

Abstract
Lentiviral vectors (LVs) are increasingly employed in gene and cell therapy. Standard laboratory production of LVs is not easily scalable, and research-grade LVs often contain contaminants that can interfere with downstream applications. Moreover, purified LV production pipelines have been developed mainly for costly, large-scale, clinical-grade settings. Therefore, a standardized and cost-effective process is still needed to obtain efficient, reproducible, and properly executed experimental studies and preclinical development of ex vivo and in vivo gene therapies, as high infectivity and limited adverse reactions are important factors potentially influencing experimental outcomes also in preclinical settings. We describe here an optimized laboratory-scale workflow whereby an LV-containing supernatant is purified and concentrated by sequential chromatographic steps, obtaining biologically active LVs with an infectious titer and specific activity in the order of 109 transducing unit (TU)/mL and 5 × 104 TU/ng of HIV Gag p24, respectively. The purification workflow removes >99% of the starting plasmid, DNA, and protein impurities, resulting in higher gene transfer and editing efficiency in severe combined immunodeficiency (SCID)-repopulating hematopoietic stem and progenitor cells (HSPCs) ex vivo, as well as reduced activation of inflammatory responses ex vivo and in vivo as compared to TU-matched, laboratory-grade vectors. Our results highlight the value of accessible purified LV production for experimental studies and preclinical testing.
Keywords: Lentiviral vectors, purification process, manufacturing, gene therapy, ex vivo, in vivo, hematopoietic stem cells, innate immunity
Graphical Abstract
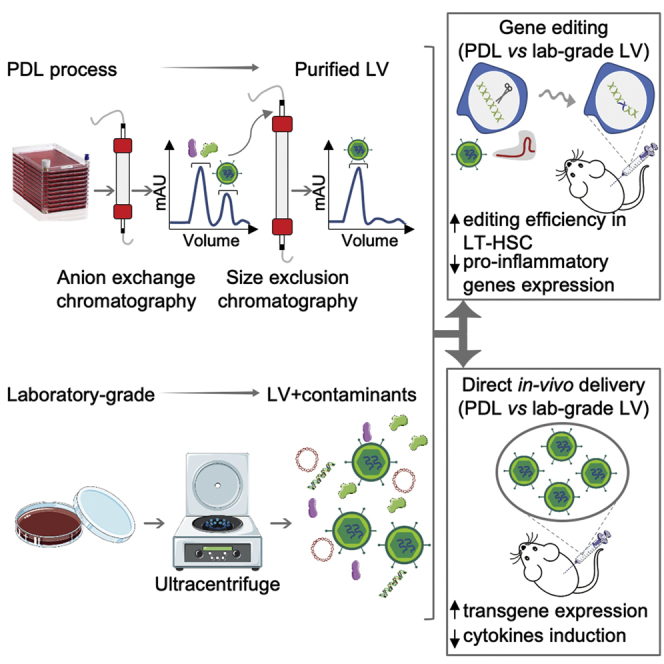
Lentiviral vectors (LVs) are powerful gene-transfer tools routinely exploited for distinct research and clinical applications. LVs produced in most research laboratories contain contaminants that can generate confounding effects in experimental studies. Soldi et al. describe a laboratory-scale workflow for purified LV production, highlighting enhanced gene-editing efficiency and diminished inflammatory responses.
Introduction
Lentiviral vectors (LVs) have become the benchmark for ex vivo gene and cell therapy applications due to their ability to efficiently and permanently integrate in the host cell genome with low risk of genotoxicity.1,2 In particular, LV-based gene therapy and precise gene editing in hematopoietic stem/progenitor cells (HSPCs) are becoming promising strategies to treat inherited monogenic diseases.3 Moreover, LVs have been efficiently used to transduce the liver in mice, dogs, and nonhuman primates (NHPs), supporting their use for in vivo gene therapy applications targeting the liver.4
Laboratory-scale production of LV is typically achieved by ultracentrifugation of filtered culture medium from human HEK293T cells transiently cotransfected with a combination of 3 or more packaging and one transfer vector plasmid. This approach is not easily scalable, and research-grade LVs often contain several types of contaminating molecules, derived from both culture media and producer cells, such as residual transfection plasmids, that might be toxic to target cells, impacting on the transduction efficiency ex vivo5 and eliciting an innate immune response when injected in vivo.6, 7, 8 To overcome these adverse effects, purification of LV is mandatory for clinical applications that require high-titer and high-quality stocks, thus increasing efficacy and safety. Furthermore, a standardized process leads to a well-characterized and reproducible product that is important for alleviating interfering and confounding effects in experimental studies and suitably performing preclinical development of ex vivo and in vivo gene therapies. Over the past several years, considerable progress has been done in the field of vector design and production/purification workflow and reviewed9, 10, 11, 12, 13, 14, 15. Purification is usually achieved by anion exchange chromatography. The eluted material is further concentrated, and finally, gel filtration (GF) chromatography is used for exchange to the release buffer. However, the outsourcing of this process to dedicated manufacturers is expensive and thus difficult to sustain in the context of explorative and preclinical studies. In an effort to produce high-titer and high-quality LVs for research-based applications, we here optimized a medium-scale workflow that can be used to purify and concentrate high-quality LVs that are suitable for a number of preclinical applications. The workflow can be adopted in a research-grade laboratory and features a flexible design in which individual steps can be adjusted and re-scaled (e.g., the volume of supernatant-containing LV particles to be loaded on the anion exchange resin), according to downstream applications.
In order to assess the benefits of reducing contaminants in purified LV preparations, we administered systemically purified or laboratory-grade (lab-grade) LVs to mice and analyzed transgene expression, as well as cytokine induction. Furthermore, we compared purified versus nonpurified LVs in HSPC gene transfer and gene editing efficiency, as well as on the induction of proinflammatory responses ex vivo and engraftment of genetically engineered cells in vivo.
Results
Optimizing the Upstream Phase of Medium-Scale Production of LVs
We first focused on the upstream phase, examining some experimental variables affecting the production of LVs, to increase transfection efficiency and thus maximize the titer of the vector preparations. LV-containing supernatant was produced by calcium phosphate-mediated transient transfection of adherent HEK293T cells,1 in Cell Factory 10-tray stacks (CF10), using the standard 3rd-generation system comprised of a vector transfer plasmid and plasmids encoding for the HIV Gag-Pol gene, the HIV rev gene, and the vesicular stomatitis virus envelope glycoprotein (VSV.G). The cell-seeding density and the amount of plasmids needed to produce LV were optimized starting from protocols that were previously developed for clinical purposes.16 The contribution of pAdVAntage in the pool of plasmids and sodium butyrate addition after transfection were evaluated. Sodium butyrate is known to activate the human cytomegalovirus (CMV) promoter/enhancer and the long terminal repeat-directed expression of HIV,17,18 thus enhancing viral production during transfection,19, 20, 21 whereas pAdVAntage increases LV yield through inhibiting double-stranded (ds) RNA recognition and consequent PKR activation.22 Of note, pAdVAntage has not been approved yet for clinical use. Here, we tested if its positive effect on LV production was maintained upon culture escalation to CF10. After 14–16 h post-transfection, the cell culture medium was replaced with a fresh one (with or without 1 mM sodium butyrate). The addition of both pAdVAntage and sodium butyrate was beneficial for the production of high-titer LV (Figure S1A). However, the concentration of sodium butyrate above 2 mM slightly decreased the yield and infectivity of the LV preparations (data not shown). With the use of the conditions determined above, the optimal time for harvesting LV after transfection was investigated. The standard double harvest,23 whereby collection is performed at 24 and 48 h postmedium exchange, was compared in a pilot experiment with a single collection at 30 h postmedium exchange. After the first collection, the medium was replaced with a fresh one. The overall vector yield was higher in the double collection at 24 and 48 h compared to the single collection at 30 h, whereas infectivity was strongly reduced (Figure S1B). Moreover, the amount of total proteins or proteins derived from the producer HEK293T cells, herein termed host cell proteins (HCPs), in the final LV stock produced with double collection was higher (0.73 mg and 857.9 ng per 108 transducing unit [TU], respectively) than in the preparations obtained with a single collection at 30 h (0.31 mg and 158.3 ng per 108 TU, respectively), indicating that the harvesting protocol impacts on the amount of contaminating proteins. Based on these results, a single harvest at 30 h postmedium exchange was chosen as the best condition to obtain purified LV stocks, as it offers less handling of the cells, whereas increasing infectivity and purity. However, double collection at 24 and 48 h remains a valid alternative to be employed according to downstream applications. Additionally, we tested the effect of distinct types of sera on transfection efficiency to consistently produce high-titer LV and minimize impurities. Three distinct sera were tested: γ-irradiated South America (SA), γ-irradiated New Zealand (NZ), and non-γ-irradiated SA. Of note, distinct batches of sera from the same supplier may yield different LV production capacity, as previously described.24 Therefore, we advise testing serum batches before committing on large-scale LV production. We measured the infectious titer, physical particles, and infectivity of LV-containing supernatant from independent productions. We found that the titer of the unconcentrated LV stock that was produced using non-γ-irradiated SA serum was overall higher than those obtained from other two γ-irradiated sera (Figure 1A, left panel). The higher titer was associated with a higher p24 concentration (Figure 1A, middle panel), resulting in a nearly comparable infectivity between γ- and the non-γ-irradiated sera (Figure 1A, right panel). This result indicates that the type of serum affects the amount of LV produced upon HEK293T transfection.
