

Abstract
The ongoing COVID-19 pandemic caused by the coronavirus, SARS-CoV-2, has already caused in excess of 1.25 million deaths worldwide, and the number is increasing. Knowledge of the host transcriptional response against this virus and how the pathways are activated or suppressed compared to other human coronaviruses (SARS-CoV, MERS-CoV) that caused outbreaks previously can help in the identification of potential drugs for the treatment of COVID-19. Hence, we used time point meta-analysis to investigate available SARS-CoV and MERS-CoV in-vitro transcriptome datasets in order to identify the significant genes and pathways that are dysregulated at each time point. The subsequent over-representation analysis (ORA) revealed that several pathways are significantly dysregulated at each time point after both SARS-CoV and MERS-CoV infection. We also performed gene set enrichment analyses of SARS-CoV and MERS-CoV with that of SARS-CoV-2 at the same time point and cell line, the results of which revealed that common pathways are activated and suppressed in all three coronaviruses. Furthermore, an analysis of an in-vivo transcriptomic dataset of COVID-19 patients showed that similar pathways are enriched to those identified in the earlier analyses. Based on these findings, a drug repurposing analysis was performed to identify potential drug candidates for combating COVID-19.
Keywords: Coronavirus, SARS-CoV-2, Transcriptome, Meta-analysis, Drug repurposing, Drug candidates
Abbreviations: COVID-19, Coronavirus disease-2019; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; SARS-CoV, Severe acute respiratory syndrome; MERS-CoV, Middle east respiratory syndrome
Graphical abstract
Highlights
-
•
SARS-CoV-2 mostly alters the host pathways similar to SARS-CoV and MERS-CoV.
-
•
Many innate immune pathways were activated upon all three H-CoVs infection in-vitro.
-
•
Glutathione metabolism and mitochondrial pathways were suppressed upon infection in-vitro.
-
•
COVID-19 patients show altered ribosomes and mitochondrial pathways.
-
•
Drug repositioning analysis identified potential drug candidates against COVID-19.
1. Introduction
Infectious diseases have always posed a threat to humanity, and new diseases continue to emerge. An “arms race” between the pathogens and the hosts has resulted in sporadic outbreaks, epidemics, and sometimes pandemics in the past, the present-day, and may also do so in the future. Among all infectious pathogens, ribonucleic acid (RNA) viruses have the supremacy [1]. They are infamous for their high mutation rate that leads to the acquisition of zoonotic potential, enhanced virulence, and evolvability [2]. Although enormous scientific and technological advances have been made in the study of these viruses, it remains a challenging task to deal with and ultimately stop the rapid evolution of viruses that can result in pandemics. The ongoing coronavirus (SARS-CoV-2, also known as novel COVID-19) pandemic is a perfect example of this challenge.
To date, seven coronaviruses have been found to infect human beings. Among them, HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU are the prevalent strains of coronavirus that can lead to mild respiratory illness [3,4]. In 2002, there was a human coronavirus (HCoV) outbreak in China, Severe acute respiratory syndrome (SARS), which caused fatal respiratory illness. Named severe acute respiratory syndrome coronavirus (SARS-CoV) [[5], [6], [7], [8]], this CoV killed around 750 people [9]. Ten years later, in 2012, there was another coronavirus outbreak, Middle East respiratory syndrome (MERS), this time in countries in the Middle East. This HCoV was named Middle East respiratory syndrome coronavirus (MERS-CoV) [10] and killed about 866 people [11]. Very recently, in December 2019, a novel HCoV outbreak was reported in the city of Wuhan in the Hubei Province of China. Identified as SARS-CoV-2 and widely known as coronavirus disease 2019 (COVID-19), the outbreak caused by this HCoV is very severe in comparison with the previous HCoV outbreaks. The virus spread quickly across almost all of the continents of the world and was declared a pandemic in March 2020 [12,13]. As of November 8, 2020, SARS-CoV-2 has infected over 50 million people, and the death toll has surpassed 1.25 million [14]. Although the mortality rate for COVID-19 is considered to be lower in comparison with MERS (34.4%) [11] and SARS (9.6%) [9], SARS-CoV-2 infects rapidly and has killed far more people in a short period.
It is, therefore, crucial to gain knowledge about the host transcriptome response of these three coronaviruses to better understand their pathogenesis with the aim of finding ways to repurpose existing drugs to combat this disease. It is known that SARS-CoV-2 uses the receptor angiotensin-converting enzyme (ACE) 2 in the same way as SARS-CoV to gain entry into human cells, whereas MERS-CoV infects cells through the dipeptidyl-peptidase 4 (DPP4) receptor [15]. Nevertheless, all three viral infections have similar clinical features, including fever, dry cough, and dyspnea. In addition, patients exhibit similar chest radiograph abnormalities, lowered total lymphocytes, increased lactate dehydrogenase, prolonged prothrombin rate, and cytokine storms [[16], [17], [18], [19]]. Also, early reports revealed that in SARS patients, there is a pulmonary infection and severe lung damage associated with elevated pro-inflammatory cytokines in serum (IL-1β, TNF-α, IL6, IL-8, IL-12, IFN-γ, IP10, MCP1, and many others) [20]. Meanwhile, other studies showed that MERS-CoV infection elicits type-I and type-II interferon responses and Th1 and Th17 cytokine profiles (IFN-γ, TNF-α, IL-15, and IL-17) [21]. A similar cytokine profile has also been reported in COVID-19 patients, where there is increased IL1β, IFN-γ, IP10, and MCP1, probably culminating in activated Th1 responses. These reports suggest that infection followed by a cytokine storm might be the probable reason for disease severity. Moreover, comorbid factors such as hypertension and diabetes are also shared by all three of these infections [22,23]. Hence, it is essential to find out whether unique or similar pathways are activated or suppressed by SARS-CoV and MERS-CoV in order to relate and compare these findings to those on the current pandemic-causing coronavirus, SARS-CoV-2. As the race for vaccines and therapeutics intensifies, it has become even more important to identify the underlying molecular mechanisms behind SARS-CoV-2 infection in order to design potential drugs or vaccine candidates.
Transcriptome meta-analysis is a powerful tool for identifying the underlying genes or signaling pathways as well as discovering the biomarkers for various diseases. Due to the increasing availability of microarrays and transcriptome high-throughput sequencing data, it is expected that the meta-analysis of different datasets under the same disease and control conditions will reveal novel pathways and genes with greater accuracy. Traditional approaches for drug development take years and involve tremendous cost and, moreover, more than 90% of drugs fail in the early development phases due to lack of efficacy and severe off-target effects [[24], [25], [26]]. On the other hand, the recently emerged genomics-based drug discovery approach utilizes databases such as the Broad Institute Connectivity Map database, which catalogs numerous approved drugs and the gene expression signatures of small molecules, in order to identify potential drugs that can be repurposed to different disease stages by using bioinformatic and computational approaches [27,28]. At the time of writing, there is still no proper drug or vaccine available for COVID-19, and many vaccine trails are still in clinical trials. Thus transcriptome-guided drug repurposing based on the SARS-CoV-2 infected transcriptome signature provides a ray of hope on the already approved list of potential drugs that can be used for the treatment of COVID-19.
In this study, first, we explored the host transcriptome responses upon SARS-CoV-2 infection with those of the two previous outbreak-related pathogenic coronaviruses, SARS-CoV, and MERS-CoV, through conducting a meta-analysis at different time points. The main rationale behind this meta-analysis was to identify the significant genes and associated pathways that are dysregulated at different time points of infection. As mentioned above, SARS-CoV and MERS-CoV share many common symptoms with SARS-CoV-2 infections. Hence, the transcriptomic responses of these two viruses were compared with those of SARS-CoV-2 in order to identify the common activated and suppressed pathways across all three of these contagious HCoV infections. Next, we cross-validated our in-vitro data observations with the results of the pathway analysis of in-vivo sequencing data derived from SARS-CoV-2 positive patients. This comparison revealed many pathways to be dysregulated upon all the three HCoVs. Then, we used a pipeline named cogena, which is based on the clustering of the coexpressed genes in the disease state, to identify potential drugs that can be repurposed for COVID-19 treatment. Thus, this study not only reveals potential drug candidates for COVID-19 treatment but also shows that specific pathways are altered similarly by different pathogenic coronaviruses. This study, therefore, provides valuable insights on the pathogenesis of human coronaviruses and novel drug targets to tackle future coronavirus outbreaks.
2. Methods
2.1. Time point transcriptome meta-analysis of SARS-CoV infection
In order to collect the transcriptome data of SARS-CoV infection, we queried the gene expression omnibus (GEO) database with the terms “SARS CoV,” “SARS CoV AND Homo sapiens”. We screened the datasets in a way that the infection was performed in a human cell line at different time points. We screened for the eligible datasets as per preferred reporting items for systematic reviews and meta-analyses (PRISMA) guidelines (Table S1). As of April 10, 2020, 6 datasets were eligible for further analysis, as per PRISMA guidelines. ( Table 1 ).
Table 1.
SARS-CoV datasets used for meta-analysis.
| GSE ID | Platform | Time points(hours) | Cell line used | |
|---|---|---|---|---|
| GSE47960 | GPL6480 | 12, 24, 36, 48, 60, 72 | Human airway epithelium cells (HAE) | |
| GSE47961 | GPL6480 | 24, 48, 60, 72 | Human airway epithelium cells (HAE) | |
| GSE47962 | GPL6480 | 12, 24, 36, 48, 60, 72 | Human airway epithelium cells (HAE) | |
| GSE37827 | GPL6480 | 12, 24, 36, 48, 60, 72 | Calu-3 | |
| GSE33267 | GPL4133 | 12, 24, 36, 48, 60, 72 | Calu-3 | |
| GSE17400 | GPL570 | 12, 24, 48 | Bronchial epithelial cell line 2B4 |
Using the GEOquery R package, the expression data of all these datasets were downloaded[29]. All these studies had corresponding triplicate time-matched mock and infected samples for each time point. The sample groups which were infected other than SARS-CoV were removed. We segregated expression data of each time point of all the datasets, and meta-analysis was performed time point-wise using the web tool NetworkAnalyst[30,31]. All the expression data were log2 transformed, if not performed, followed by quantile normalization. All the probe Ids were converted into their corresponding Entrez gene IDs for uniformity. In the case of multiple probes mapping to the same gene, the mean of their expression values was considered.
Limma R package was used to find the differentially expressed genes between the mock and SARS-CoV groups with a cutoff, adjusted p-value 0.05, and logFC 1.0 [32]. The integrity of the data was tested. The major challenge in the integration of multiple microarray data is the batch effect that arose due to the different microarray platforms, cell lines, design of the study [33]. The normalized datasets were subjected to the ComBat, an empirical Bayes method in-built in this tool, NetworkAnalyst [34]. It reduced the study-specific batch effects and confounding factors due to non-biological variation. The batch adjustment was visually examined through principal component analysis (PCA) plots.
For combining different datasets for the increased statistical power, we combined the p-values from each dataset using - Fisher's method (−2*∑Log(p)) with a significant level threshold of 0.05 [35,36]. The procedure was repeated for all individual time points- 12hr, 24hr, 36hr, 48hr, 60hr, 72hr, common across the selected studies.
2.2. Time point transcriptome meta-analysis of MERS-CoV infection
For MERS-CoV transcriptome data, we queried the GEO database with the terms “MERS CoV”, “MERS CoV AND Homo sapiens”. We screened the datasets in such a way that the infection was performed in a human cell line at different time points, and screening was performed as per PRISMA guidelines (Table S2). 12 datasets were eligible for further analysis (Table 2 ). All the expression data files of microarray were downloaded through the GEOquery R package and were segregated into different time points with matched mock controls [29]. Using NetworkAnalyst, all the datasets are log2 transformed if necessary and quantile normalized. Batch effects were adjusted by ComBat by visualizing the PCA plots. Limma R package was used to find the differentially expressed genes between the mock and MERS-CoV groups with a cutoff, adjusted p-value 0.05, and logFC 1.0 for each dataset. Since there is a high number of datasets available, we have selected the random effect model after performing Cochran's Q-test with an effect size cutoff 0.05, which basically calculates effect size across different studies based on metaMA Package. The DEGs of each time point meta-analysis were selected based on the p-value threshold 0.05 based on the random effect model.
Table 2.
