene transfer. Proc. Natl. Acad. Sci. USA. 2006;103:18249–18254. doi: 10.1073/pnas.0607218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang X., Hartley A.V., Yin Y., Herskowitz J.H., Lah J.J., Ressler K.J. AAV2 production with optimized N/P ratio and PEI-mediated transfection results in low toxicity and high titer for in vitro and in vivo applications. J. Virol. Methods. 2013;193:270–277. doi: 10.1016/j.jviromet.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.