

Summary
Regulation of glucose homeostasis is a fundamental process to maintain blood glucose at a physiological level, and its dysregulation is associated with the development of several metabolic diseases. Here, we report on a zebrafish mutant for Aldo-keto-reductase 1a1b (akr1a1b) as a regulator of gluconeogenesis. Adult akr1a1b−/− mutant zebrafish developed fasting hypoglycemia, which was caused by inhibiting phosphoenolpyruvate carboxykinase (PEPCK) expression as rate-limiting enzyme of gluconeogenesis. Subsequently, glucogenic amino acid glutamate as substrate for gluconeogenesis accumulated in the kidneys, but not in livers, and induced structural and functional pronephros alterations in 48-hpf akr1a1b−/− embryos. Akr1a1b−/− mutants displayed increased nitrosative stress as indicated by increased nitrotyrosine, and increased protein-S-nitrosylation. Inhibition of nitrosative stress using the NO synthase inhibitor L-NAME prevented kidney damage and normalized PEPCK expression in akr1a1b−/− mutants. Thus, the data have identified Akr1a1b as a regulator of gluconeogenesis in zebrafish and thereby controlling glucose homeostasis.
Subject Areas: Human Metabolism, Molecular Genetics
Graphical Abstract
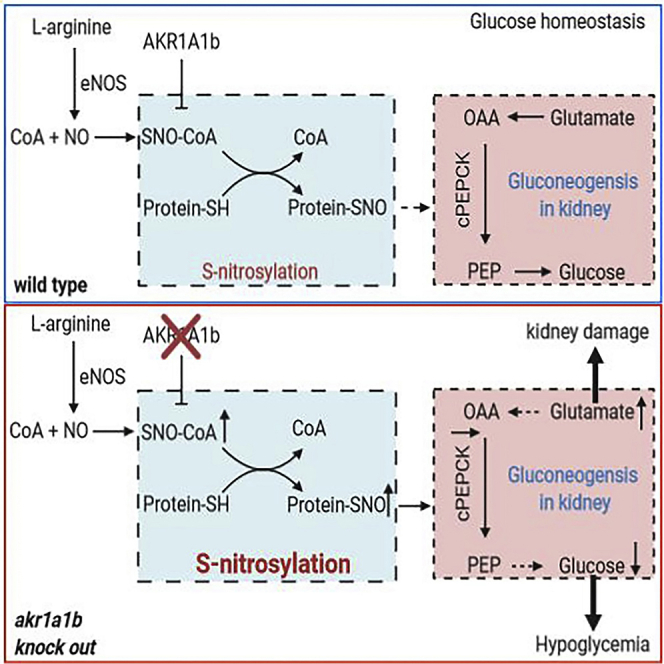
Highlights
-
•
Adult akr1a1b−/− mutant zebrafish develop fasting hypoglycemia
-
•
Loss of Akr1a1b inhibits renal phosphoenolpyruvate carboxykinase (PEPCK) expression
-
•
Accumulation of glucogenic amino acid glutamate alters the kidney in akr1a1b mutants
-
•
Akr1a1b regulates gluconeogenesis via protein-S-nitrosylation
Human Metabolism; Molecular Genetics
Introduction
Regulation of glucose homeostasis is a fundamental process to maintain blood glucose at a physiological level. Glucose is a major energy substrate for several organs, which includes but is not limited to the brain and red blood cells, and dysregulation of glucose homeostasis can lead to several metabolic diseases, such as diabetes, obesity, dyslipidemia, and cardiovascular diseases. Blood glucose concentrations are determined by the glucose uptake from the intestine, glucose-metabolizing or -producing pathways, and hormones including insulin and glucagon. Major glucose-depriving pathways are glycolysis, glycogenesis, and lipogenesis, whereas glucose-producing pathways are glycogenolysis and gluconeogenesis (Alsahli and Gerich, 2017; Petersen et al., 2017).
Gluconeogenesis produces glucose and uses mainly lactate, glycerol, or glucogenic amino acids, including alanine and glutamate as substrates (Yip et al., 2016). It has long been postulated that the liver is the sole source of glucose generation, but this hypothesis was disapproved by observations that the mammalian renal cortical tissue can make gluconeogenesis in vitro, in animals and in humans (Benoy and Elliott, 1937; Bergman and Drury, 1938; Bjorkman et al., 1979; Drury et al., 1950). Interestingly, later studies have identified a substrate preference for gluconeogenesis in the liver for alanine, whereas the kidney prefers glutamate (Stumvoll et al., 1999). Gluconeogenesis shows a reciprocal regulation with glycolysis and is regulated on different levels. One regulation relates to glucagon-induced gene expression of key regulating enzymes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase, whereas allosteric regulation of fructose-1,6-bisphosphatase is mediated by fructose-2,6-bisphosphate, AMP, and ADP. Increasing evidence supports the hypothesis that the kidney plays a vital role in glucose homeostasis via gluconeogenesis, especially in the prolonged fasting state. Aberrant renal gluconeogenesis is associated with the development of type 2 diabetes mellitus (Bjorkman et al., 1980; Meyer et al., 2002), and increased renal glucose release by up to 300% in patients with type 2 diabetes mellitus have been reported (Meyer et al., 1998). Thus, renal gluconeogenesis is essential for glucose homeostasis in physiology.
Akr1A1 belongs to the Aldo-keto reductase (Akr) superfamily, which has a function in the reduction of carbonyl-containing compounds to their corresponding alcohols (Singh et al., 2015). One additional prominent member of the family is Akr1b1, which regulates in addition to its carbonyl-detoxifying function glucose homeostasis by converting glucose into sorbitol (Brownlee, 2001). Recent data have identified a function for mouse Akr1A1 acting as an S-nitroso-CoA reductase. Akr1a1 knockout mice displayed increased protein-S-nitrosylation and were protected from acute kidney injury, which was mediated by balancing glycolysis by regulating the S-nitrosylation of pyruvate kinase M2 (Zhou et al., 2019).
