

Studies performed in chimpanzees at the National Institutes of Health (NIH) demonstrated a significant difference in ALT levels during acute hepatitis of different viral etiologies, with a hierarchy in the extent of liver damage according to the infecting virus: the highest level was in HDV superinfection, followed by infection with a precore HBV mutant, HBV/HDV coinfection, and, lastly, wild-type HBV infection. Our study demonstrates that both the virus and host are important in disease pathogenesis and offers new insights into their roles. We found that distinct cytokine profiles were associated with disease severity and clinical outcome. In particular, resolution of classic acute hepatitis B (AHB) correlated with a predominant Th1 response, whereas HBV/HDV coinfection showed a predominant proinflammatory response. Severe AHB and HDV superinfection showed a restricted cytokine profile and no evidence of Th1 response. The lack of cytokines associated with adaptive T-cell responses toward the precore HBV mutant and HDV superinfection argues in favor of a direct cytopathic effect of these viruses.
KEYWORDS: chimpanzees, pathogenesis, hepatitis B virus, precore HBV mutant, wild-type HBV, hepatitis D virus, cytokines, viral kinetics, hepatitis
ABSTRACT
Historical studies conducted in chimpanzees gave us the opportunity to investigate the basis for the different severities of liver damage and disease outcome associated with infection with wild-type hepatitis B virus (HBV) versus a precore HBV mutant, HBV/hepatitis D virus (HDV) coinfection, and HDV superinfection. Weekly samples from 9 chimpanzees were studied for immune responses by measuring plasma levels of 29 cytokines in parallel with alanine aminotransferase (ALT) levels and viral kinetics. Comparison of classic acute hepatitis B (AHB) with severe or progressive AHB and HBV/HDV coinfection or superinfection identified distinct cytokine profiles. Classic AHB (mean ALT peak, 362 IU/liter) correlated with an early and significant induction of interferon alpha-2 (IFN-α2), IFN-γ, interleukin-12 p70 (IL-12 p70), and IL-17A. In contrast, these cytokines were virtually undetectable in severe AHB (mean ALT peak, 1,335 IU/liter), characterized by significant elevations of IL-10, tumor necrosis factor alpha (TNF-α), and MIP-1β. In progressive AHB (mean ALT peak, 166 IU/liter), there was a delayed and lower-magnitude induction of cytokines. The ALT peak was also delayed (mean, 23.5 weeks) compared to those of classic (13.5 weeks) and severe AHB (7.5 weeks). HBV/HDV coinfection correlated with significantly lower levels of IFN-α2, IFN-γ, and IL-17A, associated with the presence of multiple proinflammatory cytokines, including IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, and IL-15. Conversely, HDV superinfection induced the highest ALT peak (1,910 IU/liter) and was associated with a general suppression of cytokines. Our data demonstrate that the most severe liver damage, caused by an HBV precore mutant and HDV, correlated with restricted cytokine expression and lack of Th1 response, raising the question of whether these viruses are directly cytopathic.
INTRODUCTION
Five major human hepatitis viruses, designated hepatitis A virus (HAV), hepatitis B virus (HBV), hepatitis C virus (HCV), hepatitis D virus (HDV), and hepatitis E virus (HEV), have been identified as being responsible for the vast majority of acute hepatitis in humans. Although all hepatitis viruses can induce acute hepatitis, the levels of alanine aminotransferase (ALT) and the extent of liver damage vary greatly not only among hepatitis viruses but also among individuals infected by the same virus. Pathogenesis studies support the concept that hepatitis viruses are noncytopathic and suggest that the disease they cause is immune mediated (1–3). However, several aspects of disease pathogenesis remain to be elucidated. One of the major challenges for pathogenesis studies continues to be the lack of efficient in vitro systems to grow primary virus isolates or a reliable small-animal model (4–6). None of these viruses can be transmitted to small laboratory animals, and they generally infect only nonhuman primates and, in some cases, only chimpanzees.
Historical studies performed at the National Institutes of Health (NIH) in chimpanzees demonstrated a significant difference in ALT levels during acute hepatitis of different viral etiologies, with the lowest peak (136 IU/liter) in progressive acute hepatitis B (AHB) and the highest in HDV superinfection (1,910 IU/liter). However, striking differences in the degree of necroinflammation were also observed in chimpanzees infected with the same virus. Infection with a precore HBV mutant, associated with acute liver failure (ALF) in humans, induced the most severe acute hepatitis so far observed in chimpanzees experimentally infected with HBV (7). The basis for the difference in the degree of liver damage caused by a precore HBV mutant versus the wild-type HBV is presently unknown. We recently demonstrated that in chimpanzees infected with a precore HBV mutant, hepatocellular damage at the time of ALT peak (up to 1,468 IU/liter) was associated with limited inflammatory infiltrates and the presence of B cells (8), although the B-cell infiltration was less extensive than that seen in HBV-associated acute liver failure (9). Thus, liver pathology data showed a remarkable difference between severe AHB and classic AHB, as typically observed following infection with the wild-type HBV, where the ALT peak usually coincided with an extensive inflammatory reaction, especially T-cell infiltration (8). Another clinical observation derived from these historical studies was the remarkably high degree of necroinflammation following HBV/HDV coinfection, or even higher after HDV superinfection. However, the mechanisms whereby HDV, a defective RNA virus that requires the obligatory helper function provided by HBV for viral assembly and transmission (10, 11), induces the most severe form of acute hepatitis ever documented in the chimpanzee model and in humans (12) remain unknown.
Access to a large series of archival samples from chimpanzee studies conducted at the NIH provided us with a unique opportunity to study the immunological and virological bases for the differences in severity and outcome of acute infection caused by HBV and HDV. The availability of weekly serial plasma or serum samples collected from the day of infection throughout the course of the disease from chimpanzees experimentally infected with HBV alone or with HBV/HDV coinfection or HDV superinfection allowed us to investigate the cytokine profiles and to correlate their patterns with viral kinetics, disease severity, and clinical outcome to get new insights into disease pathogenesis.
RESULTS
Alanine aminotransferase levels in acute HBV and HDV infection in chimpanzees.
Different degrees of liver damage were observed during the clinical course of acute resolving hepatitis B in 4 chimpanzees, as revealed by their ALT levels (Fig. 1 and Table 1). The two animals inoculated with the wild-type HBV (subtype ayw) (13, 14) developed classic AHB, with a mean (± standard errors of the mean [SEM]) ALT peak of 362 ± 25 IU/liter. In contrast, the other two animals that were inoculated with a precore HBV mutant, associated with ALF in humans (7), developed an unusually severe AHB, with ALT levels reaching nearly 1,468 IU/liter in one animal. The acute hepatitis seen in these two animals was characterized by the highest serum ALT peak (mean ± SEM, 1,335 ± 133 IU/liter) ever seen in a chimpanzee infected with wild-type HBV in our laboratory. In 22 chimpanzees infected with wild-type HBV strains (R. H. Purcell, unpublished data), the mean (±SEM) ALT peak was 613 ± 49 IU/liter. Based on the difference in ALT values, the acute hepatitis caused by a precore HBV mutant was defined as severe AHB. Notably, the ALT peaks occurred earlier (6 and 9 weeks) in severe AHB than in classic AHB (13 and 14 weeks) (Fig. 2A and B and Table 1). The levels of HBV DNA peaks were comparable among the four animals (Fig. 2 and Table 1), approaching 7.4 log10 IU/ml (±0.7). Instead, in the two animals that developed progressive AHB, the ALT peaks showed the lowest values (mean ± SEM, 166 ± 30 IU/liter) (Fig. 2C and Table 1) and were significantly delayed (week 23.5) compared to those of the other forms of AHB. Progressive AHB was accompanied by sustained high levels of HBV DNA (mean ± SEM, 8.5 ± 1.1 log10 IU/ml) for up to 20 weeks before the ALT peak (Fig. 2C). In HBV/HDV coinfection, the ALT levels were nearly double the levels observed in classic AHB (mean ± SEM, 622 ± 221 IU/liter) (Fig. 1 and Table 1). However, the highest ALT peak ever observed in chimpanzee studies was seen in the HDV superinfection (1,910 IU/liter) (Fig. 1 and Table 1).