Figure 1.

Optimization of Lentiviral Vector (LV) Production
(A) LV-containing supernatant infectious titer (transducing unit [TU]/mL; left panel), physical particles (ng p24/mL; middle panel), and infectivity (TU/ng p24; right panel) using SA γ-irradiated serum (n = 5), NZ γ-irradiated serum (n = 2), and SA non-γ-irradiated serum (n = 9). Data are plotted as mean with SD (n ≥ 3) or as mean with range (n = 2). (B) Anion exchange chromatographic profile of LV-containing supernatant produced using different serums. Arrows indicate LV peak. Asterisk indicates contaminating high peak. (C) Anion exchange chromatographic profile of medium supplemented with different sera. Black dashed box indicates samples subjected to MS analysis.
By comparing chromatographic separation of LV-containing supernatants, we noticed the presence of an unexpected, very high peak (Figure 1B; asterisk) near the vector peak (Figure 1B; arrows) when using γ-irradiated SA serum. In order to assess if the unexpected high peak was derived from contaminants present in the serum, cell culture media containing 10% of either γ-irradiated SA or NZ serum were subjected to chromatographic separation. We found the same peak in the cell culture medium containing γ-irradiated SA serum (Figure 1C), matching the previously identified contaminant peak in the virus chromatographic separation. We then performed mass spectrometry (MS) analysis of proteins present in both the cell culture media (SA and NZ) and their respective eluted peaks (Figure S1C). Both cell culture media were similar in terms of protein identity and abundance (Figures S1D and S1E), with the same top 5% of proteins that accounted for around 90% of the total protein abundance. On the other hand, the protein content in the eluted peak fraction was higher in the SA than in the NZ serum (Figure S1F), in agreement with the high peak of contaminants observed in Figure 1B, whereas the overall number of distinct, detected proteins was similar (Figure S1G). Protein identity and relative abundance were different between the two types of sera in the eluted peaks (Figure S1H). This result may suggest that the same proteins from distinct sera batches might bind with different efficiency to the chromatographic columns and thus be eluted at different rates during LV purification. This may depend on distinct variables in the serum batch, such as chemical and physical parameters, as well as biological molecules (e.g., DNA, RNA, exosomes, lipids, etc.).
In order to check the impact of distinct types of sera on LV purity, we carried a complete purification workflow, reported below, using the above-described sera. Based on the fact that assays measuring total protein concentration will also detect proteins present in the LV particles, we decided also to measure HCPs and residual DNA. In agreement with the results obtained above, the LV stock produced using the non-γ-irradiated SA serum contained the lowest levels of DNA, as well as total and HCPs, followed by the γ-irradiated NZ (non-γ-irradiated SA: 0.22 μg of DNA, 0.03 mg of total proteins, and 66.99 ng of HCPs per 108 TU; γ-irradiated NZ: 0.27 μg of DNA, 0.07 mg of total proteins, and 84.31 ng of HCPs per 108 TU). LV stocks produced using γ-irradiated SA serum contained the highest level of total and HCPs (0.31 mg and 158.3 ng per 108 TU, respectively). Together, these results prompted us to adopt in our workflow for LV production the non-γ-irradiated SA serum. Moreover, these observations suggest that serum may affect the efficiency of binding of distinct contaminants to the chromatographic columns, hence highlighting the importance of optimizing the upstream transfection process, including serum type and batch, to obtain high-titer and high-quality LV stocks.
Optimization of the Downstream Purification Process for LV Production
Concentration and purification of LVs are needed to increase purity and potency while reducing toxicity for preclinical and clinical applications. To get rid of contaminating DNA (derived from plasmids and lysed producer cells) and proteins (derived from serum and producer cells), we set up a medium-scale process-development laboratory (PDL) protocol to be applied for research-level LV production by modifying and scaling down protocols previously developed for clinical purposes.16,25 The general experimental design of the PDL LV downstream process was characterized mainly by three phases: an initial capture of the vector from a clarified LV-containing supernatant that leads to elimination of major contaminants, an intermediate purification of an eluted vector that results in concentrating LV, and a final step aiming at removing trace contaminants and impurities, generating a biologically active and safe product (Figure 2A). In detail, LV-containing supernatant (~6 L) was first clarified through membrane filters of decreasing pore size (0.8 to 0.45 μm) to remove cell debris and large aggregates and then loaded to anion exchange chromatography at a constant flow rate of 7–8 mL/min (depending on the starting volume) overnight at 5°C–10°C. After washing with a low-salt concentration buffer using an amount of 2.5 column volume (CV), the vector particles bound to the column were eluted with a linear salt gradient from 0 to 1 M NaCl (100%) at 5 mL/min in a volume of 300 mL. Figure 2B shows a typical elution profile after diethylaminoethyl (DEAE) capturing. Consistently, two peaks were obtained, where the first one, containing weakly bounded proteins, appeared at about 10% of salt concentration, and the second one, containing the vector particles, appeared at 20%–25% of salt concentration (~250 mM) (Figure 2B, black line). To reduce the high-salt concentration that could negatively impact the infectivity of the vector particles, a one-to-one dilution of the LV sample with phosphate-buffered saline (PBS) was performed immediately after elution. The diluted LV was subsequently concentrated by a tangential flow filtration (TFF) system to ~10% of GF CV. Cassette and hollow fiber (100 kDa molecular weight cutoff [MWCO]) were compared. The results indicated that the performance of the two systems was comparable, since reasonably similar and high-vector recovery was obtained (Figure S2A). Benzonase treatment was performed twice, at 16 U/mL and 50 U/mL, respectively, in the presence of 2 mM MgCl2 for 4 h at 4°C, before and after the capturing step, to digest contaminant DNA. Finally, GF chromatography was employed as a polishing step to efficiently remove all contaminants smaller than the pore size of the chromatographic resin and to allow buffer exchange. Figure 2C shows a typical elution profile after GF separation, where the LV was eluted at a flow rate of 2 mL/min in a volume of ~15 mL, achieving a final ~500-fold volume concentration from the starting cell medium harvest. The majority of LV was eluted in the first two fractions (#1–2), whereas only a minor fraction (~7%) was present in the nearby fraction (#3). Vector particle quantification and SDS-PAGE analysis of collected fractions during the entire run (Figures 2C and S2B) confirmed that LV was eluted in the first ~0.3 CV. The vector was finally filtered with 0.2 μm membranes in order to eliminate the risk of microbial contamination in the final product. To determine vector recovery, fractions from different purification steps were analyzed for viral particle quantification, as well as transduction efficiency. Results indicated highly reproducible and consistent LV recovery in consecutive runs for each step (Figure 2D). The capturing step retrieved 46% ± 13% of the loaded vector (vector peak), whereas 12% ± 6% was lost during loading and washing phases (flowthrough [FT]). Only 16% ± 8% was lost in nearby fractions during DEAE separation. The remaining vector fraction lost during the purification process (lost in run = ~26%) is likely explained by incomplete elution from the DEAE column and/or vector that remains stuck in the fluidics system. After TFF concentration and polishing steps, 90% ± 19% and 88% ± 16% of the vector particles were recovered, respectively (Figure 2D). Based on several test runs, we obtained an overall process yield (overall recovery) in the range of 20% to 40% (Figure 2E). Beside the initial DEAE, the most relevant loss was experienced in the final step of filtration (Figure 2E), especially for vectors with higher titer, probably due to adsorption of the vector to the sterilizing membrane or to the presence of aggregates. Moreover, the choice of serum may impact purification yield, since distinct sera seemed to influence vector titer and filtration recovery. Although the irradiation is one of the differences among the sera tested, we have no data to ascribe that serum irradiation affects the filtration recovery or overall vector production. Additional experiments would be needed to identify which factors in the serum influence filtration recovery. Importantly, the quality of the vector was maintained during the purification workflow, since no significant drop in infectivity was observed when LV stocks were compared with LV-containing supernatants (Figure 2F).
Figure 2.

