

Abstract
Alzheimer's disease (AD) is a major cause of dementia, disability, and death in the elderly. Despite recent advances in our understanding of the basic biological mechanisms underlying AD, we do not know how to prevent it, nor do we have an approved disease‐modifying intervention. Both are essential to slow or stop the growth in dementia prevalence. While our current animal models of AD have provided novel insights into AD disease mechanisms, thus far, they have not been successfully used to predict the effectiveness of therapies that have moved into AD clinical trials. The Model Organism Development and Evaluation for Late‐onset Alzheimer's Disease (MODEL‐AD; www.model-ad.org) Consortium was established to maximize human datasets to identify putative variants, genes, and biomarkers for AD; to generate, characterize, and validate the next generation of mouse models of AD; and to develop a preclinical testing pipeline. MODEL‐AD is a collaboration among Indiana University (IU); The Jackson Laboratory (JAX); University of Pittsburgh School of Medicine (Pitt); Sage BioNetworks (Sage); and the University of California, Irvine (UCI) that will generate new AD modeling processes and pipelines, data resources, research results, standardized protocols, and models that will be shared through JAX's and Sage's proven dissemination pipelines with the National Institute on Aging–supported AD Centers, academic and medical research centers, research institutions, and the pharmaceutical industry worldwide.
Keywords: Alzheimer's disease, animal models, LOAD, MRI, Open Science, PET, preclinical
1. INTRODUCTION
Evidence suggests that Alzheimer's disease (AD), the most common dementing disorder of late life, is the third leading cause of death in the United States. 1 An estimated 5.8 million Americans currently have AD and approximately 700,000 individuals over the age of 65 will die with AD in 2019, with another 18.5 million individuals acting as unpaid caregivers for those afflicted by the disease. 2 Tragically, the progression of the disease is lengthy and there is currently no effective treatment.
The AD brain exhibits unique pathological alterations, including filamentous inclusions of the microtubule‐associated protein tau in neuronal cell bodies and processes; extracellular deposits of amyloid beta (Aβ) in senile plaques and within the walls of leptomeningeal/cerebral vessels; marked neuroinflammation and activation of innate immune cells; and synaptic and neuronal cell loss. Alois Alzheimer identified plaques and tangles in a patient with presenile dementia in 1906, but despite decades of research, the precise relationship among plaques, tangles, and dementia remains unknown. Clues to understanding the biological pathways underlying these pathological processes have been provided by genetic studies of human AD.
AD is generally classified as early‐onset (EOAD) or late‐onset (LOAD), based on factors including age of onset and genetic markers. The majority of cases of EOAD are caused by mutations in the amyloid precursor protein (APP) and presenilin (PSEN1 and PSEN2) genes, but EOAD accounts for only a small fraction of the total AD cases. Unlike EOAD, genetic susceptibility to LOAD is more complex with variations in many genes significantly associated with increased risk of varying degree. The greatest genetic risk factor for LOAD in the human population is the ε4 allele of apolipoprotein E (APOE), which accounts for ≈30% of risk. More recently, next generation sequencing determined that the R47H variation in triggering receptor expressed on myeloid cells 2 (TREM2) also conferred increased risk for AD. 3 With an increased odds ratio for carriers second only to APOEε 4 carriers, TREM2R47H is the second greatest known genetic risk factor for LOAD. To date, more than 20 other genetic loci have been associated with LOAD by genome‐wide association studies (GWAS), albeit these individually confer a small increase in risk (between 1% and 3%). Candidate genes in these loci fall into a variety of pathways including cholesterol trafficking, inflammation, and endosomal recycling suggesting that AD is caused by perturbations in multiple biological processes. However, the mechanisms by which individual or combinations of genetic risk variants contribute to AD risk, onset, and progression are not known. This knowledge gap is severely hampering the development of treatments for LOAD.
Based upon EOAD studies, multiple approaches toward Aβ‐directed therapies have been developed and tested in clinical trials, including active and passive Aβ immunization, 4 γ‐ and β‐secretase inhibitors, γ‐secretase modulators, 5 and Aβ aggregation inhibitors. Notably, these strategies have thus far failed in AD clinical trials, although some of these trials are continuing and showing some promise. 6 There are numerous potential explanations as to why these clinical trials have failed, including stage of disease targeted, mechanism of delivery, suitability of the patient population, effective engagement of target and off‐target effects, suitability of the target, face and construct validity of the animal models, and others. 7 , 8 However, the exact reasons for failures of the clinical trials remain to be established. A recommendation of the National Institute on Aging (NIA) Alzheimer's Research Summit in 2015 was to develop and characterize novel animal models of AD that would facilitate the development of novel AD therapies, using genetics and systems biology to inform animal model development and subsequent pre‐clinical drug testing.