MERS-CoV datasets used for meta-analysis.
| GSE ID | Platform | Time points(hours) | Cell line used |
|---|---|---|---|
| GSE81909 | GPL13497 | 0,12,24,36,48 | Primary human airway epithelial cell |
| GSE65574 | GPL13497 | 0,7,12,24 | Calu-3 |
| GSE79172 | GPL13497 | 0,3,7,12,24 | Primary human dendritic cell |
| GSE79218 | GPL13497 | 0,12,24,36,48 | Primary human microvascular endothelial cell |
| GSE79458 | GPL13497 | 0,12,24,36,48 | Primary human fibroblast |
| GSE86528 | GPL13497 | 0,12,24,36,48 | Primary human fibroblasts |
| GSE86529 | GPL13497 | 0,12,24,36,48 | Primary human fibroblasts |
| GSE86530 | GPL13497 | 0,12,24,36,48 | Primary human microvascular endothelial cell |
| GSE100496 | GPL13497 | 0,12,24,36,48 | Primary human fibroblasts |
| GSE100504 | GPL13497 | 0,12,24,36,48 | Primary human airway epithelial cell |
| GSE100509 | GPL13497 | 0,12,24,36,48 | Primary human microvascular endothelial cell |
| GSE56677 | GPL17077 | 3,7,18,24 | Calu-3 |
2.3. Functional enrichment analysis of DEGs
The significant differential expressed genes (DEGs) at each meta-analysis were subjected to over-representation analysis, which includes the Kyoto Encyclopedia of Genes and Genomes (KEGG)-pathway analysis and gene ontology enrichment analysis using the R package clusterProfiler [37] with adjusted p-value cutoff less than 0.05. The relation between the DEGs at each time points was visualized through circle plots, venn diagram, upset plots by using Metascape[38], and Intervene[39] tools. The dot plots were generated using clusterProfiler, and the number of entries to be displayed were based on the maximum visibility and readability of the plots. Geneset enrichment analysis (GSEA)[40] was performed using clusterProfiler by inputting gene names along with the logFC values. The genes were sorted according to their log fold change, and the number of permutations was set as 10000 with minimum gene set size 3, maximum gene set size 800, and p-value cutoff = 0.05 to identify the significant pathways. The activated and suppressed pathways were calculated based on the normalized enrichment score in each GSEA analysis.
2.4. Individual microarray and RNA-seq datasets differential expression analysis
The expression data of micro-arrays GSE33267 and GSE65574 were obtained using the R package, GEOquery [41,42]. After log transformation and quantile normalization, only 24-h time point samples were selected, and differential expression analysis was performed using Limma R package with a significance cut-off adjusted p-value 0.05 and logFC 1.0 to identify the differentially expressed genes (DEGs). For RNA-seq datasets, the raw read counts of GSE147507 [43] – Calu-3 cell line samples alone were extracted from GREP2 Rpackage [44], and they were subjected to Trimmed mean of M-values (TMM)-normalization and differential expression analysis was performed using R package EdgeR[45] with a cutoff - adjusted p-value less than 0.05 and logFC greater than 1 to identify the DEGs.
For GSE152075 [46], we downloaded the raw read counts data provided in the GEO database, and TMM-normalized and differential expression analysis was performed using the R package EdgeR with a significance threshold adjusted p-value 0.05 and logFC greater 1.5 to find the differentially expressed genes (DEGs).
2.5. Drug repurposing analysis
The normalized gene expression data of DEGs obtained after differential expression analysis were subjected to drug repurposing analysis using cogena R package[47]. The clustering parameter used was partition around medoids (PAM), and the number of clusters was determined so that a smaller number of genes are found in each cluster, and the same pathways were not shared by many clusters. The drug candidates were found by querying the downregulated cluster genes against the upregulated signature of connectivity-map (c-map) database [48] and upregulated cluster against the downregulated signature c-map signature.
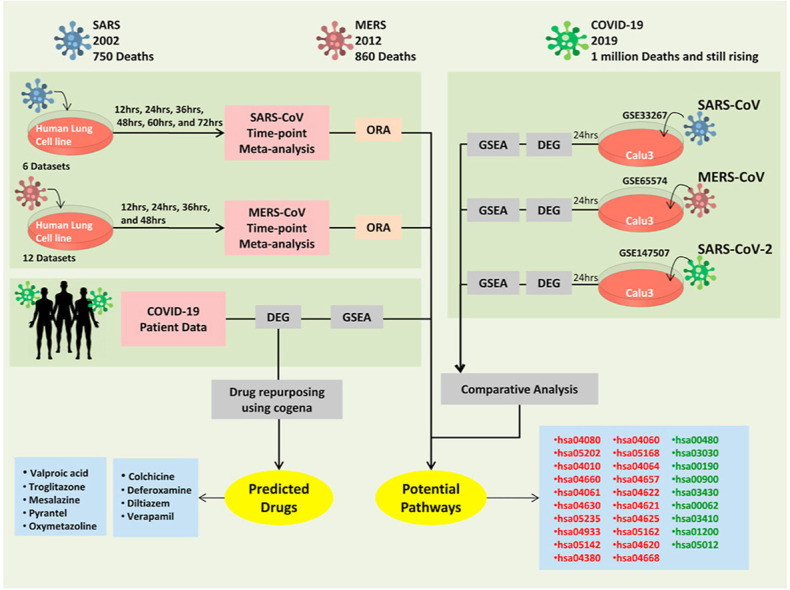
All the analyses were done using the R 3.6 environment and the tool NetworkAnalyst. The overall schematic of our approach to identify the potential drugs and pathways associated with contagious human coronaviruses were shown in Fig. 1().
Fig. 1.

Schematic workflow for the comparison of different disease-causing coronaviruses transcriptional responses and drug repurposing analysis to identify novel drugs.
Meta-analysis of SARS-CoV and MERS-CoV transcriptome data of each time point separately and significant genes were identified, and pathway enrichment analysis was performed, and the important pathways were identified. SARS-CoV, MERS-CoV, SARS-CoV-2 transcriptome data from three different studies performed in Calu-3 cell line at 24 h were analyzed to find significant genes, and GSEA analysis was performed to identify the common activated and suppressed pathways. In-vivo data of COVID-19 patient transcriptome was analyzed to check similar pathways that were enriched as observed in-vitro. The significant genes and their expression were subjected to drug repurposing analysis using a cogena workflow to identify potential drugs.
SARS-CoV-Severe Acute Respiratory Syndrome coronavirus; MERS-CoV- Middle East Respiratory Syndrome coronavirus; SARS-CoV-2 – Severe Acute Respiratory Syndrome coronavirus; GSEA- Gene set enrichment analysis; COVID-19-Coronavirus disease-2019.
3. Results
3.1. Integrated meta-analysis of transcriptome data of SARS-CoV infected cells identifies different signaling pathways enriched at different time points
To understand the molecular basis of the pathogenesis of SARS-CoV, first, we investigated the transcriptomic response of SARS-CoV-infected cells through a meta-analysis of the data at different time points (Fig. 1). From a search of the gene expression omnibus (GEO) database for the transcriptome datasets related to SARS-CoV infection in human cell lines, we selected six datasets for the analysis, as listed in Table 1. Using the GEOquery R package, the gene expression data of each microarray dataset were obtained. The matched mock and infected samples at different time point −12 h, 24 h, 36 h, 48 h, 60 h, and 72 h post SARS-CoV infection—were segregated, and the meta-analysis was performed at each time point. At each time point, the gene expression data of each dataset were log2 transformed and quantile normalized. Then, the normalized datasets were subjected to ComBat, an empirical Bayes method that is inbuilt in the NetworkAnalyst tool. This was done to reduce study-specific batch effects and confounding factors due to non-biological variation. The batch adjustment was visually examined through the use of principal component analysis (PCA) plots for each time point meta-analysis (Fig. 2 A–E).
Fig. 2.

Time point wise meta-analysis of SARS-CoV transcriptome datasets to identify differential pathways.
The common datasets for each time point were selected among screened datasets for meta-analysis. After normalization and bath adjustment using ComBat, the separation of samples at each time point 12 h,24 h,36 h,48 h,60 h,72 h (2A-F) is achieved by PCA. Differentially expressed genes in each dataset were calculated using the limma R package with a cutoff of logFC greater than 1, and the adjusted p-value less than 0.05. All the datasets at each time point were integrated using Fisher's method. The total number of significantly upregulated and downregulated genes common and unique between different time points were visualized through upset plots (2H,2J). The Over-representation analysis (ORA) for the KEGG pathway for upregulated (2G) and downregulated genes (2I) at each time points were visualized through dot plots. SARS-CoV-Severe Acute Respiratory Syndrome coronavirus; PCA – Principal component analysis; KEGG-Kyoto Encyclopedia of Genes and Genomes.
Differential expression analysis of the individual datasets of the mock and SARS-CoV groups was performed using the Limma R package with a cutoff adjusted p-value of 0.05 and logFC 1.0. Meta-analysis was performed at each time point by combining the p-values of multiple datasets using Fisher's method (−2*∑Log(p)) with a significant level threshold of 0.05.
The meta-analysis results indicated that 423, 4860, 9281, 10457, 10419, and 10147 genes were significantly dysregulated in the mock-infected and SARS-CoV-infected groups at 12 h, 24 h, 36 h, 48 h, 60 h, and 72 h, respectively. Among these, 276, 2175, 3634, 4249, 4281, and 4412 genes were upregulated, and 147, 2685, 5647, 6208, 6138, and 5735 genes were significantly downregulated at 12 h, 24 h, 36 h, 48 h, 60 h, and 72 h, respectively (Table S3). The commonalities and differences in the genes that were upregulated and downregulated at each time point were visualized through the use of upset plots see (Fig. 2H and J), respectively, and through the circos plots. (Fig. S1).
The upregulated genes at each time point were subjected to an over-representation analysis (ORA) for enriched pathways - a Kyoto Encyclopedia of Genes and Genomes (KEGG) (hereinafter, ORA-KEGG pathway analysis) at a cutoff adjusted p-value of less than 0.05 and their relations and differences were visualized through the use of a dot plot (Fig. 2G), in which the size of the dot represents the gene ratio of the particular pathway, and the color represents the significance level based on the adjusted p-value. The immune-related pathways, such as the tumor necrosis factor (TNF) signaling pathway, legionellosis pathway, and interleukin (IL-17) signaling pathway, only became enriched at 12 h. These results suggest that there are very few transcriptomic changes at 12 h of SARS-CoV infection, which is also evident from the PCA plot (Fig. 2A). The pathways related to the innate immune system such as the TNF signaling pathway, RIG-I-like receptor signaling pathway, Janus kinase/signal transducers and activators of transcription (JAK-STAT) signaling pathway, chemokine signaling pathway, interleukin (IL)-17 signaling pathway, influenza A pathway, nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling pathway, and mitogen-activated protein kinase (MAPK) signaling pathway were significantly enriched from 24 h post infection until 72 h post infection. Besides these immune pathways, other pathways such as advanced glycation end products (AGE)-receptor for advanced glycation end products (RAGE) signaling in diabetic complications pathway, osteoclast differentiation pathway, growth hormone synthesis, secretion, and cascade pathway, and apoptosis pathway were significantly upregulated 24 h post infection. At 36 h post infection, pathways such as the erythroblastic oncogene B (ErbB) signaling pathway and Hippo signaling pathway were upregulated until 72 h post infection. At later stages after 48 h of infection, adaptive immune pathways such as the antigen processing and presentation pathway and inflammatory signaling pathway associated genes were upregulated until 72 h post infection. Thus, it is evident that the innate immune pathways and chemokine signaling pathways were upregulated readily 24 h post infection, and at later stages, 48 h post infection, inflammation pathways, and adaptive immune processes and cell cycle-related signaling pathways were significantly enriched, in addition to the antiviral innate immune pathways.
The downregulated genes at each time point were subjected to an ORA-KEGG pathway analysis at a cutoff adjusted p-value of less than 0.05, and their relations and differences were visualized through the dot plot (Fig. 2I). The results of the ORA showed that none of the pathways were significantly downregulated at 12 h. Pathways such as the lysosome, cell cycle, glutathione metabolism, valine, leucine, and isoleucine metabolism pathways were significantly downregulated after 24 h until 72 h. Most of the metabolic pathways, such as carbon metabolism, oxidative phosphorylation, propoanate metabolism, fatty acid metabolism, and steroid biosynthesis pathways, were significantly downregulated after 36 h. In addition, pathways such as glycolysis, glyoxylate and dicarboxylate metabolism, TCA cycle, and pentose pathways were also significantly downregulated after 36 h. These results suggest that most of the metabolic pathways were downregulated upon SARS-CoV infection and that these pathways may, therefore, play a crucial role in the pathogenesis of SARS-CoV infection.
Gene ontology analysis for the Biological processes (GO-BP) of upregulated genes at 12-h time point revealed the enrichment of cell cycle-related processes such as chromosome segregation, mitotic nuclear division, and regulation of nuclear division, as well as the enrichment of antiviral innate immune genes such as those involved in type-I interferon signaling processes (Figs. S1–C). After 24 h, the innate immune pathways were significantly enriched, as observed in the ORA-KEGG pathway analysis. Similarly, gene ontology analysis of downregulated genes enriches for biological processes such as those related to the cell cycle, metabolic process and catabolic processes, and protein targeting to the endoplasmic reticulum (Figs. S1–D).
Thus, the meta-analysis identified previously known innate immune pathways as well as many metabolic pathways that have not been well studied with respect to coronavirus infections.
3.2. Integrated meta-analysis of transcriptome data of MERS-CoV infected cells identifies different signaling pathways enriched at different time points
Next, to gain further insights into the pathogenesis of fatal and contagious human coronaviruses (HCoVs), we investigated the transcriptomic response to MERS-CoV, which caused an outbreak in 2015 and resulted in a higher mortality rate than SARS-CoV. After searching the GEO database for transcriptome datasets related to MERS-CoV infection in human cell lines, we selected 12 datasets for the analysis, as listed in Table 2. The matched mock and infected samples at different time points—12 h, 24 h, 36 h, and 48 h post MERS-CoV infection—were segregated, and a meta-analysis was performed at each time point (Fig. 1). Since a larger number of datasets were available for this coronavirus, we applied the random effect model after performing Cochran's Q-test, which calculates the effect size across different studies based on the metaMA Package, and thus revealed high-confidence differentially expressed genes (DEGs).