Different from other animals, two separate akr1a1 genes exist in zebrafish, namely, akr1a1a and akr1a1b, whose functions have not been studied yet. The zebrafish has been established as a model for diabetes research in the past decade (Heckler and Kroll, 2017; Jorgens et al., 2015; Lodd et al., 2019; Lou et al., 2020; Schmohl et al., 2019; She et al., 2018; Wiggenhauser et al., 2020), because glucose homeostasis in zebrafish is very similar to that humans and other mammals and alterations in glucose homeostasis are associated with organ damage, including the kidney, eyes, and nerves (Olsen et al., 2010). Therefore, this study aimed to generate an akr1a1b knockout animal model and evaluate the function of Akr1a1b in glucose homeostasis and organ function in embryonic, larval, and adult zebrafish. The study has identified Akr1a1b as a regulator of gluconeogenesis and shows how altered gluconeogenesis damages the kidney.
Results
Expression of akr1a1b in Zebrafish and Generation and Validation of akr1a1b−/− Mutants
Previous studies showed akr1a1 expression in virtually every tissue in human and mice (Fagerberg et al., 2014; Yue et al., 2014), with the highest expression being in the kidney tubular system (Scotcher et al., 2016). Yet, the expression of akr1a1b in zebrafish has not been analyzed. Thus, we investigated early developmental stages from 24 to 120 hours post fertilization (hpf) and organs from adult zebrafish for akr1a1b expression using quantitative RT-PCR (Table 1). Akr1a1b was abundantly expressed throughout embryonic and larval stages (Figure 1A) and also in all analyzed adult organs with the highest expression being in livers (Figure 1B). As mouse akr1a1 is highly expressed in the kidney tubular system (Zhou et al., 2019), we assessed Akr1a1b expression in adult zebrafish kidneys by immunohistochemical staining and demonstrated a similar localization in the tubular system as described in mice (Figure 1C). Last, gene sequence alignment of Akr1a1 across different species showed that zebrafish Akr1a1a shares a 60% amino acid similarity with human Akr1a1, and a 58.1% similarity with mouse Akr1a1. For zebrafish Akr1a1b the similarity is 70.5% with human Akr1a1 and 69.3% with mouse Akr1a1 (Figure S1).
Table 1.
Primers Used for RT-qPCR
| Gene | Sequencing |
|---|---|
| akr1a1b | CGTCTCTATTAAAAACTCTGAAAGACC |
| AAGGGGTATCGCCTCGTT | |
| Cpepck | ATCACGCATCGCTAAAGAGG |
| CCGCTGCGAAATACTTCTTC | |
| gls b | GGATATGGAGCAGCGTGATT |
| CTCATCCATTGGTGTGTTGC | |
| glud 1a | CCGGTATAACCTTGGGCTGG |
| CTCGGGTCTGCGTGGATAAG | |
| glut 1a | TGACCGGCCCATACGTTTTC |
| ATCATCTCGGTTATATTTATCTGCC | |
| glut 2 | GCAGAAGAACCCTCACTC |
| TCTCCGCCACAATAAACC | |
| sglt 1 | TGGAACGCTCTGGTTGTTGT |
| TAGATGCGGATTCGCTGACC | |
| sglt 2 | ATGAGTCGGGTGCTTTCTGG |
| ATGGCGCAGGGTAAAGACAA | |
| b2m | ACTGCTGAAGAACGGACAGG |
| GCAACGCTCTTTGTGAGGTG | |
| β-actin | ACGGTCAGGTCATCACCATC |
| TGGATACCGCAAGATTCCAT |
Figure 1.

Expression of akr1a1b in Zebrafish Development and in Adult Organs
(A and B) (A) Akr1a1b is ubiquitously expressed in zebrafish development (relatively compared with 24 hpf, n = 4, mean ± SD) and (B) in all analyzed organs of adult zebrafish (relatively compared with heart, n = 5, mean ± SD).
(C) In adult zebrafish kidney, immunohistochemistry revealed high Akr1a1b expression in renal tubules (arrows). Box shows kidney immunostaining with secondary antibody only. Expression of genes in (A and B) was determined by RT-qPCR and normalized to β-actin.
∗∗∗p < 0.001, p value was calculated by one-way ANOVA. Scale bars, 50 μm. See also Figure S1.
To address the physiological role of akr1a1b in zebrafish development and physiology, we have established a permanent knockout model using CRISPR-Cas9 technology. Following the injection of the gRNA together with Cas9 RNA targeting exon 4 of the zebrafish akr1a1b gene (Figure S2A), two different frameshift mutants were identified and used for the subsequent studies, including a 17-bp insertion in the Tg(wt1b:EGFP) reporter line labeling the embryonic pronephros (Perner et al., 2007) and a 23-bp deletion in the Tg(fli1:EGFP) reporter line labeling endothelial cells (Lawson and Weinstein, 2002) (Figure 2A). Akr1a1b−/− larvae showed a strong decrease of total Akr enzyme activity (Figure 2C), and Akr1a1b protein expression in adult livers was utterly abolished (Figure 2B). Activities of other carbonyl-detoxifying enzymes as potential compensatory mechanism, including glyoxalase 1 (Glo1) and aldehyde dehydrogenase (ALDH) enzymes, were not altered (Figures S2B and S2C). Interestingly, concentration of the dicarbonyl methylglyoxal (MG) (Figure 2D), but not of 3-deoxyglucosone, and glyoxal (Figures S2D and S2E) as reactive metabolites leading to Advanced Glycation Endproducts (Brownlee, 2001), was significantly increased in 96-hpf akr1a1b−/− larvae. Together, the data have proved the successful generation and validation of akr1a1b mutant zebrafish, which only shows an increase for MG.
Figure 2.

Generation and Validation of akr1a1b-/- Zebrafish Mutants
(A) CRISPR-Cas9 technology was used to establish akr1a1b knockout zebrafish. Schematic depiction of wild-type akr1a1b target sequence and two identified frameshift mutations and their corresponding chromatograms including a 17-bp insertion (Δ+17) in the Tg(wt1b:EGFP) reporter line and a 23-bp deletion (Δ-23) in the Tg(fli1:EGFP) reporter line. Red dashed boxes indicate start of genomic alterations.