Over the past two decades, multiple groups, including our own, have focused on developing and characterizing genetically engineered rodent models of EOAD. These models have provided key insights into genes implicated in human AD and how they lead to some neuropathological abnormalities observed in AD with a focus on Aβ and tau. 9 While current models have provided critical information on biological mechanisms underlying Aβ and tau pathology, there have been a number of confounds that have limited their utility, particularly for preclinical studies assessing potential AD therapies. 7 First, existing animal models have focused on EOAD, although it remains unclear whether the relatively uncommon EOAD cases and the more common LOAD cases proceed through identical disease mechanisms. Second, most mouse models of Aβ pathology do not exhibit extensive neurodegeneration. 10 , 11 Exceptions include the 5xFAD mouse model of Aβ pathology, which exhibits regional‐specific neurodegeneration and the hTau mouse model of tau pathology. 13 , 14 , 15 Third, to date, no single EOAD model exhibits both Aβ and tau pathology, although the 3xTg mouse model, which in addition to EOAD mutations also contains a mutation in tau associated with frontotemporal dementia, develops both plaques and hyperphosphorylated tau (Oddo et al., 2003). 16 Fourth, most existing models significantly and ectopically overexpress the relevant transgenes to observe AD pathologies within a mouse's lifespan, which may introduce non‐physiologic effects which do not reflect human disease progression. Fifth, although many mouse models of Aβ and tau pathology exhibit age‐related behavioral abnormalities, it has proven difficult to relate these deficits to specific impairments observed in human AD. 8 Sixth, many models were generated on hybrid genetic backgrounds and could not be maintained in uniform genetic backgrounds due to premature lethality and seizures observed in many models, 17 , 18 , 19 , 20 , 21 , 22 making them difficult to use for preclinical studies. Seventh, the use of many models has been restricted due to legal constraints. 23 Eighth, most studies have not examined the various animal models in a side‐by‐side manner to directly assess reproducibility. Ninth, few mouse models of AD have been carefully examined for age‐related alterations in biomarkers and brain imaging abnormalities across the lifespan to relate to those observed in human AD. 8 Finally, preclinical therapeutic testing conducted in current models and with traditional behavioral tests have failed to predict clinical efficacy for cognitive improvement in human clinical trials; however, the mouse models predicted Aβ lowering without improvements in cognition. 7 Therefore, a critical need exists to generate multiple new models of AD, particularly LOAD, given that recent genetic and systems biology studies of LOAD suggest that different pathways may contribute to disease pathogenesis from those observed in EOAD.
To meet this essential need, Model Organism Development and Evaluation for Late‐onset Alzheimer's Disease (MODEL‐AD; www.model-ad.org) was established by the NIA to (1) identify novel combinations of genetic variants that increase risk for LOAD, (2) develop new animal models for LOAD including humanized Aβ and tau models that recapitulate key hallmarks of the human disease, and (3) develop robust preclinical testing pipelines, and identify and test novel therapeutic agents (Figure 1). Despite failures in most of the clinical trials for AD, key recent advances provide renewed optimism that treatments for AD will be developed. First, dedication of new funds for research and development targeting AD specifically has enabled the establishment and coordination of multi‐institutional and inter‐disciplinary precompetitive consortia to identify, characterize, and deliver new therapies to the clinic by 2025. In addition to the Alzheimer's Disease Research Centers (ADRCs) that are providing patient samples and clinical data, other key consortia include MODEL‐AD, the Accelerating Medicines Partnerships–Alzheimer's Disease (AMP‐AD), the Alzheimer's Disease Neuroimaging Initiative (ADNI), the Alzheimer's Disease Sequencing Project (ADSP), the Molecular Mechanisms of the Vascular Etiology of Alzheimer's Disease (M2OVE‐AD), and the Target Enablement to Accelerate Therapy Development for Alzheimer's Disease (TREAT‐AD) Consortium. Many of these consortia are focused on accelerating the process to identify novel therapeutic targets, moving these targets forward, testing in preclinical models, and ultimately to delivering therapies to human AD patients (Figure 2). Second, many groups, including our own, are developing novel computational approaches to interrogate large‐scale datasets to understand complex genetic disorders including AD. Third, recent advances in manipulating genomes, particularly the development of gene editing by clustered regularly interspaced short palindromic repeats (CRISPR) has accelerated and reduced the cost of introducing human relevant variants in model organisms. Last, imaging technologies in both humans and model organisms allow for more accurate assessment of particularly early stages of AD.
FIGURE 1.