Optimization of LV Purification
(A) Overview of the purification workflow. (B) Linear gradient elution profile obtained after loading of the LV-containing supernatant on a column packed with the DEAE anion exchanger. Zoomed box shows the eluted peaks (black line, conductivity in mS/cm; green line, percentage of buffer B [PBS + 1 M NaCl]; blue and red lines, absorbance at 280 and 260 nm (mAU), respectively; on the x axis, changes in volume during elution). (C) Chromatographic profile of LV after gel filtration (GF) chromatography (blue and red lines, absorbance at 280 and 260 nm [mAU], respectively). In the box, the recovery after elution is shown as percentage of physical particles. Data are plotted as mean with SD (n ≥ 3) or as mean with range (n < 3). #1–#5 indicate the fractions collected during the GF run. (D) Physical particles calculated as percentage of recovery for different steps of the downstream process. Data are plotted as mean with SD (n = 19 for DEAE and GF, n = 16 for TFF, and n = 11 for filtration and overall recovery step). (E) Infectious titer (TU/mL, plotted on left y axis) and physical particles calculated as percentage of recovery (plotted on right x axis). In group A, only one vector was produced with non-γ-irradiated serum. Data are plotted as mean with SD (total n = 11). (F) Infectivity of the particles of the LV-containing supernatant and purified LV. Significance was assessed with a Wilcoxon matched-pairs signed-rank test. (G) Laboratory-grade (lab-grade) and purified (PDL) LV transduction efficiencies in mobilized peripheral blood (mPB)-CD34+ HSPCs measured by flow cytometry 5 days post-transduction (left) and by ddPCR 14 days post-transduction (right) (mean ± SEM; lab grade n = 3; PDL n = 3).
Characterization of Purified LVs
The optimized PDL process was applied to five LV productions, obtaining vector stocks with titers spanning from 4.8 × 108 to 3.0 × 109 TU/mL and an infectivity ranging from 1.3 × 104 to 8.3 × 104 TU per ng of HIV Gag p24 (mean = 5.2 × 104 TU/ng of p24) (Table 1). Besides assessing the LV titer and infectivity, we also characterized the vector batches in terms of various contaminants to determine product purity and safety. For this aim, vector stocks were analyzed for host (HEK293T) cell DNA and proteins, residual plasmid content, endotoxin levels, and aggregates. The downstream process led to the removal of total DNA and protein contaminants in the range of 97.6% to 99.7% (mean = 99.18%) and 99.95%, respectively (Table 1). In the final product, the protein amount was less than 0.2 mg per 108 TU, which is far below the current specification level reported for clinical-grade LV.26 Total HCPs derived from the producer cells were determined using a HEK293-specific ELISA kit, and the removal was in the range of 99.96% to 99.97% (mean = 99.97%) (Table 1). The final purified vectors contain 0.22–0.86 μg of DNA and 48.4–163.1 ng of HCP per 108 TU (Table 2), which is in line with current specification criteria for clinical-grade vectors.25 In addition, the presence of contaminating plasmid was investigated, assessing the amount of VSV.G encoding DNA. For this purpose, we developed a method based on digital droplet (dd) PCR. The linearity of quantification, the specificity of primers, and the reliability of results were assessed (Figures S2C–2CE). Specifically, the specificity was tested using the Gag-Pol packaging plasmid as a negative control (Figure S2D). To confirm the reliability of results, a known amount of VSV.G plasmid was spiked into the LV samples (positive product control [PPC]), obtaining a spike recovery in the range of 70%–130% (Figure S2E). 2.4 × 106–2.3 × 107 copies of VSV.G per 108 TU of purified vector were detected by using this method (Table 2). Similar concentrations of plasmid were previously observed in LVs employed in clinical studies.25 The purified vectors displayed lower levels of total DNA, which includes plasmid and genomic DNA, than vectors obtained using lab-grade protocol, based on the use of 150 cm2 dishes and ultracentrifugation-based concentration (Tables 2 and 3). These results confirmed that the purification process was able to increase LV purity, reducing total and plasmid DNA by 1- and 2-log-fold, respectively (Table 3). The endotoxin levels, measured using the Endosafe-PTS (Portable Test System),27 were in the range of 0.02–0.08 (endotoxin unit [EU]) for 108 TU. Moreover, particle concentration and aggregates, which could impact LV transduction, stability, and biodistribution when administered in vivo systemically, were measured. The purified LV stocks contained only a minor fraction of aggregates, as appreciated by the biphasic size distribution (Figure S2F; left panel) and the cumulative particle concentration (Figure S2F; right panel). The peak at ~430 nm in diameter accounted for less than 5% (mean = 3.8%) in comparison to the peak at ~130–140 nm in diameter (Figure S2G).
Table 1.
Summary of the Biological Activity and Quality of LV Stocks
| Result (Mean) n = 5 | |
|---|---|
| Viral titer (TU/mL) | 1.6E+09 |
| Particles (μg p24/mL) | 32.4 |
| Infectivity (TU/ng p24) | 5.2E+04 |
| Total DNA content (μg/mL) | 4.7 |
| Plasmid DNA (VSV.G) (copies/mL) | 7.9E+07 |
| Host cell protein (HCP) content (ng/mL) | 1175.7 |
| Endotoxin (EU/mL) | 0.6 |
| Volume (mL) | 12.0 |
| Concentration (fold from start volume) | 514 |
| Overall DNA reduction (%) | 99.18 |
| Overall protein reduction (%) | 99.95 |
| Overall HCP reduction (%) | 99.97 |
Table 2.
Measures of Specific Contaminant in Purified LV Stocks
| Impurity | LV Batches (n = 5) per 10E+08 TU |
|---|---|
| Total DNA content (μg) | 0.22–0.86 |
| Plasmid DNA (VSV.G) (copies) | 2.4E+06–2.3E+07 |
| HCP content (ng) | 48.4–163.1 |
| Endotoxin (EU) | 0.02–0.08 |
Data are normalized per 108 TU.
Table 3.
Measures of Specific Contaminant in Vector Produced in 150 cm2 Dishes and Concentrated by Ultracentrifugation
| Impurity | PGK.GFP LV (n = 2) per 10E+08 TU |
|---|---|
| Total DNA content (μg) | 2.0–3.4 |
| Plasmid DNA (VSV.G) (copies) | 1.2–2.0E+09 |
| HCP content (ng) | 20.1–60.7 |
| Endotoxin (EU) | – |
Values are normalized per 108 TU.
To assess the biological activity of purified LV stocks, transduction efficiencies were compared to those of a lab-grade PGK.GFP LV stock in the clinically relevant mobilized peripheral blood (mPB)-derived CD34+ HSPCs. The percentage of GFP+ cells (Figure 2G; left panel) and vector copy number (VCN) per genome (Figure 2G; right panel) was measured. Comparable transduction efficiencies were observed for purified and lab-grade vectors at nonsaturating and saturating doses (Figure 2G).
Overall, these results demonstrate that our medium-scale PDL process extensively removes contaminants from the LV stocks without compromising ex vivo transduction efficiencies in primary HSPCs. In agreement, the quality of our LV stocks was in line with what was achieved with clinical-grade purification procedures,10,15,16,28 indicating that this optimized medium-scale process produces LV stocks that are suitable for both ex vivo and in vivo applications, ensuring vector safety and efficacy.
Comparison of Cytokine Depletion in Purified versus Lab-Grade LV Stocks
The transgene proteins encoded by LVs accumulate in producer cells and their medium during LV production. Upon ultracentrifugation, some of these products might precipitate with the LV particles, thus remaining in the final LV stocks. The presence of LV-encoded transgene products becomes a limitation when molecules that can interfere with downstream applications, such as cytokines, are produced. LV-encoded cytokines present in LV stocks can negatively affect transduction efficiency as well as the survival and phenotype of the target cells. In this regard, it has been previously demonstrated that in vivo administration of LVs to mice induces a rapid and transient cytokine response that may impair transduction rates and amplify unwanted immune responses.29 Moreover, interferon (IFN)-α-exposed HSPCs display lower transduction rates and engraftment capacity than unexposed HSPCs.30 In order to assess if our purification workflow lowers the concentration of LV-encoded cytokines in the LV stocks, we produced and delivered in vivo to mice an IFN-α-encoding LV (IFN-α LV) as a case study (Figure 3A) and paradigmatic example, as IFN-α-expressing LVs have been successfully developed for tumor-targeting gene therapies.31,32
Figure 3.

Cytokine Depletion in Purified LV Stocks
(A) TU/mL expressed as 108 TU/mL of lab-grade or purified (PDL) IFN-α encoding LVs (n = 1 LV preparation [prep]). (B) IFN-α level in input, flowthrough (FT), protein-containing peak, PDL, and lab-grade LV stocks (n = 1 LV prep). (C) IFN-α level in the plasma of mice treated with lab-grade or PDL LV stocks (mean ± SEM; lab-grade vector n = 7 mice/group; PDL vector n = 9 mice/group; Mann-Whitney test). (D) Vector copy number (VCN) detected in the liver of mice 3 months after lab-grade or PDL LV delivery (mean ± SEM; lab-grade vector n = 7 mice/group; PDL vector n = 9 mice/group; Mann-Whitney test). (E and F) TU/mL expressed as 108 TU/mL of lab-grade or PDL ORFless LV (E) after LV production or (F) after adjustment before delivery to mice (n = 1 LV prep). (F) Technical triplicates are shown. (G) Volcano plot showing for each reported cytokine the adjusted (adj) p values, calculated as in (H), and the associated fold change for each cytokine in lab-grade versus PDL LV (red, p values < 0.05). (H) Time course analysis showing the expression level of the indicated cytokines in the plasma of mice treated with lab-grade or PDL LV delivered systemically (lab-grade LV, black line, n = 5 mice/group; PDL LV, violet line, n = 5 mice/group; vehicle, PBS, gray area, n = 3 mice/group; Mann-Whitney test of areas under the curve from 0 to 24 h, ∗adj p value < 0.05).