(B) Western blot for Akr1a1b expression in zebrafish livers showed absence of Akr1a1b protein in the 17-bp insertion (Δ+17) and in the 23-bp deletion mutant (Δ-23), respectively, which validates the akr1a1b knockout zebrafish model. Beta-actin served as loading control.
(C) Δ+17 akr1a1b-/- zebrafish larvae at 96 hpf showed a strong descend of Akr enzyme activity (n = 3 clutches with 50 larvae, mean ± SD).
(D) Δ+17/Δ-23 akr1a1b-/- larvae at 96 hpf have increased MG concentrations (n = 9 clutches with 50 larvae).
∗p < 0.05, ∗∗∗p < 0.001, p value was calculated by t test. See also Figure S2.
Akr1a1b Knockout in Zebrafish Caused Alterations of the Embryonic Pronephros and of Adult Kidneys
As Akr1a1b is prominently expressed in zebrafish renal tubules (Figure 1C), we hypothesized a significant effect for akr1a1b knockout on kidney development and kidney function in zebrafish. The zebrafish pronephros starts to develop at 16 hpf and reach their full functionality at 96 hpf. Structurally, at 48 hpf, the zebrafish pronephros are simply composed of two nephrons and functionally consist of two blood-filtering glomeruli, although leaky at this time; two proximal and distal tubules; and the pronephric duct (Drummond and Davidson, 2010). In the morphological analysis in Tg(wt1b:EGFP) line, compared with the akr1a1b+/+ embryos at 48 hpf, akr1a1b−/− embryos displayed an enlarged glomerulus, where the length was substantially increased from 88.6 ± 10.8 μm in the akr1a1b+/+ to 106.3 ± 19.1 μm in akr1a1b−/− mutants. The pronephric neck was significantly shortened to 74.9 ± 19.3 μm in akr1a1b−/− embryos compared with 97.9 ± 14.6 μm in the akr1a1b+/+ group (Figures 3A and 3B). Furthermore, to evaluate whether renal functionality was affected by the knockout of akr1a1b, we assessed the pronephric ultrafiltration in zebrafish larvae. Upon sinus venous injection of the 70-kDa Texas Red dextran, a significantly increased loss of fluorescence in akr1a1b−/− (the ratios are 0.516 ± 0.079 and 0.364 ± 0.079 at 24 and 48 hpf, respectively) compared with the akr1a1b+/+ larvae (the ratios are 0.621 ± 0.090 and 0.427 ± 0.076 at 24 and 48 hpi, respectively) was observed (Figure 3C). Thus, the result indicated an altered glomerular filtration rate in the akr1a1b knockout zebrafish. Based on the findings in akr1a1b−/− embryos, we performed a histological and electron microscopy analysis of adult akr1a1b−/− kidneys to address whether the pronephros developmental impairments in embryos persisted into adult zebrafish (Figure 4). In most adult akr1a1b−/− kidneys, a deposition of diastase-resistant periodic acid-Schiff-positive hyaline droplets, putatively lysosomes, with a mild to a moderate amount and a small to medium size within the epithelium of proximal tubules was found (Figures 4A and 4B). In contrast, the kidneys of akr1a1b+/+ animals only have scattered and mostly small droplets (Figures 4A and 4B), which were valued as a physiological finding as known from other animal species (Decker et al., 2012; Sato et al., 2005). As livers showed the highest akr1a1b expression (Figure 1B), we also analyzed the histology of adult akr1a1b−/− livers (Figure 4C). Yet, the overall morphology was not altered in this organ. Taken together, knockout of akr1a1b in zebrafish altered the embryonic pronephros and adult kidney, but other organs, including the liver, appeared normal.
Figure 3.

Akr1a1b Knockout in Zebrafish Caused Alterations of the Embryonic Pronephros
(A) Compared with akr1a1b+/+ embryos (n = 33, mean ± SD) at 48 hpf, Δ+17 akr1a1b-/- mutants (n = 21, mean ± SD) displayed an enlarged glomerulus (encircled) and shortened tubular neck (asterisk). (B) Representative pronephros images of 48hpf old akr1a1b+/+ and Δ+17 akr1a1b-/- embryos.
(C) A significant increased loss of fluorescence in Δ+17akr1a1b-/- mutants (n = 18, mean ± SD) was observed when compared with akr1a1b+/+ larvae (n = 21, mean ± SD), which indicated an altered glomerular filtration rate in akr1a1b-/- mutants.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, p value was calculated by t test. Scale bar: 0.1 mm.
Figure 4.

Akr1a1b Knockout Altered Adult Zebrafish Kidneys, but Livers Remained Normal
(A) Periodic acid-Schiff (PAS) staining and (B) electron microscopy (EM) showed deposition of diastase-resistant PAS-positive hyaline droplets, putatively lysosomes (red arrows), within the epithelium of proximal tubules in Δ-23 akr1a1b-/- kidneys. Akr1a1b+/+ kidneys only have scattered small droplets. Quantification of 19 akr1a1b-/- EM images revealed 9 images with “+” and 10 images with “++” PAS-positive hyaline droplets. All 20 akr1a1b+/+ EM images were scored as “0.”
(C) Adult Δ-23 akr1a1b-/- livers stained by PAS were unaltered.
Scale bars: 20 μm in (A and C) and 500 nm (B).
Glutamate Accumulated in akr1a1b Knockout Zebrafish and Caused Pronephros Alterations
To investigate the underlying mechanisms of the pronephric and renal abnormalities, we performed metabolic profiling, including analysis of several amino acids, glycolytic and Krebs cycle intermediates, fatty acids, and adenosines in akr1a1−/− and akr1a1b+/+ zebrafish larvae at 96 hpf. Most intermediates were not significantly changed except for 2-keto glutaric acid and citric acid (Figure S3). Yet, interestingly, we found increased concentrations of two glucogenic amino acids, namely, glutamate and alanine (Figure 5A).