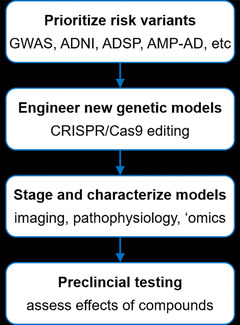
Workflow for creating and testing novel animal models of late‐onset Alzheimer's disease
FIGURE 2.
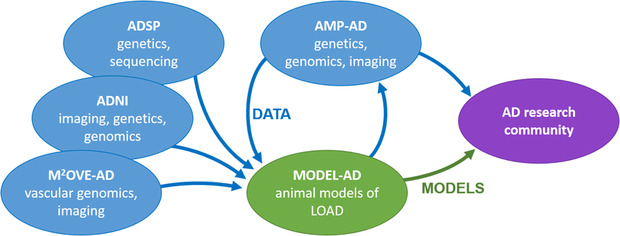
Role of the MODEL‐AD center in the system of NIH‐funded consortia created to discover new treatments for Alzheimer's disease. AD, Alzheimer's disease; ADNI, Alzheimer's Disease Neuroimaging Initiative; ADSP, Alzheimer's Disease Sequencing Project; AMP‐AD, Accelerating Medicines Partnerships–Alzheimer's Disease; MODEL‐AD, Model Organism Development and Evaluation for Late‐onset Alzheimer's Disease; M2OVE‐AD, Molecular Mechanisms of the Vascular Etiology of Alzheimer's Disease.
2. THE OVERARCHING PRINCIPLES OF MODEL‐AD
The major aims of MODEL‐AD are to design, develop, characterize, and distribute models for LOAD, and establish robust preclinical pipelines for testing new therapies. Our strategy will be to generate new rodent models, initially in the mouse. Mice will be engineered using CRISPR and other traditional methods to carry combinations of human variants identified using computational analyses of human datasets made available from AMP‐AD, M2OVE‐AD, ADSP, ADNI, and other sources. New models will be “staged’’ to precisely define phenotypes and the relevance to human AD. Human relevant‐outcome measures particularly in vivo imaging, blood biomarkers, and transcriptional profiling, as well as traditional phenotyping methods including neuropathology, biochemistry, electrophysiology, and behavioral assays. Importantly, all models and data will be made available for distribution. MODEL‐AD will permit these mice to be distributed without imposing any additional legal or licensing restrictions on these models or the use of data generated.
MODEL‐AD is a collaboration among Indiana University (IU); University of California, Irvine (UCI); The Jackson Laboratory (JAX); University of Pittsburgh (Pitt); and Sage Bionetworks. Each institute provides unique strengths that include a proven track record of translational neuroscience research and the 26‐year‐old Indiana Alzheimer's Disease Center at IU; 35‐year‐old UCI Alzheimer's Disease Research Center; 35‐year‐old University of Pittsburgh Alzheimer's Disease Research Center; more than eight decades of model production, phenotyping, and distribution (JAX); and a mission of open data curation and dissemination (Sage). An administrative core, steering committee, and external advisory board ensure the aims and milestones of MODEL‐AD are met. To maximize uptake of all resources created by MODEL‐AD (mice, data, protocols, etc) all data will be made available via the AD Knowledge Portal hosted on the Sage Synapse platform (https://adknowledgeportal.synapse.org/), thereby expanding on an established data resource for the AD research community. All mice will be made available through the JAX AD Mouse Model Resource (www.jax.org/ad-repository).
3. PRODUCTION, VALIDATION, AND DISSEMINATION OF NEW MODELS FOR LOAD
A primary goal of MODEL‐AD is to produce novel models for LOAD, and extensively characterize them using human‐relevant and translatable outcome measures. New models will be assessed side‐by‐side with prominent existing models (eg, 5xFAD, APP/PS1, 3xTg‐AD, and hTau). Wherever possible, models will be based on human‐relevant genetic variants. Models with “humanized” alleles of loci including APP and MAPT (TAU), generated via genome engineering, will be used as a platform to introduce additional risk alleles. For example, the MODEL‐AD consortium have already generated humanized Aβ models, which express human non‐mutated Aβ in the fully natural context of the endogenous mouse APP gene. The resulting line, designated hAβ‐KI mouse model, produces human Aβ at physiological levels in all cell types that normally express APP, and it does so without the addition of any FAD mutations or overexpression of APP or its metabolites. Therefore, this innovative mouse overcomes many confounding variables affecting the traditional models of AD and may provide a much more physiologically relevant understanding of the underlying mechanisms driving AD pathology by closely recapitulating the pathological cascade of events that occurs in the majority of human AD patients. Furthermore, this new model has also been engineered to permit the conditional ablation of the humanized APP gene, by incorporating LoxP sites flanking the Aβ sequence. By the virtue of these novel features, the hAβ‐KI mouse model can be used to address multiple previously inaccessible questions surrounding the involvement of Aβ in the pathogenesis of AD and multiple relevant genetic variants will be incorporated into this new model. The hAβ‐KI model was originally characterized on a mixed C57BL/6J (B6J) and C57BL/6N (B6N) genetic background but will also be available on B6J and B6N inbred lines. The IU/Jax/Pitt MODEL‐AD Center has also created a humanized Aβ knock‐in model without the loxP sites, on a C57BL6J background.