By comparing IFN-α concentration among input, FT, protein peak, and purified PDL LV stock, we observed a strong depletion of the cytokine in the latter, whereas most of it was found in the FT (Figure 3B). LV stocks obtained through the PDL workflow contained 10-fold lower IFN-α than lab-grade LVs, 0.68 ng/mL compared to 7.08 ng/mL. These data suggest that the PDL purification method strongly reduces contaminating transgene protein in LVs encoding for cytokines or other soluble proteins, thus alleviating concerns of interfering with downstream applications. To test this hypothesis, we delivered 3 × 108 TUs of either purified or lab-grade IFN-α-encoding LVs intravenously (i.v.) to 6-week-old mice. After 8 days from LV administration, we measured the concentration of IFN-α released from transduced cells in the plasma. A significantly higher concentration of IFN-α was observed in the plasma of mice treated with purified versus lab-grade LVs (Figure 3C; p < 0.01), suggesting that PDL LV yields a better functional output in terms of transgene expression at comparable administered LV doses. Similar total VCNs were observed in the liver for both groups 3 months after infusion (Figure 3D). Note that total liver VCN is not representative of the transduction efficiency in different liver cells, as previously shown.4 This result indicates that PDL-purified LVs are more efficient than nonpurified, lab-grade ones when employed to deliver immune-activating cytokines in vivo.
Purified LVs Delivered Systemically Induce Lower Cytokine Levels Than Lab-Grade Counterparts
We assessed whether vector purification impacts on the induction of the inflammatory response observed upon systemic delivery of LVs. To this aim, we employed an LV lacking any protein-coding transgene (open reading frame [ORF]less LV) to avoid transgene-related immune responses. The lab-grade ORFless LV showed higher TU concentration than the purified, PDL, counterpart (Figure 3E). We then adjusted the LV concentrations to deliver the same quantity of TUs to each experimental group (Figure 3F). We collected plasma from the treated mice and analyzed the induction of a selected panel of cytokines, including chemokines, at different time points after LV delivery. Most of the analyzed cytokines showed a trend of increased production in the lab-grade compared to the PDL LV-treated group (Figure 3G). We also analyzed the kinetics of each individual cytokine (Figures 3H and S3). Among the cytokines upregulated in the lab-grade compared to the PDL group, CCL2, CCL3, CCL4, and CXCL1 showed the highest expression 3 h after LV delivery, and their concentration in the plasma decreased at later time points. All of the analyzed cytokine levels were similar to the vehicle by day 7 postdelivery, indicating that the inflammatory event observed upon LV delivery was transient. In summary, systemic delivery of LVs induces an innate response with release of cytokines, which might impair transgene expression, reduce transduction, and cause transient toxicity. Purified LV stocks trigger less proinflammatory pathways, thus potentially limiting these adverse events that could interfere with experimental readouts.
Comparison of Ex Vivo and In Vivo Gene Editing Efficiency Using Purified and Lab-Grade LV Stocks
Precise gene editing in the context of HSPC transplantation is an attractive strategy for treating genetic diseases. However, the limited efficiency of homology-directed repair (HDR) in primitive HSPCs negatively influences the yield of corrected cells, thus affecting the feasibility of gene editing strategies for diseases in which a low yield of edited cells is not therapeutically effective. We explored how the quality of an integrase-defective LV (IDLV) donor DNA template contributes to gene editing efficiency. Lab-grade and purified IDLV stocks were tested ex vivo in human cord blood (CB)-derived CD34+ HSPCs, employing a previously optimized protocol for gene editing based on CRISPR-Cas9 and where the transduction enhancer cyclosporin H (CsH) and GSE56, a p53 inhibitor, were used to boost IDLV transduction and curb vector and editing-induced p53 signaling during the electroporation step, respectively.30,33,34 The two compounds were used in combination, since we have recently demonstrated that GSE56 alone does not increase editing efficiency but leads to an improved engraftment and higher clonality of edited cells upon transplantation in vivo.35 To this aim, after 2 days of prestimulation, HSPCs were transduced in the presence or absence of CsH, with a donor template-carrying IDLV, which harbors a GFP cassette. After 24 h post-transduction, HSPCs were electroporated with a Cas9 ribonucleoprotein (RNP) targeting the AAVS1 locus in the presence or absence of GSE56. In agreement with previous reports, delivery of the purified IDLV donor in the presence of CsH significantly increased gene editing efficiency in all CD34+ HSPC subpopulations (Figure 4A). This increase was even more pronounced in the most primitive CD34+CD133+CD90+ fraction (Figure 4A). Moreover, purified IDLV tended to reach higher editing efficiencies when compared to the lab-grade counterpart in the presence of CsH (Figure 4A). The highest rate of targeted integration by HDR was achieved using the purified IDLV donor in combination with CsH without alterations in the overall fraction of edited alleles (Figure 4B). No significant changes in the relative composition of the hematopoietic subpopulations were observed between lab-grade and purified IDLV donors in vitro, suggesting that both vectors do not affect the survival and growth of the more primitive cells (Figure 4C). A comparable p21 mRNA induction was detected at 24 h after electroporation, indicating that both IDLV stocks trigger similar p53 activation (Figure 4D; top left panel), in agreement with previous reports.34 However, purified IDLV triggered lower expression of proinflammatory genes (Figure 4D), suggesting vector purification may help dampen potentially harmful signaling induced during HSPC gene editing.
Figure 4.

CB-CD34+ Gene Editing In Vitro
(A) Percentage of edited cells at the AAVS1 locus measured within the indicated subpopulations 3 days after editing with lab-grade or purified (PDL) IDLV donor (mean ± SEM; lab-grade vector n = 3; PDL vector n = 5; Mann-Whitney test; ∗p ≤ 0.05, ∗∗p ≤ 0.01). (B) Percentage of homology-directed repair (HDR) and non-homologous end joining (NHEJ) 3 days after editing with lab-grade or purified (PDL) IDLV donor measured in vitro (mean ± SEM; lab-grade vector n = 2; PDL vector n = 5). (C) Subpopulation composition of edited human CB-CD34+ cells measured by flow cytometry 3 days after electroporation (mean ± SEM; lab grade n = 3; PDL n = 5). (D) Fold-change expression of p21 and some proinflammatory genes relative to untreated [UT] at 24 h after electroporation of CB-derived HSPCs (mean ± SEM; lab grade n = 3; PDL n = 5–4). CsH and GSE56 have been used in all experiments before IDLV transduction and during electroporation, respectively.
To assess if the improvement in gene editing efficiency was maintained in vivo, edited HSPCs (Figure S4A) were transplanted into non-obese diabetic (NOD)-severe combined immunodeficiency (SCID)-interleukin 2 receptor γ−/− (IL-2Rγ−/−) (NSG) mice and followed for their engraftment and repopulation capacity over time (Figure 5A). No difference in colony-forming capacity was observed between the lab-grade and purified IDLV donor, with similar numbers of myeloid and erythroid colonies (Figure 5B). Although we obtained comparable short- and long-term human cell engraftment (Figure 5C), a significantly higher fraction of HDR-edited cells within the human CD45+ cells was reached in PB with a purified IDLV-mediated donor delivery (Figure 5D; p < 0.05). A trend for a higher fraction of HDR-edited cells was also observed, although not statistically significant, in the bone marrow (BM) (Figure 5E) and in the myeloid, lymphoid, and progenitor cell populations from the BM (Figures 5F and 5G), in line with data obtained in the PB and consistent with the higher rates of HDR measured in the ex vivo culture. At the experimental endpoint, a tendency to higher content of edited human cells was maintained in the hematopoietic organs, especially in the spleen, of mice transplanted with HSPCs edited in the presence of purified IDLV as a donor template without any major difference in engraftment (Figure S4B). This increase was observed for all of the different cell compartments in the hematopoietic organs analyzed without major alteration in lineage composition (Figures S4C and S4D). Altogether, these data indicated that purified IDLV as a donor template enhances genome editing efficiency and diminishes the proinflammatory response in human HSPCs. In particular, a major benefit was observed in vivo where a significantly higher fraction of HDR-edited cells was engrafted, thus highlighting the importance of high-quality vectors to increase safety and efficacy of gene editing approaches.
Figure 5.