Figure 5.

Glutamate Accumulation in akr1a1b-/- Mutants Damaged the Kidneys
(A) Primary metabolites were measured in 96-hpf larvae by gas chromatography-mass spectrometry analysis and showed a significant increase for glutamate and alanine in Δ+17 akr1a1b-/- larvae (n = 3 clutches with 50 larvae, mean ± SD).
(B) Glutamate accumulated in adult Δ+17/Δ-23 akr1a1b-/- zebrafish kidneys at 3 and 18 h postprandial (2 h postprandial: n = 7 in akr1a1b+/+, n = 8 in akr1a1b-/-; 3 h postprandial: n = 9 in akr1a1b+/+, n = 6 in akr1a1b-/-; 18 h postprandial: n = 10 in both groups, mean ± SD).
(C) Enlarged glomerulus and shortened tubular neck length in 48-hpf wild-type zebrafish embryos treated with glutamate (n = 46 in control group; n = 50 in 0.1 mM group; n = 49 in 1 mM group, mean ± SD). (D) Representative pronephros images of glutamate treated 48hpf embryos. Glomeruli are encircled.
(E) A significant increased loss of fluorescence in 1 mM glutamate-treated wild-type larvae both at 24 and 48 hpi (n = 26 in control group; n = 21 in 1 mM group, mean ± SD).
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; p value in (A, B, and E) was calculated by t test, p value in (C) was calculated by one-way ANOVA, Scale bar, 0.1 mm. See also Figures S3, S4, and S5.
To test the hypothesis that elevated concentrations of glutamate and alanine may cause the observed pronephros phenotype in akr1a1b−/− mutants (Figure 3), Tg(wt1b:EGFP) embryos were incubated for 48 h in medium containing increasing concentrations of glutamate and alanine, and subsequently, the pronephros was analyzed. In zebrafish treated with 0.1 and 1 mM glutamate, the glomerular length was increased to 89.6 ± 10.9 μm and 91.8 ± 9.8 μm, respectively, whereas it was 82.6 ± 9.1 μm in egg water. The pronephric neck was shortened to 100.3 ± 14.5 μm and 88.8 ± 15.5 μm, respectively, whereas it was 111.3 ± 12.6 μm in egg water (Figures 5C and 5D). In addition, wild-type zebrafish larvae treated with 1 mM glutamate showed a significantly increased loss of fluorescence after 70-kDa Texas Red dextran injection (the ratios are 0.513 ± 0.084 and 0.350 ± 0.052 at 24 and 48 hpi, respectively), compared with larvae treated with egg water (the ratios are 0.620 ± 0.107 and 0.432 ± 0.052 at 24 and 48 hpi, respectively) (Figure 5E). In zebrafish treated with 5 mM alanine, the glomerular length was altered to 75.8 ± 8.5 μm and 87.1 ± 6.1 μm, respectively, whereas it was 77.6 ± 11.3 μm in control conditions. The pronephric neck was shortened to 127.2 ± 15.6 μm and 105.4 ± 9.3 μm, respectively, whereas it was 131.4 ± 22.0 μm in egg water (Figure S4). In conclusion, although both amino acids induced pronephric alterations, glutamate (0.1 mM) was more detrimental than alanine (5 mM). These results suggest glutamate as the main damaging factor of pronephric changes in akr1a1b−/− zebrafish embryos.
Next, we aimed to verify whether the accumulation of glutamate and alanine persisted into adult zebrafish. Therefore we measured concentrations of both amino acids in the kidneys and livers at 2, 3, and 18 h postprandial. Glutamate was significantly enriched in akr1a1−/− adult zebrafish kidneys at 3 h postprandial stage, to 2.90 ± 0.29 nmol/mg tissue, and at 18 h postprandial stage, to 6.63 ± 1.29 nmol/mg tissue; meanwhile, it was 1.96 ± 0.15 nmol/mg tissue and 3.14 ± 0.37 nmol/mg tissue in akr1a1b+/+ zebrafish kidneys, respectively (Figure 5B). In contrast, the concentration of alanine was unchanged in kidneys as it was for glutamate and alanine in adult livers (Figure S5). Thus, increased concentrations of glutamate were only identified in akr1a1b−/− kidneys.
Altered Gluconeogenesis in akr1a1b−/− Zebrafish
Glutamate and alanine are glucogenic amino acids serving as primary substrates for gluconeogenesis in kidneys and livers, respectively. The accumulation of glutamate in akr1a1b−/− embryos and in adult akr1a1b−/− kidneys led to the hypothesis of dysfunctional gluconeogenesis, specifically in akr1a1b−/− kidneys. Glucose synthesis by gluconeogenesis in the human kidney accounts for approximately 20% of total glucose production in the body in the postabsorptive state (Gerich et al., 2001), and alterations of glucose synthesis may affect glucose homeostasis. To test if glucose homeostasis is altered in adult akr1a1b−/− mutants, we measured blood glucose concentrations of adult zebrafish at 2, 3, and 18 h postprandial and we found hypoglycemia after overnight fasting with blood glucose concentrations of 26.12 ± 5.50 mg/dL in akr1a1b−/− zebrafish significantly lower compared with 34.29 ± 15.66 mg/dL in akr1a1b+/+ zebrafish (Figure 6A). Yet, blood glucose levels 2 h and 3 h after feeding were not changed in akr1a1b−/− zebrafish indicating physiological glucose metabolism after glucose uptake. Thus, the data suggest inhibition of gluconeogenesis in akr1a1b−/− mutants, especially in kidneys, because renal gluconeogenesis accounts for the major glucose production in the prolonged fasting stage.
Figure 6.