The MODEL‐AD consortium has also created an allelic series of APOE variants (ε4, ε3, ε2; JAX IDs 27894, 29018, and 29017, respectively) as well as mice carrying combinations of hAβ, APOE ε 4, and Trem2R47H on the B6J genetic background. Humanized MAPT alleles will be incorporated as soon as possible. Female and male mice from these platform strains are being characterized up to 24 months of age using an extensive set of human‐relevant assays. Additional AD‐relevant genetic variants and “humanized alleles’’ are being incorporated into the platform strains. Improved methods of in vitro fertilization will be used to accelerate production of animal models with different combinations of risk factors. MODEL‐AD aims to generate at least 50 new models for LOAD. Information on all available models can be found on the MODEL‐AD website (https://www.model-ad.org). Although models will be initially generated on the B6J background, there is a growing appreciation that alternative or even multiple genetic backgrounds will need to be considered to maximize the relevance of mouse models to human LOAD (Onos et al., 2019; Neuner et al., 2019). 24 , 25 Work is under way within MODEL‐AD to prioritize the most appropriate genetic backgrounds.
A major limitation in generating new models has been the lack of specific putative genetic variants in LOAD‐relevant loci to precisely engineer into the mouse genome. However, advances in genomics, computation, and imaging are providing large data resources to mine (eg, ADNI, AMP‐AD, M2OVE‐AD, ADSP, and the International Genomics of Alzheimer's Project). These data will be leveraged to identify and prioritize candidate variants for animal models. Our initial strategy is to use recent sequencing studies to aid in prioritizing variants in existing GWAS loci, including genes such as ABCA7, CR1, and BIN1. Although the majority of GWAS loci have small effect sizes, our hypothesis is that they will work in concert with other variants to increase risk and should be assessed in a sensitized genetic context. We then assess the mouse genome for sequence and functional homology to ensure the resulting model will faithfully reflect the genetics of LOAD. Once existing loci have been assessed, we will incorporate novel candidate genes and variants that may arise from the rapidly expanding efforts to understand the genetics of AD. We expect that these efforts will include modeling multiple non‐coding variants and integrating data from expression quantitative trait locus (eQTL) studies and efforts to annotate the regulatory genome such as the ROADMAP (Real‐World Outcomes Across the Alzheimer's Disease Spectrum for Better Care: Multi‐Modal Data Access Platform) and ENCODE (Encyclopedia of DNA Elements) projects for both mouse and human. The expanding availability of quantitative traits related to AD pathology from ADNI, eQTL, and other functional studies will enable greater statistical power and phenotypic resolution. Importantly, such use of advanced computational strategies will facilitate the ability to infer epistatic and pleiotropic networks of genes that can aid in prioritizing polygenic animal models.
Many of the new models will be initially characterized using a primary screening approach that prioritizes the most promising models for more extensive phenotyping using human‐relevant outcome measures. In some cases, the same strains will be characterized independently at multiple sites (eg, IU, JAX, and UCI) using standardized protocols to ensure reproducibility of key AD‐relevant phenotypes. This extensive phenotyping will occur at multiple ages, up to 24 months of age, in male and female mice and, in addition to more traditional phenotyping assays (eg, behavior, biochemistry, and neuropathology), will include relevant in vivo positron emission tomography (PET)/magnetic resonance (MR) imaging with autoradiography validation of tracer compounds, blood and cerebrospinal fluid (CSF) biomarkers, synaptic physiology analyses (eg, basal synaptic transmission, long‐term potentiation, axon excitability, and transmitter release kinetics), and molecular profiling by RNA sequencing. Genomic data will be systematically compared to analogous human data from the AMP‐AD Consortium to identify the specific disease‐related pathways and modules modified in each strain (Pandey et al., 2019; Johnson et al., 2018; Logsdon et al., 2019). 26 , 27 , 28 We have developed a new NanoString nCounter Mouse AD panel to specifically assess modifications of LOAD‐associated transcriptome modules that will be used in primary screening of all new mouse strains.