In Vivo Gene Editing
(A) Schematic representation of the gene editing protocol for human CB-derived CD34+ cells comparing lab-grade to purified (PDL) donor IDLV in the presence of CsH. (B) Number of myeloid and erythroid colony-forming units (CFUs) assessed in vitro 2 weeks after plating. (C) Percentage of human CD45+ cells in the PB of NSG mice transplanted with HSPCs edited following the experimental scheme (A) (mean ± SEM; lab grade n = 5; PDL n = 4). (D) Percentage of gene editing by HDR measured within human cells in the PB of mice over time (mean ± SEM; lab grade n = 5; PDL n = 4; Mann-Whitney test; ∗p ≤ 0.05, ∗∗p ≤ 0.01). (E) Percentage of human gene-edited cells and editing efficiency measured in the BM of mice 20 weeks post-transplantation. (F) Percentage of the indicated subpopulations measured within grafted human cells in the BM of mice. (G) Editing efficiency measured at FACS in CD34+ HSPCs, CD19+ B cells, and CD33+ myeloid cells from the BM of mice in (F), 20 weeks post-transplantation.
Discussion
We have described here the medium-scale production and purification of LVs for research-based in vivo and ex vivo gene therapy approaches and highlighted relevant benefits such as a platform provides for preclinical testing of gene therapy protocols. Concentrated, highly purified, and biologically active LV ensures a safe and efficient vehicle for gene delivery in preclinical studies. The PDL workflow that we have optimized here is similar to a large-scale protocol previously reported;16,25 however, both the upstream and the downstream phases were adapted for research-scale production. In spite of variable yield, the implemented process can reproducibly produce a purified and biologically active LV with a final titer in the order of 109 TU/mL and high infectivity (~5 × 104 TU/ng p24). An overall recovery of 20%–40% was usually achieved, in line with published clinical-grade methods in which multistep protocols for purification and concentration are used.16,28 PDL-purified LVs transduced to a similar extent as lab-grade counterparts the poorly permissive HSPCs that require high-concentration and high-infectivity stocks for robust gene transfer, as observed in LV-based clinical trials.25,36 Infectivity of our LV stocks is also compatible with a targeted genome editing procedure applied in preclinical studies, where clinical-grade purified vectors are employed.37 In addition, our protocol significantly lowered the levels of residual contaminants from cellular, medium, or plasmid origin, which in the context of LV-based gene therapy, could exacerbated innate immune sensing.38 Indeed, DNA could activate innate immune responses that can be potentially harmful to HSPCs, leading to exhaustion and functional deficits after long-term activation.39 Additionally, reduction of plasmid DNA as a residual VSV.G plasmid16 in the LV stock could prevent the risk of transferring plasmid DNA to transduced cells.40 In agreement, purified LVs performed better compared to lab-grade counterparts in the context of gene editing with a diminished activation of inflammatory responses and an increase in edited cells, with a consistently higher engraftment of engineered cells in vivo. The engraftment capacity of HSPCs has been shown to be affected by activation of the p53-mediated DNA damage response (DDR) by gene therapy vectors.34 Of note, activation of the p53-mediated DDRs was not different between lab-grade and purified vectors, in line with previous work showing that both clinical-grade and lab-grade LV activate this pathway to a similar extent.34 In agreement, no differences in engraftment capacity were observed. Interestingly, purified LV yielded a higher frequency of edited HSPCs in vivo. This could be related to a lower induction of inflammatory pathways that potentially affect HDR or survival of edited cells. Indeed, reduction in contaminants, such as plasmid DNA in purified LV stocks, may prevent detection by cytosolic DNA sensors and consequent activation of proinflammatory pathways.41, 42, 43 Moreover, increasing evidence suggests that an active interplay between inflammatory pathways and DNA repair machinery exists, as cells that undergo DNA damage can activate innate immune signaling by noncanonical pathways involving IFI16, STING,42,43 and nuclear cGAS, which has been reported to play a role in DNA repair and tumorigenesis.44
The benefits of using purified LV in research-grade preclinical studies were also highlighted by the diminished content of immune-activating toxic proteins (e.g., cytokines) in purified compared to lab-grade LV stocks, avoiding possible acute immune responses and tissue damage. Of note, a purified LV encoding for IFN-α delivered systemically drove transgene expression to higher levels than the same ultracentrifuged LV. This observation suggests that IFN-α, as other molecules that may trigger it, such as plasmids and serum-derived contaminants in the ultracentrifuged LV stocks, may negatively impact transgene expression thus limiting the efficacy of systemically delivered LVs. Additionally, reduction of serum proteins in the purified LVs may lower the load of immunogens,7 which negatively influence the outcome of gene transfer treatments, in particular, in the context of in vivo LV delivery.45 Our study also suggests that a different abundance of specific serum proteins might compete with vector capturing during chromatographic separation or coelute with vector particles, increasing variability and interfering with the purity and yield of vector stocks. Based on this observation, employment of serum-free media for LV production may be an attractive option,46, 47, 48 also for safety reasons. In fact, serum might be a source of animal-derived contaminants in the harvested supernatant during LV production.
It has been reported that LVs delivered systemically can induce cytokines in NHPs.4 We found that cytokine expression can also be observed in mice upon systemic LV delivery, even in the presence of a construct lacking a transgene-encoding cassette. Chemokines were immediately upregulated after LV delivery, whereas other cytokines, such as IL-10 or IL-12p40, were upregulated in a second phase (6 h), likely as a consequence of the recruitment and activation of inflammatory cells. Importantly, purified LV triggered lower cytokine induction than the nonpurified counterpart, suggesting that nonpurified LVs trigger inflammatory responses that may confound the readouts of experiments employing LVs systemically. Indeed, LV-induced inflammation may lead to less-reproducible experiments and erroneous interpretations of results. Therefore, the usage of purified LVs should be preferred for systemic LV delivery to curb cytokine production and induction of inflammation in the recipient.
Altogether, the reduction of contaminants in LV stocks achieved by the workflow described here is similar and in some cases, even better compared to values reported for clinical-grade vector preparations.15,16,25 This ensures high-quality vectors similar to those required in a clinical setting, thus allowing faithful modeling of expected outcomes in preclinical studies. Further efforts will be focused on optimizing the filtration step and increasing the pore size or the area of filtrating membrane to reduce the loss of LV while maintaining high infectivity. In addition, strategies, such as affinity-based isolation of vector particles, could further increase LV purification yield, as recently described for the capture of cTag8-expressing viral particles.49 Finally, this platform could be further developed toward a more versatile workflow, compatible also with other transfection protocols (e.g., polyethylenimine) and viral vectors.
Overall, our work provides a comprehensive description of how to set up a medium-scale LV production and purification pipeline for research-grade preclinical testing and highlights the advantages that such vectors provide over standard lab-grade preparations in preclinical gene therapy studies.
Materials and Methods
Cells and Culture Media
HEK293T were grown in Iscove’s modified Dulbecco’s medium (IMDM) without (w/o) phenol red (Corning) supplemented with 10% (v/v) fetal bovine serum (FBS; Carlo Erba) and 1% penicillin-streptomycin (Pen/Strep) (Lonza). Cell concentration was assessed by counting cells with trypan blue after cell detachment. Human CD34+ HSPCs were isolated through positive magnetic bead selection, according to the manufacturer’s instructions (Miltenyi) from umbilical CB collected upon informed consent from healthy volunteers, according to the Institutional Ethical Committee-approved protocol (TIGET01). All cells were maintained in a 5% CO2 humidified atmosphere at 37°C.
Plasmid
The HIV-derived LV particles were produced using a different type of transfer plasmid, third-generation packaging plasmids pGag-Pol and pRSV-REV, and the envelope protein plasmid encoding for VSV.G. In addition, the pAdVAntage plasmid was used. All plasmids were bought from Nature Technology. For IDLV production for gene editing, the packaging plasmid pMDLg/pRRE was substituted with pMD.Lg/pRRE.D64VInt,50 and a transfer plasmid containing homologies for the AAVS1 locus and comprising a PGK.GFP reporter cassette51 was used.
LV Production
The lab-grade LVs were produced by calcium phosphate transient transfection as previously described.4 For CF10 (Corning), HEK293T cells were seeded in CF10 at a concentration of 1.08 × 104 cells/cm2 in IMDM without phenol red containing 10% FBS and 1% Pen/Strep (day 0). Medium was changed 3–4 h before transfection (day 3), and cells were transfected by a calcium phosphate method. After 14–16 h post-transfection, the medium was replaced with a fresh one, supplemented with sodium butyrate at 1 mM final concentration. Supernatant was harvested 30 h after medium exchange. The LV-containing supernatant was filtered and clarified through 5 μm and 0.8–0.45 μm filters (Sartorius), respectively, and collected into a 5- to 10-L bag. The supernatant was treated with Benzonase (Merck) at a final concentration of 16 U/mL for 4 h at 4°C prior to anion exchange chromatography.