Inhibition of Gluconeogenesis Led to Hypoglycemia in Overnight Fasted akr1a1b-/-Zebrafish
(A) Blood glucose was measured at different time points after feeding in adult akr1a1b+/+ and Δ+17/Δ-23 akr1a1b-/- zebrafish and found hypoglycemia after overnight fasting in akr1a1b-/- zebrafish (2 h postprandial: n = 23 in akr1a1b+/+, n = 20 in akr1a1b-/-; 3 h postprandial: n = 14 in akr1a1b+/+, n = 14 in akr1a1b-/-; 18 h postprandial: n = 31 in akr1a1b+/+, n = 16 in akr1a1b-/-, mean ± SD).
(B) Schematic depiction of gluconeogenesis and how glucogenic amino acid glutamate serves as substrate. PEPCK: phosphoenolpyruvate carboxykinase; PEP: phosphoenolpyruvate; OAA: oxaloacetate; α-KG: α-ketoglutarate; GLUD1: glutamate dehydrogenase 1.
(C) Loss of cytosolic cPEPCK expression in adult Δ+17/Δ-23 akr1a1b-/- kidneys 3 and 18 h after feeding (2 h postprandial: n = 6 in akr1a1b+/+, n = 6 in akr1a1b-/-; 3 h postprandial: n = 9 in akr1a1b+/+, n = 8 in akr1a1b-/-; 18 h postprandial: n = 8 in akr1a1b+/+, n = 7 in akr1a1b-/-, mean ± SD).
(D) Loss of cytosolic cPEPCK expression in adult Δ+17/Δ-23 akr1a1b-/- livers 3 h after feeding (2 h postprandial: n = 7 in akr1a1b+/+, n = 7 in akr1a1b-/-; 3 h postprandial: n = 7 in akr1a1b+/+, n = 7 in akr1a1b-/-; 18 h postprandial: n = 5 in akr1a1b+/+, n = 4 in akr1a1b-/-, mean ± SD). cPEPCK expression was analyzed by RT-qPCR and normalized to b2m.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, p value was calculated by t test. See also Figure S6.
Gluconeogenesis is regulated by different mechanisms. Among the genes involved in gluconeogenesis regulation, the critical rate-limiting enzyme is PEPCK (Figure 6B), whose activity is directly related by its mRNA abundance (Quinn and Yeagley, 2005). Thus, we compared PEPCK expression (Table 1) in livers and kidneys between akr1a1b+/+ and akr1a1b−/− adult zebrafish after 2, 3, and 18 h postprandial. In addition, important genes (Table 1) involved in glutamate metabolism and glucose transportation, including glutamate dehydrogenase 1 (glud 1), Glutaminase (gls), glut 1 (glucose transporter 1), glut 2 (glucose transporter 2), sglt 1 (sodium dependent glucose co-transporter 1), and sglt 2 (sodium dependent glucose co-transporter 2), in the kidney were also measured (Figure S6). It was found that PEPCK expression 2 h after feeding remained unaltered in akr1a1b−/− livers and kidneys, whereas 3 h and 18 h postprandial, PEPCK expression in akr1a1b−/− kidneys was significantly reduced (Figures 6C and 6D). Together, the data indicated that akr1a1b deficiency in zebrafish blocks PEPCK expression in kidneys leading to inhibition of gluconeogenesis, accompanied by hypoglycemic episodes and renal glutamate accumulation promoting kidney alterations.
Akr1a1b Regulates S-Nitrosylation
A recent study in mice has proved that Akr1a1 mediates glucose metabolism by inhibiting S-nitrosylation of glycolytic enzymes (Zhou et al., 2019). Thus, we hypothesized an altered S-nitrosylation as the upstream mechanism regulating kidney malformation in akr1a1b−/− mutants. First, we determined nitrotyrosine as a marker for nitrosative stress in 96-hpf akr1a1b−/− larvae and found a strong increase (Figure 7A). Second, SNO (S-nitrosylated protein) levels were measured by western blot, revealing that SNOs were highly increased in kidneys of akr1a1b−/− zebrafish (Figure 7B). These data pointed to an increased stimulation of S-nitrosylation in akr1a1b−/− zebrafish.
Figure 7.

NO-Dependent S-Nitrosylation Regulated Gluconeogenesis and Pronephros Development in akr1a1b−/− Mutants
(A) Nitrotyrosine was measured by ultrahigh-performance liquid chromatography-mass spectrometry, and was increased in 96-hpf Δ-23 akr1a1b-/- larvae (n = 3 clutches with 50 larvae, mean ± SD).
(B) Western blots show increased S-nitrosylated proteins (SNOs) in adult Δ-23 akr1a1b-/- kidneys (n = 2).
(C) Inhibition of NO-dependent S-nitrosylation by L-NAME in Δ+17 akr1a1b-/- zebrafish 48-hpf embryos rescued the altered pronephros (n = 27 in akr1a1b+/+ group; n = 27 in akr1a1b+/+& L-NAME group; n = 34 in akr1a1b-/- group; n = 25 in akr1a1b-/-& L-NAME group, mean ± SD).
(D) Representative pronephros images of 48hpf akr1a1b+/+ and Δ+17 akr1a1b-/- embryos treated with L-NAME. Glomeruli are encircled.
(E) cPEPCK expression was regulated by S-nitrosylation. Inhibition of S-nitrosylation by L-NAME treatment increased cPEPCK expression in akr1a1b+/+ and Δ-23 akr1a1b-/- encircled larvae at 96 hpf and normalized cPEPCK expression in akr1a1b-/- encircled larvae as measured by RT-qPCR (n = 10 in all groups, mean ± SD).
∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, p value in (A) was calculated by t test, p values of (C and E) were calculated by one-way ANOVA. Scale bar, 0.1 mm. See also Figure S7.