The results of the meta-analysis showed that 1552, 8682, 9301, and 9318 genes were significantly dysregulated in the mock-infected and MERS-CoV infected groups with an adjusted p-value cutoff of less than 0.05, at 12 h, 24 h, 36 h, and 48 h, respectively (Table S4). Among these, 1026, 4037, 4038, and 4116 genes were upregulated, and 526, 4645, 5263, and 5202 genes were downregulated at 12 h, 24 h, 36 h, and 48 h, respectively. The significantly upregulated and downregulated genes obtained from each time point were visualized through circos plots (Fig. 3 A and C) and venn diagrams (Fig. 3B and D).
Fig. 3.

Time point wise meta-analysis of MERS-CoV transcriptome datasets to identify differential pathways.
The common datasets for each time point were selected among the screened datasets for meta-analysis. After normalization and differential expression analysis of each datasets using limma with significance threshold logFC greater than 1 and adjusted p-value less than 0.05, the batch adjustment was performed. A random effect model was chosen to integrate the data after performing Cochran's Q-test with a significance threshold of less than 0.05. Such meta-analysis was performed at 12 h, 24 h, 36 h, 48 h time points. Significant upregulated and downregulated genes between different time points obtained through meta-analysis were visualized through circos plot (3A and 3C) and venn diagram (3B and 3D), respectively. The Overrepresentation analysis for the KEGG pathway for upregulated (3E) and downregulated (3F) genes for enriched pathways at each time points was visualized through dot plots.
MERS-CoV- Middle East Respiratory Syndrome coronavirus; KEGG-Kyoto Encyclopedia of Genes and Genomes.
Then, an ORA-KEGG analysis was performed for the upregulated and downregulated genes at each time point with a cutoff of an adjusted p-value of less than 0.05 and visualized through the use of the dot plot (Fig. 3E and F). After 12 h of infection, many genes were differentially expressed compared to SARS-CoV, where MERS-CoV upregulates innate immune pathways such as the MAPK signaling pathway, TNF signaling pathway, and NFκB signaling pathway at 12 h. After 24 h, most of the innate immune antiviral pathways were upregulated, as observed for SARS-CoV. However, notably, these pathways were activated earlier in MERS-CoV as compared to SARS-CoV. As regards the downregulated genes, the ORA-KEGG analysis showed that the pathways related to Parkinson's disease (PD), Huntington's disease, Alzheimer's disease, oxidative phosphorylation, thermogenesis, and glutamate metabolism were significantly downregulated across all time points. Moreover, it should be noted that most of the genes related to the metabolic pathways that were significantly downregulated in the SARS-CoV infection, such as the carbon metabolism, oxidative phosphorylation, propoanate metabolism, and fatty acid metabolism pathways, were enriched for the downregulated genes in MERS-CoV too. Furthermore, pathways such as the glyoxylate and dicarboxylate metabolism, TCA cycle, and pentose pathways that were also significantly downregulated in SARS-CoV infection were also significantly downregulated in the MERS-CoV infection as well. It is also important to note that the glutathione metabolism pathway was downregulated at almost all of the time points in both the SARS-CoV and MERS-CoV infection.
The GO-BP analysis of the upregulated and downregulated genes at each time point with a cutoff adjusted p-value of less than 0.05 revealed that the p38 MAPK cascade was enriched among the upregulated genes at all time points in the MERS-CoV infection. Histone modification, acetylation, innate immune pathways, cytokine production, ribosome biogenesis, RNA transport, and viral responses were also observed to be upregulated after 24 h. However, the interferon pathways, which are critical for the antiviral response, were not much upregulated in MERS-CoV as compared to the SARS-CoV infection. Furthermore, the electron transport chain and other cellular respiratory processes and energy-generating processes, as well as metabolic processes, were significantly downregulated (Fig. S2).
Overall, we found that similar pathways, including innate immune system-related pathways, and metabolic pathways such as glutathione metabolism were significantly dysregulated in both the SARS-CoV and MERS-CoV infections. Also, there were differences in the time points of activation of several pathways, and the type 1 interferon response also varied between both coronavirus infections. These results revealed the different as well as the commonly disturbed pathways, which may be ideal targets for therapeutics and drug development.
3.3. Comparative gene set enrichment analysis of SARS-CoV-2, SARS-CoV, and MERS-CoV reveals crucial pathways for coronavirus pathogenesis
We also sought to investigate and better understand the SARS-CoV-2 transcriptomic response by identifying its similarities and differences with the transcriptomic responses of SARS-CoV and MERS-CoV. This approach was adopted as it was envisaged that it would help us to identify the common and specific features of SARS-CoV-2 that have enabled it to cause a pandemic as compared to the two previous strains that caused comparatively less spread. To this end, we searched the GEO database for transcriptomic profiles related to SARS-CoV-2. As of April 10, 2020, we obtained only the GSE147507 dataset, which is an RNA-sequencing dataset. That only one dataset was identified is not surprising, considering that we are continually developing our knowledge about this novel virus. Using the GSE147507 dataset, we decided to perform experiments on four lung cell lines, namely, Calu-3, NHBE, A549, and ACE2-overexpressed A549, which were infected with SARS-CoV-2 for 24 h.
Then, differential expression analysis was performed for SARS-CoV-2, and the results were visualized through a PCA plot (Fig. 4 A). To compare the transcriptomic changes at 24 h post infection of SARS-CoV-2 with those of MERS-CoV and SARS-CoV, we selected the dataset results for the Calu-3 cell line due to the availability of the transcriptome data for all three Human coronavirus infection in the same cell line. (Fig. 4B). Another important reason for the selection of Calu-3 cell line data for the comparative analysis is that it is the predominant cell line that is used for in-vitro studies of coronaviruses, and it has been shown that this cell line is highly susceptible to infection by SARS-CoV, MERS-CoV and SARS-CoV-2 [[49], [50], [51]]. This analysis focused on MERS-CoV(GSE65574), SARS-CoV(GSE37827) and SARS-CoV-2 (GSE147507). For MERS-CoV (GSE65574) and SARS-CoV (GSE37827), 24-h mock and infected samples were obtained, log2 transformed, and quantile normalized. Then, DEGs were calculated using the Limma R package, and this enabled us to identify the DEGs for both the virus infection (Fig. S3). For SARS-CoV-2 (GSE147507), (TMM)-normalized counts were obtained using the R package, GREP2, and differential expression analysis was performed using the EdgeR package. The subsequent ORA-KEGG pathway analysis showed that similar pathways were enriched to those that were observed to undergo enrichment in the meta-analysis, and there were no pathways that were enriched for the SARS-CoV downregulated genes (Fig. S3).
Fig. 4.

Comparison of Gene set enrichment analysis of SARS-CoV-2, SARS-CoV, and MERS-CoV transcriptomic response to Calu-3 cell line at 24 h identifies common activated and suppressed pathways:
Differential expression analysis of GSE147507 (SARS-CoV-2) was performed, and they were visualized through the PCA plot (4A). The transcriptome datasets of the Calu-3 cell line infected by SARS-CoV, MERS-CoV, and SARS-CoV-2 at 24 h time point were analyzed for comparison. GSE33267 for SARS-CoV, which is a microarray-based transcriptome dataset, GSE65574 for MERS-CoV, which is a microarray dataset, and GSE147507 for SARS-CoV-2, an RNA-seq transcriptomic data (Calu-3) was analyzed at 24 h. The Schematic of comparative Gene set enrichment analysis of three different virus-infected profiles in the same Calu-3 cell line is shown in (4B). Gene set enrichment analysis KEGG pathway of MERS-CoV (4C), SARS-CoV (4D), and SARS-CoV-2 (4E) were visualized through dot plots for significantly activated and suppressed pathways in each of virus infections.
SARS-CoV-Severe Acute Respiratory Syndrome coronavirus; MERS-CoV- Middle East Respiratory Syndrome coronavirus; SARS-CoV-2 – Severe Acute Respiratory Syndrome coronavirus; KEGG-Kyoto Encyclopedia of Genes and Genomes.
We also conducted a gene set enrichment analysis (GSEA) as it is a powerful method that can be used to identify whether a set of genes shows statistically concordant differences between two biological states. Hence, GSEA for the KEGG pathway for SARS-CoV (Fig. 4C), MERS-CoV (Fig. 4D), and SARS-CoV-2 (Fig. 4E) was performed to identify the significantly enriched pathways that were activated and suppressed in the case of each coronavirus infection (Table S5). From a comparison of the results, we found that 29 pathways were enriched in all three HCoVs infections at 24 h (Table 3 ). Among these, 20 pathways were activated, and nine pathways were suppressed after viral infection.
Table 3.
Common pathways dysregulated in SARS-CoV, MERS-CoV and SARS-CoV-2 infection.
| ID | Description | NES_SARS-CoV | NES_MERS-CoV | NES_SARS-CoV-2 |
|---|---|---|---|---|
| hsa00480 | Glutathione metabolism | −2.298910091 | −2.176969599 | −1.540403682 |
| hsa03030 | DNA replication | −1.958624185 | −2.306080464 | −2.33524989 |
| hsa00190 | Oxidative phosphorylation | −1.756224352 | −1.755329437 | −2.070679952 |
| hsa00900 | Terpenoid backbone biosynthesis | −1.655287551 | −1.710323926 | −2.110362992 |
| hsa03430 | Mismatch repair | −1.629671576 | −1.878065092 | −1.742815129 |
| hsa00062 | Fatty acid elongation | −1.621082916 | −2.303986236 | −1.832364297 |
| hsa03410 | Base excision repair | −1.515543072 | −1.934350218 | −2.13217846 |
| hsa01200 | Carbon metabolism | −1.471887044 | −2.329863517 | −2.028882929 |
| hsa05012 | Parkinson disease | −1.354013742 | −1.797668907 | −1.874990083 |
| hsa04080 | Neuroactive ligand-receptor interaction | 1.315559522 | 1.48141617 | 1.711103112 |
| hsa05202 | Transcriptional misregulation in cancer | 1.850616761 | 1.352428496 | 1.861600109 |
| hsa04010 | MAPK signaling pathway | 2.000048224 | 1.303268048 | 1.680859916 |
| hsa04660 | T cell receptor signaling pathway | 2.012926776 | 1.425834322 | 1.875550834 |
| hsa04061 | Viral protein interaction with cytokine and cytokine receptor | 2.055214707 | 1.92521804 | 2.321081622 |
| hsa04630 | JAK-STAT signaling pathway | 2.086423404 | 1.824222172 | 2.331190225 |
| hsa05235 | PD-L1 expression and PD-1 checkpoint pathway in cancer | 2.144907843 | 1.471589071 | 1.707746178 |
| hsa04933 | AGE-RAGE signaling pathway in diabetic complications | 2.178445053 | 1.448078598 | 2.172532258 |
| hsa05142 | Chagas disease (American trypanosomiasis) | 2.186347695 | 1.578187068 | 2.043611249 |
| hsa04380 | Osteoclast differentiation | 2.235233404 | 1.434292735 | 2.106658166 |
| hsa04060 | Cytokine-cytokine receptor interaction | 2.278204288 | 2.032755857 | 2.576990971 |
| hsa05168 | Herpes simplex virus 1 infection | 2.316383304 | 1.39516878 | 2.10152662 |
| hsa04064 | NF-kappa B signaling pathway | 2.32014547 | 1.766160539 | 2.262514526 |
| hsa04657 | IL-17 signaling pathway | 2.362340271 | 1.502799399 | 2.267902173 |
| hsa04622 | RIG-I-like receptor signaling pathway | 2.382563829 | 1.843020397 | 2.14365696 |
| hsa04621 | NOD-like receptor signaling pathway | 2.386470137 | 1.539298358 | 2.30220384 |
| hsa04625 | C-type lectin receptor signaling pathway | 2.400224233 | 1.417470732 | 2.177013108 |
| hsa05162 | Measles | 2.485373463 | 1.550968138 | 2.015915756 |
| hsa04620 | Toll-like receptor signaling pathway | 2.508984953 | 1.571493312 | 2.303162043 |
| hsa04668 | TNF signaling pathway | 2.738121175 | 1.703456341 | 2.424995068 |
NES-Normalized enrichment score.
Most of the pathways that were activated upon infection by these three HCoVs were found to be related to the innate immune system, such as the TNF signaling pathway, toll-like receptor pathways, retinoic acid-inducible gene-I(RIG-I)-like receptors pathway, and the nucleotide-binding oligomerization domain (NOD)-like receptor signaling pathway, as well as other immune pathways such as the T-cell receptor signaling pathway. Other non-immune pathways, such as the AGE-RAGE signaling pathway in diabetic complications, neuroactive ligand-receptor interaction pathway, and the osteoclast differentiation pathway, were activated upon these coronavirus infections. This finding is in accordance with the meta-analysis results we obtained for SARS-CoV and MERS-CoV at 24 h, and these pathways are upregulated until the later stages of infection.