Capturing of LV by Anion Exchange Chromatography
Lentivirus particles were captured by anion exchange chromatography, using DEAE-650C resin (Tosoh Bioscience). A XK26/40 column (GE Healthcare) was packed under flow using AKTA Avant 150 (GE Healthcare) at 25 mL/min (bed height = 35–40 cm). Once the packing was completed, the efficiency test was performed, evaluating the number of theoretical plates per meter packed bed (N/m) and the asymmetry factor (As). After column equilibration with PBS (Corning), clarified and digested supernatant was loaded overnight (O/N) in the refrigerated cart at constant flow of 7–8 mL/min. After linear gradient elution (0% to 100% Buffer B) at a constant flow of 5 ± 0.5 mL/min with PBS containing 1 M NaCl (100% Buffer B), the vector-containing eluate was immediately diluted 1 to 1 with PBS. Finally, the column was washed with PBS containing 1 M NaCl to regenerate the column and to remove stronger bounded impurities. The column was kept at 10°C through a refrigerator thermostatic bath during the run. Before and after each run, a sanitization step was performed, employing HCl 0.1 M, NaOH 0.5 M, and NaOH 0.1 M (storage buffer). The column was stored at 4°C to prevent microbial growth until further runs.
LV Concentration
The eluted vector was further digested with Benzonase at the final concentration of 50 U/mL for 2 h at 4°C and in the meantime, concentrated approximately 30- to 40-fold by TFF using a hollow fiber cartridge (GE Healthcare) or VivaFlow cassette (Sartorius) of 100 kDa MWCO and 50 cm2 surface area. The VivaFlow cassette was previously washed with water and equilibrated with PBS, and the concentration was performed setting the feed pressure indicator to 1–1.5 bar that corresponds to a permeate flow rate of approximately 5 mL/min. A hollow fiber was washed and equilibrated as the VivaFlow cassette, and the concentration was instead performed with the following parameters: feed flow = 75 mL/min, transmembrane pressure (TMP) = 0.3 bar, flux LMH = 30–35, and the permeate flow rate = approximately 3 mL/min.
Polishing of LV by GF and LV Storage
GF was performed using Sepharose 6FF resin (GE Healthcare). The XK16/70 column (GE Healthcare) was packed under flow using AKTA Avant 150 at 10 mL/min (bed height = 60–65 cm). Once the packing was completed, the efficiency test was performed, evaluating the N/m and the As. After column equilibration with PBS, concentrated vector was loaded and eluted at constant flow of 2 mL/min using PBS. The column was kept at 10°C through a refrigerator thermostatic bath during the run. Before and after each run, a sanitization step was performed, employing NaOH 1.0 M and NaOH 0.1 M (storage buffer). The column was stored at 4°C to prevent microbial growth until further runs.
The final LV stocks were subjected to sterile microfiltration using a 0.2-μm polyethersulfone (PES) filter (Sartorius), aliquoted, and stored at −80°C until further analysis.
LV Titration and p24
For the LV titer, 100,000 HEK293T cells were transduced with serial dilutions in the presence of polybrene (8 μg/mL). For LV-GFP, cells were analyzed by flow cytometry 5 days after transduction, and the infection titer, expressed as TU/mL, was calculated using the formula TU/mL = ([%GFP+ cells/100] × 100,000 × [1/dilution factor]). For all other LVs, genomic DNA was extracted 10–14 days after transduction, using the Maxwell 16 Cell DNA Purification Kit (Promega), following the manufacturer’s instructions. VCN was determined by ddPCR, starting from 15 ng of template DNA using primers (HIV Fw: 5′-TACTGACGCTCTCGCACC-3′; HIV Rv: 5′-TCTCGACGCAGGACTCG-3′) and a probe (FAM 5′-ATCTCTCTCCTTCTAGCCTC-3′) against the primer binding-site region of LV. The amount of endogenous human DNA was quantified by a primer/probe set against the TAF7 gene (commercially available: Thermo Fisher Scientific; Rh02916247_s1). The PCR was performed with each primer (900 nM) and the probe (250 nM) following the manufacturer’s instructions (Bio-Rad), read with a QX200 reader, and analyzed with QuantaSoft software (Bio-Rad). The infectious titer was expressed as TU/mL and calculated as VCN × number of cells (100,000) × (1/dilution factor). Specifically, VCN = (copies of genome-integrated LV/copies of genomic DNA, TAF7) × 2 (HEK293T considered as diploid cells). As a positive control, a CEM cell line stably carrying four vector integrants (VCN = 4) was used. LV physical particles were measured by HIV-1 Gag 24 antigen immunocapture assay (PerkinElmer) following the manufacturer’s instructions. LV-specific infectivity was calculated as the ratio between the infectious titer and physical particles. LV particle concentration and size were measured by using multiangle dynamic light scattering (MADLS) technology using Zetasizer Ultra (Malvern Panalytical) following the manufacturer’s instructions.
Protein and DNA Contaminant Test
Total protein content was measured using the Bradford protein colorimetric assay (Bio-Rad). Total DNA content was measured with Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen) using a high-range standard curve from 1 μg to 1 ng of DNA/mL. The total HCP level was measured by a HEK293 HCP ELISA assay kit with a LOQ (limit of quantitation) of ~2 ng/mL and 200 ng/mL as threshold (Cygnus Technologies, Southport, NC). As PPC, the HCP standard has been spiked into each LV sample at 50 ng/mL final concentration. Due to the type of nonlinear curve describing the relationship between concentration and absorbance values, the curve was modeled as absorbance = a × concentration/(b + concentration), and it was estimated with the nonlinear regression implemented in the function nlsLM of the R package minpack.lm. The absorbance (λ = 450 nm) values of standards (after “blank” subtraction [samples diluent]) were used to estimate a reference curve (Figure S2H). The concentrations of the samples were then retrieved from the estimated reference curve by using their absorbance values after the subtraction of the “blank value.” The results were considered reliable when the spike recovery was in the range of 70%–130%. For each test, LV samples, the standard curve, and controls were analyzed in duplicate, obtaining a CV% less than 10%. The IFN-α level in the LV stocks and intermediate states of the purification was quantified by ELISA assay using the IFN-α high-sensitivity kit from PBL Assay Science (catalog number: 42115-1) following the manufacturer’s instructions. Samples were measured in technical duplicates in the following dilutions: 1/50, 1/250, and 1/1,250.
Endotoxin Test
The endotoxin level was determined by the Endosafe PTS system using a single use cartridge with sensitivity of 0.005–0.5 EU/mL. Each cartridge consists of two sample channels and two spiked channels. Each reservoir contains a specific amount of Limulus amebocyte lysate (LAL) reagent, synthetic chromogenic substrate, control standard endotoxin (CSE), and buffer uniformly imbedded in the cartridge. Acceptance criteria for the test were the following: samples and spike in CV% between replicates less than 20% and spike recovery in the range of 50%–200%.
Residual Plasmid Quantification
VSV.G DNA copies were determined by ddPCR on LV stocks using primer/probe sets against the VSV.G DNA (Fw: 5′-CAGTCCATCCGATCCTTCAC-3′, Rv: 5′-TCCGTCACAGTTGCATATCC-3′, and probe: FAM 5′-GCTGAATCCAGGCTTCCCTCCT-3′). The PCR was performed with each primer (900 nM) and the probe (250 nM) following the manufacturer’s instructions (Bio-Rad), read with the QX200 reader and analyzed with QuantaSoft software (Bio-Rad). LV stocks heated at 56°C for 45 min were used. As PPC, ~6,000 molecules of the VSV.G plasmid were spiked into each LV sample. LV samples and controls were analyzed in duplicate, obtaining a CV% less than 10%. The results were considered reliable when the spike recovery was in the range of 70%–130%.
In Vivo LV Delivery and Cytokine Quantification
LVs were delivered i.v. through tail-vein injection to 5-week-old female C57BL/6 mice (purchased from Charles River). A dose of 3 × 108 TU/mice solved in PBS was used in all of the experiments. Plasma was isolated by centrifugating the blood samples in heparin-coated tubes at 850 g × 10 min and collecting the supernatant. IFN-α levels in the plasma were quantified by ELISA assay using the IFN-α high-sensitivity kit from PBL Assay Science (catalog number: 42115-1) following the manufacturer’s instruction. The concentrations of cytokines and chemokines in mouse plasma were determined by a magnetic beads-based multiplex immunoassay for 24 analytes (Bio-Plex mouse 23-Plex, group I; mixed with single-plex mouse IL-18, group II; Bio-Rad), following the manufacturer’s instructions and acquired by MAGPIX Multiplex Reader (Luminex). All animal procedures were performed according to protocols approved by the Animal Care and Use Committee of the Ospedale San Raffaele (Institutional Animal Care and Use Committee [IACUC] 1098) and communicated to the Ministry of Health and local authorities according to the Italian law.