Additional phenotyping for selected lines will also include proteomics, metabolomics, and microbiome characterization. Assays are designed to complement existing and forthcoming data from human studies, and will systematically align the phenotypes of each mouse model with corresponding human data. For example, early gene expression signatures that appear in mouse models may be present in human brain samples, providing evidence for pathway dysfunction in LOAD. The identification of such signatures in human subpopulations may further discriminate between heterogeneous etiologies within the human population. These analyses will link precise genetic variation in the mouse model with pathological outcomes that contribute to LOAD, which can be further assessed in human carriers of the homologous variants. Staged mouse cohorts can potentially clarify temporal ordering of transcriptomic modifications that have accumulated in human decedent cases. Such signals of disease progression will be correlated with imaging and other molecular phenotypes to find candidate biomarkers for early disease. Furthermore, our analysis will help refine key disease markers present in human data that may be confounded by phenotypic variation unrelated to LOAD. Importantly, these human/mouse comparisons will provide critical data to both determine the most appropriate models to use in preclinical studies and the novel targets to test as therapies for LOAD.
4. THE NEED FOR AN ACCESSIBLE VALIDATED PRECLINICAL TESTING PIPELINE
Historically, preclinical screening of test compounds for AD used behavioral endpoints in rodent models as the primary screen owing to a falsely perceived ease of conducting these experiments and relative high throughput. 8 Moreover, the rodent models used did not necessarily have construct validity for AD, and experiments often evaluated the ability of a test compound to reverse an acute pharmacological deficit (eg, scopolamine‐induced memory deficit) in wild‐type or normal animals, and frequently only in males. Other screens evaluated the ability of the test compound to normalize a behavioral phenotype, and these studies rarely used biomarkers or other clinically translational endpoints. Young or naïve wild‐type animals were often used in place of aging animals, with the rationale that aged animals were costly and would minimize throughput. Critically, in many cases pharmacokinetic (PK) and pharmacodynamic data (PD) in AD models at biologically and pathologically relevant ages have not been evaluated and when performed they were conducted in a single sex without neuropathology.
MODEL‐AD is comprehensively addressing many identified concerns. The consortium represents a unique opportunity to standardize practices and provide an established pipeline for preclinical testing for the AD community while interrogating mechanisms of action that failed in clinic for AD via a back‐translational approach. The Preclinical Testing Core (PTC) has established a streamlined preclinical testing strategy with go/no‐go decision points allowing critical and unbiased assessments of potential therapeutic agents (Figure 3).
FIGURE 3.
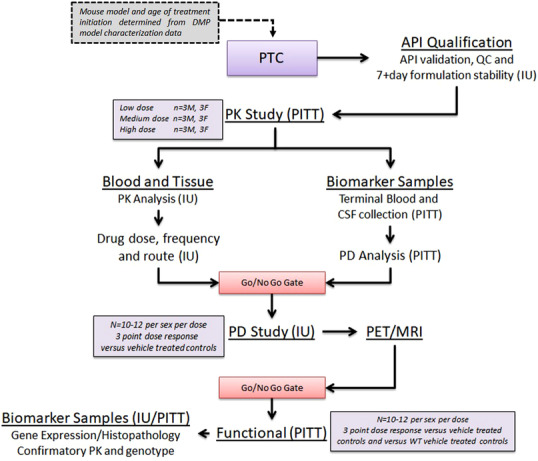
Workflow for testing compounds through the Preclinical Testing Core. API, active pharmaceutical ingredient; IU, Indiana University; MRI, magnetic resonance imaging; PET, positron emission tomography; PITT, University of Pittsburgh School of Medicine; PK, pharmokinetic; QC, quality control.
The primary screen includes (1) an initial qualification of the active pharmaceutical ingredient (API) of the compound to be tested, (2) drug formulation optimization for dosing in mice, (3) drug stability assessment in that formulation, and (4) multi‐dose in vivo pharmacokinetics for the determination of appreciable blood and target tissue activity in the disease model and at the pathologically and disease‐relevant age in both sexes. Quantification of PK parameters for the parent compound will use standard moment theory (non‐compartmental) methods and parameters. 29 Using Cl/F and Vd/F as initial parameter estimates, nonlinear mixed effect analyses 30 , 31 , 32 will be performed. Specifically, to meet the screening criteria for this go/no‐go decision, test articles need to have low Cl/F, which affords less than or equal to twice daily dosing, good blood–brain barrier penetration supporting appreciable exposure in brain tissue, appreciable brain retention, and low serum protein binding. In the absence of this, compounds will not move forward. Provided the compound meets the “go” criteria, the PK data (eg, Cmax, Cl/F, Vd/F) will be used to inform PK/PD modeling to develop the dosing paradigm for the secondary screen where appropriate disease models at the pathological ages achieves target brain exposure levels to evaluate disease‐ (or symptom‐) modifying effects.