To investigate whether evaluated S-nitrosylation may account for kidney alterations observed in akr1a1b−/− embryos, akr1a1b+/+ and akr1a1b−/− embryos were treated with 20 μM L-NAME, a known inhibitor of nitric oxide formation (Figure S7). It was found that inhibition of S-nitrosylation by L-NAME treatment in akr1a1b−/− embryos normalized the alterations of the pronephros at 48 hpf (Figures 7C and 7D). In addition, we analyzed if PEPCK expression is regulated by S-nitrosylation and therefore determined PEPCK expression in 96-hpf akr1a1b−/− larvae after L-NAME treatment. PEPCK expression was increased in 96-hpf L-NAME-treated akr1a1b+/+ larvae, whereas PEPCK expression in akr1a1b−/− larvae compared with akr1a1b+/+ larvae was slightly decreased, but could be significantly increased and normalized to akr1a1b+/+ levels after L-NAME treatment (Figure 7E). Altogether, the data have proved that altered gluconeogenesis and glutamate accumulation in akr1a1b−/− zebrafish caused by enriched SNOs accounted for the underlying kidney abnormalities.
Discussion
In this study, we have established zebrafish Akr1a1b as a regulator of gluconeogenesis by regulating PEPCK expression and thereby maintaining glucose homeostasis. Loss of akr1a1b leads to hypoglycemia and glutamate accumulation, which severely damages the pronephros, and this process is regulated by NO-dependent S-nitrosylation.
The most important finding of the study is the identification of a regulatory mechanism of how glucose homeostasis is controlled. Although glucose is an essential energy substrate for the body with, e.g., the human brain requiring 130 g glucose a day (Cunnane et al., 2011), continuously increased or decreased blood glucose levels lead to severe metabolic diseases. Diabetes, defined by hyperglycemia, is the most common metabolic disease leading to several organ alterations (Roglic and World Health Organization, 2016), and glucose-induced metabolic changes have been identified as one crucial damaging mechanism (Borg et al., 2011; Diabetes et al., 1993; Duckworth et al., 2009; UKPDS, 1998). In contrast, severe hypoglycemia is also a life-threatening condition, accompanied by heart racing, nausea, trembling, and sweating (Tesfaye and Seaquist, 2010) and can lead to greater risk of cardiovascular events (Zinman et al., 2018), seizure (Buckingham et al., 2008), coma, and death (Zoungas et al., 2010). Hypoglycemic conditions are typically seen in diabetic patients induced by inappropriate insulin usage but may also be caused by other metabolic conditions. These observations strongly suggest that maintaining physiological glucose homeostasis is a fundamental process and highlights zebrafish Akr1a1b as a regulator of this process. The function of the Akr gene family has mostly been mysterious, and only a few genes have functionally been analyzed in more detail thus far. Akr1b1 has attracted most attention in the past because it can convert glucose to sorbitol, has an increased activity during diabetes, and inhibition of Akr1b1 has been suggested as a promising approach to prevent diabetic complications (Brownlee, 2001). Yet, although preclinical studies were promising, later clinical trials failed (Singh et al., 2015). A recent study in mice highlighted a function for another Akr family member, namely, Akr1a1. It was shown that Akr1a1 has an S-nitroso-CoA reductase activity, and akr1a1 knockout mice were protected from acute kidney injury (Zhou et al., 2019).
Interestingly, Akr1a1 has been suggested as a regulator of glucose homeostasis in mice by regulating S-nitrosylation of pyruvate kinase 2 (PKM2) and thereby affecting its activity. In detail, S-nitrosylation of PKM2 blocked its activity and subsequently pyruvate production (Zhou et al., 2019). In contrast to mice and humans, the zebrafish genome contains only a few akr family members, but for akr1a1, two genes, namely, akr1a1a and akr1a1b are present in the zebrafish genome. In this study, Akr1a1b was identified as a regulator of gluconeogenesis, which regulates NO-dependent S-nitrosylation. Thus, although mouse Akr1a1 and zebrafish Akr1a1b both regulate glucose homeostasis, they act on reciprocal pathways, namely, glycolysis by Akr1a1 and gluconeogenesis by Akr1a1b. This leads now to the major question, if the other zebrafish akr1a1 gene, namely, akr1a1a, is the functional homolog to mouse and human akr1a1 and which gene in mouse and human is the functional counterpart to zebrafish akr1a1b? The opposite regulation of glucose homeostasis by mouse Akr1a1 regulating glycolysis and zebrafish Akr1a1b regulating gluconeogenesis suggests the concept that in both species opposite regulators exist and these unknown regulators must now be identified in future experimental studies.
The second important observation of the study is the identification of zebrafish Akr1a1b as a regulator S-nitrosylation and subsequently controlling PEPCK expression as the key regulatory enzyme in gluconeogenesis and that the inhibition of gluconeogenesis not only leads to hypoglycemic conditions but also leads to glutamate accumulation in embryos and adult akr1a1b−/− kidneys and alters the embryonic pronephros and adult kidney. The data now have three important implications. First, it suggests the Akr family as a therapeutic target to block gluconeogenesis as it would be beneficial to prevent hyperglycemic conditions in diabetes. Alternatively, activation of the Akr enzymes could prevent severe hypoglycemia thereby preventing coma and death. Second, it shows that altered regulations of evolutionarily conserved pathways, such as gluconeogenesis, can lead to severe organ damage in the body. In akr1a1b−/− kidneys, we found an accumulation of glutamate, and we have proved in zebrafish embryos that glutamate treatment damaged the pronephros. Thus, prolonged altered concentrations of physiological metabolites, such as the amino acid glutamate, damage organs. Last, the data in akr1a1b−/− zebrafish have further proved the different substrate preference for gluconeogenesis occurring in liver and kidney. Although in akr1a1b−/− embryos, both glucogenic amino acids alanine and glutamate, accumulated and damaged the embryonic pronephros, glutamate accumulation as the substrate for gluconeogenesis only occurred in adult kidneys. In contrast, alanine as the preferred substrate for gluconeogenesis in the liver remained normal.
In summary, the data have identified a mechanism of how glucose homeostasis is controlled. The Akr gene family and specifically the akr1a1 genes have a major function in maintaining physiological blood glucose concentrations in different species and thereby controlling the important equilibrium between providing glucose-derived energy and preventing glucose-induced organ damage.