HSPC Transduction and Gene Editing
For transduction, human mPB-derived HSPCs were maintained in CellGro medium (CellGenix) containing a cocktail of cytokines: 60 ng/mL IL-3, 100 ng/mL thrombopoietin (TPO), 300 ng/mL stem cell factor (SCF), and 300 ng/mL FLT-3L (all from PeproTech). Cells were then transduced with the indicated dose of vectors for 14–15 h in the same cytokine-containing medium. After transduction, cells were washed and maintained in serum-free medium, supplemented with cytokines, as above, until the reading of the percentage of positive cells by fluorescence-activated cell sorting (FACS), after which, they were maintained in IMDM, supplemented with 10% FBS, 25 ng/mL recombinant human (rh)SCF, 5 ng/mL rhIL-6, 25 ng/mL rhFlt-3, and 5 ng/mL rhTPO for another 7 days before analysis of VCNs. For gene editing, human CB-derived HSPCs were cultured in serum-free StemSpan medium (STEMCELL Technologies), supplemented with Pen (100 IU/mL), Strep (100 μg/mL), 100 ng/mL rhSCF, 20 ng/mL rhTPO, 100 ng/mL rhFlt-3 ligand, and 20 ng/mL rhIL-6 (all from PeproTech) 16 to 24 h prior to transduction. For gene editing experiments in human HSPC, 106 CD34+ cells/mL were stimulated in serum-free StemSpan medium (STEMCELL Technologies), supplemented with Pen, Strep, glutamine, 1 μM SR-1 (BioVision Technologies), 50 μM UM171 (STEMCELL Technologies), 10 μM PGE2 added only at the beginning of the culture (Cayman Chemical), and human early-acting cytokines (SCF 100 ng/mL, Flt-3L 100 ng/mL, TPO 20 ng/mL, and IL-6 20 ng/mL; all purchased from PeproTech).37 Transduction with an IDLV containing homology arms for the AAVS1 locus and comprising a PGK.GFP reporter cassette51 was performed at MOI 100, after 2 days of prestimulation with early-acting cytokines. After 24 h from IDLV transduction, cells were washed with PBS and electroporated (P3 Primary Cell 4D-Nucleofector X Kit, program EO-100; Lonza) with 1.25 μM of RNPs. RNPs were assembled by incubating at 1:1.5 molar ratio spCas9 protein (Integrated DNA Technologies) with synthetic cr:tracrRNA (Integrated DNA Technologies) for 10 min at 25°C. Electroporation enhancer (Integrated DNA Technologies) was added prior to electroporation, according to the manufacturer’s instructions. Genomic sequences recognized by the guide RNAs (gRNAs) are the following: 5′-TCACCAATCCTGTCCCTAGtgg-3′ for AAVS1 locus. Gene editing efficiency was measured from cultured cells in vitro 3 days after electroporation. For AAVS1 edited cells, editing by HDR was quantified by flow cytometry measuring the percentage of cells expressing the GFP marker.
Colony-Forming Cell (CFC) Assay and NSG Mice
CFC assays were performed by plating 8 × 102 human HSPCs transduced in the presence of the different compounds in a methylcellulose-based medium (Methocult GF4434; STEMCELL Technologies). 15 days later, colonies were scored by light microscopy for colony numbers and morphology as erythroid or myeloid and were collected both as a pool and picked as a single colony and lysed for molecular analysis to evaluate transduction efficiencies with clinical-grade LVs. Female NSG mice were purchased from Jackson Laboratory. All animal procedures were performed according to protocols approved by the Animal Care and Use Committee of the Ospedale San Raffaele (Institutional Animal Care and Use Committee [IACUC] 782) and communicated to the Ministry of Health and local authorities according to Italian law. After gene editing, 2–5 × 105 cells were infused into the tail vein of sublethally irradiated 8- to 10-week-old NSG mice (radiation dose: 200 cGy for mice weighing 18–25 g and 220 cGy for mice above 25 g of weight).
Statistical Analysis
Statistical analyses were performed after consulting with professional statisticians at the San Raffaele University Center for Statistics in the Biomedical Sciences (CUSSB). When normality assumptions were not possible to verify, nonparametric statistical tests were preferred. When analyzing cytokines over time, the area under the curve from time point 0 to 24 h was calculated and used to perform statistical analysis, as indicated in figure legends. Number and type of measurements, p values, and statistical tests are indicated for each figure.
Author Contributions
M.S. optimized and performed the experiments (downstream LV purification and quality controls) with help of L.S.S. (upstream LV production), analyzed the data, and wrote the manuscript. G.U. performed gene editing experiments. T.K. and A.A. generated and analyzed the data on IFN-α LV and cytokine concentration in plasma. I.C. helped L.S.S. in LV production and provided technical support. P.C. and M.B. participated at the initial phase of downstream set-up. P.M.V.R. supported statistical analysis. A.L. and A.C. provided critical input. L.N. provided intellectual and critical input and edited the manuscript. M.L.S. contributed to writing the manuscript, analyzed the data, and helped in the supervision of the study, and A.K.-R. supervised the study, analyzed the data, and wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We would like to thank Annapaola Andolfo and Cinzia Magagnotti from the ProMeFa facility (IRCCS San Raffaele Scientific Institute, Milan) for MS data acquisition; Luigi Gianolli for giving us the authorization to use the MCS instrument at the Department of Nuclear Medicine, San Raffaele Hospital; Maria Grazia Minotti and Cristina Monterisi for help with MCS usage; Tiziana Plati for help in setting the VSV.G ddPCR quantification; and Margherita Neri, Francesca Bellintani, and Simona La Seta Catamancio (MolMed S.p.A.) for helpful exchanges to set up the PDL workflow. This work was supported by the European Research Council (ERC-CoG 819815-ImmunoStem) and the Telethon Foundation (TELE20-C3) to AKR.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.10.009.
Supplemental Information
References
- 1.Naldini L., Blömer U., Gage F.H., Trono D., Verma I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naldini L., Blömer U., Gallay P., Ory D., Mulligan R., Gage F.H., Verma I.M., Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 3.Naldini L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol. Med. 2019;11:e9958. doi: 10.15252/emmm.201809958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milani M., Annoni A., Moalli F., Liu T., Cesana D., Calabria A., Bartolaccini S., Biffi M., Russo F., Visigalli I. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci. Transl. Med. 2019;11:eaav7325. doi: 10.1126/scitranslmed.aav7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamada K., McCarty D.M., Madden V.J., Walsh C.E. Lentivirus vector purification using anion exchange HPLC leads to improved gene transfer. Biotechniques. 2003;34:1074–1078. doi: 10.2144/03345dd04. 1080. [DOI] [PubMed] [Google Scholar]
- 6.Baekelandt V., Claeys A., Eggermont K., Lauwers E., De Strooper B., Nuttin B., Debyser Z. Characterization of lentiviral vector-mediated gene transfer in adult mouse brain. Hum. Gene Ther. 2002;13:841–853. doi: 10.1089/10430340252899019. [DOI] [PubMed] [Google Scholar]
- 7.Baekelandt V., Eggermont K., Michiels M., Nuttin B., Debyser Z. Optimized lentiviral vector production and purification procedure prevents immune response after transduction of mouse brain. Gene Ther. 2003;10:1933–1940. doi: 10.1038/sj.gt.3302094. [DOI] [PubMed] [Google Scholar]
- 8.Scherr M., Battmer K., Eder M., Schüle S., Hohenberg H., Ganser A., Grez M., Blömer U. Efficient gene transfer into the CNS by lentiviral vectors purified by anion exchange chromatography. Gene Ther. 2002;9:1708–1714. doi: 10.1038/sj.gt.3301848. [DOI] [PubMed] [Google Scholar]
- 9.Segura M.M., Kamen A., Garnier A. Downstream processing of oncoretroviral and lentiviral gene therapy vectors. Biotechnol. Adv. 2006;24:321–337. doi: 10.1016/j.biotechadv.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Schweizer M., Merten O.W. Large-scale production means for the manufacturing of lentiviral vectors. Curr. Gene Ther. 2010;10:474–486. doi: 10.2174/156652310793797748. [DOI] [PubMed] [Google Scholar]
- 11.McCarron A., Donnelley M., McIntyre C., Parsons D. Challenges of up-scaling lentivirus production and processing. J. Biotechnol. 2016;240:23–30. doi: 10.1016/j.jbiotec.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 12.Wolf M.W., Reichl U. Downstream processing of cell culture-derived virus particles. Expert Rev. Vaccines. 2011;10:1451–1475. doi: 10.1586/erv.11.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segura M.M., Mangion M., Gaillet B., Garnier A. New developments in lentiviral vector design, production and purification. Expert Opin. Biol. Ther. 2013;13:987–1011. doi: 10.1517/14712598.2013.779249. [DOI] [PubMed] [Google Scholar]
- 14.Nestola P., Peixoto C., Silva R.R., Alves P.M., Mota J.P., Carrondo M.J. Improved virus purification processes for vaccines and gene therapy. Biotechnol. Bioeng. 2015;112:843–857. doi: 10.1002/bit.25545. [DOI] [PubMed] [Google Scholar]