FIG 1.
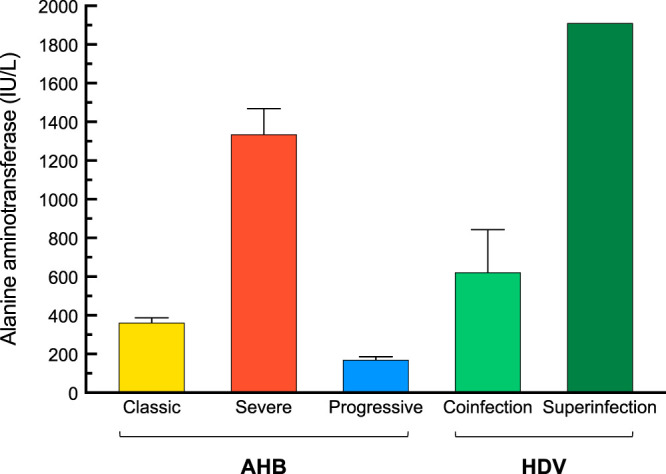
Alanine aminotransferase (ALT) levels in chimpanzees acutely infected with HBV alone, with HBV/HDV coinfection, and with HDV superinfection. AHB, acute hepatitis B.
TABLE 1.
Biochemical and virologic features of acute infection with HBV and HDV in chimpanzeesa
| Disease/inocula | No. | Mean HBV DNA peakb | Mean HDV RNA peakb (HBV DNA) | Mean HBcrAg peakb | Interval (wk) from viremia to ALT peak | Mean ALT peak | Seroconversion wk (anti-HBs) |
|---|---|---|---|---|---|---|---|
| Classic AHB, HBV wild-type (ayw) | 2 | 7.5 ± 1.4 | 7.7 ± 1.5 | 2 | 362 ± 25 | 22.0 ± 4.0 | |
| Severe AHB, precore HBV mutant | 2 | 7.4 ± 0.9 | 7.0 ± 0.1 | 1 | 1,335 ± 133 | 11.5 ± 2.5 | |
| Progressive AHB, HBV MS-2 strain (ay) or challenge pool (ayw) | 2 | 8.6 ± 1.0 | 9.1 ± 0.2 | 6 | 166 ± 30 | ND | |
| HBV/HDV coinfection, HBV MS-2 strain (ay), HDV NK or CH infectious pool | 2 | 6.4 ± 2.0 (8.3 ± 1.1) | 7.4 ± 0.9 | 1 | 622 ± 221 | 11.0 ± 2.0 | |
| HDV superinfection, HBV pool (ayw), CH infectious pool | 1 | 8.8 (9.5) | 8.4 | 2 | 1,910 | ND |
AHB, acute hepatitis B; ALT, alanine aminotransferase; HBcrAg, hepatitis B core antigen; anti-HBs, antibodies to hepatitis B surface antigen; ND, not detectable.
The mean HBV DNA peaks, mean HBcrAg peaks, and mean HDV RNA peaks are expressed in log10 units. The results are represented as means ± SEM.
FIG 2.

Biochemical and virological course in four chimpanzees with acute resolving hepatitis B and in two chimpanzees with acute progressing hepatitis B. AHB, acute hepatitis B; HBsAg, hepatitis B surface antigen; anti-HBs, antibodies to hepatitis B surface antigen.
Viral kinetics and serology in acute HBV and HDV infection in chimpanzees.
The kinetics of HBV DNA (peak, 7.78 ± 0.57 log10 IU/ml) and hepatitis B core-related antigen (HBcrAg) (peak, 7.62 ± 0.55 log10 U/ml) paralleled each other in acute hepatitis B and, regardless of the outcome, were similar in classic, severe, and progressive AHB (Fig. 3). In classic AHB, both HBV DNA and HBcrAg were detected as early as 1 week postinoculation, and both markers peaked near week 8 postinoculation (Fig. 3A). HBV DNA and HBcrAg levels waned after the ALT peaks, eventually becoming undetectable, concomitant with hepatitis B surface antigen (HBsAg) clearance and the appearance of antibodies to hepatitis B surface antigen (anti-HBs). Similarly, in severe AHB, both HBV DNA and HBcrAg appeared early, within week 1 in CH1410 and week 3 in CH1420, but at variance with classic AHB, their peaks occurred noticeably earlier, at weeks 5 and 8 in CH1410 and CH1420, respectively, just before the ALT peak in both animals (Fig. 3B). A hallmark of severe AHB was the lack of detectable hepatitis B e antigen (HBeAg), with the exception of a single time point in CH1420. This finding was consistent with previous observations in clinical studies in which the lack of HBeAg was associated with infection by a precore HBV mutant and fulminant hepatitis B (15–18). In both animals with severe AHB, antibodies to hepatitis B e antigen (anti-HBe) appeared concomitant with the ALT peak. In progressive AHB, HBV DNA and HBcrAg became detectable within 3 weeks postinoculation in both animals, reached their highest levels around weeks 11 and 13, respectively, and persisted at high levels thereafter (Fig. 3C). One of the most distinctive features of progressive AHB was a significantly delayed ALT peak in both animals compared to those of classic and severe AHB (Fig. 2). HBsAg appeared later, at week 7, along with HBeAg. No changes in viral kinetics were seen during the chronic phase, and these animals remained HBsAg and HBeAg positive throughout the observation period. One of these animals (CH1133) was subsequently infected by HDV (superinfection pattern).
FIG 3.
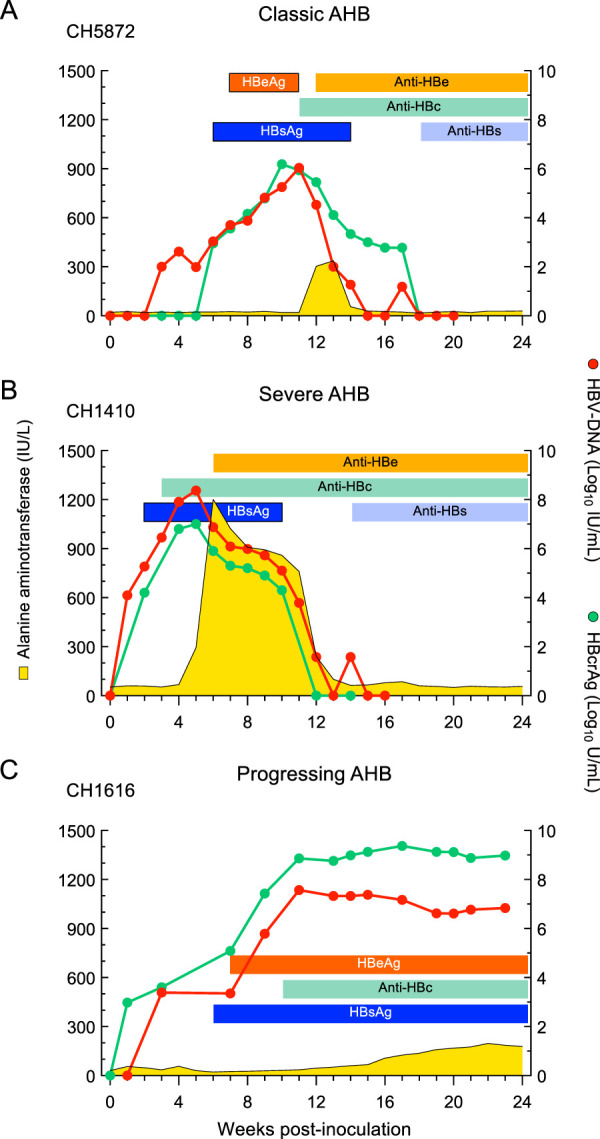
Biochemical and virological course of representative chimpanzees with acute classic or severe acute resolving hepatitis B or progressive acute hepatitis B. AHB, acute hepatitis B; HBeAg, hepatitis B e antigen; anti-HBe, antibodies to hepatitis B e antigen; anti-HBc, antibodies to hepatitis B core antigen; HBsAg, hepatitis B surface antigen; anti-HBs, antibodies to hepatitis B surface antigen.