On the other hand, the pathways for glutathione metabolism, carbon metabolism, oxidative phosphorylation, terpenoid backbone biosynthesis, fatty acid elongation, DNA replication, and PD were some of the significantly suppressed pathways. Pathways such as glutathione metabolism, carbon metabolism, and oxidative phosphorylation were downregulated at most of the time points in the meta-analysis also. Hence these pathways may play an important role in disease progression as well as virus replication. Glutathione levels are downregulated in many viral infections, not just in HCoV infections, and a reduction in glutathione levels has been found to be associated with increased cytokine response or cytokine storm [52]. Another important observation is that glutathione levels are very low in patients with acute respiratory distress syndrome (ARDS) and sepsis [53], hypertension [54], diabetes [52], cardiovascular diseases [55], and thrombosis [56]. All of these diseases are comorbid factors in patients with SARS-CoV-2 infection and can ultimately lead to high susceptibility and, eventually, fatality in these patients. Hence, we strongly speculate that drugs targeted at enhancing glutathione metabolism may reduce the severity of SARS-CoV-2 infection, particularly in patients with metabolic diseases. This speculation is supported by prior research that reported that inducing the level of glutathione in the later stage of the human immunodeficiency virus (HIV) infection inhibited replication [57].
We also found that pathways such as butanoate metabolism, d-glutamine and d-glutamate metabolism, arginine, and proline metabolism were suppressed in the case of SARS-CoV-2 alone. The meta-analysis results showed that these pathways were downregulated at different time points at later stages in MERS-CoV and SARS-CoV infections (Table S6). These results suggest that SARS-CoV-2 infection enriches mostly similar pathways to those enriched by SARS-CoV and MERS-CoV, but the transcriptional response of SARS-CoV-2 seems to be higher as compared to SARS-CoV. Hence common pathways would be ideal targets for drug discovery for the COVID-19 pandemic and possibly for any future coronavirus outbreaks or pandemics.
3.4. Analysis of COVID-19 patient transcriptome reveals similar pathways enriched, and drug repurposing analysis identifies potential drug candidates
In addition, we sought to extend our observations by analyzing the in-vivo transcriptome data of human COVID-19 patients. We were curious to know whether the common pathways that we observed in the SARS-CoV and MERS-CoV meta-analyses, as well as those that were revealed in the comparative GSEA analysis at the 24-h time point for all three pathogenic HCoVs (SARS-CoV, MERS-CoV, SARS-CoV-2), were similarly enriched in SARS-CoV-2- infected patients. For this purpose, we selected GSE152075, in which high-throughput RNA-sequencing of the nasal swab samples of 54 healthy subjects and 430 SARS-CoV-2 positive (POS) patients was performed [46]. After TMM- normalization, differential gene expression analysis was performed through EdgeR. The separation of the two different groups, healthy and POS patients, was visualized through a PCA plot (Fig. 5 A). In addition, GSEA was performed to identify the activated and suppressed pathways (Fig. 5B, Table S7).
Fig. 5.

GSEA and Coexpression Gene enrichment (Cogena) analysis of SARS-CoV-2 positive patients' transcriptome identify potential pathways and drug candidates.
GSE152075, high throughput transcriptome sequencing data obtained from SARS-CoV-2 positive (POS) patients’ nasal swabs were re-analyzed. The Segregation of Healthy and SARS-CoV-2 positive (POS) group was visualized through the PCA plot (5A). The Differential expression analysis was performed using EdgeR with significance threshold logFC>1.5 and adjusted p-value less than 0.05. GSEA analysis identified the activated and suppressed pathways in SARS-CoV-2 positive patients and visualized through the dot plot (5B). The Normalized expression of significant genes was subjected to cogena analysis, and different clusters were visualized through heatmap (5C). KEGG gene enrichment analysis for each cluster was visualized through the dot plot (5D). Drug repurposing analysis of the selected cluster identified potential drug candidates, which were visualized along with their enrichment score (5E).
PCA-Principal component analysis; SARS-CoV-2 – Severe Acute Respiratory Syndrome coronavirus; KEGG-Kyoto Encyclopedia of Genes and Genomes.
The results indicated that most of the innate immune pathways such as the toll-like receptor signaling, NOD-like receptor signaling, RIG-I-like signaling pathway, JAK-STAT signaling, chemokine signaling, and antiviral pathways as well as other pathways such as the neuroactive ligand-receptor interaction and olfactory transduction pathways were activated in the POS group, as observed in the in-vitro data analysis. In addition, there was activation of pathways such as complement and coagulation cascades and platelet activation in the SARS-CoV-2 positive patients, and the activation was more significant than that observed in the in-vitro data. Also, suppressed pathways such as PD, oxidative phosphorylation, carbon metabolism, glutathione metabolism, and thermogenesis were found to be suppressed in the POS group as in the in-vitro data analysis. The most significant suppression that was identified in the POS group occurred in the ribosome-associated pathways. Thus, we would suggest that these pathways are crucial for HCoV pathogenesis and associated comorbid effects.
In light of the foregoing, drugs that can reverse the above-described pathogenic signature of SARS-CoV-2 will be more effective in the treatment of COVID-19 disease. To test this hypothesis, we used the cogena pipeline, which identifies the coexpressed genes in a diseased group that differs from those in a healthy group. The basic assumption behind this tool is that coexpressed genes tend to have a similar function at the cellular level. Therefore, we subjected the normalized expression data of 1214 significant genes at above logFC 1.5 and at less than an adjusted p-value of 0.05 to cogena analysis. The number of clusters was adjusted so that the same pathways were not enriched for the same cluster. The DEGs were separated into eight clusters (Fig. 5C) using PAM clustering. Clusters 1, 2, 5, and 8 consisted of the downregulated DEGs in the POS samples, and clusters 3, 4, 6, and 7 contained the upregulated DEGs in the POS samples. A KEGG pathway analysis was performed on each cluster (Fig. 5D).
The results showed that Cluster 1 enriched pathways such as hypertrophic cardiomyopathy, cell adhesion molecules, cardiac muscle contraction, oxidative phosphorylation, Huntington's disease, and Alzheimer's disease. Cluster 2 enriched mainly the ribosome pathway. Cluster 3 enriched neuroactive ligand-receptor interaction. Cluster 4 enriched many innate immune pathways as well as chemokine signaling pathways. Cluster 5 did not enrich any pathway. Cluster 6 enriched pathways such as type 1 diabetes, graft versus host diseases, and complement and coagulation cascades. Cluster 7 enriched pathways related to chemokine signaling. Cluster 8 enriched the ribosome, oxidative phosphorylation as well as the neurological diseases pathways and cardiac muscle contraction.
Cluster 8 was selected for the drug repurposing analysis because it attained a higher enrichment score as compared to the other seven clusters (Fig. 5E). Most of the genes in this cluster are involved in the ribosome pathway and in the mitochondrial pathways, including oxidative phosphorylation that are crucial for the pathogenesis of neurological diseases such as PD, Huntington's disease, and Alzheimer's disease. It was recently hypothesized that ribosomal proteins exert antiviral activity by interacting with virus proteins, especially RPL9 (a protein in cluster 8 in our study), which binds with the rabies virus phosphoprotein and exerts antiviral activity [58]. Many of the HCoV proteins bind with many of the proteins in cluster 8. Hence it has been proposed that ribosomal proteins can be targeted for vaccine candidates [59]. Consequently, we performed a drug repurposing analysis to find drugs that can reverse the altered effect of these proteins. The results of this analysis showed that most of the drugs identified exert antiviral activity. Identified drugs such as Colchicine, Deferoxamine, Diltiazem, and verapamil are already undergoing clinical trials for use in the treatment of COVID-19 [[60], [61], [62], [63]]. Other identified drugs such as valproic acid, metformin, troglitazone, mesalazine, pyrantel, and oxymetazoline are promising drugs whose antiviral activity has already been reported [[64], [65], [66], [67]] and therefore show potential for use in COVID-19 treatment. The genes that were coexpressed in cluster 8 are listed in Table S9. All of the drugs predicted by this cluster are listed in Table 4 and depicted in Fig. 5E.
Table 4.
Drug repositioning using cogena.
| Drug | Mode of Action | Antiviral activity |
|---|---|---|
| Dexverapamil | R-enantiomer of calcium channel blocker verapamil | No |
| Deferoxamine | Iron-chelator | Yes |
| Diltiazem | Hyper tension drug | Yes |
| Gw-8510 | CDK-2 inhibitor | No |
| Colchicine | Anti-gout medicine | Yes |
| Paclitaxel | Anti-cancer drug | Yes |
| Nilutamide | Prostate cancer drug | No |
| Valproic acid | Anti-epilepsy drug | Yes |
| Metformin | Anti-diabetic drug | Yes |
| Verapamil | Anti-hypertensive drug | Yes |
| Mesalazine | Inflammatory bowel disease drug | Yes |
| Troglitazone | Anti-diabetic drug | Yes |
| Pyrantel | Anti-parasitic drug | Yes |
| Prilocaine | Anaesthic drug | No |
| Oxymetazoline | Anti-inflammatory drug | Yes |
| Cefotiam | Anti-biotic | Yes |
| Pentetrazol | Respiratory stimulant | No |
| Oxolinic acid | Antibiotic | Yes |
| Nitrofural | Antibiotic | No |
Most of the drugs that we identified through this approach exert antiviral activity. In addition, they are related to the treatment of comorbid factors such as diabetes, hypertension, and thrombosis. Many of these drugs are currently undergoing clinical trials involving COVID-19 patients. The drugs identified for the other clusters are listed inFig. S4 Table S8.
4. Discussion
The current SARS-CoV-2 pandemic is one of the biggest challenges to human health and poses a huge problem for scientists working in the various disciplines of biomedical sciences such as immunology, virology, vaccine biology, drug discovery and so on. Our study identified the crucial genes and pathways that are activated or suppressed at different time points upon infection with two pathogenic coronaviruses, SARS-CoV and MERS-CoV, through a meta-analysis of publicly available transcriptome data. We compared the transcriptome response of SARS-CoV-2 with SARS-CoV and MERS-CoV at 24 h post infection in a similar cell line to identify the common activated and suppressed pathways through GSEA. A comparison of the global GSEA of SARS-CoV, MERS-CoV, and SARS-CoV-2 in the same cell line and at the same time point revealed the common activated and suppressed signaling pathways that are crucial for the pathogenesis of HCoVs. Thus, more studies on these pathways and their contribution to pathogenesis may be helpful in understanding and controlling future coronavirus outbreaks. Finally, we identified some potential drug candidates that may be efficacious against SARS-CoV-2 based on co-expression and differential gene expression analyses of COVID-19 patient samples.
Many transcriptome meta-analysis studies have performed functional enrichment analysis by considering both the upregulated-regulated and downregulated DEGs in the same list. In this meta-analysis, the approach of conducting an enrichment analysis of the upregulated and downregulated genes separately using a hypergeometric distribution model proved to be more powerful in identifying important pathways [68] at different time points. Our time point-wise meta-analysis of eligible datasets revealed the genes that were dysregulated at each time point post infection. The advantage of this approach is that it can identify the involvement of several genes and pathways at different stages of infection. In line with a previous report [69], our meta-analysis showed that SARS-CoV induced much fewer transcriptomic changes as compared to MERS-CoV at early time points. Also, the ORA through KEGG pathway analysis and analysis of the gene ontology-biological processes revealed the pathways that were enriched at each time points for significantly upregulated and downregulated genes. The results indicated that immune pathways such as IL-17 signaling [18,21,69], TNF-signaling [70], MAPK signaling [71,72], RIG-I-like receptor signaling [73,74], NOD-like receptor signaling [75], toll-like receptor signaling [76], NFκB signaling [69], cytokine-cytokine receptor interaction, and C-type lectin receptor signaling [77] were all enriched for upregulated genes at most of the time points. These pathways have already been associated with coronavirus pathogenesis in previous studies and were therefore also expected to be activated upon SARS-CoV-2 infection, and indeed, our meta-analysis revealed that these pathways were significantly enriched and activated upon infection. On the other hand, the results showed that metabolic pathways such as PD, glutathione metabolism, TCA cycle, oxidative phosphorylation, thermogenesis, valine, leucine, and isoleucine degradation were some of the pathways that were enriched for downregulated genes in both SARS-CoV and MERS-CoV across most of the time points in our meta-analysis. Most of these pathways were also enriched for downregulated genes upon SARS-CoV-2 infection at 24 h (Fig. 4C).