Limitations of the Study
In our study, we have generated akr1a1b mutant zebrafish, and we demonstrate that Akr1a1b regulates glucose homeostasis by controlling gluconeogenesis. Yet, the function of the gene homolog of akr1a1b in zebrafish, named akr1a1a, has so far been unknown, and it remains elusive what potential role it may have in glucose metabolism regulation. Furthermore, as zebrafish Akr1a1b has overlapping, but also different mechanisms when compared with mouse Akr1A1 in glucose metabolisms, and the mouse/human genome consists of several other akr genes, it has so far been unknown which mouse/human gene is the homolog to zebrafish Akr1a1b. Last, it must be further analyzed in detail how exactly Akr1a1b regulates cPEPCK expression.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Jens Kroll (jens.kroll@medma.uni-heidelberg.de).
Materials Availability
The anti-Akr1a1b antibody and both akr1a1b zebrafish mutants generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study. For Akr1a1b amino acid sequence, please refer to: https://www.uniprot.org/.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The study was supported by grants from Deutsche Forschungsgemeinschaft (CRC 1118 and IRTG 1874/2 DIAMICOM) and China Scholarship Council (CSC). The authors thank the Metabolomics Core Technology Platform of the Excellence cluster “CellNetworks” (Heidelberg University), the Deutsche Forschungsgemeinschaft (grant ZUK 40/2010-3009262) for support with ultra-performance liquid chromatography-based metabolite quantification, Prof. Dr. Ilse Hofmann & Claudia Tessmer from the CF Unit Antibodies from DKFZ Heidelberg, and Dr. Stefan Hillmer, Electron Microscopy Core Facility of Heidelberg University. The authors also acknowledge the support of the Zebrafish Core Facility of Medical Faculty Mannheim.
Author Contributions
X.L. performed experiments, analyzed data, and wrote the manuscript. F.S., H.Q., K.B., C.T.T., G.P., N.V., T.P., I.H., J.M., and T.F. performed experiments and analyzed data. R.H. and P.P.N. gave conceptual advice. J.K. conceived and designed the study and wrote the manuscript. J.K. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Declaration of Interests
The authors report no conflict of interest.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101763.
Supplemental Information
References
- Alsahli M., Gerich J.E. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 2017;133:1–9. doi: 10.1016/j.diabres.2017.07.033. [DOI] [PubMed] [Google Scholar]
- Benoy M.P., Elliott K.A.C. The metabolism of lactic and pyruvic acids in normal and tumour tissues V. Synthesis of carbohydrate. Biochem. J. 1937;31:1268–1275. doi: 10.1042/bj0311268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman H., Drury D.R. The relationship of kidney function to the glucose utilization of the extra abdominal tissues. Am. J. Phys. 1938;124:279–284. [Google Scholar]
- Bjorkman O., Felig P., Wahren J. Gluconeogenesis by the human-kidney - unique stimulatory effect of fructose. Clin. Res. 1979;27:A409. [Google Scholar]
- Bjorkman O., Felig P., Wahren J. The contrasting responses of splanchnic and renal glucose output to gluconeogenic substrates and to hypoglucagonemia in 60-H-fasted humans. Diabetes. 1980;29:610–616. doi: 10.2337/diab.29.8.610. [DOI] [PubMed] [Google Scholar]
- Borg R., Kuenen J.C., Carstensen B., Zheng H., Nathan D.M., Heine R.J., Nerup J., Borch-Johnsen K., Witte D.R., Grp A.S. HbA(1c) and mean blood glucose show stronger associations with cardiovascular disease risk factors than do postprandial glycaemia or glucose variability in persons with diabetes: the A1C-Derived Average Glucose (ADAG) study. Diabetologia. 2011;54:69–72. doi: 10.1007/s00125-010-1918-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Buckingham B., Wilson D.M., Lecher T., Hanas R., Kaiserman K., Cameron F. Duration of nocturnal hypoglycemia before seizures. Diabetes Care. 2008;31:2110–2112. doi: 10.2337/dc08-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane S., Nugent S., Roy M., Courchesne-Loyer A., Croteau E., Tremblay S., Castellano A., Pifferi F., Bocti C., Paquet N. Brain fuel metabolism, aging, and Alzheimer's disease. Nutrition. 2011;27:3–20. doi: 10.1016/j.nut.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker J.H., Dochterman L.W., Niquette A.L., Brej M. Association of renal tubular hyaline droplets with lymphoma in CD-1 mice. Toxicol. Pathol. 2012;40:651–655. doi: 10.1177/0192623311436184. [DOI] [PubMed] [Google Scholar]
- Diabetes C., Complications Trial Research G., Nathan D.M., Genuth S., Lachin J., Cleary P., Crofford O., Davis M., Rand L., Siebert C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- Drummond I.A., Davidson A.J. Zebrafish kidney development. Method Cell Biol. 2010;100:233–260. doi: 10.1016/B978-0-12-384892-5.00009-8. [DOI] [PubMed] [Google Scholar]
- Drury D.R., Wick A.N., Mackay E.M. Formation of glucose by the kidney. Am. J. Phys. 1950;163:655–661. doi: 10.1152/ajplegacy.1950.163.3.655. [DOI] [PubMed] [Google Scholar]
- Duckworth W., Abraira C., Moritz T., Reda D., Emanuele N., Reaven P.D., Zieve F.J., Marks J., Davis S.N., Hayward R. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- Fagerberg L., Hallstrom B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteomics. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerich J.E., Meyer C., Woerle H.J., Stumvoll M. Renal gluconeogenesis - its importance in human glucose homeostasis. Diabetes Care. 2001;24:382–391. doi: 10.2337/diacare.24.2.382. [DOI] [PubMed] [Google Scholar]