In HBV/HDV coinfection, the kinetics of HBV DNA and HBcrAg levels resembled that of classic AHB in both of the chimpanzees studied (Fig. 4A and B). HBV DNA first appeared within 1 week of inoculation along with HBcrAg. In CH1309 and CH1441, HBV DNA peaked at weeks 5 and 10, i.e., several weeks before the ALT peak, which occurred at weeks 9 and 12, respectively. In contrast to the early appearance of HBV DNA, HDV RNA became detectable several weeks later, at weeks 5 and 10, and then increased rapidly to reach the highest levels of replication concomitant with the ALT peak. Both HBV DNA and HDV RNA levels diminished rapidly after the ALT peak, and HBsAg became negative concomitant with anti-HBs seroconversion. A striking difference was the magnitude of the ALT levels, which in HDV coinfection were twice as high as those in classic AHB (Table 1). CH1133, one of the two chimpanzees that developed progressive AHB, gave us the unique opportunity to study the viral kinetics before and during the time of HDV superinfection throughout the course of the disease (Fig. 4C). This animal, which was initially infected with wild-type HBV, developed progressive AHB that was characterized by a modest and delayed ALT peak (136 IU/liter, at week 22). At the time of HDV superinfection, which occurred 18 months after the first HBV inoculation, the animal was HBsAg and HBeAg positive with persistently high levels of HBV DNA and HBcrAg. HDV superinfection induced the highest ALT peak ever observed in chimpanzee studies, reaching 1,910 IU/ml (Fig. 4C and Table 1). As seen in HBV/HDV coinfection, the HDV RNA peak coincided with the ALT peak, in contrast to the pattern seen for HBV DNA, which always preceded the ALT peak. The HDV RNA peak was associated with either a dramatic decrease in HBV DNA, leading to its clearance in coinfection, or to a decrease of 3 logs in HDV superinfection, confirming the suppression of HBV replication by HDV RNA (10, 19, 20). After the ALT peak, this animal remained HBeAg negative with persisting high levels of HBV DNA and HBcrAg, as well as HDV RNA, albeit at lower levels (Fig. 4C).
FIG 4.
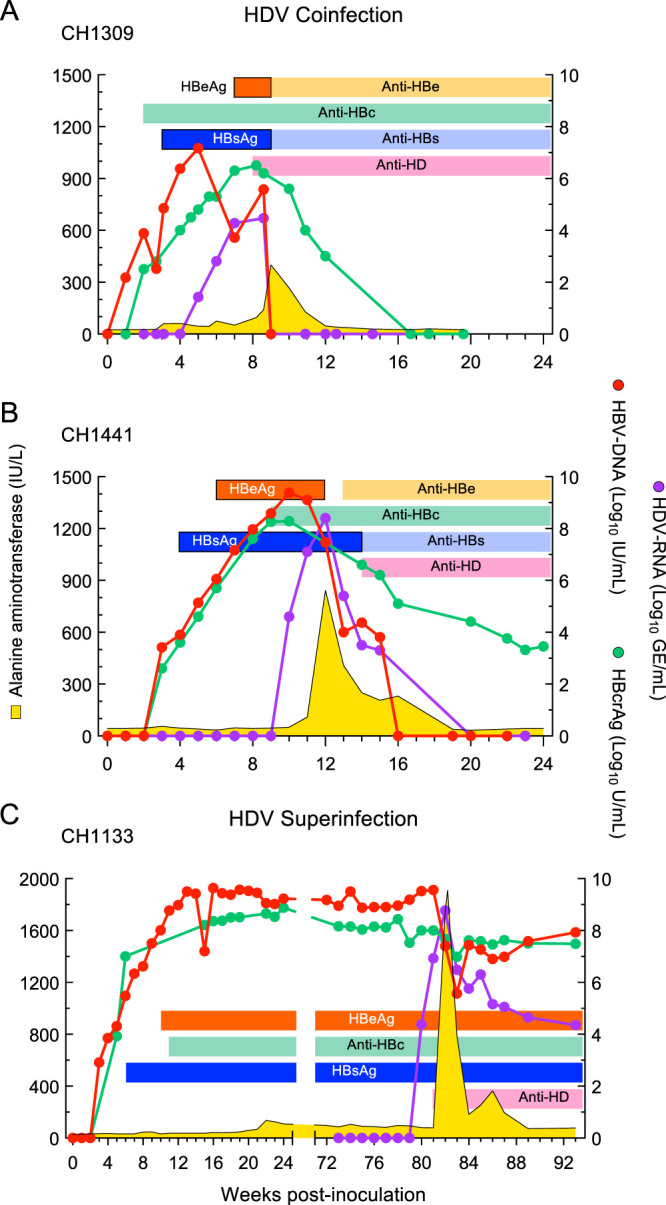
Clinical, serological, and virological course of acute resolving HBV/HDV infection in two chimpanzees and HDV superinfection in a chimpanzee chronically infected by HBV. HBeAg, hepatitis B e antigen; Anti-HBe, antibodies to hepatitis B e antigen; anti-HBc, antibodies to hepatitis B core antigen; HBsAg, hepatitis B surface antigen; anti-HBs, antibodies to hepatitis B surface antigen; anti-HD, antibodies to hepatitis delta antigen (HDAg).
Cytokine and chemokine levels in acute HBV and HDV infection in chimpanzees.
Since viable cellular samples were not available from this unique historical collection, we studied the long-term profile of 29 cytokines and chemokines (see Table S1 in the supplemental material) in serial plasma or serum samples taken at weekly or biweekly intervals throughout the observation period. Statistical analysis for the comparison of all cytokines at different time points between classic AHB and the other forms of acute hepatitis, including severe AHB, progressive AHB, and HBV/HDV coinfection, is reported in Tables S2, S3, and S4.
Members and functional features of the cytokines/chemokines analyzed in this study. Download Table S1, PDF file, 0.1 MB (139.2KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Statistical comparisons of cytokine levels within the indicated time intervals between classic and severe acute hepatitis B (AHB). Download Table S2, PDF file, 0.07 MB (68.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Our longitudinal study allowed us to identify distinct cytokine patterns according to disease severity and clinical outcome of acute HBV infection (Fig. 5). We found that classic resolving AHB was associated with the induction of multiple cytokines and chemokines with distinct kinetics. Comparison of the magnitude and kinetics of the cytokine levels revealed differences between classic and severe AHB (Table S2). Classic AHB correlated with significantly elevated levels of 12 cytokines compared to only 3 cytokines in severe AHB. The rise in viremia within the first 3 weeks postinoculation seen in classic AHB (Fig. 2) was found to be associated with elevated levels of interferon alpha-2 (IFN-α2), interleukin-5 (IL-5), IL-7, IL-8, and IL-17A, followed by a second wave appearing within 4 to 6 weeks and comprising IFN-γ, IL-10, and IL-12 p70, followed by IL-1RA and epidermal growth factor (EGF) during weeks 10 to 12 (Fig. 5). Thus, our data indicate that classic AHB correlated with an early and significant induction of IFN-α2 as well as IFN-γ, IL-12 p70, and IL-17A. In contrast to classic AHB, severe AHB was associated with virtually undetectable IFN-α2, IFN-γ, IL-12 p70, and IL-17A at any time point from baseline throughout the course of the disease. Compared to classic AHB, severe AHB was characterized by significantly elevated levels of IL-10, tumor necrosis factor alpha (TNF-α), and MIP-1β (Fig. 5). There was an early induction of IL-10 within 1 to 3 weeks of infection, which persisted at high levels, peaked concomitantly with the ALT peak, and rapidly decreased thereafter, returning to baseline values at week 13. TNF-α was already detectable at baseline but increased significantly to reach the highest levels concomitantly with the ALT peak and persisted at significantly higher levels throughout the course of the disease (Fig. 5 and Table S2). The levels of IL-2, IL-4, and IL-6 were consistently higher in severe than in classic AHB, but the difference did not reach statistical difference due to a wide range of value distribution.