The secondary screen evaluates target engagement and disease‐modifying activity of the test compound at multiple dose levels, in both males and females in the disease model at the pathologically relevant age using non‐invasive in vivo PET/MRI as a pharmacodynamic readout of cerebral changes in metabolism (18F‐FDG), cerebral blood flow (64Cu‐PTSM), Aβ deposition (18F‐AV45), or tau deposition (18F‐AV1451). PET and MRI images will be coregistered, 33 and mapped to stereotactic mouse brain coordinates 34 and volume of interest (VOI) extracted from a mouse brain atlas. Each VOI will be analyzed for standardized uptake value (SUV) or %ID/g according to published methods. 35 At the completion of the study, blood samples are collected and processed for plasma to confirm PK from the primary screen. To permit secondary confirmation for PET and autoradiography studies, tissue sections will be immunostained with Aβ, tau, or neuroinflammation antibodies.
Plasma concentrations across animal models and dose levels will be combined with PET, autoradiography, and secondary confirmation data for PK/PD model analysis (ie, direct effect, indirect response, signal transduction, etc) will be assessed for each compound. 36 Only after demonstrating target engagement will test compounds move to the tertiary functional assessment.
Tertiary screening will evaluate both dose response curve and dose range regiments (acute/chronic, route of administration and pretreatment time) to determine disease‐modifying effects of the test compound to normalize a disease‐related functional phenotype. Tertiary screening will include assessments of cognition (eg, working memory) and activity measures (eg, locomotor activity, motor coordination) to identify whether the dose range perceived to improve a functional (ie, memory) deficit is without any side effects that confound the interpretation of the data or suggest a limited therapeutic window. Importantly, the PTC is well aware of the translational limitations of the behavioral assays historically used to predict cognitive improvement in mouse models (eg, water maze, fear conditioning, novel object recognition). In this respect it is important to point out that behavioral outcome measures are limited and only being used as tertiary screens after target engagement has been confirmed in the secondary screens, described above. Further, in lieu of a large battery of behavioral assays for cognitive outcome measures which have for all intents and purposes failed to translate to the clinic, the PTC will use improved translational assays such as electroencephalogram as functional outcome measures. 45 , 46 At the conclusion of the tertiary screen, plasma and brain samples are used to confirm PK and brain samples are also sent for post‐treatment transcriptomics analysis.
This approach is innovative, first by establishing this standardized, streamlined preclinical screening strategy, which has been validated, and provides access to these resources including standard operating procedures, all raw data along with negative and positive findings, and hands‐on training opportunities to the AD research community.
5. SCREENING THE OPTIMAL PHARMACEUTICAL FOR ALZHEIMER'S DISEASE (STOP‐AD)
The PTC supports preclinical screening of test compounds nominated by the greater research community through its streamlined preclinical screening strategy in mouse models developed and characterized by the Disease Modeling Project (DMP). Researchers can apply through the STOP‐AD portal (www.STOPADportal.synapse.org). Compounds selected for screening will be conducted within the PTC labs at Indiana University and the University of Pittsburgh. Submitters are required to provide detailed data and information about the compound they wish to nominate. A review panel consisting of experts in pharmacology, pharmacokinetics, neuroscience, animal model systems, behavioral pharmacology, preclinical imaging, genetics, and AD will provide a composite score based on novelty of science, relevance of target for AD, chemical properties of the compound including optimal drug‐like properties, and quality of the data available for assessment. Selected compounds will be best matched to a MODEL‐AD mouse model based on mechanism of action and relevant PD endpoint. Access to this rigorous testing pipeline is innovative and a major benefit to researchers that may not otherwise have the resources available to conduct comprehensive in vivo PK, PD, preclinical imaging, and functional evaluations of their test compounds. Consequently, the PTC resources are being leveraged by the NIA's newly funded Alzheimer's Centers for the Discovery of New Medicines (TREAT‐AD), which will facilitate advancing compounds with novel mechanisms of action from early drug discovery stages to preclinical efficacy testing.