- Heckler K., Kroll J. Zebrafish as a model for the study of microvascular complications of diabetes and their mechanisms. Int. J. Mol. Sci. 2017;18:2002. doi: 10.3390/ijms18092002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgens K., Stoll S.J., Pohl J., Fleming T.H., Sticht C., Nawroth P.P., Hammes H.P., Kroll J. High tissue glucose alters intersomitic blood vessels in zebrafish via methylglyoxal targeting the VEGF receptor signaling cascade. Diabetes. 2015;64:213–225. doi: 10.2337/db14-0352. [DOI] [PubMed] [Google Scholar]
- Lawson N.D., Weinstein B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002;248:307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- Lodd E., Wiggenhauser L.M., Morgenstern J., Fleming T.H., Poschet G., Buttner M., Tabler C.T., Wohlfart D.P., Nawroth P.P., Kroll J. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight. 2019;4:e126154. doi: 10.1172/jci.insight.126154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou B., Boger M., Bennewitz K., Sticht C., Kopf S., Morgenstern J., Fleming T., Hell R., Yuan Z., Nawroth P.P. Elevated 4-hydroxynonenal induces hyperglycaemia via Aldh3a1 loss in zebrafish and associates with diabetes progression in humans. Redox Biol. 2020;37:101723. doi: 10.1016/j.redox.2020.101723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C., Dostou J.M., Welle S.L., Gerich J.E. Role of human liver, kidney, and skeletal muscle in postprandial glucose homeostasis. Am. J. Physiol. endocrinol. Metab. 2002;282:E419–E427. doi: 10.1152/ajpendo.00032.2001. [DOI] [PubMed] [Google Scholar]
- Meyer C., Stumvoll M., Nadkarni V., Dostou J., Mitrakou A., Gerich J. Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J. Clin. Invest. 1998;102:619–624. doi: 10.1172/JCI2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen A.S., Sarras M.P., Intine R.V. Limb regeneration is impaired in an adult zebrafish model of diabetes mellitus. Wound Repair Regen. 2010;18:532–542. doi: 10.1111/j.1524-475X.2010.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner B., Englert C., Bollig F. The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev. Biol. 2007;309:87–96. doi: 10.1016/j.ydbio.2007.06.022. [DOI] [PubMed] [Google Scholar]
- Petersen M.C., Vatner D.F., Shulman G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017;13:572–587. doi: 10.1038/nrendo.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn P.G., Yeagley D. Insulin regulation of PEPCK gene expression: a model for rapid and reversible modulation. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005;5:423–437. doi: 10.2174/156800805774912962. [DOI] [PubMed] [Google Scholar]
- Roglic G., World Health Organization . World Health Organization; 2016. Global Report on Diabetes. [Google Scholar]
- Sato S., Kitamura H., Ghazizadeh M., Adachi A., Sasaki Y., Ishizaki M., Inoue K., Wakamatsu K., Sugisaki Y. Occurrence of hyaline droplets in renal biopsy specimens: an ultrastructural study. Med. Mol. Morphol. 2005;38:63–71. doi: 10.1007/s00795-004-0272-1. [DOI] [PubMed] [Google Scholar]
- Schmohl F., Peters V., Schmitt C.P., Poschet G., Buttner M., Li X., Weigand T., Poth T., Volk N., Morgenstern J. CNDP1 knockout in zebrafish alters the amino acid metabolism, restrains weight gain, but does not protect from diabetic complications. Cell. Mol. Life Sci. 2019;76:4551–4568. doi: 10.1007/s00018-019-03127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotcher D., Jones C., Posada M., Rostami-Hodjegan A., Galetin A. Key to opening kidney for in vitro-in vivo extrapolation entrance in health and disease: Part I: in vitro systems and physiological data. AAPS J. 2016;18:1067–1081. doi: 10.1208/s12248-016-9942-x. [DOI] [PubMed] [Google Scholar]
- She J., Yuan Z., Wu Y., Chen J., Kroll J. Targeting erythropoietin protects against proteinuria in type 2 diabetic patients and in zebrafish. Mol. Metab. 2018;8:189–202. doi: 10.1016/j.molmet.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Kapoor A., Bhatnagar A. Oxidative and reductive metabolism of lipid-peroxidation derived carbonyls. Chem. Biol. Interact. 2015;234:261–273. doi: 10.1016/j.cbi.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumvoll M., Perriello G., Meyer C., Gerich J. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int. 1999;55:778–792. doi: 10.1046/j.1523-1755.1999.055003778.x. [DOI] [PubMed] [Google Scholar]
- Tesfaye N., Seaquist E.R. Neuroendocrine responses to hypoglycemia. Ann. N. Y. Acad. Sci. 2010;1212:12–28. doi: 10.1111/j.1749-6632.2010.05820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UKPDS Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- Wiggenhauser L.M., Qi H., Stoll S.J., Metzger L., Bennewitz K., Poschet G., Krenning G., Hillebrands J.L., Hammes H.P., Kroll J. Activation of retinal angiogenesis in hyperglycemic pdx1 (−/−) zebrafish mutants. Diabetes. 2020;69:1020–1031. doi: 10.2337/db19-0873. [DOI] [PubMed] [Google Scholar]
- Yip J., Geng X., Shen J., Ding Y. Cerebral gluconeogenesis and diseases. Front. Pharmacol. 2016;7:521. doi: 10.3389/fphar.2016.00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.L., Zhang R., Anand P., Stomberski C.T., Qian Z., Hausladen A., Wang L., Rhee E.P., Parikh S.M., Karumanchi S.A. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature. 2019;565:96–100. doi: 10.1038/s41586-018-0749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinman B., Marso S.P., Christiansen E., Calanna S., Rasmussen S., Buse J.B., LEADER Publication Committee on behalf of the LEADER Trial Investigators Hypoglycemia, cardiovascular outcomes, and death: the LEADER experience. Diabetes Care. 2018;41:1783–1791. doi: 10.2337/dc17-2677. [DOI] [PubMed] [Google Scholar]
- Zoungas S., Patel A., Chalmers J., de Galan B.E., Li Q., Billot L., Woodward M., Ninomiya T., Neal B., MacMahon S. Severe hypoglycemia and risks of vascular events and death. N. Engl. J. Med. 2010;363:1410–1418. doi: 10.1056/NEJMoa1003795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. For Akr1a1b amino acid sequence, please refer to: https://www.uniprot.org/.