FIG 5.



Distinct cytokine profiles correlated with disease severity and clinical outcome. Cytokine profiles in chimpanzees with classic or severe acute resolving or progressive hepatitis B, and acute resolving HBV/HDV coinfection, are shown. The circulating cytokine levels were quantified at weekly or biweekly intervals in each of the 8 animals after inoculation using a multiplex assay. The data, expressed in picograms per milliliter, represent the results from 3 sequential weeks at each time point (t1-3, t4-6, t7-9, t10-12, t13-15, t16-18, and t19-21) and are shown in a box-and-whisker plot, in which the median of the values for each time point is indicated by a horizontal line, with the 25th and 75th percentiles shown at the top and bottom of each box; the maximum and the minimum values are indicated by the whiskers. In each graph, the gray triangle represents the time point of the ALT peak, and the height is proportional to the mean value of the peak in each group. In progressive AHB, the archived samples available for weeks 4 to 6 were insufficient for cytokine profiling. AHB, acute hepatitis B.
In progressive AHB, which was characterized by a delayed ALT peak preceded by sustained high levels of HBV DNA and HBcrAg (Fig. 3C), there was a delayed and lower-magnitude induction of cytokines, such as IFN-α2, IFN-γ, and IL-12 p70, and IL-17A was virtually undetectable (Fig. 5 and Table S3). In this form of AHB, the levels of the entire cytokine profile ranged from undetectable to very low levels for the first 15 weeks, with the only exception of IL-6 levels, which were significantly higher than those of classic AHB starting from week 13 throughout the course of the disease. Due to the delayed ALT peak, which occurred after a mean of 23.5 weeks, and the lack of resolution of HBV infection in these animals, progressive AHB correlated with a late increase of several cytokines (between weeks 16 and 18 as well as 19 and 21) at a time when they tended to disappear in classic AHB, where the levels returned to baseline by weeks 19 to 21 (Fig. 5 and Table S3). Thus, progressive AHB was associated with very low to undetectable circulating cytokine levels for over 15 weeks, although the levels of HBV DNA were persistently high (over 8 log10 IU/ml) for more than 20 weeks before the ALT peak and remained high thereafter, in parallel with the establishment of chronic HBV infection.
Statistical comparisons of cytokine levels within the indicated time intervals between classic and progressive acute hepatitis B (AHB). Download Table S3, PDF file, 0.1 MB (68.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
We next analyzed the cytokine profile in HBV/HDV coinfection, which documented significant differences in the levels of 19 circulating cytokines compared to classic AHB (Fig. 5 and Table S4). We found that HBV/HDV coinfection correlated with lower levels of IFN-α2 and IFN-γ and virtually undetectable IL-12 p70 and IL-17A throughout the course of the disease. Compared to classic AHB, the difference reached statistical significance for IFN-γ (from weeks 7 to 12), IL-17A (from weeks 1 to 6), and IL-8 (during weeks 1 to 3) (Fig. 5 and Table S4). In HBV/HDV coinfection, IL-6 was the first and only cytokine with significantly elevated levels within the first 3 weeks postinfection. However, multiple cytokines showed elevated levels in successive waves: the first, within 4 to 6 weeks, included EGF, IL-2, IL-4, IL-10, and IL-15; the second, within 7 to 9 weeks, included IL-1α, IL-1β, IL-5, and IL-12 p40; and the third included EGF within 10 to 12 weeks and eotaxin, MCP-1, and TNF-α within 13 to 15 weeks (Table S4). Some of the cytokines were transiently elevated, while others were sustained at elevated levels for a longer time. The latter included IL-4, IL-6, and IL-15. Interestingly, most of the elevated cytokines appeared soon after the detection of HDV RNA, which was preceded by HBV DNA by 3 to 8 weeks, which suggests that this cytokine storm was triggered by HDV rather than by HBV.
Statistical comparisons of cytokine levels within the indicated time intervals between classic and HBV/HDV coinfection. Download Table S4, PDF file, 0.1 MB (67.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
We had the opportunity to analyze the systemic cytokine profile in the pattern of HDV superinfection as well. Strikingly, the circulating cytokine and chemokine profiles in HDV superinfection, which induced the highest ALT peak (1,910 IU/liter) ever seen in experimental chimpanzee studies, were almost completely silent (Fig. 6). Among the 29 cytokines tested, the only exceptions were IL-10, which showed two peaks, the first coinciding with the ALT peak and the second, of similar magnitude, immediately after the ALT peak, and IL-1RA, which showed a distinct peak 4 weeks after the ALT peak. The levels of MCP-1 and MIP-1β started high but showed 2 spikes, the first concomitant with the major ALT peak, and the second, 3 weeks later, concomitant with the second, lower-magnitude ALT peak. TNF-α levels were present and persisted at low levels with minor fluctuations from the baseline. Similar to severe AHB, in HDV superinfection, IFN-α2, IFN-γ, IL-12, and IL-17A were not detectable at any time point from baseline throughout the course of the disease, and, in contrast to HBV/HDV coinfection, in HDV superinfection we did not detect significant elevations of multiple proinflammatory cytokines, such as IL-1-α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-13, and IL-15. In all 9 chimpanzees with different forms of acute hepatitis B and D included in this study, the ALT peak coincided with a peak of IP-10, likely reflecting necroinflammation (Fig. 7). No significant difference in the levels of IP-10 was observed among the different forms of disease.
FIG 6.


Cytokine profiles in HDV superinfection of a chimpanzee chronically infected by HBV. The graphs represent the longitudinal analysis of circulating cytokine/chemokine levels. The red line represents the plasma/serum values of each cytokine, while the gray areas represent the ALT levels.
FIG 7.

Levels of circulating IP-10 (CXCL10) in chimpanzees with acute classic or severe acute resolving hepatitis B or progressive acute hepatitis B, HBV/HDV coinfection, and HDV superinfection. In each graph, the red line represents the plasma/serum IP-10 values of each animal at different time points, while the gray area shows the ALT levels. AHB, acute hepatitis B.
DISCUSSION
Historical studies conducted in chimpanzees at the NIH in the 1980s and 1990s provided us with a unique opportunity to investigate the basis for the different severities of liver damage and disease outcome associated with infection with wild-type HBV versus a precore HBV mutant, HBV/HDV coinfection, and HDV superinfection. Our study demonstrates that both the host and the virus play a role in determining disease severity and outcome. The importance of the host is illustrated by the different disease outcomes that we observed among the 4 animals that were inoculated with the wild-type HBV, two of which developed classic resolving AHB and two progressive AHB. The most peculiar feature that differentiated these two forms was the magnitude and timing of the ALT peak, which was significantly lower and delayed in progressive compared to classic AHB (mean, 23.5 week versus 13 weeks). In contrast, the shortest incubation time with the highest ALT peak (mean, 7.5 weeks) was observed in the two chimpanzees inoculated with the HBV precore mutant associated with ALF (7), illustrating the importance of viral factors in HBV pathogenesis. The severity of the disease was independent of the inoculum size, since one of these two animals received about 6 log10 more viral genome equivalents than the other. Indeed, both animals developed an equally severe form of AHB, which suggests that the precore HBV mutant carries an inherently higher pathogenicity that is independent of the host and the inoculum size. In a recent study (8), we characterized the intrahepatic antibody response in the same two chimpanzees by using phage display library and next-generation sequencing (NGS) technologies and compared the results with those seen in classic AHB and in HBV-associated ALF in humans (8). We found that severe AHB was characterized by a limited inflammatory reaction at the time of the ALT peak relative to the degree of hepatocellular damage, associated with B-cell infiltration, albeit less extensive than that in HBV-associated ALF (8). Thus, severe AHB shares a closer resemblance to ALF than to classic AHB.