We also compared the SARS-CoV-2 signature at 24 h post infection with the signatures of SARS-CoV and MERS-CoV in the same cell line, Calu-3, in different studies as there was no time point-wise transcriptome data available for SARS-CoV-2. Gene set enrichment analysis was performed to identify the activated and suppressed pathways in each type of infection. The results revealed that a few pathways were significantly dysregulated upon infection with all three of these HCoVs, but the association of these pathways with the respective infections is not well studied or understood. The AGE-RAGE signaling in diabetic complications pathway was one such pathway that was upregulated and activated upon infection by each of the three HCoVs, and thus this pathway may play an important role in the pathogenesis of SARS-CoV-2 infection. The RAGE is an inflammatory pattern recognition receptor that can recognize damage-associated molecular patterns (DAMPs) such as HMGB1 and S100s [78]. The AGE, which is formed as a result of non-enzymatic glycation of proteins at prolonged oxidative stress, binds to RAGE and, in turn, activates the extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) signaling pathway, leading to activation of pro-inflammatory nuclear factor (NFκB) [79]. Furthermore, AGE-RAGE signaling activation has been associated with cardiovascular diseases such as diabetes [80] and hypertension [81,82], as well as inflammatory diseases [83], ARDS [84], sepsis [85], and thrombosis [86], which are all co-morbidities of SARS-CoV-2 infection. The RAGE expression seems to be upregulated during inflammation, and knockdown of RAGE has been shown to protect mice from influenza-induced mortality [87]. However, studies related to this pathway and its association with HCoV pathogenesis remain scarce. Nevertheless, a recent work hypothesized that RAGE could act as a biomarker for the severity of COVID-19 [88]. Another study has shown that heightened susceptibility to gram-negative bacterial infection and hyper-inflammation in diabetes patients is activated by RAGE signaling [89]. Hence more studies are needed to understand the mechanism of this pathway activation upon HCoV infection and how it leads to comorbid mortality.
Another important observation is that glutathione metabolism was suppressed in all three pathogenic HCoV infections, and in the time point meta-analysis also it was enriched for downregulated genes at almost all time points in both SARS-CoV and MERS-CoV. Glutathione (γ-glutamylcysteinylglycine, GSH) is a ubiquitous antioxidant sulfhydryl containing tripeptide and is produced in most mammalian cells [[90], [91], [92]]. Glutathione exists in the cell in oxidized (GSSG) and reduced (GSH) states. The primary role of glutathione is to eliminate reactive oxygen species (ROS) and scavenge free radicals. The potent source of ROS, hydrogen peroxide, is detoxified by glutathione peroxidase by using GSH as the substrate. This results in a reduction in the ratio of hydrogen peroxide to water by linking two GSH molecules together via a disulfide bridge to form oxidized glutathione (GSSG). Then, GSSG is converted back to GSH by glutathione reductase utilizing nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor. This glutathione redox balance can be disturbed if there is excess production of ROS [93]. Moreover, glutathione regulates many other cellular functions such as a cofactor for several antioxidant enzymes as well as the regeneration of vitamins C and E, and it is also vital to mitochondrial function and maintenance of mitochondrial DNA and so on.
Several studies have indicated that the intracellular redox balance is altered during the establishment of a viral infection [94,95]. Hence shifting the GSH/GSSG redox toward the oxidizing state (oxidative stress) can activate several signaling pathways, including the MAPK, NFκB activation, protein kinase B, protein phosphatases 1 and 2A, apoptosis signal-regulated kinase 1, calcineurin, and c-Jun N-terminal kinase pathways, thereby increasing apoptosis, reducing cell proliferation and increasing cytokine levels, leading to tissue damage [96,97]. Thus, oxidative stress and low GSH levels play a key role in the pathogenesis of viral infections and diseases such as cancer [98,99], Alzheimer's disease [100], PD [101], liver diseases [102,103], cystic fibrosis [[102], [103], [104]], HIV [105], influenza [106], HSV-1, [107] rhinovirus [108], cardiovascular diseases [54], stroke, and diabetes [52]. Another interesting finding is that GSH levels also drop in patients with ARDS and sepsis [53].
Glutathione, the major antioxidant in fighting oxidative stress, has been found to inhibit various viral infections including influenza [109], dengue [110], and HIV [111]. Glutathione has also been reported to enhance the vitamin D regulatory and glucose metabolism genes and to increase 25-hydroxy-vitamin D levels in the blood [112]. Moreover, a recent study that used oral glutathione derivatives to block NFκB and subsequent cytokine storm syndrome in two patients suffering from COVID-19 indicated that this approach could have potential as a novel therapy to fight the pandemic[113].
Our results also indicated that the olfactory transduction pathway is significantly activated upon MERS-CoV and SARS-CoV-2 infection, as well as in COVID-19 patients. The involvement of this pathway in SARS-CoV-2 pathogenesis needs to be explored further because SARS-CoV-2 patients have been found to exhibit anosmia and olfactory dysfunction [[114], [115], [116]], and those with olfactory dysfunction as an early symptom recover relatively early from COVID-19 [117,118]. Recently, a report was published that showed that olfactory receptor neurons initiate ultra-rapid antiviral innate immunity against rhabdovirus after binding with its surface glycoprotein in a zebra-fish model [119]. This raises the prospect that those who experience olfactory dysfunction during the early stages of SARS-CoV-2 infection may have better antiviral immunity, and thus, their recovery rate is high from COVID-19. People in the older age group who have fewer olfactory receptor neurons may have a suppressed early response, which may lead to this group experiencing higher disease severity. However, more studies will be needed to justify this speculation.
Our analysis also showed that a similar neuroactive ligand-receptor interaction pathway was activated in all three pathogenic HCoVs, and is in line with the neurological implications of SARS-CoV-2 infection that have recently been raised [120]. We also found that the osteoclast differentiation pathway was activated in these three HCoVs. It has already been shown that osteoclast differentiation is induced by the SARS-CoV 3a/XI accessory protein with the enhancement of NFκB activity leading to excessive inflammation [121]. It has also been reported that the programmed death-ligand-1/programmed death-1(PD-L1/PD-1) checkpoint pathway in cancer is activated upon infection by all three pathogenic HCoVs. In addition, a fall in total lymphocyte counts in severe COVID-19 patients is common and associated with mortality [122]. Furthermore, over-expression of exhaustion markers such as PD-1 on the surface of T-cells has been observed in COVID-19 severe patients [123]. Blockage of PD-1 or PD-L1 restored the loss of T-cells and regulated cytokine production. Hence the use of immune checkpoint inhibitors has recently been proposed for COVID-19 treatment [124].
We found that oxidative phosphorylation was another pathway that was suppressed upon infection by the three HCoVs. It has recently been proposed that upon SARS-CoV-2 infection, there is a faltering of oxidative photophosphorylation, and the TCA cycle, leading to the shutdown of melatonin, which may ultimately cause a cytokine storm [125]. We also observed most of this pathway's genes were downregulated in our time point meta-analysis of SARS-CoV and MERS-CoV as well as in the transcriptome dataset derived from SARS-CoV-2 positive nasal swabs. A recent unpublished pre-print showed that Nsp12 and Nsp13 proteins of SARS-CoV-2 bear a mitochondrial recognition signal and the possibility of causing mitochondrial dysfunction [126]. Hence further studies that explore the mitochondrial pathways and their association with HCoV infections and cytokine storm is encouraged. Mitochondrial dysfunction has been reported in COVID-19 [127,128] as well as in SARS [129]. Moreover, it has been shown that there is a significant upregulation of mitochondrial genes in the patients who have recovered from SARS [130].
Our results also indicated that the PD pathway was suppressed in the case of all three HCoVs. It is known that PD is associated with mitochondrial dysfunction [131]. Besides, viral infections are considered to be a risk factor for PD [132]. Although there are no studies that prove the association between PD and HCoV infections, one study detected antibodies against four coronavirus antigens, including two HCoVs, in the cerebrospinal fluid of PD patients and a possible correlation between HCoV infection and PD [133]. Following the proposed dual-hit theory for PD [134], it may be that SARS-CoV-2 may also have the potential to increase PD risk in the future, especially considering its neuro-invasive potential [135]. It has already been shown in a comorbid factor analysis that SARS-CoV infection is correlated with PD [136]. A controversial hypothesis has also been put forward that the 1918 influenza pandemic increased the risk of PD and those who were born or who were young during the pandemic had a higher risk of developing PD as compared to those born before 1888 or after 1924 [[137], [138], [139], [140], [141]]. In accordance with our analysis findings, it was recently reported that the COVID-19 patients developed PD after infection [142] and the speculation that the infected individuals may be at the increased risk of developing PD in the future [143]. Although these findings seem dubious, future studies should focus on the continuous monitoring of COVID-19 recovered patients to determine the long-term deleterious effects of SARS-CoV-2 on neurological disorders such as PD, Alzheimer's disease, and Huntington's disease, the pathways of which were found in our analysis to be dysregulated upon HCoV infections.
In an attempt to validate our in-vitro observations by comparing them with in-vivo COVID-19 infected patient samples, we reanalyzed the transcriptome datasets derived from the nasal swabs taken from SARS-CoV-2 positive patients and compared them with their corresponding controls. The GSEA of both datasets revealed that most of the pathways that were activated or suppressed were similar to those observed in our analysis using the Calu-3 cell line data.
We also employed the cogena workflow to evaluate potential drugs based on the coexpressed genes among the DEGs. Several studies have utilized this workflow to predict potential drug candidates from patient transcriptome datasets [144,145]. In a recently published study [145], after performing differential analysis, drug repurposing was performed based on the cogena workflow in a reanalysis of transcriptome data of the BALF-fluid obtained from 10 COVID-19 patients and 20 healthy subjects. Although that study employed the same pipeline for drug repurposing analysis as ours, we selected a study with a higher number of COVID-19 positive samples (430 COVID-19 positive patients) and healthy controls (54 COVID-19 negative healthy subjects), which allows more reliable prediction. The potential drug candidates that were identified in each cluster were expected to reverse the pathogenic disease state and can be further recommended for repurposing.
Among the predicted drugs identified in our drug repurposing analysis, Verapamil and its R-enantiomer, Dexverapamil, are calcium channel blockers that are already approved for therapeutic use in the treatment of high blood pressure and heart diseases, which are among the comorbid factors of COVID-19. It has already been shown that Verapamil at higher doses can inhibit filoviruses such as Ebola virus in-vitro [146]. Also, it has been reported that Verapamil can inhibit the influenza virus by disrupting the calmodulin-dependent intracellular activities necessary for viral assembly [147,148]. There is an ongoing clinical trial to investigate its role in treating COVID-19 patients [60]. Another calcium channel blocker in the same group, Diltiazem, has also been reported to exert anti-viral activity [148] and is also reported to be an inhibitor of NFκB signaling that can reduce excessive cytokine production [149] which is the most significant cause of COVID-19 associated deaths. The use of Diltiazem for COVID-19 treatment is being under clinical trials [62].
Other drugs that were predicted by our study include deferoxamine, which is an iron-chelator drug that has been shown to inhibit HIV replication [150], and its effect on COVID-19 patients is being tested in an ongoing clinical trial. Another drug with potential is colchicine, which is used to treat gout and Bechet's disease, and a clinical trial is underway to test its effect on COVID-19 patients [63]. Paclitaxel, which is basically a chemotherapy agent, is another drug that our analysis predicted could be useful in the fight against COVID-19. It has also been reported to have antiviral activity against HIV [151]. However, its use against HCoV infections is yet to be explored.
Valproic acid, an epilepsy drug, is another promising drug for the treatment of COVID-19 infection that was predicted by the drug repurposing analysis. Its antiviral activity has already been reported [64], and it can be used in the treatment of ARDS and can reduce cytokine storm by inhibiting the activation of NFκB signaling [152,153]. Moreover, a recent unpublished study showed that the metabolite of valproic acid Co-enzyme A (CoA) has the potential to bind with the non-structural protein-12 (nsp-12) of SARS-CoV-2 [154]. However, further studies are needed to evaluate its potential in treating COVID-19 infection.
The results of our analysis also identified metformin as a potential drug candidate. This drug is widely used to treat type 2 diabetes, which is also one of the important comorbid factors associated with COVID-19 disease. It has recently been shown that Metformin can reduce the complications and severity of COVID-19 in patients through its ability to reduce the C-reactive protein (CRP) and TNF-A levels [65,155,156]. Troglitazone is another antidiabetic drug that can be used for COVID-19 treatment. Troglitazone has been shown to inhibit the hepatitis-B virus [66] and has immunomodulatory effects on cytokine production [157]. It has also been shown to significantly reduce IL-6 levels [158].
Among the other drugs predicted by the same cluster in our analysis was Mesalazine. This drug is mainly used in the treatment of inflammatory bowel diseases [159], and it has been reported to reduce mortality when used in combination with neuraminidase inhibitor and results in better recovery from H5N1 influenza in infected mice. It would, therefore, be interesting to study its role in treating COVID-19 infection [160]. Another drug was Pyrantel, which is used to treat parasitic worm infection. It has been reported that pyrantel can inhibit Chikungunya virus nsp1 cap protein [161]. A further drug, oxymetazoline, which is basically a decongestant used in nasal sprays, has been reported to resolve inflammatory effects mediated by neutrophils [162] and has also been shown to reduce replication of the human rhinovirus [67]. Hence, it may be a promising drug for mediating the severity of COVID-19 disease. On the other hand, Cefotiam is a broad-spectrum antibiotic whose antiviral role has not been much studied. However, a recent study identified the potential of cefotiam to target TMPRSS2, and hence it may have an important role to play in interfering with SARS-CoV-2 binding [163]. Lastly, oxalinic acid is a quinolone antibiotic that can inhibit bacterial DNA gyrase enzyme. It has been shown that oxalinic acid can inhibit human polyomavirus (BKV) [164], but its association with HCoV pathogenesis is yet to be explored.