6. SUMMARY AND OUTLOOK
Optimizing the return from the next generation of animal models for AD will require combining contemporary genetics, computational biology, and genetic engineering to create a range of models that faithfully recapitulate the disease. MODEL‐AD is configured to fully exploit contemporary resources to create the next generation of LOAD models and, moving forward, will be augmented with novel information and technologies. The expanded efforts to quantify proteins and metabolites in addition to transcriptomes by AMP‐AD can be readily reproduced in mouse models. This will allow multiscale molecular comparisons between novel models and human LOAD cohorts, possibly identifying subpopulations of AD patients with distinct neuropathology and/or genetic etiology. Such efforts can be greatly enriched with the integration of functional information from other model systems, such as induced pluripotent stem cell culture and fly populations. Furthermore, using inbred genetic models enables a broader study of how genetic backgrounds alter AD risk factors, both through gene–gene and gene–environment interactions. The recent proliferation of genetically complex mouse populations (eg, Diversity Outbred, Heterogeneous Stock) and inbred lines (eg, Collaborative Cross, BXD) provide the resources to systematically study how susceptibility to neurodegeneration varies by strain and, in mapping populations, specific genetic factors. Alignment of multiscale phenotypes (molecular, histological, and behavioral) of these models to human data will likely identify key pathways and processes that drive neuropathology and LOAD. Of note, while much of LOAD is driven by the interaction between aging and genetics, there is a considerable influence of environmental factors, much of which will not be modeled or explored by this consortium. However, MODEL‐AD will provide the next generation of mouse models to the research community where the impact of environment, such as diet, stress, social isolation/environmental enrichment, the microbiome, and other influences can be determined and subsequently used to develop animal models of LOAD in additional species.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Oblak AL, Forner S, Territo PR, et al. Model organism development and evaluation for late‐onset Alzheimer's disease: MODEL‐AD. Alzheimer's Dement. 2020;6:e12110 10.1002/trc2.12110
REFERENCES
- 1. James BD, Leurgans SE, Hebert LE, Scherr PA, Yaffe K, Bennett DA. Contribution of Alzheimer disease to mortality in the United States. Neurology. 2014;82(12):1045‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alzheimer's A 2019 https://www.alz.org/media/documents/alzheimers-facts-and-figures-2019-r.pdf
- 3. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368(2):117‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lannfelt L, Relkin NR, Siemers ER. Amyloid‐ss‐directed immunotherapy for Alzheimer's disease. J Intern Med. 2014;275(3):284‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6(2):99‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shineman DW, Basi GS, Bizon JL, et al. Accelerating drug discovery for Alzheimer's disease: best practices for preclinical animal studies. Alzheimers Res Ther. 2011;3(5):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bales KR. The value and limitations of transgenic mouse models used in drug discovery for Alzheimer's disease: an update. Expert Opin Drug Discov. 2012;7(4):281‐297. [DOI] [PubMed] [Google Scholar]
- 9. Onos KD, Sukoff Rizzo SJ, Howell GR, Sasner M. Toward more predictive genetic mouse models of Alzheimer's disease. Brain Res Bull. 2016;122:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Irizarry MC, Mcnamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age‐related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56(9):965‐973. [DOI] [PubMed] [Google Scholar]
- 11. Irizarry MC, Soriano F, Mcnamara M, et al. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17(18):7053‐7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oakley H, Cole SL, Logan S, et al. Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129‐10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86(3):582‐590. [DOI] [PubMed] [Google Scholar]
- 14. Andorfer C. Cell‐cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25(22):5446‐5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age‐dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29(34):10741‐10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003a;24(8):1063‐1070. [DOI] [PubMed] [Google Scholar]
- 17. Carlson GA, Borchelt DR, Dake A, et al. Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum Mol Genet. 1997;6(11):1951‐1959. [DOI] [PubMed] [Google Scholar]
- 18. Chishti MA, Yang D‐S, Janus C, et al. Early‐onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276(24):21562‐21570. [DOI] [PubMed] [Google Scholar]
- 19. Moechars D, Lorent* K, Van Leuven F. Premature death in transgenic mice that overexpress a mutant amyloid precursor protein is preceded by severe neurodegeneration and apoptosis. Neuroscience. 1999;91(3):819‐830. [DOI] [PubMed] [Google Scholar]
- 20. Moechars D, Dewachter I, Lorent K, et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274(10):6483‐6492. [DOI] [PubMed] [Google Scholar]