In this study, we also analyzed three chimpanzees infected with HDV, two HBV/HDV coinfected and one HDV superinfected. We found that HDV superinfection of a chronic HBsAg carrier induced the highest ALT peak ever observed in chimpanzee studies, whereas in HBV/HDV coinfection the ALT peaks were considerably higher than that in classic AHB but lower than that in severe AHB caused by the precore mutant. Altogether, these data establish a hierarchy in the extent of liver damage according to the infecting virus: highest in HDV superinfection, followed by infection with the precore HBV mutant, HBV/HDV coinfection, and, lastly, wild-type HBV infection. We found that the kinetics of serum HBV DNA and HBcrAg did not distinguish among the different forms of hepatitis B and D. However, the similar kinetics that we observed between HBV DNA and HBcrAg indicate that HBcrAg is a very sensitive marker of HBV DNA replication. Others have reported similar findings, namely, that HBcrAg correlates positively with serum HBV DNA levels and with HBV cccDNA (21, 22). However, much remains to be done before the clinical usefulness of this assay is definitively established (23).
In HBV/HDV-coinfected chimpanzees, although there were two distinct peaks of viral replication several weeks apart, the first due to HBV and the second to HDV RNA, the acute hepatitis presented as a single peak concomitant with the peak of HDV RNA replication. However, two distinct peaks have been reported in other chimpanzee studies (24). Whether monophasic or biphasic hepatitis reflects different mechanisms of disease pathogenesis remains to be elucidated. The setting of HDV superinfection offered important insights into the role of viral and host factors in disease pathogenesis. The significant difference seen in the ALT peaks (136 versus 1,910 IU/liter) in the same animal suggests that the magnitude of liver damage can differ significantly according to the infecting virus and that the virus, not the host, was responsible for the enhanced liver damage in CH1133. It also underscores that the liver damage does not solely reflect the immune response of the host, raising the question of whether HDV is directly cytopathic, since high levels of HBV DNA were present throughout the course of the disease despite nearly normal ALT values for a long time before HDV superinfection. Consistent with our hypothesis (25), using a mouse model of severe acute HBV/HDV infection Usai et al. demonstrated that hepatic expression of HDAg resulted in the induction of severe liver damage.
To investigate the basis for the difference in the extent of liver damage following acute HBV and HDV infection, we evaluated virus-elicited immune responses by studying the long-term profile of 29 cytokines in serial plasma or serum samples taken weekly or biweekly over a period of 24 weeks. Our aim was to define the cytokine profile of classic AHB and to compare it with severe AHB, progressive AHB, and HBV/HDV coinfection or superinfection. Despite extensive investigations over 5 decades of HBV research, the immunopathogenesis of HBV infection is still incompletely understood. HBV is considered a noncytopathic virus, and several lines of evidence suggest that viral clearance is T-cell mediated and associated with hepatocyte killing by cytotoxic T lymphocytes (CTL) as well as by noncytopathic antiviral mechanisms (1, 14, 26–28). Moreover, limited data exist on the pathogenesis of the liver damage caused by HBV precore mutants or by HBV/HDV coinfection. Our collection of longitudinal samples allowed us to identify distinct cytokine profiles during the acute phase of HBV infection, which correlated with disease severity and clinical outcome. In classic AHB, the early interaction between HBV and innate immunity has not been fully elucidated. Previous studies performed by microarray analysis using liver specimens from chimpanzees infected with HBV failed to detect genes related to the innate immunity (29). An early suppressive effect of HBV on the innate immune response was also reported using an in vitro model (30) as well as in patients with acute HBV infection (31). However, other in vitro and in vivo studies have shown that HBV can trigger a rapid innate immune response (32–34). The availability of very early time points after virus inoculation allowed us to document an early induction of IFN-α2, suggesting that HBV does trigger an early innate immune response in classic AHB, along with IFN-γ, IL-12 p70, and IL-17A. All these cytokines were virtually undetectable at baseline and peaked very early, rapidly decreasing thereafter, prior to the ALT peak and the beginning of viral clearance. The fact that in infected chimpanzees IFN-α2 was detected very early, within the first 3 weeks, and then rapidly decreased may explain the discrepant results reported in the literature on the induction of innate responses by HBV, especially in human studies where the date of HBV infection can rarely be determined (31, 35). Although IFN-γ, IL-12 p70, and IL-17A are typically associated with Th1 responses and effective virus-specific CD4+ and CD8+ T-cell immunity (14, 26), their detection early in the course of HBV infection raises the question of whether cells associated with innate immunity, such as NK cells, γδ T cells, monocytes, and neutrophils (1), contribute to the increased circulating cytokine levels during the early phase of infection. NK and NKT cells are abundant in liver tissue, making up 30 to 40% of intrahepatic lymphocytes, and might constitute the main source of cytokine production during the initial phase of HBV infection (36).
Comparison of the cytokine profiles in classic and severe AHB caused by an HBV precore mutant demonstrated that the magnitude of liver damage in severe AHB correlated with a distinct cytokine profile. Strikingly, severe AHB was associated with a silent cytokine profile with respect to IFN-α2, IFN-γ, IL-12 p70, and IL-17A at all time points throughout the course of the disease. Interestingly, this silent cytokine profile in the same animal at the time of the ALT peak (week 6) correlated with a limited inflammatory infiltrate within the liver and a positive staining for caspase 3 activation, a marker of hepatocyte death (8). In contrast, caspase 3 staining was negative in classic AHB infected with the wild-type HBV, where at the time of ALT peak there was an extensive inflammatory infiltrate, particularly of T cells (8). Thus, the difference in the necroinflammatory reaction strongly suggests that the mechanisms of disease pathogenesis induced by the precore mutant differ from that induced by the wild-type HBV. At variance with classic AHB, severe AHB was associated with a significant induction of TNF-α within 4 to 6 weeks that persisted throughout the course of the disease, along with transient elevations of IL-10 and MIP-1β. TNF-α has been shown to trigger antiviral responses, but it can also enhance hepatocyte damage (37). Likewise, the association of IL-10 with acute symptomatic HBV infection was previously reported (31). In severe AHB, the elevation of IL-10 occurred earlier and more abundantly than that in classic AHB. Interestingly, IL-10 has been shown to promote tissue damage by increasing the viability of CTL (38). However, in severe AHB we observed an early increase of IL-10 without concomitant production of IFN-γ and other cytokines related to CTL activity. These observations indicate that liver damage in severe AHB is due to the intervention of other cells, including B cells and monocytes, neutrophils, and NK and NKT cells. Further studies are necessary to investigate the expression of these cytokines inside the liver, the organ where the disease occurs. A distinctive cytokine profile was also associated with progressive AHB, which was characterized by the presence of very low to undetectable levels of cytokines over the first 15 weeks, followed by modest elevations of multiple cytokines just before the ALT peak. The only cytokine that was significantly elevated compared to classic AHB was IL-6, starting at week 13 and persisting throughout the course of the disease. Thus, in progressive AHB there were very low to undetectable circulating cytokine levels in spite of persistently high levels of viremia since the very first weeks postinoculation, with values of HBV DNA higher than 8 log10 IU/ml. In a recent study conducted in acute resolving versus acute progressing hepatitis B in humans, Yoshio et al. found that resolving hepatitis was associated with higher levels of CXCL9, CXCL10, CXCL11, CXCL13, and IL-21 at ALT peak (39). They also showed that levels of the same cytokines were remarkably lower during a flare of hepatitis in chronic HBV infection, which is characterized by an exhaustion of HBV-specific T cells (40).