In sum, through our multipronged approach, we identified potential drugs for COVID-19 treatment as well as the crucial pathways that are dysregulated upon infection by contagious HCoVs, which cause outbreaks. However, this study has its limitations. First, the time point meta-analysis was constrained to an extent by the availability of the data as well as the number of time points post infection. For SARS-CoV, a relatively smaller number of datasets with more common time points were analyzed as compared to MERS-CoV. Also, due to the varied number of datasets included in each meta-analysis required the use of different statistical methods to integrate the datasets. In addition, different meta-analysis approaches were adopted for SARS-CoV and MERS-CoV due to the differences in the number of samples as well as the number of datasets used in the analysis. However, we found that most of the meta-analysis pathway results at the 24-h time point coincided with those for the individual datasets that were reanalyzed for comparison in the Calu-3 cell line. Second, due to the non-availability of appropriate transcriptome datasets related to other HCoVs that are not so pathogenic and did not cause outbreaks, such as HCoV-OC43 and HCoV-229E, we cannot say with certainty that the pathways we identified are unique to pathogenic HCoVs such as SARS-CoV, MERS-CoV, and SARS-CoV-2. Another important limitation is that we were unable to validate the identified drugs by in-vitro experiments due to the prevailing conditions in the country, which made it challenging to get to the laboratory.
Nevertheless, this study has revealed many important pathways and their associations which have not previously been studied in relation to HCoV infections. More studies on these pathways may be beneficial for the design of suitable drugs and the treatment of the current COVID-19 pandemic as well as future CoV outbreaks. It is hoped that the potential drug candidates identified herein may be further validated and proposed for clinical studies and, if promising results are forthcoming, we expect that this pandemic will come to an end soon.
5. Conclusion
Through time point meta-analysis, this study revealed that crucial pathways were dysregulated upon SARS-CoV and MERS-CoV infection. Most of these pathways were also found to be dysregulated by SARS-CoV-2, the cause of the current COVID-19 pandemic. We validated these findings by means of an in-vivo analysis of the transcriptome response of COVID-19 patients. Subsequently, we identified potential pathways and the drugs to be targeting these pathways for the treatment using co-gene expression and drug repurposing analysis.
CRediT authorship contribution statement
Pandikannan Krishnamoorthy: Conceptualization, Data curation, Formal analysis, Validation, Writing - original draft, Writing - review & editing, Project administration. Athira S. Raj: Data curation, Writing - original draft, Writing - review & editing. Swagnik Roy: Writing - review & editing. Nachimuthu Senthil Kumar: Writing - review & editing. Himanshu Kumar: Conceptualization, Validation, Writing - original draft, Writing - review & editing, Project administration, Supervision.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge all the contributors of Gene expression profiling datasets we used in this study for making it available public for reuse.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2020.104123.
Funding
This work is supported by the Indian Institute of Science Education and Research (IISERB) Bhopal. P.K. is supported by (IISER)- Bhopal Institutional fellowship; A.R. is supported by CSIR fellowship.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
PRISMA flow chart for SARS-CoV meta-analysis
PRISMA flow chart for MERS-CoV meta-analysis
Significant genes of SARS-CoV at each time point after meta-analysis.
Significant genes of MERS-CoV at each time point after meta-analysis.
Common pathways enriched by GSEA KEGG analysis for SARS-CoV, MERS-CoV, SARS-CoV-2 Calu-3 -24 hours datasets.
Potential pathways common for SARS-CoV, MERS-CoV, and SARS-CoV-2.
GSEA KEGG analysis results for SARS-CoV-2 positive patients compared with the healthy patient samples.
Drugs enriched in other clusters in cogena analysis.
Genes enriched at cluster 8 subjected to Drug Repositioning analysis.
Figure S1.

Gene ontology analysis of significant genes obtained through SARS-COV meta-analysis at each time points.
Circos plot denoting a relation between different genes upregulated (A) and downregulated(B) significantly at each time points after meta-analysis. Gene Ontology analysis of the Biological process for each time point for upregulated (C) and downregulated (D) significant genes after meta-analysis were visualized through dot plots
Figure S2.

Gene ontology analysis of significant genes obtained through MERS-COV meta-analysis at each time points.
The Gene Ontology analysis for biological processes at each time points for the genes upregulated (A) and (B) downregulated obtained through meta-analysis were visualized through dot plots
Figure S3.

ORA analysis of Calu-3 datasets of SARS-CoV, MERS-CoV, and SARS-CoV-2 at 24 hours post infection
Differential expression analysis of GSE33267 (SARS-CoV), GSE65574 (MERS-COV), GSE147507 (SARS-CoV-2) was performed individually, and they were visualized through PCA plots (A), (B), (C), respectively. The significant genes with logFC greater than 1 and adjusted p-value less than 0.05 were subject to overrepresentation analysis for KEGG pathways enrichment. SARS-CoV upregulated genes enriched pathways were visualized through the dot plot (C). Significantly downregulated genes did not enrich any pathways. MERS-CoV upregulated, and down-regulated genes pathways enrichment was visualized through the dot plots (D) and (E), respectively. SARS-CoV-2 upregulated and downregulated genes pathway enrichment was visualized through dot plots (F) and (G), respectively.
Figure S4.

Drug repurposing analysis for all clusters of co-expressed genes
The Potential drug candidates targeting the clusters 4 (A),5(B),1(C),2(E)were visualized along with their enrichment scores.
References
- 1.Carrasco-Hernandez R., Jacome R., Lopez Vidal Y., Ponce de Leon S. Are RNA viruses candidate agents for the next global pandemic? A review. ILAR J. 2017;58(3):343–358. doi: 10.1093/ilar/ilx026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duffy S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018;16(8) doi: 10.1371/journal.pbio.3000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kin N., Miszczak F., Lin W., Gouilh M.A., Vabret A., Consortium E. Genomic analysis of 15 human coronaviruses OC43 (HCoV-OC43s) circulating in France from 2001 to 2013 reveals a high intra-specific diversity with new recombinant genotypes. Viruses. 2015;7(5):2358–2377. doi: 10.3390/v7052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin Y., Wunderink R.G. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology. 2018;23(2):130–137. doi: 10.1111/resp.13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drosten C., Gunther S., Preiser W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 6.Fouchier R.A., Kuiken T., Schutten M. Aetiology: koch's postulates fulfilled for SARS virus. Nature. 2003;423(6937):240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ksiazek T.G., Erdman D., Goldsmith C.S. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 8.Zhong N.S., Zheng B.J., Li Y.M. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet. 2003;362(9393):1353–1358. doi: 10.1016/S0140-6736(03)14630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.WHO) WHO . 2003. Summary of Probable SARS Cases with Onset of Illness from 1 November 2002 to 31 July 2003. [Google Scholar]
- 10.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 11.WHO) WHO . 2020. MERS Situation Update, January. [Google Scholar]
- 12.Wu F., Zhao S., Yu B. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q., Guan X., Wu P. Early transmission dynamics in wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020;382(13):1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.WHO) WHO . 2020. Coronavirus Disease (COVID-2019) Situation Reports. [Google Scholar]
- 15.Hoffmann M., Kleine-Weber H., Schroeder S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi H., Han X., Jiang N. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect. Dis. 2020;20(4):425–434. doi: 10.1016/S1473-3099(20)30086-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee N., Hui D., Wu A. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348(20):1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 18.Huang C., Wang Y., Li X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Z., McGoogan J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese center for disease control and prevention. J. Am. Med. Assoc. 2020;323(13) doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 20.Wong C.K., Lam C.W., Wu A.K. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004;136(1):95–103. doi: 10.1111/j.1365-2249.2004.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahallawi W.H., Khabour O.F., Zhang Q., Makhdoum H.M., Suliman B.A. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine. 2018;104:8–13. doi: 10.1016/j.cyto.2018.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan W.J., Liang W.H., Zhao Y. Comorbidity and its impact on 1590 patients with COVID-19 in China: a nationwide analysis. Eur. Respir. J. 2020;55(5) doi: 10.1183/13993003.00547-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badawi A., Ryoo S.G. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus (MERS-CoV): a systematic review and meta-analysis. Int. J. Infect. Dis. 2016;49:129–133. doi: 10.1016/j.ijid.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiMasi J.A., Hansen R.W., Grabowski H.G. The price of innovation: new estimates of drug development costs. J. Health Econ. 2003;22(2):151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 25.Sirota M., Dudley J.T., Kim J. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med. 2011;3(96):96ra77. doi: 10.1126/scitranslmed.3001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chong C.R., Sullivan D.J., Jr. New uses for old drugs. Nature. 2007;448(7154):645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 27.Xue H., Li J., Xie H., Wang Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018;14(10):1232–1244. doi: 10.7150/ijbs.24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang G., Li J., Wang P., Li W. Comb Chem High Throughput Screen; 2017. A Review of Computational Drug Repositioning Approaches. [DOI] [PubMed] [Google Scholar]
- 29.Davis S., Meltzer P.S. GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–1847. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- 30.Zhou G., Soufan O., Ewald J., Hancock R.E.W., Basu N., Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47(W1):W234–W241. doi: 10.1093/nar/gkz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia J., Fjell C.D., Mayer M.L., Pena O.M., Wishart D.S., Hancock R.E. INMEX--a web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 2013;41:W63–W70. doi: 10.1093/nar/gkt338. Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ritchie M.E., Phipson B., Wu D. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cahan P., Rovegno F., Mooney D., Newman J.C., St Laurent G., 3rd, McCaffrey T.A. Meta-analysis of microarray results: challenges, opportunities, and recommendations for standardization. Gene. 2007;401(1–2):12–18. doi: 10.1016/j.gene.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 35.Chang L.C., Lin H.M., Sibille E., Tseng G.C. Meta-analysis methods for combining multiple expression profiles: comparisons, statistical characterization and an application guideline. BMC Bioinf. 2013;14:368. doi: 10.1186/1471-2105-14-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhodes D.R., Barrette T.R., Rubin M.A., Ghosh D., Chinnaiyan A.M. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Canc. Res. 2002;62(15):4427–4433. [PubMed] [Google Scholar]
- 37.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Y., Zhou B., Pache L. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10(1):1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan A., Mathelier A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinf. 2017;18(1):287. doi: 10.1186/s12859-017-1708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian A., Tamayo P., Mootha V.K. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Menachery V.D., Mitchell H.D., Cockrell A.S. MERS-CoV accessory ORFs play key role for infection and pathogenesis. mBio. 2017;8(4) doi: 10.1128/mBio.00665-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sims A.C., Tilton S.C., Menachery V.D. Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J. Virol. 2013;87(7):3885–3902. doi: 10.1128/JVI.02520-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–1045 e1039. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahi N.A., Najafabadi M.F., Pilarczyk M., Kouril M., Medvedovic M. GREIN: an interactive web platform for Re-analyzing GEO RNA-seq data. Sci. Rep. 2019;9(1):7580. doi: 10.1038/s41598-019-43935-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lieberman NAP, Peddu V, Xie H. In vivo Antiviral Host Response to SARS-CoV-2 by Viral Load. Sex, and Age. bioRxiv. 2020 doi: 10.1371/journal.pbio.3000849. 2020.2006.2022.165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia Z., Liu Y., Guan N., Bo X., Luo Z., Barnes M.R. Cogena, a novel tool for co-expressed gene-set enrichment analysis, applied to drug repositioning and drug mode of action discovery. BMC Genom. 2016;17:414. doi: 10.1186/s12864-016-2737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamb J., Crawford E.D., Peck D. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 49.Cagno V. SARS-CoV-2 cellular tropism. Lancet Microb. 2020;1(1):e2–e3. doi: 10.1016/S2666-5247(20)30008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tseng C.T., Tseng J., Perrone L., Worthy M., Popov V., Peters C.J. Apical entry and release of severe acute respiratory syndrome-associated coronavirus in polarized Calu-3 lung epithelial cells. J. Virol. 2005;79(15):9470–9479. doi: 10.1128/JVI.79.15.9470-9479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chu H., Chan J.F., Yuen T.T. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: an observational study. Lancet Microb. 2020;1(1):e14–e23. doi: 10.1016/S2666-5247(20)30004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan K.S., Lee K.O., Low K.C. Glutathione deficiency in type 2 diabetes impairs cytokine responses and control of intracellular bacteria. J. Clin. Invest. 2012;122(6):2289–2300. doi: 10.1172/JCI57817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pacht E.R., Timerman A.P., Lykens M.G., Merola A.J. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. 1991;100(5):1397–1403. doi: 10.1378/chest.100.5.1397. [DOI] [PubMed] [Google Scholar]