- 15.Merten O.W., Hebben M., Bovolenta C. Production of lentiviral vectors. Mol. Ther. Methods Clin. Dev. 2016;3:16017. doi: 10.1038/mtm.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merten O.W., Charrier S., Laroudie N., Fauchille S., Dugué C., Jenny C., Audit M., Zanta-Boussif M.A., Chautard H., Radrizzani M. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum. Gene Ther. 2011;22:343–356. doi: 10.1089/hum.2010.060. [DOI] [PubMed] [Google Scholar]
- 17.Soneoka Y., Cannon P.M., Ramsdale E.E., Griffiths J.C., Romano G., Kingsman S.M., Kingsman A.J. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 1995;23:628–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laughlin M.A., Chang G.Y., Oakes J.W., Gonzalez-Scarano F., Pomerantz R.J. Sodium butyrate stimulation of HIV-1 gene expression: a novel mechanism of induction independent of NF-kappa B. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995;9:332–339. [PubMed] [Google Scholar]
- 19.Gasmi M., Glynn J., Jin M.J., Jolly D.J., Yee J.K., Chen S.T. Requirements for efficient production and transduction of human immunodeficiency virus type 1-based vectors. J. Virol. 1999;73:1828–1834. doi: 10.1128/jvi.73.3.1828-1834.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karolewski B.A., Watson D.J., Parente M.K., Wolfe J.H. Comparison of transfection conditions for a lentivirus vector produced in large volumes. Hum. Gene Ther. 2003;14:1287–1296. doi: 10.1089/104303403322319372. [DOI] [PubMed] [Google Scholar]
- 21.Sena-Esteves M., Tebbets J.C., Steffens S., Crombleholme T., Flake A.W. Optimized large-scale production of high titer lentivirus vector pseudotypes. J. Virol. Methods. 2004;122:131–139. doi: 10.1016/j.jviromet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 22.Pernod G., Fish R., Liu J.W., Kruithof E.K. Increasing lentiviral vector titer using inhibitors of protein kinase R. Biotechniques. 2004;36:576–578. doi: 10.2144/04364BM02. 580. [DOI] [PubMed] [Google Scholar]
- 23.Follenzi A., Naldini L. HIV-based vectors. Preparation and use. Methods Mol. Med. 2002;69:259–274. [PubMed] [Google Scholar]
- 24.Powers J.M., Trobridge G.D. Effect of fetal bovine serum on foamy and lentiviral vector production. Hum. Gene Ther. Methods. 2013;24:307–309. doi: 10.1089/hgtb.2013.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 26.Dropulic B., Schonely K., Slepushkin V., Lu X., Andre K., Boehmer J., Bengtson K., Doub M., Cohen R., Berlinger D. QC Release Testing of an HIV-1 Based Lentiviral Vector Lot and Transduced Cellular Product. Bioprocessing J. 2003;2:39–47. [Google Scholar]
- 27.Gee A.P., Sumstad D., Stanson J., Watson P., Proctor J., Kadidlo D., Koch E., Sprague J., Wood D., Styers D. A multicenter comparison study between the Endosafe PTS rapid-release testing system and traditional methods for detecting endotoxin in cell-therapy products. Cytotherapy. 2008;10:427–435. doi: 10.1080/14653240802075476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bandeira V., Peixoto C., Rodrigues A.F., Cruz P.E., Alves P.M., Coroadinha A.S., Carrondo M.J. Downstream processing of lentiviral vectors: releasing bottlenecks. Hum. Gene Ther. Methods. 2012;23:255–263. doi: 10.1089/hgtb.2012.059. [DOI] [PubMed] [Google Scholar]
- 29.Brown B.D., Sitia G., Annoni A., Hauben E., Sergi L.S., Zingale A., Roncarolo M.G., Guidotti L.G., Naldini L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood. 2007;109:2797–2805. doi: 10.1182/blood-2006-10-049312. [DOI] [PubMed] [Google Scholar]
- 30.Petrillo C., Thorne L.G., Unali G., Schiroli G., Giordano A.M.S., Piras F., Cuccovillo I., Petit S.J., Ahsan F., Noursadeghi M. Cyclosporine H Overcomes Innate Immune Restrictions to Improve Lentiviral Transduction and Gene Editing In Human Hematopoietic Stem Cells. Cell Stem Cell. 2018;23:820–832.e9. doi: 10.1016/j.stem.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Escobar G., Barbarossa L., Barbiera G., Norelli M., Genua M., Ranghetti A., Plati T., Camisa B., Brombin C., Cittaro D. Interferon gene therapy reprograms the leukemia microenvironment inducing protective immunity to multiple tumor antigens. Nat. Commun. 2018;9:2896. doi: 10.1038/s41467-018-05315-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Escobar G., Moi D., Ranghetti A., Ozkal-Baydin P., Squadrito M.L., Kajaste-Rudnitski A., Bondanza A., Gentner B., De Palma M., Mazzieri R., Naldini L. Genetic engineering of hematopoiesis for targeted IFN-α delivery inhibits breast cancer progression. Sci. Transl. Med. 2014;6:217ra3. doi: 10.1126/scitranslmed.3006353. [DOI] [PubMed] [Google Scholar]
- 33.Schiroli G., Conti A., Ferrari S., Della Volpe L., Jacob A., Albano L., Beretta S., Calabria A., Vavassori V., Gasparini P. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell. 2019;24:551–565.e8. doi: 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piras F., Riba M., Petrillo C., Lazarevic D., Cuccovillo I., Bartolaccini S., Stupka E., Gentner B., Cittaro D., Naldini L., Kajaste-Rudnitski A. Lentiviral vectors escape innate sensing but trigger p53 in human hematopoietic stem and progenitor cells. EMBO Mol. Med. 2017;9:1198–1211. doi: 10.15252/emmm.201707922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrari S., Jacob A., Beretta S., Unali G., Albano L., Vavassori V., Cittaro D., Lazarevic D., Brombin C., Cugnata F. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0551-y. Published online June 29, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schiroli G., Ferrari S., Conway A., Jacob A., Capo V., Albano L., Plati T., Castiello M.C., Sanvito F., Gennery A.R. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017;9:eaan0820. doi: 10.1126/scitranslmed.aan0820. [DOI] [PubMed] [Google Scholar]
- 38.Kajaste-Rudnitski A., Naldini L. Cellular innate immunity and restriction of viral infection: implications for lentiviral gene therapy in human hematopoietic cells. Hum. Gene Ther. 2015;26:201–209. doi: 10.1089/hum.2015.036. [DOI] [PubMed] [Google Scholar]
- 39.King K.Y., Goodell M.A. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011;11:685–692. doi: 10.1038/nri3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sastry L., Xu Y., Cooper R., Pollok K., Cornetta K. Evaluation of plasmid DNA removal from lentiviral vectors by benzonase treatment. Hum. Gene Ther. 2004;15:221–226. doi: 10.1089/104303404772680029. [DOI] [PubMed] [Google Scholar]
- 41.Wu J., Sun L., Chen X., Du F., Shi H., Chen C., Chen Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Unterholzner L., Dunphy G. cGAS-independent STING activation in response to DNA damage. Mol. Cell. Oncol. 2019;6:1558682. doi: 10.1080/23723556.2018.1558682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunphy G., Flannery S.M., Almine J.F., Connolly D.J., Paulus C., Jonsson K.L., Jakobsen M.R., Nevels M.M., Bowie A.G., Unterholzner L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol. Cell. 2018;71:745–760.e5. doi: 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu H., Zhang H., Wu X., Ma D., Wu J., Wang L., Jiang Y., Fei Y., Zhu C., Tan R. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. 2018;563:131–136. doi: 10.1038/s41586-018-0629-6. [DOI] [PubMed] [Google Scholar]
- 45.Tuschong L., Soenen S.L., Blaese R.M., Candotti F., Muul L.M. Immune response to fetal calf serum by two adenosine deaminase-deficient patients after T cell gene therapy. Hum. Gene Ther. 2002;13:1605–1610. doi: 10.1089/10430340260201699. [DOI] [PubMed] [Google Scholar]
- 46.Bauler M., Roberts J.K., Wu C.C., Fan B., Ferrara F., Yip B.H., Diao S., Kim Y.I., Moore J., Zhou S. Production of Lentiviral Vectors Using Suspension Cells Grown in Serum-free Media. Mol. Ther. Methods Clin. Dev. 2019;17:58–68. doi: 10.1016/j.omtm.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Segura M.M., Garnier A., Durocher Y., Ansorge S., Kamen A. New protocol for lentiviral vector mass production. Methods Mol. Biol. 2010;614:39–52. doi: 10.1007/978-1-60761-533-0_2. [DOI] [PubMed] [Google Scholar]
- 48.Ansorge S., Lanthier S., Transfiguracion J., Durocher Y., Henry O., Kamen A. Development of a scalable process for high-yield lentiviral vector production by transient transfection of HEK293 suspension cultures. J. Gene Med. 2009;11:868–876. doi: 10.1002/jgm.1370. [DOI] [PubMed] [Google Scholar]
- 49.Mekkaoui L., Parekh F., Kotsopoulou E., Darling D., Dickson G., Cheung G.W., Chan L., MacLellan-Gibson K., Mattiuzzo G., Farzaneh F. Lentiviral Vector Purification Using Genetically Encoded Biotin Mimic in Packaging Cell. Mol. Ther. Methods Clin. Dev. 2018;11:155–165. doi: 10.1016/j.omtm.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lombardo A., Genovese P., Beausejour C.M., Colleoni S., Lee Y.L., Kim K.A., Ando D., Urnov F.D., Galli C., Gregory P.D. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 51.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.