Very limited information exists on the pathogenesis of HDV disease and on the specific role of HBV and HDV. The complex interplay between these two viruses represents an additional challenge due to the lack of suitable animal models. Our analysis of historical chimpanzee studies demonstrated that the cytokine profile of acute HBV/HDV coinfection was distinct from that seen in classic AHB. We found that the increase in HBV DNA viremia was associated with remarkably lower levels of IFN-α2, IFN-γ, IL-12 p70, and IL-17A, whereas the subsequent increase in HDV RNA was associated with successive waves of elevated cytokines and chemokines, including, but not limited to, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12 p40, IL-15, and, later, MCP-1 and TNF-α. Thus, our results define a highly proinflammatory profile that was not seen in classic AHB, although both forms exhibited similarly high levels of serum HBV DNA. Since the monophasic ALT peak coincided with the HDV RNA peak, our data suggest that the pathogenesis of the liver damage differs between HBV/HDV coinfection and classic AHB, with a marked Th2 and innate immune response in the former compared to a predominant Th1 response in classic AHB. In contrast, HDV superinfection was characterized by the highest ALT peak but, surprisingly, a nearly silent cytokine profile. Of the 29 cytokines tested, we found that only IL-10, MCP-1, and MIP-1β showed changes concomitant with the ALT peaks, whereas IFN-α2, IFN-γ, IL-12 p70, and IL-17A were undetectable. Whether HDV enacts mechanisms that suppress the systemic activation of cytokines in the setting of acute superinfection remains to be elucidated. Further studies are needed to investigate whether, in the superinfection setting, high titers of HBsAg exert an immunoregulatory function by inducing tolerance, which may favor a silent cytokine profile, as has been suggested for chronic HBV infection (40, 41). The natural history of chronic hepatitis D is the superinfection of a chronic HBsAg carrier. However, to the best of our knowledge, there are no data on the cytokine profile during the acute phase of HDV superinfection in humans, and the data are also very limited for the chronic phase. In a recent study performed at a single time point in patients with chronic hepatitis D, the levels of IFN-γ and IL-12 p70 did not differ compared to those of controls, whereas IL-17A, IL-10, CXCL9, and TNF-α were found to be increased (42). Moreover, it has been shown that circulating CD4+ and CD8+ T cells from patients with chronic hepatitis D express low levels of IFN-γ and TNF-α upon in vitro activation (43).
In conclusion, despite the inherent limitations with historical chimpanzee studies to address the cellular sources of circulating cytokines in acute HBV and HDV infection, our data documented marked differences in the cytokine profiles that correlated with disease severity and clinical outcome. Pathogenesis studies support the notion that HBV and HDV are noncytopathic, but several aspects of disease pathogenesis remain to be elucidated. Our data challenge this concept and raise the question of whether an HBV precore mutant and HDV exert their damage through a direct cytopathic effect as well. Studies are under way to elucidate the role of other immune correlates of viral clearance in severe AHB and the mechanisms of disease pathogenesis in the setting of HBV/HDV coinfection and HDV superinfection, which are important not only to expand our understanding of HBV and HDV immunobiology but also to devise new therapeutic strategies.
MATERIALS AND METHODS
Ethics statement.
The animals were handled according to humane use and care guidelines specified by law and approved by the Animal Care and Use Committees of the National Institute of Allergy and Infectious Diseases.
Design of the study.
Weekly serum or plasma samples from nine chimpanzees, which were experimentally infected with HBV (6 animals) and HDV (3 animals) and monitored throughout the course of the disease, were included in this retrospective study. Animals represented several independent prior studies and were inoculated with well-characterized experimental preparations. HBV strains included the MS-2 strain, a challenge pool of wild-type HBV, subtype ayw, genotype D (13), a monoclonal HBV, subtype ayw, genotype D (14), and a precore mutant virus (genotype C) recovered from a case of fulminant hepatitis B (7). The HDV study inocula were HDV sera from a Japanese patient (N.K.) and chimpanzee passage infectious pools (44). The two animals (CH5872 and CH1627) that received approximately 8.3 log10 copies/ml of wild-type (serotype ayw) HBV developed classic AHB. Resolving acute hepatitis B, in both the classic and severe form, was defined by the clearance of HBsAg with the appearance of anti-HBs within 24 weeks from the time of inoculation.
Chimpanzees inoculated with the precore HBV mutant received 10−1 (CH1410) and 10−7 (CH1420) dilutions, respectively, of the index serum, which had an estimated titer of 8.0 log10 copies/ml. Thus, CH1410 received about 7 log10 copies and CH1420 about 10 copies. Both CH1410 and CH1420 developed severe AHB. Two chimpanzees, CH1133 and CH1616, were infected with the MS-2 strain (ay) and an HBV challenge pool (subtype ayw, genotype D), respectively. Both developed progressing AHB, which was defined by the persistence of HBsAg for more than 24 weeks after inoculation and lack of anti-HBs seroconversion. CH1133 was subsequently superinfected with HDV. Both HBV/HDV-coinfected animals (CH1309 and CH1441) received the MS-2 strain of HBV (as described above) 2 weeks before the HDV (CH1309) inoculum and 1 week after HDV (CH1441).
Serology.
Serum ALT was measured in serial serum samples as described previously (45, 46). Hepatitis markers for HBV (HBsAg, anti-HBs, and anti-HBc) were tested with commercial assays from Abbott (North Chicago, IL) and for HDV (anti-HDV) using an assay from DiaSorin (Saluggia, Italy) by following all manufacturer recommendations.
HBV core-related antigen.
HBcrAg titers were determined using a Lumipulse G HBcrAg assay with the Lumipulse G1200 analyzer (Fujirebio, Tokyo, Japan). The test has a 4-log dynamic range (3.0 to 7.0 log10 U/ml) and a lower detection limit of 2.6 log10 U/ml. Samples that fell outside the upper limit of detection were diluted and retested to obtain results within the linear analysis range.
NAT.
Serum RNA or DNA was extracted using commercial methods (QIAamp viral RNA minikit or QIAamp DNA minikit; Qiagen) from approximately 140 to 200 μl of serum or plasma. Real-time quantitative PCR methods were performed with cycling conditions based on the manufacturer's (ABI) recommendations and TaqMan chemistry. All the nucleic acid test (NAT) procedures featured a minimum 6-log dynamic range standard with lower detection limits approaching 50 to 200 copies, depending on the targeted amplicons. Appropriate negative and no template controls were included in all test runs.
HBV DNA TaqMan.
Serum HBV DNA was tested with techniques described previously (45). Briefly, extracted total DNA was assayed with TaqMan fast advanced master mix (ThermoFisher) and primers/MGB-probe directed near the 5′ end of the S gene.
HDV RNA TaqMan.
Serum HDV RNA was tested by real-time quantitative PCR, as described previously (47), in a one-step RT-PCR with TaqMan RNA-to-CT one-step kit chemistry, random hexamers, and primers/MGB-probe spanning nucleotides 894 to 971.
Multiplex cytokine analysis.
Serial plasma or serum samples were tested using a highly sensitive magnetic bead human cytokine kit (29-plex; Millipore). The Milliplex custom kit included cytokine targets EGF, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, IFN-α2, IFN-γ, IL-10, IL-12 p40, IL-12 p70, IL-13, IL-15, IL-17A, IL-1RA, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, TNF-α, TNF-β, and vesicular endothelial growth factor and chemokines eotaxin (CCL11), IL-8 (CXCL8), IP-10 (CXCL10), MCP-1 (CCL2), MIP-1α (CCL3), and MIP-1β (CCL4). The dynamic range, for all of the analytes in the assay, was 5 to 10,000 pg/ml and required 25 μl of serum or plasma. The samples were tested in duplicate along with relevant controls and standards. All manufacturer recommendations were followed in the performance of these assays. Data were acquired in real time via a Bioplex 200 (Bio-Rad) instrument, and Bioplex analysis software was employed for data fitting using a 5-parameter logistic curve.