- 54.Chaves F.J., Mansego M.L., Blesa S. Inadequate cytoplasmic antioxidant enzymes response contributes to the oxidative stress in human hypertension. Am. J. Hypertens. 2007;20(1):62–69. doi: 10.1016/j.amjhyper.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 55.Damy T., Kirsch M., Khouzami L. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PloS One. 2009;4(3) doi: 10.1371/journal.pone.0004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin R.C., Mahoney C.E., Coleman Anderson L. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation. 2011;123(18):1963–1973. doi: 10.1161/CIRCULATIONAHA.110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herzenberg L.A., De Rosa S.C., Dubs J.G. Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. U. S. A. 1997;94(5):1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y., Dong W., Shi Y. Rabies virus phosphoprotein interacts with ribosomal protein L9 and affects rabies virus replication. Virology. 2016;488:216–224. doi: 10.1016/j.virol.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 59.Rofeal M., El-Malek F.A. Ribosomal proteins as a possible tool for blocking SARS-COV 2 virus replication for a potential prospective treatment. Med. Hypotheses. 2020;143:109904. doi: 10.1016/j.mehy.2020.109904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amiodarone or verapamil in COVID-19 hospitalized patients with symptoms. https://ClinicalTrials.gov/show/NCT04351763
- 61.Application of desferal to treat COVID-19. https://ClinicalTrials.gov/show/NCT04333550
- 62.Hydroxychloroquine or diltiazem-niclosamide for the treatment of COVID-19. https://ClinicalTrials.gov/show/NCT04372082
- 63.Colchicine coronavirus SARS-CoV2 trial (COLCORONA) https://ClinicalTrials.gov/show/NCT04322682
- 64.Vazquez-Calvo A., Saiz J.C., Sobrino F., Martin-Acebes M.A. Inhibition of enveloped virus infection of cultured cells by valproic acid. J. Virol. 2011;85(3):1267–1274. doi: 10.1128/JVI.01717-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo P., Qiu L., Liu Y. Metformin treatment was associated with decreased mortality in COVID-19 patients with diabetes in a retrospective analysis. Am. J. Trop. Med. Hyg. 2020;103(1):69–72. doi: 10.4269/ajtmh.20-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fukano K., Tsukuda S., Oshima M. Troglitazone impedes the oligomerization of sodium taurocholate cotransporting polypeptide and entry of hepatitis B virus into hepatocytes. Front. Microbiol. 2018;9:3257. doi: 10.3389/fmicb.2018.03257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koelsch S., Tschaikin M., Sacher F. Anti-rhinovirus-specific activity of the alpha-sympathomimetic oxymetazoline. Arzneimittelforschung. 2007;57(7):475–482. doi: 10.1055/s-0031-1296635. [DOI] [PubMed] [Google Scholar]
- 68.Hong G., Zhang W., Li H., Shen X., Guo Z. Separate enrichment analysis of pathways for up- and downregulated genes. J. R. Soc. Interface. 2014;11(92):20130950. doi: 10.1098/rsif.2013.0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Josset L., Menachery V.D., Gralinski L.E. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. mBio. 2013;4(3) doi: 10.1128/mBio.00165-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alosaimi B., Hamed M.E., Naeem A. MERS-CoV infection is associated with downregulation of genes encoding Th1 and Th2 cytokines/chemokines and elevated inflammatory innate immune response in the lower respiratory tract. Cytokine. 2020;126:154895. doi: 10.1016/j.cyto.2019.154895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem. Biophys. Res. Commun. 2004;319(4):1228–1234. doi: 10.1016/j.bbrc.2004.05.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kindrachuk J., Ork B., Hart B.J. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015;59(2):1088–1099. doi: 10.1128/AAC.03659-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu X., Pan J., Tao J., Guo D. SARS-CoV nucleocapsid protein antagonizes IFN-beta response by targeting initial step of IFN-beta induction pathway, and its C-terminal region is critical for the antagonism. Virus Gene. 2011;42(1):37–45. doi: 10.1007/s11262-010-0544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Versteeg G.A., Bredenbeek P.J., van den Worm S.H., Spaan W.J. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology. 2007;361(1):18–26. doi: 10.1016/j.virol.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nieto-Torres J.L., Verdia-Baguena C., Jimenez-Guardeno J.M. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–339. doi: 10.1016/j.virol.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Totura A.L., Whitmore A., Agnihothram S. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. 2015;6(3):e00638–615. doi: 10.1128/mBio.00638-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao X., Chu H., Wong B.H. Activation of C-type lectin receptor and (RIG)-I-like receptors contributes to proinflammatory response in Middle East respiratory syndrome coronavirus-infected macrophages. J. Infect. Dis. 2020;221(4):647–659. doi: 10.1093/infdis/jiz483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Y., Wu R., Tian Y. RAGE/NF-kappaB signaling mediates lipopolysaccharide induced acute lung injury in neonate rat model. Int. J. Clin. Exp. Med. 2015;8(8):13371–13376. [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y., Liang C., Liu X. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-kappaB pathways. Atherosclerosis. 2010;208(1):34–42. doi: 10.1016/j.atherosclerosis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 80.Kosmopoulos M., Drekolias D., Zavras P.D., Piperi C., Papavassiliou A.G. Impact of advanced glycation end products (AGEs) signaling in coronary artery disease. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2019;1865(3):611–619. doi: 10.1016/j.bbadis.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 81.McNulty M., Mahmud A., Feely J. Advanced glycation end-products and arterial stiffness in hypertension. Am. J. Hypertens. 2007;20(3):242–247. doi: 10.1016/j.amjhyper.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 82.Prasad K. AGE-RAGE stress in the pathophysiology of pulmonary hypertension and its treatment. Int. J. Angiol. 2019;28(2):71–79. doi: 10.1055/s-0039-1687818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reynaert N.L., Gopal P., Rutten E.P.A., Wouters E.F.M., Schalkwijk C.G. Advanced glycation end products and their receptor in age-related, non-communicable chronic inflammatory diseases; Overview of clinical evidence and potential contributions to disease. Int. J. Biochem. Cell Biol. 2016;81(Pt B):403–418. doi: 10.1016/j.biocel.2016.06.016. [DOI] [PubMed] [Google Scholar]
- 84.Jabaudon M., Berthelin P., Pranal T. Receptor for advanced glycation end-products and ARDS prediction: a multicentre observational study. Sci. Rep. 2018;8(1):2603. doi: 10.1038/s41598-018-20994-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hofer S., Uhle F., Fleming T. RAGE-mediated inflammation in patients with septic shock. J. Surg. Res. 2016;202(2):315–327. doi: 10.1016/j.jss.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 86.Takenaka K., Yamagishi S., Matsui T., Nakamura K., Imaizumi T. Role of advanced glycation end products (AGEs) in thrombogenic abnormalities in diabetes. Curr. Neurovascular Res. 2006;3(1):73–77. doi: 10.2174/156720206775541804. [DOI] [PubMed] [Google Scholar]
- 87.van Zoelen M.A., van der Sluijs K.F., Achouiti A. Receptor for advanced glycation end products is detrimental during influenza A virus pneumonia. Virology. 2009;391(2):265–273. doi: 10.1016/j.virol.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rojas A., Gonzalez I., Morales M.A. SARS-CoV-2-mediated inflammatory response in lungs: should we look at RAGE? Inflamm. Res. 2020;69(7):641–643. doi: 10.1007/s00011-020-01353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nielsen T.B., Pantapalangkoor P., Yan J. Diabetes exacerbates infection via hyperinflammation by signaling through TLR4 and RAGE. mBio. 2017;8(4) doi: 10.1128/mBio.00818-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pizzorno J. Glutathione! Integr. Med. (Encinitas) 2014;13(1):8–12. [PMC free article] [PubMed] [Google Scholar]
- 91.Meister A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988;263(33):17205–17208. [PubMed] [Google Scholar]
- 92.Anderson M.E., Meister A. Transport and direct utilization of gamma-glutamylcyst(e)ine for glutathione synthesis. Proc. Natl. Acad. Sci. U. S. A. 1983;80(3):707–711. doi: 10.1073/pnas.80.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morris D., Khurasany M., Nguyen T. Glutathione and infection. Biochim. Biophys. Acta. 2013;1830(5):3329–3349. doi: 10.1016/j.bbagen.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 94.Beck M.A., Handy J., Levander O.A. The role of oxidative stress in viral infections. Ann. N. Y. Acad. Sci. 2000;917:906–912. doi: 10.1111/j.1749-6632.2000.tb05456.x. [DOI] [PubMed] [Google Scholar]
- 95.Burckhardt C.J., Greber U.F. Redox rescues virus from ER trap. Nat. Cell Biol. 2008;10(1):9–11. doi: 10.1038/ncb0108-9. [DOI] [PubMed] [Google Scholar]
- 96.Sen C.K. Cellular thiols and redox-regulated signal transduction. Curr. Top. Cell. Regul. 2000;36:1–30. doi: 10.1016/s0070-2137(01)80001-7. [DOI] [PubMed] [Google Scholar]
- 97.Janssen-Heininger Y.M., Poynter M.E., Baeuerle P.A. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radic. Biol. Med. 2000;28(9):1317–1327. doi: 10.1016/s0891-5849(00)00218-5. [DOI] [PubMed] [Google Scholar]
- 98.Traverso N., Ricciarelli R., Nitti M. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell Longev. 2013;2013:972913. doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Balendiran G.K., Dabur R., Fraser D. The role of glutathione in cancer. Cell Biochem. Funct. 2004;22(6):343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 100.Saharan S., Mandal P.K. The emerging role of glutathione in Alzheimer's disease. J. Alzheimers Dis. 2014;40(3):519–529. doi: 10.3233/JAD-132483. [DOI] [PubMed] [Google Scholar]
- 101.Sofic E., Lange K.W., Jellinger K., Riederer P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson's disease. Neurosci. Lett. 1992;142(2):128–130. doi: 10.1016/0304-3940(92)90355-b. [DOI] [PubMed] [Google Scholar]
- 102.Burgunder J.M., Lauterburg B.H. Decreased production of glutathione in patients with cirrhosis. Eur. J. Clin. Invest. 1987;17(5):408–414. doi: 10.1111/j.1365-2362.1987.tb01135.x. [DOI] [PubMed] [Google Scholar]
- 103.Shaw S., Rubin K.P., Lieber C.S. Depressed hepatic glutathione and increased diene conjugates in alcoholic liver disease. Evidence of lipid peroxidation. Dig. Dis. Sci. 1983;28(7):585–589. doi: 10.1007/BF01299917. [DOI] [PubMed] [Google Scholar]
- 104.Roum J.H., Borok Z., McElvaney N.G. Glutathione aerosol suppresses lung epithelial surface inflammatory cell-derived oxidants in cystic fibrosis. J. Appl. Physiol. 1985;87(1):438–443. doi: 10.1152/jappl.1999.87.1.438. [DOI] [PubMed] [Google Scholar]
- 105.Buhl R., Jaffe H.A., Holroyd K.J. Systemic glutathione deficiency in symptom-free HIV-seropositive individuals. Lancet. 1989;2(8675):1294–1298. doi: 10.1016/s0140-6736(89)91909-0. [DOI] [PubMed] [Google Scholar]
- 106.Nencioni L., Iuvara A., Aquilano K. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. Faseb. J. 2003;17(6):758–760. doi: 10.1096/fj.02-0508fje. [DOI] [PubMed] [Google Scholar]
- 107.Palamara A.T., Perno C.F., Ciriolo M.R. Evidence for antiviral activity of glutathione: in vitro inhibition of herpes simplex virus type 1 replication. Antivir. Res. 1995;27(3):237–253. doi: 10.1016/0166-3542(95)00008-a. [DOI] [PubMed] [Google Scholar]
- 108.Papi A., Contoli M., Gasparini P. Role of xanthine oxidase activation and reduced glutathione depletion in rhinovirus induction of inflammation in respiratory epithelial cells. J. Biol. Chem. 2008;283(42):28595–28606. doi: 10.1074/jbc.M805766200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cai J., Chen Y., Seth S., Furukawa S., Compans R.W., Jones D.P. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 2003;34(7):928–936. doi: 10.1016/s0891-5849(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 110.Tian Y., Jiang W., Gao N. Inhibitory effects of glutathione on dengue virus production. Biochem. Biophys. Res. Commun. 2010;397(3):420–424. doi: 10.1016/j.bbrc.2010.05.108. [DOI] [PubMed] [Google Scholar]