- 21. Sebastiani G, Krzywkowski P, Dudal S, et al. Mapping genetic modulators of amyloid plaque deposition in TgCRND8 transgenic mice. Hum Mol Genet. 2006;15(15):2313‐2323. [DOI] [PubMed] [Google Scholar]
- 22. Born HA, Kim J‐Y, Savjani RR, et al. Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J Neurosci. 2014;34(11):3826‐3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Check Hayden E. Patent dispute threatens US Alzheimer's research. Nature. 2011;472(7341):20. [DOI] [PubMed] [Google Scholar]
- 24. Onos K, Uyar A, Keezer KJ, Jackson HM, Preuss C, Acklin CJ, O″Rourke R, Buchanan RA, Cossette TL, Rizzo SJS, Soto I, Carter GW, Howell GR. Enhancing face validity of mouse models of Alzheimer's disease with natural genetic variation. PLoS Genetics. 2019;15(5):e1008155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neuner SM, Heuer SE, Huentelman MJ, O'Connell KMS, Kaczorowski CC. Harnessing Genetic Complexity to Enhance Translatability of Alzheimer's Disease Mouse Models: A Path toward Precision Medicine. Neuron. 2019;101(3):399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pandey RS, Graham L, Uyar A, Preuss C, Howell GR, Carter GW. Genetic perturbations of disease risk genes in mice capture transcriptomic signatures of late‐onset Alzheimer's disease. Molecular Neurodegeneration. 2019;14(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wan YW, et al. Meta‐Analysis of the Alzheimer's Disease Human Brain Transcriptome and Functional Dissection in Mouse Models. Cell Rep. 2020;32(2):107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson ECB, Dammer EB, Duong DM, Yin L, Thambisetty M, Troncoso JC, Lah JJ, Levey AI, Seyfried NT. Deep proteomic network analysis of Alzheimer's disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener. 2018;13:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Veng‐Pedersen P. Noncompartmentally‐based pharmacokinetic modeling. Adv Drug Deliv Rev. 2001;48(2‐3):265‐300. [DOI] [PubMed] [Google Scholar]
- 30. Beal S, Sheiner LB, Boeckmann R, Bauer RJ. NONMEM User's Guides. (1989‐2009). Ellicot City, MD: Icon Development Solutions; 2009. [Google Scholar]
- 31. Friedman AN, Strother M, Quinney SK, et al. Measuring the glomerular filtration rate in obese individuals without overt kidney disease. Nephron Clin Pract. 2010;116(3):c224‐c234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model‐based drug development. CPT Pharmacometrics Syst Pharmacol. 2012;1(9):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Studholme C, Hill DLG, Hawkes DJ. Automated three‐dimensional registration of magnetic resonance and positron emission tomography brain images by multiresolution optimization of voxel similarity measures. Med Phys. 1997;24(1):25‐35. [DOI] [PubMed] [Google Scholar]
- 34. Paxinos G, Franklin K. Paxinos and Franklin's the Mouse Brain in Stereotaxic Coordinates. Academic Press; 2012:. 360. [Google Scholar]
- 35. Dandekar M, Tseng JR, Gambhir SS. Reproducibility of 18F‐FDG microPET studies in mouse tumor xenografts. J Nucl Med. 2007;48(4):602‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mager DE, Wyska E, Jusko WJ. Diversity of mechanism‐based pharmacodynamic models. Drug Metab Dispos. 2003;31(5):510‐518. [DOI] [PubMed] [Google Scholar]
- 37. Galvan V, Gorostiza OF, Banwait S, et al. Reversal of Alzheimer's‐like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci U S A. 2006;103(18):7130‐7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Galvan V, Zhang J, Gorostiza OF, et al. Long‐term prevention of Alzheimer's disease‐like behavioral deficits in PDAPP mice carrying a mutation in Asp664. Behav Brain Res. 2008;191(2):246‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ryan KA, Pimplikar SW. Activation of GSK‐3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J Cell Biol. 2005;171(2):327‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65(11):1509‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pike KE, Savage G, Villemagne VL, et al. Beta‐amyloid imaging and memory in non‐demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130(pt 11):2837‐2844. [DOI] [PubMed] [Google Scholar]
- 42. Wisse LEM, Butala N, Das SR, et al. Suspected non‐AD pathology in mild cognitive impairment. Neurobiol Aging. 2015;36(12):3152‐3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Monsell SE, Kukull WA, Roher AE, et al. Characterizing Apolipoprotein E epsilon4 carriers and noncarriers with the clinical diagnosis of mild to moderate Alzheimer Dementia and minimal beta‐Amyloid peptide plaques. JAMA Neurol. 2015;72(10):1124‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Castellani RJ, Lee H‐G, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol. 2006;111(6):503‐509. [DOI] [PubMed] [Google Scholar]
- 45. Silverman JL, Nithianantharajah J, Der‐Avakian A, Young JW, Sukoff Rizzo SJ. Lost in translation: At the crossroads of face validity and translational utility of behavioral assays in animal models for the development of therapeutics. Neurosci Biobehav Rev. 2020;116:452‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sukoff Rizzo SJ, Masters A, Onos KD, Quinney S, Sasner M, Oblak A, Lamb BT, Territo PR. MODEL‐AD consortium. Improving preclinical to clinical translation in Alzheimer's disease research. Alzheimers Dement (N Y). 2020;6(1):e12038. [DOI] [PMC free article] [PubMed] [Google Scholar]