Statistical analysis.
Differences in the plasma/serum levels of 29 cytokines/chemokines were calculated by using the values obtained from 3 sequential weeks at each time point (t1-3, t4-6, t7-9, t10-12, t13-15, t16-18, and t19-21) between classic AHB and the other forms of acute hepatitis B, including severe AHB, progressive AHB, and HBV/HDV coinfection, and were assessed by Mann-Whitney U test. Statistical analysis was performed using Prism 8 version 8.3.0 (GraphPad software). A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENT
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
This article is a direct contribution from Paolo Lusso, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Robert Perrillo, Hepatology Division, Baylor University Medical Center, and Thomas Schall, Chemocentrix Inc.
Citation Engle RE, De Battista D, Danoff EJ, Nguyen H, Chen Z, Lusso P, Purcell RH, Farci P. 2020. Distinct cytokine profiles correlate with disease severity and outcome in longitudinal studies of acute hepatitis B virus and hepatitis D virus infection in chimpanzees. mBio 11:e02580-20. https://doi.org/10.1128/mBio.02580-20.
REFERENCES
- 1.Chisari FV, Ferrari C. 1995. Hepatitis B virus immunopathogenesis. Annu Rev Immunol 13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 2.Shin SY, Jeong SH, Sung PS, Lee J, Kim HJ, Lee HW, Shin EC. 2016. Comparative analysis of liver injury-associated cytokines in acute hepatitis A and B. Yonsei Med J 57:652–657. doi: 10.3349/ymj.2016.57.3.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas HC. 1990. The hepatitis B virus and the host response. J Hepatol 11(Suppl 1):S83–S89. doi: 10.1016/0168-8278(90)90170-V. [DOI] [PubMed] [Google Scholar]
- 4.Hu J, Lin YY, Chen PJ, Watashi K, Wakita T. 2019. Cell and animal models for studying hepatitis B virus infection and drug development. Gastroenterology 156:338–354. doi: 10.1053/j.gastro.2018.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerin JL. 2001. Animal models of hepatitis delta virus infection and disease. Ilar J 42:103–106. doi: 10.1093/ilar.42.2.103. [DOI] [PubMed] [Google Scholar]
- 6.Wieland SF. 2015. The chimpanzee model for hepatitis B virus infection. Cold Spring Harb Perspect Med 5:a021469. doi: 10.1101/cshperspect.a021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogata N, Miller RH, Ishak KG, Purcell RH. 1993. The complete nucleotide sequence of a pre-core mutant of hepatitis B virus implicated in fulminant hepatitis and its biological characterization in chimpanzees. Virology 194:263–276. doi: 10.1006/viro.1993.1257. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, Engle RE, Shen CH, Zhao H, Schuck PW, Danoff EJ, Nguyen H, Nishimura N, Bock KW, Moore IN, Kwong PD, Purcell RH, Govindarajan S, Farci P. 2020. Distinct disease features in chimpanzees infected with a precore HBV mutant associated with acute liver failure in humans. PLoS Pathog 16:e1008793. doi: 10.1371/journal.ppat.1008793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z, Diaz G, Pollicino T, Zhao H, Engle RE, Schuck P, Shen CH, Zamboni F, Long Z, Kabat J, De Battista D, Bock KW, Moore IN, Wollenberg K, Soto C, Govindarajan S, Kwong PD, Kleiner DE, Purcell RH, Farci P. 2018. Role of humoral immunity against hepatitis B virus core antigen in the pathogenesis of acute liver failure. Proc Natl Acad Sci U S A 115:E11369–E11378. doi: 10.1073/pnas.1809028115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizzetto M, Canese MG, Gerin JL, London WT, Sly DL, Purcell RH. 1980. Transmission of the hepatitis B virus-associated delta antigen to chimpanzees. J Infect Dis 141:590–602. doi: 10.1093/infdis/141.5.590. [DOI] [PubMed] [Google Scholar]
- 11.Taylor JM. 2012. Virology of hepatitis D virus. Semin Liver Dis 32:195–200. doi: 10.1055/s-0032-1323623. [DOI] [PubMed] [Google Scholar]
- 12.Farci P, Niro GA. 2012. Clinical features of hepatitis D. Semin Liver Dis 32:228–236. doi: 10.1055/s-0032-1323628. [DOI] [PubMed] [Google Scholar]
- 13.Ogata N, Ostberg L, Ehrlich PH, Wong DC, Miller RH, Purcell RH. 1993. Markedly prolonged incubation period of hepatitis B in a chimpanzee passively immunized with a human monoclonal antibody to the determinant of hepatitis B surface antigen. Proc Natl Acad Sci U S A 90:3014–3018. doi: 10.1073/pnas.90.7.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. 2003. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 77:68–76. doi: 10.1128/jvi.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milich D, Liang TJ. 2003. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 38:1075–1086. doi: 10.1053/jhep.2003.50453. [DOI] [PubMed] [Google Scholar]
- 16.Carman WF, Fagan EA, Hadziyannis S, Karayiannis P, Tassopoulos NC, Williams R, Thomas HC. 1991. Association of a precore genomic variant of hepatitis B virus with fulminant hepatitis. Hepatology 14:219–222. doi: 10.1002/hep.1840140203. [DOI] [PubMed] [Google Scholar]
- 17.Terazawa S, Kojima M, Yamanaka T, Yotsumoto S, Okamoto H, Tsuda F, Miyakawa Y, Mayumi M. 1991. Hepatitis B virus mutants with precore-region defects in two babies with fulminant hepatitis and their mothers positive for antibody to hepatitis B e antigen. Pediatr Res 29:5–9. doi: 10.1203/00006450-199101000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Liang TJ, Hasegawa K, Rimon N, Wands JR, Ben-Porath E. 1991. A hepatitis B virus mutant associated with an epidemic of fulminant hepatitis. N Engl J Med 324:1705–1709. doi: 10.1056/NEJM199106133242405. [DOI] [PubMed] [Google Scholar]
- 19.Rizzetto M. 2009. Hepatitis D: thirty years after. J Hepatol 50:1043–1050. doi: 10.1016/j.jhep.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Pollicino T, Raffa G, Santantonio T, Gaeta GB, Iannello G, Alibrandi A, Squadrito G, Cacciola I, Calvi C, Colucci G, Levrero M, Raimondo G. 2011. Replicative and transcriptional activities of hepatitis B virus in patients coinfected with hepatitis B and hepatitis delta viruses. J Virol 85:432–439. doi: 10.1128/JVI.01609-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong DK, Seto WK, Cheung KS, Chong CK, Huang FY, Fung J, Lai CL, Yuen MF. 2017. Hepatitis B virus core-related antigen as a surrogate marker for covalently closed circular DNA. Liver Int 37:995–1001. doi: 10.1111/liv.13346. [DOI] [PubMed] [Google Scholar]
- 22.Wong DK, Tanaka Y, Lai CL, Mizokami M, Fung J, Yuen MF. 2007. Hepatitis B virus core-related antigens as markers for monitoring chronic hepatitis B infection. J Clin Microbiol 45:3942–3947. doi: 10.1128/JCM.00366-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Halewijn GJ, Geurtsvankessel CH, Klaasse J, van Oord GW, de Knegt RJ, van Campenhout MJ, Boonstra A, van der Eijk AA. 2019. Diagnostic and analytical performance of the hepatitis B core related antigen immunoassay in hepatitis B patients. J Clin Virol 114:1–5. doi: 10.1016/j.jcv.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Purcell RH, Satterfield WC, Bergmann KF, Smedile A, Ponzetto A, Gerin JL. 1987. Experimental hepatitis delta virus infection in the chimpanzee. Prog Clin Biol Res 234:27–36. [PubMed] [Google Scholar]