- 111.Palamara A.T., Perno C.F., Aquaro S., Bue M.C., Dini L., Garaci E. Glutathione inhibits HIV replication by acting at late stages of the virus life cycle. AIDS Res. Hum. Retrovir. 1996;12(16):1537–1541. doi: 10.1089/aid.1996.12.1537. [DOI] [PubMed] [Google Scholar]
- 112.Jain S.K., Parsanathan R., Achari A.E., Kanikarla-Marie P., Bocchini J.A., Jr. Glutathione stimulates vitamin D regulatory and glucose-metabolism genes, lowers oxidative stress and inflammation, and increases 25-hydroxy-vitamin D levels in blood: a novel approach to treat 25-hydroxyvitamin D deficiency. Antioxidants Redox Signal. 2018;29(17):1792–1807. doi: 10.1089/ars.2017.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Horowitz R.I., Freeman P.R., Bruzzese J. Efficacy of glutathione therapy in relieving dyspnea associated with COVID-19 pneumonia: a report of 2 cases. Respir. Med. Case Rep. 2020;30:101063. doi: 10.1016/j.rmcr.2020.101063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tong J.Y., Wong A., Zhu D., Fastenberg J.H., Tham T. The prevalence of olfactory and gustatory dysfunction in COVID-19 patients: a systematic review and meta-analysis. Otolaryngol. Head Neck Surg. 2020;163(1):3–11. doi: 10.1177/0194599820926473. [DOI] [PubMed] [Google Scholar]
- 115.Butowt R., Bilinska K. SARS-CoV-2: olfaction, brain infection, and the urgent need for clinical samples allowing earlier virus detection. ACS Chem. Neurosci. 2020;11(9):1200–1203. doi: 10.1021/acschemneuro.0c00172. [DOI] [PubMed] [Google Scholar]
- 116.Giacomelli A., Pezzati L., Conti F. Self-reported olfactory and taste disorders in SARS-CoV-2 patients: a cross-sectional study. Clin. Infect. Dis. 2020;71(15):889–890. doi: 10.1093/cid/ciaa330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yan C.H., Faraji F., Prajapati D.P., Ostrander B.T., DeConde A.S. Self-reported olfactory loss associates with outpatient clinical course in COVID-19. Int. Forum Allergy Rhinol. 2020;10(7):821–831. doi: 10.1002/alr.22592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lechien J.R., Chiesa-Estomba C.M., De Siati D.R. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur. Arch. Oto-Rhino-Laryngol. 2020;277(8):2251–2261. doi: 10.1007/s00405-020-05965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sepahi A., Kraus A., Casadei E. Olfactory sensory neurons mediate ultrarapid antiviral immune responses in a TrkA-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 2019;116(25):12428–12436. doi: 10.1073/pnas.1900083116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Acharya A., Kevadiya B.D., Gendelman H.E., Byrareddy S.N. SARS-CoV-2 infection leads to neurological dysfunction. J. Neuroimmune Pharmacol. 2020;15(2):167–173. doi: 10.1007/s11481-020-09924-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Obitsu S., Ahmed N., Nishitsuji H. Potential enhancement of osteoclastogenesis by severe acute respiratory syndrome coronavirus 3a/X1 protein. Arch. Virol. 2009;154(9):1457–1464. doi: 10.1007/s00705-009-0472-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhou F., Yu T., Du R. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Diao B., Wang C., Tan Y. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19) Front. Immunol. 2020;11:827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Di Cosimo S., Malfettone A., Perez-Garcia J.M. Immune checkpoint inhibitors: a physiology-driven approach to the treatment of coronavirus disease 2019. Eur. J. Canc. 2020;135:62–65. doi: 10.1016/j.ejca.2020.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Reiter R.J., Sharma R., Ma Q., Dominquez-Rodriguez A., Marik P.E., Abreu-Gonzalez P. Melatonin inhibits COVID-19-induced cytokine storm by reversing aerobic glycolysis in immune cells: a mechanistic analysis. Med. Drug Discov. 2020:100044. doi: 10.1016/j.medidd.2020.100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ray U., Begum F. 2020. Proteins Nsp12 and 13 of SARS-CoV-2 Have Mitochondrial Recognition Signal: A Connection with Cellular Mitochondrial Dysfunction and Disease Manifestation. Preprints. [Google Scholar]
- 127.Singh K.K., Chaubey G., Chen J.Y., Suravajhala P. Decoding SARS-CoV-2 hijacking of host mitochondria in pathogenesis of COVID-19. Am. J. Physiol. Cell Physiol. 2020;319(2):C258–C267. doi: 10.1152/ajpcell.00224.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Saleh J., Peyssonnaux C., Singh K.K., Edeas M. Vol. 54. Mitochondrion; 2020. Mitochondria and Microbiota dysfunction in COVID-19 pathogenesis; pp. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shi C.S., Qi H.Y., Boularan C. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014;193(6):3080–3089. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shao H., Lan D., Duan Z. Upregulation of mitochondrial gene expression in PBMC from convalescent SARS patients. J. Clin. Immunol. 2006;26(6):546–554. doi: 10.1007/s10875-006-9046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Park J.S., Davis R.L., Sue C.M. Mitochondrial dysfunction in Parkinson's disease: new mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 2018;18(5):21. doi: 10.1007/s11910-018-0829-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Olsen Laura K., Dowd E., McKernan Declan P. A role for viral infections in Parkinson's etiology? Neuronal Signal. 2018;2(2) doi: 10.1042/NS20170166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fazzini E., Fleming J., Fahn S. Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson's disease. Mov. Disord. 1992;7(2):153–158. doi: 10.1002/mds.870070210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mori I. Viremic attack explains the dual-hit theory of Parkinson's disease. Med. Hypotheses. 2017;101:33–36. doi: 10.1016/j.mehy.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 135.De Felice F.G., Tovar-Moll F., Moll J., Munoz D.P., Ferreira S.T. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the central nervous system. Trends Neurosci. 2020;43(6):355–357. doi: 10.1016/j.tins.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Moni M.A., Lio P. Network-based analysis of comorbidities risk during an infection: SARS and HIV case studies. BMC Bioinf. 2014;15:333. doi: 10.1186/1471-2105-15-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Helmich R.C., Bloem B.R. The impact of the COVID-19 pandemic on Parkinson's disease: hidden sorrows and emerging opportunities. J. Parkinsons Dis. 2020;10(2):351–354. doi: 10.3233/JPD-202038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Martyn C.N. Infection in childhood and neurological diseases in adult life. Br. Med. Bull. 1997;53(1):24–39. doi: 10.1093/oxfordjournals.bmb.a011603. [DOI] [PubMed] [Google Scholar]
- 139.Martyn C.N., Osmond C. Parkinson's disease and the environment in early life. J. Neurol. Sci. 1995;132(2):201–206. doi: 10.1016/0022-510x(95)00148-u. [DOI] [PubMed] [Google Scholar]
- 140.Takahashi M., Yamada T. Viral etiology for Parkinson's disease--a possible role of influenza A virus infection. Jpn. J. Infect. Dis. 1999;52(3):89–98. [PubMed] [Google Scholar]
- 141.Victorino D.B., Guimaraes-Marques M., Nejm M., Scorza F.A., Scorza C.A. COVID-19 and Parkinson’s disease: are we dealing with short-term impacts or something worse? J. Parkinsons Dis. 2020;10(3):899–902. doi: 10.3233/JPD-202073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cohen M.E., Eichel R., Steiner-Birmanns B. A case of probable Parkinson's disease after SARS-CoV-2 infection. Lancet Neurol. 2020;19(10):804–805. doi: 10.1016/S1474-4422(20)30305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Beauchamp L.C., Finkelstein D.I., Bush A.I., Evans A.H., Barnham K.J. Parkinsonism as a third wave of the COVID-19 pandemic? J. Parkinsons Dis. 2020;10(4):1343–1353. doi: 10.3233/JPD-202211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kwak M.S., Lee H.H., Cha J.M., Shin H.P., Jeon J.W., Yoon J.Y. Novel candidate drugs in anti-tumor necrosis factor refractory Crohn's diseases: in silico study for drug repositioning. Sci. Rep. 2020;10(1):10708. doi: 10.1038/s41598-020-67801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jia Z., Song X., Shi J., Wang W., He K. Transcriptome-based drug repositioning for coronavirus disease 2019 (COVID-19) Pathog. Dis. 2020;78(4) doi: 10.1093/femspd/ftaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gehring G., Rohrmann K., Atenchong N. The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J. Antimicrob. Chemother. 2014;69(8):2123–2131. doi: 10.1093/jac/dku091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nugent K.M., Shanley J.D. Verapamil inhibits influenza A virus replication. Arch. Virol. 1984;81(1–2):163–170. doi: 10.1007/BF01309305. [DOI] [PubMed] [Google Scholar]
- 148.Fujioka Y., Nishide S., Ose T. A sialylated voltage-dependent Ca(2+) channel binds hemagglutinin and mediates influenza A virus entry into mammalian cells. Cell Host Microbe. 2018;23(6):809–818. doi: 10.1016/j.chom.2018.04.015. e805. [DOI] [PubMed] [Google Scholar]
- 149.Gilmore T.D., Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25(51):6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 150.Georgiou N.A., van der Bruggen T., Oudshoorn M., Nottet H.S., Marx J.J., van Asbeck B.S. Inhibition of human immunodeficiency virus type 1 replication in human mononuclear blood cells by the iron chelators deferoxamine, deferiprone, and bleomycin. J. Infect. Dis. 2000;181(2):484–490. doi: 10.1086/315223. [DOI] [PubMed] [Google Scholar]
- 151.Ryang J., Yan Y., Song Y., Liu F., Ng T.B. Anti-HIV, antitumor and immunomodulatory activities of paclitaxel from fermentation broth using molecular imprinting technique. Amb. Express. 2019;9(1):194. doi: 10.1186/s13568-019-0915-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ichiyama T., Okada K., Lipton J.M., Matsubara T., Hayashi T., Furukawa S. Sodium valproate inhibits production of TNF-alpha and IL-6 and activation of NF-kappaB. Brain Res. 2000;857(1–2):246–251. doi: 10.1016/s0006-8993(99)02439-7. [DOI] [PubMed] [Google Scholar]
- 153.Liao W.I., Chien W.C., Chung C.H. Valproic acid attenuates the risk of acute respiratory failure in patients with subarachnoid hemorrhage. QJM. 2018;111(2):89–96. doi: 10.1093/qjmed/hcx199. [DOI] [PubMed] [Google Scholar]
- 154.Bhavesh N.S., Patra A. Preprints; 2020. Virtual screening and molecular dynamics simulation suggest valproic acid Co-A could bind to SARS-CoV2 RNA depended RNA polymerase. [DOI] [Google Scholar]
- 155.Sharma S., Ray A., Sadasivam B. Metformin in COVID-19: a possible role beyond diabetes. Diabetes Res. Clin. Pract. 2020;164:108183. doi: 10.1016/j.diabres.2020.108183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bramante C., Ingraham N., Murray T. medRxiv; 2020. Observational Study of Metformin and Risk of Mortality in Patients Hospitalized with Covid-19. 2020.2006.2019.20135095. [DOI] [Google Scholar]
- 157.Giorgini A.E., Beales P.E., Mire-Sluis A., Scott D., Liddi R., Pozzilli P. Troglitazone exhibits immunomodulatory activity on the cytokine production of activated human lymphocytes. Horm. Metab. Res. 1999;31(1):1–4. doi: 10.1055/s-2007-978686. [DOI] [PubMed] [Google Scholar]
- 158.Marion-Letellier R., Butler M., Dechelotte P., Playford R.J., Ghosh S. Comparison of cytokine modulation by natural peroxisome proliferator-activated receptor gamma ligands with synthetic ligands in intestinal-like Caco-2 cells and human dendritic cells--potential for dietary modulation of peroxisome proliferator-activated receptor gamma in intestinal inflammation. Am. J. Clin. Nutr. 2008;87(4):939–948. doi: 10.1093/ajcn/87.4.939. [DOI] [PubMed] [Google Scholar]
- 159.Iacucci M., de Silva S., Ghosh S. Mesalazine in inflammatory bowel disease: a trendy topic once again? Can. J. Gastroenterol. 2010;24(2):127–133. doi: 10.1155/2010/586092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zheng B.J., Chan K.W., Lin Y.P. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc. Natl. Acad. Sci. U. S. A. 2008;105(23):8091–8096. doi: 10.1073/pnas.0711942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Feibelman K.M., Fuller B.P., Li L., LaBarbera D.V., Geiss B.J. Identification of small molecule inhibitors of the Chikungunya virus nsP1 RNA capping enzyme. Antivir. Res. 2018;154:124–131. doi: 10.1016/j.antiviral.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Beck-Speier I., Oswald B., Maier K.L., Karg E., Ramseger R. Oxymetazoline inhibits and resolves inflammatory reactions in human neutrophils. J. Pharmacol. Sci. 2009;110(3):276–284. doi: 10.1254/jphs.09012FP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kim J., Zhang J., Cha Y. Advanced bioinformatics rapidly identifies existing therapeutics for patients with coronavirus disease-2019 (COVID-19) J. Transl. Med. 2020;18(1):257. doi: 10.1186/s12967-020-02430-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Portolani M., Pietrosemoli P., Cermelli C. Suppression of BK virus replication and cytopathic effect by inhibitors of prokaryotic DNA gyrase. Antivir. Res. 1988;9(3):205–218. doi: 10.1016/0166-3542(88)90004-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PRISMA flow chart for SARS-CoV meta-analysis
PRISMA flow chart for MERS-CoV meta-analysis
Significant genes of SARS-CoV at each time point after meta-analysis.
Significant genes of MERS-CoV at each time point after meta-analysis.
Common pathways enriched by GSEA KEGG analysis for SARS-CoV, MERS-CoV, SARS-CoV-2 Calu-3 -24 hours datasets.
Potential pathways common for SARS-CoV, MERS-CoV, and SARS-CoV-2.
GSEA KEGG analysis results for SARS-CoV-2 positive patients compared with the healthy patient samples.
Drugs enriched in other clusters in cogena analysis.
Genes enriched at cluster 8 subjected to Drug Repositioning analysis.