- 25.Usai C, Maestro S, Camps G, Olague C, Suarez-Amaran L, Vales A, Aragon T, Hommel M, Aldabe R, Gonzalez-Aseguinolaza G. 2020. TNF-alpha inhibition ameliorates HDV-induced liver damage in a mouse model of acute severe infection. JHEP Rep 2:100098. doi: 10.1016/j.jhepr.2020.100098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertoletti A, Ferrari C. 2016. Adaptive immunity in HBV infection. J Hepatol 64:S71–S83. doi: 10.1016/j.jhep.2016.01.026. [DOI] [PubMed] [Google Scholar]
- 27.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science 284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 28.Guidotti LG, Chisari FV. 2000. Cytokine-mediated control of viral infections. Virology 273:221–227. doi: 10.1006/viro.2000.0442. [DOI] [PubMed] [Google Scholar]
- 29.Wieland S, Thimme R, Purcell RH, Chisari FV. 2004. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A 101:6669–6674. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luangsay S, Gruffaz M, Isorce N, Testoni B, Michelet M, Faure-Dupuy S, Maadadi S, Ait-Goughoulte M, Parent R, Rivoire M, Javanbakht H, Lucifora J, Durantel D, Zoulim F. 2015. Early inhibition of hepatocyte innate responses by hepatitis B virus. J Hepatol 63:1314–1322. doi: 10.1016/j.jhep.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, Lascar RM, Brown D, Gilson RJ, Tedder RJ, Dusheiko GM, Jacobs M, Klenerman P, Maini MK. 2009. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 137:1289–1300. doi: 10.1053/j.gastro.2009.06.054. [DOI] [PubMed] [Google Scholar]
- 32.Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, Self SG, Borrow P. 2009. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol 83:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoneda M, Hyun J, Jakubski S, Saito S, Nakajima A, Schiff ER, Thomas E. 2016. Hepatitis B virus and DNA stimulation trigger a rapid innate immune response through NF-kappaB. J Immunol 197:630–643. doi: 10.4049/jimmunol.1502677. [DOI] [PubMed] [Google Scholar]
- 34.Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, Watanabe T, Iijima S, Sakurai Y, Watashi K, Tsutsumi S, Sato Y, Akita H, Wakita T, Rice CM, Harashima H, Kohara M, Tanaka Y, Takaoka A. 2015. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42:123–132. doi: 10.1016/j.immuni.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 35.Keating SM, Heitman JD, Wu S, Deng X, Stramer SL, Kuhns MC, Mullen C, Norris PJ, Busch MP. 2014. Cytokine and chemokine responses in the acute phase of hepatitis B virus replication in naive and previously vaccinated blood and plasma donors. J Infect Dis 209:845–854. doi: 10.1093/infdis/jit563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura K, Kakimi K, Wieland S, Guidotti LG, Chisari FV. 2002. Activated intrahepatic antigen-presenting cells inhibit hepatitis B virus replication in the liver of transgenic mice. J Immunol 169:5188–5195. doi: 10.4049/jimmunol.169.9.5188. [DOI] [PubMed] [Google Scholar]
- 37.Valaydon Z, Pellegrini M, Thompson A, Desmond P, Revill P, Ebert G. 2016. The role of tumour necrosis factor in hepatitis B infection: Jekyll and Hyde. Clin Transl Immunol 5:e115. doi: 10.1038/cti.2016.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fioravanti J, Di Lucia P, Magini D, Moalli F, Boni C, Benechet AP, Fumagalli V, Inverso D, Vecchi A, Fiocchi A, Wieland S, Purcell R, Ferrari C, Chisari FV, Guidotti LG, Iannacone M. 2017. Effector CD8(+) T cell-derived interleukin-10 enhances acute liver immunopathology. J Hepatol 67:543–548. doi: 10.1016/j.jhep.2017.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshio S, Mano Y, Doi H, Shoji H, Shimagaki T, Sakamoto Y, Kawai H, Matsuda M, Mori T, Osawa Y, Korenaga M, Sugiyama M, Mizokami M, Mita E, Katayama K, Tanaka J, Kanto T. 2018. Cytokine and chemokine signatures associated with hepatitis B surface antigen loss in hepatitis B patients. JCI Insight 3:e122268. doi: 10.1172/jci.insight.122268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrari C. 2015. HBV and the immune response. Liver Int 35(Suppl 1):121–128. doi: 10.1111/liv.12749. [DOI] [PubMed] [Google Scholar]
- 41.Op den Brouw ML, Binda RS, van Roosmalen MH, Protzer U, Janssen HL, van der Molen RG, Woltman AM. 2009. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: a possible immune escape mechanism of hepatitis B virus. Immunology 126:280–289. doi: 10.1111/j.1365-2567.2008.02896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Townsend EC, Zhang GY, Ali R, Firke M, Moon MS, Han MAT, Fram B, Glenn JS, Kleiner DE, Koh C, Heller T. 2019. The balance of type 1 and type 2 immune responses in the contexts of hepatitis B infection and hepatitis D infection. J Gastroenterol Hepatol 34:764–775. doi: 10.1111/jgh.14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schirdewahn T, Grabowski J, Owusu Sekyere S, Bremer B, Wranke A, Lunemann S, Schlaphoff V, Kirschner J, Hardtke S, Manns MP, Cornberg M, Wedemeyer H, Suneetha PV. 2017. The Third signal cytokine interleukin 12 rather than immune checkpoint inhibitors contributes to the functional restoration of hepatitis D virus-specific T cells. J Infect Dis 215:139–149. doi: 10.1093/infdis/jiw514. [DOI] [PubMed] [Google Scholar]
- 44.Purcell RH, Satterfield WC, Bergmann KF, Smedile A, Ponzetto A, Gerin JL. 1987. Experimental hepatitis delta virus infection in the chimpanzee, p 27–36. In Rizzetto M, Gerin JL, Purcell RH (ed), The hepatitis delta virus and its infection. Alan R. Liss, New York, NY. [PubMed] [Google Scholar]
- 45.Engle RE, Bukh J, Alter HJ, Emerson SU, Trenbeath JL, Nguyen HT, Brockington A, Mitra T, Purcell RH. 2014. Transfusion-associated hepatitis before the screening of blood for hepatitis risk factors. Transfusion 54:2833–2841. doi: 10.1111/trf.12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karmen A, Wroblewski F, Ladue JS. 1955. Transaminase activity in human blood. J Clin Investig 34:126–131. doi: 10.1172/JCI103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diaz G, Engle RE, Tice A, Melis M, Montenegro S, Rodriguez-Canales J, Hanson J, Emmert-Buck MR, Bock KW, Moore IN, Zamboni F, Govindarajan S, Kleiner DE, Farci P. 2018. Molecular signature and mechanisms of hepatitis D virus-associated hepatocellular carcinoma. Mol Cancer Res 16:1406–1419. doi: 10.1158/1541-7786.MCR-18-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Members and functional features of the cytokines/chemokines analyzed in this study. Download Table S1, PDF file, 0.1 MB (139.2KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Statistical comparisons of cytokine levels within the indicated time intervals between classic and severe acute hepatitis B (AHB). Download Table S2, PDF file, 0.07 MB (68.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Statistical comparisons of cytokine levels within the indicated time intervals between classic and progressive acute hepatitis B (AHB). Download Table S3, PDF file, 0.1 MB (68.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Statistical comparisons of cytokine levels within the indicated time intervals between classic and HBV/HDV coinfection. Download Table S4, PDF file, 0.1 MB (67.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.